

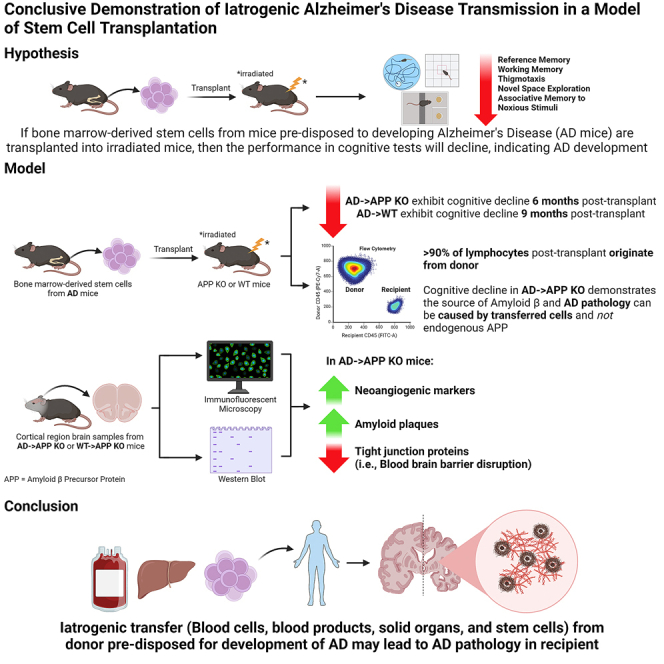
Summary
The risk of iatrogenic disease is often underestimated as a concern in contemporary medical procedures, encompassing tissue and organ transplantation, stem cell therapies, blood transfusions, and the administration of blood-derived products. In this context, despite the prevailing belief that Alzheimer’s disease (AD) manifests primarily in familial and sporadic forms, our investigation reveals an unexpected transplantable variant of AD in a preclinical context, potentially indicating iatrogenic transmission in AD patients. Through adoptive transplantation of donor bone marrow stem cells carrying a mutant human amyloid precursor protein (APP) transgene into either APP-deficient knockout or normal recipient animals, we observed rapid development of AD pathological hallmarks. These pathological features were significantly accelerated and emerged within 6–9 months post transplantation and included compromised blood-brain barrier integrity, heightened cerebral vascular neoangiogenesis, elevated brain-associated β-amyloid levels, and cognitive impairment. Furthermore, our findings underscore the contribution of β-amyloid burden originating outside of the central nervous system to AD pathogenesis within the brain. We conclude that stem cell transplantation from donors harboring a pathogenic mutant allele can effectively transfer central nervous system diseases to healthy recipients, mirroring the pathogenesis observed in the donor. Consequently, our observations advocate for genomic sequencing of donor specimens prior to tissue, organ, or stem cell transplantation therapies, as well as blood transfusions and blood-derived product administration, to mitigate the risk of iatrogenic diseases.
Keywords: Alzheimer’s disease, CNS disorders, iatrogenic diseases, organ transplant, blood transfusion, stem-cell transplant, prion diseases, protein misfolding, mouse behaviour analysis, megakaryocytes derived amyloid beta
Graphical abstract
Highlights
-
•
Identifies a transplantable form of Alzheimer’s disease (AD) in a preclinical model
-
•
Demonstrates rapid development of AD pathology post bone marrow transplantation
-
•
Reveals β-amyloid burden outside CNS contributes to AD
-
•
Suggests genomic sequencing of donors to avoid iatrogenic diseases
In a groundbreaking study, the corresponding author and colleagues uncover a transplantable form of Alzheimer’s disease (AD), challenging the existing understanding of the disease. Through preclinical experiments, they show that transplanting bone marrow cells harboring a mutant amyloid precursor protein can rapidly induce AD pathology in recipients. This significant finding highlights the potential for iatrogenic transmission in AD, suggesting the need for comprehensive genomic screening of donors in tissue, organ, and stem cell transplantations to prevent the inadvertent transfer of central nervous system diseases.
Introduction
The utilization of blood-derived products, blood transfusions, platelet transfusions, organ transplantation, and bone marrow transplantation, as well as emerging personalized cell and tissue-based therapies such as adoptive transfer of induced pluripotent stem (iPS) cells, represents critical clinical interventions. Nonetheless, there exist looming concerns regarding their safety. Recently, it has been proposed that Alzheimer’s disease (AD) is a prion-like disease where pathology can be transmitted in a prion-like manner; for example, the transfer of a misfolded protein is enough to cause disease in the recipient. The idea that misfolded β-amyloid (Aβ) peptides are proteinaceous infectious particles has previously been reported where brain extracts of AD patients were able to induce cerebral amyloidosis when transferred into mouse and primate models (Baker et al., 1993; Eisele et al., 2010; Meyer-Luehmann et al., 2006; Stohr et al., 2012). Reports of the transfer of AD pathology in humans have been associated with the use of contaminated human cadaveric pituitary-derived growth hormone (c-hGH). Patients who had received the contaminated c-hGH died of iatrogenic Creutzfeldt-Jakob disease (CJD), but, upon investigation, it was reported that these patients also had substantial immunoreactive Aβ and increased numbers of microvessels in the brain (Jaunmuktane et al., 2015a, 2015b; b), which is a pathology associated with AD, not CJD. Subsequently, some batches of c-hGH, to which CJD patients with Aβ pathology were exposed, were found to have substantial levels of Aβ40, Aβ42, and tau proteins (Purro et al., 2018). However, in the case of these Aβ seeding studies, the assumption is that the endogenous Aβ of the recipient contributes to the disease in a misfolded-cascading process. Regardless, the ability of transmission of disease raises concern for many of the cell-based therapies, including bone marrow transplantation or treatment with cord blood or human embryonic stem cells. Currently, many countries approve the use of bone marrow and stem cells as cancer therapies (EuroStemCell, 2020a; b; National_Stem_Cell_Foundation_of_Australia and StemCells_Australia, 2015; Ontario_Institute_for_Regenerative_Medicine, 2020; US_Food_&_Drug_Administration, 2020), and there is much research into their use for central nervous system (CNS) disorders, including AD (Zuroff et al., 2017), Parkinson’s disease (Daadi et al., 2012; Kikuchi et al., 2017; Kim et al., 2002), multiple sclerosis (Freedman et al., 2010; Karussis et al., 2010), and spinal cord injury (Jin et al., 2019; Sahni and Kessler, 2010).
The traditional understanding of AD centers on the accumulation of Aβ in the brain. Previously, it was thought that this Aβ primarily originated from the brain itself (Goedert, 1987; Mita et al., 1989). However, recent research suggests that Aβ species in the bloodstream can cross the blood-brain barrier (BBB), significantly contributing to AD pathology (Bu et al., 2018; Chen et al., 2017; Deane et al., 2003; Murphy and LeVine, 2010; Zlokovic et al., 1993, 1994). These findings, predominantly based on models with an intact amyloid precursor protein (APP) gene in the recipients, do not conclusively determine whether Aβ generated within the CNS parallels the process observed in prion diseases, nor do they fully explore alternative pathways, such as those involving inflammation and inflammatory mediators, that could influence the accumulation of endogenous Aβ in the CNS.
Our previous data demonstrate that Aβ provides a pro-angiogenic signal for brain vasculature and that Aβ may subsequently enter the brain by diffusion after the BBB is disrupted when tight junctions disappear during cell division (Biron et al., 2011, 2013; Dickstein et al., 2006; Jefferies et al., 2013; Singh et al., 2017; Ujiie et al., 2003). The primary potential source of peripheral APP contributing to this mechanism are platelets that originate from megakaryocytes formed from hematopoietic stem cells (HSCs) (Evin et al., 2003; Li et al., 1995). Upon platelet activation, β-secretase enzyme activity increases in their membranes, resulting in the production of soluble Aβ species (Chen et al., 1995). Although platelets originating from HSCs are the major source of peripheral Aβ, other sources such as skin fibroblasts, skeletal muscles, and cerebrovascular smooth muscle cells can also produce Aβ (Deane and Zlokovic, 2007).
To date, none of the preceding studies have endeavored to fulfill Koch’s postulates for establishing the microbiological etiology of infection and disease. In this study, we address whether the pathological hallmarks of AD can be transferred by repopulation of the HSC compartments of wild-type or an APP-deficient knockout mouse model with donor bone marrow cells (BMCs) harboring the Swedish mutant human APP gene (APPKM670/671NL) linked to early-onset familial AD.
Other than prion diseases or transmissible spongiform encephalopathies (TSEs), this appears to be the first definitive report that cellular transplantation can adoptively transfer a disease of the CNS, and this occurrence is independent of endogenous Aβ. Furthermore, to mimic the clinical setting, we also examined whether AD pathology can be established in APP wild-type (WT) animals after the adoptive transfer of BMCs that contain the mutated, disease-causing APP gene and discovered our observations have a direct correlation to clinical scenarios where iatrogenic disease may be a risk.
This study provides new insights into the role of peripheral APP in the development of AD, challenging our existing understanding. Moreover, it underscores the sobering implications for the iatrogenic transfer of other diseases.
Results
Successful bone marrow transfer from AD mice and WT littermates into APP-knockout recipients and from AD mice into WT recipients
To establish that transplantation of bone marrow and HSC can cause AD pathology, we (1) transferred bone marrow from AD and WT mice, both of which were positive for the lymphocyte common antigen cluster of differentiation (CD) 45.1, into APP-knockout (APP KO) mice that were positive for the lymphocyte common antigen CD45.2.; and (2) transferred BMCs from CD45.2-positive AD donors to CD45.1-positive WT mice.
After long-term repopulation (60 days), we assessed the presence of the CD45.2+ donor BMCs in APP KO mice. We found that recipient APP KO mice exhibited significant CD45.1+ donor lymphocytes. Similarly, recipient WT mice exhibited significant CD45.2 + donor lymphocytes. The representative fluorescence-activated cell sorting (FACS) graph shows (Figure 1) that the majority of the lymphocyte population was derived from the donor cells (91.6%), and only a small residual population (6.8%) of CD45.2 cells survived irradiation. Negative control for FACS analysis of successful reconstitution of donor BMC population in the recipient is illustrated in Figure S1. This was consistent across the different groups. Figure S2 shows the summary statistics of the percentage of lymphocytes from donors and recipients. The percentage of donor lymphocytes is higher than the threshold set for a successful transplant (90%), indicating a successful reconstitution of AD or WT donor BMCs in (1) irradiated APP KO recipients or Tg2576 donor BMCs and (2) irradiated WT recipients (p < 0.0001). For both figures, freshly isolated peripheral blood mononuclear cells (PBMCs) from mice were stained with isotype-specific CD45.1 and CD45.2 antibodies and analyzed by flow cytometry. An unpaired t test was used to calculate significance.
Figure 1.

Successful reconstitution of the donor BMC population in the recipient
(A) Representative overlaid scatterplot of splenocytes from a CD45.1+ donor (blue, AD or WT) and a CD45.2+ recipient mouse (red, APP KO) before bone marrow reconstitution.
(B) Representative scatterplot of PBMCs from a CD45.2+ APP KO mouse, reconstituted with CD45.1+ bone marrow from a donor mouse (AD or WT), 2 months post transplant. The recipient APP KO mouse is 91.5% CD45.1 positive, indicating successful reconstitution.
Behavior assessment of mice
AD→APP KO was cognitively impaired 6 months after transplantation, as compared to B6/SJL.BM→APP KO. AD→APP KO mice performed poorly in the open-field test, spending significantly more time exploring (Figure 2A, p = 0.0002) and making more entries (Figure 2C, p < 0.0001) in the central region of the field as compared to the WT→APP KO mice. No significant difference was seen in the distance traveled (Figure 2B) between the two groups. Figure 2D shows the track plots of animals representative of the groups. Cognitively aware mice show a high percentage of alternation (spatial awareness), as seen in WT→APP KO mice. AD→APP KO had low spatial awareness when tested on the Y maze, with a significantly lower percentage of alternation (POA) than WT→APP KO mice (Figure 2E, p < 0.0001) The POA was not confounded by total distance traveled or the total number of arm entries (Figure S3.). A significantly lower percentage of freezing in the contextual fear conditioning test indicated that the AD→APP KO had poorer associative memory than WT→APP KO mice (Figure 2F). Figures 2G and 2H show the latency time and the total number of memory errors made in the radial arm water maze (RAWM). AD→APP KO mice had diminished working and reference memories, showing no learning over the 5-day test, as compared to WT→APP KO mice. This study had data pooled from two different trials where n = 14 for WT mice and n = 18 for AD mice unless otherwise stated. No significant difference was observed in behavior with respect to sex. An unpaired t test was used to calculate significance.
Figure 2.

Bone marrow transfer from AD mice causes cognitive deficits in APP KO recipient mice
APP KO recipient mice with a successful reconstitution of donor BMCs were assessed for their cognitive status, using tests designed for the analysis of different aspects of memory. The data were pooled from two different trials unless stated otherwise and are represented as the mean ± standard deviation.
(A–D) Open-field test (OFT). (A) APP KO mice that received BMCs from WT mice (WT→APP KO) spent significantly less time in the center of the field compared to APP KO mice that received BMCs from AD mice (AD→APP KO). (B) There was no significant difference in the distance traveled in the open field between WT→APP KO mice and AD→APP KO mice. (C) AD→APP KO mice have a significantly higher number of entries in the central squares of the field than WT→APP KO mice. (D) Representative track plots from the OFT. WT→APP KO mice spend less time exploring the open center of the test arena while AD→APP KO mice explore the entire field indiscriminately. Each plot in the figure represents the track from a different mouse. (A–C) An unpaired t test was used to calculate statistical significance (p ≤ 0.05). Data from two studies were pooled; AD→APP KO, n = 8 males, n = 10 females; and WT→APP KO, n = 7 males, n = 7 females.
(E) Spontaneous alternation (Y maze) test. A significantly high POA was observed in the WT→APP KO mice versus the AD→APP KO mice. An unpaired t test was used to calculate statistical significance (p ≤ 0.05). AD→APP KO, n = 8 males, n = 10 females; and WT→APP KO, n = 7 males, n = 7 females.
(F) Contextual fear conditioning test (FC). WT→APP KO mice exhibited high freezing percentages compared to AD→APP KO mice. Due to technical issues, data for this test were collected only from one trial. An unpaired t test was used to calculate statistical significance (p ≤ 0.05). Data for this test are only from the first trial due to issues with equipment at the time of the experiment. AD→APP KO, n = 4 males, n = 4 females; and WT→APP KO, n = 3 males, n = 3 females.
(G and H) RAWM. Both (G) latency time and (H) the total number of errors were measured. WT→APP KO mice showed a significant decrease in the latency time and decreased number of errors when comparing results from test day 1 and test day 5. No significant differences between test day 1 and test day 5 were seen in the AD→APP KO. A paired t test was used to calculate significance (p ≤ 0.05). AD→APP KO, n = 8 males, n = 10 females; and B6/SJL.BM→APP KO, n = 7 males, n = 7 females.
A second study was conducted to see if a bone marrow transplant from AD mice into irradiated WT mice could establish AD pathology. A significantly high POA was observed in WT mice as compared to AD animals in the Y maze, (Figure 3A, p = 0.0002). AD→WT mice were shown to have no significant difference in POA as compared to AD mice; however, they showed a significantly lower POA as compared to the WT mice (Figure 3A, p = 0.0002). Data are representative of the following: AD→WT, n = 8 (five males, three females); and WT, n = 11 (six males, five females); and AD, n = 13 (six males, seven females). An ordinary one-way ANOVA with Tukey’s multiple comparisons test was used to calculate statistical significance. In the RAWM (Figure 3), WT mice showed a significantly lower latency time (Figure 3B, p = 0.0351) and number of errors (Figure 3C, p = 0.0114) on day 5 compared to day 1. Although AD→WT animals showed a significantly lower latency time (Figure 3B, p = 0.0097), like the AD mice, they showed no significant difference in the number of errors between test day 1 and test day 5 (Figure 3C). AD showed no difference in the latency time (Figure 3B) between day 1 and day 5. The data in Figure 3B and 3C are expressed as the mean of the latency time or the total number of errors in a single trial per day by individual animals in each group. Since the difference was being noted on day 1 versus day 5 within each group a paired t test was used to calculate significance (p ≤ 0.05).
Figure 3.

Bone marrow transfer from AD mice causes cognitive deficits in certain aspects of memory in WT recipient mice
(A) Y maze. A significantly high POA was observed in WT mice as compared to AD animals. AD→WT mice were tested and shown to have no significant difference in percentage alternation as compared to AD mice; however, they showed a significantly lower POA as compared to the WT mice. Data is shown as the mean ± SD and is representative of AD→WT, n = 8 (five males, three females) and WT; n = 11 (six males, five females); and AD, n = 13 (6 males, 7 females). An ordinary one-way ANOVA with Tukey’s multiple comparisons test was used to calculate statistical significance (p ≤ 0.05).
(B and C) RAWM. Both: (B) latency time and (C) the total number of errors were measured. B6/SJL control mice showed a significantly lower latency time and number of errors on day 5 compared to day 1. Although AD→WT animals showed a significantly lower latency time, like the AD mice, they showed no significant difference in the number of errors between test day 1 and test day 5. AD mice showed no difference in the latency time between day 1 and day 5. The data in (B) and (C) are expressed as the mean of the latency time or the total number of errors in a single trial per day by individual animals in each group. Since the difference was being noted on day 1 versus day 5 within each group, a paired t test was used to calculate significance (p ≤ 0.05).
AD pathology was seen through molecular and histological analysis of the mice receiving AD bone marrow transplants
Figures 4A and 4B show representative images of immunofluorescence of the pre-frontal cortex for each antibody and both animal groups, AD→APP KO and WT→APP KO mice. Human Aβ can be seen in the AD→APP KO mice but not in the WT→APP KO mice. A significant increase in the neo-angiogenic vessels, shown by endoglin (CD105), can be seen in the brains of AD→APP KO mice but not in the brains of the WT→APP KO mice. A qualitative confocal image shows that CD105 and Aβ colocalize with each other. The tight junction protein (TJP), occludin, is reduced in AD→APP KO brains compared to WT→APP KO, which has a continuous pattern of occludin expression resembling WT mice (Figure 4B). Western blot analysis shows that there is a presence of the Aβ protein in the brain homogenates of AD→APP KO mice but no detectable Aβ expression in the brains of WT→APP KO mice (Figure 4C).
Figure 4.

AD pathology is seen in the brains of APP KO mice reconstituted with AD bone marrow
(A) Micrographs show the immunostaining and confocal imaging of the cortical region of WT→APP KO and AD→APP KO mouse brains. The expression levels of CD105 (green), DAPI (cyan), Aβ (red), and CD31 (blue) are shown where Aβ and CD105 are significantly higher in AD→APP KO mice compared to WT→APP KO mice. Data are shown as mean ± SD of percentage area of expression. Unpaired Student’s t test was used to calculate significance (p ≤ 0.05).
(B) Qualitative IHC shows the expression of CD105 (green), Aβ (red), and occludin (blue). The occludin expression is inverse to that of CD105 and Aβ, and Aβ is seen co-localizing with CD105.
(C) Qualitative western blotting analysis indicates the presence of mutant human APP protein in AD→APP KO animals and an absence of it in the WT→APP KO. VEGFa is expressed higher in AD→APP KO (n = 5) compared to WT→APP KO (n = 4). Expression levels of TJP, ZO1, are higher in WT→APP KO (n = 7) compared to AD→APP KO (n = 8). Blots shown here are from pooled samples from the animals in the groups to be representative of the group. Histograms show expression levels of proteins in individual animals. Unpaired Student’s t test was used to calculate significance (p ≤ 0.05).
To examine whether the presence of this Aβ had any functional implications on the vasculature, we looked at the expression of vascular endothelial growth factor a (VEGFa), a factor that is crucial for angiogenesis initiation, and, to assess the effect of Aβ transferred via HSC transplant on the BBB integrity of the recipient, the TJP, zona occludens 1 (ZO1) expression levels were analyzed (Figure 4C). The western blot analysis showed higher expression of the VEGFa and lower expression of ZO1 in the brain homogenates of the AD→APP KO bone marrow compared to the WT→APP KO mice (Figure 4C). This implies not only that there is a transfer Aβ to the recipient mouse brain but also that this Aβ can induce AD pathology in mice that have no endogenous APP expression in their brains or indeed any other organ.
To assess the effect of a bone marrow transplant from AD mice into WT mice as a direct translation for a clinical setting, we looked at the establishment of amyloid plaques generated by the mutant human APP gene. Figure 5 shows micrographs that are representative of the cortical region of mouse brains. Congo red staining showed that Tg2576 mice had significantly higher plaques than the WT mice (p = 0.0002). The Congo red staining also showed the establishment of amyloid plaques in AD→WT mice, which were significantly higher as compared to the WT animals (p = 0.0105) and similar to the levels seen in AD mice. An ordinary one-way ANOVA was used to calculate significance (p ≤ 0.05).
Figure 5.

Presence of amyloid plaques in AD→WT mice suggests establishment of AD pathology in WT mice through a bone marrow transplant
(A) Micrographs are representative of the cortical region of mouse brains.
(B) Congo red staining indicates the establishment of amyloid plaques in AD→WT mice, which is seen to be significantly higher as compared to WT and similar to the levels seen in AD animals. Each data point in the histograms corresponds to data from individual mice in each group, where n = 6 for WT, n = 8 for AD, and n = 8 for AD→WT. Data is represented as mean ± SD. Ordinary one-way ANOVA was used to calculate significance (p ≤ 0.05).
Discussion
This study challenges the prevailing understanding that AD is exclusively familial or sporadic. Using a preclinical model, we demonstrate a transplantable form of AD, suggesting potential iatrogenic transmission in AD patients. Here we specifically address the potential of bone marrow stem cell transplants in the transmission of AD in a familial model of this deadly disease. We found that transferring bone marrow from AD mice into APP KO mice resulted in cognitive impairment as well as Aβ extracellular deposits and BBB dysfunction. In particular, we demonstrated that AD→APP KO chimeric mice accumulate the human Aβ in the brain originating exclusively from the donor show increased expression of the neoangiogenic marker CD105, and show a decreased expression of the TJPs occludin and ZO1. Our data demonstrate that transplantation of bone marrow, containing HSCs, from a donor mouse that overproduces a mutant human APP, can transfer AD pathology in a recipient animal that does not synthesize endogenous APP. Furthermore, the rate of disease formation in the transplantable model is much more rapid than in the AD mice as it is established in the AD bone marrow recipients in 6 months compared to 12 months in the AD transgenic mice (Biron et al., 2011, 2013; Dickstein et al., 2006; Hsiao et al., 1996; Jefferies et al., 2013; Singh et al., 2021; Ujiie et al., 2003).
There is indeed evidence that transplant recipients, including those of solid organs and HSCs, have a higher incidence of various neurological diseases. For solid organ transplant recipients, CNS complications are relatively common and include both focal and diffused neurologic deficits. These complications can be due to a range of causes, including infections, drug toxicity, cerebrovascular events, metabolic disorders, and cancer. Notably, the incidence of neurological complications in kidney transplant recipients is reported to be around 10%–21%, which is higher than in the general population. Factors contributing to these complications include the need for multiple drugs, decreased cellular immunity, accelerated atherosclerotic disease, and frequent metabolic abnormalities (Meena et al., 2020; Potluri et al., 2014; van den Bogaart et al., 2022; Wright and Fishman, 2014).
In the specific context of hematopoietic stem cell transplantation (HSCT) for pediatric acute lymphoblastic leukemia, neurological complications, both acute and long term, are also common and contribute to significant morbidity and mortality. The incidence of neurotoxicity in children following HSCT ranges from 11% to 59%. A post mortem study of 180 HSCT recipients, including both adults and children, found that 90% had evidence of CNS abnormality, and, in 17% of cases, this was the cause of death. Furthermore, outcomes of HSCT are poorer for patients who develop acute neurotoxicity. The majority of studies on the neurological effects of HSCT in children have focused on acute neurotoxicity, but, as more children undergoing HSCT for acute lymphoblastic leukemia become long-term survivors, understanding the long-term neurological consequences of this treatment modality is increasingly important. Radiotherapy and chemotherapy, as parts of HSCT treatment, can add to neurological injury and have profound effects on brain maturation and cognitive function (Gabriel et al., 2021).
The APP KO mice do not produce endogenous Aβ in any tissue or organ. If current theories relating to the causation of AD were correct, for example, that brain-derived Aβ causes AD pathology, then the reconstitution of the APP KO mice with AD HSCs should not have induced AD pathology. The findings in this study establish that HSC-derived Aβ can initiate vascular pathology leading to BBB disruption and eventual AD pathology and the findings in this study establish that peripherally derived Aβ can initiate vascular pathology and BBB disruption.
The Swedish mutation version of the human amyloid precursor transgene expression is driven by the prion promoter that drives expression in a variety of tissues, including blood cells, and we assume this is the source of the APP originating from the donor HPC cell (PRNP Gene - Prion Protein and Gene Cards, 2023; Asante et al., 2002a, 2002b; Levine et al., 2009). A significant amount of the peripheral Aβ is found in circulating platelets (Chen et al., 1995; Matsubara et al., 2002) that originate from megakaryocytes. In the AD mice (Tg2576), bone marrow and HSCs carrying the human mutant APP are known to overproduce Aβ in megakaryocytes (Starke et al., 2005). Aβ generated from activated platelets is known to exist in a soluble form (Chen et al., 1995; Matsubara et al., 2002) and this property may enable them to be transported from another location in the body into the CNS through the cerebral vessels. Another way for the soluble Aβ to enter the CNS is via the platelets circulating in the cerebral vessels (Inyushin et al., 2017; Kucheryavykh et al., 2017). Tight junctions, which are present in the endothelial layer of the cerebral vessels, have high amounts of collagen, a platelet activator, in their vicinity, especially the basement membrane of cerebral vessels (Xu et al., 2019). This collagen may lead to the activation of the platelets and release of the Aβ into the cerebral vessels. The released Aβ activates cell division and vasculogenesis leading to the breakdown in BBB integrity, allowing Aβ to freely cross the BBB and enter the CNS by diffusion where it deposits in amyloid plaques (Biron et al., 2011, 2013; Dickstein et al., 2006; Jefferies et al., 2013; Singh et al., 2017; Ujiie et al., 2003). The receptor for advanced glycation products (RAGE) is a multi-ligand receptor that also regulates the entry of peripheral Aβ to the brain. It binds soluble Aβ in the nanomolar range, and its expression is upregulated by the presence of Aβ and is also implicated in BBB breakdown and AD pathogenesis (Deane et al., 2003; Yan et al., 1996). Another possibility of transfer of Aβ into the CNS could be as a result of the physiological age-related disruption of the BBB. This breakdown could permit the transfer of the soluble Aβ into the brain (Montagne et al., 2015). Once AD pathology initiates, it can trigger further vasculogenesis and breakdown of the BBB. However, several other types of blood cells also express APP in the Tg2576 mouse; therefore, future studies are needed in order to address this.
One of the potential outcomes of this study is to spur the field to move away from the conventional central dogma of AD pathology, which states that the accumulation of brain-derived Aβ, specifically produced by neurons, is the cause of the disease. This study demonstrates the contribution Aβ, generated outside the brain, in establishing the disease. This may provide an opportunity for the development of new biomarkers for AD. The production of AD pathology in AD→APP KO mice also supports the concept that AD can be transferred from individual to individual, resulting in the discovery of a transplantable form of AD that is distinct from the sporadic or familial forms of the disease. This suggests the importance of biomarker screening and sequencing the genomes of candidate cell, tissue, and organ donors for deleterious disease-associated alleles to eliminate the potential for the transfer of disease before utilizing them as donors for any transplantation therapies including tissue and organ transplantation as well as bone marrow transplants, blood transfusion, or stem cell-based therapies.
However, it is important to acknowledge the limitations of this work. While the results from the study using the Tg2576 AD model and APP KO mice provide valuable insights, caution must be exercised when translating these findings to human stem cell transplant recipients. The Tg2576 model, with mutant APP expression under the control of the hamster prion protein promoter, generates a high level of pathology not typically observed in human familial AD patients. The use of a heterologous promoter and recipients completely deficient in APP, a condition rare in humans, adds further methodological layers that may not directly reflect conditions in humans, though it does establish, for the first time, that exclusively donor derived mutant APP is necessary and sufficient to cause disease in recipients.
Given these differences, the study’s findings may not directly correspond to the clinical experience of stem cell transplantation and its potential neurological implications. The adoption of a murine model may not fully capture the complexity of AD in humans, and variations in the immune response and genetic factors between species should be acknowledged. Furthermore, the observed pathological features, while resembling AD, may not precisely replicate the multifaceted nature of the disease in humans. Notably, the form of AD studied here is familial, which may limit the transferability of results to humans, as familial forms are rarer than sporadic ones. However, the additional observation in this study demonstrating the transfer of the AD phenotype from Tg2576 AD model bone marrow donors into healthy WT recipients lessens the chance that our data will not find its human sequelae.
It is also important to recognize the broader context of transplant-related neurological complications. As shown by the higher incidence of neurological diseases in transplant recipients, both in solid organ and hematopoietic stem cell transplant settings, there is a clear indication that transplant procedures can lead to significant CNS complications. These include a wide array of issues stemming from infections, drug toxicities, metabolic abnormalities, vascular and immunologic events, and the primary disease itself. Thus, while the specific findings of the study might not directly translate to human cases, they do contribute to a growing body of knowledge about the potential neurological risks associated with various types of transplantation. This information is crucial for the development of strategies to mitigate these risks and improve patient outcomes in both pediatric and adult populations.
In conclusion, the underestimation of iatrogenic disease risk in contemporary medical practices, including tissue and organ transplantation, stem cell therapies, blood transfusions, and blood-derived product administration, has begun to be addressed here. Contrary to prevailing beliefs regarding AD occurring solely in familial or sporadic forms, our study reveals an unexpected transplantable form of AD in a preclinical model, suggesting potential iatrogenic transmission in AD patients. Adoptive transplantation of donor BMCs harboring a mutant human APP transgene into both APP-deficient and healthy WT recipient animals resulted in the rapid development of AD pathological hallmarks. These included compromised BBB integrity, increased cerebral vascular neoangiogenesis, elevated brain-associated Aβ levels, and cognitive impairment that is accelerated and occurs within 6–9 months post transplantation. Moreover, our findings suggest that Aβ accumulation originating externally to the CNS contributes to AD pathology.
While the study provides valuable insights, further research involving human subjects is essential to validate the applicability of these findings to clinical settings. However, given the findings of this study, the proposed recommendation for genomic screening of donor specimens before transplantation therapies or the transfer of blood-derived products necessitates careful deliberation.
Experimental procedures
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Wilfred A. Jefferies.
Materials availability
This study did not generate new unique reagents.
Data and code availability
Data and code availability can be provided upon request.
Mice
Key animal nomenclature:
AD mice or Tg2576: transgenic mice, heterozygous for the mutant human APP gene.
WT: WT littermates of Tg2576 mice, homozygous for the normal endogenous APP gene.
APP KO: mice homozygous for the APP gene being knocked out.
AD→APP KO: irradiated APP KO mice that have been reconstituted with BMCs from AD mice.
WT→APP KO: irradiated APP KO mice that have been reconstituted with BMCs from WT mice.
AD→WT: irradiated WT mice that have been reconstituted with BMCs from AD mice.
The Tg2576 AD model mouse expresses the K670N/M671L Swedish mutation of the APP (Hsiao et al., 1995, 1996) under the control of the hamster prion protein promoter. Tg2576 mice are always heterozygous for the transgene as the homozygous offspring is not viable. These are on a B6.SJL background where B6 represents the C57BL/6 mice and the SJL is Swiss Jim Lambert mice, and the colony at the University of British Columbia is occasionally refreshed with B6.SJL mice purchased from Taconic Biosciences (B6.SJL-Ptprca/BoyAiTac; catalog #4007-F). Homozygous APP KO (APP KO) mice (B6.129S7-Apptm1Dbo/J; catalog #004133, Jackson Labs, USA) on a C57BL/6 background (Zheng et al., 1995) were fed standard lab chow and water ad libitum and kept under a 12-h light/dark cycle. All protocols and procedures involving the care and use of animals in these studies were reviewed and approved by the UBC Animal Care Committee under the guidelines of the Canadian Council of Animal Care.
Flow cytometric analysis of lymphocytes
CD45 is a type 1 transmembrane protein that is present on all differentiated hematopoietic cells and is a useful marker for determining bone marrow transplant efficiency (Omilusik et al., 2011). C57Bl/6 mice are genetically CD45.2, whereas SJL mice are CD45.1, so all AD and WT littermates were tested for CD45 alleles and only those that were CD45.1 were used as donors; all APP KO recipients were CD45.2 (on C57Bl/6 background). Briefly, 50–100 μL of blood was collected into EDTA-coated tubes and lymphocytes were isolated after red blood cell (RBC) lysis. Isolated lymphocytes were then incubated with antibodies against CD45.2 (FITC anti-mouse CD45.2 antibody; BioLegend; 109805) and CD45.1(PE/Cy7 anti-mouse CD45.1 Antibody (Biolegend; 110729) in FACS buffer (phosphate-buffered saline [PBS], 0.5%–1% BSA or 5%–10% FBS). The samples were analyzed for CD45 type using a BD LSRII (BD Biosciences). Lymphocytes were gated based on size and shape, and gating was consistent among all samples. The representative graph was normalized to mode, which allows the visualization of the relative percentage of a cell population expressing the marker fluorescence and compensates for the visual difference in cell count. FlowJo software (Treestar) was used to analyze flow cytometry data.
Bone marrow transfer: Experimental setup
Two transplant studies were conducted:
-
(1)
AD mice and their WT littermates were used as donors to transplant bone marrow into APP KO mice. Twelve-month-old AD and WT of both sexes were used as donors.
-
(2)
The second study consisted of the bone marrow transfer of AD mice into WT animals. Twelve-month-old AD mice of both sexes were used as donors.
For the collection of BMCs from donor mice, the proximal ends of the tibia and femur were cut off and placed in a 0.5-mL microfuge tube with the bottom cut off, and this setup was placed inside a 2-mL centrifuge tube. The tubes were centrifuged at 2000 rpm for 5 s. The bone marrow pellet was resuspended in culture medium and treated with RBC lysis buffer (10× buffer: NH4Cl [ammonium chloride] 8.02 g, NaHCO3 [sodium bicarbonate)] 0.84 g, EDTA [disodium] 0.37 g, quantum satis to 100 mL with Millipore water). The working solution of the lysis buffer was used at 1×. The cells were then centrifuged and resuspended in PBS and cells were counted with the Bio-Rad TC20 automated cell counter.
For the first study, 6- to 8-week-old APP KO recipient mice of both sexes were X-irradiated at 900 Rads. Then 2 × 106 donor BMCs were injected via the tail vein into each recipient mouse on the same day. One donor mouse per sex and genotype was used per two sex-matched recipient mice. APP KO mice (n = 5 female and n = 5 male) received BMCs from n = 3 AD mice of each sex. APP KO mice (n = 4 male and n = 4 female) received BMCs from n = 2 WT mice of each sex. This transplant experiment was repeated with the same conditions and number of mice of both sexes for both recipient and donors. It was observed that two mice from both the groups (i.e., those that received AD and WT BMCs) in one of the trials either died randomly or had to be euthanized after reaching the humane endpoint. This occurred immediately after the transplant, which could indicate procedural issues in those animals. No relation to sex was observed. N is indicated in the figure legend of each experiment.
Two months post injections, the mice were assessed for a successful bone marrow reconstitution by FACS analysis of lymphocyte populations. The donors used were positive for CD45.1, whereas the recipients were CD45.2 positive. After reconstitution, the recipients were checked to see the percentage of lymphocytes that were CD45.1 positive. A reconstitution was considered successful if 90% or more of the cells in the recipient were CD45.1 positive.
For the second study, 6- to 8-week-old WT mice underwent a similar irradiation protocol as stated in the first study. n = 3 female and n = 7 male WT mice received BMCs from sex-matched AD. Similar to the first study, the successful bone marrow reconstitution was established by FACS analysis of lymphocyte populations 2 months post transplant. The donor mice were either CD45.2 positive or double-positive for CD45.1 and CD45.2.
Behavioral testing
Open-field test
The mice were allowed to explore a plexiglass chamber with dark-colored walls and a light source focused in the center one at a time for 5 min. The path traveled and the time spent in the center or periphery of the field were tracked and recorded (Singh et al., 2021). The setup for the test is described in the supplemental experimental procedures.
Spontaneous alternation (Y maze)
The test for novelty exploration using spatial and working memory was conducted where the spatial acquisition phase comprised a 1-day trial with the mice tracked while moving freely through the three arms of the Y maze during an 8-min session. The movements were tracked by a computer tracking system (ANY-maze, Stoelting). The performance was gauged by the POAs, which was calculated as the total number of alternations × 100/(total number of arm entries − 2) (Singh et al., 2021). The minimum number of arm entries needed for a mouse to be included in the analysis was five entries (International Mouse Phenotyping Consortium). The setup for the test is described in the supplemental experimental procedures.
Contextual fear conditioning
Contextual fear conditioning was used to determine associative working memory. One at a time, the animals were kept in a chamber with a steel-grid floor with a shock generator. On day 1, they were allowed to explore freely for 5 min, during which, at the 180th second, they received a foot shock of 0.50–0.80 mA for 3 s. On day 2, they were placed in the chamber for 4 min, with no noxious stimuli. The mice were monitored for movement and freezing behavior was recorded using computer software (Limelight, ActiMetrics, Wilmette, IL, USA) (Singh et al., 2021). The setup for the test is described in the supplemental experimental procedures.
RAWM
The RAWM consists of eight swim paths (arms) extending out of an open central area, with an escape platform located at the end of any of four alternate arms called the goal arms (Penley et al., 2013). The mice were given 60 s to locate one of the four escape platforms. With each trial, the platform that was used to escape was removed. These trials were conducted over 5 days. The latency to reach the platform for each trial and the arm entries were recorded manually. Performance of memory and learning was gauged each day based on the latency time and the total number of errors (Singh et al., 2021). The setup for the test is described in the supplemental experimental procedures.
Tissue preparation
After the behavior studies, the animals were terminally anesthetized with ketamine/xylazine (150 mg/kg; 10 mg/kg) and perfused with PBS for 5 min at a 5 mL/min flow rate. Brains were removed, and one hemisphere was fixed with 4% paraformaldehyde (PFA) for histology and stored at 4°C and the other hemisphere was flash-frozen on dry ice for biochemical studies and stored at −80°C until they were ready to be analyzed. Flash-frozen mouse brain hemispheres were homogenized mechanically using a Dounce homogenizer (Wheaton, 7-mL Tissue Grinder, Dounce) in 1× PBS solution containing 1× Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific; 78440) to prevent protein degradation, and then centrifuged at 14,000 rpm for 10 min to separate the tissue into a soluble fraction, containing cytosolic proteins, and a pellet. The pellet was resuspended in 2% sodium dodecyl sulfate (SDS) solution in distilled H2O and then centrifuged at maximum speed for 30 min. The supernatant from this treatment contained membrane-bound proteins.
Immunoblotting analysis
For western blot analysis, the fractional homogenate samples (cytosolic and membrane bound) were prepared in the sample buffer (100 mM Tris-Cl [pH 6.8], 4% [w/v] SDS, 0.2% [w/v] bromophenol blue, 20% [v/v] glycerol, and 200 mM dithiothreitol [DTT]). Samples were then electrophoresed on 10% SDS-PAGE gels and the proteins were transferred to a nitrocellulose membrane. Immunoblotting was performed using primary antibodies against CD105/endoglin (0.25 μg/mL, R&D systems; AF1097), Aβ (1:1,000, 6E−10; BioLegend 39320), anti-ZO1 (2 μg/mL, Thermo Fisher Scientific; 61-7300), anti-GAPDH antibody (1:100, Abcam; ab181602), anti-actin (1:1000, Santa Cruz; sc1615 and anti-VEGFa (1 μg/mL, Abcam; ab46154). After the overnight incubating with primary antibodies at 4°C, the membranes were washed three times with 1× PBS solution for 15 min each and then incubated with the secondary antibodies (1:10,000) for 1 h at room temperature followed by washing three times with 1× PBS. The signal intensities on the membrane were imaged using the Odyssey infrared image system (LICOR), and relative levels of immunoreactivity were analyzed by the Image Studio Lite Software. The blots shown are representative of n = 6 per group.
Immunofluorescence and confocal imaging
The PFA fixed hemispheres were processed for immunofluorescent microscopy for various proteins of interest: CD105 (R&D Systems; 15 μg/mL, AF1097); Aβ, 1–16 (1 μg/mL, BioLegend; 803004); TJP, occludin (1:200, Abcam; ab31721); and CD-31 (1:50, Abcam; ab28364) as described in Singh et al., 2021.
The cortical region of the mouse brain was analyzed for markers of interest involved in AD pathology. The image acquisition was done using the Olympus FV-10i confocal microscope with a high-resolution Olympus 60×/1.4 oil-immersion objective lens (Olympus, Tokyo, Japan). For 3D image dataset acquisition, the excitation beam was first focused at the maximum signal intensity focal position within the brain tissue sample and the appropriate exposure times were selected to avoid pixel saturation. A series of 2D images (z stack) were taken at a step size of 1 μm. The beginning and end of the 3D stack were set based on the signal level degradation. The series of images taken were saved in the Olympus software. The Volocity software (PerkinElmer) was then used to process the series of images that were taken and generate a 3D reconstruction of the tissue.
Congo red staining
In addition, immunofluorescence analysis was carried out for the AD, WT and AD → WT mice, where the PFA-fixed mouse hemibrains were embedded in paraffin, cut into 4-μm sections, and collected onto charged slides. For the Congo red staining, the brain sections were immersed in 2.5% NaCl in 80% ethanol, followed by incubation in 0.2% Congo red for 20 min, and slides were mounted with a Fluoromount and were scanned in the fluorescent module using a Tetramethylrhodamine (TRITC) filter at 20× in an Olympus VS-120 slide scanner (Evident, Tokyo, Japan). One section of 4-μm thickness was stained with Congo red for each animal. Image acquisition of the entire area of the 4-μm-thick hemibrain section was done and measurement of the Congo red-stained areas was performed using the Area Quantification FL V2.3.4 module from HALO Image Analysis Platform V3.6.4134 (Indica Labs, USA). Each data point in the histograms corresponds to data from individual mice in each group where n = 6 for WT, n = 8 for AD, and n = 8 for AD→WT.
Statistical analysis
The data are presented as the mean ± standard deviation. The statistical analyses were done with the help of the GraphPad Prism software using the unpaired or paired Student’s t test when comparing two groups, which is specified in the figure legends, and a two-way ANOVA test for multiple comparisons with a Bonferroni test to correct for the multiple comparisons. The sample size for each experiment is indicated in the figure legend.
Acknowledgments
We thank Drs. Dara Dickstein and Hitesh Arora for their helpful discussions and comments on the manuscript; and Dr. Hitesh Arora, Sarah Dada, Wing Yan Leung, Wenjing Xia, Dr. Eliana Al Haddad, Dr. Giorgia Caspani, Dr. Jay Young, and Emmanuel Garrovillas for technical assistance. We recognize the following funding sources for their financial assistance with this study: W.A.J. was funded by the Canadian Institutes of Health Research (CIHR) operating grant (MOP-133635) and a grant from the W. Garfield Weston Foundation/Weston Brain Institute (RR161038); C.S.B.S. was supported by a Centre for Blood Research (UBC) Graduate Student Award; F.F. was the recipient of a DOC Fellowship of the Austrian Academy of Science and the Dmitry Apel Memorial Scholarship; and donations to the laboratory of W.A.J. through the Sullivan Urology Foundation at Vancouver General Hospital (https://www.urologyfoundation.ca).
The authors wish to acknowledge the contribution of Vivian Bradaschia and Kyle Roberton in the Pathology Core at The Center for Phenogenomics (Toronto, Canada) for the Congo red staining, digital pathology, and image analysis.
Author contributions
W.A.J conceived the study. C.S.B.S., L.M., C.G.P., and W.A.J. designed research. C.S.B.S., K.M.J., L.M., and F.F. performed research. C.S.B.S., F.F., S.K., A.M., and W.A.J. analyzed data. C.S.B.S. and W.A.J. wrote the paper. C.S.B.S., C.G.P., and W.A.J. edited the paper.
Declaration of interests
Authors hold equity in the start-up company, Cava Healthcare, which possesses intellectual property related to these findings. This had no role in the study design, data collection, analysis or interpretation of data, or in the writing of the paper.
Published: March 28, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2024.02.012.
Supplemental information
References
- PRNP Gene - Prion Protein . In: GeneCards, editor. 2023. (GeneCards the Human Gene Database)). [Google Scholar]
- Asante E.A., Gowland I., Linehan J.M., Mahal S.P., Collinge J. Expression Pattern of a Mini Human PrP Gene Promoter in Transgenic Mice. Neurobiol. Dis. 2002;10:1–7. doi: 10.1006/nbdi.2002.0486. [DOI] [PubMed] [Google Scholar]
- Asante E.A., Linehan J.M., Desbruslais M., Joiner S., Gowland I., Wood A.L., Welch J., Hill A.F., Lloyd S.E., Wadsworth J.D.F., Collinge J. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J. 2002;21:6358–6366. doi: 10.1093/emboj/cdf653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker H.F., Ridley R.M., Duchen L.W., Crow T.J., Bruton C.J. Evidence for the experimental transmission of cerebral beta-amyloidosis to primates. Int. J. Exp. Pathol. 1993;74:441–454. [PMC free article] [PubMed] [Google Scholar]
- Biron K.E., Dickstein D.L., Gopaul R., Fenninger F., Jefferies W.A. Cessation of neoangiogenesis in Alzheimer's disease follows amyloid-beta immunization. Sci. Rep. 2013;3:1354. doi: 10.1038/srep01354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron K.E., Dickstein D.L., Gopaul R., Jefferies W.A. Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer's disease. PLoS One. 2011;6 doi: 10.1371/journal.pone.0023789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu X.L., Xiang Y., Jin W.S., Wang J., Shen L.L., Huang Z.L., Zhang K., Liu Y.H., Zeng F., Liu J.H., et al. Blood-derived amyloid-beta protein induces Alzheimer's disease pathologies. Mol. Psychiatr. 2018;23:1948–1956. doi: 10.1038/mp.2017.204. [DOI] [PubMed] [Google Scholar]
- Chen G.F., Xu T.H., Yan Y., Zhou Y.R., Jiang Y., Melcher K., Xu H.E. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017;38:1205–1235. doi: 10.1038/aps.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M., Inestrosa N.C., Ross G.S., Fernandez H.L. Platelets are the primary source of amyloid beta-peptide in human blood. Biochem. Biophys. Res. Commun. 1995;213:96–103. doi: 10.1006/bbrc.1995.2103. [DOI] [PubMed] [Google Scholar]
- Daadi M.M., Grueter B.A., Malenka R.C., Redmond D.E., Jr., Steinberg G.K. Dopaminergic neurons from midbrain-specified human embryonic stem cell-derived neural stem cells engrafted in a monkey model of Parkinson's disease. PLoS One. 2012;7 doi: 10.1371/journal.pone.0041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R., Du Yan S., Submamaryan R.K., LaRue B., Jovanovic S., Hogg E., Welch D., Manness L., Lin C., Yu J., et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- Deane R., Zlokovic B.V. Role of the blood-brain barrier in the pathogenesis of Alzheimer's disease. Curr. Alzheimer Res. 2007;4:191–197. doi: 10.2174/156720507780362245. [DOI] [PubMed] [Google Scholar]
- Dickstein D.L., Biron K.E., Ujiie M., Pfeifer C.G., Jeffries A.R., Jefferies W.A. A{beta} peptide immunization restores blood-brain barrier integrity in Alzheimer disease. Faseb. J. 2006;20:426–433. doi: 10.1096/fj.05-3956com. [DOI] [PubMed] [Google Scholar]
- Eisele Y.S., Obermüller U., Heilbronner G., Baumann F., Kaeser S.A., Wolburg H., Walker L.C., Staufenbiel M., Heikenwalder M., Jucker M. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science. 2010;330:980–982. doi: 10.1126/science.1194516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EuroStemCell . 2020. Cord Blood Stem Cells: Current Uses and Future Challenges. [Google Scholar]
- EuroStemCell . 2020. Medicine and Stem Cells. [Google Scholar]
- Evin G., Zhu A., Holsinger R.M.D., Masters C.L., Li Q.X. Proteolytic processing of the Alzheimer's disease amyloid precursor protein in brain and platelets. J. Neurosci. Res. 2003;74:386–392. doi: 10.1002/jnr.10745. [DOI] [PubMed] [Google Scholar]
- Freedman M.S., Bar-Or A., Atkins H.L., Karussis D., Frassoni F., Lazarus H., Scolding N., Slavin S., Le Blanc K., Uccelli A., MSCT Study Group The therapeutic potential of mesenchymal stem cell transplantation as a treatment for multiple sclerosis: consensus report of the International MSCT Study Group. Mult. Scler. 2010;16:503–510. doi: 10.1177/1352458509359727. [DOI] [PubMed] [Google Scholar]
- Gabriel M., Hoeben B.A.W., Uhlving H.H., Zajac-Spychala O., Lawitschka A., Bresters D., Ifversen M. A Review of Acute and Long-Term Neurological Complications Following Haematopoietic Stem Cell Transplant for Paediatric Acute Lymphoblastic Leukaemia. Front. Pediatr. 2021;9 doi: 10.3389/fped.2021.774853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M. Neuronal localization of amyloid beta protein precursor mRNA in normal human brain and in Alzheimer's disease. EMBO J. 1987;6:3627–3632. doi: 10.1002/j.1460-2075.1987.tb02694.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K., Chapman P., Nilsen S., Eckman C., Harigaya Y., Younkin S., Yang F., Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hsiao K.K., Borchelt D.R., Olson K., Johannsdottir R., Kitt C., Yunis W., Xu S., Eckman C., Younkin S., Price D., et al. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron. 1995;15:1203–1218. doi: 10.1016/0896-6273(95)90107-8. [DOI] [PubMed] [Google Scholar]
- Inyushin M.Y., Sanabria P., Rojas L., Kucheryavykh Y., Kucheryavykh L. Abeta Peptide Originated from Platelets Promises New Strategy in Anti-Alzheimer's Drug Development. BioMed Res. Int. 2017;2017 doi: 10.1155/2017/3948360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaunmuktane Z., Mead S., Ellis M., Wadsworth J.D.F., Nicoll A.J., Kenny J., Launchbury F., Linehan J., Richard-Loendt A., Walker A.S., et al. Erratum: Evidence for human transmission of amyloid-beta pathology and cerebral amyloid angiopathy. Nature. 2015;526:595. doi: 10.1038/nature15704. [DOI] [PubMed] [Google Scholar]
- Jaunmuktane Z., Mead S., Ellis M., Wadsworth J.D.F., Nicoll A.J., Kenny J., Launchbury F., Linehan J., Richard-Loendt A., Walker A.S., et al. Evidence for human transmission of amyloid-beta pathology and cerebral amyloid angiopathy. Nature. 2015;525:247–250. doi: 10.1038/nature15369. [DOI] [PubMed] [Google Scholar]
- Jefferies W.A., Price K.A., Biron K.E., Fenninger F., Pfeifer C.G., Dickstein D.L. Adjusting the compass: new insights into the role of angiogenesis in Alzheimer's disease. Alzheimer's Res. Ther. 2013;5:64. doi: 10.1186/alzrt230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M.C., Medress Z.A., Azad T.D., Doulames V.M., Veeravagu A. Stem cell therapies for acute spinal cord injury in humans: a review. Neurosurg. Focus. 2019;46:E10. doi: 10.3171/2018.12.FOCUS18602. [DOI] [PubMed] [Google Scholar]
- Karussis D., Karageorgiou C., Vaknin-Dembinsky A., Gowda-Kurkalli B., Gomori J.M., Kassis I., Bulte J.W.M., Petrou P., Ben-Hur T., Abramsky O., Slavin S. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch. Neurol. 2010;67:1187–1194. doi: 10.1001/archneurol.2010.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi T., Morizane A., Doi D., Magotani H., Onoe H., Hayashi T., Mizuma H., Takara S., Takahashi R., Inoue H., et al. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson's disease model. Nature. 2017;548:592–596. doi: 10.1038/nature23664. [DOI] [PubMed] [Google Scholar]
- Kim J.H., Auerbach J.M., Rodríguez-Gómez J.A., Velasco I., Gavin D., Lumelsky N., Lee S.H., Nguyen J., Sánchez-Pernaute R., Bankiewicz K., McKay R. Dopamine neurons derived from embryonic stem cells function in an animal model of Parkinson's disease. Nature. 2002;418:50–56. doi: 10.1038/nature00900. [DOI] [PubMed] [Google Scholar]
- Kucheryavykh L.Y., Dávila-Rodríguez J., Rivera-Aponte D.E., Zueva L.V., Washington A.V., Sanabria P., Inyushin M.Y. Platelets are responsible for the accumulation of beta-amyloid in blood clots inside and around blood vessels in mouse brain after thrombosis. Brain Res. Bull. 2017;128:98–105. doi: 10.1016/j.brainresbull.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine S., Saltzman A., Levy E., Ginsberg S.D. Systemic pathology in aged mouse models of Down's syndrome and Alzheimer's disease. Exp. Mol. Pathol. 2009;86:18–22. doi: 10.1016/j.yexmp.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.X., Evin G., Small D.H., Multhaup G., Beyreuther K., Masters C.L. Proteolytic processing of Alzheimer's disease beta A4 amyloid precursor protein in human platelets. J. Biol. Chem. 1995;270:14140–14147. doi: 10.1074/jbc.270.23.14140. [DOI] [PubMed] [Google Scholar]
- Matsubara E., Shoji M., Murakami T., Abe K., Frangione B., Ghiso J. Platelet microparticles as carriers of soluble Alzheimer's amyloid beta (sAbeta) Ann. N. Y. Acad. Sci. 2002;977:340–348. doi: 10.1111/j.1749-6632.2002.tb04836.x. [DOI] [PubMed] [Google Scholar]
- Meena P., Bhargava V., Rana D., Bhalla A., Gupta A. An Approach to Neurological Disorders in a Kidney Transplant Recipient. Kidney360. 2020;1:837–844. doi: 10.34067/KID.0002052020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M., Coomaraswamy J., Bolmont T., Kaeser S., Schaefer C., Kilger E., Neuenschwander A., Abramowski D., Frey P., Jaton A.L., et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- Mita S., Schon E.A., Herbert J. Widespread expression of amyloid beta-protein precursor gene in rat brain. Am. J. Pathol. 1989;134:1253–1261. [PMC free article] [PubMed] [Google Scholar]
- Montagne A., Barnes S.R., Sweeney M.D., Halliday M.R., Sagare A.P., Zhao Z., Toga A.W., Jacobs R.E., Liu C.Y., Amezcua L., et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296–302. doi: 10.1016/j.neuron.2014.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Mouse Phenotyping Consortium Y-Maze Protocol - IMPReSS. https://www.mousephenotype.org/impress/ProcedureInfo?action=list&procID=955
- Murphy M.P., LeVine H., 3rd Alzheimer's disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010;19:311–323. doi: 10.3233/JAD-2010-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National_Stem_Cell_Foundation_of_Australia, and StemCells_Australia . National Stem Cell Foundation of Australia; 2015. The Australian Stem Cell Handbook: What You Should Know about Stem Cell Therapies: Now and in the Future. [Google Scholar]
- Omilusik K., Priatel J.J., Chen X., Wang Y.T., Xu H., Choi K.B., Gopaul R., McIntyre-Smith A., Teh H.S., Tan R., et al. The Ca(v)1.4 calcium channel is a critical regulator of T cell receptor signaling and naive T cell homeostasis. Immunity. 2011;35:349–360. doi: 10.1016/j.immuni.2011.07.011. [DOI] [PubMed] [Google Scholar]
- Ontario_Institute_for_Regenerative_Medicine . 2020. For Patients: About Stem Cell Therapies. [Google Scholar]
- Penley S.C., Gaudet C.M., Threlkeld S.W. Use of an eight-arm radial water maze to assess working and reference memory following neonatal brain injury. J. Vis. Exp. 2013;4:50940. doi: 10.3791/50940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potluri K., Holt D., Hou S. In: Handbook of Clinical Neurology. Biller J., Ferro J.M., editors. Elsevier; 2014. Chapter 84 - Neurologic complications in renal transplantation; pp. 1245–1255. [DOI] [PubMed] [Google Scholar]
- Purro S.A., Farrow M.A., Linehan J., Nazari T., Thomas D.X., Chen Z., Mengel D., Saito T., Saido T., Rudge P., et al. Transmission of amyloid-beta protein pathology from cadaveric pituitary growth hormone. Nature. 2018;564:415–419. doi: 10.1038/s41586-018-0790-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni V., Kessler J.A. Stem cell therapies for spinal cord injury. Nat. Rev. Neurol. 2010;6:363–372. doi: 10.1038/nrneurol.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh C., Pfeifer C.G., Jefferies W.A. InTechOpen; 2017. Pathogenic Angiogenic Mechanisms in Alzheimer's Disease: Chapter 6. [Google Scholar]
- Singh C.S.B., Choi K.B., Munro L., Wang H.Y., Pfeifer C.G., Jefferies W.A. Reversing pathology in a preclinical model of Alzheimer's disease by hacking cerebrovascular neoangiogenesis with advanced cancer therapeutics. EBioMedicine. 2021;71 doi: 10.1016/j.ebiom.2021.103503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starke R., Harrison P., Mackie I., Wang G., Erusalimsky J.D., Gale R., Massé J.M., Cramer E., Pizzey A., Biggerstaff J., Machin S. The expression of prion protein (PrP(C)) in the megakaryocyte lineage. J. Thromb. Haemostasis. 2005;3:1266–1273. doi: 10.1111/j.1538-7836.2005.01343.x. [DOI] [PubMed] [Google Scholar]
- Stöhr J., Watts J.C., Mensinger Z.L., Oehler A., Grillo S.K., DeArmond S.J., Prusiner S.B., Giles K. Purified and synthetic Alzheimer's amyloid beta (Abeta) prions. Proc. Natl. Acad. Sci. USA. 2012;109:11025–11030. doi: 10.1073/pnas.1206555109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S._Food_&_Drug_Administration . 2020. Approved Cellular and Gene Therapy Products. [Google Scholar]
- Ujiie M., Dickstein D.L., Carlow D.A., Jefferies W.A. Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation. 2003;10:463–470. doi: 10.1038/sj.mn.7800212. [DOI] [PubMed] [Google Scholar]
- van den Bogaart L., Lang B.M., Rossi S., Neofytos D., Walti L.N., Khanna N., Mueller N.J., Boggian K., Garzoni C., Mombelli M., et al. Central nervous system infections in solid organ transplant recipients: Results from the Swiss Transplant Cohort Study. J. Infect. 2022;85:1–7. doi: 10.1016/j.jinf.2022.05.019. [DOI] [PubMed] [Google Scholar]
- Wright A.J., Fishman J.A. Central Nervous System Syndromes in Solid Organ Transplant Recipients. Clin. Infect. Dis. 2014;59:1001–1011. doi: 10.1093/cid/ciu428. [DOI] [PubMed] [Google Scholar]
- Xu L., Nirwane A., Yao Y. Basement membrane and blood-brain barrier. Stroke Vasc. Neurol. 2019;4:78–82. doi: 10.1136/svn-2018-000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S.D., Chen X., Fu J., Chen M., Zhu H., Roher A., Slattery T., Zhao L., Nagashima M., Morser J., et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- Zheng H., Jiang M., Trumbauer M.E., Sirinathsinghji D.J., Hopkins R., Smith D.W., Heavens R.P., Dawson G.R., Boyce S., Conner M.W., et al. beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell. 1995;81:525–531. doi: 10.1016/0092-8674(95)90073-x. [DOI] [PubMed] [Google Scholar]
- Zlokovic B.V., Ghiso J., Mackic J.B., McComb J.G., Weiss M.H., Frangione B. Blood-brain barrier transport of circulating Alzheimer's amyloid beta. Biochem. Biophys. Res. Commun. 1993;197:1034–1040. doi: 10.1006/bbrc.1993.2582. [DOI] [PubMed] [Google Scholar]
- Zlokovic B.V., Martel C.L., Mackic J.B., Matsubara E., Wisniewski T., McComb J.G., Frangione B., Ghiso J. Brain uptake of circulating apolipoproteins J and E complexed to Alzheimer's amyloid beta. Biochem. Biophys. Res. Commun. 1994;205:1431–1437. doi: 10.1006/bbrc.1994.2825. [DOI] [PubMed] [Google Scholar]
- Zuroff L., Daley D., Black K.L., Koronyo-Hamaoui M. Clearance of cerebral Abeta in Alzheimer's disease: reassessing the role of microglia and monocytes. Cell. Mol. Life Sci. 2017;74:2167–2201. doi: 10.1007/s00018-017-2463-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data and code availability can be provided upon request.