

Significance
The spread of tumor cells in the brain is difficult to remove via surgery, and these most aggressive brain cancers are difficult to treat through systemic medications due to the off-target effects and the inability of most medications to treat both the tumor at the peripheral site and at the brain. Further to this, primary cancer cell’s ability to adapt and share metabolic substrates with other cells in the tumor microenvironment results in metabolic plasticity. Therefore, understanding altered metabolic pathways in migrated tumor cells in the brain and development of therapeutics tailored to plasticity of primary and brain tumor cells may provide clinically relevant therapeutic options for this deadly disease.
Keywords: altered metabolism, Sirtuin 3, chemoresistance, metabolic plasticity
Abstract
Brain metastasis of advanced breast cancer often results in deleterious consequences. Metastases to the brain lead to significant challenges in treatment options, as the blood–brain barrier (BBB) prevents conventional therapy. Thus, we hypothesized that creation of a nanoparticle (NP) that distributes to both primary tumor site and across the BBB for secondary brain tumor can be extremely beneficial. Here, we report a simple targeting strategy to attack both the primary breast and secondary brain tumors utilizing a single NP platform. The nature of these mitochondrion-targeted, BBB-penetrating NPs allow for simultaneous targeting and drug delivery to the hyperpolarized mitochondrial membrane of the extracranial primary tumor site in addition to tumors at the brain. By utilizing a combination of such dual anatomical distributing NPs loaded with therapeutics, we demonstrate a proof-of-concept idea to combat the increased metabolic plasticity of brain metastases by lowering two major energy sources, oxidative phosphorylation (OXPHOS) and glycolysis. By utilizing complementary studies and genomic analyses, we demonstrate the utility of a chemotherapeutic prodrug to decrease OXPHOS and glycolysis by pairing with a NP loaded with pyruvate dehydrogenase kinase 1 inhibitor. Decreasing glycolysis aims to combat the metabolic flexibility of both primary and secondary tumors for therapeutic outcome. We also address the in vivo safety parameters by addressing peripheral neuropathy and neurobehavior outcomes. Our results also demonstrate that this combination therapeutic approach utilizes mitochondrial genome targeting strategy to overcome DNA repair–based chemoresistance mechanisms.
Patients with common solid tumors often spread to the central nervous system (CNS) (1). Most common solid cancers of the lung, breast, and skin metastasize to the brain and cause a brain tumor (2, 3). Tumor metastasis to the CNS affects survival, neurocognition, speech, coordination, and quality of life of these patients. A solid cancer which serves as a source to cause brain cancer is breast cancer, where up to 10% of breast cancer patients present with metastasis at diagnosis and up to 30% of individuals with stage IV breast cancer develop brain tumors (1, 2). Breast cancer is known to have a wide window of relapse, due in large part to dormant cancer cells (3) and when breast cancers recur, they are usually metastatic (4). These brain tumors which spread throughout the brain are difficult to remove via surgery. Aggressive brain cancers are difficult to treat through systemic medications due to off-target effects and the inability of these medications to accumulate in the brain navigating the highly regulated blood–brain barrier (BBB). The primary treatment options to manage these brain tumors include whole brain radiotherapy (5), stereotactic radiosurgery, surgery, and chemotherapy. None of these available treatment options can remove the invaded brain tumors effectively thus, improving quality of life and overall survival for patients with such brain tumors need alternative strategies (6). The cancer cells which are growing in a peripheral organ such as breast adapt and share metabolic substrates with stromal and immune cells in the tumor microenvironment which can drive these cells to become metabolically plastic thereby allowing these cells to migrate, survive, and thrive in the brain. Key to providing clinically relevant and meaningful therapeutic options for this deadly disease includes understanding altered but shared metabolic pathways by these tumor cells when in the peripheral organ and in the brain to design therapeutics tailored to the metabolic plasticity. This increased metabolic plasticity manifests in brain tumors originating from a peripheral tumor such as breast cancer through higher glycolytic and tricarboxylic acid cycle flux, increased ability to switch between oxidative phosphorylation (OXPHOS) and glycolysis (7). This increased plasticity of brain tumors and the differences between peripheral and brain tumor necessitate the development of technologies that can target tumors at both the locations. Free drug combinations attacking different metabolic targets are unable to be delivered to a peripheral breast tumor and invaded brain tumors due to the inability of small molecule-based drugs to cross the BBB, inability to choose the correct ratio of the drugs, varied pharmacokinetics of the combinatory molecules, and different distribution property of individual drugs. Therefore, the creation of a nanoparticle (NP) that distributes to both the peripheral tumor site and across the BBB in the brain for invaded brain tumors can be extremely beneficial.
Our conventional understanding describes tumor cells as having dysfunctional mitochondria, but increasing evidence suggests that the mitochondria of cancer cells can utilize glycolysis, OXPHOS, a combination of both, or switch between these two pathways (8). This switch between the metabolic phenotypes or metabolic plasticity is usually in response to external perturbations, tumor progression, and/or migration of tumor cells (9–11). Under metabolic phenotypic changes, mitochondria in cancer cells shift from glycolysis to increased levels of fatty acid beta oxidation (FAO), often continuing to shift between the two to maintain proliferation throughout changing environmental conditions (12, 13). Recent studies demonstrated that inhibition of FA synthase, a multienzyme protein that catalyzes FA synthesis, reduced metastasis of human epidermal growth factor receptor 2 positive breast tumor in the brain (14). Due to metabolic plasticity, a peripheral tumor such as the cancer at the breast and formation of a tumor site at the brain could switch from the FAO back to glycolytic phenotype when fat utilization is inhibited. Thus, we hypothesized that a suitably designed combination of FAO and glycolysis inhibitors that can be administered with a BBB penetrating NP in a spatiotemporal manner to address the timing of metabolic switching along with a chemotherapeutic component can potentially eliminate the hybrid metabolic phenotype for maximum reduction of triple negative breast cancer (TNBC) and their progression toward brain metastasis (Fig. 1).
Fig. 1.
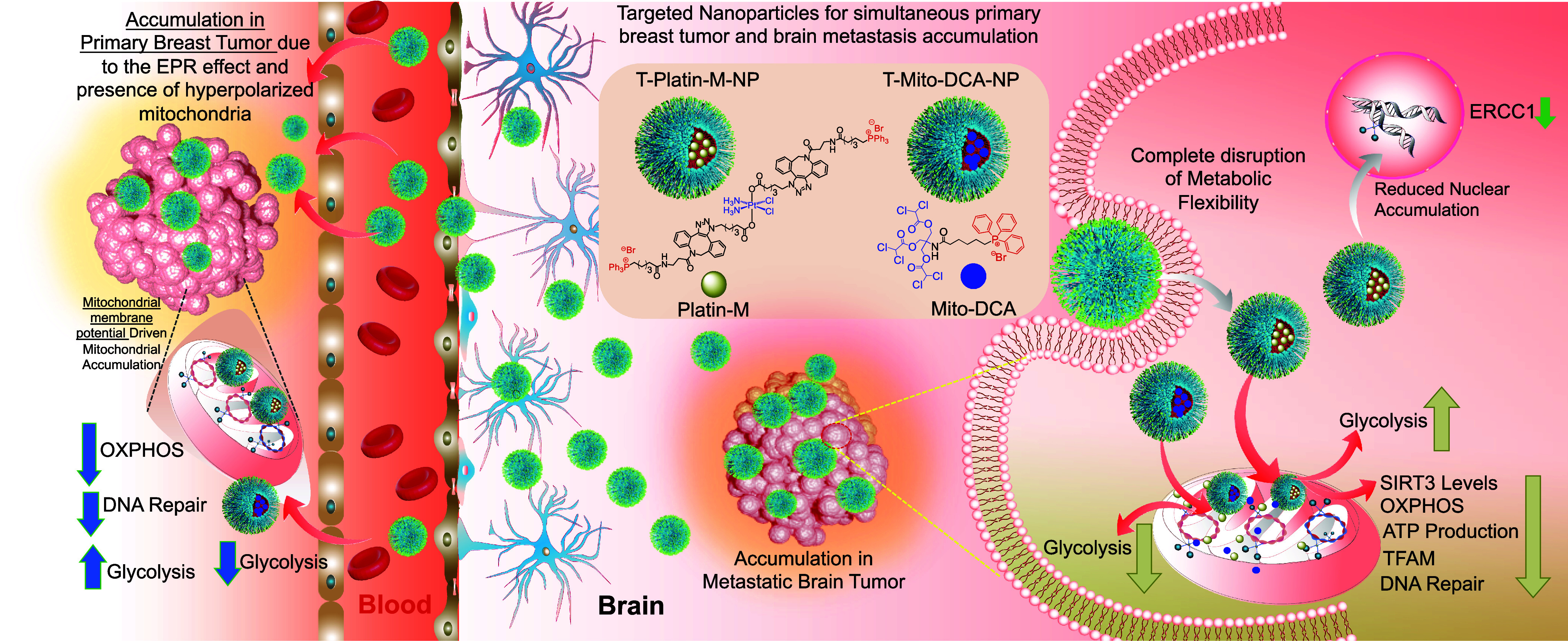
Schematic description of nanotherapeutic technology for treating primary breast tumor and the secondary metastatic tumor in the brain using two brain accumulating NPs one with a chemotherapeutic platinum prodrug and the other with a glycolysis inhibitor to tackle metabolic flexibility by simultaneous disruption of OXPHOS and glycolysis and to overcome repair-induced resistance by attacking mitochondrial DNA.
Mitochondrial DNA (mtDNA) plays an integral role in driving cancer to a metastatic stage. Close proximity of mtDNA to reactive oxygen species (ROS) in the mitochondrial lumen makes these DNA vulnerable to oxidative damage resulting in mutated mtDNA (15–17). In FAO utilizing tumors, this method of energy production can be a major source of increased net ROS and mutated mtDNA. Mutated mtDNA is found in brain tumors and studies support the correlation between somatic mtDNA mutations and cancer progression (18–20). Thus, targeting mtDNA with a chemotherapeutic incorporated in a NP is an attractive alternative. Cisplatin is a purely inorganic compound that has Food and Drug Administration approval for an appreciable number of cancer types (21–23). Cisplatin’s therapeutic efficacy arises from nuclear DNA (nDNA) damaging activity by forming interstrand and intrastrand cross-links (24). Resistance associated with prolonged cisplatin therapy arises from efficient DNA repair by the nucleotide excision repair (NER) (25, 26) in the nucleus. In addition, cisplatin is found to cause widespread peripheral neuropathy due to its off-target effects. Targeting mtDNA with cisplatin-based therapy can be a promising way to manage resistance. The absence of efficient DNA repair machinery in the mitochondria and increased mtDNA mutation in secondary brain tumor gives a strong rationale in routing cisplatin to the mitochondrial lumen. We recently reported a cisplatin prodrug, Platin-M (27). In our current work using this prodrug on TNBC cells indicated the ability of Platin-M to inhibit mitochondrial Sirtuin 3 (SIRT3) which regulates the acetylation levels of metabolic enzymes and modulates FAO (28). We further supplemented our observation by conducting complementary in vitro and in vivo studies documenting the ability of Platin-M to inhibit FAO and subsequent switch of TNBC cells to glycolysis (Fig. 1). In a chemotherapeutic combination of Platin-M and mitochondrion-targeted dichloroacetate (DCA), a glycolytic inhibitor, Mito-DCA (29, 30), metabolic adaptation was prevented and resulted chemotherapeutic effects by utilizing the cisplatin component of the prodrug (Fig. 1). Thus, we hypothesized that a combination of Platin-M and Mito-DCA in a BBB penetrating as well as primary tumor distributing NP can be an effective way to manage the tumors at a peripheral site and at the brain.
Over the years, we have been meticulously focused on understanding biological membrane crossing ability and mechanism of action of a biocompatible polymeric NP using poly(lactic-co-glycolic acid) (PLGA)-block (b)-polyethyleneglycol (PEG) functionalized with a terminal triphenylphosphonium (TPP) cation with efficient ability to cross the BBB and mitochondrial double membrane, the two impermeable biological membranes. The ability of this polymer to result in NPs which can accumulate in the brain and tumor can be extremely important in studying brain tumors (31–34). The TPP cation in PLGA-b-PEG-TPP polymer takes advantage of the substantial hyperpolarized mitochondrial membrane potential (Δψm) across the inner mitochondrial membrane to efficiently associate with the mitochondria. In rodent and dog models, an optimized formulation of targeted NP (T-NP) was found to accumulate in the brain and mostly inside the glia cells with hyperpolarized Δψm (33–36). The nature of the mitochondrion-targeted, BBB-penetrating NP along with the enhanced permeability and retention effect, allow for simultaneous targeting and drug delivery to the hyperpolarized mitochondrial membrane of cancer cells at the primary site in addition to secondary brain metastases. Thus, we hypothesized that attacking metastatic brain tumor cells using this BBB-penetrating NP to deliver combination therapeutics utilizing Platin-M and Mito-DCA to disrupt metabolic adaptability and initiating chemotherapeutic effects by forming repair-resistant Pt adducts with mtDNA can provide meaningful therapeutic option considering the clinical settings of brain tumors (Fig. 1). In this report, we demonstrate the mechanism of action and efficacious tackling of both primary breast and brain metastasis when Platin-M and Mito-DCA are delivered with the brain-accumulating polymeric NP (T-NP) by disrupting the metabolic adaptability of the proliferating cells at both the tumor sites (Fig. 1).
Results and Discussion
Effects of T-Platin-M-NP on Whole Genome and mtDNA.
A mitochondria-targeted Pt(IV)-prodrug of cisplatin, Platin-M, was constructed by appending two mitochondria targeting TPP cations in the axial positions (27, 35, 37). T-NPs loaded with Platin-M, T-Platin-M-NPs, were synthesized by nanoprecipitation using PLGA-b-PEG-TPP, 30% PLGA-COOH, and 10% TPP-(CH2)5-COOH to give the highest brain accumulation of NPs (SI Appendix, Fig. S1 for characterization); the NT-Platin-M-NPs were prepared from 10% TPP-(CH2)5-COOH, 30% PLGA-COOH, and PLGA-b-PEG-OH (SI Appendix, Fig. S1 for characterization). To understand the effect of Platin-M-NP on mitochondrial gene expression, we first evaluated the mitochondrial accumulation ability of Platin-M and T-Platin-M-NPs in brain metastatic breast cancer MDA-MB-231-BR cells treated with cisplatin, Platin-M, NT-Platin-M-NP, or T-Platin-M-NP. We observed that the cells treated with either Platin-M or T-Platin-M-NP showed higher platinum (Pt) accumulation in the mitochondrial fraction compared to the respective cytosolic or nuclear fraction (SI Appendix, Fig. S2A). Binding ability of cisplatin to mtDNA enhanced when delivered in the form of its prodrug, Platin-M and the corresponding nano-formulations (SI Appendix, Fig. S2B). Comparatively, the cells treated with T-Platin-M-NP showed higher mtDNA adducts than cisplatin-treated cells (SI Appendix, Fig. S2B). Cisplatin adducts in the nDNA was higher than cisplatin-mtDNA adducts in the cells treated with cisplatin (SI Appendix, Fig. S2B). NT-Platin-M-NPs showed reduced levels of mtDNA and nDNA adduct formation. Radiation treatment was used as a positive control for increased TFAM.
To understand the effects of T-NP-delivered Platin-M on the mitochondrial genome, we first evaluated the expressions of excision repair cross-complementation group 1 (ERCC1) and mitochondrial transcription factor A (TFAM) after cisplatin, Platin-M, NT-Platin-M-NP, or T-Platin-M-NP treatment in TNBC cell line MDA-MB-231 (Fig. 2A) and the corresponding brain seeking MDA-MB-231-BR cells (Fig. 2B). ERCC1 is a nuclear gene encoding the protein ERCC1 which is involved in the NER pathway. In MDA-MB-231-BR, Western blot data suggested that cisplatin treatment increases ERCC1 expression; however, T-Platin-M-NP did not have a significant effect on ERCC1 expression (Fig. 2 A and B and SI Appendix, Fig. S3 for quantification). TFAM is a mitochondrial gene encoding the protein TFAM which is involved in mtDNA replication, transcription, and genome integrity (38). Our data also suggested that in both the cell lines, T-Platin-M-NP inhibited the expression of TFAM as compared to control and cisplatin (Fig. 2 A and B and SI Appendix, Fig. S3 for quantification). Cisplatin did not have any significant effect on mitochondrial protein TFAM in MDA-MB-231 cells, but marginally increased expression in MDA-MB-231-BR cells. We then carried out RNA sequencing analyses in MDA-MB-231-BR cells after treatment with cisplatin or T-Platin-M-NP to understand the overall impact on the genome (SI Appendix, Fig. S4 for whole genome cluster heat map and SI Appendix, Fig. S5 for principal component analyses) (39). Analyses indicated that T-Platin-M-NP treatment affected more genes compared to cisplatin (Fig. 2 C and D). We found out that T-Platin-M-NP down-regulated 4,198 genes and up-regulated 4,074 genes of 8,272 differentially expressed genes (DEGs). Cisplatin, in total, 4,303 DEGs, 2,092 genes were down-regulated, and 2,211 genes were up-regulated. The Venn diagram representation of DEGs by cisplatin and T-Platin-M-NP are shown in Fig. 2E. There are 1,158 nuclear-encoded mitochondrial genes that have been reported (40). T-Platin-M-NP differentially expressed 341 nuclear-encoded mitochondrial genes; in contrast, cisplatin affected 187 genes (Fig. 2E). Additionally, 53 nuclear-encoded mitochondrial genes involved in OXPHOS and energy production were differentially expressed by T-Platin-M-NP. T-Platin-M-NP also affected 27 nuclear-encoded mitochondrial genes responsible for mitochondrial ribosome assembly and gene translation. A greater number of mitochondrial genes were regulated in case of T-Platin-M-NP treatment (Fig. 2F). Effects on the genes associated with NER because of treatment was significantly different in case of T-Platin-M-NP treatment compared to cisplatin (Fig. 2G). Gene set enrichment analysis (GSEA) on the genes involved in the NER pathway of Kyoto encyclopedia of genes and genomes (KEGG) databases revealed enrichment scores (ES) of less than zero (ES < 0) for T-Platin-M-NP group vs. cisplatin group and T-Platin-M-NP group vs. control group which suggest that NER-related genes are down-regulated with T-Platin-M-NP treatment (SI Appendix, Fig. S6). Several DNA repair genes such as XRCC1, XRCC3 were up-regulated in cisplatin-treated cells compared to the T-Platin-M-NP-treated group suggesting that the cisplatin-induced nDNA damage is undergoing repair (Fig. 2G). We also observed downregulation of BReast CAncer gene 1 BRCA1 in the NP-treated group compared to cisplatin, indicating repair pathway operative in the cisplatin-treated cells. Overall, we observed greater effects of T-Platin-M-NP treatment on the mitochondrial gene and the ability of T-Platin-M-NP to avoid upregulation of NER (SI Appendix, Fig. S6).
Fig. 2.
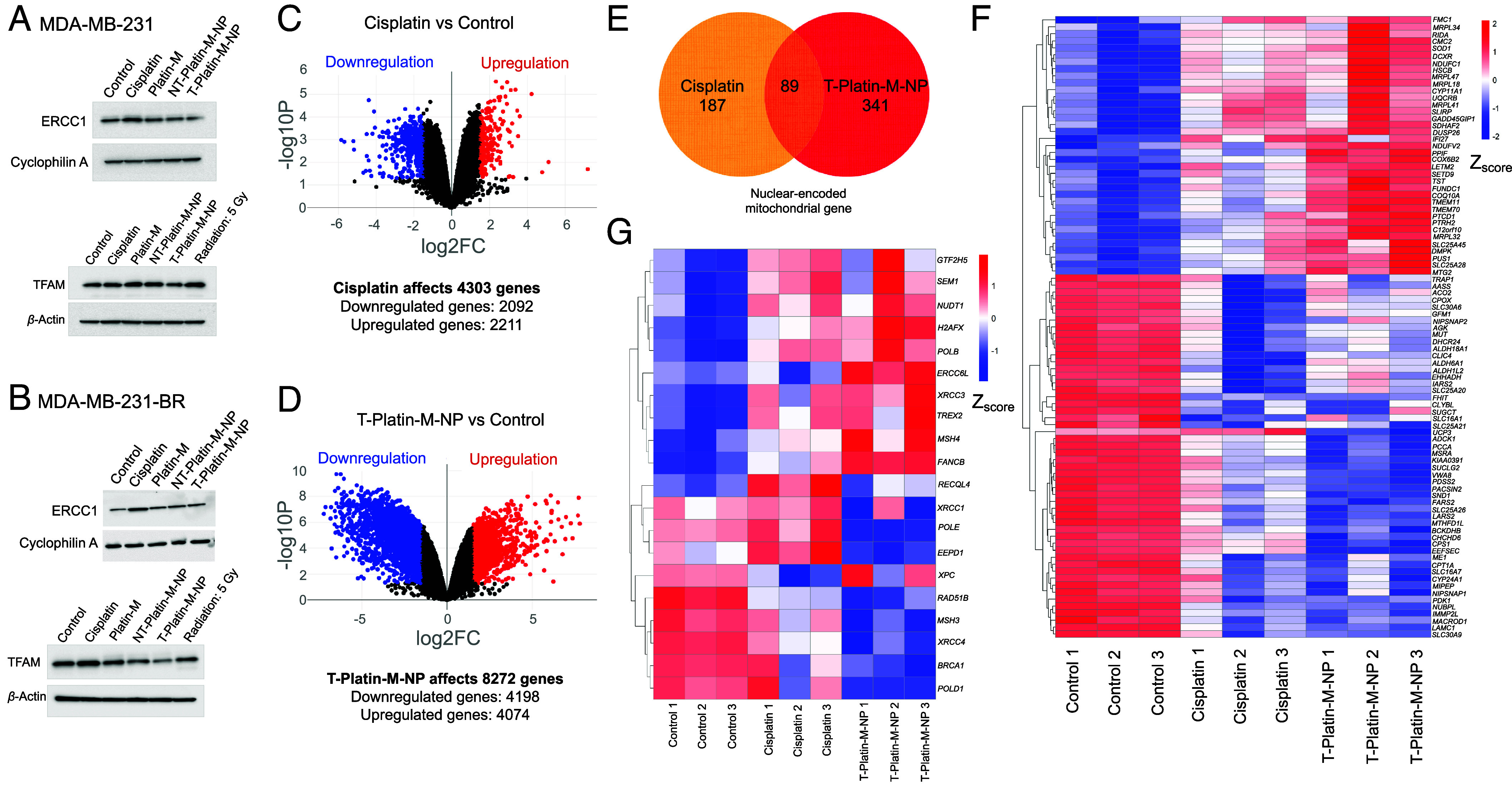
Effects of T-Platin-M-NP on whole genome. Western blot analyses in (A) MDA-MB-231 and (B) MDA-MB-231-BR cells showing the effects of treatments on ERCC1 and TFAM expressions. Volcano plots showing the changes in total number of genes when compared between (C) control and cisplatin-treated groups and (D) control and T-Platin-M-NP-treated groups. (E) Venn diagram illustrating the total number of nuclear-encoded mitochondrial genes affected by cisplatin and T-Platin-M-NP treatments. Cisplatin affected 187 genes and T-Platin-M-NP affected 341 genes, out of which 89 genes were common between both the treatments. (F) Gene clustering heatmap showing the differential gene expression of the 89 genes that are common in cisplatin- and T-Platin-M-NP-treated groups. (G) Gene clustering heatmap showing the differential gene expression of the genes involved in the NER pathway after treatments with cisplatin and T-Platin-M-NP in MDA-MB-231-BR cells.
Effects of T-Platin-M-NP on Mitochondrial Respiration.
Analyses of MDA-MB-231-BR cells upon treatment with T-Platin-M-NPs indicated that several genes related to OXPHOS were down-regulated (Fig. 3A). GSEA on the genes involved in the OXPHOS pathway revealed a negative ES with leading edge subset found toward the down-regulated genes for T-Platin-M-NP vs. cisplatin and T-Platin-M-NP vs. control which suggested decreased OXPHOS and mitochondrial translation on T-Platin-M-NP treatment (SI Appendix, Fig. S7). RNAseq data indicated that T-Platin-M-NP significantly down-regulated the expressions of MT-ND2, MT-ND3, MT-ND4, MT-ND4L, MT-ND5, and MT-ND6 coding for complex I; gene MT-CYB coding for complex III; gene MT-CO1, MT-CO2 and MT-CO3 coding for complex IV and gene MT-ATP6 and MT-ATP8 coding for complex V (Fig. 3A). Additionally, T-Platin-M-NP down-regulated the expression of mitochondrial genes such as MT-TP, MT-TF and up-regulated MT-TM, MT-RNR1, and MT-RNR2 which are necessary for mtDNA translation (Fig. 3A). Cisplatin up-regulated the genes involved in OXPHOS and mtDNA translation (Fig. 3A). We thus set out to understand the mechanism of action of T-Platin-M-NP and investigated how the treatment with the NP alters cellular metabolism using MDA-MB-231 and MDA-MB-231-BR cells. Analyses of mitochondrial OXPHOS in these cells upon treatment with T-Platin-M-NP resulted in complete disruption of cellular respiration (Fig. 3B and SI Appendix, Fig. S8). Analyses of substrate preferences indicated that both the primary and metastatic cells rely on FAO (41) to a greater extent for their adenosine triphosphate (ATP) production compared to glucose or glutamine (Fig. 3C and SI Appendix, Fig. S9). The observations in MDA-MB-231 and MDA-MB-231-BR cell lines were further validated in another TNBC cell line HCC1806. Detailed analyses of mitochondrial metabolic pathways revealed that these cells are less dependent on glycolysis (SI Appendix, Fig. S10) and utilize mitochondrial OXPHOS (SI Appendix, Fig. S10). Further analyses of substrate utilization for mitochondrial OXPHOS indicated that these cells are flexible in terms of their substrate utilization compared to the MDA-MB-231 and MDA-MB-231-BR cells; HCC1806 cells utilize lipid, glucose, and glutamine to some extent for energy production (SI Appendix, Fig. S11). Treatment of HCC1806 cells by Platin-M or its NPs demonstrated significant reduction of oxygen consumption during mitochondrial OXPHOS which resulted in lesser ATP production compared to cisplatin (SI Appendix, Fig. S12). Thus, it can be concluded that Platin-M and its T-NP have prominent effects on TNBC cell lines which utilize mitochondrial OXPHOS for energy production and growth.
Fig. 3.
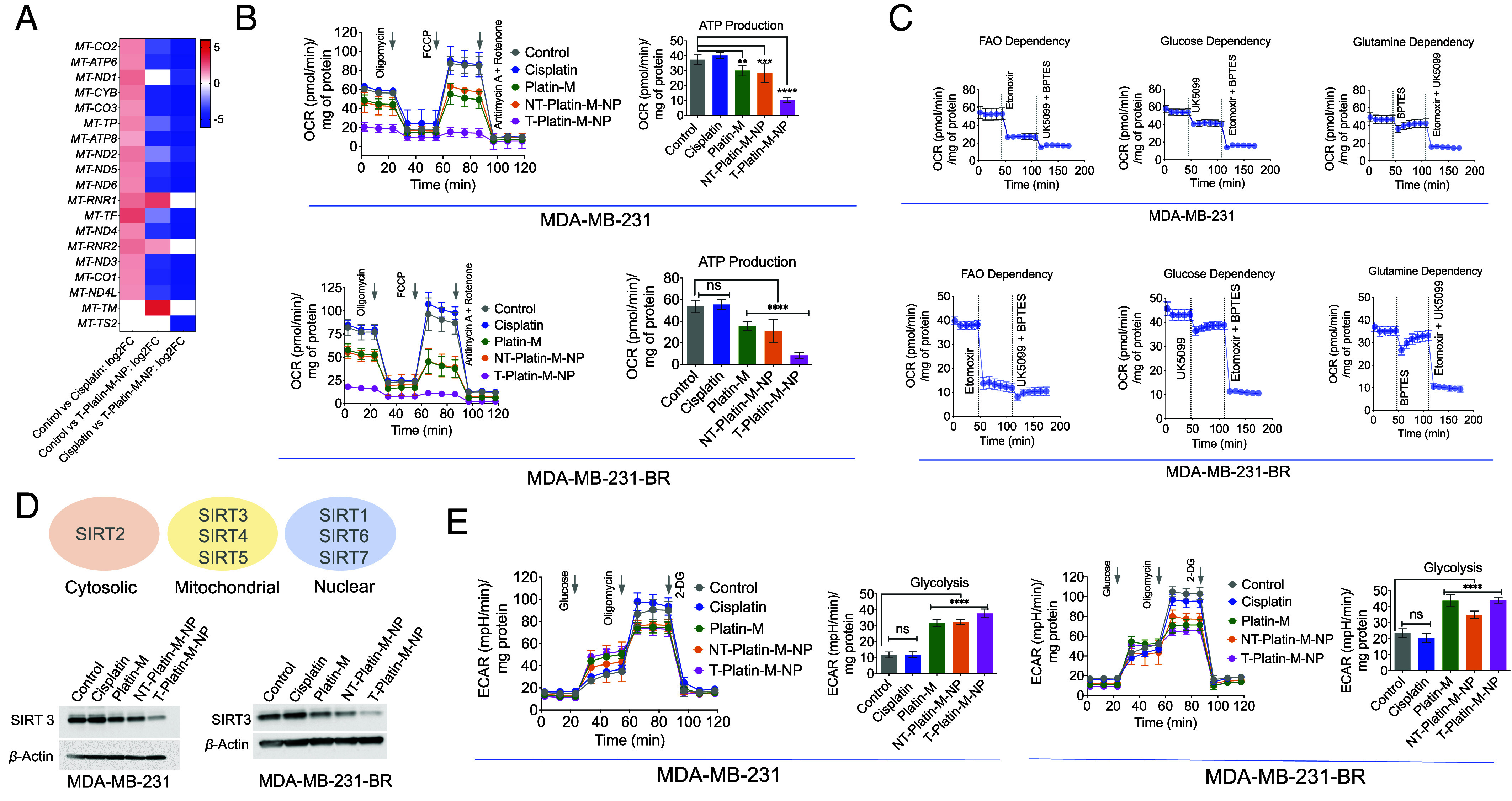
Effect of T-Platin-M-NP on the mtDNA and metabolic pathway inhibition. (A) Heatmap showing the differential mitochondrial OXPHOS gene expression based on the log2 fold change in comparison to control and cisplatin; control and T-Platin-M-NP; and cisplatin and T-Platin-M-NP. Cisplatin treatment resulted in upregulation of the mitochondrial gene and T-Platin-M-NP down-regulated the mitochondrial genes. When compared with respect to cisplatin, T-Platin-M-NP treatment showed a downregulation of the mitochondrial genes. (B) Mitostress analyses performed in MDA-MB-231 and MDA-MB-231-BR cells showing significant reduction in ATP production upon treatment with T-Platin-M-NP. (C) Fuel flex analyses showing to understand the dependence of MDA-MB-231 and MDA-MB-231-BR cells on fatty acid oxidation, glucose oxidation, or glutamine oxidation. (D) Western blot analyses in MDA-MB-231 and MDA-MB-231-BR cells showing the change in expression of SIRT3 upon treatment with T-Platin-M-NP. Schematic representation showing the localization of SIRTs 1-7. (E) Glycostress analyses performed in MDA-MB-231 and MDA-MB-231-BR cells showing significant increase in glycolysis upon treatment with Platin-M and T-Platin-M-NPs. Statistical analyses were carried out by a one-way ANOVA with multiple analysis with an alpha value of 0.05.
Effects of T-Platin-M-NP on SIRT3.
Assembling the observations together from gene analyses and mitochondrial metabolic pathways together, we wanted to explore whether Platin-M has any specific effect on a particular pathway which dominates in mitochondrial OXPHOS. Since mitochondrial sirtuins (SIRTs) play important roles during metabolic adaptation, we carried out western blot analyses for SIRT1-7 using MDA-MB-231-BR cells in presence Platin-M, T/NT-Platin-M-NPs, or cisplatin. Among seven mammalian sirtuins, SIRT3, SIRT4, and SIRT5 are in the mitochondria; SIRT2 is in the cytosol, and SIRT1, SIRT6, and SIRT7 are nuclear localized (Fig. 3 D, Top). Two independent sets of experiments using MDA-MB-231-BR cells upon treatment indicated that Platin-M and its NPs significantly reduce the levels of SIRT3 (SI Appendix, Fig. S13). We thus set out to investigate SIRT3 which is actively involved in mitochondrial respiration using both MDA-MB-231 and MDA-MB-231-BR cell lines (Fig. 3 D, Bottom and SI Appendix, Fig. S14 for quantification). Analyses of SIRT3 in MDA-MB-231 and MDA-MB-231-BR cells indicated that T-Platin-M-NP treatments lowered the level of SIRT3 significantly (Fig. 3D and SI Appendix, Fig. S14). Thus, we believe that the inhibition of SIRT3 by T-Platin-M-NPs results in disruption of FAO driven OXPHOS. We then investigated whether these cells have any plasticity to switch to any other metabolism when OXPHOS is diminished by T-Platin-M-NPs. It was observed that Platin-M and T-Platin-M-NP increased glycolysis in MDA-MB-231-BR and MDA-MB-231 cells (Fig. 3E). Cisplatin did not show such effect. Thus, this finding led us to hypothesize that T-Platin-M-NP treatment should be combined with a glycolysis inhibitor to account for metabolic plasticity.
Efficacy of T-Platin-M-NP when Combined with a Glycolysis Inhibitor and Ability to Distribute in Peripheral Breast Tumor and the Brain.
To test the ability of the T-Platin-M-NP to distribute in both primary tumor and site of metastasis, the brain; xenograft tumors were formed in BALB/c nude mice using MDA-MB-231-BR cells (Fig. 4A). In this experiment, we combined T-Platin-M-NP with a glycolysis inhibitor 2-Deoxy-D-Glucose (2-DG) to disrupt the metabolic plasticity when the cells switch to glycolysis upon FAO inhibition by Platin-M. Cisplatin was found to distribute in only the primary tumor site, but T-Platin-M-NP alone or in combination with 2-DG was found to distribute in both the primary tumor and the brain, indicating the BBB-penetrating abilities of the T-NP (Fig. 4B). A total of 80% of the animals treated with T-Platin-M-NP demonstrated tumor volume reduction compared to the saline-treated group and 40% of the mice treated with T-Platin-M-NP + 2-DG demonstrated significant tumor reduction (Fig. 4C). No significant reduction in mice body weight was observed, indicating the treatments did not cause excess toxicity (Fig. 4D). Heterogeneity of tumors is a hallmark of tumor evolution and cancer progression and to understand whether the overall trends are still conclusive, we conducted RNA sequencing analyses (39). RNA seq analyses of the tumors indicated that OXPHOS genes such as COX11 and COX7C showed increased expression after cisplatin treatment as compared to lower expression following T-Platin-M-NP treatment with or without 2-DG (Fig. 4E). Glycolysis gene PDK4 was up-regulated following T-Platin-M-NP treatment, likely because the cancer cells with reduced OXPHOS were naturally shifting to glycolysis (Fig. 4F). For DNA repair genes, cisplatin treatment was found to increase expression of several NER-associated genes such as FANCL, XPA, and NBN; NP-treated or NP- and 2-DG-treated samples did not show such increases (Fig. 4 G and H). To understand the effect of T-Platin-M-NP in vivo on SIRT3 and ERCC1, we carried out Western blot analyses on the tumor tissues (Fig. 4 I and J). These data suggested that the expression of SIRT3 is inhibited greatly in most animals by T-Platin-M-NP and T-Platin-M-NP + 2-DG. Cisplatin treatment increased ERCC1 (Fig. 4I), whereas T-Platin-M-NP and T-Platin-M-NP + 2-DG decreased ERCC1 expression (Fig. 4J). We want to stress that even though there is heterogeneity within each group, when taken as a whole, we can see that the activity of T-Platin-M-NP when combined with 2-DG is evident in bringing changes which correspond to a combined metabolic disruption and mtDNA damage.
Fig. 4.
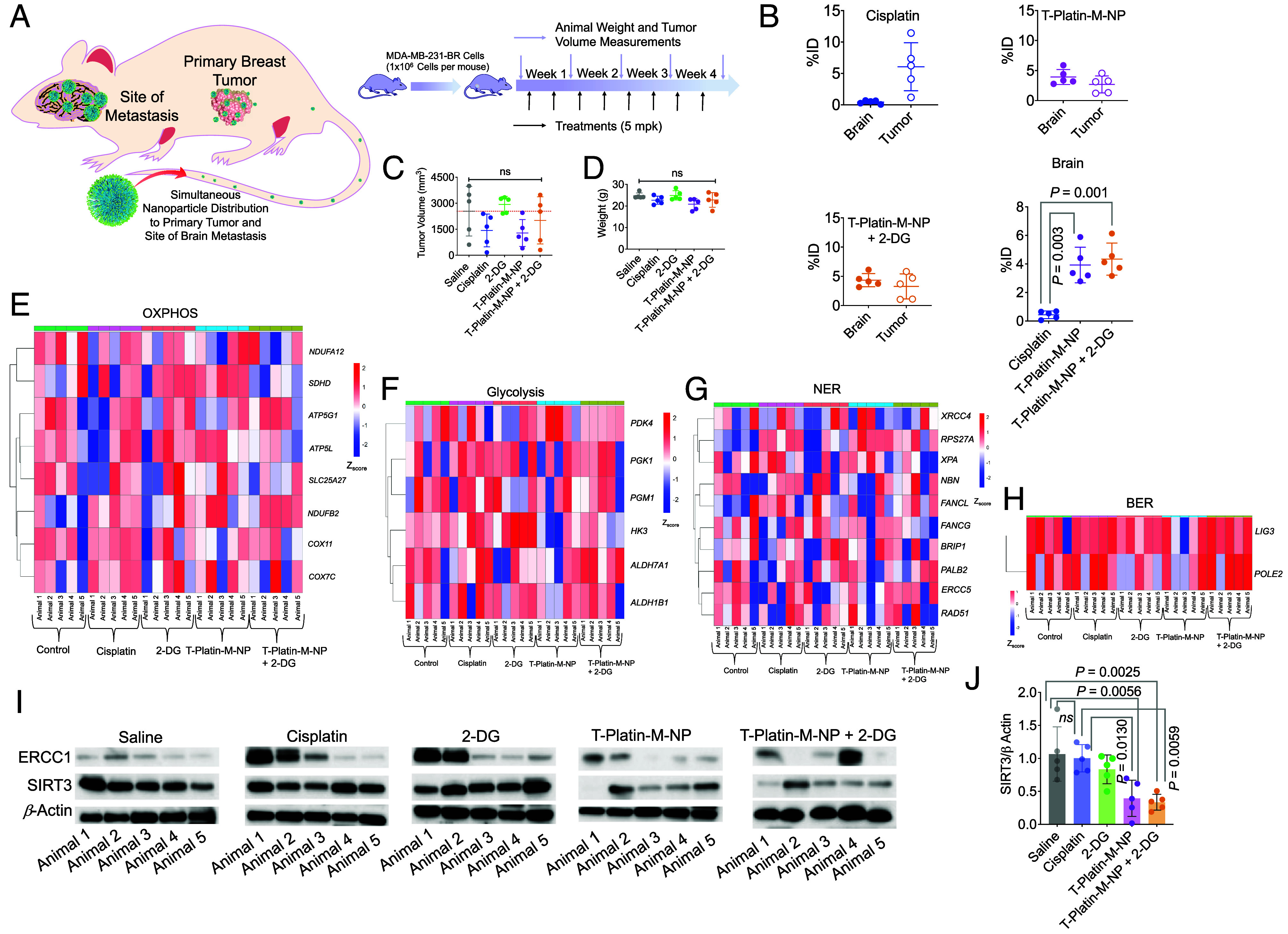
Efficacy of T-Platin-M-NP in combination with a glycolysis inhibitor and ability to distribute in primary breast tumor and the site of the brain. (A) Schematic representation showing the experimental design with n = 5 mice per group. (B) Scatter plot showing the percent injected dose (%ID) of cisplatin, T-Platin-M-NP, and T-Platin-M-NP + 2-DG in the brain and primary tumor site following treatment. A comparison in brain distribution between cisplatin, T-Platin-M-NP, and T-Platin-M-NP + 2-DG is also shown. Statistical analyses were carried out using one way ANOVA with multiple comparison. (C) Tumor volume (mm3) and (D) body weight among the different treatment groups. Statistical analysis for (C) and (D) was carried out by one-way ANOVA with an alpha value of 0.05. Gene cluster heatmap showing expression of various genes related to (E) OXPHOS, (F) glycolysis, (G) NER, and (H) BER pathway in tumor samples. (I) Western blot analyses of tumor samples for studying the change in expression of ERCC1 and SIRT3. (J) Quantification of the expression of SIRT3 with respect to β-actin using the western blot. Statistical analysis was carried out by one-way ANOVA with an alpha value of 0.05.
Toxicity of Multidose T-Platin-M-NP in Normal Mice.
Analyses of cisplatin-induced neuronal toxicity were carried out in a cohort of C57BL/6 mice. Experimental details can be found in SI Appendix, Fig. S15A. Regular monitoring of the body weight indicated that none of the treatment groups resulted in weight loss of the animals (SI Appendix, Fig. S15B). Blood biochemical analyses showed that Na, K, Cl, total CO2, total protein, albumin, glucose, P, Mg, cholesterol, and blood urea nitrogen and blood cell counts did not show any significant changes with T-Platin-M-NP treatment (SI Appendix, Fig. S15 C and D). The alanine aminotransferase, aspartate transaminase, and bilirubin levels were increased in the cisplatin-treated group indicating elevated hepatotoxicity due to cisplatin; however, T-Platin-M-NP alone and in combination with 2-DG did not show any significant changes in the respective levels (SI Appendix, Fig. S15C). SIRT2 plays a role in protecting neurons following cisplatin treatment (42). Western blot analyses were performed to determine the levels of expression of SIRT1-7 levels in the brain tissue samples (SI Appendix, Fig. S15E for western blot and SI Appendix, Fig. S15F for quantification). We observed that two of the three mice treated with cisplatin showed increased levels of SIRT2, an effect not seen following treatment with 2-DG or T-Platin-M-NP alone or in combination, indicating cisplatin may be impacting these neurons and causing toxicity. In addition, there were no global changes in mitochondrial SIRT proteins, including SIRT3, which is tied to OXPHOS, because of the T-Platin-M-NPs. While previous data suggested that the T-Platin-M-NPs decrease SIRT3 expression and inhibit OXPHOS in the tumors, these data support the idea that the T-Platin-M-NPs are particularly targeted to the tumors and do not cause changes to SIRT3 or OXPHOS in the whole brain. Tissue samples from organs were characterized by histological studies (SI Appendix, Fig. S15G). Since the NPs accumulate in the brain and liver; Pt-based drugs show toxicity parameters related to liver and kidney; and mitochondria targeted delivery vehicles often accumulate in the heart; we analyzed liver, kidney, brain, and heart. Analyses of these organs indicated no significant changes related to any signs of toxicity from T-Platin-M-NPs (SI Appendix, Fig. S15G).
Combating Metabolic Plasticity Using Combination of T-Platin-M-NP and T-Mito-DCA-NP.
Our observations indicated that Platin-M and its T-NPs inhibit mitochondrial SIRT3 which modulates the rate acetylation of metabolic enzymes and thereby regulates FAO (28). The in vivo xenograft study indicated the benefit of combining T-Platin-M-NP with a glycolysis inhibitor such as 2-DG. Physicochemical characteristics of 2-DG do not allow its incorporation inside a NP system for targeted delivery to the brain and to the tumor to tackle breast cancer brain metastasis. Thus, we then explored the option to combine T-Platin-M-NPs with a mitochondrion-targeted glycolytic inhibitor, Mito-DCA (29, 30) by creating T-Mito-DCA-NPs (SI Appendix, Fig. S16). The stability profiles of T-Platin-M-NPs and T-Mito-DCA-NPs were studied by storing freshly prepared suspension of the respective NPs and storing at 4 °C for a period of 10 d. Analyses of NPs’ diameters, surface charge by zeta potential, and morphology by transmission electron microscopy indicated that the particles remain monodisperse with suitable surface charge through this period (SI Appendix, Fig. S17 A–C). To have an insight on payload release kinetics, we conducted release of Platin-M from T-Platin-M-NPs and Mito-DCA from T-Mito-DCA-NPs under physiological conditions of pH 7.4 and keeping the temperature at 37 °C (SI Appendix, Fig. S17D). Analyses of released Platin-M by performing Pt-based inductively coupled plasma mass spectrometry (ICP-MS) analyses on the remaining T-Platin-M-NPs and release of Mito-DCA by high performance liquid chromatography (HPLC) indicated that both the NPs release their payloads in a controlled fashion.
Simultaneous Targeting of Peripheral and Brain Tumors by T-Platin-M-NP in a Dual Tumor Model.
We created a dual tumor mouse model of MDA-MB-231-BRLuc cells where the peripheral breast tumor was implanted in the right flank of the mice via subcutaneous injection. Once these tumors had grown and were palpable, the brain tumor was implanted in the brain orthotopically via stereotactic intracranial injection (Fig. 5A). Once both the peripheral and tumor at the brain were observed (Fig. 5B, representative images from three animals), the mice were randomly divided to study the distribution of T-Platin-M-NP (SI Appendix, Fig. S18). The investigative groups were saline (n = 5), cisplatin (n = 5) at a dosage of 5 mg/kg, and a combination of T- Platin-M-NP + T-Mito-DCA-NP (n = 4) at a dosage of 10 mg/kg with respect to Platin-M and 30 mg/kg with respect to Mito-DCA, respectively. T-Mito-DCA-NPs were injected 24 h post T-Platin-M-NP injection. All the test articles were injected via tail iv route. With the aim of accurately quantifying the amount of NP at the brain tumor site, we inoculated MDA-MB-231-BR cells that were transfected with firefly luciferase. Presence of luciferase label on the tumor cells enabled sorting of these cells from the whole brain suspension. We performed flow cytometric cell sorting of the tumor cells using anti-firefly luciferase antibody to specifically tag the MDA-MB-231-BRLuc cells. Once the luciferase-tagged tumor cells and the remaining brain cells were sorted, the NP accumulation was measured by ICP-MS. Sorting of the tumor cells from the whole brain was confirmed by using luciferase antibody tagged MDA-MB-231-BRLuc positive control cells and the cell population at the similar region from all treatment groups was collected as the tumor cells (Fig. 5C). MDA-MB-231-BRLuc cells without the luciferase antibody served as the negative control (Fig. 5C). We quantified %Pt in the luciferase-tagged brain tumor cells, remaining brain cells, peripheral tumor, and across all the major organs of the animals (SI Appendix, Fig. S19 for complete biodistribution pattern). Our data showed that T-Platin-M-NPs distributed both at the peripheral tumor site as well as in the brain tumor cells and throughout the rest of the brain to attack the invaded tumor cells which will be present under clinical settings (Fig. 5D).
Fig. 5.

Distribution of T-Platin-M-NPs in a dual tumor model. (A) Schematic representation showing the experimental design where biodistribution of T-Platin-M-NPs was studied in female BALB/c nude mice that were inoculated with MDA-MB-231-BRLuc cells subcutaneously at the right flank followed by secondary tumor implantation in the brain through stereotactic intracranial injection. Animals were randomly divided into 3 groups: saline (n = 5), cisplatin (n = 5), and polytherapy (n = 4). Organs were harvested for biodistribution post treatment. Animals in the saline group received 100 µL saline injection, those in the cisplatin group were administered with 5 mg/kg cisplatin, and animals in the polytherapy group received a combination of T-Platin-M-NP at 10 mg/kg dosage with respect to Platin-M and T-Mito-DCA-NP at 30 mg/kg with respect to Mito-DCA. Here, T-Mito-DCA-NP was administered 24 h post T-Platin-M-NP administration. The tail i.v. route was used for administering all the test articles. All major organs were harvested 24 h post the terminal dosage of T-Mito-DCA-NP. (B) Representative IVIS images showing the simultaneous appearance of primary tumor and brain tumor demarcated using red dotted circles. (C) Cell sorting performed using flow cytometry to sort the brain tumor initiating MDA-MB-231-BRLuc cells from the remaining brain cells to quantify the (D) %injected dose of platinum (Pt) in cisplatin- and T-Platin-M-NPs + T-Mito-DCA-NPs-treated mice. Biodistribution was quantified in sorted brain tumor cells, the remaining brain tissue, and in primary tumor. The statistical significance was determined using the unpaired t test and the data was represented as mean ± SD. Sorting of the tumor specific cells from the whole brain tissue was confirmed by using luciferase antibody in the whole brain suspension prior to sorting. For each of the three groups, the cell population which stained positive for luciferase was collected as brain tumor cells for further biodistribution analyses. The residual suspension was used for analyzing biodistribution in the remaining brain tissue. Luciferase-tagged MDA-MB-231-BRLuc cells were used as positive control and unstained cells were used as negative control. (E) Representative images showing the analyses of BBB integrity by using tumor-containing brain via immunofluorescence for tight junction protein ZO-1 in CD31 expressing brain endothelial cells and comparison with nude mice without any tumor confirming that the BBB remains intact after intracranial tumor implantation. (F) Quantification of ZO-1 in CD31 positive brain endothelial cells. Statistical analyses were performed using the unpaired t test.
To make sure that the BBB integrity was not compromised due to the stereotactic intracranial tumor implantation, brain tissues were analyzed for ZO-1 protein in the brain microvascular endothelial cells within the brain coronal sections from the tumor-containing mice and compared with BALB/c nude mice without tumor (Fig. 5E). The brain endothelial cells were marked with CD31 antibody. Quantification of ZO-1 intensity only in CD31 positive brain endothelial cells indicated a comparable ZO-1 expression in brain sections of tumor-containing mice with that seen in nontumor nude animals suggesting that postorthotopic implantation, the BBB integrity remains unaffected (Fig. 5F and SI Appendix, Fig. S20).
T-Platin-M-NP Inhibits Cellular Migration and Disrupts Actin Organization.
Cellular migration is an important step of the metastasis cascade, allowing cancer cells to metastasize to distant organs. Using a wound healing assay, we evaluated whether T-Platin-M-NP is capable of inhibiting cancer cell migration. Our data revealed that cisplatin inhibited cellular migration by 36% while Platin-M and T-Platin-M-NP inhibited 46% and 58%, respectively in MDA-MB-231-BR cells (SI Appendix, Figs. S21A and S22, for quantification). In MDA-MB-231 cells, cisplatin, Platin-M, and T-Platin-M-NP inhibited cellular migration by 43%, 51%, and 68%, respectively (SI Appendix, Figs. S21A and S22 for quantification). Actin reorganization is essential for cancer cells to migrate (43). We observed that cisplatin, Platin-M, and T-Platin-M-NP disrupt actin organization of MDA-MB-231-BR and MDA-MB-231 cells (SI Appendix, Fig. S21B). Furthermore, from RNAseq data of treated MDA-MB-231-BR cells, we cataloged the genes involved in cellular migration affected by cisplatin and T-Platin-M-NP. It was observed that cisplatin down-regulated 25 genes involved in cellular migration while T-Platin-M-NP down-regulated 35 genes (SI Appendix, Fig. S21C). Cellular adhesion and interaction with extracellular matrix components of surrounding tissue is a prerequisite for cancer invasion, migration, and metastasis formation. Adhesion assay using Matrigel indicated that both in MDA-MB-231 and MDA-MB-231-BR cells, pretreatment with cisplatin, Platin-M, or T-Platin-M-NP inhibited cellular adhesion (SI Appendix, Fig. S23, for quantification). For mechanistic analyses, we evaluated the effects on integrins, the heterodimeric transmembrane cell adhesion receptors which are crucial for cell adhesion to extracellular matrices, invasion, cellular migration and cancer metastasis (44). There are reports documenting potential roles of integrin β5 and β1 in breast carcinoma cell adhesion and migration (45, 46). Our analyses indicated that treatment with Platin-M and T-Platin-M-NP decreased integrin β5 in MDA-MB-231 and MDA-MB-231-BR cells but expression of integrin β1 remains unchanged (SI Appendix, Fig. S21 D and E).
Efficacy of T-Platin-M-NP and T-Mito-DCA-NP Combination in Dual Tumor Model.
To investigate the efficacy of the combinatorial therapeutic strategy to tackle tumors at a peripheral site and at the brain, we developed a dual tumor mouse model, expressing primary tumor at the right flank and secondary tumor in the brain (Fig. 6A). The mice were randomly divided to perform the therapeutic efficacy of T-Platin-M-NP + T-Mito-DCA-NP (SI Appendix, Fig. S24). We would like the reader to know that this kind of dual tumor models are technically challenging to develop as the uncertainty remains about animal survival since some animals develop so aggressive tumors that they will not survive. Thus, some of these studies needed to be staggered to have data from significant number of animals. In one experiment, our analyses of the NP treated animals through IVIS imaging revealed that the combination therapeutic NP-treated group showed significant reduction in both primary and secondary tumor proliferation (Fig. 6B). Quantification of both primary and secondary tumors using bioluminescence values from the tumor region of interest throughout the course of the study indicated that the animals which were treated with combination therapeutic NP experience less tumor burden at both the sites (Fig. 6C). Since most of the animals which were treated with saline did not survive, through two sets of experiments, we were able to analyze three animals from the saline-treated group and seven animals from the NP-treated group. We observed tumor reduction for both primary and secondary tumors in the NP-treated group compared to saline (Fig. 6D). Out of the seven animals that were treated with the combinatorial NPs, 3 animals survived past 32 d post treatment initiation whereas the saline-treated animals did not survive more than 19 d post treatment (Fig. 6E). The NP-treated group thus showed significantly increased survival (P = 0.0016) until day 32 post treatment (Fig. 6E) with no significant change in body weight (g) from predose through all the weeks of treatment regimen (Fig. 6F). Analyses of activity of cisplatin using the dual tumor model in another set of experiment documented no significant ability of cisplatin to reduce both the tumors compared to saline (SI Appendix, Fig. S25).
Fig. 6.
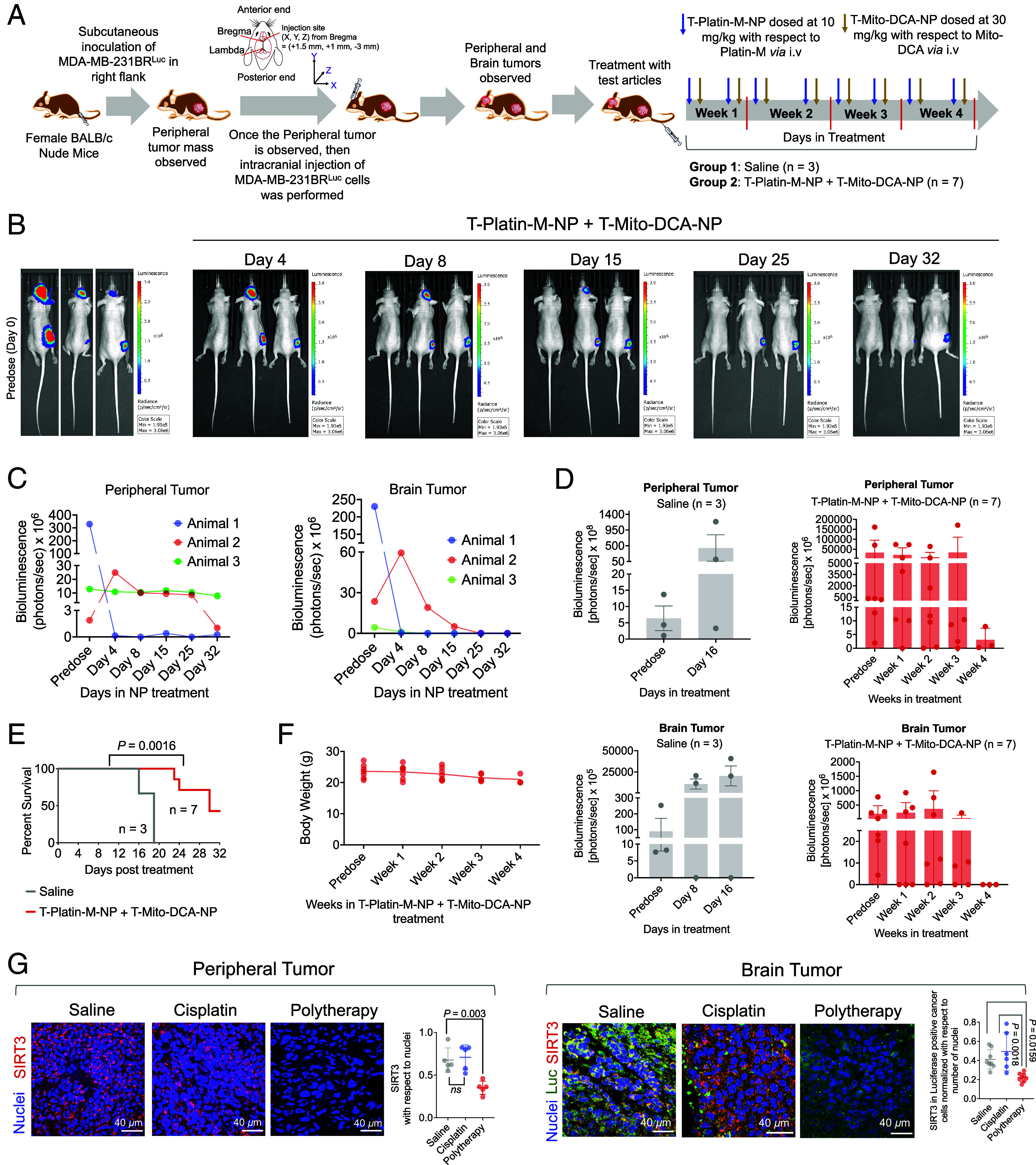
Combinatory effects of T-Platin-M-NP and T-Mito-DCA-NP in a dual tumor model. (A) A schematic diagram of the experimental details. (B) A representative comparison of three animals from the NP-treated group by IVIS imaging predose and at different time points post treatment with NP as well as on the terminal day of the study. (C) Comparison of the primary and secondary tumor volumes in terms of luminescence from those three animals. (D) Tumor volumes of primary breast cancer and secondary brain tumor from saline-treated and combination therapeutic–treated groups using two sets of experiments. (E) Kaplan–Meier analysis of survival comparison between saline and NP-treated groups. The statistical significance was determined by the log-rank (Mantel-Cox) test. (F) Body weight (g) distribution of all animals in the combinatorial NP treated group from predose through all the weeks of treatment regimen. (G) Representative immunofluorescence images illustrating the SIRT3 expression in peripheral tumor and brain tumor sections from saline-, cisplatin-, and polytherapy-treated groups. For the peripheral tumor sections, the relative change of SIRT3 expression was quantified with respect to nuclei. For the brain tumor sections, the mean intensity of SIRT3 was quantified in luciferase positive cancer cells with respect to number of nuclei. Ordinary one-way ANOVA was used to determine the statistical differences between the groups for both peripheral and brain tumor tissues.
Using the peripheral and brain tumor tissue samples from the above study, we performed ex vivo immunofluorescence analyses to evaluate the effect of polytherapy, which is the combination of T-Platin-M-NP and T-Mito-DCA-NP and compared the activity against saline- or cisplatin-treated samples. Polytherapy significantly reduced SIRT3 expression in both peripheral and brain tumor sections compared to saline and cisplatin highlighting its ability to perform mitochondrial OXPHOS inhibition in vivo at both the tumor sites (Fig. 6G and SI Appendix, Fig. S26 for peripheral tumor and SI Appendix, Fig. S28 for brain tumor). It is crucial to note that, since we used MDA-MB-231-BRLuc cells in the orthotopic tumor implantation, our focus was to specifically investigate SIRT3 in the luciferase expressing cells by using anti-luciferase antibody in the brain samples and the quantification indicated significantly less number of luciferase expressing tumor cells and reduced SIRT3 intensity in these cells in the polytherapy-treated group (Fig. 6G and SI Appendix, Fig. S28). Our analyses also included changes in expression of SIRT1, SIRT5, and SIRT6 in the peripheral tumor tissue with polytherapy (SI Appendix, Fig. S26). A reduction in SIRT6 suggested reduced tumor growth as augmented SIRT6 is a characteristic of an aggressive tumor growth. We further evaluated the expression of succinate dehydrogenase (SDHA), an OXPHOS-related protein and HKII, to probe the impact of sequentially administering T-Mito-DCA-NP 24 h post T-Platin-M-MP administration in inhibiting the metabolic plasticity in the peripheral tumor (SI Appendix, Fig. S27) as well as in brain tumor sections (SI Appendix, Fig. S28). We observed a significantly reduced expression of SDHA in both tumor sections and HKII in the brain tumor tissues by polytherapy (SI Appendix, Fig. S27).
Treatment Mediated Effects on Genomic Landscape of the Brain Tumor.
In our earlier study using the flank xenograft model as depicted in Fig. 4, we evaluated the genomic profiles of the flank tumor mimicking the primary tumor site after treatment. Thus, in this segment, we analyzed the genomic profile of the tumor site at the brain to understand the effects of the combination therapeutic NP treatment, abbreviated as polytherapy in Figs. 7 and 8 (39). Hierarchical indexing for spliced alignment of transcripts enabled us to map the differential expression in the polytherapy-treated group compared to saline or cisplatin (Fig. 7A). Our analyses of the mitochondrial genome indicated polytherapy mediated disruption of mitochondrial genes compared to the saline-treated group. On the contrary, these genes were up-regulated upon cisplatin treatment indicating participation of mitochondria for allowing cells to escape cell death in response to cisplatin (Fig. 7B). This result further strengthens our hypothesis that allowing cisplatin to access mtDNA using a mitochondria accumulating Pt(IV) prodrug such as Platin-M has the ability to overcome resistance. In depth analyses of FAO-related genes indicated significant downregulation of important genes such as ACADS, ACADSD, ACADVL, ECl1, ECl2, HADH, HADHA, and HADHB in polytherapy-treated tumors compared to saline; all these genes were up-regulated in cisplatin-treated animals (Fig. 7C). These genomic alteration data were further supported by our previous results which indicated that Platin-M induced suppression of SIRT3 leads to decreased fatty acid oxidation. Since our polytherapy contained Mito-DCA as a glycolytic inhibitor, we further analyzed the genes for glycolysis from the brain tumor samples (Fig. 7D). The main target gene of DCA is PDK1 and this gene was down-regulated in polytherapy-treated groups compared to saline or cisplatin (Fig. 7D). Most of the other genes related to glycolysis were down-regulated in the NP-treated group; few important ones are HK1, HK2, PDHB, PDHX, and PDHA1. Thus, the data presented in Fig. 7 C and D supported that the combination therapy is fully functional at controlling the metabolic plasticity of the secondary tumor at the brain site. Further analyses of the genes related to five complexes of the ETC indicated that most of these genes were down-regulated in the polytherapy-treated group in comparison with saline- or cisplatin-treated groups (Fig. 7E). The ability of the combination therapeutic NP in modulating metabolic plasticity was further reflected in apoptosis-inducing capacity of the treatment as shown in Fig. 7F; several proapoptotic genes were up-regulated and antiapoptotic genes were down-regulated in the NP-treated group (Fig. 7F).
Fig. 7.
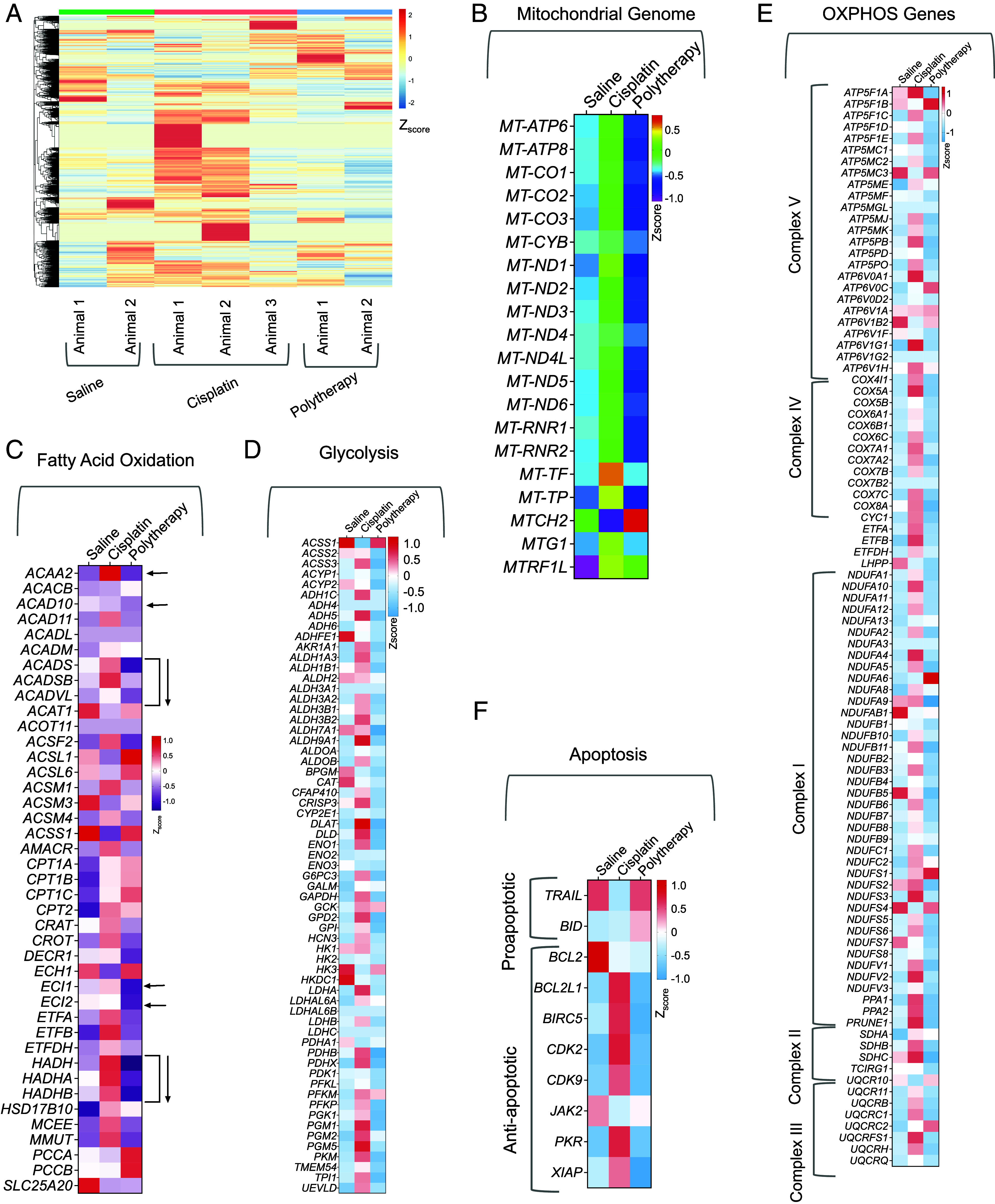
Genomic analyses of brain tumors for effects of polytherapy on tumor metabolism. (A) Hierarchical clustering of all genes from all the animals treated with saline, cisplatin, or combination therapeutic NPs (abbreviated as polytherapy). Gene expression profiles of (B) mitochondrial genome, (C) fatty acid oxidation–related genes, (D) glycolysis-related genes, (E) OXPHOS-related genes, and (F) apoptotic genes from the different treatment groups compared to saline.
Fig. 8.
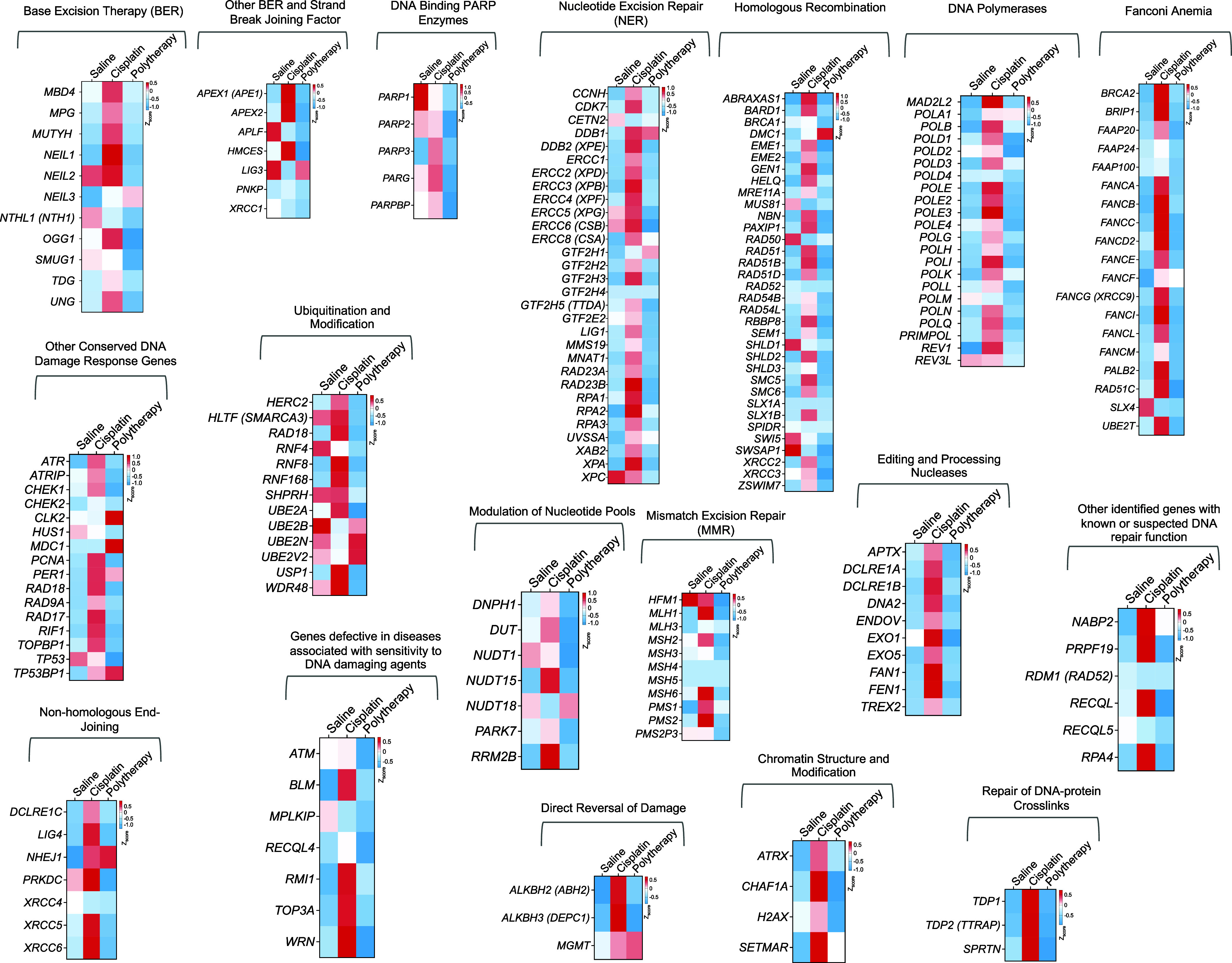
Effects on DNA repair pathways. Heat map representing the genes related to different DNA repair pathways including base excision repair, NER, homologous recombination, nonhomologous end joining, mismatch excision repair, and other genes helping in the processing of DNA damage repair. These include genes involved in strand break and joining, ubiquitination and modification, chromatin structure modification, DNA polymerases, DNA binding PARP enzymes, genes related in Fanconi anemia, and editing and processing nucleases.
Combination Therapeutic NP Mediated mtDNA Damage Evades DNA Repair.
Proficiency or deficiency of DNA repair pathways operative in different cancer types often drive resistance or sensitivity of a particular cancer toward cisplatin-based therapy. As a main defense toward cisplatin activity on cancer cells, the cells gear up their NER pathways against the DNA damage occurred by cisplatin binding. Thus, over the years several studies were conducted where cisplatin was combined with inhibitors of different DNA repair pathways (47–51). Instead of combining cisplatin with inhibitors of DNA repair pathways, we hypothesized that delivering cisplatin prodrug to the mitochondria can potentially overcome repair-induced resistance by forming mtDNA-Pt adducts that cannot be recognized by DNA-repair mechanisms. Human breast cancer brain metastases samples demonstrate overexpression of homologous recombinant genes BARD1 and RAD51 compared with the matched primary tumors (52). We thus analyzed the genes involved in different repair pathways in the tumors from the brain and compared the expressions between combination NP treatment with saline or cisplatin (Fig. 8). Our analyses indicated that a significant number of DNA repair genes in the cisplatin-treated group was up-regulated which can contribute to cisplatin resistance; in contrast, most of these repair machinery genes were either significantly down-regulated or remained unchanged in the polytherapy-treated group (Fig. 8).
Neurocognitive Parameters after Repeated T-NP Administration.
We further focused on whether the simultaneous primary and brain-distributing T-NPs have any impact on the neurocognitive parameters of normal mice in comparison to cisplatin treatment. We followed a treatment regimen where we repeatedly administered the T-NPs at the same dosage (300 mg/kg) that we inject for the combinatorial NP efficacy study. We also administered cisplatin at 5 mg/kg dosage biweekly. We performed an open-field test to monitor whether the animals are experiencing any anxiety-like symptoms. All the five mice that were treated with T-NPs showed no signs of anxiety-like behavior which is evident from their path trace that showed the mice comfortably explore the entire region of the unknown open field box (SI Appendix, Fig. S29A). All 5 mice in the cisplatin-treated group showed a tendency to be near the walls of the open field box suggesting they are experiencing anxiety (SI Appendix, Fig. S29A). The quantification of the path in SI Appendix, Fig. S29B suggested impaired locomotor activity in cisplatin-treated mice which is evident by the reduced number of beam breaks in each 5-min bin-blocks compared to the T-NPs. We also performed the tail suspension test in these mice to study depression-like behavioral pattern. Our results suggested no depression-like behavior from either cisplatin or T-NPs (SI Appendix, Fig. S29C). We performed a Y-maze test where we noticed a significant elapse in the time spent exploring the novel “N” arm of the maze in our T-NPs treated mice compared to the cisplatin-treated mice which reveals that the mice in the T-NP-treated group does not lose the enthusiasm toward exploring new areas (SI Appendix, Fig. S29D). We further conducted foot placement study to measure stride length and stride width of animals which received either cisplatin or T-NPs (SI Appendix, Figs. S29 E–G and S30). This study can potentially provide information on impairment in the locomotor activity of the mice. For each animal, the distance in millimeters between the hind limbs of the successive foot placements and the distance in millimeters between the left and right hind limbs were measured as the stride length and the stride width, respectively (SI Appendix, Figs. S29E and S30). The stride width was measured as the distance in millimeters between the left and right hind limbs (SI Appendix, Fig. S29E). We observed no significant disruption or impairment in the locomotion parameters in the T-NP-treated mice compared to saline (SI Appendix, Fig. S29 E–G). Taken together, the different behavioral studies revealed that although these NPs enter the brain, but the nanoparticles have no deleterious effects on the neurocognitive behaviors of the animals at the doses used in our studies.
In this work, we employed a biodegradable and biocompatible polymeric NP for simultaneous targeting and drug delivery to the hyperpolarized mitochondrial membranes of cancer cells at the peripheral site in addition to secondary brain tumor. Attacking metastatic brain tumor cells using this BBB-penetrating NP system and delivering combination therapeutics to disrupt metabolic adaptability was achieved to manage metastatic breast cancer. Secondary brain tumors are resistant toward commonly used chemotherapeutics such as cisplatin, often due to enhanced repair of Pt-nDNA adducts using NER. Our results demonstrated that targeting mtDNA has the potential to efficiently block mtDNA-induced cancer progression without possibility of repair since mitochondria lack NER machinery. The results presented in this manuscript are significant due to the following: 1) as it features a BBB-penetrating and mitochondrial targeting NP allowing for dual targeting and drug delivery to both primary tumor and secondary brain metastasis sites; 2) utilizes a mitochondrion-targeted cisplatin prodrug to overcome NER-based chemoresistance mechanisms; 3) uses brain-accumulating NPs which have no significant association with neurons and thus can lower cisplatin-induced peripheral neuropathy; 4) combats the metabolic flexibility of brain metastases through the pairing of the cisplatin prodrug to inhibit OXPHOS with glycolytic inhibitors. Thus, the delivery of combination therapeutic prodrugs to attack metabolic plasticity and mutated repair-resistant mtDNA using this BBB-penetrating NP can provide a therapeutic strategy to combat breast cancer brain metastasis.
Animals.
In this study, we used 4 to 5 wk old female BALB/c nude, 4 to 8 wk old female BALB/c Albino, and 13 wk old C57BL/6 male mice from Jackson Laboratory. Animal housing, surgical procedure including all the in vivo experimental protocols performed in this work were executed once it received prior approval from the Institutional Animal Care and Use Committee of University of Miami, Miller School of Medicine. Animal care and handling was followed in accordance with “The Guide for the Care and Use of Laboratory Animals” of American Association for Accreditation of Laboratory Animal Care, Animal Welfare Act, and other applicable federal and state guidelines. The animals were given free access to normal chow diet and water supply throughout the timeline of the study.
Materials and Methods
A detailed account of the chemicals, materials, instruments, methodologies followed for synthesis of NPs, experimental protocols for all in vitro and in vivo studies, and all additional data with relevance as cited in the context of the discussion in the main manuscript are provided in SI Appendix.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
S.D. acknowledges the financial support from Sylvester Comprehensive Cancer Center, the NCI funded Sylvester Comprehensive Cancer Center support grant 1P30CA240139, Bankhead Coley Cancer Research grant (8BC10), and Sylvester bridge funding award. We thank Dr. Joan Massagué from Memorial Sloan Kettering Cancer Center, for providing the MDA-MB-231-BR cells. We thank Dr. Eléonore Beurel and Dr. Eva M. Medina-Rodriguez for their help with the behavioral studies in mice. We thank Anuj Shah and Dr. Kolishetti for their critical comments on the manuscript.
Author contributions
S.D. designed research; A.A., S.S., M.Z.K., B.S., and A.S. performed research; A.A., M.Z.K., and B.S. contributed new reagents/analytic tools; A.A., S.S., M.Z.K., B.S., A.A.K., and S.D. analyzed data; S.D. provided resources and supervised the research; and A.A., S.S., M.Z.K., B.S., A.A.K., A.S., and S.D. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix. The sequencing data presented in this study have been deposited in the GEO repository under accession codes the RNA sequence data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE264575 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE264575) (39).
Supporting Information
References
- 1.Posner J. B., Management of brain metastases. Rev. Neurol. 148, 477–487 (1992). [PubMed] [Google Scholar]
- 2.Eichler A. F., Loeffler J. S., Multidisciplinary management of brain metastases. Oncologist 12, 884–898 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Wong J., Hird A., Kirou-Mauro A., Napolskikh J., Chow E., Quality of life in brain metastases radiation trials: A literature review. Curr. Oncol. 15, 25–45 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailleux C., Eberst L., Bachelot T., Treatment strategies for breast cancer brain metastases. Br. J. Cancer 124, 142–155 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khuntia D., Brown P., Li J., Mehta M. P., Whole-brain radiotherapy in the management of brain metastasis. J. Clin. Oncol. 24, 1295–1304 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Kodack D. P., Askoxylakis V., Ferraro G. B., Fukumura D., Jain R. K., Emerging strategies for treating brain metastases from breast cancer. Cancer Cell 27, 163–175 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simoes R. V., et al. , Metabolic plasticity of metastatic breast cancer cells: Adaptation to changes in the microenvironment. Neoplasia 17, 671–684 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dang C. V., Links between metabolism and cancer. Genes Dev. 26, 877–890 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeBerardinis R. J., Lum J. J., Hatzivassiliou G., Thompson C. B., The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Ward P. S., Thompson C. B., Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 21, 297–308 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jia D., et al. , Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. U.S.A. 116, 3909–3918 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Birch-Machin M. A., The role of mitochondria in ageing and carcinogenesis. Clin. Exp. Dermatol. 31, 548–552 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Tokarz P., Blasiak J., Role of mitochondria in carcinogenesis. Acta. Biochim. Pol. 61, 671–678 (2014). [PubMed] [Google Scholar]
- 14.Ferraro G. B., et al. , Fatty acid synthesis is required for breast cancer brain metastasis. Nat. Cancer 2, 414–428 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chatterjee A., Mambo E., Sidransky D., Mitochondrial DNA mutations in human cancer. Oncogene 25, 4663–4674 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Smiraglia D. J., Kulawiec M., Bistulfi G. L., Gupta S. G., Singh K. K., A novel role for mitochondria in regulating epigenetic modification in the nucleus. Cancer Biol. Ther. 7, 1182–1190 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wallace D. C., et al. , Sequence analysis of cDNAs for the human and bovine ATP synthase beta subunit: Mitochondrial DNA genes sustain seventeen times more mutations. Curr. Genet. 12, 81–90 (1987). [DOI] [PubMed] [Google Scholar]
- 18.Shay J. W., Werbin H., Are mitochondrial DNA mutations involved in the carcinogenic process? Mutat. Res. 186, 149–160 (1987). [DOI] [PubMed] [Google Scholar]
- 19.Baggetto L. G., Role of mitochondria in carcinogenesis. Eur. J. Cancer 29A, 156–159 (1992). [DOI] [PubMed] [Google Scholar]
- 20.Cavalli L. R., Varella-Garcia M., Liang B. C., Diminished tumorigenic phenotype after depletion of mitochondrial DNA. Cell Growth Differ. 8, 1189–1198 (1997). [PubMed] [Google Scholar]
- 21.Rosenberg B., Vancamp L., Trosko J. E., Mansour V. H., Platinum compounds—A new class of potent antitumour agents. Nature 222, 385–386 (1969). [DOI] [PubMed] [Google Scholar]
- 22.Shah A. S., Surnar B., Kolishetti N., Dhar S., Intersection of inorganic chemistry and nanotechnology for the creation of new cancer therapies. Acc. Mater. Res. 3, 283–296 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dhar S., Lippard S. J., “Current status and mechanism of action of platinum-based anticancer drugs” in Bioinorganic Medicinal Chemistry, Alessio E. Ed. (Wiley-VCH, Weinheim, Germany, 2011) pp. 79–95. [Google Scholar]
- 24.Jamieson E. R., Lippard S. J., Structure, recognition, and processing of cisplatin-DNA adducts. Chem. Rev. 99, 2467–2498 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Zamble D. B., Mu D., Reardon J. T., Sancar A., Lippard S. J., Repair of cisplatin–DNA adducts by the mammalian excision nuclease. Biochemistry 35, 10004–10013 (1996). [DOI] [PubMed] [Google Scholar]
- 26.Moggs J. G., Szymkowski D. E., Yamada M., Karran P., Wood R. D., Differential human nucleotide excision repair of paired and mispaired cisplatin-DNA adducts. Nucleic Acids Res. 25, 480–491 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marrache S., Pathak R. K., Dhar S., Detouring of cisplatin to access mitochondrial genome for overcoming resistance. Proc. Natl. Acad. Sci. U.S.A. 111, 10444–10449 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirschey M. D., et al. , SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pathak R. K., Marrache S., Harn D. A., Dhar S., Mito-DCA: A mitochondria targeted molecular scaffold for efficacious delivery of metabolic modulator dichloroacetate. ACS Chem. Biol. 9, 1178–1187 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kolb D., et al. , Metabolic modulation of the tumor microenvironment leads to multiple checkpoint inhibition and immune cell infiltration. ACS Nano 14, 11055–11066 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Marrache S., Dhar S., Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proc. Natl. Acad. Sci. U.S.A. 109, 16288–16293 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marrache S., Tundup S., Harn D. A., Dhar S., Ex vivo programming of dendritic cells by mitochondria-targeted nanoparticles to produce interferon-gamma for cancer immunotherapy. ACS Nano 7, 7392–7402 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Marrache S., Pathak R. K., Dhar S., “Formulation and optimization of mitochondria-targeted polymeric nanoparticles”, in Mitochondrial Medicine, Methods in Molecular Biology, Weissig V., Edeas M.. Eds. (Springer, New York, 2015), vol. 1265, pp. 103-112. [DOI] [PubMed] [Google Scholar]
- 34.Surnar B., et al. , Nanotechnology-mediated crossing of two impermeable membranes to modulate the stars of the neurovascular unit for neuroprotection. Proc. Natl. Acad. Sci. U.S.A. 115, E12333–E12342 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feldhaeusser B., et al. , Evaluation of nanoparticle delivered cisplatin in beagles. Nanoscale 7, 13822–13830 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Surnar B., et al. , Brain-accumulating nanoparticles for assisting astrocytes to reduce human immunodeficiency virus and drug abuse-induced neuroinflammation and oxidative stress. ACS Nano 15, 15741–15753 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pathak R. K., McNitt C. D., Popik V. V., Dhar S., Copper-free click-chemistry platform to functionalize cisplatin prodrugs. Chemistry 20, 6861–6865 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larsson N. G., et al. , Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 18, 231–236 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Ashokan A., et al. , Simultaneous targeting of peripheral and brain tumors with therapeutic nanoparticle to disrupt metabolic adaptability at both sites. NCBI Gene Expression Omnibus (GEO). https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE264575. Deposited 22 April 2024. [DOI] [PMC free article] [PubMed]
- 40.S. E. Calvo, K. R. Clauser, V. K. Mootha, MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 44, D1251–D1257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalathil A., et al. , New pathway for cisplatin prodrug to utilize metabolic substrate preference to overcome cancer intrinsic resistance. ACS Cent. Sci. 9, 1297–1312 (2023), 10.1021/acscentsci.3c00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang M., Du W., Acklin S., Jin S., Xia F., SIRT2 protects peripheral neurons from cisplatin-induced injury by enhancing nucleotide excision repair. J. Clin. Invest. 130, 2953–2965 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Svitkina T., The actin cytoskeleton and actin-based motility. Cold Spring Harb. Perspect. Biol. 10, a018267 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hamidi H., Ivaska J., Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 18, 533–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nistico P., Di Modugno F., Spada S., Bissell M. J., beta1 and beta4 integrins: From breast development to clinical practice. Breast Cancer Res. 16, 459 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bianchi-Smiraglia A., Paesante S., Bakin A. V., Integrin beta5 contributes to the tumorigenic potential of breast cancer cells through the Src-FAK and MEK-ERK signaling pathways. Oncogene 32, 3049–3058 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Helleday T., Petermann E., Lundin C., Hodgson B., Sharma R. A., DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 8, 193–204 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Neher T. M., Bodenmiller D., Fitch R. W., Jalal S. I., Turchi J. J., Novel irreversible small molecule inhibitors of replication protein A display single-agent activity and synergize with cisplatin. Mol. Cancer Ther. 10, 1796–1806 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jordheim L. P., et al. , Small molecule inhibitors of ERCC1-XPF protein-protein interaction synergize alkylating agents in cancer cells. Mol. Pharmacol. 84, 12–24 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Inoue A., et al. , A small molecule inhibitor of monoubiquitinated Proliferating Cell Nuclear Antigen (PCNA) inhibits repair of interstrand DNA cross-link, enhances DNA double strand break, and sensitizes cancer cells to cisplatin. J. Biol. Chem. 289, 7109–7120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chapman T. M., et al. , Catechols and 3-hydroxypyridones as inhibitors of the DNA repair complex ERCC1-XPF. Bioorg Med. Chem. Lett. 25, 4097–4103 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Mishra A. K., Dormi S. S., Turchi A. M., Woods D. S., Turchi J. J., Chemical inhibitor targeting thereplication protein A-DNA interaction increases the efficacy of Pt-based chemotherapy in lung and ovarian cancer. Biochem. Pharmacol. 93, 25–33 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix. The sequencing data presented in this study have been deposited in the GEO repository under accession codes the RNA sequence data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE264575 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE264575) (39).