

Abstract
Since Friedrich Wöhler’s groundbreaking synthesis of urea in 1828, organic synthesis over the past two centuries has predominantly relied on the exploration and utilization of chemical reactions rooted in two-electron heterolytic ionic chemistry. While one-electron homolytic radical chemistry is both rich in fundamental reactivities and attractive with practical advantages, the synthetic application of radical reactions has been long hampered by the formidable challenges associated with the control over reactivity and selectivity of high-energy radical intermediates. To fully harness the untapped potential of radical chemistry for organic synthesis, there is a pressing need to formulate radically different concepts and broadly applicable strategies to address these outstanding issues. In pursuit of this objective, researchers have been actively developing metalloradical catalysis (MRC) as a comprehensive framework to guide the design of general approaches for controlling over reactivity and stereoselectivity of homolytic radical reactions. Essentially, MRC exploits the metal-centered radicals present in open-shell metal complexes as one-electron catalysts for homolytic activation of substrates to generate metal-entangled organic radicals as the key intermediates to govern the reaction pathway and stereochemical course of subsequent catalytic radical processes. Different from the conventional two-electron catalysis by transition metal complexes, MRC operates through one-electron chemistry utilizing stepwise radical mechanisms.
Keywords: metalloradical catalysis, radical reaction, cyclopropanation, aziridination, C–H functionalization
Graphical Abstract

1. Introduction
Homolytic one-electron radical chemistry, which enjoys a complementary profile of reactivity and selectivity to heterolytic two-electron ionic chemistry, has recently garnered significant attention in the realm of organic synthesis.[1] In addition to possessing a set of fundamental reactions such as radical addition, radical substitution, atom abstraction, and radical scission, radical chemistry exhibits a number of attractive features. For example, radical reactions can proceed at fast rates under mild and neutral conditions in a broad spectrum of solvents, including water. Additionally, they are less sensitive to electronic and steric properties of substrates and can tolerate common functional groups. Furthermore, neutral radical species have inherent propensity to undergo homolytic reactions in a cascade fashion, permitting the rapid construction of complex molecular structures in a single operation. Moreover, the hallmark radical reactions of H-atom abstraction and β-scission provide general pathways for the homolytic activation of ubiquitous C(sp3)–H and C(sp3)–C(sp3) bonds in organic molecules. These reactions have the potential to redefine the state-of-the-art in organic synthesis and offer innovative pathways for designing efficient and concise synthetic strategies from abundantly accessible starting materials. Despite these remarkable merits, the synthetic promises of radical reactions have been long hindered by the formidable challenges associated with the control of reactivity and selectivity.[2] These challenges arise from the highly diverse and often unselective nature of radical reactions, typically giving rise to a complex mixture of products. Notably, controlling the enantioselectivity of radical reaction has proven particularly challenging due to facile inversion between two prochiral faces of the trivalent radical intermediates. To overcome these inherent issues, metalloradical catalysis (MRC) has emerged as a fundamentally new strategy to harness the potential of homolytic radical reactions for stereoselective organic synthesis.[3–6] The evolution of MRC as a strategy for controlling the reactivity and selectivity of homolytic radical reactions marks a significant advancement in the field of catalysis. This innovative approach was initially inspired by pioneering studies on the unique reactivities of rhodium(II) porphyrin complexes [Rh(II)(Por)].[7] These studies unveiled the ability of monomeric [Rh(II)(Por)], functioning as d7-metalloradicals, to perform one-electron activation of O2 and CO, generating [(Por)Rh(II)(O2)•] and [(Por)Rh(II)(CO)•] radical species, respectively.[8–9] Moreover, these Rh(II)-based metalloradicals demonstrated remarkable proficiency in undergoing radical addition to alkenes and alkynes, resulting in the formation of β-Rh(III)-alkyl and -alkenyl radicals.[10–12] Exceptionally, they also displayed the capability to homolytically activate H2 and CH4 through a linear four-centered transition state, enabling the cleavage of strong H–H and C(sp3)–H bonds.[13–15] These groundbreaking observations of one-electron activation modes by metalloradicals laid the groundwork for a new paradigm in metal-based catalysis, suggesting vast potential for MRC in achieving unprecedented control over radical processes. This historical context provides a foundation for understanding the current development and future directions of MRC, illuminating its role as a transformative tool in synthetic chemistry.
This minireview introduces the underlying concept of MRC and illustrate the key steps in its general catalytic cycle. Furthermore, several well-defined metalloradical systems are discussed to exemplify the operational mechanisms of MRC and highlight their synthetic applications in important organic transformations. While this minireview does not provide an exhaustive overview of all MRC systems, it aims to serve as a source of inspiration for future research directions in MRC and to stimulate further exploration of its applications as innovative synthetic methods for molecular construction.
1.1. Metalloradicals and Metalloradical Catalysis
Metalloradicals (LnM•) are a type of open-shell transition-metal complexes with the unpaired d-electron(s) to demonstrate radical-like homolytic reactivities. Different from the typical two-electron chemistry exhibited by most close-shell transition-metal complexes, metalloradicals engage in one-electron chemistry with a distinct reactivity and selectivity profile that arises from the interaction between the metal’s frontier SOMO (singly occupied molecular orbital) and the substrates. Irrespective of their oxidation state, all transition metals in principle have the ability to form metalloradical complexes. While complexes of metal ions with an odd number of d-electrons can be readily classified as metalloradicals, the potential for metal ion complexes with an even number of d-electrons to function as metalloradicals also exists. This potential hinges on the specific spin state that comes into play. Beyond being governed by the d-electronic configuration and spin state of the metal center, the reactivity and selectivity of metalloradical complexes can be precisely tailored or significantly modified by the surrounding ligand environment, enabling fine-tuning of their one-electron homolytic properties. Despite the typically high energy and reactivity associated with metalloradicals, there are notable examples of stable metalloradicals. One such example is the family of cobalt(II) complexes of porphyrins. These well-demonstrated metalloradicals exhibit remarkable stability in their solid forms, even when exposed to open air for an extended period without noticeable oxidation. However, in solution, they retain the ability to homolytically activate substrates and display radical-like reactivity.
Metalloradical catalysis (MRC)[16] harnesses the potential of metal-centered radicals within metalloradical complexes as one-electron catalysts for homolytic activation of substrates to generate metal-entangled organic radicals as key intermediates to direct the reaction pathways and influence the stereochemical outcomes of subsequent catalytic radical processes. Conventional catalysis employing close-shell transition metal catalysts typically follows two-electron concerted ionic mechanisms, which involve elementary reactions such as oxidative addition, migratory insertion, β-hydride elimination and reductive elimination. In comparison, MRC with the use of metalloradical catalysts operates through one-electron stepwise radical mechanisms that comprise homolytic reactions, including radical addition, atom abstraction, radical scission and radical substitution. Unlike the concerted ionic mechanism seen in conventional catalysis with closed-shell transition metal catalysts, MRC relies on these stepwise radical processes to achieve the desired reactivity and selectivity in the catalytic transformations. In the general mechanism of MRC (Scheme 1), the process typically initiates with the homolytic activation of organic substrates, referred to as radical precursors or metalloradicophiles, by metalloradical catalysts LnM•. This activation step generates the corresponding organic radical intermediates LnM–X•(I) that are intricately associated or covalently bonded with the metal complex. The intermediates I, known as metal-entangled organic radicals, play a pivotal role in the catalytic process. While the mode of activation may vary, the initial step of metalloradical activation shares a common characteristic: the transfer of the original radical character from the metal center to a nonmetal atom site within the metalloradicophile. Depending on the choice of metalloradicophiles, this radical translocation catalytically generates different metal-entangled organic radicals I, such as α-metalloalkyl radicals LnM–(R1)-(R2)C•, α-metalloaminyl radicals LnM–(R1)N•, and α-metalloxyl radicals LnM–O• (Scheme 1). Notably, the generation of these metal-supported carbon-, nitrogen- and oxygen-centered radicals occur without the need for traditional radical initiators or the utilization of photolysis through light or electrolysis powered by electricity, showcasing the practicality and sustainability of MRC as an innovative approach. While these metal-entangled organic radicals I possess the ability to undergo common radical reactions similar to free organic radicals, they are distinct in that they are no longer “free” radicals. Instead, they engage in the subsequent reactions with reactants under the influence of the metal complex through various interactions such as covalent bonding or noncovalent interactions, resulting in the selective formation of new metal-entangled organic radicals, denoted as LnM–Y•(II). Importantly, the radical character in intermediate II does not necessarily translocate to the α-position of the metal center and can be at different sites within the organic substrates, depending on the specific reaction conditions and the nature of the reactants involved. This regulated reactivity governed by the metal complex allows for precise control over the reaction outcomes and enables the formation of specific organic radicals tailored to the desired products. Due to the relatively weaker nature of M–Y (Y=CR2, NR or O) bonds compared to C–Y bonds, homolytic radical substitution (SH1 or SH2), which is inherently difficult in free radical reaction, becomes an efficient pathway to turn over the catalytic cycle, forming the product while regenerating the metalloradical catalysts LnM•. For metal-entangled organic radicals II where the radical is located at the β-position of the metal center, radical β-scission can function as an alternative pathway to facilitate the turnover of MRC and lead to product formation. In contrast to traditional catalysis, where the metal center typically undergoes metal oxidation changes in increments of two units per step (Mn→Mn+2→Mn), MRC exhibits a distinctive oxidation state modulation by a single unit per step (Mn→Mn+1→Mn) in the catalytic cycle (Scheme 1).
Scheme 1.
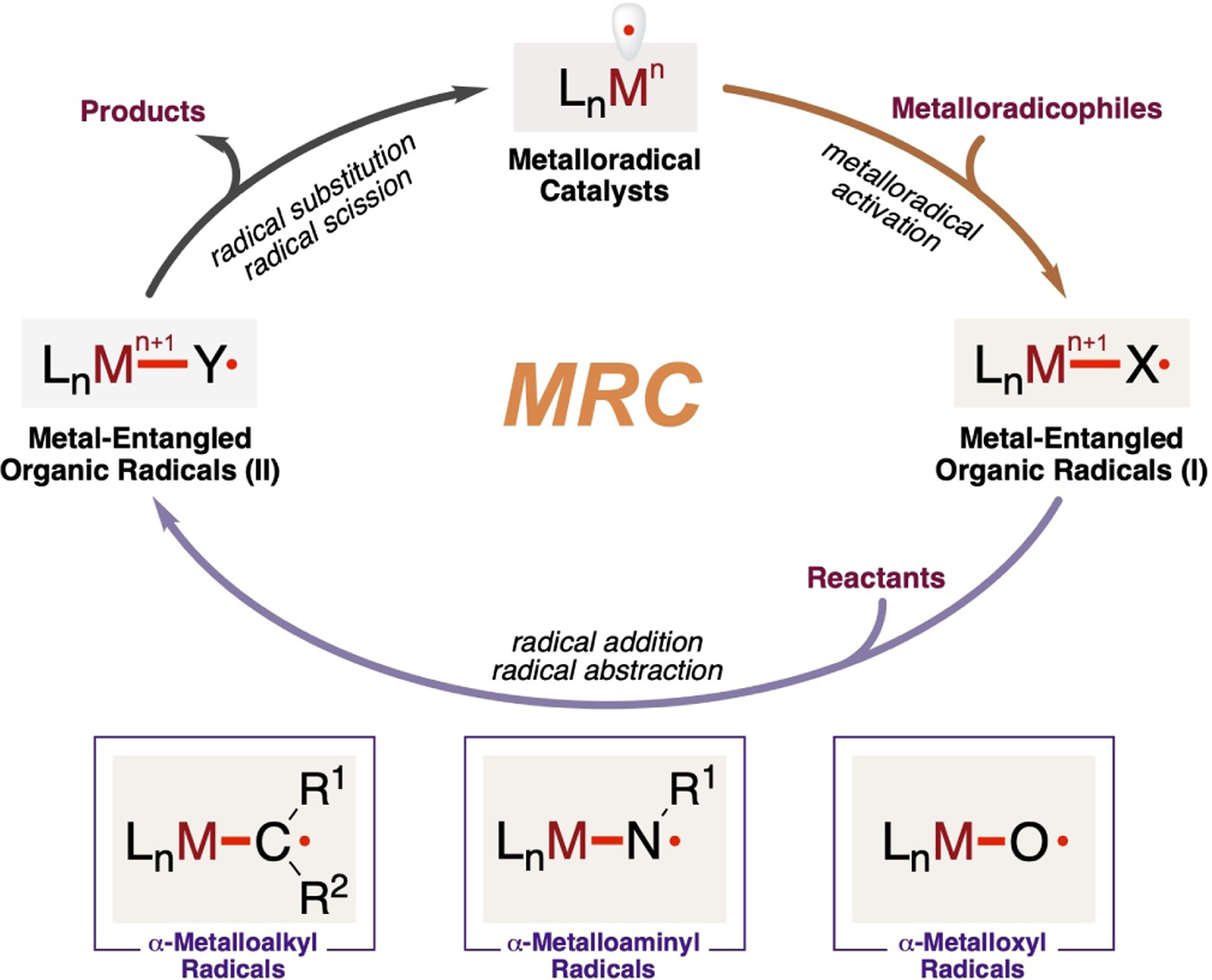
General catalytic mechanism of metalloradical catalysis.
1.2. Metalloporphyrins as Versatile Metalloradical Catalysts
In principle, the broad range of metalloradicals encompassing various combinations of metal centers and supporting ligands hold the potential to function as metalloradical catalysts in MRC. Due to the unique ligand environment and metal coordination modes (Scheme 2), metalloporphyrins with open-shell electronic configurations offer a general platform for the development of metalloradical catalysts in the context of MRC. As a family of versatile tetradentate ligands with four nitrogen donor atoms, porphyrins can form complexes with virtually all transition metal ions. With the electron-rich π-conjugated aromatic system, porphyrins can stabilize metal ions at different oxidation states with varied electronic structures, including the formation of open-shell metalloporphyrins to function as metalloradicals. The choice of metal ion in metalloporphyrin-based metalloradicals can be tailored to achieve specific MRC reactivity profile. Due to the planar macrocyclic structure of porphyrin ligands, metalloporphyrins have well-defined coordination geometries where the cis-coordination sites are fully occupied by the four nitrogen donor atoms. In contrast to traditional metal-based catalytic processes that typically rely on cis-coordination, MRC operates through a distinct stepwise radical mechanism that can circumvent the need for cis-coordination (Scheme 1). In fact, the absence of cis-coordination is advantageous in MRC. The unique metal coordination mode of metalloporphyrins allows for the avoidance of potential side reactions associated with cis-coordination. As a result, MRC by metalloporphyrin-based metalloradical catalysts can achieve enhanced catalytic efficiency and selectivity. Furthermore, it has been well demonstrated that the chemical reactivity of metalloporphyrins can be precisely modulated by incorporating peripheral substituents with diverse electronic, steric, and stereochemical environments onto the aromatic ring structure of the porphyrin ligand (Scheme 2). This systematic tuning of metalloporphyrins allows for fine control over reactivity and selectivity, enabling tailored catalytic performance for specific applications. By strategically designing porphyrin ligands with desired peripheral substituents, the scope and versality of metalloporphyrin-based MRC can be expanded for a wide-range of chemical transformations. Moreover, metalloporphyrins exhibit remarkable thermal stability and exceptional coordination strength due to the macrocyclic chelation effect of the aromatic ligands. Once the metal ion is incorporated into the macrocyclic ring, its dissociation becomes exceedingly difficult, if not practically impossible, under a wide range of reaction conditions. These exceptional robustness and reliability contribute to prolonged catalyst lifetime and prevent metal contamination in the reaction products. The elimination of metal ion contamination is particularly significant in pharmaceutical applications of metal-catalyzed processes, where purity and product integrity are of utmost importance. Collectively, these intrinsic advantages position open-shell metalloporphyrins as an exceptional class of metalloradical catalysts for facilitating homolytic radical reactions through MRC.
Scheme 2.
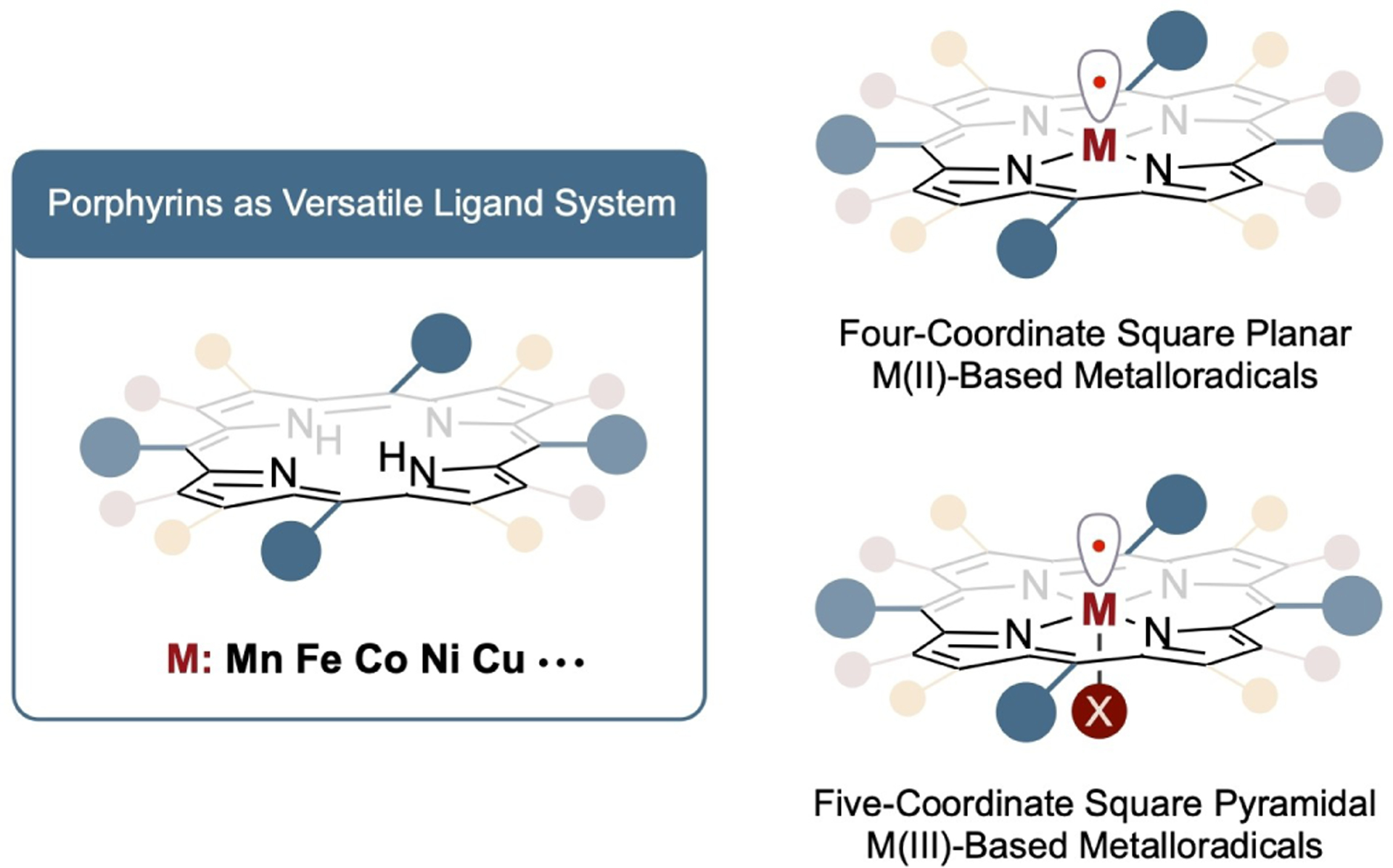
Metalloporphyrins: general platform for metalloradical catalysts.
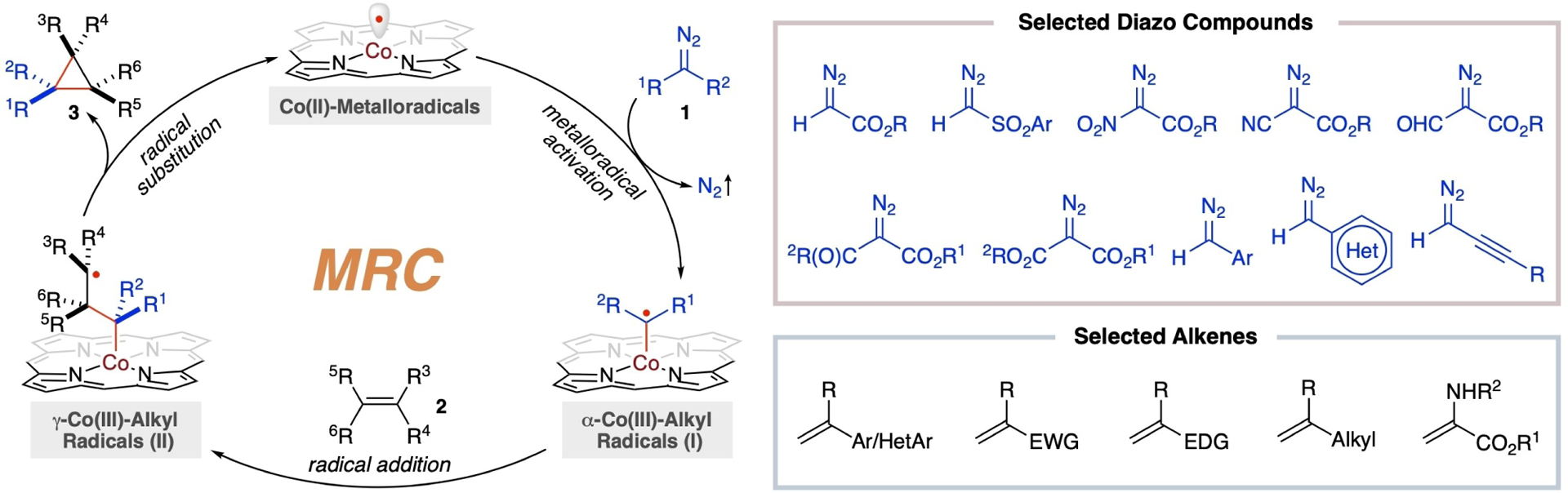
2. MRC by Cobalt Complexes of Porphyrins
Porphyrins have the ability to form stable complexes with cobalt ion at different accessible oxidations, including +1, +2 and +3, with +2 oxidation state being the most prevalent. The cobalt(II) complexes of porphyrins [Co(Por)], as stable 15e-metalloradicals with well-defined low spin d7 electronic configuration of , [17] have been demonstrated as effective metalloradical catalysts for facilitating a wide range of homolytic radical reactions through the operation of MRC. In Co(II)-based MRC, it has been shown that [Co(Por)] metalloradicals can homolytically activate both carbon-based metalloradicophiles, such as diazo compounds, and nitrogen-based metalloradicophiles, such as organic azides, generating the corresponding α-Co(III)-alkyl radicals and α-Co(III)-aminyl radicals, respectively (Scheme 3). Mechanistic studies have provided extensive support for the generation of both α-Co(III)-alkyl radicals and α-Co(III)-aminyl radicals. Electron paramagnetic resonance (EPR) spectroscopy has successfully detected the α-Co(III)-alkyl radicals and α-Co(III)-aminyl radicals based on their characteristic signals.[18–31] High-resolution mass spectrometry (HRMS) with ESI ionization has identified the α-Co(III)-aminyl radicals. [20–22,24,26–27,29–30] Direct trapping of α-Co(III)-alkyl radicals with stable radical 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) has been accomplished, and the resulting bis-TEMPO-trapped products have been characterized through X-ray crystallography.[21–23,25,27–28,31–37] The involvement of α-Co(III)-alkyl radicals during catalysis has been further supported by the introduction of a H-atom source, resulting in the formation of α-H-trapped product through a sequence of H-atom abstraction and radical substitution by the radical intermediates.[25] In addition, the radical dimerization of α-Co(III)-allyl radicals has been evidenced by the formation of a dimeric Co(III) porphyrin complex upon the use of ethyl styryldiazoacetate as the metalloradicophile.[38] Collectively, these comprehensive studies, combined with DFT calculations,[18–19,24–29,31,36–37,39–41] provide compelling evidence for the generation of α-Co(III)-alkyl radical and α-Co(III)-aminyl radical intermediates.
Scheme 3.
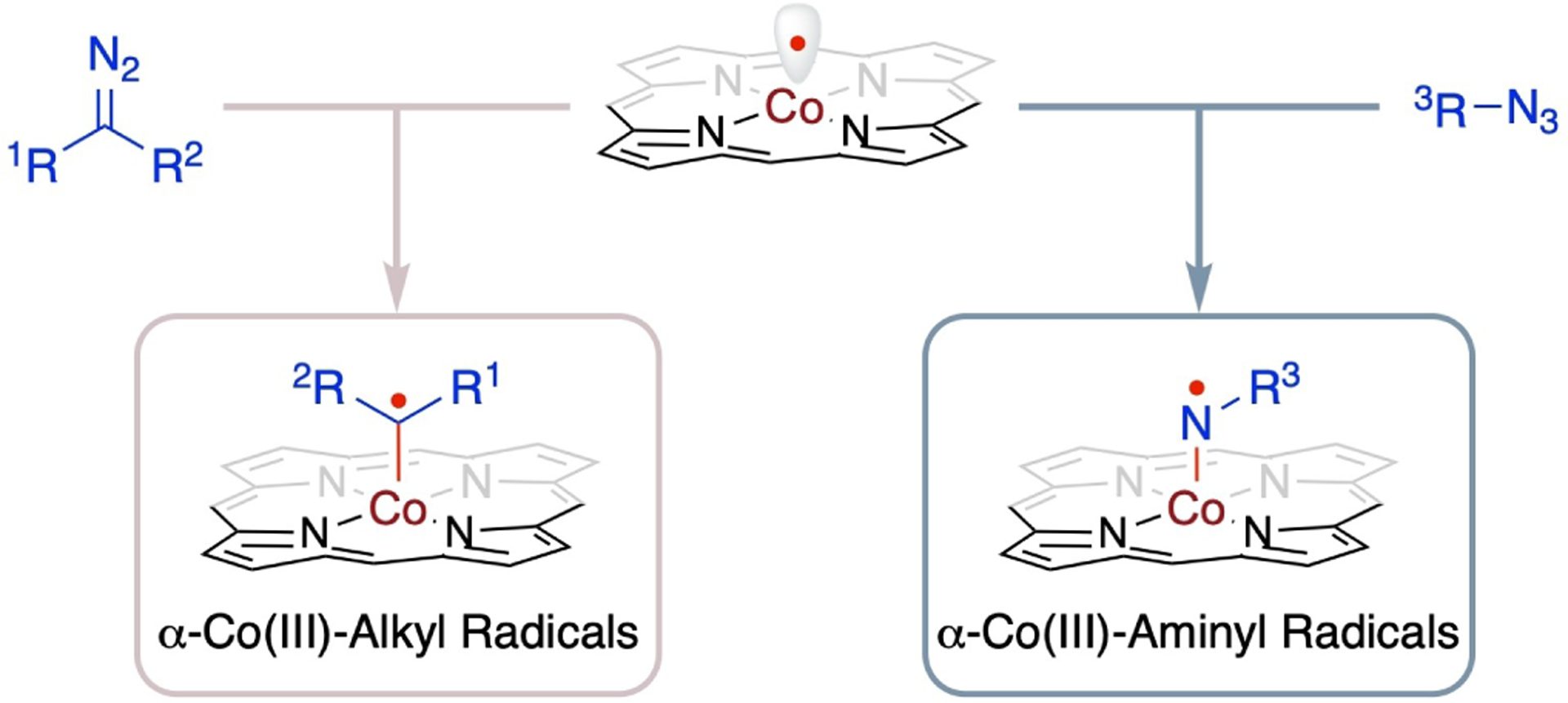
Generation of α-Co(III)-alkyl radicals and α-Co(III)-aminyl radicals.
The successful development of Co(II)-based MRC for controlling homolytic radical reactions is intricately connected to the innovative design and effective synthesis of porphyrins as the supporting ligands. In the pursuit of achieving enantioselective radical reactions through MRC, a family of D2-symmetric chiral amidoporphyrins, [H2(D2-Por*)], with pocket-like structures has emerged as a versatile ligand platform to effectively support Co(II)-based metalloradical catalysts, [Co(D2-Por*)], for catalyzing different organic transformations.[42] Several generations of [H2(D2-Por*)] with diverse steric, electronic, and chiral environments have been modularly designed and efficiently constructed from two readily available building blocks of aldehydes and chiral amides through a general synthetic scheme that is based on Pd-catalyzed quadruple amidation of tetrabromoporphyrin synthons.[42–45] As a representative example of first-generation [H2(D2-Por*)], 3,5-DitBu-Chen-Phyrin, [H2(P1)], can be synthesized in high yield on multigram scale in a practical manner from the commercially available building blocks of (S)-2,2-dimethylcyclopropanecarboxamide and 3,5-di-tert-butylbenzaldehyde (Scheme 4).[42] By taking advantage of the general effectiveness of first-generation metalloradical catalysts such as [Co(P1)] in catalyzing olefin cyclopropanation with excellent control of both diastereoselectivity and enantioselectivity,[42,46–47] an iterative approach has been demonstrated to utilize the resulting highly-enantioenriched cyclopropanecarboxamides bearing two contiguous stereocenters on the three-membered ring structure as new chiral building blocks for the construction of second-generation [H2(D2-Por*)] such as 3,5-DitBu-Tao(tBu)Phyrin [H2(P2)].[44–45] More recently, new-generation [H2(D2-Por*)] has been designed to adopt a cavity-like structure by installing bridges across the two chiral amide units of both sides of the porphyrin plane.[45] For example, 3,5-DitBu-Hu(C6)Phyrin [H2(P3)] and 3,5-DitBu-Hu(C8)Phyrin [H2(P4)] containing C6- and C8-alkyl bridges, respectively, (Scheme 4) can be efficiently constructed in a modular fashion from second-generation 3,5-DitBu-Tao(tBu)Phyrin [H2(P2)] by a five-step synthesis featuring double ring-closing olefin metathesis in high overall yields. In addition to offering a new dimension for fine-tuning steric, electronic and chiral environments, the bridged amidoporphyrins gain enhanced capability for hydrogen-bonding and other noncovalent interactions due to the rigidification of the chiral amide units. Typically, the Co(II) complexes of D2-symmetric chiral amidoporphyrins, [Co(D2-Por*)], can be readily prepared in high yields from the reaction of [H2(D2-Por*)] with CoCl2 in the presence of 2,6-lutidine as the base on scales ranging from hundred milligrams to multigrams. The Co(II)-based metalloradicals [Co(D2-Por*)] have demonstrated as excellent chiral catalysts for diverse asymmetric radical transformations.
Scheme 4.
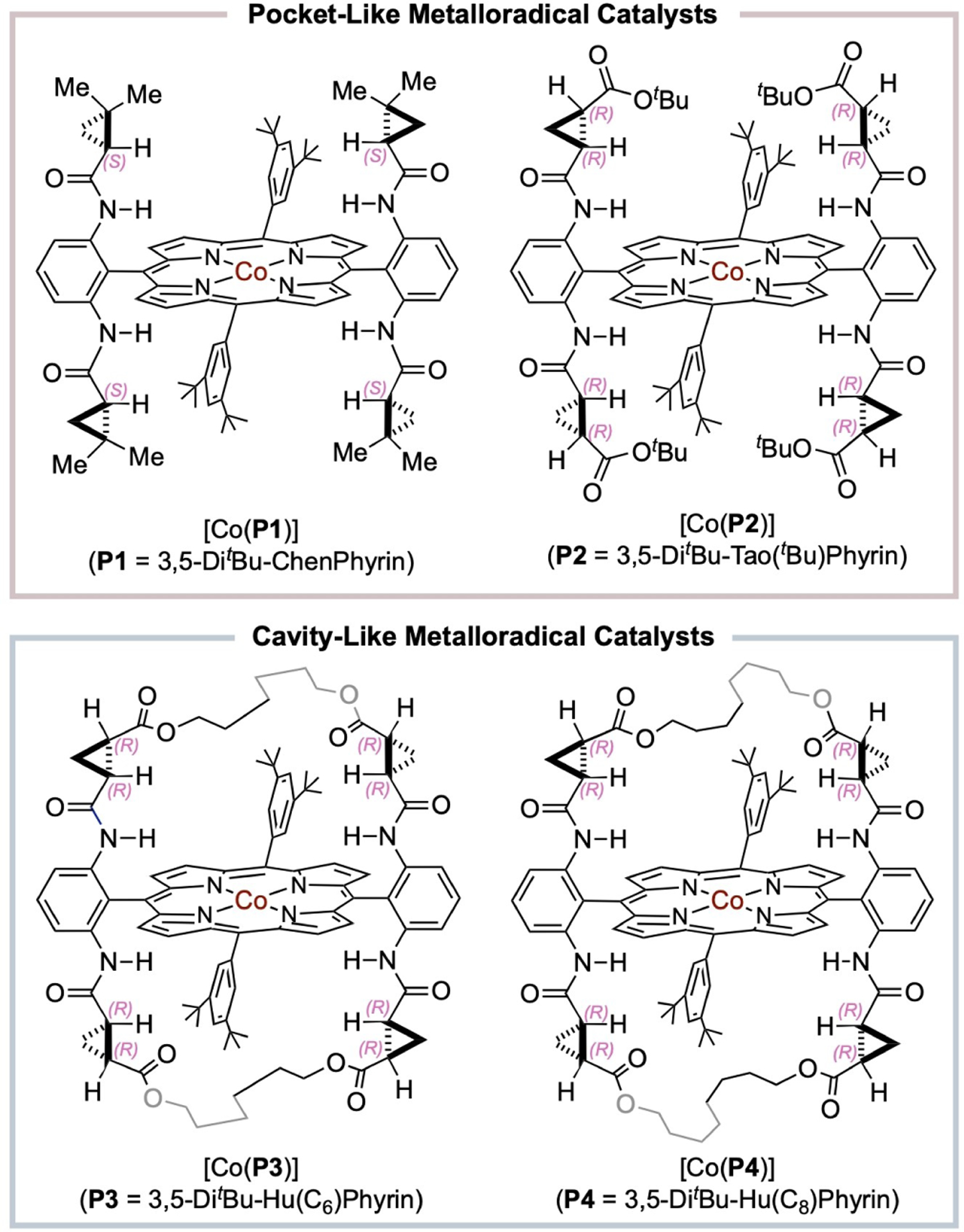
Representative examples of Co(II) complexes of D2-symmetric chiral amidoporphyrins with tuneable pocket- and cavity-like environments.
2.1. Radical Cyclopropanation of Alkenes
As the first application of Co(II)-based MRC for stereoselective radical transformations, [Co(Por)] metalloradicals have been demonstrated to exhibit an exceptional capability for homolytic activation of diazo compounds 1, generating previously-unknown α-Co(III)-alkyl radicals I (Scheme 5).[4] This one-electron metalloradical activation mode, which typically occurs under mild conditions, stands in stark contrast to the two-electron activation mode by closed-shell transition metal complexes, such as the extensively-studied Rh2-based systems, known for generating the well-established metallocarbenes. Beyond the commonly-used diazoacetates, different classes of diazo compounds, including acceptor/H-substituted, acceptor/acceptor-substituted and donor/H-substituted diazo compounds, have been identified as viable metalloradicophiles. This versatility allows for the metalloradical activation process to furnish the corresponding α-Co(III)-bonded C-centered organic radicals I, featuring different combinations of R1 and R2 substituents (Scheme 5).[4] Functioning akin to free alkyl radicals, these initially-generated metal-entangled alkyl radicals I have been shown to be kinetically competent to undergo radical addition to alkenes 2, resulting in the formation of γ-Co(III)-alkyl radicals II while forging one C–C bond. Both enthalpically and entropically, these subsequently-formed γ-Co(III)-alkyl radicals II favor intramolecular radical substitution over intermolecular radical addition to another alkene molecule. This preference facilitates another C–C bond formation and the cleavage of the weaker Co–C bond, leading to 3-exo-tet cyclization to construct three-membered cyclopropanes 3 while regenerating the Co(II)-metalloradical catalysts. In contrast to the concerted mechanism involving metallocarbene intermediates that forms two C–C bonds in a single step, the Co(II)-based metalloradical system for olefin cyclopropanation controls enantioselectivity and diastereoselectivity separately in two successive steps of C–C bond formation. Owing to the reduced sensitivity of radical reactions to electronic properties and steric hindrances, the metalloradical cyclopropanation system, operating via the stepwise radical pathway, is applicable to a wide range of alkene substrates, including electron-deficient olefins, which are known as challenging substrates in the electrophilic metallocarbene systems.
Scheme 5.
General mechanism for radical olefin cyclopropanation with diazo compounds via Co(II)-based MRC. Reprinted from ref. [4] with permission from Elsevier.
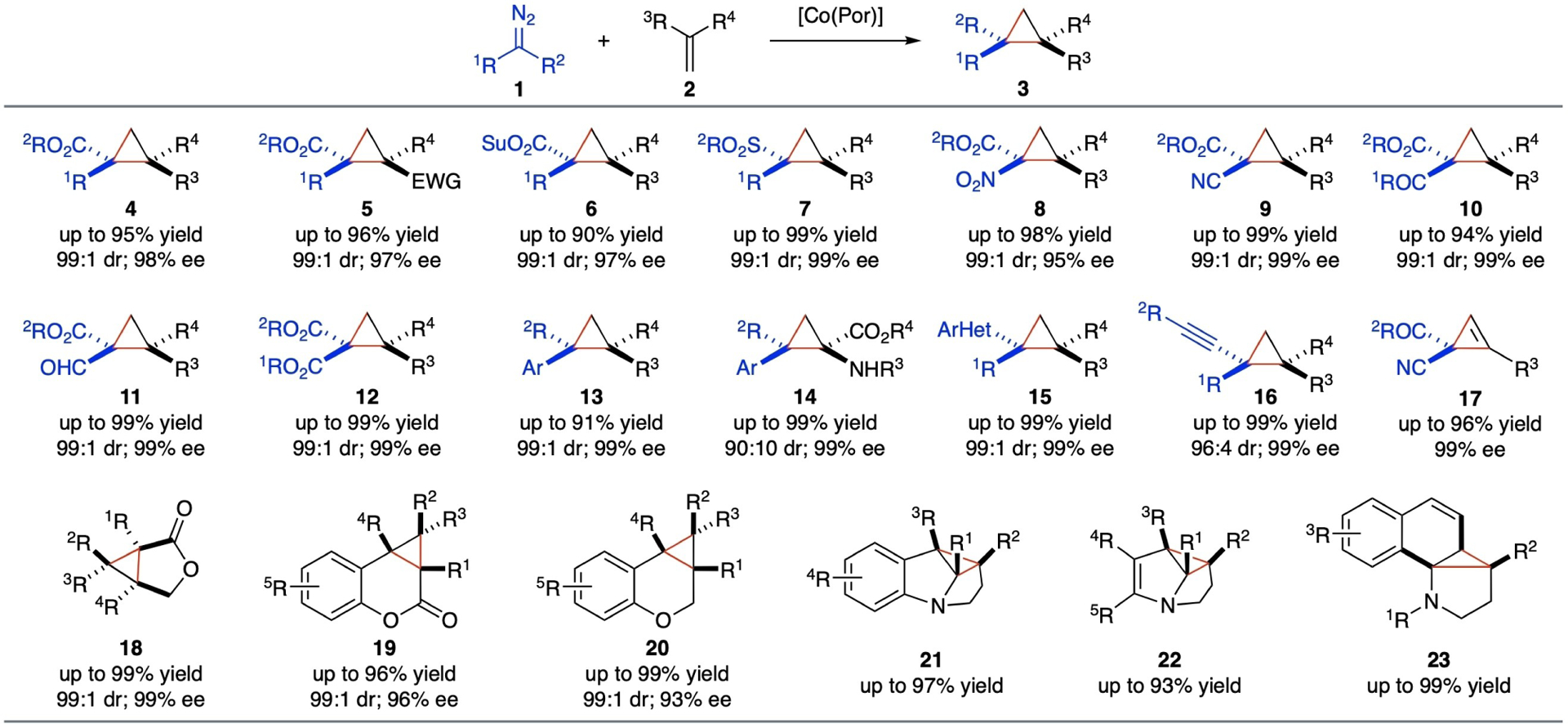
With the support of D2-symmetric chiral amidoporphyrin ligand platform, [Co(D2-Por*)] have proved to be one of the most general catalytic systems for asymmetric olefin cyclopropanation. The Co(II)-based metalloradical system can effectively activate different classes of diazo compounds 1 for asymmetric cyclopropanation of various types of alkenes 2, resulting in the high-yielding formation of three-membered cyclopropanes 3 with both high diastereoselectivity and enantioselectivity. A variety of chiral cyclopropanes containing diverse functionalities have been synthesized by Co(II)-catalyzed radical asymmetric cyclopropanation (Scheme 6), including cyclopropaneesters (4),[42,46,48–49] 1,2-cyclopropanediesters (5),[47] succinimidyl cyclopropaneesters (6),[50] cyclopropyl sulfones (7),[43] cyclopropane α-nitroesters (8),[51] cyclopropane α-cyanoesters (9),[52] 1,1-cyclopropane-ketoesters (10),[53] 1,1-cyclopropaneformylesters (11),[54] 1,1-cyclopropanediesters (12),[29] 1,2-diphenyl cyclopropanes (13),[55] cyclopropyl α-amino acids (14),[25] heteroaryl cyclopropanes (15),[36,56] and alkynyl cyclopropanes (16).[28] Furthermore, it has been shown that the resulting α-Co(III)-alkyl radical intermediates I from metalloradical activation of diazo compounds by [Co(D2-Por*)] were also capable to undergo radical addition to alkynes under mild conditions for asymmetric cyclopropenation, leading to the construction of enantioenriched cyclopropenes (17).[16] In addition to the intermolecular radical cyclization, [Co(D2-Por*)] have demonstrated catalytic capacity for intramolecular radical bicyclization of diazo compounds bearing alkene functionalities to construct multi-functionalized bicyclic compounds in high yields with high stereoselectivities, such as 3-oxabicyclo[3.1.0]hexan-2-ones (18),[44,57] cyclopropane-fused chromanones (19),[31] and cyclopropane-fused chromanes (20).[31] Moreover, [Co(Por)] have shown to be effective in catalyzing intramolecular bicyclization for dearomatization of arenes and heteroarenes such as indoles, pyrroles and naphthalenes, forming highly-strained polycyclic N-heterocycles 21–23 in good to high yields.[58–59]
Scheme 6.
Selected examples of chiral cyclopropanes from catalytic radical cyclopropanation, cyclopropenation and bicyclization via Co(II)-based MRC.
2.2. Radical Aziridination of Alkenes
While diazo compounds are effective carbon-based metalloradicophiles, organic azides represent a potent class of nitrogen-based metalloradicophiles. Studies have demonstrated that [Co(Por)] metalloradicals can homolytically activate a range of organic azides 24 under mild conditions, leading to the formation of α-Co(III)-aminyl radicals III upon the release of nitrogen gas (Scheme 7).[40] Like α-Co(III)-alkyl radicals I, the metal-entangled aminyl radicals III, which possess radical character on the α-nitrogen atom adjacent to the cobalt center, can undergo radical addition to different types of alkenes 2, delivering γ-Co(III)-alkyl radicals IV by breaking a relatively weak C–C π-bond while forging one stronger σ-C–N bond. Instead of undergoing intermolecular radical addition again to another alkene molecule, the resulting metal-entangled alkyl radical intermediates IV, which bear radical character on the γ-carbon atom from the cobalt center, favor both kinetically and thermodynamically to proceed intramolecular radical substitution (3-exo-tet radical cyclization) to form another C–N bond while breaking the weaker Co–N bond, resulting in the selective production of the aziridines 25 with the regeneration of the Co(II)-metalloradicals. This stepwise radical mechanism by the Co(II)-based metalloradical system is noteworthy because it provides a contemporary pathway for olefin aziridination that is fundamentally different from the established concerted ionic mechanism by electrophilic metallonitrene systems. Considering that radical reaction is generally less sensitive to electronic and steric requirements as well as reaction conditions, the metalloradical approach for aziridination is expected to enjoy broad scopes of both organic azides and alkenes, including typically challenging electron-deficient olefins.
Scheme 7.
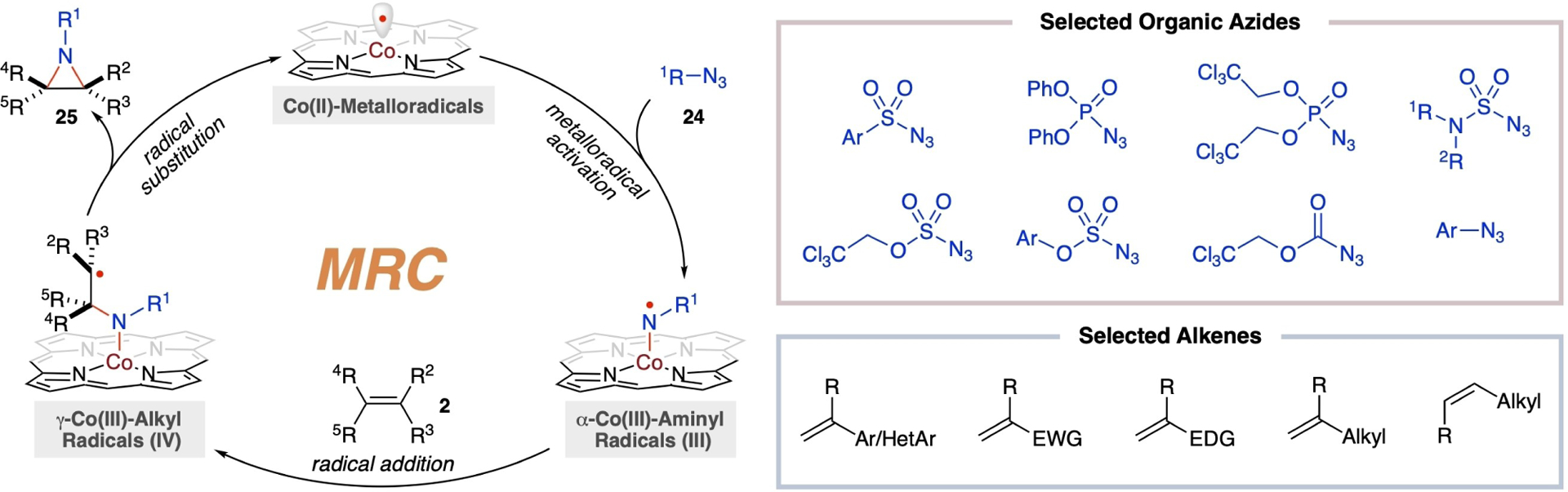
General mechanism of radical aziridination of alkenes with organic azides via Co(II)-based MRC and selected examples of substrates.
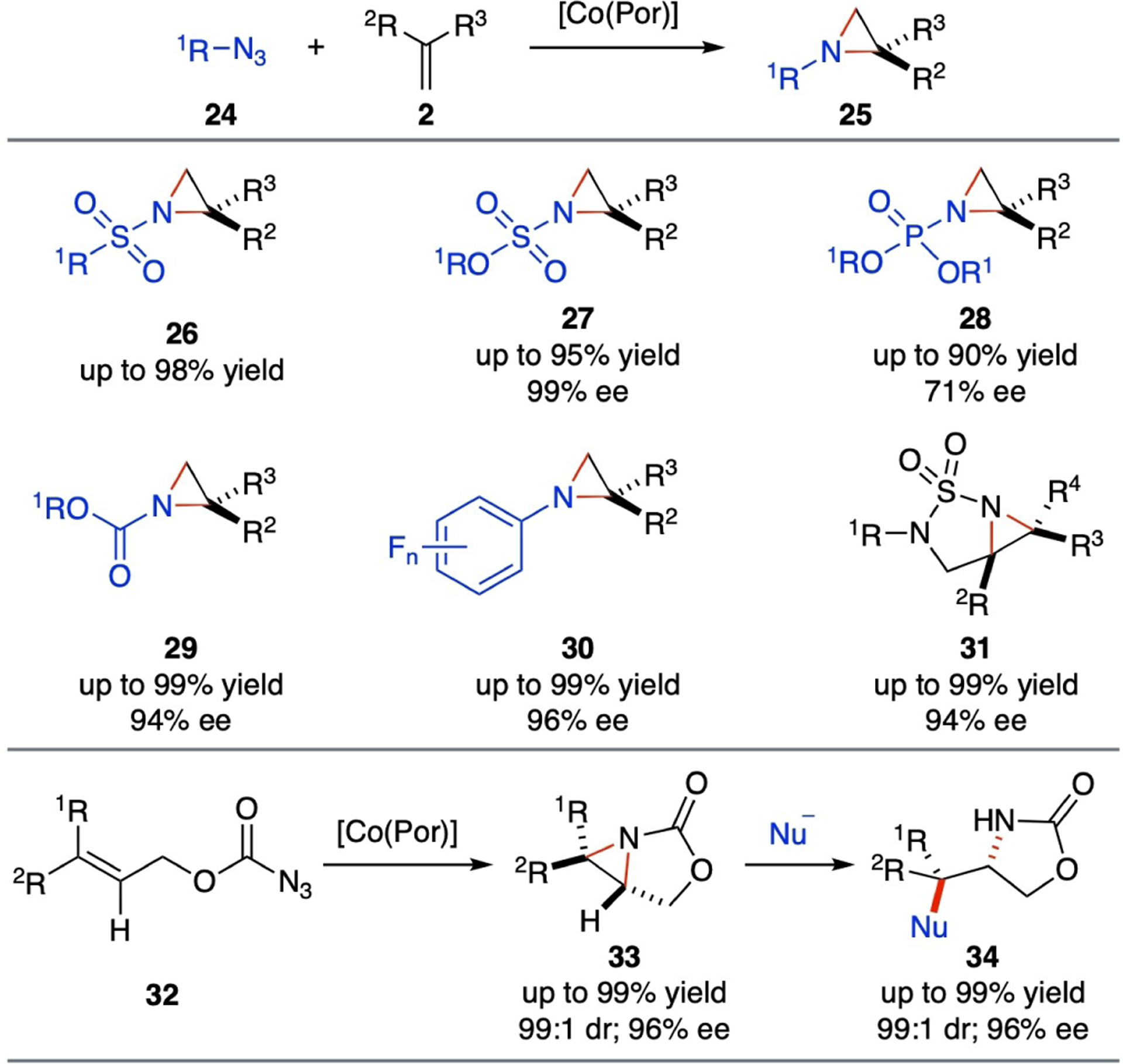
By leveraging the support of the porphyrin ligand platform, the Co(II)-based metalloradical system homolytically activates a variety of organic azides bearing substituents with different electronic properties, enabling the stereoselective construction of three-membered heterocycles (Scheme 8), such as N-sulfonylated aziridines (26),[60–61] N-trichloroethoxysulfonyl (Tces) aziridines (27),[62–63] N-phosphorus-substituted aziridines (28),[64–66] N-carbonyl aziridines (29),[24] and N-fluoroaryl aziridines (30)[67–68] in typically high yields with high enantioselectivities. Additionally, [Co(D2-Por*)] complexes have been shown to catalyze asymmetric radical bicyclization of allylic sulfamoyl azides with varied electronic and steric properties through an intramolecular radical aziridination pathway. This process allows for the stereoselective construction of highly-strained 2-sulfonyl-1,3-diazabicyclo[3.1.0]hexane structures (31) under mild conditions in excellent yields with high stereoselectivities.[69–70]
Scheme 8.
Selected examples of chiral aziridines from catalytic radical aziridination and bicyclization via Co(II)-based MRC.
The resulting enantioenriched [3.1.0]bicyclic aziridines 31 have been showcased as useful chiral synthons for the preparation of valuable chiral 1,2-diamines and 1,3-diamines. Furthermore, this Co(II)-based metalloradical system effectively activates allyl azidoformates (32) for asymmetric radical bicyclization, affording chiral [3.1.0]bicyclic aziridines (33) in high yields with excellent diastereoselectivities and enantioselectivities.[71] The enantioenriched oxazolidinones 33 resulting from this catalytic process have been demonstrated to undergo stereospecific ring-opening reactions with different nucleophiles, producing chiral oxazolidinones (34) and vicinal amino alcohols in high yields while retaining the original enantiopurity.
2.3. Radical Alkylation of C(sp3)–H Bonds
In addition to participating in radical addition to alkenes for catalytic cyclopropanation (Scheme 5), α-Co(III)-alkyl radicals I, generated from homolytic activation of diazo compounds 1 by Co(II)-based metalloradicals, can also function as kinetically viable intermediates to undergo hydrogen atom abstraction (HAA) from C(sp3)–H bonds in substrates 35, offering a radical approach for catalytic C–H alkylation (Scheme 9). In contrast to the concerted mechanism of C–H insertion characteristic of Fisher-type electrophilic metallocarbenes in existing catalytic systems, C–H alkylation via Co(II)-MRC proceeds with a distinctive, stepwise radical mechanism featuring two metal-entangled radicals as pivotal intermediates: α-Co(III)-alkyl radicals I and ∞-Co(III)-alkyl radicals V. The mechanism involves H-atom abstract by the initially-generated intermediate I from C–H substrate 35 and radical substitution by the subsequently-formed intermediate V to yield alkylation product 36 while regenerating the Co(II)-metalloradical catalysts. Rather than traversing a single, congested high-energy transition state that requires simultaneous bond-breaking and bond-forming events, the Co(II)-based metalloradical system executes C–H bond cleavage and C–C bond formation in two discrete steps. These steps typically have lower activation barriers, which is why the Co(II)-based metalloradical system for C–H alkylation generally operates under mild conditions with rapid reaction rates. It also boasts a wide substrate scope, exhibiting less sensitivity to electronic and steric demands. Furthermore, the cobalt(II)-based metalloradical system for C–H alkylation leverages the bond dissociation energy (BDE) of C–H bonds as a critical determinant in modulating the reactivity and selectivity amongst various C–H bonds in substrates, an effect that can be fine-tuned with ligand design.
Scheme 9.
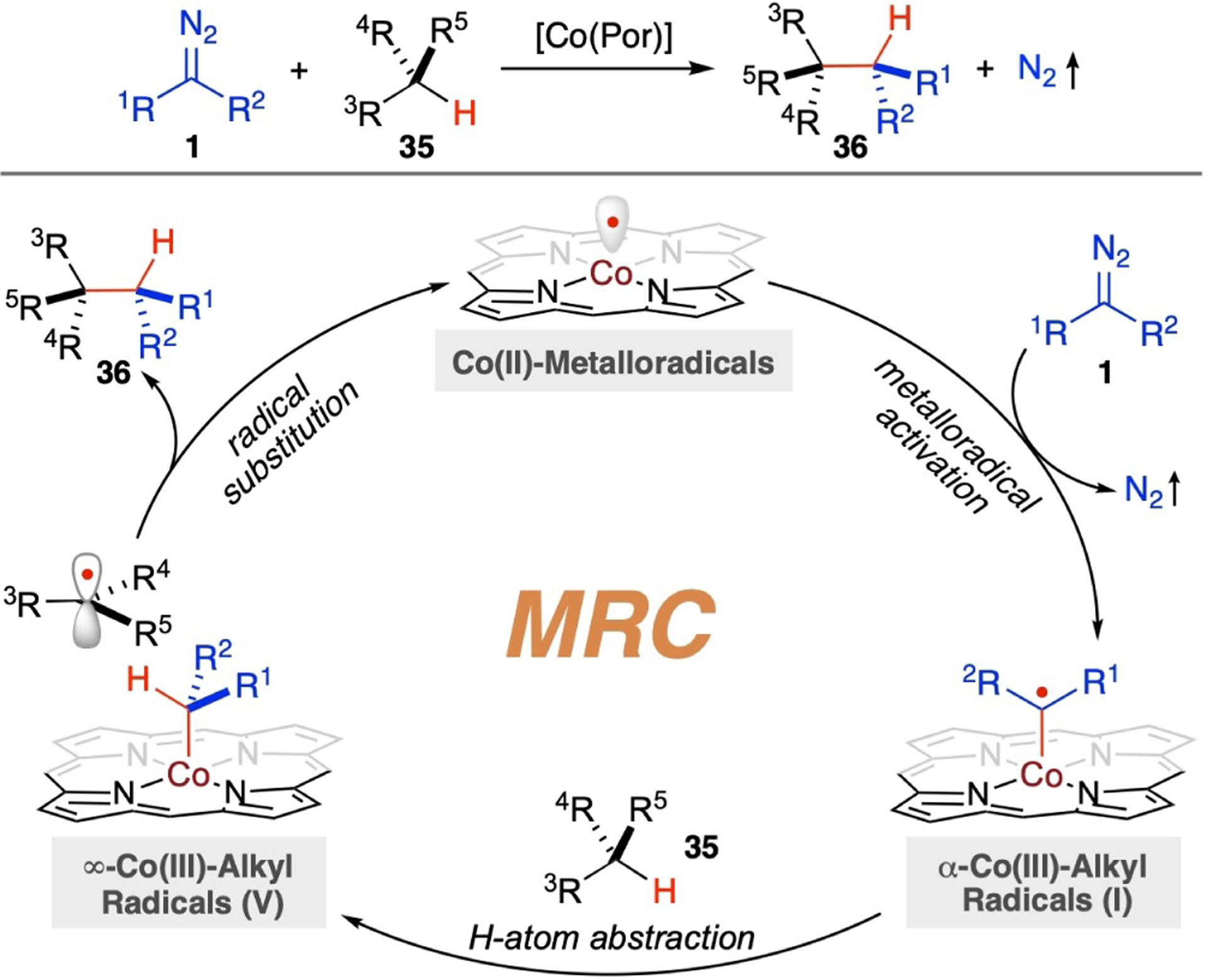
General mechanism for radical C–H alkylation via Co(II)-based MRC.
While metalloradical systems for intermolecular C–H alkylation are yet to be developed, Co(II)-catalyzed C–H alkylation of diazo compounds has proven successful in intramolecular reactions, enabling the stereoselective synthesis of a variety of cyclic compounds (Scheme 10). The [Co(D2-Por*)] complexes have been notably effective in catalyzing 1,5-C–H alkylation of acceptor/acceptor-substituted diazo compounds, delivering 5-membered sulfolane derivatives (37) in high yields with excellent stereoselectivities.[72] Similarly, aliphatic diazo compounds have been utilized as metalloradicophiles for intramolecular radical 1,5-C–H alkylation by [Co(D2-Por*)], leading to the effective construction of 2-substituted pyrrolidines (38), oxolanes (39), thiolanes (40), 3-substituted pyrrolidines (41) and cyclopentanes (42) with effective control of stereoselectivities.[32] Furthermore, [Co(D2-Por*)] have been shown to be equally effective in activating aryldiazomethanes as metalloradicophiles for intramolecular 1,5-C–H alkylation, furnishing chiral indolines (43) in high yields with high enantioselectivities.[34,73] Beyond five-membered rings, the Co(II)-based metalloradical system has also demonstrated the ability to construct six-membered structures through intramolecular C–H alkylation of donor-substituted diazo compounds via 1,6-H-atom abstraction. It has been reported that 6-membered cyclic compounds such as 1,2-dihydronaphthalenes (44)[74] and piperidines (45)[75] with various functionalities can be synthesized in good to high yields. In contrast, 1,4-C–H radical alkylation has been largely undeveloped due to the challenging nature of 1,4-H-atom abstraction, both entropically and enthalpically. However, recent advancement has shown that [Co(D2-Por*)] can effectively catalyze asymmetric 1,4-C–H alkylation of α-aryldiazoketones by facilitating both 1,4-H-atom abstraction and ensuing 4-exo-tet radical cyclization, leading to the high-yielding formation of cyclobutanones (46) with high diastereoselectivities and enantioselectivities.[27]
Scheme 10.
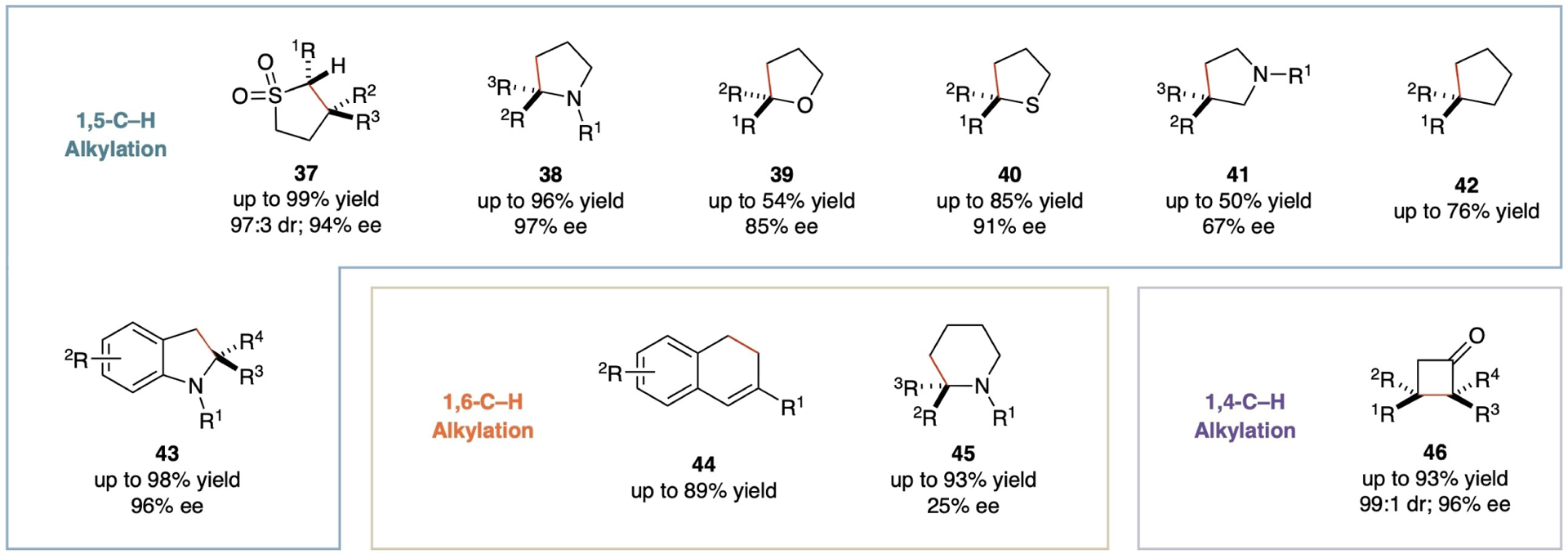
Selected examples of carbocyclic and heterocyclic compounds from catalytic intramolecular radical C–H alkylation via Co(II)-based MRC.
2.4. Radical Amination of C(sp3)–H Bonds
Similar to α-Co(III)-alkyl radical intermediate I in catalytic radical C–H alkylation (Scheme 9), α-Co(III)-aminyl radicals III, which arise from homolytic activation of organic azides 24 by Co(II)-based metalloradicals, can act as kinetically competent intermediates to undergo H-atom abstraction (HAA) from C(sp3)–H bonds in substrates 47, resulting in the generation of ∞-Co(III)-alkyl radicals VI as the second key metal-entangled radical intermediates (Scheme 11). Owing to the lower strength and higher polarity of the Co–N bond relative to the C–N bond, the subsequent radical substitution of the alkyl radical at the nitrogen atom in intermediate VI is both thermodynamically favorable and kinetically facile, leading to effective C–N bod formation to yield amination product 48 while regenerating the Co(II)-metalloradical catalyst. The stepwise radical pathway, marked by the sequential translocation of radical character from the Co-center to the N-center and then to the C-center, enables catalytic C–H amination with organic azides via Co(II)-MRC (Scheme 11). This method provides a general strategy for the direct transformation of the otherwise inert C(sp3)–H bonds in organic molecules into valuable amine functionalities, with nitrogen gas as the sole byproduct. Utilizing the support of D2-symmetric chiral amidoporphyrins, which offer tailored environments to optimize noncovalent attractive interactions, the Co(II)-based metalloradical system has the potential to control reactivity as well as various types of selectivities, including site-selectivity and enantioselectivity.
Scheme 11.
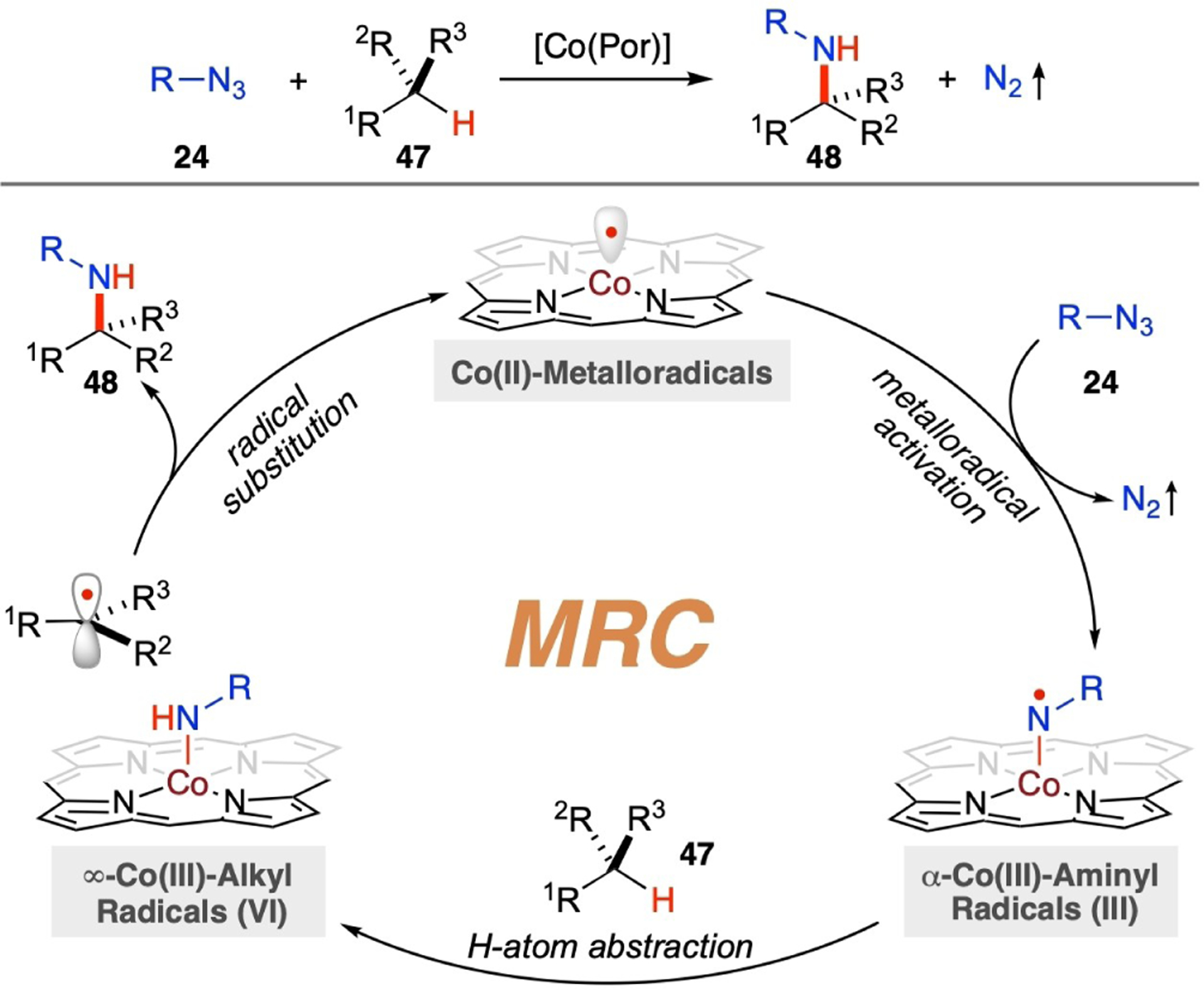
General mechanism for radical C–H amination via Co(II)-based MRC.
Both intramolecular and intermolecular radical C–H amination via Co(II)-MRC have been successfully implemented. In the intramolecular context, Co(II)-catalyzed C–H amination emerges as a potentially general approach for the stereoselective construction of N-heterocyclic compounds from organic azides (Scheme 12). The initial demonstration of this methodology involved intramolecular C–H amination using arylsulfonyl azides to synthesize 5-membered benzosultams.[76] Subsequently, phosphoryl azides have also been effectively activated by [Co(Por)] metalloradicals. This process facilitates intramolecular radical amination of C(sp3)–H bonds proceeding via 1,6- and 1,7-H-atom abstraction, leading to the construction of six-membered cyclophosphoramidates (49) and seven-membered cyclophosphoramidates (50) in excellent yields.[77] With the support of D2-symmetric chiral amidoporphyrin ligand platform, [Co(D2-Por*)] have proven to be highly effective in activating sulfamoyl azides for asymmetric intramolecular radical 1,6-amination of wide-ranging C(sp3)–H bonds, including benzylic, allylic, propargylic, and even electron-deficient C–H bonds, resulting in high-yielding formation of six-membered heterocycles 51–53 with excellent enantioselectivities.[35,78–81] These enantioenriched cyclic sulfamide compounds can be stereospecifically transformed into valuable chiral 1,3-diamines with the retention of the original enantiopurity. By taking advantage of facile radical racemization, the Co(II)-based metalloradical system with the support of a suitable D2-symmetric chiral amidoporphyrin ligand has been recently demonstrated with an unusual ability to catalyze enantioconvergent 1,6-C(sp3)–H amination of sulfamoyl azides containing racemic tertiary C-(sp3)–H bonds, giving rise to the stereoselective formation of six-membered chiral cyclic sulfamides (54) in excellent yields with high control of the newly-generated quaternary stereocenters.[23] Furthermore, with the support of new-generation HuPhyrin chiral ligand with tunable cavity environments, Co(II)-based MRC has enabled the development of effective catalytic systems for enantiodivergent radical 1,5-C–H amination of sulfamoyl azides.
Scheme 12.
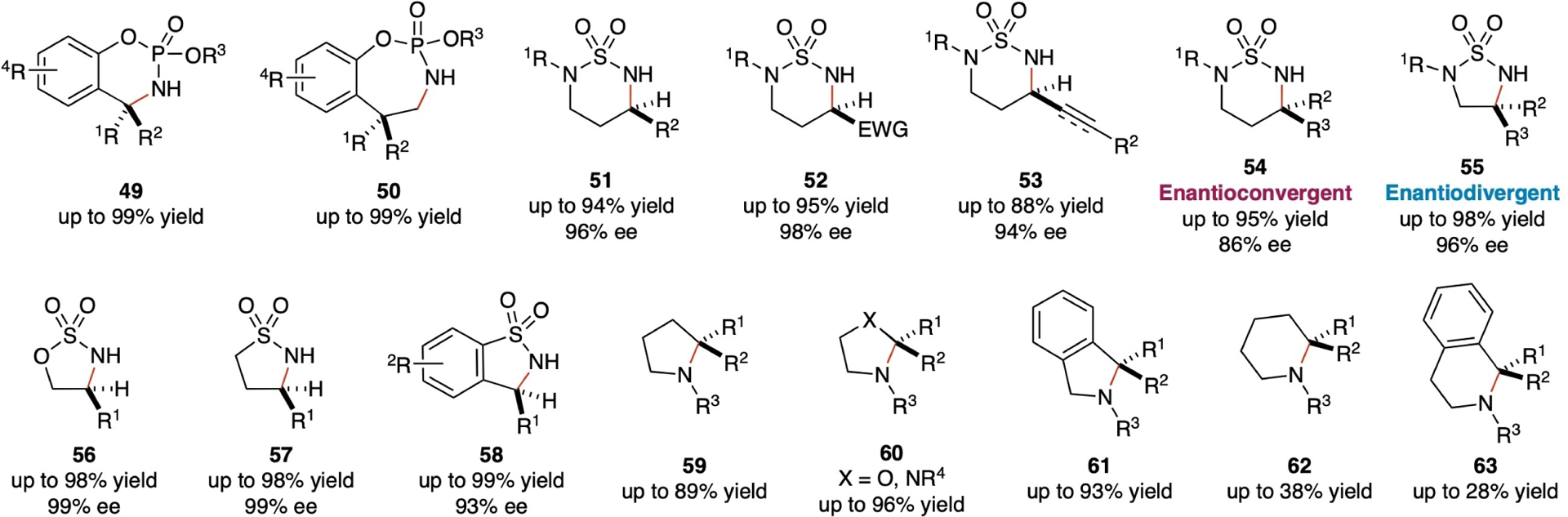
Selected examples of N-heterocyclic compounds from catalytic intramolecular radical C–H amination via Co(II)-based MRC.
Two optimal [Co(HuPhyin)] catalysts, which differ only by the length of the distal bridge and the position of the remote nonchiral substituents, have been identified as the optimal catalysts for the enantiodivergent amination, providing the access to both enantiomers of the strained five-membered cyclic sulfamides (55) in high yields with high enantioselectivities.[41,82] In addition to sulfamoyl azides, the Co(II)-based metalloradical system for 1,5-C(sp3)–H amination has been further expanded with the use of alkoxysulfonyl azides as the metalloradicophiles, leading to the high-yielding production of five-membered chiral cyclic sulfamidates (56) with excellent diastereoselectivities and enantioselectivities.[30] The resulting enantioenriched cyclic sulfamidates 56 can subsequently undergo stereospecific ring-opening reactions with various types of nucleophiles, providing a streamlined approach for the synthesis of diverse chiral amines bearing a wide range of β-functionalities from alcohols. As an effort to develop asymmetric version of the previously-reported system by [Co(TPP)],[76] it has been reported that [Co(D2-Por*)] are more effective metalloradical catalysts and can catalyze enantioselective radical 1,5-C–H amination of sulfonyl azides under milder conditions.[21] In addition to arylsulfonyl azides, the Co(II)-catalyzed asymmetric system is also applicable to the more challenging alkylsulfonyl azides with different types of C–H bonds, resulting in the productive formation of five-membered cyclic sulfonamides (57) and benzofused cyclic sulfonamides (58) with high enantioselectivities.[21] Additionally, Co(II)-based metalloradical system has demonstrated its capability to activate aliphatic azides for intramolecular 1,5-C–H amination and 1,6-C–H amination. This has led to the synthesis of a variety of N-heterocyclic compounds. Notable examples include the production of pyrrolidines (59), 1,3-oxazolidines (60), imidazolidines (60), isoindolines (61), piperidines (62), and tetrahydroisoquinolines (63), all of which have been synthesized in moderate to high yields.[83] Utilizing a chiral porphyrin as the supporting ligand, moderate enantioselectivities (up to 46% ee) were attained for the 1,5-C–H amination of a specific substrate.
Compared to its intramolecular counterpart, Co(II)-based intermolecular radical amination of C–H bonds remains less developed due to its inherent challenges in controlling reactivity and stereoselectivity in the intermolecular radical process. To date, there have been only a few reports on Co(II)-based nonasymmetric intermolecular C–H amination, and even fewer on their asymmetric variants (Scheme 13). It has been reported that both bromamine-T and arylsulfonyl azides can serve as suitable metalloradicophiles for [Co(Por)]-catalyzed intermolecular radical amination of benzylic C–H bonds, producing N-sulfonyl amines (64) in good yields.[84–85] Additionally, [Co(Por)] metalloradical catalysts have also demonstrated with the ability to activate carbonyl azides and aryl azides for amination of benzylic C–H bonds, delivering the corresponding amines (65 and 66) in moderate to high yields.[85–86] Furthermore, aryl azides have been shown to be viable metalloradicophiles for Co(II)-based intermolecular amination of allylic C–H bonds, leading to the formation of allylic amines (67) in good yields.[68] There have been two recent reports on Co(II)-based metalloradical systems for asymmetric intermolecular radical C–H amination. With the support of D2-symmetric chiral amidoporphyrin ligands, it has been shown that [Co(D2-Por*)] can homolytically activate fluoroaryl azides for enantioselective amination of benzylic C–H bonds, affording chiral amine derivatives (68) in high yields with high enantioselectivities.[22] The Co(II)-catalyzed intermolecular C–H amination, which operates under mild conditions with the C–H substrates as the limiting reagent, exhibits a broad substrate scope and high chemoselectivity. It offers an attractive methodology for stereoselective synthesis of chiral α-amino acid derivatives directly from readily available carboxylic esters.[22] More recently, metalloradical catalysts [Co(D2-Por*)] have been demonstrated to have the capability of catalyzing intermolecular allylic C–H amination with fluoroaryl azides.[37] The Co(II)-based metalloradical system can aminate allylic C–H bonds in a wide range of trisubstituted alkenes under mild conditions, leading to stereoselective synthesis of chiral allylic amines (69) such as α-tertiary amino acid derivatives. Besides chemoselectivity and regioselectivity, the Co(II)-based radical amination can concurrently control diastereoselectivity and enantioselectivity while enabling multiple stereochemical convergences at the same time. As a result, the catalytic C–H amination methodology can directly employ an isomeric mixture of alkenes as the substrates.[37] In addition to benzylic and allylic C–H bonds, Co(II)-based metalloradical system can also utilize fluoroaryl azides for intermolecular radical amination of aldehydic C(sp2)–H bonds in aldehydes (70), delivering the corresponding N-fluoroaryl amides (71) in good to excellent yields.[33]
Scheme 13.
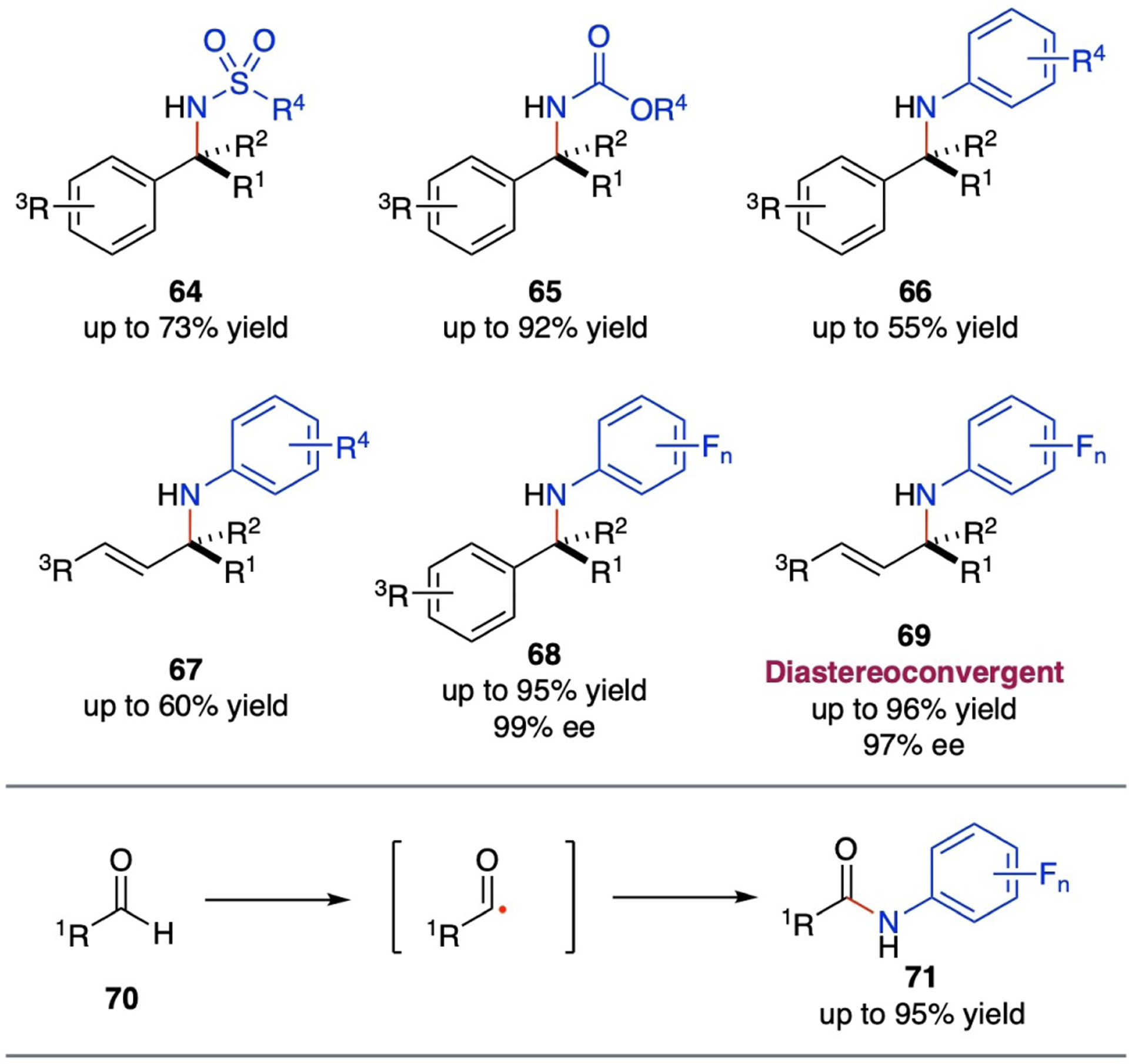
Selected examples of amine compounds from catalytic intermolecular radical C–H amination via Co(II)-based MRC.
2.5. Radical Cyclization Reactions
Radical cascade cyclization offers an attractive strategy for rapid construction of polycyclic structures through the efficient formation of multiple bonds in a single operation, all under mild conditions and with a broad tolerance for diverse functionalities. Although it is highly attractive to the synthesis of natural products and pharmaceutical compounds, radical cascade cyclization with the use of free organic radicals has encountered longstanding challenges with the control over reactivity and enantioselectivity. Through the introduction of metal-entangled organic radicals, such as α-Co(III)-alkyl radicals I and α-Co(III)-aminyl radicals III, as the controlling elements, Co(II)-based MRC has the potential to offer a general approach for controlling both reactivity and enantioselectivity as well as other types of selectivities in radical cascade cyclization. Indeed, Co(II)-based metalloradical system has already been successfully applied to radical cyclization reactions for the efficient construction of diverse cyclic compounds (Scheme 14). Significant examples include (i) the synthesis of polyfunctionalized furans (72) and 6H-furo[2,3-b]pyran-6-ones (73) from the reaction of alkynes with α-diazocarbonyls involving a sequence of tandem radical additions and subsequent radical β-scission;[87] (ii) the formation of 2H-chromenes (74) from the reaction of alkynes with in situ-generated α-(o-hydroxyaryl)diazomethanes involving a sequence of tandem radical additions and subsequent H-atom abstraction;[88] and (iii) the creation of indolizines (75) from the reaction of alkynes with in situ-generated α-heteroaryldiazomethanes involving a sequence of tandem radical additions and subsequent radical β-scission.[56,89] An exemplary case of asymmetric catalysis features the use of [Co(D2-Por*)] for catalyzing radical cascade cyclization of 1,6-enynes with α-cyanodiazoacetates.[26] This reaction proceeds under mild conditions and allows for stereoselective construction of multisubstituted cyclopropane-fused tetrahydrofurans (76 and 77). These products feature three contiguous stereogenic centers, including two quaternary carbon centers, and are formed in high yields with excellent control of both diastereoselectivities and enantioselectivities.[26] The Co(II)-based asymmetric cascade bicyclization is characterized by a sequence of tandem radical additions followed by radical β-scission, engaging a relay of multiple metal-entangled organic radical intermediates, including α-, β-, γ-, and ɛ-metalloalkyl radicals. Furthermore, Co(II)-based metalloradical system for radical cascade cyclization has been demonstrated as a powerful approach to the efficient synthesis of multifunctionalized eight-membered ring structures. Examples include dibenzocyclooctenes (78),[90] monobenzocyclooctadienes (79),[91] and 1H-benzo[c]oxocines (80),[92] all of which have been synthesized in good to excellent yields. These structures result from intramolecular cyclization reactions of α-aryldiazomethane derivatives generated in situ. The process encompasses a sequence of metalloradical activation, 1,6-H-atom abstraction, and radical substitution, demonstrating the system’s capability for intricate molecular construction.
Scheme 14.
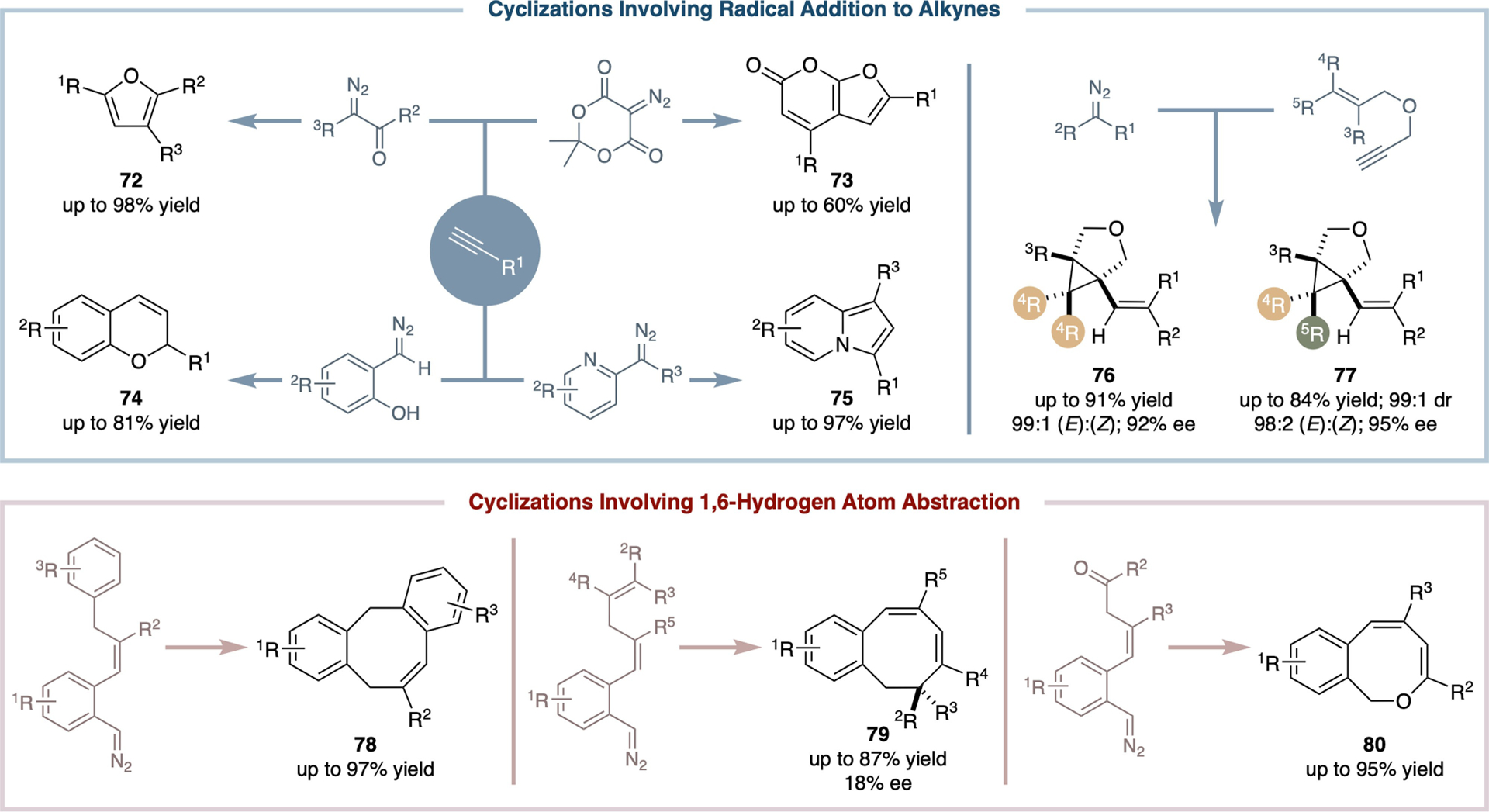
Selected examples of cyclic compounds from catalytic radical cyclization reactions via Co(II)-based MRC.
3. MRC by Iron Complexes of Porphyrins
Porphyrins are among the most versatile ligands known for complexation with transition metal ions. Metalloporphyrins, involving nearly every transition metal ion, have been synthesized and can stabilize these metal ions at various oxidation states with diverse electronic structures. This include the formation of open-shell metalloporphyrins that may function as metalloradicals. So far, Co(II) complexes of porphyrins have been at the forefront of advancements in MRC. To expand the scope of MRC beyond the use of Co(II)-based systems, there have been some recent progress on exploring other open-shell metalloporphyrins as metalloradical catalysts for controlling homolytic radical reactions. Particularly intriguing is the exploration of iron complexes of porphyrins as viable candidates for MRC. The attractiveness of iron stems not only from its affordability, low toxicity and high nature abundance but also from its ability to form open-shell complexes with porphyrins in various oxidation states, most notably in +2 (ferrous) and +3 (ferric) oxidation states.
3.1. Fe(II)-Based Metalloradical Catalysis
The Fe(II) complexes of porphyrins, characterized by their d6 electronic configuration, are known to exhibit multiple spin states, which are typically classified as high-spin, low-spin, or occasionally intermediate-spin state. The specific spin state of an Fe(II) complex of porphyrin depends on a range of factors, including the nature of the porphyrin ring, the strength of the ligand field, and the coordination environment of the iron ion. Due to their high sensitivity to air, Fe(II) complexes of porphyrins are often prepared in situ from their stable Fe(III) counterparts via reduction and directly used without isolation. Recent studies have demonstrated that Fe(II) complexes of porphyrins can function as effective metalloradical catalysts, facilitating organic transformations through a stepwise radical mechanism, in a manner similar to that of Co(II)-based MRC.
It has been shown that Fe(II) complexes of porphyrins, generated in situ from the reduction of Fe(III) complexes with zinc dust, can effectively activate 1,2,3,4-tetrazoles, which serve as the surrogates for organic azides, to generate the corresponding α-Fe(III)-aminyl radicals (Scheme 15).[93–95] These α-Fe(III)-aminyl radicals are capable of engaging radical addition to both C=C and C≡C bonds, as well as abstracting hydrogen atoms from a variety of C–H bonds, leading to catalytic cyclization and amination processes via a stepwise radical pathway (Scheme 16). For instance, the Fe(II)-based metalloradical system has been applied to intermolecular denitrogenative annulation of 1,2,3,4-tetrazoles with alkynes, where radical addition to C≡C bonds is a key step, giving rise to the general synthesis of functionalized imidazopyridines (81) in good to high yields.[96] Moreover, Fe(II)-based MRC has proven to be an effective method for catalytic intramolecular cyclization, enabling the high-yielding construction of multi-substituted α-carbolines (82),[97] indoles (83),[98] and 7-azaindoles (83),[98] respectively, through a stepwise radical mechanism. Beyond radical cyclization, the Fe(II)-based metalloradical system has shown considerable versatility for both intramolecular and intermolecular radical amination of C–H bonds. With the support of porphyrin ligands, the in situ-generated Fe(II) catalysts can effectively activate and facilitate intramolecular radical 1,5-C–H amination of 2-azidopyridine derivatives that contain different types of C–H bonds, including primary, secondary, and tertiary C–H bonds as well as benzylic C–H bonds, resulting in the formation of 7-azaindolines (84) in generally high yields.[99] The Fe(II)-based metalloradical system has also been applied to intramolecular radical 1,6-C–H amination, as exemplified by the efficient production of six-membered heterocyclic structures 85–87 in high yields.[97–99] An instance of intramolecular radical 1,7-C–H amination has been reported, offering a streamlined approach for the synthesis of seven-membered heterocycle 88 in a promising yield.[97] Further expanding its scope, the Fe(II)-based metalloradical system has been explored to activate 1,2,3,4-tetrazoles for catalyzing intermolecular radical amination of benzylic C–H bonds. This approach enables the synthesis of 2-pyridine-substituted benzylamines (89) in good to high yields,[100] underscoring the adaptability and potency of Fe(II)-based MRC for diverse C–H amination reactions.
Scheme 15.
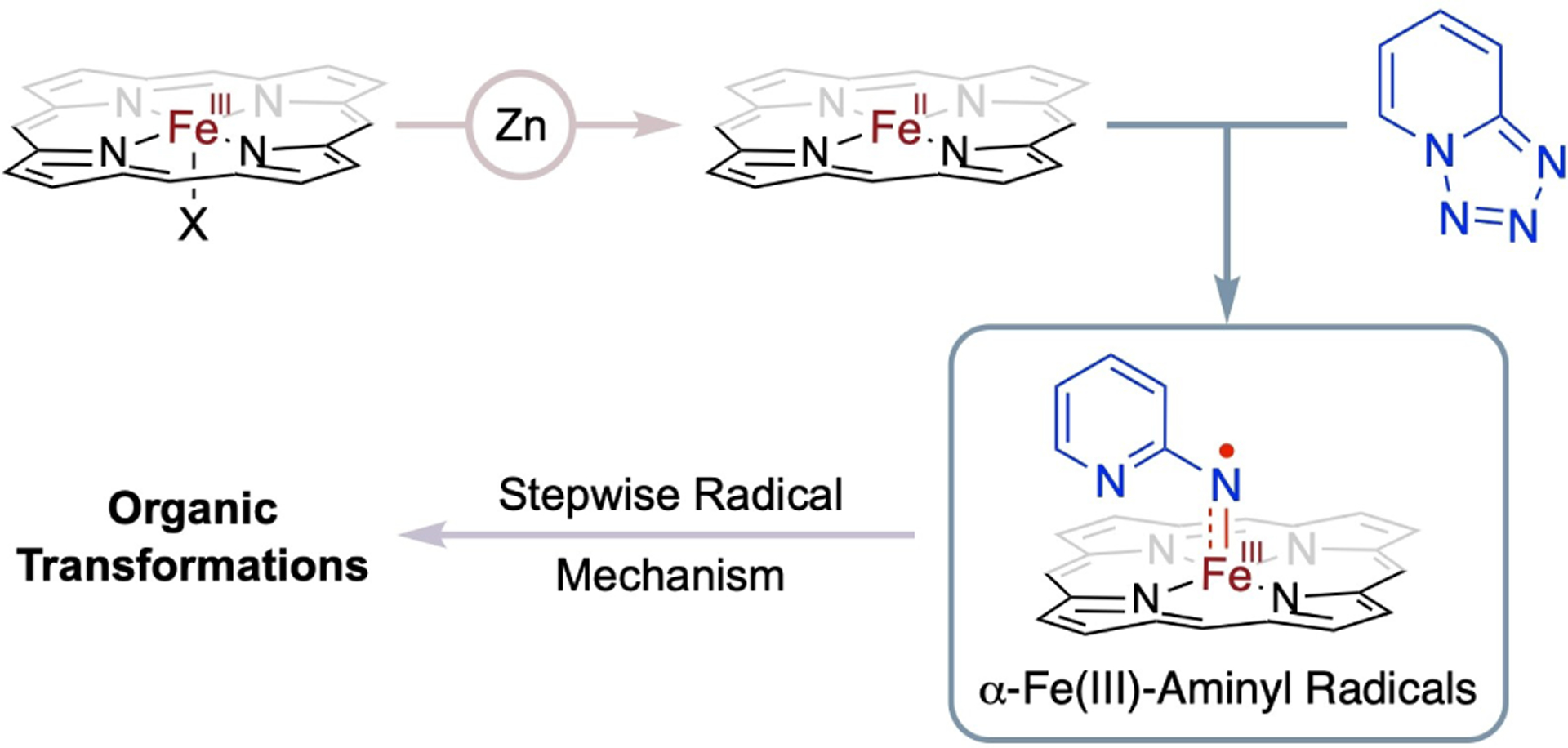
Fe(II)-based MRC involving α-Fe(III)-aminyl radicals generated from activation of tetrazoles by in situ-reduced Fe(II) complexes of porphyrins.
Scheme 16.
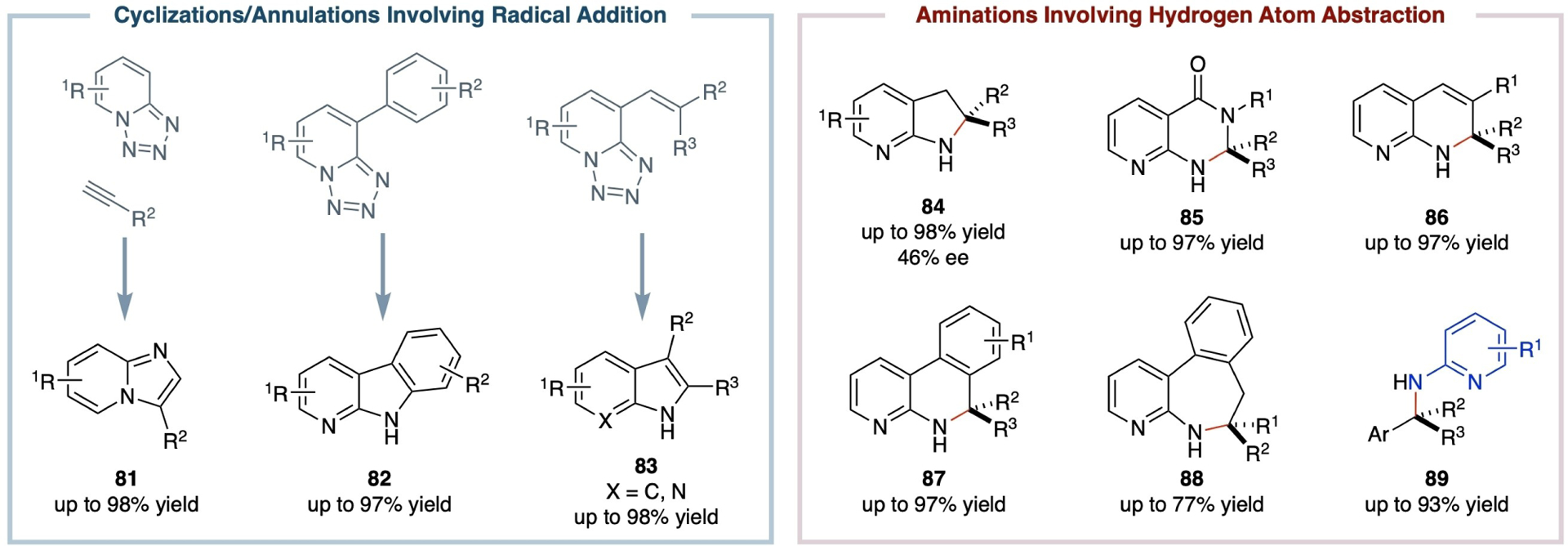
Selected examples of N-heterocyclic and amine compounds from catalytic radical cyclization reactions via Fe(II)-based MRC.
3.2. Fe(III)-Based Metalloradical Catalysis
With their d5 electronic configuration, five-coordinate Fe-(III) complexes of porphyrins with an axial ligand X, denoted as [Fe(Por)X], constitute a distinct family of stable 15e-metalloradicals. These Fe(III) complexes can exist in multiple spin states, ranging from low-spin to intermediate-spin to high-spin states. Until quite recently, [Fe(Por)X] complexes have not been definitely recognized as true catalysts in catalytic transformations. It was commonly assumed that [Fe(Por)X] complexes would undergo in situ reduction to the four-coordinate Fe(II) complexes [Fe(Por)], which would then act as the active catalysts, irrespective of the presence of a reducing agent. However, recent studies have revealed that [Fe(Por)X] complexes can indeed function as potent metalloradical catalysts for radical cyclopropanation of alkenes. This marks the first establishment for the operation of Fe(III)-based MRC (Scheme 17).[101] Supported by an optimal D2-symmetric chiral amidoporphyrin ligand, the Fe(III)-based metalloradical system operates under mild conditions and can effect homolytic activation of in situ-generated α-trifluoromethyldiazomethane. This enables the cyclopropanation of a wide range of alkenes, forming trifluoromethyl-substituted cyclopropanes in high yields with both high diastereoselectivities and enantioselectivities. Furthermore, this Fe(III)-based metalloradical system has been shown to be broadly effective in activating various classes of diazo compounds for asymmetric radical cyclopropanation. This Fe(III)-based catalytic system presents a potentially general and practically sustainable methodology for stereoselective synthesis of the valuable three-membered cyclopropanes. Through experimental investigations, complemented by detailed computational studies, have shed light on the details of the underlying stepwise radical mechanism. This mechanism features two unprecedented metal-entangled organic radicals as the key intermediates: α-Fe(IV)-alkyl radicals (VII) and γ-Fe(IV)-alkyl radicals (VIII) (Scheme 17). This work has laid a solid foundation and provided a mechanistic framework for the future exploration of new stereoselective radical transformations via Fe(III)-based MRC.
Scheme 17.
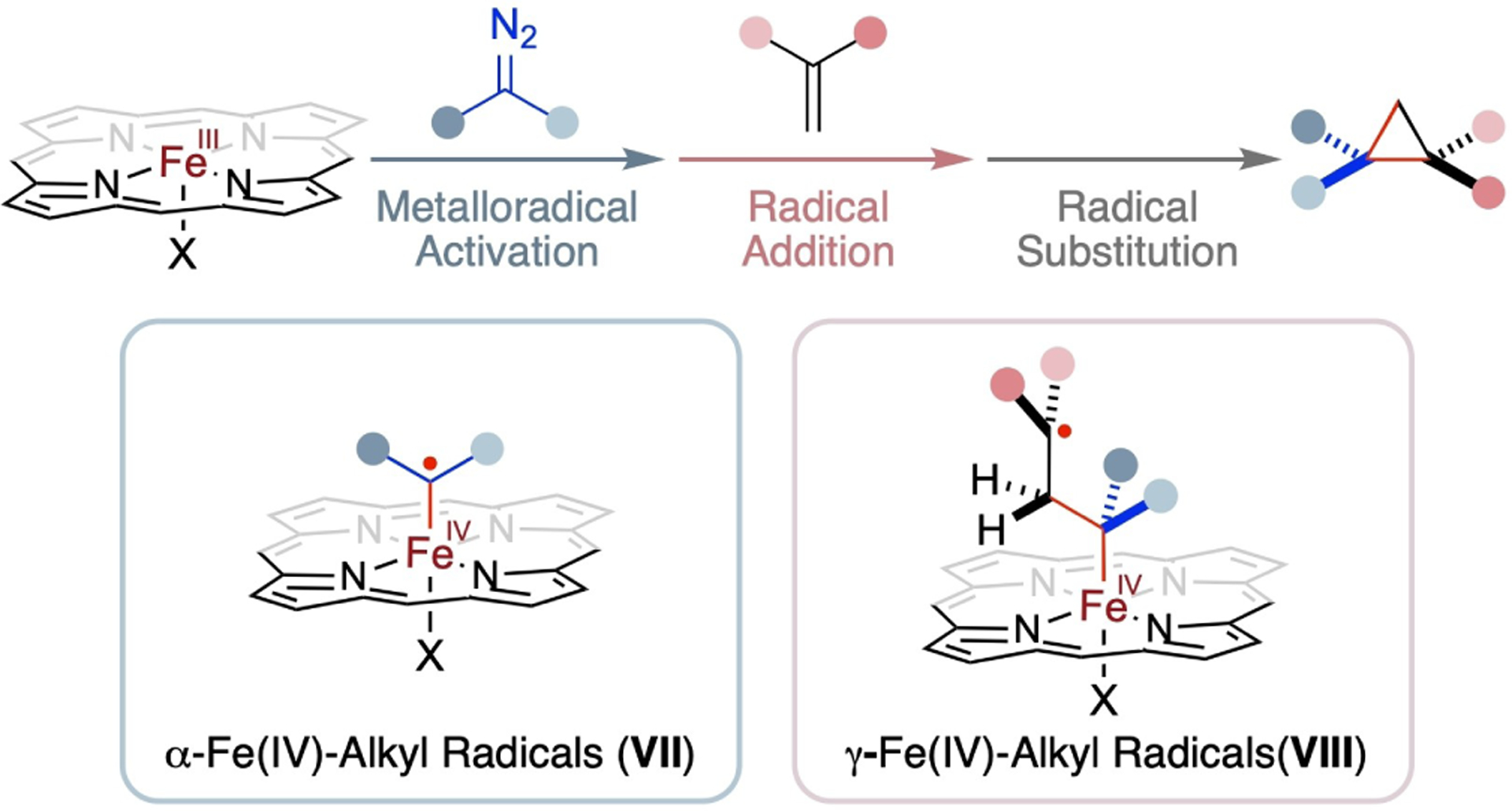
Radical cyclopropanation of alkenes with diazo compounds via Fe(III)-based MRC involving α-Fe(IV)-alkyl radicals and γ-Fe(IV)-alkyl radicals.
4. MRC by Other Metalloradical Catalysts
In principle, any open-shell transition metal complex with unpaired d-electron(s) has the potential to function as a metalloradical catalyst, promoting homolytic radical reactions via metalloradical catalysis. Beyond the above well-characterized examples of MRC using Co(II), Fe(II), and Fe(III) complexes of porphyrins, there exist other catalytic systems that may also be mechanistically classified as metalloradical catalysis or as a hybrid of metalloradical catalysis and traditional metal catalysis. Notable examples include (i) Ti(III)-based catalytic system that has been employed for radical transformations involving epoxides,[102–112] cyclopropanes,[113–114] and aziridines;[115] (ii) Cu(I)-based catalytic system that has found applications in asymmetric radical cross-couplings,[116–118] alkene difunctionalization,[119–120] and C–H functionalization;[121–123] (iii) Ni(I)-based catalytic system that has been utilized in enantioselective radical cross-couplings of alkyl halides;[124–125] and (iv) Fe(III)-based catalytic system that has been applied in radical C–H amination of organic azides.[126] Moreover, well-characterized metalloradicals such as Rh(II) complexes of porphyrins show great promise as potential metalloradical catalysts that may lead to the discovery of new types of catalytic radical transformations.[127–130] These examples highlight the extensive scope and adaptability of MRC in modern chemical research.
5. Summary and Outlook
Metalloradical catalysis (MRC) has emerged as a general approach for controlling the reactivity and selectivity of homolytic radical reactions. By employing metalloradicals as one-electron catalysts, MRC offers a strategic framework to harness the vast potential of homolytic radical chemistry in the realm of organic synthesis. Leading the way with Co(II) complexes of porphyrins, several well-characterized metalloradical systems have been developed, enabling the facilitation of homolytic radical reactions via MRC. Among various substrates, diazo compounds and organic azides have been recognized as potent carbon and nitrogen metalloradicophiles, respectively, which can be homolytically activated by metalloradical catalysts. This metalloradical activation results in the formation of α-metalloalkyl radicals and α-metalloaminyl radicals. As pivotal intermediates in MRC, these metal-entangled organic radicals are finely modulated and precisely governed by the metal center and its surrounding ligand framework, while retaining the kinetic competence for participating in standard radical reactions. This dynamic allows for catalytic radical processes with enhanced control over reactivity and selectivity. Supported by chiral ligands, such as D2-symmetric chiral amidoporphyrins that provide specifically tailored steric, electronic and chiral environments, asymmetric metalloradical catalytic systems have been successfully developed, enabling diverse organic transformations, including olefin cyclopropanation, olefin aziridination, C–H alkylation, C–H amination, and different types of cyclization reactions. Owing to their distinct stepwise radical mechanism, these metalloradical-based catalytic processes exhibit desirable reactivity and selectivity profiles characteristic of homolytic radical reactions, thereby greatly enhancing the scope and efficiency of organic synthesis.
Despite its considerable progress, MRC is still in its formative stages of development, reminiscent of the early days of traditional metal catalysis in the 1950s and 1960s. The future of MRC is filled with exciting challenges, immense promise and untapped potential. New catalytic transformations involving α-metalloalkyl radicals and α-metalloaminyl radicals as the key intermediates are on the horizon, propelled by the effective metalloradical catalysts based on Co(II), Fe(II) and Fe(III) complexes of porphyrins. These advancements are anticipated to arise from continuous innovation in catalyst design, particularly through the engineering of tailored ligands. A critical area for exploration will be the identification of potent oxygen metalloradicophiles that can be effectively activated by metalloradical catalysts. This will pave the way for the routine generation of the as-yet-uncharacterized α-metalloxyl radicals, leading to asymmetric radical processes like olefin epoxidation, C–H hydroxylation, and other related reactions. The development of innovative metalloradical catalysts, supported by diverse ligands and based on other metal ions, is likely to unveil new modes of metalloradical activation, expanding the range of metalloradicophiles for MRC. This expansion will give rise to metal-entangled organic radicals beyond α-metalloalkyl, α-metalloaminyl, and α-metalloxyl radicals, unlocking a vast territory of research exploration. In addition to broadening applicability of MRC in diverse organic transformations, the continued developments in MRC are poised to uncover transformative reactions that could redefine the state-of-the-art in organic synthesis.
Acknowledgements
We are grateful for financial support by NIH (R01-GM132471 and R01-GM102554) and NSF (CHE-2154885). We also thank the financial support by NIH (S10-OD026910) and NSF (CHE-2117246) for the purchases of NMR spectrometers at Magnetic Resonance Center of Boston College and by NIH (S10-OD030360) for the purchase of X-ray single crystal diffractometer at X-ray Crystallography Center of Boston College.
Biographies

Wan-Chen Cindy Lee hails from Taiwan and completed her Ph.D. study at Boston College under the supervision of Professor Peter Zhang. During her graduate studies, Dr. Lee developed Co(II)-based catalytic systems for enantioselective radical cyclization and bicyclization reactions. She also conducted extensive mechanistic investigation on Fe(III)-based metalloradical catalysis and its synthetic applications for stereoselective construction of organic molecules. She is currently pursuing postdoctoral research at Cornell University. Dr. Lee’s research revolves around organic synthesis with a particular emphasis on asymmetric catalysis.

Peter Zhang received his Ph.D. from the University of Pennsylvania and pursued postdoctoral research at Massachusetts Institute of Technology. He started his independent career at the University of Tennessee in 2001 and later transitioned to the University of South Florida in 2006. In 2015, Dr. Zhang joined Boston College. His research has been dedicated to formulating metalloradical catalysis (MRC) as a fundamental concept and general strategy to guide the development of metalloradical systems for controlling the reactivity and selectivity of homolytic radical reactions and exploring their applications in organic synthesis.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- [1].Curran DP, Porter NA, Giese B, Stereochemistry of Radical Reactions: Concepts, Guidelines, and Synthetic Applications, John Wiley & Sons, Weinheim; New York, 2008. [Google Scholar]
- [2].Mondal S, Dumur F, Gigmes D, Sibi MP, Bertrand MP, Nechab M, Chem. Rev 2022, 122, 5842–5976. [DOI] [PubMed] [Google Scholar]
- [3].Lu HJ, Zhang XP, Chem. Soc. Rev 2011, 40, 1899–1909. [DOI] [PubMed] [Google Scholar]
- [4].Lee W-CC, Zhang XP, Trends Chem 2022, 4, 850–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wang X, Zhang XP, Catalytic Radical Approach for Selective Carbene Transfers via Cobalt(II)-Based Metalloradical Catalysis, in Transition Metal-Catalyzed Carbene Transformations, Wiley, 2022, pp. 25–66. [Google Scholar]
- [6].Lee W-CC, Zhang XP, Trends Chem. 2024, 6, 95–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Thompson SJ, Brennan MR, Lee SY, Dong GB, Chem. Soc. Rev 2018, 47, 929–981. [DOI] [PubMed] [Google Scholar]
- [8].Wayland BB, Newman AR, J. Am. Chem. Soc 1979, 101, 6472–6473. [Google Scholar]
- [9].Sherry AE, Wayland BB, J. Am. Chem. Soc 1989, 111, 5010–5012. [Google Scholar]
- [10].Ogoshi H, Setsune J, Yoshida Z, J. Am. Chem. Soc 1977, 99, 3869–3870. [DOI] [PubMed] [Google Scholar]
- [11].Paonessa RS, Thomas NC, Halpern J, J. Am. Chem. Soc 1985, 107, 4333–4335. [Google Scholar]
- [12].Wayland BB, Feng YP, Ba SJ, Organometallics 1989, 8, 1438–1441. [Google Scholar]
- [13].Wayland BB, Ba S, Sherry AE, Inorg. Chem 1992, 31, 148–150. [Google Scholar]
- [14].Sherry AE, Wayland BB, J. Am. Chem. Soc 1990, 112, 1259–1261. [Google Scholar]
- [15].Wayland BB, Ba S, Sherry AE, J. Am. Chem. Soc 1991, 113, 5305–5311. [Google Scholar]
- [16].The term “metalloradical catalysis”, its abbreviation “MRC”, and the associated term “metalloradical catalyst” were initially introduced in the following publication: Cui X, Xu X, Lu HJ, Zhu SF, Wojtas L, Zhang XP, J. Am. Chem. Soc 2011, 133, 3304–3307. [DOI] [PubMed] [Google Scholar]
- [17].Wayland BB, Sherry AE, Bunn AG, J. Am. Chem. Soc 1993, 115, 7675–7684. [Google Scholar]
- [18].Dzik WI, Xu X, Zhang XP, Reek JNH, de Bruin B, J. Am. Chem. Soc 2010, 132, 10891–10902. [DOI] [PubMed] [Google Scholar]
- [19].Lyaskovskyy V, Suarez AIO, Lu HJ, Jiang HL, Zhang XP, de Bruin B, J. Am. Chem. Soc 2011, 133, 12264–12273. [DOI] [PubMed] [Google Scholar]
- [20].Goswami M, Lyaskovskyy V, Domingos SR, Buma WJ, Woutersen S, Troeppner O, Ivanovic-Burmazovic I, Lu HJ, Cui X, Zhang XP, Reijerse EJ, DeBeer S, van Schooneveld MM, Pfaff FF, Ray K, de Bruin B, J. Am. Chem. Soc 2015, 137, 5468–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hu Y, Lang K, Li CQ, Gill JB, Kim I, Lu HJ, Fields KB, Marshall M, Cheng QG, Cui X, Wojtas L, Zhang XP, J. Am. Chem. Soc 2019, 141, 18160–18169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jin LM, Xu P, Xie JJ, Zhang XP, J. Am. Chem. Soc 2020, 142, 20828–20836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lang K, Li CQ, Kim I, Zhang XP, J. Am. Chem. Soc 2020, 142, 20902–20911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Riart-Ferrer X, Sang P, Tao JR, Xu H, Jin LM, Lu HJ, Cui X, Wojtas L, Zhang XP, Chem 2021, 7, 1120–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lee W-CC, Wang DS, Zhang CZ, Xie JJ, Li B, Zhang XP, Chem 2021, 7, 1588–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang CZ, Wang DS, Lee W-CC, McKillop AM, Zhang XP, J. Am. Chem. Soc 2021, 143, 11130–11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xie JJ, Xu P, Zhu YL, Wang JY, Lee W-CC, Zhang XP, J. Am. Chem. Soc 2021, 143, 11670–11678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ke J, Lee W-CC, Wang XX, Wang Y, Wen X, Zhang XP, J. Am. Chem. Soc 2022, 144, 2368–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang JY, Xie JJ, Lee W-CC, Wang DS, Zhang XP, Chem Catal. 2022, 2, 330–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lang K, Hu Y, Lee W-CC, Zhang XP, Nat. Syn 2022, 1, 548–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lee W-CC, Wang JY, Zhu YL, Zhang XP, J. Am. Chem. Soc 2023, 145, 11622–11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang Y, Wen X, Cui X, Zhang XP, J. Am. Chem. Soc 2018, 140, 4792–4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jin LM, Lu HJ, Cui Y, Lizardi CL, Arzua TN, Wojtas L, Cui X, Zhang XP, Chem. Sci 2014, 5, 2422–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wen X, Wang Y, Zhang XP, Chem. Sci 2018, 9, 5082–5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Li CQ, Lang K, Lu HJ, Hu Y, Cui X, Wojtas L, Zhang XP, Angew. Chem. Int. Ed 2018, 57, 16837–16841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang XX, Ke J, Zhu YL, Deb A, Xu YJ, Zhang XP, J. Am. Chem. Soc 2021, 143, 11121–11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Xu P, Xie J, Wang DS, Zhang XP, Nat. Chem 2023, 15, 498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lu HJ, Dzik WI, Xu X, Wojtas L, de Bruin B, Zhang XP, J. Am. Chem. Soc 2011, 133, 8518–8521. [DOI] [PubMed] [Google Scholar]
- [39].Belof JL, Cioce CR, Xu X, Zhang XP, Space B, Woodcock HL, Organometallics 2011, 30, 2739–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Suarez AIO, Jiang HL, Zhang XP, de Bruin B, Dalton Trans. 2011, 40, 5697–5705. [DOI] [PubMed] [Google Scholar]
- [41].Lang K, Torker S, Wojtas L, Zhang XP, J. Am. Chem. Soc 2019, 141, 12388–12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chen Y, Fields KB, Zhang XP, J. Am. Chem. Soc 2004, 126, 14718–14719. [DOI] [PubMed] [Google Scholar]
- [43].Zhu SF, Ruppel JV, Lu HJ, Wojtas L, Zhang XP, J. Am. Chem. Soc 2008, 130, 5042–5043. [DOI] [PubMed] [Google Scholar]
- [44].Xu X, Lu HJ, Ruppel JV, Cui X, de Mesa SL, Wojtas L, Zhang XP, J. Am. Chem. Soc 2011, 133, 15292–15295. [DOI] [PubMed] [Google Scholar]
- [45].Hu Y, Lang K, Tao JR, Marshall MK, Cheng QG, Cui X, Wojtas L, Zhang XP, Angew. Chem. Int. Ed 2019, 58, 2670–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chen Y, Zhang XP, J. Org. Chem 2007, 72, 5931–5934. [DOI] [PubMed] [Google Scholar]
- [47].Chen Y, Ruppel JV, Zhang XP, J. Am. Chem. Soc 2007, 129, 12074–12075. [DOI] [PubMed] [Google Scholar]
- [48].Penoni A, Wanke R, Tollari S, Gallo E, Musella D, Ragaini F, Demartin F, Cenini S, Eur. J. Inorg. Chem 2003, 1452–1460. [Google Scholar]
- [49].Fantauzzi S, Gallo E, Rose E, Raoul N, Caselli A, Issa S, Ragaini F, Cenini S, Organometallics 2008, 27, 6143–6151. [Google Scholar]
- [50].Ruppel JV, Gauthier TJ, Snyder NL, Perman JA, Zhang XP, Org. Lett 2009, 11, 2273–2276. [DOI] [PubMed] [Google Scholar]
- [51].Zhu SF, Perman JA, Zhang XP, Angew. Chem. Int. Ed 2008, 47, 8460–8463. [DOI] [PubMed] [Google Scholar]
- [52].Zhu SF, Xu X, Perman JA, Zhang XP, J. Am. Chem. Soc 2010, 132, 12796–12799. [DOI] [PubMed] [Google Scholar]
- [53].Xu X, Zhu SF, Cui X, Wojtas L, Zhang XP, Angew. Chem. Int. Ed 2013, 52, 11857–11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Xu X, Wang Y, Cui X, Wojtas L, Zhang XP, Chem. Sci 2017, 8, 4347–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wang Y, Wen X, Cui X, Wojtas L, Zhang XP, J. Am. Chem. Soc 2017, 139, 1049–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Roy S, Das SK, Chattopadhyay B, Angew. Chem. Int. Ed 2018, 57, 2238–2243. [DOI] [PubMed] [Google Scholar]
- [57].Ruppel JV, Cui X, Xu X, Zhang XP, Org. Chem. Front 2014, 1, 515–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Reddy AR, Hao F, Wu K, Zhou CY, Che CM, Angew. Chem. Int. Ed 2016, 55, 1810–1815. [DOI] [PubMed] [Google Scholar]
- [59].Wang HX, Zhou CY, Che CM, Adv. Synth. Catal 2017, 359, 2253–2258. [Google Scholar]
- [60].Ruppel JV, Jones JE, Huff CA, Kamble RM, Chen Y, Zhang XP, Org. Lett 2008, 10, 1995–1998. [DOI] [PubMed] [Google Scholar]
- [61].Gao GY, Harden JD, Zhang XP, Org. Lett 2005, 7, 3191–3193. [DOI] [PubMed] [Google Scholar]
- [62].Subbarayan V, Ruppel JV, Zhu S, Perman JA, Zhang XP, Chem. Commun 2009, 4266–4268. [DOI] [PubMed] [Google Scholar]
- [63].Subbarayan V, Jin LM, Cui X, Zhang XP, Tetrahedron Lett. 2015, 56, 3431–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Gao GY, Jones JE, Vyas R, Harden JD, Zhang XP, J. Org. Chem 2006, 71, 6655–6658. [DOI] [PubMed] [Google Scholar]
- [65].Jones JE, Ruppel JV, Gao GY, Moore TM, Zhang XP, J. Org. Chem 2008, 73, 7260–7265. [DOI] [PubMed] [Google Scholar]
- [66].Tao JR, Jin LM, Zhang XP, Beilstein J Org. Chem 2014, 10, 1282–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Jin LM, Xu X, Lu HJ, Cui X, Wojtas L, Zhang XP, Angew. Chem. Int. Ed 2013, 52, 5309–5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Caselli A, Gallo E, Fantauzzi S, Morlacchi S, Ragaini F, Cenini S, Eur. J. Inorg. Chem 2008, 3009–3019. [Google Scholar]
- [69].Jiang HL, Lang K, Lu HJ, Wojtas L, Zhang XP, Angew. Chem. Int. Ed 2016, 55, 11604–11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Xu H, Wang D-S, Zhu Z, Deb A, Zhang XP, Chem 2024, 10, 283–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Jiang HL, Lang K, Lu HJ, Wojtas L, Zhang XP, J. Am. Chem. Soc 2017, 139, 9164–9167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Cui X, Xu X, Jin LM, Wojtas L, Zhang XP, Chem. Sci 2015, 6, 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Karns AS, Goswami M, de Bruin B, Chem. Eur. J 2018, 24, 5253–5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].te Grotenhuis C, Das BG, Kuijpers PF, Hageman W, Trouwborst M, de Bruin B, Chem. Sci 2017, 8, 8221–8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lankelma M, Olivares AM, de Bruin B, Chem. Eur. J 2019, 25, 5658–5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ruppel JV, Kamble RM, Zhang XP, Org. Lett 2007, 9, 4889–4892. [DOI] [PubMed] [Google Scholar]
- [77].Lu HJ, Tao JR, Jones JE, Wojtas L, Zhang XP, Org. Lett 2010, 12, 1248–1251. [DOI] [PubMed] [Google Scholar]
- [78].Lu HJ, Jiang HL, Wojtas L, Zhang XP, Angew. Chem. Int. Ed 2010, 49, 10192–10196. [DOI] [PubMed] [Google Scholar]
- [79].Lu HJ, Jiang HL, Hu Y, Wojtas L, Zhang XP, Chem. Sci 2011, 2, 2361–2366. [Google Scholar]
- [80].Lu HJ, Hu Y, Jiang HL, Wojtas L, Zhang XP, Org. Lett 2012, 14, 5158–5161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lu HJ, Li CQ, Jiang HL, Lizardi CL, Zhang XP, Angew. Chem. Int. Ed 2014, 53, 7028–7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Lu HJ, Lang K, Jiang HL, Wojtas L, Zhang XP, Chem. Sci 2016, 7, 6934–6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kuijpers PF, Tiekink MJ, Breukelaar WB, Broere DLJ, van Leest NP, van der Vlugt JI, Reek JNH, de Bruin B, Chem. Eur. J 2017, 23, 7945–7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Harden JD, Ruppel JV, Gao GY, Zhang XP, Chem. Commun 2007, 4644–4646. [DOI] [PubMed] [Google Scholar]
- [85].Lu HJ, Subbarayan V, Tao JR, Zhang XP, Organometallics 2010, 29, 389–393. [Google Scholar]
- [86].Ragaini F, Penoni A, Gallo E, Tollari S, Gotti CL, Lapadula M, Mangioni E, Cenini S, Chem. Eur. J 2003, 9, 249–259. [DOI] [PubMed] [Google Scholar]
- [87].Cui X, Xu X, Wojtas L, Kim MM, Zhang XP, J. Am. Chem. Soc 2012, 134, 19981–19984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Paul ND, Mandal S, Otte M, Cui X, Zhang XP, de Bruin B, J. Am. Chem. Soc 2014, 136, 1090–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Zhang ZY, Gevorgyan V, Org. Lett 2020, 22, 8500–8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].te Grotenhuis C, van den Heuvel N, van der Vlugt JI, de Bruin B, Angew. Chem. Int. Ed 2018, 57, 140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Zhou MH, Lankelma M, van der Vlugt JI, de Bruin B, Angew. Chem. Int. Ed 2020, 59, 11073–11079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Zhou MH, Wolzak LA, Li ZR, de Zwart FJ, Mathew S, de Bruin B, J. Am. Chem. Soc 2021, 143, 20501–20512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Roy S, Das SK, Khatua H, Das S, Chattopadhyay B, Acc. Chem. Res 2021, 54, 4395–4409. [DOI] [PubMed] [Google Scholar]
- [94].Das SK, Roy S, Chattopadhyay B, Angew. Chem. Int. Ed 2023, 62, e202210912. [DOI] [PubMed] [Google Scholar]
- [95].Das S, Ehlers AW, Patra S, de Bruin B, Chattopadhyay B, J. Am. Chem. Soc 2023, 145, 14599–14607. [DOI] [PubMed] [Google Scholar]
- [96].Roy S, Khatua H, Das SK, Chattopadhyay B, Angew. Chem. Int. Ed 2019, 58, 11439–11443. [DOI] [PubMed] [Google Scholar]
- [97].Das SK, Das S, Ghosh S, Roy S, Pareek M, Roy B, Sunoj RB, Chattopadhyay B, Chem. Sci 2022, 13, 11817–11828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Roy S, Das SK, Khatua H, Das S, Singh KN, Chattopadhyay B, Angew. Chem. Int. Ed 2021, 60, 8772–8780. [DOI] [PubMed] [Google Scholar]
- [99].Das SK, Roy S, Khatua H, Chattopadhyay B, J. Am. Chem. Soc 2020, 142, 16211–16217. [DOI] [PubMed] [Google Scholar]
- [100].Khatua H, Das S, Patra S, Das SK, Roy S, Chattopadhyay B, J. Am. Chem. Soc 2022, 144, 21858–21866. [DOI] [PubMed] [Google Scholar]
- [101].Lee W-CC, Wang DS, Zhu Y, Zhang XP, Nat. Chem 2023, 15, 1569–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Nugent WA, Rajanbabu TV, J. Am. Chem. Soc 1988, 110, 8561–8562. [Google Scholar]
- [103].Rajanbabu TV, Nugent WA, J. Am. Chem. Soc 1994, 116, 986–997. [Google Scholar]
- [104].Gansäuer A, Pierobon M, Bluhm H, Angew. Chem. Int. Ed 1998, 37, 101–103. [Google Scholar]
- [105].Gansäuer A, Lauterbach T, Bluhm H, Noltemeyer M, Angew. Chem. Int. Ed 1999, 38, 2909–2910. [DOI] [PubMed] [Google Scholar]
- [106].Gansauer A, Rinker B, Pierobon M, Grimme S, Gerenkamp M, Muck-Lichtenfeld C, Angew. Chem. Int. Ed 2003, 42, 3687–3690. [DOI] [PubMed] [Google Scholar]
- [107].Gansäuer A, Rinker B, Pierobon M, Grimme S, Gerenkamp M, Mück-Lichtenfeld C, Angew. Chem. Int. Ed 2003, 42, 3687–3690. [DOI] [PubMed] [Google Scholar]
- [108].Gansauer A, Fan CA, Keller F, Keil J, J. Am. Chem. Soc 2007, 129, 3484–3485. [DOI] [PubMed] [Google Scholar]
- [109].Gansäuer A, Fleckhaus A, Lafont MA, Okkel A, Kotsis K, Anoop A, Neese F, J. Am. Chem. Soc 2009, 131, 16989–16999. [DOI] [PubMed] [Google Scholar]
- [110].Gansauer A, Shi L, Otte M, J. Am. Chem. Soc 2010, 132, 11858–11859. [DOI] [PubMed] [Google Scholar]
- [111].Mühlhaus F, Weißbarth H, Dahmen T, Schnakenburg G, Gansäuer A, Angew. Chem. Int. Ed 2019, 58, 14208–14212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Yao CB, Dahmen T, Gansauer A, Norton J, Science 2019, 364, 764–767. [DOI] [PubMed] [Google Scholar]
- [113].Hao W, Harenberg JH, Wu XY, MacMillan SN, Lin S, J. Am. Chem. Soc 2018, 140, 3514–3517. [DOI] [PubMed] [Google Scholar]
- [114].Robinson SG, Wu XY, Jiang BY, Sigman MS, Lin S, J. Am. Chem. Soc 2020, 142, 18471–18482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Hao W, Wu XY, Sun JZ, Siu JNC, MacMillan SN, Lin S, J. Am. Chem. Soc 2017, 139, 12141–12144. [DOI] [PubMed] [Google Scholar]
- [116].Jiang SP, Dong XY, Gu QS, Ye L, Li ZL, Liu XY, J. Am. Chem. Soc 2020, 142, 19652–19659. [DOI] [PubMed] [Google Scholar]
- [117].Dong XY, Li ZL, Gu QS, Liu XY, J. Am. Chem. Soc 2022, 144, 17319–17329. [DOI] [PubMed] [Google Scholar]
- [118].Cheng YF, Yu ZL, Tian Y, Liu JR, Wen HT, Jiang NC, Bian JQ, Xu GX, Xu DT, Li ZL, Gu QS, Hong X, Liu XY, Nat. Chem 2023, 15, 395–404. [DOI] [PubMed] [Google Scholar]
- [119].Li ZL, Fang GC, Gu QS, Liu XY, Chem. Soc. Rev 2020, 49, 32–48. [DOI] [PubMed] [Google Scholar]
- [120].Gu QS, Li ZL, Liu XY, Acc. Chem. Res 2020, 53, 170–181. [DOI] [PubMed] [Google Scholar]
- [121].Zhang W, Wang F, McCann SD, Wang D, Chen P, Stahl SS, Liu G, Science 2016, 353, 1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Wang F, Chen P, Liu G, Acc. Chem. Res 2018, 51, 2036–2046. [DOI] [PubMed] [Google Scholar]
- [123].Li J, Zhang Z, Wu L, Zhang W, Chen P, Lin Z, Liu G, Nature 2019, 574, 516–521. [DOI] [PubMed] [Google Scholar]
- [124].Wang Z, Yin H, Fu GC, Nature 2018, 563, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Huo H, Gorsline BJ, Fu GC, Science 2020, 367, 559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Liu YG, Wei JH, Che CM, Chem. Commun 2010, 46, 6926–6928. [DOI] [PubMed] [Google Scholar]
- [127].Zhang X-X, Wayland BB, J. Am. Chem. Soc 1994, 116, 7897–7898. [Google Scholar]
- [128].Zhang X-X, Parks GF, Wayland BB, J. Am. Chem. Soc 1997, 119, 7938–7944. [Google Scholar]
- [129].Chan KS, Li XZ, Dzik WI, de Bruin B, J. AM. Chem. Soc 2008, 130, 2051–2061. [DOI] [PubMed] [Google Scholar]
- [130].Chan YW, Chan KS, J. Am. Chem. Soc 2010, 132, 6920–6922. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.