

Summary
Human alveolar macrophages are a unique myeloid subset critical for understanding pulmonary diseases and are difficult to access. Here, we present a protocol to generate human alveolar macrophage-like (AML) cells from fresh peripheral blood mononuclear cells or purified monocytes. We describe steps for cell isolation, incubation in a defined cocktail of pulmonary surfactant and lung-associated cytokines, phenotype analysis, and validation with human alveolar macrophages. We then detail procedures for quality control and technical readouts for monitoring microbial response.
For complete details on the use and execution of this protocol, please refer to Pahari et al.1 and Neehus et al.2
Subject areas: cell biology, cell culture, cell isolation, flow cytometry, immunology
Graphical abstract
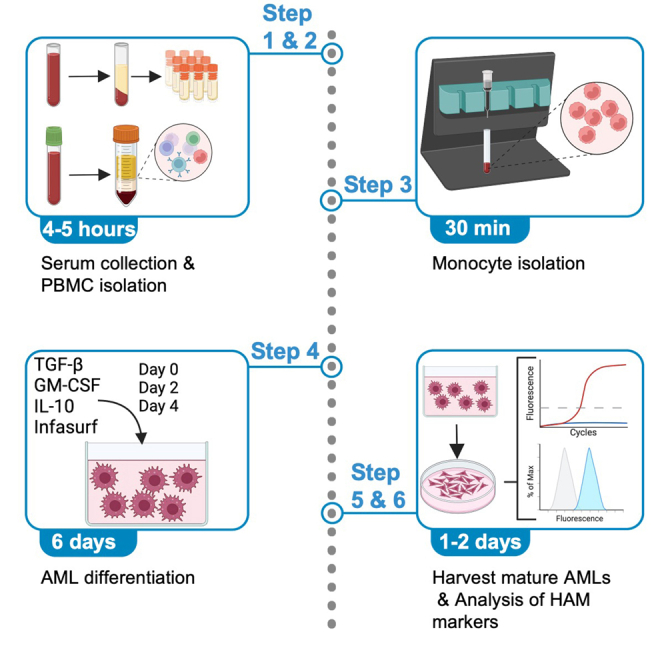
Highlights
-
•
Peripheral blood mononuclear cell (PBMC)/monocyte isolation from human blood
-
•
Alveolar macrophage-like (AML) cell cocktail treatment and differentiation
-
•
AML cell phenotype analysis by real-time qPCR and flow cytometry
-
•
Suitable model to study lung infections and inflammatory disorders
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Human alveolar macrophages are a unique myeloid subset critical for understanding pulmonary diseases and are difficult to access. Here, we present a protocol to generate human alveolar macrophage-like (AML) cells from fresh peripheral blood mononuclear cells or purified monocytes. We describe steps for cell isolation, incubation in a defined cocktail of pulmonary surfactant and lung-associated cytokines, phenotype analysis, and validation with human alveolar macrophages. We then detail procedures for quality control and technical readouts for monitoring microbial response.
Before you begin
Every year, millions of people lose their lives due to respiratory disorders, both infectious and inflammatory. Alveoli in the lower respiratory tract that exchange gases must balance the fight against infections or inhaled particulates with minimizing damage to self-tissue. Alveolar macrophages (AMs) play a crucial role in this balance by being the first immune cells to encounter airborne pathogens and environmental particles.3,4 However, there is currently no easily accessible in vitro model of human AMs (HAMs) which creates a roadblock for comprehensively studying this cell population in health and disease states. Thus, there is an unmet need for cost-effective methods to generate sufficient numbers of primary human cells that closely resemble a HAM phenotype, which is especially important for translational studies and clinical benefits to humans. To address this need, we have developed a novel model for generating human Alveolar Macrophage-Like (AML) cells. This model protocol includes differentiating peripheral blood monocytes in vitro using a defined cocktail of lung components that mimic the alveolar environment. This model, requiring only venipuncture (or access to a blood bank for buffy coats), is significantly less costly than performing a bronchoalveolar lavage to recover HAMs, yields more AML cells relative to HAMs recovered from a single individual and requires less time than iPSC-derived macrophages. AML cells cannot be generated by individual components of the cocktail. However, AML cells can be maintained in culture with the cocktail, retaining the alveolar macrophage phenotype for an extended period of time.5 Researchers have also used this model to study M. tuberculosis1,6 and SARS-CoV-21 infection, and polycystic lung disease.2 The expectation is that this AML model will significantly advance respiratory biology research.
Institutional permissions
The following text refers to the isolation of human peripheral blood mononuclear cells (PBMCs) or purified monocytes to differentiate AML cells from healthy adult donors. The isolation was carried out following a Texas Biomed-approved institutional review board (IRB) protocol (20170315HU). All donors provided informed, written consent for these studies. Please note that laboratories must acquire permission from their relevant institutions to conduct this study, including use of human blood products under biosafety level 2 conditions.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human CD64-FITC (1:50 dilution) | BioLegend | Cat# 305006 RRID:AB_314490 |
| Mouse IgG1 κ isotype control-FITC (1:50 dilution) | BioLegend | Cat# 400108 RRID:AB_396090 |
| Human CD163-PE (1:50 dilution) | eBioscience | Cat# 12-1639-42 RRID:AB_1963570 |
| Mouse IgG1 κ isotype control-PE (1:50 dilution) | eBioscience | Cat# 12-4714-82 RRID:AB_470060 |
| Human CD206-BV421 (1:50 dilution) | BioLegend | Cat# 321126 RRID:AB_2563839 |
| Mouse IgG1 κ isotype control-BV421 (1:50 dilution) | BioLegend | Cat# 400158 RRID:AB_11150232 |
| Human MARCO-PE (1:50 dilution) | eBioscience | Cat# 12-5447-42 RRID: AB_2762430 |
| Mouse IgG3 isotype control-PE (1:50 dilution) | eBioscience | Cat#12-4742-42 RRID: AB_10733011 |
| Human CD36-PE (1:50 dilution) | BD Biosciences | Cat# 555455 RRID: AB_395848 |
| Mouse IgM κ isotype control-PE (1:50 dilution) | BD Biosciences | Cat# 555584 RRID: AB_395960 |
| Human TruStain FcX (Fc receptor blocking solution) (1:50 dilution) | BioLegend | Cat#422302 RRID: AB_2818986 |
| Cell staining buffer | BioLegend | Cat#420201 |
| Anti-human CD14-PerCP-Cy5.5 (1:20 dilution) | Calibre Scientific | Cat# CAPR-103465 |
| Human MARCO APC (1:20 dilution) | eBioscience | Cat# 17-5447-42 RRID: AB_2762440 |
| Mouse IgG3, κ isotype control- Alexa Fluor 647 (1:20 dilution) | BioLegend | Cat# 401322 RRID: AB_10683446 |
| 7-AAD viability staining solution | BioLegend | Cat#420404 |
| Biological samples | ||
| Heparinized whole blood | This manuscript | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| GMP recombinant human GM-CSF (carrier-free) | BioLegend BD Biosciences |
Cat# 572914 Cat# 550068 |
| Recombinant human TGF-β1 (carrier-free) | BioLegend | Cat# 781804 |
| Recombinant human IL-10 | BD Biosciences | Cat# 554611 |
| Infasurf/CUROSURF | ONY Biotech, Inc. and Chiesi Farmaceutici, S.p.A, hospital systems | N/A |
| RPMI 1640 medium, GlutaMAX supplement | Gibco/Invitrogen, Thermo Fisher Scientific | Cat# 11875-093; Cat# 61870044 |
| Human AB serum | Sigma-Aldrich | Cat# H4522 |
| Lymphoprep | STEMCELL Technologies | Cat# 07851 |
| Ficoll-Paque PLUS | GE Healthcare/Sigma | Cat# 17-1440-03 |
| Heparin sodium, 1,000 U/mL | Nationwide Medical Surgical/Sagent Pharmaceuticals | Cat #25021040010 |
| Gibco cell dissociation buffer | Thermo Fisher Scientific | Cat# 13-150-016 |
| 0.9% sodium chloride, irrigation | Abbott Laboratories | |
| DPBS 1x, no calcium, no magnesium | Thermo Fisher Scientific | Cat# 14190169 |
| Critical commercial assays | ||
| Classical monocyte isolation kit | Miltenyi | Cat# 130-117-337 |
| EasySep human monocyte isolation kit | STEMCELL technologies | Cat# 19359 |
| Direct-zol RNA microprep kit | Zymo Research | Cat# R2062 |
| High-capacity RNA-to-cDNA kit | Applied Biosystems | Cat# 4387406 |
| TaqMan fast universal PCR master mix (2X), no AmpErase UNG | Thermo Fisher Scientific | Cat# 4352042 |
| Oligonucleotides | ||
| GUSB | Thermo Fisher Scientific | Cat# 1702016 |
| SPI1 | Thermo Fisher Scientific | Cat# Hs02786711_m1 |
| MRC1 | Thermo Fisher Scientific | Cat# Hs00267207_m1 |
| PPARG | Thermo Fisher Scientific | Cat# Hs01115513_m1 |
| ACTB | Thermo Fisher Scientific | Cat# Hs01060665_g1 |
| Software and algorithms | ||
| Prism software 9.3.1 | GraphPad by Dotmatics | N/A |
| FlowJo 10.8.1 | BD Biosciences | N/A |
| ViiA 7 software | Thermo Fisher Scientific | N/A |
| BioRender | Institution subscription | AV26P8RJCI, II26NWLUOR, FE26NWLKOB |
| ImageJ | National Institutes of Health | ImageJ-win64 |
| Other | ||
| LS columns | Miltenyi | Cat# 130-042-401 |
| Nunc 96-well polystyrene conical bottom MicroWell plates | Thermo Fisher Scientific | Cat# 249570 |
| QuadroMACS separator | Miltenyi | Cat# 130-090-976 |
| EasySep magnet | STEMCELL Technologies | Cat# 18000 |
| 50 mL Falcon tubes | Corning | Cat# 352098 |
| 15 mL Falcon tubes | Thomas Scientific | Cat# 12-565-269 |
| BD LSRFortessa X-20 | BD Biosciences | N/A |
| BD FACSymphony | BD Biosciences | N/A |
| ViiA 7 real-time PCR system | Thermo Fisher Scientific | N/A |
| Applied Biosystems 7500 real-time PCR system | Applied Biosystems | N/A |
| Beckman Allegra 6R centrifuge | Beckman Coulter | N/A |
| Beckman Allegra X-15R centrifuge | Beckman Coulter | N/A |
| Motic AE2000 inverted biological microscope with Moticam | Motic | N/A |
| Glass tubes for serum | Corning (Fisher) | Cat# 8422-50 (05-588-5B) |
| Teflon jar 2 fL. Oz/60 mL (holds 7–8 mL volume) | Savillex | Cat# 100-0060-01 |
| Teflon wells lids, 53 mm Closure | Savillex | Cat# 600-053-01 |
| Teflon jar 4 fL. Oz/120 mL (holds 13–15 mL volume) | Savillex | Cat# 100-0120-01 |
| Teflon well lids, 70 mm closure | Savillex | Cat# 600-070-01 |
| Steriflip-GP sterile centrifuge tube top filter unit | Millipore/Sigma | Cat# SCGP00525 |
| Falcon round-bottom polypropylene tubes | Falcon | Cat# 352063 |
| Falcon round-bottom polystyrene tubes with cell strainer snap cap, 5 mL | Falcon | Cat# 352235 |
| Falcon 96-well, non-treated, U-shaped-bottom microplate | Falcon | Cat# 351177 |
Reagents and supplies for monocyte collection and isolation.
Materials and equipment
This section outlines the required materials for preparation of the "ALL cocktail" components used for AML differentiation.
Preparation of Infasurf (surfactant), GM-CSF, TGF-β, and IL-10 culture medium
| Reagent | Final concentration | Final volume |
|---|---|---|
| Infasurf (35 μg/µL) | 100 μg/mL | 1 mL |
| GM-CSF (10 μg/µL) | 10 ng/mL | 1 mL |
| TGF-β (5 μg/µL) | 5 ng/mL | 1 mL |
| IL-10 (5 μg/µL) | 5 ng/mL | 1 mL |
Note: Infasurf is a surfactant that requires proper storage and handling. It should not be diluted or reconstituted. To redistribute the contents of the vial, gently swirl it, but avoid shaking it. It is normal for the suspension to have visible flecks and foam at the surface.
Alternative: Users may use CUROSURF (poractant alfa) (80 μg/mL, final concentration) as an alternative surfactant component to generate AML cells.
CRITICAL: The main 3 mL stock vial of Infasurf should be kept upright and refrigerated between 2°C to 8°C until it expires. It is recommended to make small aliquots for one or two uses. Before adding to the Teflon wells, warm the aliquot at 37°C for 5 min and mix it slowly. The stock of unopened cytokine vials should be stored at −80°C until expiration. Reconstitute cytokines with toxin-free, sterile PBS 1x + 1% BSA and create working vials at a concentration of GM-CSF (10 μg/mL), TGF-β (5 μg/mL) and IL-10 (5 μg/mL). For a longer shelf-life, cytokines (GM-CSF, TGF-β, and IL-10) can be stored at a temperature of −20°C for up to six months or keep them at −80°C for long-term storage (until expired). Prepare 50 μL reconstituted cytokine aliquots. To ensure the quality of the protein, it is recommended to avoid repeated cycles of freezing and thawing. It is recommended to thaw working vials on ice before adding them into the Teflon wells on Day 0, 2, and 4 during AML differentiation. Once thawed, the used aliquots can be stored at 4°C for a maximum of 7 days.
-
•
Clean and autoclave Teflon wells.
Step-by-step method details
Serum collection
Timing: Day 0 (∼150 min)
This step involves the collection of autologous serum from freshly isolated blood, which is required during AML differentiation in culture. Autologous serum provides the necessary nutrients, growth factors, and cytokines for macrophage survival and differentiation. It is derived from the same individual as the cells, ensuring compatibility and thus reducing the risk of immune reactions.
-
1.
Collect blood from a healthy human donor by venipuncture: collect blood in syringes (or other similar collection tube) prefilled with sodium heparin for cell isolation as well as in anticoagulant-free collection tubes for serum isolation.
-
2.For serum isolation, add blood from anticoagulant-free syringes or collection tubes without heparin very slowly to sterile glass tubes.
-
a.Keep the cap on loosely.
-
a.
Note: Do not fill blood completely in the glass tubes, leave a gap ∼ thumb width wide at the top.
-
3.
Let sit at temperature 20–30°C inside the Biosafety Cabinet (BSC) for ∼60 min to form a clot.
-
4.Using a 1 mL sterile plastic pipette, rim once around clot with the pipette to separate the clot from the glass.Note: Careful not to disturb the clot.
-
a.Tightly replace the cap.
-
a.
-
5.
Transfer the tube to 4°C (on ice or in the fridge) for another 60 min (can be longer) to retract the clot in the tube with serum separated on top.
-
6.
Centrifuge tubes at 464 × g 4°C for 15 min with a low brake (deceleration = 3 may be used).
-
7.Using a 10 mL pipette, carefully withdraw serum (yellow, upper layer) into a 50 mL tube.
-
a.Prepare 1 mL and 4 mL aliquots in sterile cryovials and store them at −80°C.
-
a.
Note: Filter through a 0.2-micron filter (Steriflip, Cat# SCGP00525). If a portion of serum is contaminated with blood clot, repeat the centrifugation process and separate to collect excess serum. Gelatinous clots can be gently pressed to the side of the tube with a sterile cotton Q tip or sterile 1 mL serological pipette to release excess serum, but fibrous material must be removed completely. Using this protocol will result in approximately 40% of the blood obtained to consist of isolated serum.
Alternative: We recommend autologous serum. We do not recommend serum from different individuals for the experiment. If the users do not have access to autologous serum, it is recommended to use commercially obtained human AB serum. Human serum from type AB donors is commonly used in cell culture since it lacks antibodies against the A and B blood-type antigens, reducing immunoreactivity against human cells. We do not recommend fetal bovine serum (FBS) for AML experiments to avoid cellular activation (a major confounder).
Human peripheral blood mononuclear cell (PBMC) isolation from human donors
This step involves isolating human peripheral blood mononuclear cells (PBMCs) from freshly collected, heparinized blood. The cells are then cultured in Teflon wells to differentiate into human Alveolar Macrophage-Like (AML) cells.
-
8.
After blood collection from the human donor, slowly add 20 mL of heparinized blood to a 50 mL tube containing 15 mL saline solution (0.9%) or PBS 1x (35 mL total volume) (Figure 1).
Note: 1 mL of blood will generate approximately 1–2 × 106 PBMCs. The number of tubes required for each experiment depends on the size of the experiment.
-
9.Take up 14 mL of Ficoll-Hypaque and swiftly place the pipette tip to the bottom of blood and saline mix.
-
a.Slowly deliver Ficoll to bottom of tube (no faster than 0.5–1 mL / sec) layering the Ficoll under the saline/blood mixture.
-
a.
Note: Gently remove the pipette from the tube and cap the tube tightly.
Alternatives: 35 mL saline/blood mixture can also be layered carefully on top of 14 mL Ficoll-Hypaque solution in a 50 mL tube. PBMC isolation can also be performed using Lymphoprep solution as an alternative to Ficoll-Hypaque solution.
-
10.
Centrifuge the tube at 20–30°C at 404 × g for 40 min, no brake (deceleration = 0).
-
11.Aspirate all but 5–10 mL of the upper yellow plasma layer.
-
a.With a sterile 10 mL pipette, remove buffy coat into a fresh 50 mL conical tube.CRITICAL: With care not to disturb the buffy coat layer.Note: The PBMC layer is a cloudy band (“buffy coat”) at the plasma-Ficoll interface. There is room for ∼2 buffy coats per tube (∼30 mL buffy coat from 2 blood tubes).CRITICAL: Keep tubes on ice from this step.
-
b.Dilute buffy coat amount about 1:2 with RPMI 1640 4°C up to 50 mL mark.
-
a.
-
12.Centrifuge at 464 × g 4°C for 10 min no brake (deceleration = 0).
-
a.Aspirate supernatant.
-
a.
Note: Leave a little media, not to disturb pellet and resuspend the pellet (or pellets from different tubes) in a minimal volume of RPMI 1640 (1–2 mL) and combine them into one tube. Rinse the bottom of the individual buffy coat-containing tubes with RPMI 1640. Make the final volume of the combined buffy coats up to ∼50 mL with RPMI 1640.
-
13.
Centrifuge again at 132 × g 4°C for 10 min no brake (deceleration = 0).
-
14.
Remove media and resuspend cells in RPMI 1640 (∼35 mL, depending on the number of buffy coats combined).
-
15.Count cells.
-
a.Add 50 μL cells to 950 μL RPMI 1640 (1:20 dilution), add 10 μL to hemocytometer, let settle for 1 min and count.
-
b.Calculate number of cells harvested.
-
a.
Note: Should have 0%–5% red blood cells (RBCs) or platelets. To avoid errors in counting and account for any remove dead cells (should be minimal), we conservatively suggest calculating 10% loss of the cells.
-
16.
Bring cell suspension to 2 × 106 PBMC cells/mL in RPMI 1640 plus 10% autologous serum collected in step-7, “serum collection” section.
-
17.
Place 28 million PBMCs/purified monocytes (see below) in 14 mL volume in big Teflon wells (Savillex, 4 fL. Oz. / 120 mL, Cat# 100-00120-01).
Alternative: Small Teflon wells (Savillex, 2 fL. Oz. / 60 mL, Cat# 100-0060-01): 14 million cells in 7 mL volume with 10% autologous serum.
-
18.
Place the sterile Teflon wells in an ethanol-cleaned pan and incubate at 37°C/5% CO2 for 6 days to allow for differentiation of cocktail-treated monocytes into AML cells (see below).
Alternative: Another method to isolating PBMCs from individual donors is to utilize PBMCs derived from commercially obtained fresh buffy coats from a blood bank.
Note: The advantage for this approach is that no IRB is required. The disadvantage is that the storage time of the buffy coats is not certain. Only use freshly obtained buffy coats. It is up to the users to decide which approach to take and standardize them based on their individual lab protocols.
Note: Cryopreserved PBMCs can be tried but with caution due to the likelihood of reduced viability.
Figure 1.

PBMC isolation
Peripheral blood is layered over Ficoll-Hypaque and subjected to density cushion centrifugation. Post centrifugation, blood components are separated into four layers containing plasma, buffy coat (lymphocytes, monocytes and few platelets and eosinophils), Ficoll-Hypaque, granulocytes and erythrocytes. Created with Biorender.com.
Isolating human monocytes from PBMCs
This method involves isolating monocytes (CD14+) from human PBMCs and then culturing them in Teflon wells to differentiate AML cells.
-
19.
Monocytes can be isolated by EasySep Human Monocyte Isolation Kit, Stem cell, Cat#19359, for processing 1 × 109 cells by negative selection as per the manufacturer’s instructions (https://cdn.stemcell.com/media/files/pis/10000011612-PIS_00.pdf).
Note: Recommended Medium EasySep Buffer (Stem cell, Cat# 20144) and RoboSep Buffer (Stem cell, Cat# 20104).
-
20.
Place purified monocytes in the sterile Teflon wells (as above for PBMCs, 2 × 106 cells/mL) in an ethanol-cleaned pan.
-
21.
Incubate at 37°C/5% CO2 for 6 days to allow for differentiation of cocktail-treated monocytes into AML cells (see below).
Alternative: Monocyte isolation can be performed using the classical monocyte isolation kit (Miltenyi Biotec, Cat# 130-117-337) according to the manufacturer’s instructions (https://static.miltenyibiotec.com/asset/150655405641/document_tk75vb9vrt3i15vt07s7qg8g1p?content-disposition=inline). Additional equipment such as LS columns (Miltenyi Biotec, Cat# 130-042-401) and the QuadroMACS Separator (Miltenyi Biotec, Cat# 130-090-976) are required.
Purity assessment
Isolated cells should be checked for purity.
-
22.
For purity assessment of monocytes (CD14+) by flow cytometry, use anti-human CD14 antibody at a 1:20 dilution (Figure 2).
Note: For detailed flow cytometry staining see section “analyses of AML cells for HAM markers” step 38 below.
Figure 2.

CD14 surface staining of isolated monocytes
Representative flow cytometry staining on isolated monocytes to assess purity based on CD14 expression.
Treatment of PBMCs or purified monocytes with ALL cocktail to generate AML cells
This step involves differentiating AML cells in Teflon wells by treating PBMCs or purified monocytes with surfactant and lung-associated cytokines.
-
23.
Add Infasurf (calfactant, natural bovine surfactant; 100 μg/mL, final concentration) to the Teflon wells on days 0, 2, 4 for the AML group.
Alternative: Users have the option to use CUROSURF (poractant alfa) (80 μg/mL, final concentration) as an alternative surfactant component to generate AML cells. Infasurf and CUROSURF can be directly ordered from ONY Biotech, Inc. and Chiesi Farmaceutici, S.p.A, respectively. Alternatively, surfactants can be obtained found the local hospital pharmacy.
-
24.Add GM-CSF (10 ng/mL, final concentration), TGF-β (5 ng/mL, final concentration) and IL-10 (5 ng/mL, final concentration) to the Teflon wells on days 0, 2, 4 for the AML group.
-
a.Mix gently using a 1 mL pipette or gently rock to mix (Figure 3).
-
a.
Note: This completes “ALL cocktail” (Infasurf, GM-CSF, TGF-β, IL-10).
-
25.
Close the Teflon wells and incubate at 37°C/5% CO2 for 6 days to allow for differentiation (Figure 4).
Note: Fluid in the “ALL treated” groups will be a little turbid due to surfactant in the culture. It is not required to add or remove any amount of media while undergoing cocktail treatment.
Note: One can use untreated monocyte-derived macrophages (UT MDM, autologous serum and RPMI 1640 only) for comparison.
Figure 3.

Schematic representation of the AML cell differentiation protocol
PBMCs are isolated from heparinized blood and are either used directly or undergo monocyte isolation prior to AML cell differentiation. Cells can be cultured without the Infasurf, GM-CSF, TGF-β and IL-10 (ALL cocktail) to generate untreated (UT) MDMs or with the ALL cocktail to generate AML cells. Created with Biorender.com.
Figure 4.

Upregulation of MARCO during AML cell differentiation
(A) Monocytes were differentiated in the presence of ALL cocktail for six days. ALL cocktail treatment and flow cytometric analyses were performed on the indicated days.
(B) Representative flow cytometry staining for CD14 and MARCO shows increased MARCO expression during AML differentiation. The highest expression of MARCO is visible after 6 days of ALL cocktail treatment.
AML cell isolation for subsequent analyses
This section outlines the step-by-step process for isolating AML cells following differentiation for subsequent assays and analysis.
-
26.
Tighten lids and place Teflon wells on ice (pushed halfway into ice) for 30 min.
-
27.Collect cells in suspension from Teflon wells using 10 mL or 25 mL serological pipette into a 50 mL conical tube.Note: Angle Teflon well on ice during cell collection. Place RPMI 1640 media on ice.
-
a.Wash each Teflon well 3–4 times (for complete cell de-attachment) with chilled RPMI 1640 (∼4 mL per wash).Note: Pool washes into the same tube - should see cell layer detach (“slimy” liquid)- DO NOT touch the wells with the pipette to avoid scratches in the Teflon wells.
-
a.
-
28.
Centrifuge tubes at 132 × g 4°C for 10 min no brake.
-
29.Aspirate the supernatant, leaving a little media above the pellet.
-
a.Resuspend each pellet with 1–2 mL RPMI 1640.
-
b.Pool all cells into one tube, rinse the tubes with RPMI 1640,
-
c.Bring the volume up to 10 mL and add 5%–10% autologous serum to the final volume.
-
a.
-
30.Count cells on a hemocytometer (1:20 dilution). Small, round cells are lymphocytes, and larger cells are MDM or AML cells. AML cells are rounder and have a smaller footprint (Figure 5).
-
a.Total cell count/mL= (Q1 + Q2 + Q3 + Q4)/4 × 20 (dilution) × 104 (dilution factor).
-
b.Calculate percent MDM or AML cells: (number of AML counted) / (total number counted) = percent AML.
-
a.
Note: Expect 10%–15% macrophages from PBMCs. When developing the design of experiments, predict 10%–20% loss of cells out of the Teflon wells.
-
31.
Add the required volume of serum (10% final) and RPMI 1640 to the cell suspension to generate 4 × 106 cells/mL.
-
32.
Dispense cells per well in a tissue culture plate as indicated below assuming that MDMs or AML cells are ∼10% of PBMCs (see Table 1).
Note: These are the calculations for AML cells derived from PBMCs (Total cells). AML cells derived from purified monocytes can be used assuming ∼90% purity. If AML cells are generated from purified monocytes, then calculate the AML cell number as indicated in the table under “after adherence”.
-
33.
Incubate cells in tissue culture wells at 37°C for 1.5–2 h at 37°C/5% CO2 to allow for adherence.
-
34.Gently aspirate wells in the corner of the well using a 1 mL pipette.
-
a.Wash each well 3x with pre-warmed (37°C) RPMI 1640 to remove lymphocytes (in the case of PBMCs).Note: Can do more washes if necessary to remove most/all lymphocytes (check under the microscope every time).
-
b.Slowly add pre-warmed RPMI 1640 to the corner of the well each time.Note: If the experiment does not proceed on day 6, then, after the removal of lymphocytes, add a fresh ALL cocktail to the well after the adherence period.
-
a.
-
35.
Proceed with the experiment.
Note: For flow cytometry, Western blot, immunohistochemistry and some other assays, it is necessary to remove the cells from the monolayer. Consider using a cell dissociation protocol such as Gibco cell dissociation buffer, enzyme-free, Hanks' Balanced Salt Solution (Gibco, Cat# 13–150-016).
Figure 5.

Morphology of monocytes after 6 days of various treatments
Monocytes were cultured for six days and either (A) left untreated, (B) treated with Infasurf, (C) treated with GM-CSF, TGF-β and IL-10 or (D) treated with Infasurf, GM-CSF, TGF-β and IL-10. Only treatment with Infasurf, GM-CSF, TGF-β and IL-10 (ALL cocktail) led to the characteristic rounded morphology. Scale bar 20 μm.
Table 1.
Cell number determination in cell culture plates
| Plate types | Descriptions |
|---|---|
| 96 well plate | Cell culture volume 150 μL per well. Total cells 0.5 × 106, after adherence ∼ 0.5 × 105 MDM or AML cells |
| 24 well plate | Cell culture volume 500 μL per well. Total cells 2 × 106, after adherence ∼ 2 × 105 MDM or AML cells |
| 12 well plate | Cell culture volume 1.25 mL per well. Total cells 5 × 106, after adherence ∼ 5 × 105 MDM or AML cells |
| 6 well plate | Cell culture volume 2.5 mL per well. Total cells 10 × 106, after adherence ∼ 1 × 106 MDM or AML cells |
| 4 well plate | Cell culture volume 6 mL per well. Total cells 24 × 106, after adherence ∼ 2.4 × 106 MDM or AML cells |
| 100 × 20 mm petri dish | Cell culture volume 12 mL per well. Total cells 48 × 106, after adherence ∼ 4.8 × 106 MDM or AML cells |
Analyses of AML cells for HAM markers
This section outlines various immunological assays for characterizing AML cells using markers specific for Human Alveolar Macrophages (HAM). Analyze successful differentiation of AML cells by morphology, real-time qPCR or flow cytometry. The article by Pahari et al. 2023 provides an extensive AML characterization.1
-
36.
After AML cell isolation, carefully analyze the isolated cells under the microscope.
Note: AML cells show a distinct, more rounded morphology (similar to plated HAMs) when compared to untreated MDM (UT MDMs). Rounded morphology of AML cells can only be achieved after six days of ALL cocktail treatment.
-
37.For mRNA expression analysis, RNA can be isolated from AML cells or UT MDMs (as negative control).Note: For RNA isolation, use Trizol or another commercially available kit for small number of cells Direct-zol RNA Microprep kit or Direct-zol RNA Miniprep Kit (Zymo Research, Cat# R2062, #R2052). https://files.zymoresearch.com/protocols/_r2060_r2061_r2062_r2063_direct-zol_rna_microprep.pdf
-
a.Transcribe 1 μg of RNA with the High-Capacity RNA-to-cDNA Kit (ABI, Cat# 4387406).Note: Perform real-time qPCR with the TaqMan Fast Universal PCR Master Mix in combination with appropriate probes (e.g. targeting GUSB, PPARG, SPI1 and MRC1).Note: AML cells show high expression of PPARG, SPI1 and MRC1 when compared to UT MDMs. (Figure 6A).
-
a.
-
38.For flow cytometry analysis, harvest AML cells or UT MDMs (as a control) using Gibco cell dissociation buffer.
-
a.Wash cells with PBS 1x. Carefully aspirate and discard the solution.
-
b.Add appropriate volume of Gibco cell dissociation buffer (e.g., 1 mL per well of a 6-well plate).
-
c.Incubate for 15 min at 20–30°C and confirm dissociation under the microscope. Tap the culture vessel against the palm of your hand to dislodge cells.CRITICAL: On average, it takes approximately 15 min for monolayers of AML cells to dissociate in enzyme-free cell dissociation buffer. However, the dissociation time for cells can vary and it may take up to 20 min, depending on the donor. Some donors have stronger cell adherence, which requires more time for the cells to detach. It is important to confirm dissociation by checking under the microscope. Improper dissociation and incorrect handling can adversely affect cell viability and yield.
-
d.Add at least 2x volume of prechilled culture medium and collect cells in a 15 mL Falcon tube.
-
e.Rinse culture dish with culture medium to collect all the cells.
-
f.Centrifuge cells for 5 min at 463 × g at 4°C.
-
g.Resuspend cells in 1 mL of prechilled fluorescence-activated cell sorting (FACS) staining buffer (PBS 1x + 2% BSA or PBS 1x + 2% FBS) or Cell Staining Buffer (BioLegend, Cat# 420201) and count.
-
h.Transfer a minimum of 0.2–5 × 105 AMLs to round-bottom polypropylene tubes (Falcon, Cat#352063) or 96 well U-bottom plates (Falcon, Cat# 351177) for staining.Note: Prepare one well/condition plus additional wells with cells or beads for compensation controls, if needed.
-
i.Raise volume to 1 mL/200 μL with FACS buffer and centrifuge tube/plate at 250 g for 10 min at 4°C.
-
j.Re-suspend cells in add human TruStain FcX (1:50 dilution) up to 0.5 million cells in 50 μL FACS buffer, mix and incubate at 20–30°C for 15 min before staining with antibody of interest.CRITICAL: Use a blocking solution such as Human TruStain FcX (Fc Receptor Blocking Solution) to block the FC receptors and prevent non-specific binding of antibodies to the macrophages. It is not necessary to wash cells between these blocking and immunostaining steps.
-
k.Resuspend cells in 50 μL FACS buffer containing the appropriate antibodies or corresponding isotypes (Table 2).CRITICAL: When analyzing multiple fluorochromes simultaneously, prepare appropriate single-color controls for compensation setup. Users can utilize multi-color, non-overlapping antibodies in the same tubes.
-
l.Incubate plate for 30 min at 4°C in the dark.
-
m.Wash cells twice with FACS buffer and resuspend in 200–300 μL of FACS buffer containing a viability dye such as 7AAD.
-
n.Incubate cells at 20–30°C for 5–10 min before acquiring them on a flow cytometer immediately (e.g., Fortessa X-20, BD FACSymphony).Alternative: Users can fix stained cells by resuspending them in 200 μL of 2% paraformaldehyde in FACS buffer for 8 min at 20–30°C, followed by the addition of 1 mL FACS buffer. Centrifuge the cells at 250 × g for 10 min. Some antibodies work better without fixation.
-
o.Resuspend the cells in 300 μL of FACS buffer and filter them through a round-bottom polystyrene tubes with a cell strainer snap cap (Falcon, Cat#352235).Note: Close the cap of the FACS tube and store it at 4°C before acquiring them on a flow cytometer.CRITICAL: It is crucial to filter cells before FACS acquisition. If cell clumps, debris, and aggregates are not removed from the sample, this may cause clogging of the instrument, reduce the purity of the population, and even damage the machine. Filtration ensures a clean and homogeneous sample, preventing clogging and damage to the machine, and yielding precise and reliable results.Note: AML cells are characterized by high expression of CD64, CD206, MARCO and CD163 when compared to untreated MDMs and low expression of CD36 (Figure 6B).
-
a.
Figure 6.

Characterization of AML cells after 6 days of differentiation
(A) qRT-PCR data of AML cells or untreated (UT) MDMs for characteristic HAM genes. Data are expressed as mean ± SD of 2 biological replicates.
(B) Representative flow cytometry staining of AML cells and UT MDMs for selected markers.
Table 2.
Suggested antibodies for AML cell phenotyping by flow cytometry
| Antibody | Dilution | Isotype |
|---|---|---|
| Human CD64-FITC | 1:50 | Mouse IgG1 κ Isotype control-FITC |
| Human CD163-PE | 1:50 | Mouse IgG1 κ Isotype control-PE |
| Human CD206-BV421 | 1:50 | Mouse IgG1 κ Isotype control-BV421 |
| Human MARCO-PE | 1:50 | Mouse IgG3 Isotype control-PE |
| Human CD36-PE | 1:50 | Mouse IgM κ Isotype control-PE |
Expected outcomes
Studying alveolar macrophages can provide valuable insights into the mechanisms underlying respiratory diseases, such as asthma, cystic fibrosis, chronic obstructive pulmonary disease, tuberculosis and other respiratory diseases such as COVID-19, and polycystic lung disease. Alveolar macrophages play a critical role in the immune defense of the lungs and are involved in the inflammatory response associated with many respiratory diseases. By studying these cells, researchers can better understand the genetic and environmental factors that contribute to these conditions. This understanding can lead to the development of more effective treatments for respiratory diseases, benefiting millions of people worldwide.
The human Alveolar Macrophage-Like (AML) cell model allows researchers to simulate human lung conditions in vitro, leading to more accurate results and potential breakthroughs in the field. In addition to the conditions noted above, the AML model can help assess how aging affects alveolar macrophages, including their ability to respond to pathogens and maintain a healthy alveolar space. Lastly, this model can aid in clinical studies, including the response to new therapies and vaccines in clinical cohorts.
Limitations
To ensure that the AML cell phenotype optimally mimics lung-associated conditions, it is essential to maintain the optimum concentration of individual lung-associated components and ensure that they are functional. These components need to be continually added to maintain the cell phenotype over time. Recall that HAMs in vivo are also chronically exposed to these components.
Human donor variability needs to be considered when assessing the AML response, requiring adequate sample size to properly analyze results. AML cells are also sensitive to mechanical stressors, so it’s crucial to handle them with care during the isolation and handling processes. The washing steps should be done carefully to avoid loss of the cell monolayer.
Finally, the viability and behavior of the cells can also be affected by factors such as temperature, humidity, and pH levels. To ensure consistent and reliable results, it’s essential to maintain strict quality control measures and standardize experimental conditions in generating the AML cells.
Troubleshooting
Problem 1
Inefficient cell isolation: Related to the sections “human peripheral blood mononuclear cell (PBMC) isolation from human donors” (steps 4, 5), “AML cell isolation for subsequent analyses” (steps 26, 27, 29), materials and equipment: “Clean and autoclave Teflon wells: CRITICAL”). This most commonly results from incomplete removal of the buffy coat layer, incomplete removal from the Teflon wells, lack of maintaining cold buffers and tubes to avoid cell adherence and careful attention to staying away from aspirating or pipetting completely down to cell pellets or over cells in tissue culture wells.
Potential solutions
-
•
Ensure complete PBMC acquisition from Ficoll-Hypaque by removing fluid slightly below the buffy coat level (can mark the tube to this level). Always leave a small amount of fluid over cell pellets or monolayers and remove fluid by aspirating on the corners of the tube or tissue culture well.
-
•
Make sure the Teflon wells are embedded in ice to help with more complete cell removal. Also, use ice-cold RPMI for wash steps.
-
•
Teflon wells need to be cleaned properly in between use with mild soap followed by an alcohol cleaning to remove any contaminating endotoxin. Teflon wells should be checked carefully with each use to ensure that there are no scratches. Monocytes/macrophages can get stuck in the scratches.
Problem 2
Low cell yield: Related to the sections “AML cell isolation for subsequent analyses” (step 34) and “analyses of AML cells for HAM markers” (step 38a–c). This most commonly occurs by incomplete removal of cells from the Teflon wells or incomplete cell dissociation from monolayers.
Potential solution
-
•
Make sure to visualize tissue culture wells after each wash step during cell dissociation to ensure that monolayer cells are completely removed. AML cells are slightly less adherent than MDMs.
Problem 3
Loss of cell viability: Related to the sections “human peripheral blood mononuclear cell (PBMC) isolation from human donors” (step 18), “treatment of PBMCs or purified monocytes with ALL Cocktail to generate AML cells” (steps 23–24), “materials and equipment”. This most commonly occurs by undue delay in isolating cells from the buffy coat or leaving cells at room temperature (20–30°C) for too long. Also, commercially acquired buffy coats may not be fresh.
Potential solutions
-
•
Proceed through the protocol in a timely fashion and do not use donor blood that was collected more than 24 h before processing. Avoid use of expired reagents.
-
•
Strict aseptic technique should be followed while handling cells and proper sterilization of equipment and media should be ensured.
-
•
Ensure that all personnel involved in experiments are trained in the experimental protocols, sterile technique and proper use of biohazards and human materials.
Problem 4
Donor variability: Related to the section “Study limitations”. Donor variability is a common issue in human sample analyses, which can lead to inconsistent and unreliable results. This variability can arise due to differences in genetics, environmental factors, and other variables among individuals.
Potential solutions
-
•
Power analyses and biostatistics are critical to ensuring adequate sample size.
-
•
Additionally, careful selection and standardization of sample collection and processing methods can help to minimize variability and ensure consistent results.
-
•
Include a control in each experiment and compare test group results in individual experiments to the control group (e.g., fold or percent increase or decrease).
Resource availability
Lead contact
For any additional information and requests for resources and reagents, please contact the lead person in charge, who will be able to assist. Lead contact: Larry S. Schlesinger, Email: lschlesinger@txbiomed.org.
Technical contact
For any technical questions regarding executing this protocol, please contact Susanta Pahari, Email: spahari@txbiomed.org.
Materials availability
This manuscript indicates the availability of newly generated materials associated with this protocol, including any conditions for access. The related individual components of the cocktail are commercially available and indicated in the key resources table.
Data and code availability
The published article includes all datasets generated or analyzed during this study. The research data can be found in1 and.2
RNA-seq data can be found in the NCBI GEO database: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE188945. GEO accession: GSE188945.
Acknowledgments
The work was funded by several sources, including the National Institutes of Health (NIH) awards AI136831 and P30AI168439 to L.S.S., Texas Biomed Cowles and Forum Postdoctoral Fellowships, and the Interdisciplinary NexGen TB Research Advancement Center (IN-TRAC) Pilot Grant to S.P. Flow cytometry assay and related research in this publication were supported by the Office of the Director, NIH, award (S10OD028653). The RNA sequencing research data were generated in the Genome Sequencing Facility, supported by UT Health San Antonio., NIH-NCI P30 CA054174, NIH Shared Instrument grant (1S10OD021805-01) (S10 grant), and CPRIT Core Facility Award (RP160732). TEM was conducted at the Electron Microscopy Laboratory at UT Health San Antonio, and fluorescence microscopy imaging was conducted with instruments in the Biology Core at Texas Biomed.
A.-L.N. was supported by the Imagine Institute international PhD program, the Fondation Bettencout-Schueller, and the Fondation pour la Recherche Médicale (FDT202204015102 to A.-L.N.). The Laboratory of Human Genetics of Infectious Diseases is supported by Institut National de la Santé et de la Recherche Médicale (INSERM); Imagine Institute; the University of Paris Cité; the St. Giles Foundation; the Howard Hughes Medical Institute; The Rockefeller University; National Institute of Allergy and Infectious Diseases (R01AI095983 to J.-L.C. and J.B.); the National Center for Advancing Translational Sciences (UL1TR001866 to J.-L.C.); the Square Foundation; Grandir – Fonds de solidarité pour l’enfance; William E. Ford, General Atlantic’s Chairman and Chief Executive Officer; Gabriel Caillaux, General Atlantic’s Co-President, Managing Director, and Head of business in EMEA; the General Atlantic Foundation; Laboratoire d’Excellence Integrative – Biology of Emerging Infectious Diseases (ANR-10-LABX-62-IBEID to J.-L.C.); the French National Research Agency (ANR-10-IAHU-01 to J.-L.C.); and MAFMACRO (ANR-22-CE92-0008 to J.B.).
The graphical abstract and Figures 1 and 3 were created using Biorender.com.
Author contributions
S.P. and L.S.S. designed the studies and generated protocols. S.P. and A.-L.N. conducted the experiments, acquired the data, analyzed them, and generated figures. S.P. wrote the manuscript with input from A.-L.N. L.S.S., B.C.T., J.B., and J.-L.C. edited the manuscript and performed a critical review.
Declaration of interests
A non-provisional patent application has been filed with the number 17/657,344 for an invention titled “Cell culture media and methods for generating human Alveolar Macrophage-Like cells.” The applicant is the Texas Biomedical Research Institute, located in San Antonio, Texas, USA. The inventors listed in the application are L.S.S. and S.P., both from San Antonio, Texas, USA. For more details, please refer to the following link: https://patentcenter.uspto.gov/applications/17657344.
Contributor Information
Susanta Pahari, Email: spahari@txbiomed.org.
Larry S. Schlesinger, Email: lschlesinger@txbiomed.org.
References
- 1.Pahari S., Arnett E., Simper J., Azad A., Guerrero-Arguero I., Ye C., Zhang H., Cai H., Wang Y., Lai Z., et al. A new tractable method for generating human alveolar macrophage-like cells in vitro to study lung inflammatory processes and diseases. mBio. 2023;14 doi: 10.1128/mbio.00834-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neehus A.L., Carey B., Landekic M., Panikulam P., Deutsch G., Ogishi M., Arango-Franco C.A., Philippot Q., Modaresi M., Mohammadzadeh I., et al. Human inherited CCR2 deficiency underlies progressive polycystic lung disease. Cell. 2024;187:390–408.e23. doi: 10.1016/j.cell.2023.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morales-Nebreda L., Misharin A.V., Perlman H., Budinger G.R.S. The heterogeneity of lung macrophages in the susceptibility to disease. Eur. Respir. Rev. 2015;24:505–509. doi: 10.1183/16000617.0031-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hussell T., Bell T.J. Alveolar macrophages: plasticity in a tissue-specific context. Nat. Rev. Immunol. 2014;14:81–93. doi: 10.1038/nri3600. [DOI] [PubMed] [Google Scholar]
- 5.Papp A.C., Azad A.K., Pietrzak M., Williams A., Handelman S.K., Igo R.P., Jr., Stein C.M., Hartmann K., Schlesinger L.S., Sadee W. AmpliSeq transcriptome analysis of human alveolar and monocyte-derived macrophages over time in response to Mycobacterium tuberculosis infection. PLoS One. 2018;13 doi: 10.1371/journal.pone.0198221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arnett E., Wolff J., Leopold Wager C.M., Simper J., Badrak J.L., Ontiveros C.O., Ni B., Schlesinger L.S. Cutting Edge: Cytosolic Receptor AIM2 Is Induced by Peroxisome Proliferator-activated Receptor gamma following Mycobacterium tuberculosis Infection of Human Macrophages but Does Not Contribute to IL-1beta Release. J. Immunol. 2024;212:765–770. doi: 10.4049/jimmunol.2300418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The published article includes all datasets generated or analyzed during this study. The research data can be found in1 and.2
RNA-seq data can be found in the NCBI GEO database: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE188945. GEO accession: GSE188945.