

Abstract
Background:
CD8+ T cells (Tc2) become potent, steroid-insensitive pathogenic effector cells in experimental asthma undergoing transcriptional reprogramming to IL-13 production in the presence of IL-4. However, no studies have described the effects of hypoxia exposure on Tc2 differentiation.
Objective:
We determined the effects of hypoxia exposure on IL-13-producing CD8+ Tc2 cells.
Methods:
CD8+ transgenic OT1 cells, differentiated with IL-2 and IL-4 (Tc2 cells) were exposed to normoxia (21% O2) or hypoxia (3% O2) and IL-13 production in vitro was monitored. Following differentiation under these conditions, cells were adoptively transferred into CD8-deficient mice and lung allergic responses including airway hyperesponsiveness to inhaled methacholine were assessed. The effects of pharmacologic inhibitors of hypoxia inducible factor 1α (HIF-1α) and HIF-2α were determined as were responses in HIF-1α-deficient OT1 cells.
Results:
Under hypoxic conditioning, CD8+ Tc2 differentiation was significantly enhanced with increased numbers of IL-13+ T cells and increased production of IL-13 in vitro. Adoptive transfer of Tc2 cells differentiated under hypoxia restored lung allergic responses in sensitized and challenged CD8-deficient recipients to a greater degree than seen in recipients of Tc2 cells differentiated under normoxia. Pharmacologic inhibition of HIF-1α or genetic manipulation to reduce HIF-1α expression reduced the hypoxia-enhanced differentiation of Tc2 cells, IL-13 production, and the capacity of transferred cells to restore lung allergic responses in vivo. IL-4-dependent, hypoxia-mediated increases in HIF-1α and Tc2 differentiation were shown to be mediated through activation of JAK1/3 kinase and GATA3.
Conclusions:
Hypoxia enhances CD8+ Tc2-dependent airway hyperresponsiveness and inflammation through activation of HIF-1α. These findings coupled with the known insensitivity of CD8+ T cells to corticosteroids suggests that activation of the IL-4-HIF-1α-IL-13 axis may play a role in the development of steroid-refractory asthma.
Keywords: Asthma, CD8+ T cells, hypoxia, IL-13, HIF-1α
Graphical Abstract
Capsule summary:
Hypoxia exposure amplifies CD8+ Tc2 differentiation with increased release of IL-13 in vitro and augmentation of lung allergic responses in vivo. The increases in Tc2 differentiation under hypoxia were shown to be dependent on HIF-1α.
Introduction
Asthma is a complex clinical syndrome characterized by airway inflammation, airway hyperresponsiverness (AHR), and airflow obstruction. This complexity is underscored by the number of cell types, mediators, and pathways linked to the disease. A major advance in the treatment of asthma was achieved with the regular use of inhaled corticosteroids (ICS) as a controller medication. Nevertheless, despite diligent compliance and technique, a large percentage of patients with asthma fails to achieve control of their disease with current therapies1. A subset of patients with a limited response to ICS has been defined as severe asthma2, considered here as steroid-resistant (SR) asthma. Little is understood regarding the underlying mechanisms or pathways (endotypes) responsible for SR asthma and it is likely that many factors impede the achievement of asthma control. Defining the underlying mechanism(s) of SR asthma is a priority but few such endotypes have been well characterized3–5. One endotype, characterized by airway eosinophilia, CD4+ Th2 cytokine production, and steroid responsiveness has been best studied6. In contrast, asthma associated with prominent airway neutrophilia has been associated with steroid resistance7–9. Neutrophils, unlike eosinophils, fail to undergo apoptosis in the presence of corticosteroids9. Other cell types including mast cells and basophils, Th17 cells, and even some innate lymphocytes (ILCs) may be similarly insensitive to corticosteroids10–12, linking disease where these cells are prominent in the airways to SR asthma.
Accumulating evidence from studies of animal models and severe asthmatics has identified the prominence of a subset of CD8+ T cells in the airways and epithelium which may contribute to steroid-insensitivity and the pathophysiology of severe or fatal asthma13–15. These CD8+ T lymphocytes, unlike CD4+ T cells, are refractory to corticosteroids16 and have the capacity to undergo transcriptional reprogramming from a Tc1-IFNγ- to a Tc2-IL-13-producing lymphocyte subset17. Numbers of these CD8+ T cells correlated with reduced lung function and basement membrane thickening18–21. Moreover, increased numbers of CD8+T cells have been demonstrated in lung tissues following a fatal asthma attack22. In severe asthmatics, increased numbers of IL-13+CD8+ T cells have also been shown20. Central to CD8+ T cell reprogramming to IL-13 production was IL-4, which triggered a cascade of transcriptional events culminating in IL-13 production17.
Low oxygen tension in tissues (hypoxia) occurs in many pathological conditions including chronic inflammation and promotes expression of genes through post-translational modifications and stabilization of the a subunits (HIF-1α and HIF-2α) of hypoxia-inducible factors (HIFs)23. Although there is growing evidence that HIFs contribute to the pathophysiology of asthma24, 25, little is known about HIFs in the regulation of lung allergic immune responses. Hypoxia has often been considered to be an inhibitor of T lymphocyte function, although active and robust T cell responses occur at many hypoxic inflammatory sites26, 27. Hypoxia-related pathways have been shown to facilitate differentiation of CD8+ cytotoxic T cells leading to enhanced clearance of chronic virus infection and tumor cells28. Such findings indicate that in contrast to inhibition of some T cell functions, hypoxia exposure may enhance the functional differentiation of certain T cell subsets. Since HIF-1α expression was increased following ligation of the T cell receptor29, we determined whether hypoxia enhanced transcriptional reprogramming in CD8+ T cells through a HIF-dependent pathway. Further, we defined a role for hypoxia and HIF-1α in the enhanced reprogramming of CD8+ T cells to pathogenic IL-13-producing effector cells.
Materials and Methods
Animals
OT-1 TCR transgenic (OT-1) mice, Lck-Cre/OT-1, Lck-Cre/HIF-1αflox/OT-1 and homozygous CD8-deficient mice were bred in the animal facility at National Jewish Health (Denver, CO). OT-1 mice (C57BL/6 background) express a transgenic TCR specific for SIINFEKL peptide (OVA257–264)17· Lck-Cre/OT-1 mice were F2 progeny of Lck-Cre mice and OT-1 mice. Lck-Cre/HIF-1αflox/OT-1 mice were F2 progeny of Lck-Cre/HIF-1αfl0× mice (provided by Dr. Holger Eltzschig, University of Colorado Denver, Aurora, CO)30 and OT-1 mice. CD8-deficient mice were generated by targeting the CD8α chain gene in C57BL/6 mice31. Animal experiments in this study were conducted under a protocol approved by the Institutional Animal Care and Use Committee of National Jewish Health.
CD8+ T cell culture
CD8+ effector memory T cells were generated in vitro as described previously17, 32, 33. In brief, mononuclear cells were harvested from the spleens of OT-1 mice, followed by stimulation with 1 μg/mL SIINFEKL peptide (GenScript, Piscataway, NJ) for 1.5 hrs (20×106). Two days after culture, living cells were re-isolated using Histopaque (Sigma, St. Louis, MO) and cultured in complete RPMI medium 1640 that contained recombinant mouse IL-2 (20 ng/mL) (Peprotech, Rocky Hill, NJ) or IL-2+IL-4 (20 ng/mL) (Peprotech). For HIF inhibition experiments, DMSO, 1 μM BAY 87–2243 (Selleck Chemicals, Houston, TX) or 10 μM HIF-C2 (Xcessbio Biosciences, San Diego, CA) were added into the medium together with IL-2 or IL-2+IL-4. Cells were either cultured in a CO2 incubator in a normoxia (21% O2+5% CO2) or hypoxia chamber (3% O2+5% CO2) at 37°C. Cytokines and inhibitors were changed every day for a further 4 days of culture. During hypoxia conditioning, O2 and CO2 levels were controlled by Proox C21 (BioSpherix, Lacoma, NY). For JAK inhibition experiments, DMSO, or 25, 50, or 100 nM of the inhibitor R256 (Rigel Pharmaceuticals, South San Francisco, CA) was added to the medium together with IL-2+IL-4. R256 was shown to be a selective inhibitor of JAK1/3 signaling as described34. Cells were cultured in a CO2 incubator at 37’C and medium with cytokines and inhibitors were changed every day for a further 4 days of culture. For flow cytometry analyses, cells were restimulated with 1 μg/mL SIINFEKL in medium containing 2 μM monensin (Calbiochem, San Diego, CA) for 4 hrs. For ELISA, cells were restimulated with 1 μg/mL SIINFEKL in medium for 4 hrs at a concentration of 1×106 cells/mL.
Steroid-induced apoptosis and effects on cytokine production
To study steroid-induced cell apoptosis, thymocytes were isolated from C57BL/6 mice. Tc1 and Tc2 cells were differentiated from OT1 cells for 4 days. Cells (1×106), differentiated under normoxia or hypoxia, were cultured in the presence of 0, 10, 100, 1000 nM dexamethasone (DEX) (Sigma, St. Louis, MO) dissolved in methanol for 24 hrs. Annexin V-positive cells were detected by eBioscienceTM Annexin V Apoptosis Detection Kit APC (Thermo Scientific, Grand Island, NY) and analyzed by flow cytometry.
To study the impact of DEX on cytokine production, Tc2 cells were differentiated under normoxia or hypoxia. Cells (1×106) were incubated with 0, 1, 10, 100nM DEX dissolved in methanol for 1 hr. Cells were then restimulated with 1 μg/mL SIINFEKL in medium for 4 hrs at a concentration of 1×106 cells/mL. Supernatant IL-13 production was measured by ELISA.
RNA preparation and analyses
Total RNA was extracted from 1×106 differentiated CD8+ T cells using the NucleoSpin RNA II isolation kit (Macherey-Nagel, Düren, Germany) following the manufacturer’s protocol. Reverse transcription of total RNA (1 μg) was performed using the QuantiTect reverse transcription kit (Qiagen, Valencia, CA) following the manufacturer’s protocol. Quantitative real-time PCR was performed using the SYBR Green RT-PCR Kit and the ABI 7500 fast real-time PCR system (Applied Biosystems, Foster City, CA). Primers for GATA3 were as follows: Gata3 forward 5’-GACTCTTCCCACCCAGCAGC-3’, reverse 5’-CCATCTCGCCGCCACAG-3’. Primers for T-bet were as follows: Tbx21 forward 5’-CAACCAGCACCAGACAGAGATG-3’, reverse 5’-GACCACATCCACAAACATCCTG-3’. Primers for HIF-1α were as follows: Hif-1α forward 5’-AACGACCACTGCTAAGGCAT-3’, reverse 5’-GGCT CCTT GGAT GAGCTTT G-3’.
The determined cycle threshold (CT) reflects the number of PCR cycles required for the fluorescence signal to exceed the detection threshold, which was set to the log-linear range of the amplification curve. The differences in CT values of the gene of interest and the house keeping gene 18SrRNA were used to calculate delta CT (ΔCT) values. Relative fold changes were then calculated using the 2-ΔΔCT algorithm.
lmmunoblot analyses
CD8+ T cells (5×106) were lysed in SDS buffer containing Halt protease and phosphatase inhibitor mixture (Thermo Scientific, Grand Island, NY) and sonicated for 10 seconds. Samples were heated for denaturation at 95°C and analyzed by SDS/PAGE and transferred to nitrocellulose membranes. The membranes were blocked using buffer containing 5% (wt/vol) non-fat milk and 0.5% sodium azide in tris-buffered saline with tween (TBST) for 1 hr and incubated with rabbit anti-HIF-1α mAb (Clone D2U3T, Cell Signaling, Boston, MA) overnight at 4°C. Horseradish peroxidase-conjugated anti-rabbit lgG (GE Healthcare, Wauwatosa, WI) was used to detect HIF-1α protein. Mouse monoclonal anti-β-actin antibody (Sigma) was used as an internal control. Immunoreactive bands of Western blots were quantified by densitometric quantification of autoradiographs using lmageJ (National Institutes of Health) and expressed as relative HIF-1α normalized by β-actin.
CD8+ T cell transfection
All retroviral constructs were derived from the MSCV2.2-IRES-GFP retroviral vector (provided by Dr. J. Hagman, National Jewish Health, Denver, CO). Generation of retroviruses and infection of cells were described previously35. Briefly, Phoenix packaging cells were transfected in a 6-well plate with 20 μg retroviral vector (with GATA3 shRNA or scrambled shRNA) using 10 μL/well Lipofectamine 2000 (lnvitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Viral supernatants were collected after 24–48 hrs. On day 0, 2–3 mL of viral particles were used to transduce 1×106 CD8+ T cells. Medium with IL-2+IL-4 was changed every day for an additional 4 days. GFP+ cells were sorted on day 4 using the MoFlo flow cytometer (Beckman Coulter, Fort Collins, CO).
Flow cytometric analyses
Intracellular staining of CD8+ T cells was carried out following stimulation SIINFEKL as described above. Cells were incubated with anti-mouse CD16/CD32 (2.4G2; BD Bioscience, San Jose, CA) at 4O for 5 m inutes. After fixation with fixation buffer (BD Biosciences) and permeabilization with 0.1% saponin (Sigma), cells were then stained with APC-labeled anti-mouse IFNγ (XMG 1.2; eBioscience, San Diego, CA) and PE-labeled anti-mouse IL-13 (eBio13A; eBioscience). For staining of phosphorylated STAT6 and GATA3, CD8+ T cells differentiated in IL-2 + IL-4 for 1, 4, 24 hrs without SIINFEKL stimulation. Cells were stained with APC-labeled anti-mouse CD8a (53–6.7; eBioscience) and FITC-labled anti-mouse CD3(17A2; eBioscience) then stained with PE-labeled anti-human/mouse GATA3 (TWAJ; eBioscience) or cells were stained with FITC-labeled anti-mouse CD8a (53–6.7; eBioscience) and PE-labeled antimouse CD3 (17A2; BD Biosciences) then stained with Alexa Fluor 647-labeled antimouse STAT6 (pY641; BD Biosciences). Cell staining was analyzed using the Flowjo software (Tree Star, Ashland, OR).
Secondary allergen challenge model and adoptive transfer
The secondary challenge model was used as previously described36 to reduce the time between cell manipulation, transfer, and assay. The experimental protocol for sensitization and challenge to OVA was as described previously16, 36, with some modifications. CD8-deficient mice were sensitized with 20 μg of OVA (Calbiochem, Calbiochem, La Jolla, CA) emulsified in 1.0 mg of alum (Alumlmuject; Pierce, Rockford, IL) on days 0 and 14 by intraperitoneal injection. Mice were challenged (primary) with0.2% OVA for 20 minutes on days 21, 22, and 23 using an ultrasonic nebulizer (model NE-U07; Omron Healthcare, Kyoto, Japan). CD8+ T cells (8×105) generated in medium containing IL-2+IL-4 under 21% O2 or 3% O2 were injected intravenously into OVA-sensitized CD8-deficient mice on day 37. Two hrs after transfer, mice were challenged (secondary) with 1% OVA for 20 minutes. Airway function was measured and samples were collected on day 39. Airway function was assessed as described previously37. Briefly, changes in airway resistance (RL) of anesthetized and intubated mice were measured using a whole body plethysmography-based system with artificial ventilation in response to increasing doses of inhaled methacholine (Sigma). Data are presented as percent change from the baseline RL values after saline inhalation. Baseline RL values were not significantly different among the various groups.
BAL analyses
After measurement of airway function, lungs were lavaged via the tracheal tube with 1 mL of HBSS. The supernatants were collected, and IFNγ, IL-4, IL-5, and IL-13 (eBioscience) levels were measured by ELISA and total leukocyte numbers were counted and differentiated as described previously16,31.
Lung histology
Lungs were fixed in 10% formalin, and then embedded in paraffin16‘17‘31–33‘38 Paraffin sections (5-μm thick) were stained with PAS. Mucus-containing goblet cells were quantified. Histology analyses was performed in a blinded manner under light microscopy linked to an image system. The numbers of PAS-positive goblet cells were determined in cross-sectional areas of the airway wall. Six to ten different sections were evaluated per animal. The measurements were averaged for each animal and the mean values (±SEM) were determined for each group.
Statistical analysis
Data from in vivo experiments were from at least three independent experiments with four mice per group or where the number of replicates is indicated. Results were expressed as means±SEM. Student’s two-tailed t test was used to determine the levels of difference between the two groups. ANOVA was used to determine the levels of difference among more than three groups. Nonparametric analysis using the Mann-Whitney U test or Kruskal-Wallis test was also used to confirm that the statistical differences remained significant even if the underlying distribution was uncertain. The P values for significance were set to 0.05 for all tests.
Results
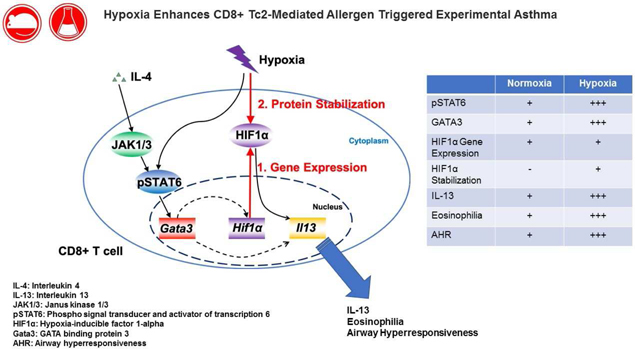
Hypoxia enhances IL-4-dependent CD8+ T cell transcriptional reprogramming
To determine the effects of hypoxia on CD8+ T cell differentiation, splenic OT-1 CD8+ T cells were differentiated in vitro for four days in the presence of IL-2 or IL-2+IL-4 under normoxic (21% O2) or hypoxic (3% O2) conditions. As previously shown17, in the presence of IL-2, CD8+ T cells were predominantly IFNγ-producers (Tc1). In the presence of IL-2 and IL-4, a marked increase in IL-13+ T cells (Tc2) was observed when cultured under normoxic conditions (Figs. 1A–1C). Following differentiation, CD8+ Tc1 and Tc2 cells remained resistant to DEX-induced apoptosis, unlike, for example, thymocytes (Fig. S1A). When cells were differentiated under hypoxia, the effect on Tc1 cells was minimal. In contrast, when Tc2 cells were differentiated under hypoxia, the number of IL-13+ cells was markedly increased and IFNγ+ cell numbers were correspondingly decreased. Following in vitro culture of these cells in the presence of IL-2+IL-4 (4 days) and antigen activation (4 hrs), the levels of IL-13 released from cells cultured under hypoxia were more than double compared to cells cultured under normoxic conditions (Fig. 1D). As shown in Figure 1, the effects of hypoxia exposure were only seen in the presence of IL-4 and were restricted to Tc2 and not Tc1 differentiation. When CD4+ T cells were differentiated under Th2 polarizing conditions, hypoxia conditioning did not enhance IL-13 production (data not shown). There was little difference between cells differentiated in 3% or 1% oxygen when numbers of IL-13+ cells or IL-13 production were examined (data not shown). As cell viability was lower under 1% oxygen conditioning, all further hypoxia experiments were carried out under 3% oxygen. When cytokine production was monitored following addition of DEX, hypoxia-treated CD8+ T cells were less affected than the normoxia-exposed group. Even at the highest dose of DEX, cytokine production was largely maintained (Fig. S1B).
Figure 1: Th1 and Th2 cytokine expression in CD8+ T cells differentiated in IL-2 or IL-2+IL-4 under normoxia or hypoxia.

CD8+ T cells were differentiated in vitro for 4 days under normoxia (21% O2) or hypoxia (3% O2) in the presence of IL-2 or IL-2+IL-4 followed by SIINFEKL re-stimulation. (A) Representative results (%) of cells staining for IFNγ and IL-13 following SIINFEKL stimulation. (B) Percentages of IL-13 single-positive cells from five independent experiments. (C) Percentages of IFNγ single-positive cells from five independent experiments. (D) Protein levels of IL-13 in 4-day cultures following differentiation in IL-2 or IL-2+IL-4 and activation by SIINFEKL. Results (means±SEM) are from 5 independent experiments. ns: not significant, **p<0.01.
Enhancement of allergen-induced airway responses following adoptive transfer of hypoxia-exposed Tc2 cells
Adoptive transfer of Tc2 cells prior to challenge of sensitized CD8-deficient recipients resulted in the full restoration of AHR, mucus hyperproduction, and airway eosinophilia17. Since hypoxia enhanced Tc2 differentiation in vitro, we compared in vivo outcomes after adoptive transfer of Tc2 cells exposed to either normoxia or hypoxia into OVA-sensitized or non-sensitized CD8-deficient mice prior to secondary allergen challenge. OVA-sensitized recipients of Tc2 cells differentiated under normoxia developed airway allergic responses including methacholine-induced AHR and airway eosinophilia (Figs. 2A and 1b). OVA-sensitized recipients of Tc2 cells differentiated under hypoxia showed significantly increased AHR and airway eosinophilia compared to OVA-sensitized recipients of Tc2 cells differentiated under normoxia. Non-sensitized recipients of Tc2 cells differentiated under normoxia did not develop allergic airway responses.
Figure 2: Adoptive transfer of hypoxia-exposed CD8+ Tc2 cells into OVA-sensitized CD8-deficient recipients enhances AHR and airway inflammation.


CD8-deficient mice were initially sensitized and challenged and received no cells (OVA/OVA), 4×105 CD8+ T cells differentiated in IL-2+IL-4 under normoxia (OVA/OVA + Tc2_Normoxia) or 4×105 CD8+ T cells differentiated in IL-2+IL-4 under hypoxia (OVA/OVA + Tc2_Hypoxia) prior to secondary challenge. As control, CD8-deficient recipients were not initially sensitized but challenged and received no cells (PBS/OVA) or 4×105 CD8+ T cells differentiated in IL-2+IL-4 under normoxia (PBS/OVA + Tc2_Normoxia) prior to secondary challenge. (A) Changes in RL were measured in response to increasing concentrations of inhaled MCh. (B) Eosinophil numbers in BALfluid. (C) Representative photomicrographs of lung histology (PAS staining): I) PBS/OVA, II) OVA/OVA, III) PBS/OVA + Tc2 normoxia, IV) OVA/OVA + Tc2 normoxia, and V) OVA/OVA + Tc2 hypoxia. (D) Quantitative analysis of goblet cells. Data for A, B, and D (means±SEM) are from 2 independent experiments with 8 mice/group. *p<0.05 compared to PBS/OVA and OVA/OVA, +p<0.05 compared to OVA/OVA + Tc2 normoxia.
Lung tissue sections were processed for histological analyses and areas stained with PAS were measured. As shown in Figures 2C and 2D, OVA-sensitized recipients of Tc2 cells differentiated under hypoxia showed marked increases in inflammatory cell accumulation and significantly increased numbers of mucus-producing goblet cells compared to OVA-sensitized recipients of Tc2 cells differentiated under normoxia.
When bronchoalvelolar lavage (BAL) fluid was assayed at 48 hrs for cytokine levels (the time AHR and airway inflammation peaked) levels of IFNγ, IL-4, IL-5, and IL-13 were similar in both sensitized recipient groups (Fig. S2). This was not surprising, as in the secondary challenge model, cytokine levels between groups were most different at 6 hrs after the last challenge36.
Hypoxia exposure leads to increased HIF-1α, pSTAT6, and GATA3 protein levels in Tc2 cells
Hypoxia inducible factors are transcription factors that facilitate cellular responses to hypoxia and play different roles depending on the specific T-cell subset examined23, 39. To determine the molecular events that accompanied IL-4-dependent Tc2 differentiation under normoxia or hypoxia, mRNA expression levels of Hif-1α, Tbx21, and Gata3 were examined. When differentiated in IL-2+IL-4, increases in Hif-1α and Gata3 and decreases in Tbx21 mRNA were detected, but no significant differences in expression levels of these genes were detectable in cells differentiated under normoxia or hypoxia for 96 hrs (Fig. S3). However, as shown in Figure 3, HIF-1α, phosphorylated signal transducer and activator of transcription 6 (pSTAT6) and GATA3 protein levels were significantly increased at 24 hrs following hypoxia exposure in Tc2 cells compared to cultures carried out under normoxia. pSTAT6 protein levels measured as early as 1 hr and 4 hrs following hypoxia exposure were not increased and increases were first detected at 24 hrs following hypoxia exposure (Fig. S4). These data indicated that hypoxia-enhanced Tc2 differentiation was associated with the upregulation of HIF-1α, pSTAT6, and GATA3 protein levels at an early time-point during differentiation.
Figure 3: Lineage-specific transcription factor protein expression in CD8+ T cells.

CD8+ T cells were differentiated in IL-2 or IL-2 plus IL-4 under normoxic or hypoxic conditioning. A, HIF-1α protein levels detected by using immunoblot analysis in differentiated CD8+ T cells. B and C, Representative flow cytometric analyses of pSTAT6 (Fig 3, B) and GATA-3 (Fig 3, C) protein expression in TC2 cells differentiated in IL-2 plus IL-4 for 24 hours. Cells were gated on the CD8+ population. Solid line, Differentiation under normoxic conditioning; dashed line, differentiation under hypoxic conditioning. Experiments were repeated 3 times with similar results.
Following binding of IL-4 to the IL-4 receptor, Janus kinase 1 (JAK1) and Janus kinase 3 (JAK3) were activated, leading to the activation and phosphorylation of STAT6 in CD8+ T cells40. To determine the events leading to IL-4-dependent regulation of HIF-1α, we added an inhibitor of JAK1 and JAK3 during CD8+ T-cell differentiation. We previously showed this inhibitor to be selective for Th2 and not Th1 or Th17 cells. When administered to allergen sensitized and challenged mice, inhibition significantly reduced the full spectrum of lung allergic responses34. As shown in Figure S5A, Tc2 differentiation as determined by numbers of IL-13+CD8+ T cells was reduced in the presence of the inhibitor in a dose-dependent manner. In parallel, Hif-1α mRNA expression was significantly reduced with increasing concentrations of the inhibitor (Fig. S5B).
As previously demonstrated, GATA3 was induced by IL-4 during CD8+ Tc2 differentiation17. To determine whether GATA3 regulates Hif-1α gene expression during Tc2 differentiation, CD8+ T cells differentiated in the presence of IL-2 and IL-4 under normoxia were transfected with a green fluorescent protein (GFP)-encoding virus containing an shRNA construct specific for mouse GATA3. A noneffective 29-mer scrambled shRNA was used as a control. Four days after virus infection and differentiation in the presence of IL-2+IL-4, GFP-positive cells were sorted and Tc2 differentiation and Hif-1α mRNA expression were assessed. Among the GFP-positive cells, the numbers of IL-13+ cells identified following antigen-restimulation were significantly lower in cells transfected with GATA3 shRNA in comparison to the scrambled shRNA (Fig. S5C). In parallel, Hif-1α gene expression was significantly lower in the GATA3 shRNA transfected, GFP-positive cells (Fig. S5D). Taken together, these data suggested that in CD8+ T cells, Hif-1α gene expression was upregulated by IL-4 through JAK1/3 and GATA3 activation.
Inhibition of HIF-1α impairs Tc2 differentiation under both normoxia and hypoxia
HIFs form a heterodimeric complex which acts as a transcriptional regulator of genes whose promoters contain hypoxia response elements (HREs)23. This complex is comprised of HIF-1β, which is constitutively expressed, and either of the HIF-1α isoforms, HIF-1α or HIF-2a23. To determine the role of HIF-1α or HIF-2α in CD8+ Tc2 differentiation, we examined the influence of two HIF inhibitors, BAY 87–2243 and HIF-C2. BAY 87–2243 targets mitochondrial complex 1 and is suggested to inhibit HIF-1α41. HIF-C2 is a potent and selective allosteric inhibitor of HIF-2α binding within the internal cavity of HIF-2a, disrupting hetero-dimerization of the full-length HIF-2 transcription factor42, 43. As shown in Figure 4A, BAY 87–2243 impaired Tc2 differentiation under normoxic and hypoxic conditioning. When culture supernatants from TCR-stimulated Tc2 cells were compared, IL-13 secretion was also markedly reduced by BAY 87–2243 (Fig. 4B). In contrast, the inhibition of HIF-2α by HIF-C2 in vitro did not affect Tc2 differentiation under either normoxic or hypoxic conditioning (Fig. 4B). These results suggested that HIF-1α but not HIF-2α played a critical role in IL-4-dependent Tc2 differentiation and the enhancement seen under hypoxic conditioning.
Figure 4: Effect of HIF inhibitors on Tc2 differentiation.

The vehicle (DMSO) or inhibitors BAY 87–2243 (BAY) or HIF-C2 were added during CD8+ T cell differentiation. (A) Results of intracellular staining for IL-13 (% positive cells) following SIINFEKL re-stimulation of cells differentiated in IL-2 or IL-2+IL-4 under normoxia or hypoxia. (B) IL-13 protein levels after SIINFEKL re-stimulation of cells differentiated in IL-2 or IL-2+IL-4 under normoxia or hypoxia. Data (means±SEM) are from 5 independent experiments. **p<0.01, compared with cells differentiated DMSO or HIF-C2 in each group.
Consequences of HIF-1α deficiency in Tc2 differentiation under hypoxic conditioning
To directly determine the role of HIF-1α in the IL-4-dependent transcriptional reprogramming of CD8+ T cells, CD8+ T cells were harvested from progeny derived from crossing Lck-Cre/HIF-1αflox mice in which T cells from OT-1 mice were rendered deficient in HIF-1α (HIF knock-out (KO)). Cells from Lck-Cre mice crossed with OT-1 mice served as (wild type, WT) controls. Cells were differentiated in the presence of IL-2 or IL-2+IL-4 under normoxia or hypoxia. As shown in Figure S6, HIF KO CD8+ T cells were not completely deficient in HIF-1α; levels were reduced by approximately 85% in T lymphocytes including CD8+ T cells isolated from HIF KO mice. HIF KO CD8+ T cells, similar to the WT controls, were fully capable of Tc1 differentiation in the presence of IL-2 when differentiated under normoxia or hypoxia (Fig. S7), further confirming the selectivity of the effects of hypoxia on Tc2 conversion and the normalcy of responses to IL-2 (Tc1) in the genetically-manipulated HIF KO mice. Under normoxia, Tc2 differentiation was not significantly altered in CD8+ T cells from HIF KO mice compared to cells from the WT controls. However, the frequency of IL-13 single-positive cells was significantly lower in HIF KO compared to WT CD8+ T cells differentiated under hypoxia (Fig. 5A). In parallel, analyses of secreted protein levels in cell culture supernatants confirmed significantly decreased IL-13 production from HIF KO Tc2 cells compared to WT Tc2 cells cultured under hypoxia (Fig. 5B). HIF-1α deficiency did not significantly alter IL-13 secretion when the cells were differentiated under normoxia. These data confirmed that the increased Tc2 differentiation seen under hypoxic conditioning was dependent on HIF-1α.
Figure 5: IL-13 expression in differentiated HIF-1-α-deficient CD8+ T cells.

CD8+ T cells from Lck-Cre/OT-1 (WT) and HIF-1α-deficient Lck-Cre/HIF-1αflox/OT-1 (HIF KO) mice were differentiated in vitro in the presence of IL-2 or IL-2 plus IL-4, followed by SIINFEKL restimulation. A, Results (percentage positive) of intracellular staining for IL-13 in cells differentiated in IL-2 plus IL-4 and SIINFEKL restimulation. B, IL-13 protein levels in cells differentiated in IL-2 or IL-2 plus IL-4 under normoxic or hypoxic conditioning after SIINFEKL restimulation. Data (means ± SEMs) are from 5 independent experiments. *P < .05.
Impaired development of allergen-induced airway responses following adoptive transfer of hypoxia-exposed HIF-1α-deficient Tc2 cells
Since hypoxia enhanced both Tc2 differentiation in vitro and allergic airway responses after adoptive transfer of Tc2 cells exposed to hypoxia into sensitized CD8-deficient mice prior to secondary allergen challenge, we compared the outcomes of transfer of WT and HIF-1α-deficient Tc2 cells differentiated under hypoxia. Recipients of WT Tc2 cells differentiated under hypoxia developed significant airway allergic responses including methacholine-induced AHR and airway eosinophilia (Figs. 6A and 6B) compared to mice that did not receive any cells. Recipients of HIF-1α-deficient Tc2 cells differentiated under hypoxia showed significantly reduced enhancement of AHR and airway eosinophilia compared to the recipients of WT Tc2 cells differentiated under hypoxia (Figs. 6A and 6B).
Figure 6: Adoptive transfer of hypoxia-exposed HIF-1α–deficient TC2 cells results in reduced restoration of lung allergic responses in CD8-deficient recipients.


CD8-deficient mice were initially sensitized and challenged and received no cells (vehicle), 4 × 105 WT CD8+ T cells differentiated in IL-2 plus IL-4 under hypoxic conditioning (WT Tc2), or 4 × 105 HIF-1α-deficient CD8+ T cells differentiated in IL-2 plus IL-4 under hypoxic conditioning (HIF KO Tc2) before secondary challenge. A, Changes in RL were measured in response to increasing concentrations of inhaled methacholine (MCh). B, Eosinophil numbers in bronchoalveolar lavage fluid. C, Representative photomicrographs of lung histology. H&E, Hematoxylin and eosin. D, Quantitative analysis of PAS+ areas. Data (means ± SEMs) are from 2 independent experiments with 8 mice per group. *P < .05 compared with vehicle and HIF KO Tc2.
Lung tissue sections were processed for analyses of histological changes and PAS+ cells. As shown in Figures 6C and 6D, recipients of HIF-1α-deficient Tc2 cells differentiated under hypoxia showed marked decreases in inflammatory cell accumulation and significantly decreased numbers of mucus-producing goblet cells compared to recipients of WT Tc2 cells differentiated under hypoxia.
Discussion
Despite the findings that increased hypoxia-related responses have been demonstrated in asthma, the specific pathways activated or mechanistic consequences have not been well defined, especially in the context of disease pathobiology. The goals of this study were to delineate the role of hypoxia-related responses in mice specifically as they pertained to CD8+ Tc2-mediated experimental asthma. We demonstrated that CD8+ T-cell differentiation to IL-13-producing Tc2 cells was enhanced by hypoxia exposure with significantly increased numbers of IL-13-producing cells and, following culture, significantly increased IL-13 production. These effects were specific to Tc2 cells in the presence of IL-4 and were not detected in Tc1 cells or CD4+ Th2 cells. In Tc2 cells differentiated under hypoxia, increased levels of HIF-1α, pSTAT6, and GATA3 protein were detected. Sensitized and challenged CD8-deficient mice which received Tc2 cells differentiated under hypoxia prior to secondary challenge developed enhanced airway reactivity to inhaled methacholine and eosinophilic airway inflammation compared to recipients of Tc2 cells differentiated under normoxia.
Hypoxia inducible factors serve as decisive transcription factors of a variety of biological responses23. HIFs are heterodimeric proteins containing one α subunit and one β subunit. While the β subunit is constitutively expressed, the α subunits are degraded by von Hippel Lindau complex but are stabilized under hypoxia23. Previous studies have demonstrated that HIF-1α may exhibit different activities in asthma44–46. The nonspecific HIF inhibitor 2-methoxyestradiol (2ME) reduced pulmonary inflammatory and airway remodeling responses to ovalbumin (OVA) in mice45. In mice heterozygous for a HIF-1α null allele, diminished lung eosinophilia in response to OVA challenge was demonstrated47. Following exposure of mice to a hypoxic environment (10% O2) during allergen challenge, the contributions of hypoxia to airway inflammation and remodeling were determined48. Although neither hypoxia nor OVA alone induced significant neutrophil influx into the lung, the combination of OVA and hypoxia induced a synergistic increase in peribronchial neutrophils and enhanced expression of HIF-1α and the CXC-family neutrophil chemokine KC. In addition, the combination of hypoxia and OVA increased eotaxin-1, peribronchial eosinophilia, lung TGF-β1 expression, and indices of airway remodeling (fibrosis and smooth muscle) compared to either stimulus alone.
To define the role of the α subunits of HIFs in the hypoxia-induced enhancement of CD8+ Tc2 cell differentiation, HIF-1α and HIF-2α inhibitors were utilized. Although there is some controversy as to the specificity of these inhibitors41–43, our data suggested that HIF-1α but not HIF-2α was essential to the enhancement of Tc2 differentiation under hypoxic conditioning. When the two inhibitors were compared, HIF-C2 a selective allosteric inhibitor of HIF-2α was shown to have no effect on Tc2 conversion in cells exposed to normoxia or hypoxia; HIF-1α inhibitor, BAY 87–2243 led to decreases in IL-13+ Tc2 cells and IL-13 production from cultured Tc2 cells. In contrast to Tc2 differentiation, Tc1 differentiation was not affected by either drug.
To confirm the importance of HIF-1α in Tc2 differentiation in the response to hypoxia, HIF-1α-deficient mice were used where HIF-1α expression was depleted exclusively in T cells. Although not completely deficient in HIF-1α protein (approximate 85% reduction), the results using differentiated CD8+ T cells from these mice established several important aspects. HIF-1α-deficient CD8+ T cells were fully capable of Tc1 differentiation in the presence of IL-2 alone. The effects of the deficiency of HIF-1α were most obvious in Tc2 cells exposed to hypoxia where the frequency of IL-13+CD8+ T cells was lower and IL-13 production was markedly reduced compared to similarly treated WT CD8+ T cells. In the model of experimental asthma, we previously showed that transferred CD8+ T cells which were exposed to an environment rich in IL-4 displayed changes at the GATA3 and IL-13 promoter indicative of transcriptional activation and increased IL-13 production17. Transfer of these cells into CD8-deficient mice fully restored all lung allergic responses17. Transfer of hypoxia-exposed, HIF-1α-deficient CD8+ T cells into CD8-deficient recipients failed to restore the full spectrum of lung allergic responses as seen following transfer of hypoxia-exposed WT CD8+ T cells. Taken together, the findings established that enhanced Tc2 differentiation under hypoxia was mediated through HIF-1α activation.
In this study, we demonstrated that when CD8+ T cells were differentiated under normoxia, HIF-1α mRNA was increased by IL-4 and that hypoxia exposure further enhanced HIF-1α protein expression in Tc2 cells. As well, increased levels of pSTAT6 and GATA3 protein were only detected in cells differentiated under hypoxia in the presence of IL-2 and IL-4, but not with IL-2 alone. IL-4 exerts the majority of its activity by signaling through STAT6. In response to IL-4 binding to its receptor, JAK1 and JAK3 are phosphorylated which in turn phosphorylates STAT640. STAT6 is then dimerized and transported to the nucleus where it regulates gene expression. In activated CD8+ T cells, IL-13 induction progressed through a series of distinct IL-4/GATA3-regulated stages characterized by gene expression and epigenetic changes 17. In vitro, IL-4 triggered the stepwise molecular conversion of CD8+ T cells from IFNγ to IL-13 production. During the initial stage, IL-4 suppressed T-bet expression and induced GATA3 expression, characterized by histone modifications and RNA polymerase II (Pol II) recruitment to the GATA3 locus. Exposure to hypoxia appears to have enhanced these molecular events including increases in levels of pSTAT6 and GATA3. Both inhibition of JAK1/3 activation, leading to reduced pSTAT6, and silencing of GATA3 reduced Tc2 differentiation and Hif-1α gene expression suggesting that HIF-1α is regulated by IL-4 downstream of JAK1/3 and GATA3 activation. It is unclear at present at what level hypoxia and HIF-1α enhanced this IL-4-JAK1/3-pSTAT6-GATA3-IL-13 cascade.
The IL-4-dependent upregulation of HIF-1α in CD8+ T cells is in contrast to reports in other cell types. In fibroblast-like synoviocytes, IL-4 did not alter levels of HIF-1α49; in macrophages, IL-4 reduced HIF-1α translation50. Thus, effects of IL-4 on HIF-1α activation appear variable and dependent on the cell type. To date, only limited numbers of studies have investigated the specific influence of hypoxic conditioning on CD8+ T cells. The mammalian target of rapamycin complex 1 (mTORC1) is suggested to be involved in the regulation of HIF-1α in effector CD8+ T cells51. Silly et al. explored the impact of hypoxia on effector CD8+ T cells cultured under 5% or 1% O252, 53. These studies revealed that oxygen deprivation did not modify CD8+ cytotoxic T lymphocyte (CTL) killing capacities but potently induced IL-10 secretion upon reactivation and dramatically decreased CTL expansion. Of note, 5% O2 was defined as “physioxia” reflecting the general levels of oxygen fraction in healthy tissues and 1% O2 as “hypoxia”, levels found in inflamed or tumor sites53. In the present study, we observed similar effects on IL-13 production following Tc2 differentiation whether cells were differentiated in 3% or 1% O2.
A critical question in all of the studies associating hypoxia with asthma, experimental or in asthmatics, is where hypoxic conditioning of cells occurs and at what levels of O2 concentration. Standard cell culture conditions (95% air and 5% CO2) expose cells to 20% O2 which is much higher than the O2 concentrations to which most immune cells are exposed to in vivo. Physioxia in different tissues varies greatly; in the skin (1.1%), in the brain (4.4%), and was as high as 14.5% in pulmonary alveoli53. Tissue oxygen concentrations are likely even lower during an inflammatory response as edema interferes with the diffusion of oxygen from the microvasculature and the infiltration of inflammatory cells results in increased O2 consumption53. The concentration of oxygen in secondary lymphoid organs is generally low54, suggesting that HIFs may be activated in lymphocytes during their normal circulation in the body. More research is needed to better understand potential effects of varying oxygen concentrations on the differentiation and survival of subsets of T lymphocytes in various target organs. Activation of the hypoxia response pathway and increases in HIF-1α can be induced without hypoxic conditioning as seen in response to inflammation. Moreover, HIF-1α activity can be associated with asthma directly44–47.
In summary, hypoxia conditioning significantly enhanced IL-4-dependent reprogramming of CD8+ T cells to IL-13 production through activation of HIF-1α. Activation of HIF-1α, STAT6, and GATA3 may be relevant not only to severe asthma, but also be present in mild and moderate asthmatics. The Tc2 cells differentiated under hypoxia were capable of enhancing allergen-induced airway inflammation and AHR. Together with the insensitivity of CD8+ T cells to corticosteroids16, activation of the IL-4-hypoxia-HIF-1α-IL-13 axis may play a role in the development of severe, steroid-refractory asthma.
Supplementary Material
Clinical implications:
Airway inflammation can trigger the hypoxia response pathway with induction of HIF-1α expression. The combination of CD8+ Tc2 cells, activation of the hypoxia response pathway and HIF-1α upregulation may contribute to steroid-refractory asthma.
Acknowledgments
The authors are grateful to Dr. Holger Eltzschig for providing the HIF-1α-deficient mice and to Diana Nabighian and Cynthia Chavez for assistance in preparation of the manuscript.
Funding Support: This work was supported by NIH grants HL-36577 and AI-77609 (to EWG) and the Joanne Siegel Fund. FN and MS were supported by the Eugene F. and Easton M. Crawford Charitable Lead Unitrust, Pohs, and Dreizzesun funds. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NHLBI or the NIH.
Abbreviations used:
- AHR
Airway hyperresponsiverness
- BAL
Bronchoalveolar lavage
- DEX
Dexamethasone
- GATA3
GATA Binding Protein 3
- HIF-1α
Hypoxia inducible factor 1α
- IFNγ
Interferon-γ
- IL
Interleukin
- JAK
Janus kinase
- KO
Knock out
- OVA
Ovalbumin
- PAS
Periodic acid-Schiff
- pSTAT6
Phospho Signal transducer and activator of transcription 6
- Tc2
Type 2 cytotoxic T cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest statement: The authors declare no relevant conflicts of interest.
References
- 1.Bel EH, Sousa A, Fleming L, Bush A, Chung KF, Versnel J, et al. Diagnosis and definition of severe refractory asthma: an international consensus statement from the Innovative Medicine Initiative (IMI). Thorax 2011; 66:910–7. [DOI] [PubMed] [Google Scholar]
- 2.Bush A, Zar HJ. WHO universal definition of severe asthma. Curr Opin Allergy Clin Immunol; 11:115–21. [DOI] [PubMed] [Google Scholar]
- 3.Brusselle G, Bracke K. Targeting immune pathways for therapy in asthma and chronic obstructive pulmonary disease. Ann Am Thorac Soc 2014; 11:S322–8. [DOI] [PubMed] [Google Scholar]
- 4.Hansbro PM, Kim RY, Starkey MR, Donovan C, Dua K, Mayall JR, et al. Mechanisms and treatments for severe, steroid-resistant allergic airway disease and asthma. Immunol Rev 2017; 278:41–62. [DOI] [PubMed] [Google Scholar]
- 5.Martin AA, Fainardi V, Saglani S. Severe therapy resistant asthma in children: translational approaches to uncover sub-phenotypes. Expert Rev Respir Med 2017; 11:867–74. [DOI] [PubMed] [Google Scholar]
- 6.Bhakta NR, Erle DJ. IL-17 and “TH2-high” asthma: Adding fuel to the fire? J Allergy Clin Immunol 2014; 134:1187–8. [DOI] [PubMed] [Google Scholar]
- 7.Alcorn JF, Crowe CR, Kolls JK. TH17 Cells in Asthma and COPD. Annual Review of Physiology 2010; 72:495–516. [DOI] [PubMed] [Google Scholar]
- 8.Wang M, Gao P, Wu X, Chen Y, Feng Y, Yang Q, et al. Impaired antiinflammatory action of glucocorticoid in neutrophil from patients with steroid-resistant asthma. Respir Res. 2016; 17:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saffar AS, Ashdown H, Gounni AS. The Molecular Mechanisms of Glucocorticoids-Mediated Neutrophil Survival. Current Drug Targets 2011; 12:556–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKinley L, Alcorn JF, Peterson A, DuPont RB, Kapadia S, Logar A, et al. TH17 Cells Mediate Steroid-Resistant Airway Inflammation and Airway Hyperresponsiveness in Mice. The Journal of Immunology 2008; 181:4089–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim HY, Umetsu DT, Dekruyff RH. Innate lymphoid cells in asthma: Will they take your breath away? Eur J Immunol 2016; 46:795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belvisi MG. Regulation of inflammatory cell function by corticosteroids. Proc Am Thorac Soc 2004; 1:207–14. [DOI] [PubMed] [Google Scholar]
- 13.Hilvering B, Hinks TSC, Stöger L, Marchi E, Salimi M, Shrimanker R, et al. Synergistic activation of pro-inflammatory type-2 CD8+ T lymphocytes by lipid mediators in severe eosinophilic asthma. Mucosal Immunol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.den Otter I, Willems LN, van Schadewijk A, van Wijngaarden S, Janssen K, de Jeu RC, et al. Lung function decline in asthma patients with elevated bronchial CD8, CD4 and CD3 cells. Eur Respir J, 2016:393–402. [DOI] [PubMed] [Google Scholar]
- 15.Oda H, Kawayama T, Imaoka H, Sakazaki Y, Kaku Y, Okamoto M, et al. Interleukin-18 expression, CD8(+) T cells, and eosinophils in lungs of nonsmokers with fatal asthma. Ann Allergy Asthma Immunol 2014; 112:23–8e1. [DOI] [PubMed] [Google Scholar]
- 16.Ohnishi H, Miyahara N, Dakhama A, Takeda K, Mathis S, Haribabu B, et al. Corticosteroids enhance CD8+ T cell-mediated airway hyperresponsiveness and allergic inflammation by upregulating leukotriene B4 receptor 1. J Allergy Clin Immunol 2008; 121:864–71e4. [DOI] [PubMed] [Google Scholar]
- 17.Jia Y, Takeda K, Han J, Joetham A, Marcus RA, Lucas JJ, et al. Stepwise epigenetic and phenotypic alterations poise CD8+ T cells to mediate airway hyperresponsiveness and inflammation. J Immunol 2013; 190:4056–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Rensen EL, Sont JK, Evertse CE, Willems LN, Mauad T, Hiemstra PS, et al. Bronchial CD8 cell infiltrate and lung function decline in asthma. Am J Respir Crit Care Med 2005; 172:837–41. [DOI] [PubMed] [Google Scholar]
- 19.Lloyd CM, Hessel EM. Functions of T cells in asthma: more than just T(H)2 cells. Nat Rev Immunol 2010; 10:838–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dakhama A, Collins ML, Ohnishi H, Goleva E, Leung DY, Alam R, et al. IL-13-producing BLT1-positive CD8 cells are increased in asthma and are associated with airway obstruction. Allergy 2013; 68:666–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho SH, Stanciu LA, Holgate ST, Johnston SL. Increased interleukin-4, interleukin-5, and interferon-gamma in airway CD4+ and CD8+ T cells in atopic asthma. Am J Respir Crit Care Med 2005; 171:224–30. [DOI] [PubMed] [Google Scholar]
- 22.Cagnoni EF, Ferreira DS, Ferraz da Silva LF, Nicoletti Carvalho Petry AL, Gomes dos Santos AB, Rodrigues Medeiros MC, et al. Bronchopulmonary lymph nodes and large airway cell trafficking in patients with fatal asthma. J Allergy Clin Immunol 2015; 135:1352–7e1–9. [DOI] [PubMed] [Google Scholar]
- 23.Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity 2014; 41:518–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fajardo I, Svensson L, Bucht A, Pejler G. Increased levels of hypoxia-sensitive proteins in allergic airway inflammation. Am J Respir Crit Care Med 2004; 170:477–84. [DOI] [PubMed] [Google Scholar]
- 25.Crotty Alexander LE, Akong-Moore K, Feldstein S, Johansson P, Nguyen A, McEachern EK, et al. Myeloid cell HIF-1αlpha regulates asthma airway resistance and eosinophil function. J Mol Med (Berl) 2013; 91:637–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011; 146:772–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar V, Gabrilovich DI. Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunology 2014; 143:512–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol 2013; 14:1173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakamura H, Makino Y, Okamoto K, Poellinger L, Ohnuma K, Morimoto C, et al. TCR Engagement Increases Hypoxia-Inducible Factor-1 Protein Synthesis via Rapamycin-Sensitive Pathway under Hypoxic Conditions in Human Peripheral T Cells. The Journal of Immunology 2005; 174:7592–9. [DOI] [PubMed] [Google Scholar]
- 30.Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci U S A 2012; 109:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyahara N, Swanson BJ, Takeda K, Taube C, Miyahara S, Kodama T, et al. Effector CD8+ T cells mediate inflammation and airway hyper-responsiveness. Nat Med 2004; 10:865–9. [DOI] [PubMed] [Google Scholar]
- 32.Miyahara N, Takeda K, Kodama T, Joetham A, Taube C, Park JW, et al. Contribution of Antigen-Primed CD8+ T Cells to the Development of Airway Hyperresponsiveness and Inflammation Is Associated with IL-13. The Journal of Immunology 2004; 172:2549–58. [DOI] [PubMed] [Google Scholar]
- 33.Miyahara N, Takeda K, Miyahara S, Taube C, Joetham A, Koya T, et al. Leukotriene B4 Receptor-1 Is Essential for Allergen-Mediated Recruitment of CD8+ T Cells and Airway Hyperresponsiveness. The Journal of Immunology 2005; 174:4979–84. [DOI] [PubMed] [Google Scholar]
- 34.Ashino S, Takeda K, Li H, Taylor V, Joetham A, Pine PR, et al. Janus kinase 1/3 signaling pathways are key initiators of TH2 differentiation and lung allergic responses. J Allergy Clin Immunol 2014; 133:1162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maier H, Colbert JF,D, Clark D, Hagman J. Activation of the early B-cell-specific mb-1 (Ig-alpha) gene by Pax-5 is dependent on an unmethylated Ets binding site. Mol Cell Biol. 2003; 23:1946–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanehiro A, Ikemura T, Makela M, Lahn M, Joetham A, Dakhama A, et al. Inhibition of Phosphodiesterase 4 Attenuates Airway Hyperresponsiveness and Airway Inflammation in a Model of Secondary Allergen Challenge. Am J Respir Crit Care Med 2001; 163:173–84. [DOI] [PubMed] [Google Scholar]
- 37.Takeda K, Haczku A, Lee JJ, Irvin CG, Gelfand EW. Strain dependence of airway hyperresponsiveness reflects differences in eosinophil localization in the lung. Am J Physiol Lung Cell Mol Physiol 2001; 281:L394–L402. [DOI] [PubMed] [Google Scholar]
- 38.Tomkinson A, Cieslewicz G, Duez C, Larson KA, Lee JJ, Gelfand EW. Temporal Association between Airway Hyperresponsiveness and Airway Eosinophilia in Ovalbumin-Sensitized Mice. Am J Respir Crit Care Med 2001; 163:721–30. [DOI] [PubMed] [Google Scholar]
- 39.Pollizzi KN, Powell JD. Integrating canonical and metabolic signaling programmes in the regulation of T cell responses. Nat Rev Immunol 2014; 14:435–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Acacia de Sa Pinheiro A, Morrot A, Chakravarty S, Overstreet M, Bream JH, Irusta PM, et al. IL-4 induces a wide-spectrum intracellular signaling cascade in CD8+ T cells. J Leukoc Biol 2007; 81: 1102–10. [DOI] [PubMed] [Google Scholar]
- 41.Ellinghaus P, Heisler I, Unterschemmann K, Haerter M, Beck H, Greschat S, et al. BAY 87–2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med 2013; 2:611–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rogers JL, Bayeh L, Scheuermann TH, Longgood J, Key J, Naidoo J, et al. Development of inhibitors of the PAS-B domain of the HIF-2alpha transcription factor. J Med Chem 2013; 56:1739–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scheuermann TH, Li Q, Ma HW, Key J, Zhang L, Chen R, et al. Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat Chem Biol 2013; 9:271–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahmad T, Kumar M, Mabalirajan U, Pattnaik B, Aggarwal S, Singh R, et al. Hypoxia response in asthma: differential modulation on inflammation and epithelial injury. Am J Respir Cell Mol Biol 2012; 47:1–10. [DOI] [PubMed] [Google Scholar]
- 45.Huerta-Yepez S, Baay-Guzman GJ, Garcia-Zepeda R, Hernandez-Pando R, Vega MI, Gonzalez-Bonilla C, et al. 2-Methoxyestradiol (2-ME) reduces the airway inflammation and remodeling in an experimental mouse model. Clin Immunol 2008; 129:313–24. [DOI] [PubMed] [Google Scholar]
- 46.Kim SR, Lee KS, Lee KB, Lee YC. Recombinant IGFBP-3 inhibits allergic lung inflammation, VEGF production, and vascular leak in a mouse model of asthma. Allergy 2012; 67:869–77. [DOI] [PubMed] [Google Scholar]
- 47.Guo J, Lu W, Shimoda LA, Semenza GL, Georas SN. Enhanced interferon-gamma gene expression in T Cells and reduced ovalbumin-dependent lung eosinophilia in hypoxia-inducible factor-1-alpha-deficient mice. Int Arch Allergy Immunol 2009; 149:98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baek KJ, Cho JY, Rosenthal P, Alexander LE, Nizet V, Broide DH. Hypoxia potentiates allergen induction of HIF-1αlpha, chemokines, airway inflammation, TGF-beta1, and airway remodeling in a mouse model. Clin Immunol 2013; 147:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Larsen H, Muz B, Khong TL, Feldmann M, Paleolog EM. Differential effects of Th1 versus Th2 cytokines in combination with hypoxia on HIFs and angiogenesis in RA. Arthritis Research & Therapy 2012; 14:R180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dehne N, Tausendschon M, Essler S, Geis T, Schmid T, Brune B. IL-4 reduces the proangiogenic capacity of macrophages by down-regulating HIF-1 alpha translation. J Leukoc Biol 2014; 95:129–37. [DOI] [PubMed] [Google Scholar]
- 51.Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med 2012; 209:2441–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vuillefroy de Silly R, Ducimetiere L, Yacoub Maroun C, Dietrich PY, Derouazi M, Walker PR. Phenotypic switch of CD8(+) T cells reactivated under hypoxia toward IL-10 secreting, poorly proliferative effector cells. Eur J Immunol 2015; 45:2263–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vuillefroy de Silly R, Dietrich PY, Walker PR. Hypoxia and antitumor CD8+ T cells: An incompatible alliance? Oncoimmunology 2016; 5:e1232236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cho SH, Raybuck AL, Stengel K, Wei M, Beck TC, Volanakis E, et al. Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature 2016; 537:234–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.