

Summary
Knowing the genes involved in quantitative traits provides an entry point to understanding the biological bases of behavior, but there are very few examples where the pathway from genetic locus to behavioral change is known. To explore the role of specific genes in fear behavior, we mapped three fear-related traits, tested fourteen genes at six quantitative trait loci (QTLs) by quantitative complementation, and identified six genes. Four genes, Lamp, Ptprd, Nptx2, and Sh3gl, have known roles in synapse function; the fifth, Psip1, was not previously implicated in behavior; and the sixth is a long non-coding RNA, 4933413L06Rik, of unknown function. Variation in transcriptome and epigenetic modalities occurred preferentially in excitatory neurons, suggesting that genetic variation is more permissible in excitatory than inhibitory neuronal circuits. Our results relieve a bottleneck in using genetic mapping of QTLs to uncover biology underlying behavior and prompt a reconsideration of expected relationships between genetic and functional variation.
Keywords: inbred mouse strains, fear conditioning, QTL mapping, quantitative complementation, single-nucleus RNA-seq, single-nucleus ATAC-seq
Graphical abstract
Highlights
-
•
Meta-analysis with 6,544 mice found 93 quantitative trait loci for fear-related behavior
-
•
Quantitative complementation testing identified six genes at six quantitative trait loci
-
•
Epigenetic analyses implicated excitatory neurons as mediating the genetic effects
-
•
Genetic variation occurs preferentially in excitatory versus inhibitory neuronal circuits
Chen et al. use quantitative complementation to identify genes at six quantitative trait loci influencing fear-related behaviors. Epigenetic analysis indicated that genetic variation is more permissible in excitatory than inhibitory neuronal circuits. Creating CRISPR knockouts in inbred strains makes it possible to identify genes at QTLs in mice.
Introduction
A major challenge in behavior genetics is to turn genetic information into mechanistic understanding of the sort that would, for example, be useful in designing new treatments for psychiatric disorders and, more generally, understanding how genetic variation leads to behavioral variation. A key step is progressing from quantitative trait locus (QTL) to gene, which has only been achieved for any complex trait, in any species, in a small number of cases.1,2 Of the 5,000 QTLs identified in rodents, less than 100 genes have been identified, almost all on the basis of correlative evidence, such as proximity of a gene to the QTL or alterations in transcript abundance, rather than by a causal test of a gene’s candidacy.2 Currently, there is no consensus on how to proceed from QTL to gene.
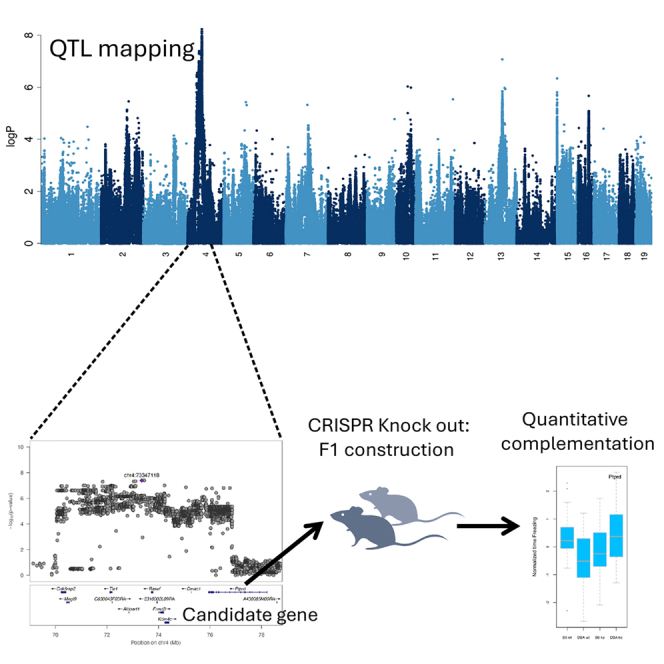
Here, we demonstrate the power of a quantitative complementation (QC) test, first applied in Drosophila3 and later shown to work in rodents,4 to directly query the causal gene impacted by the QTL. Construction and phenotyping of F1 hybrids with and without a knockout (KO) of a candidate gene, and of inbred strains with and without the KO of a candidate gene, test whether the QTL operates through the gene under investigation. By separately assaying the joint effect of QTLs (from the phenotypic difference between strains) and the effect of the mutation (from the phenotypic difference between KOs and wild type [WT]), the QC test reveals a QTL whose effect depends on the presence of the candidate gene as a significant interaction between the effect of mutation and the effect of strain. Applying the test requires access to inbred animals carrying a KO on the same genetic background as the QTL, which has been difficult to achieve when KOs were generated using homologous recombination in 129 strains or C57BL/6N. The development of the CRISPR-Cas9 technology now lifts that restriction by enabling genetic manipulation in any strain.5,6
We set out to apply the QC test to fear-related behaviors. We chose these traits because we could use extensive information available about the brain regions involved (ventral hippocampus and amygdala7,8), and their underlying circuitry,9,10,11,12 to explore where and how in the brain genetic variation results in behavioral change. We reasoned that by knowing the causal genes, we could identify changes in their expression and regulation at a single-cell level, revealing the molecular mechanisms that mediate the impact of genetic variants on traits.
Results
Generating a large-scale set of phenotypes and genotypes
Our first task was to identify QTLs involved in fear-related behaviors that ideally indicated one or a small number of genes for QC testing. We chose a hybrid mouse diversity panel (HMDP) for mapping because of its potential to deliver high resolution2 (thus increasing the chance of finding QTLs containing a small number of candidate genes) and because it includes recombinant inbreds derived from C57BL/6J, hence providing many QTL alleles on a strain readily amenable to CRISPR-Cas9 modification (the HMDP strains and numbers of animals used are listed in Table S1). We mapped variation in a conditioned fear assay in which animals were exposed to an auditory cue associated with an aversive shock. Re-exposure to the auditory cue elicits a fear response, measured as the amount of time spent freezing13,14 and referred to in this paper as FC-cue. The context in which conditioning occurs also elicits freezing (FC-context),15 and we assayed this too. We also mapped variation in an unconditioned fear assay using the elevated plus maze (EPM), whose open arms act as anxiogenic environment. We counted the number of entries into the EPM’s open arms as a measure of fear, referred to henceforth as EPM-open.
To enable us to perform a well-powered meta-analysis and robustly identify loci at high resolution, we combined our data with results from 29 publications.16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45 We implemented a rigorous pipeline to harmonize phenotypes across the multiple datasets (STAR Methods: harmonizing phenotyping data for meta-analysis), generating phenotypes on a total of 6,544 mice from seven cohorts (Tables S1 and S2).
Our next requirement was a set of genotypes at markers sufficiently dense to capture most causative variants. Short-read sequencing provides almost complete catalogs of genetic variants for sixteen inbred strains,46 which are also a resource for imputing variants in the other mapping strains.47 We carried out imputation using a hidden Markov model-based technique with proven effectiveness in inbred model organisms.48 After imputation, we obtained genotypes on 16,767,664 markers (the imputation strategy and validation results are described in STAR Methods: imputation of genotypes). For mapping, marker sets were chosen that are polymorphic within the strains of each dataset, a number varying from 4 to 16 million SNPs segregating between the inbred strains, sufficient to identify causative variants at QTLs.49,50
Genetic mapping of conditioned and unconditioned fear
The phenotypic dataset we obtained is highly structured, consisting of closely related individuals (recombinant inbreds share half their genome, like full siblings) as well as distantly related individuals. Genome-wide association was carried out with GEMMA, which implements a linear mixed-model marker association test,51,52 including a genomic relationship matrix to control for the population structure arising from the inclusion of individuals with different degrees of relatedness. We mapped each phenotype in each cohort and carried out a meta-analysis of the results using a random effects model implemented in METASOFT.53,54 We used Genome Reference Consortium Mouse Build 38 (GCA_000001635.2, mm10), and all coordinates in this paper refer to that build.
To assess significance, we used GCTA55 to simulate data for each cohort and meta-analyzed the results in the same way as we did for the real datasets. From 100,000 simulations, we obtained a 5% threshold of negative logarithm (base 10) of the association p value (logp) of 4.50, consistent with thresholds estimated by others using the same experimental design.35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56 Applying this significance threshold, we identified 14 loci for FC-context, 15 for FC-cue, and 72 for EPM-open (Table S3). At a threshold corrected for testing three phenotypes (logp = 4.97) the number of loci for EPM-open fell to 60 while remaining unchanged for the other two phenotypes. Four loci were common to FC-context and FC-cue and four were common to EPM-open and FC-cue, giving 93 unique loci. The Manhattan plots in Figure 1 show all loci that exceeded the logp threshold of 4.5.
Figure 1.

Manhattan plots for three fear-related behaviors in mice
(A–C) The data show the results of a meta-analysis of (A) 1,931 mice for cue conditioning (FC-cue), (B) 2,671 for contextual conditioning (FC-context), and (C) 1,942 for entries into the open arms of the elevated plus maze (EPM-open arms). Chromosome numbers are listed on the horizontal axis. The vertical scale is the negative logarithm (base 10) of the association p value. A horizontal dotted red line gives the location of the 5% significance threshold (from simulation).
While the genetic architecture of fear-related behaviors was highly polygenic, we found evidence for a few large-effect loci, notably on chromosomes 7 and 13 for FC-cue (Figure 1A) and chromosomes 4 and 13 for FC-context (Figure 1B). More than five times as many QTLs were identified underlying EPM-open than either of the fear-conditioning phenotypes, likely reflecting higher polygenicity of the trait (Figure 1C). The size of the QTL intervals varied considerably from 0.02 to 19.6 Mb (confidence intervals estimated by simulation57) with a median of 2.5 Mb, and each locus contained a median of 23 genes (range from 0 to 329; Table S3).
QC testing identifies six genes for fear-related behavior
Which of the genes at a locus is causal for the phenotype? To answer this for specific loci, we first chose a 2 Mb QTL at the end of chromosome 13 associated with FC-cue (logp = 7.6), previously identified in a panel of BXD recombinant inbreds,27 that includes a hyperpolarization-activated cyclic nucleotide-gated channel 1 (Hcn1). Since pharmacological blockade of HCN1 reduces freezing, Hcn1 has been proposed as the causal gene at this locus27 (although deletion of Hcn1 does not alter conditioned fear58). We used an exon-excision CRISPR-Cas9 strategy to knock out out all four annotated and one unannotated gene lying within the 95% confidence intervals of the QTL (Figure 2A) (we omitted the single-exon unannotated transcript Gm6416). We used a quantitative polymerase chain reaction to confirm that the engineered mutations altered RNA abundance. The CRISPR-Cas9 strategy and characterization of the mutants is described in STAR Methods and Tables S4 and S5.
Figure 2.

Quantitative complementation of five genes at a locus on chromosome 13 for cue fear conditioning
(A) QTL regional information from LocusZoom.59 The top part shows the association results from a meta-analysis, with the position of the highest-scoring variant annotated in purple. The vertical scale is the negative logarithm (base 10) of the association p value. The bottom section gives the location and orientation of genes at the locus. A red box identifies each gene used in the quantitative complementation tests.
(B) Design of the quantitative complementation test. Black boxes indicate the knockout (KO) allele, and strain is indicated by color, where blue is C57BL/6J and red is DBA/2J. An “X” indicates a cross between the named groups above and below the X, and an arrow points to the progeny of each cross. The four groups used in the quantitative complementation test are wild-type C57BL/6J (B6 WT), heterozygote KOs (B6 KOs), an F1 from crossing DBA/2J to C57BL/6J (D2 WT), and the F1 KO by DBA/2J (D2 KO).
(C) Results of quantitative complementation testing of four annotated genes in the region. The vertical axis is the quantile normalized duration of freezing to a cue from the fear-conditioning test. The horizontal axis lists the groups of mices used. Groups are B6.WT: WT C57BL/6J; B6.KO: heterozygote KOs on C57BL/6J; D2.WT: F1 from crossing DBA/2J to C57BL/6J; D2.KO: the F1 from crossing the KO onto DBA/2J. These genes were not significant by quantitative complementation (Table 1).
(D) Results of the quantitative complementation test for the long non-coding RNA 4933413L06Rik. The axes labels are the same as in (C). The difference between these groups yielded a significant interaction result in the quantitative complementation test (Table 1).
Each KO was subject to a QC test (Figure 2B) to find out which gene was mediating the effect of the locus on FC-cue. Four genes tested negative (Figure 2C), while one was positive (Figure 2D), which to our surprise was an unannotated long non-coding RNA (lncRNA), 4933413L06Rik. To interpret Figures 2C and 2D, consider that the first two groups in Figure 2B (C57BL/6J wild type [B6.WT] and DBA/2J wild type [DBA.WT]) measure a strain effect arising from a single copy of the D2 genome in the F1 animals (assuming strict additivity, the strain effect should be about half that found in inbred strain comparisons, consistent with our results). Any difference between the B6.WT and B6.KO groups is attributable to the presence of the KO.
The pattern of results in Figure 2C for the four genes tested looks identical: a strain effect can be seen but no discernable difference between groups carrying the KO and the WTs. By contrast, Figure 2D shows a different pattern. Animals with the KO do differ from their respective WTs, indicating that the KO influences the phenotype but with differences between the two strains. The B6.KO animals spend less time freezing than their WT siblings (B6.WT), while the DBA.KO group freezes more than their WT siblings (DBA.WT). In other words, the effect of the mutation depends on the strain background, which we assume to be due to a nearby QTL.
We tested this relationship between strain and KO by analysis of variance and found a significant interaction (p = 0.0015; Table 1), indicating that the effect of the QTL is mediated by the unannotated gene 4933413L06Rik. We found no evidence of the involvement of Hcn1, demonstrating that QC testing can unambiguously identify genes mediating the effects of QTLs.
Table 1.
Quantitative complementation testing of fourteen genes at six QTLs for fear-related behavior
| Phenotype | Gene | Strain p value | KO p value | Interaction p value | QTL location | N |
|---|---|---|---|---|---|---|
| FC-cue | Emb | 5.33E−02 | 1.20E−01 | 3.73E−01 | chr13:116.4–118.4 | 73 |
| FC-cue | Hcn1 | 8.12E−02 | 9.93E−01 | 5.02E−01 | chr13:116.4–118.4 | 100 |
| FC-cue | Mrps30 | 5.67E−05 | 4.18E−01 | 8.53E−01 | chr13:116.4–118.4 | 94 |
| FC-cue | Parp8 | 2.32E−02 | 3.94E−01 | 7.00E−01 | chr13:116.4–118.4 | 85 |
| FC-cue | 4933413L06Rika | 5.37E−01 | 6.04E−02 | 1.45E−03a | chr13:116.4–118.4 | 87 |
| FC-context | Megf9 | 1.30E−01 | 3.44E−01 | 4.69E−01 | chr4:70.1–78.2 | 48 |
| FC-context | Ptprda | 5.15E−01 | 2.69E−02 | 1.95E−04a | chr4:70.1–78.2 | 137 |
| FC-context | Psip1a | 2.68E−01 | 2.83E−02 | 1.09E−03a | chr4:78.7–85.2 | 92 |
| FC-context | Sh3gl2a | 5.43E−02 | 3.09E−01 | 3.35E−04a | chr4:78.7–85.2 | 117 |
| FC-context | Snapc3 | 8.48E−01 | 8.59E−01 | 3.00E−02 | chr4:78.7–85.2 | 82 |
| FC-context | Ttc39b | 4.52E−01 | 7.28E−01 | 2.99E−02 | chr4:78.7–85.2 | 75 |
| EPM-open | Nptx2a | 1.97E−02 | 2.27E−01 | 1.74E−03a | chr5:144.2–145.0 | 96 |
| EPM-open | Stim2 | 1.85E−03 | 1.84E−04 | 1.72E−01 | chr5:54.1–54.3 | 105 |
| EPM-open | Lsampa | 2.26E−01 | 9.06E−02 | 1.17E−03a | chr16:41.3–41.4 | 79 |
The table shows the p values of an analysis of variance testing for an interaction between strain and knockout (KO) in the four groups of the quantitative complementation test. QTL locations are given in Mb and the name of the phenotype tested. N is the total number of animals used for the quantitative complementation test. QTL location coordinates are to mouse Genome Reference Consortium Mouse Build 38 (mm10).
Results exceeding a multiple testing corrected p value of 0.0035.
We next tackled the most significant region of association for FC-context, a 15-Mb region on chromosome 4 (between 70 and 85). There were two QTLs here (70–78 and 79–85 Mb; Figure 3), together containing 46 genes. To narrow the choice of genes, we tested genes close to the most highly associated markers and with confirmed gene expression in the hippocampus and amygdala (from published sources60,61,62,63,64,65,66,67). At the first locus, the interaction p value of Ptprd exceeded a multiple correction testing threshold (p < 0.0036 for testing 14 genes listed in Table 1), while at the second two genes, Psip1 and Sh3gl3, exceeded the threshold (results shown in Figure 3 and Table 1).
Figure 3.

Quantitative complementation of four genes at two QTLs on chromosome 4 for contextual fear conditioning
(A and C) QTL regional information from LocusZoom.59 The top part shows the association results from a meta-analysis, with the position of the highest-scoring variant annotated in purple. The vertical scale is the negative logarithm (base 10) of the association p value. The bottom section gives the location and orientation of genes at the locus. A red box identifies each gene used in the quantitative complementation tests.
(B and D) Results of quantitative complementation testing of six annotated genes. The vertical axis is the quantile normalized duration of freezing to the context from the fear-conditioning test. The horizontal axis lists the groups of mice used. Groups are B6.WT: WT C57BL/6J; B6.KO: heterozygote KOs on C57BL/6J; D2.WT: F1 from crossing DBA/2J to C57BL/6J; D2.KO: the F1 from crossing the KO onto DBA/2J. Genes significant by quantitative complementation are shown in gray (results in Table 1).
Finally, we chose three loci contributing to variation in EPM-open behavior (Figure 4). The first, on chromosome 16, had a highly significant association (logp = 9.48) and lies in an intron of a cell adhesion molecule, Lsamp (limbic system-associated membrane protein), whose deletion is known to alter behavior in the EPM.68,69 We found a significant interaction (p = 0.001; Table 1), indicating that the effect of the QTL is mediated by the Lsamp gene. We tested Stim2 and Nptx2 at two other loci using the same CRISPR-Cas9 exon deletion approach. While the QC test confirmed the candidacy of Nptx2, it was negative for Stim2. Table 1 summarizes the results of the QC tests for all genes at the six loci we examined.
Figure 4.

Quantitative complementation of three genes at three QTLs for the number of entries into the open arms of the EPM
(A, C, and E) QTL regional information from LocusZoom.59 The top part shows the association results from a meta-analysis, with the position of the highest-scoring variant annotated in purple. The vertical scale is the negative logarithm (base 10) of the association p value. The bottom section gives the location and orientation of genes at the locus. A red box identifies each gene used in the quantitative complementation tests.
(B, D, and F) Results of quantitative complementation testing. The vertical axis is the quantile normalized duration of the number of entries into the open arms of the elevated plus maze. The horizontal axis lists the groups of mice used. Groups are B6.WT: WT C57BL/6J; B6.KO: heterozygote KOs on C57BL/6J; D2.WT: F1 from crossing DBA/2J to C57BL/6J; D2.KO: the F1 from crossing the KO onto DBA/2J.
Strain differences in gene expression, open chromatin, and methylation are concentrated in excitatory hippocampal neurons
One hypothesis about how QTLs act is that they alter gene expression, which in our case would manifest as a difference in the abundance of transcripts from C57BL/6J and DBA/2J genomes in relevant cell types. With six genes in hand, we set out to test this assumption. We generated single-nucleus RNA-sequencing (snRNA-seq) datasets in C57BL/6J and DBA/2J animals from the ventral hippocampus and amygdala. We identified 17 cell types in the ventral hippocampus and 22 in the amygdala with two replicates per strain per region (Figure 5, joint uniform manifold approximation and projection). The clusters included excitatory (glutamatergic) and inhibitory (GABAergic) neurons, as well as several types of non-neuronal cells that were annotated based on known marker genes. We define individual neuronal cell types based on neurotransmitter expression and region or top marker gene (for example, Exc-DG in the ventral hippocampus refers to excitatory cells in the dentate gyrus, and Exc-Tafa1 in the amygdala refers to excitatory cells marked by the gene Tafa1), and we define neuronal cell class as either excitatory or inhibitory through combining all excitatory or all inhibitory cell types, respectively (one amygdala neuronal type was excluded from both cell classes due to the expression of both excitatory and inhibitory markers). Non-neuronal cell types were not further defined due to an enrichment step for neuronal nuclei during sample processing limiting the number of non-neuronal nuclei captured.
Figure 5.

Cell-type uniform manifold approximation and projection for ventral hippocampus and amygdala in B6 and DBA from single-nucleus RNA, ATAC, and methylation sequencing. In all panels, vertical and horizontal axes represent UMAP space.
(A–C) Ventral hippocampal clusters plotted by (A) modality, (B) strain, and (C) cell-type identity.
(D–F) Amygdala neuronal clusters plotted by (D) modality, (E) strain, and (F) cell-type identity.
Given that QTL action is believed to have its effect on transcription through the alteration of the sequence at regulatory regions, we also generated single-nucleus data for CpG methylation70 and from a single-nucleus assay of transposase-accessible chromatin (snATAC-seq).71 Both epigenetic modalities can be used to identify regulatory elements, such as promoters and enhancers.72 Cell-type clusters generated from snRNA profiles were propagated to snATAC and methylation datasets through joint embedding (Figure 5). We confirmed that cell types were integrated by modality (Figures 5A and 5D), that there was no obvious strain bias for cell types (Figures 5B and 5E), and that cell types were well defined (Figures 5C and 5F). Importantly for interpreting our analyses, the three epigenetic datasets were obtained independently from different animals, with two independent replicates per modality.
Five out of six genes showed significant differences in expression between C57BL/6J and DBA/2J (significance defined as exceeding a 10% p value [logp > 1.0], adjusted for the total number of transcripts tested, from DEseq273 output; negative log2 fold change [log2FC] indicates higher expression in B6): 4933413L06Rik (in eleven cell types; median log2FC = −0.74, range = −1.63 to −0.53), Lsamp (in eleven cell types; median log2FCc = −0.21, range = −0.78 to 1.1), Psip1 (in five cell types; median log2FC = 0.41, range = 0.30 to 0.49), Nptx2 (in one cell type; log2FC = 1.82), and Ptprd (in eight cell types; median log2FC = −0.31, range = −1.24. to 1.85). Of the 36 cell types exhibiting differential expression for these genes, 31 were within the hippocampus. Different hippocampal cell types showed different degrees of overlap for differential gene expression of genes; for example, four of the five genes were differentially expressed within Exc-CA1-Galntl6 cells, while only 4933413L06Rik was differentially expressed within Exc-CA1-Camk2d cells. We noted that 22 of the 31 significant differences in the hippocampus and three out of five significant differences in the amygdala occurred in cell types categorized as excitatory neurons. Results of the DESeq2 analyses are given in Table S6.
We asked if the pattern of gene expression also held for snATAC-seq and methylation profiles, namely showing more strain differences in the hippocampus with an enrichment in excitatory neurons. To do this, we made an inventory of variable snATAC-seq and methylation sites. We counted differential ATAC sites (using a 5% threshold from adjusted p values derived from DESeq273) that contained sequence variants under the assumption that differences at these sites were more likely genetic in origin than those without such differences (more than 90% of the significantly different ATAC sites contain a sequence variant compared to 35% of non-variable sites). For the methylation data, we counted the number of fully methylated CpG sites in each cell type and divided them into those with and without mutations that disrupted methylation.
Figure 6 plots differential methylation and ATAC sites in the hippocampus and amygdala at the five QTLs where we identified causal genes. Three general observations arose from the data presented in Figure 6. First, there were more strain differences in the hippocampus than amygdala, as reflected in the greater number of dots compared to crosses. Second, at each locus, the strain differences were found primarily in excitatory neurons, in both the hippocampal and amygdala, as shown by the preponderance of red. Third, the pattern of strain differences for each modality varied between loci and genes, providing no unequivocal location or molecular signature to identify causal variants. The Ptprd locus (Figure 6A) contained a concentration of methylation, ATAC sites, and sequence differences at the 3′ end of the gene, suggesting a location for one or more causal variants here. At the Psip1 and Sh3gl2 locus (Figure 6B), strain differences were spread across the region with no indication of which might be relevant. The QTLs containing 4933413L06Rik (Figure 6C) and Nptx2 (Figure 6D) included a concentration of strain differences in methylation sites around the genes but with no obvious candidates for causative variants. One possible exception was the single ATAC site that exceeded a corrected significance threshold (logp = 6) in an intron of the Lsamp gene (Figure 6E) coinciding with a 2-kb deletion in DBA/2J. The function of this site and the consequences of its deletion are not known.
Figure 6.

ATAC-seq and methylation data at five QTLs in the ventral hippocampus and amygdala
(A–E) Data from the hippocampus are marked by dots (⋅) and data from the amygdala by crosses (+). In each image, the lowest row shows the distribution of genetic variants (black). The next two tracks, immediately below the genes and indicated by the blue background, show the location of methylation sites that coincide with a sequence variant and are thus present in only one of the two strains. These sites are colored according to the class of cell type in which they occur: red for excitatory neurons, blue for inhibitory neurons, and gray for non-neuronal cell types. Above the methylation tracks are shown ATAC sites, which differ significantly between the strains. The vertical axis shows the absolute value of log2 fold change for these ATAC sites. Sites are again colored according to the class of cell type in which they occur using the same color scheme as for methylation. The vertical positioning of the genes is only for ease of plotting and carries no meaning.
(F) Comparison of the percentage of significantly different RNA species in QTL regions and in the genome for the hippocampus. The vertical axis is the percentage of transcripts that are significantly different between the two strains, and the horizontal axis lists the cell types. Data from QTL intervals are indicated by dots colored according to class: excitatory (red), inhibitory (blue), and non-neuronal (gray). Data from the entire genome are indicated by black crosses.
The apparent enrichment of genetically mediated variation in excitatory neurons at the five QTLs containing the causal genes led us to examine whether the same was true for all the QTLs we had identified. We compared gene expression and epigenetic variation at 93 QTLs with the rest of the genome, expecting that any enrichment would be confined to, or more prominent at, the QTLs. Surprisingly, although the QTLs occupy about 0.01% of the genome, the percentage of significantly different transcripts (again defined as exceeding a 5% threshold from adjusted p values derived from DESeq273) in QTL regions was indistinguishable from the percentage of significantly different transcripts in the genome (Figure 6F), a finding replicated for methylation and snATAC-seq data (Tables S7 and S8). In other words, there was no enrichment in QTLs for genetically mediated variation in excitatory neurons; all regions of the genome contribute to this phenomenon.
Is the enrichment of genetic variants in excitatory neurons significant? It certainly seems so, but at least two confounds could produce this result. For the snATAC-seq and snRNA-seq data, identification of significant strain differences is more likely with higher read coverage, and for methylation, the more sites we identify, the higher the likelihood that some will coincide with a sequence variant. To assess the contribution of sequence coverage, we compared a null model, in which sequence coverage predicted the number of differentially expressed genes, ATAC peaks, or methylation sites with mutations, to one that additionally included cell type and found highly significant improvements in fit (all p values < 2.2E−16; Table S9), demonstrating that cell type was an important contributor, over and above the contribution of sequence coverage. A generalized linear model to predict differences also showed that sequence coverage does not account for the differences between excitatory and inhibitory neurons (Table S10).
Second, the inclusion of more excitatory cell types than inhibitory types could bias findings (eight excitatory cell types compared to three inhibitory in the hippocampus) because the addition of extra cell types increases the chances of finding variant sites. Against this, we note that in the amygdala, there were more inhibitory than excitatory cell types (eight versus six). We ran a test of this hypothesis, by comparing the contribution of class (excitatory and inhibitory) to cell type, considering that cell type was completely nested within class. We resorted to a Bayesian analysis, which revealed a significant effect of class (the 95% confidence intervals do not overlap zero) (Table S11).
Is the enrichment in excitatory cells a peculiarity of the difference between C57BL/6J and DBA/2J? We answered this question for the hippocampus using methylation data from six other strains. Using CAST/EiJ as an outgroup, we compared the number of mutations in CpG sites at excitatory and inhibitory neurons in the ventral hippocampus. Table S12 shows that for each strain, compared to CAST/EiJ, the proportion of mutated CpG sites was higher in excitatory than inhibitory neurons, supporting our finding that at least for methylation, alterations in functional elements directly attributable to genetic variation were enriched in excitatory neurons compared to other cell types. Together, these results suggest a model whereby genetic effects act on specific hubs of circuits/cell types.
Discussion
We have established the feasibility of combining mapping with QC testing for gene identification, breaking open a bottleneck in the genetic dissection of complex traits in mice, and provided a set of six genes for further mechanistic understanding of fear-related behavior. Unexpectedly, genetically mediated variation in three independent molecular analyses, methylation, snRNA-seq, and snATAC-seq, was found to occur preferentially in excitatory neurons and to be enriched in the hippocampus compared to the amygdala. These observations demonstrate the possibility of moving quickly from locus to gene, identify unexplored biology underlying fear-related behavior, and raise questions about expected relationships between genetic and functional variation.
Our findings highlight the power of unbiased genetic examination of the biological basis of behavioral variation. At a locus on chromosome 13, we made the unexpected discovery that an lncRNA (4933413L06Rik) is involved in cue fear conditioning. Little is known about 4933413L06Rik (one study found it to be differentially expressed during development of the auditory forebrain74). Our finding opens the possibility for clarifying the function of this lncRNA in fear-related behavior. Two other causal genes (Psip1, Sh3gl2) have also never been studied in the context of fear-related behaviors. We note that of the five genes with known functions, four (Lsamp, Ptprd, Nptx2, and Sh3gl2) are implicated in synapse development or function.75,76,77,78,79 Ptprd belongs to the type IIA receptor-type protein tyrosine phosphatase family of phosphatases, involved in synaptic formation and structure during development and neurogenesis.76 Lsamp is a cell surface adhesion molecule that is involved in neuritogenesis and axon guidance (and is also implicated in the maturation of serotonergic80 and thalamocortical circuits81). Nptx2 is a secreted glycoprotein that localizes to both pre- and post-synaptic compartments of excitatory synapses containing AMPA receptors and is involved with synaptogenesis82 and adult neurogenesis.83 Sh3gl2 (endophilin1), a cytoplasmic Src homology 3 domain-containing protein,84 localizes to presynaptic nerve terminals, where it functions in synaptic vesicle endocytosis85 and the regulation of exocytosis.86 The significance of these functional observations is difficult to assess, but it at least suggests that variation in fear-related behavior in part originates in synaptic development.
The QC test could serve as a gold standard for gene identification following QTL mapping in inbred mice. Following mapping, the construction of KOs is relatively straightforward, and the test requires a simple breeding protocol. We recommend powering the QC tests based on results from the mapping experiment (the effect size of the locus in the mapping population will be lower than that in QC test87), but note that our failure to detect a candidate gene at a locus on chromosome 5 could be due to low power or because we tested the wrong gene.
Our instantiation of the QC test is not the only one; alternative designs are possible. For example, in an experiment to test the candidacy of a gene at a QTL for resistance to obesity in mice, a QC test was constructed from a KO, a subcongenic containing the QTL, and their corresponding background strains (a cross giving rise to four genotypes on a uniform genetic background).88 However, it is essential to use KOs on an isogenic background so as to avoid the complication of the effect of additional QTLs that may be present on the non-isogenic DNA.
Interpretation of the QC results is subject to some qualifications. First, while a positive QC test result reveals a gene through which a QTL has its effect, it does not necessarily mean that the QTL is the one containing the candidate gene. The QC test will detect the effect of any QTL that requires the gene to be present. Typically, that QTL will be the one local to the gene tested, but that assumption is not essential for the functioning of the QC test. Second, the QC test makes no assumptions about how the QTL and KO interact. Does this matter? It may, because in the QC test, non-additive interactions are typically easier to detect than additive ones. Under an additive model, the KO removes the effect of one of the QTLs so that the phenotypic effect is reduced by the allelic effect of that QTL. In non-additive models, the presence of the KO could reduce one QTL effect to zero (recessive effect) or increase it, potentially in a multiplicative fashion. In these cases, the effect of the interaction is to augment the allelic effect over that of the additive interaction. We have some evidence that non-additive interactions predominate in our results.
QC tests results from four genes (Lsamp, Ptprd, 4933413L06Rik, and Psip1) are consistent with a non-additive interaction between QTL and KO. In each case, the phenotype of the KO on the hybrid background is larger than either the WT DBA/2J or the KO on the C57BL/6J (Figures 2, 3, and 4). One interpretation is that non-additivity is common. Another interpretation is that we failed to detect additive interactions because non-additivity is easier to detect: the larger effect attributable to the interaction confers greater power for the QC test. This may contribute to false negative findings, but it does not undermine the conclusion that the effect of the QTL is mediated by the gene. Under both additive and non-additive models, a significant result means that the effect of the QTL requires the gene.
One tantalizing corollary emerging from the analysis of gene causality in fear-related behaviors is that genetic variation may act preferentially in excitatory, rather than inhibitory, neurons. We note that any patterns of strain differences could not be attributed solely to coverage, as even downsampling our RNA-seq and ATAC-seq data to as low as 25% of the total unique molecular identifiers still resulted in substantial detection of significant strain differences (see https://dx.doi.org/10.6084/m9.figshare.25521304). We had expected that the pattern of strain differences might point to the involvement of a particular cell type in one region of the brain. Instead of finding cell-type enrichment, we observed that strain differences in three different modalities, snRNA-seq, methylation, and snATAC-seq, were more prevalent in excitatory than inhibitory neurons, particularly in the hippocampus compared to the amygdala. Moreover, we found this enrichment pattern to be true not just for the QTL regions but for the entire genome. There was little evidence of strain differences in non-neuronal tissues, although we can be less confident in this assertion because our sample was enriched for neuronal cells and may not be representative of non-neuronal variation.
What might explain this finding? One possibility is that it is specific to a comparison between C57BL/6J and DBA/2J. We think that this is unlikely and provide some evidence against this idea by running a similar analysis for methylation data in other strain comparisons, though we have not extended this observation to other modalities. Alternatively, the finding represents a preference for genetic effects in excitatory neurons. Why might this be so?
The idea that genetic effects operate preferentially in excitatory neurons fits with a neuronal circuit model where inhibitory neurons sculpt behaviors driven by excitatory neurons. Excitatory neurons are extremely diverse, resulting in high-dimensional and sparse coding,89 consistent with the expectation that such an architecture permits highly efficient information transfer.90 Consequently, encoding within excitatory networks cannot be predicted from their transcriptome alone. In contrast, activity within inhibitory neuronal networks is lower dimensional, with correlations between inhibitory cell types determined primarily by cell type, so that cells belonging to a single transcriptomic class are highly correlated.91,92,93 In this model, genetic variation is more permissible in excitatory neuronal networks than inhibitory, suggesting that disturbances of inhibitory networks are more likely to be pathological. If this interpretation is correct, then identifying genetic effects that disturb function at a circuit level may be more fruitful than looking for cell-type-specific genetic effects.
Limitations of the study
Our results are subject to some limitations. First, the results from the meta-analysis have not been replicated, so it is possible that some of the loci may be false positive. This may explain why the QC test was negative for Stim2 on chromosome 5. Second, some of the negative QC test results may be due to relatively low power to detect additive effects compared to non-additive effects. Third, we did not test every expressed transcript at every QTL. For instance, it is possible that single-exon unannotated transcripts (e.g., Gm6416, which we did not test at the chromosome 13 locus) might also contribute. However, the failure to detect the involvement of additional genes does not mitigate the main result of our experiment, namely, that the QC test can be used in a systematic fashion to identify genes mediating the QTL effect.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| NeuN-488 | Millipore Sigma | MAB377X; RRID:AB_2149209 |
| NeuN-405 | Novus Biologicals | NBP1-92693AF405 |
| Critical commercial assays | ||
| Next GEM scATAC-Seq v1.1 | 10X Genomics | PN-1000175 |
| Chromium Next GEM Automated Single Cell 3′ Library and Gel Bead Kit v3.1 | 10X Genomics | PN-100014 |
| Deposited data | ||
| snmC-seq2 data of 8 mouse strains, amygdala | This study | GEO: GSE262259 |
| snATAC-seq data of 2 mouse strains, amygdala | This study | GEO: GSE262259 |
| snRNA-seq data of 2 mouse strains, amygdala | This study | GEO: GSE262259 |
| snmC-seq2 data of 8 mouse strains, vhippocampus | Flint et al.94 | GEO: GSE245367 |
| snATAC-seq data of 2 mouse strains, vhippocampus | Flint et al.94 | GEO: GSE245367 |
| snRNA-seq data of 2 mouse strains, vhippocampus | Flint et al.94 | GEO: GSE245367 |
| CRISPR mouse KO generation | This study | https://doi.org/10.6084/m9.figshare.25521283 |
| Subsampling of single-nucleus RNA and ATAC sequencing data and significant genes detected | This study | https://doi.org/10.6084/m9.figshare.25521304 |
| Experimental models: Organisms/strains | ||
| C57BL/6J | JAX | Strain ID: 000664 |
| DBA/2J | JAX | Strain ID: 000671 |
| CAST/EiJ | JAX | Strain ID: 000928 |
| FVB/NJ | JAX | Strain ID: 001800 |
| A/J | JAX | Strain ID: 000646 |
| WSB/EiJ | JAX | Strain ID: 001145 |
| PWK/PhJ | JAX | Strain ID: 003715 |
| BALB/cJ | JAX | Strain ID: 000651 |
| C57BL/6J-Megf9-KO | This study | N/A |
| C57BL/6J-Emb- KO | This study | N/A |
| C57BL/6J-Hcn1-KO | This study | N/A |
| C57BL/6J-Ptprd-KO | This study | N/A |
| C57BL/6J-Lsamp-KO | This study | N/A |
| C57BL/6J-Nptx2-KO | This study | N/A |
| C57BL/6J-Stim2-KO | This study | N/A |
| C57BL/6J-Mrps30-KO | This study | N/A |
| C57BL/6J-Parp8-KO | This study | N/A |
| C57BL/6J-Psip1-KO | This study | N/A |
| C57BL/6J-Sh3gl2-KO | This study | N/A |
| C57BL/6J-Snapc3-KO | This study | N/A |
| C57BL/6J-Ttc39b-KO | This study | N/A |
| C57BL/6J-4933413L06Rik-KO | This study | N/A |
| Software and algorithms | ||
| Cell Ranger V6.0.2 | 10X Genomics | https://www.10xgenomics.com/support/software/cell-ranger/downloads |
| Cell Ranger ATAC V2.0.0 | 10X Genomics | https://support.10xgenomics.com/single-cell-atac/software/downloads/latest |
| Bismark V0.20.0 | Krueger and Andrews95 | https://github.com/FelixKrueger/Bismark |
| DESeq2 V1.34.0 | Love et al.73 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Seurat 4.0.5 | Stuart et al.96 | https://github.com/satijalab/seurat |
| Signac 1.5.0 | Stuart et al.97 | https://github.com/stuart-lab/signac |
| Model-based Analysis for ChIP-Seq (MACS) V3.0.0a7 | Zhang et al.98 | https://github.com/macs3-project/MACS |
| DoubletFinder V2.0.3 | McGinnis et al.99 | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| Scanpy V1.9.3 | Wolf et al.100 | https://pypi.org/project/scanpy/ |
| Harmony V0.0.9 | Korsunsky et al.101 | https://github.com/immunogenomics/harmony |
| SCTransform V0.3.2 | Hafemeister et al.102 | https://github.com/satijalab/sctransform |
| EMINIM Beta 1.01 | Kang et al.48 | http://genetics.cs.ucla.edu/eminim/index.html |
| GEMMA V0.94 | Zhou and Stephens51 | https://github.com/genetics-statistics/GEMMA |
| PLINK V1.90b3.38 | Purcell et al.103 | https://zzz.bwh.harvard.edu/plink |
| METASOFT V2.0.1 | Han and Eskin53 | http://genetics.cs.ucla.edu/meta_jemdoc/ |
| GCTA V1.93.1 | Yang et al.55 | https://yanglab.westlake.edu.cn/software/gcta |
| Wrappers for genetic mapping and quantitative complementation in inbred mouse strains | This study | https://doi.org/10.5281/zenodo.10892164 |
Resource availability
Lead contact
Further information and requests for reagents and resources should be directed to and will be fulfilled by the lead contact, Jonathan Flint (JFlint@mednet.ucla.edu).
Materials availability
All mutant KO animals are deposited as frozen sperm at the Transgenic and Genome Editing Core Facility at Augusta University. Ordering information can be found at https://doi.org/10.6084/m9.figshare.25521283.
Data and code availability
-
•
Raw and processed sequencing data generated for this study were deposited to NCBI GEO/SRA with accession number GSE245367 and GSE262259 and are publicly available at the time of publication.
-
•
Mapping data (including imputed genotypes of all mouse inbred strains and F1s, together with results from the meta-analysis of the three phenotypes) https://figshare.com/s/7fcb1eebfd9c607afd12.
-
•
Detailed CRISPR KO mouse generation for each gene, including exon targeted, sgRNA sequences, genotyping primers, and DNA gel images https://doi.org/10.6084/m9.figshare.25521283.
-
•
Knockout interaction data, including phenotypes and genotypes for all quantitative complementation tests https://figshare.com/s/cc86ed5777cb95b99cec.
-
•
Processed single nucleus epigenetic and gene expression data https://figshare.com/s/39e1c671ec0dc09d5b27.
-
•
All original code has been deposited at https://doi.org/10.5281/zenodo.10892164.
Experimental model and study participant details
Mouse strains
All experimental procedures using live animals were approved by UCLA’s Animal Care and Use Committee (protocol number ARC-2018-026). Male mice from eight inbred strains A/J, C57BL/6J, BALB/cJ, FVB/J, DBA/2J, WSB/EiJ, PWK/PhJ, and CAST/EiJ were purchased from Jackson Laboratories at 8 weeks of age and transferred to UCLA where they were kept for at least 7 days before tissue extraction. Animals were housed with ad libitum food and water in a 12 h light-dark cycle. Mutant KO animals were generated on a C57BL/6J background.
Method details
Mouse phenotyping and mapping
Published data were obtained from three sources: the Jackson Laboratory website (https://phenome.jax.org), the Gene Network website (https://genenetwork.org) and PubMed. PubMed articles were identified using search terms including ‘fear’, ‘anxiety’, ‘elevated plus maze’, and ‘fear conditioning’. This identified 29 potentially useful datasets.16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45
Harmonizing phenotyping data for meta-analysis
To harmonize the phenotypic data for the meta-analysis, we had to deal with the heterogeneity of the datasets collected. Each experiment used different protocols, different testing equipment and often reported different, though related, assays. Different studies used different names for what was supposedly the same phenotype. For example, for fear conditioning some studies reported the time freezing,26 other reported its opposite, the amount of activity.19 Some recorded 3 min of freezing, some 5 min, or more. EPM behavior was recorded in apparatus of different dimensions, under different lighting conditions, at different times of day, all of which are known to affect behavior.104
To deal with this, after downloading data and examining what was most collected, we decided to use the time freezing during a 5-min period after exposure to an altered context (contextual freezing), and the mean time spent freezing during exposure to a conditioned stimulus (most usually a tone). For the elevated plus maze we used the number of entries into the open arms. We created a data dictionary to convert the different phenotype names to a common set, and this is provided in Table S2.
Some studies also provided information about sex and age, and we tested, using a linear model, whether there was an effect on phenotypes of either. Since we found no significant effects (at a 5% threshold after correcting for multiple testing) and since some studies did not provide this information, we did not include covariates in downstream analyses.
We examined the relationship between phenotypes from different studies in two ways. First, for each strain for each study we obtained the trait mean and then calculated the correlation between the same strains in different studies. Not every study tested the same strains, so although there were 42 possible pairwise comparisons for FC-context, only 30 comparisons could be carried out. One study (Palmer5) was dropped because of insufficient overlap in the strains used with other studies. There was a considerable range in the correlations between studies (from −0.04 to 0.93), though only 13 comparisons were significant (at a p < 0.05, uncorrected for multiple testing), in part reflecting differences in the variation in the number of strains in each comparison (from 3 to 31). In some cases, a pair of studies were correlated, but not with other studies (for example for the FC-cue phenotype, study Bolivar2 is correlated with Bothe-2005 (r = 0.85) but neither study was highly correlated with others).
As a second measure of the relationship between studies, we ordered the means of each strain, and ranked them. We then applied the order for one study to another to examine the extent to which studies agreed on the order of strain means. We decided to use the pattern of correlations to select studies for the genetic analysis, requiring the presence of more than 5 strains in common, and a positive correlation greater than 0.2 (though we did not require this to be significant since). The studies we chose, together with the number of animals and the phenotype names, is given in Table S1.
Behavioral phenotyping
Mice from the Mouse Diversity Panel (HMDP) were used for the behavioral analyses. Mice (n = 700) were obtained through Jackson Laboratory at approximately 60 days old and housed for a 14-day acclimation period prior to testing. Mice were housed in groups (3–4 per cage) under a 12h/12h day/night cycle with ad libitum access to food and water. Testing was carried out between 10 a.m. and 4 p.m. Auditory background stimulus in the form of white noise (80db) was delivered through overhead speakers. All protocols conformed to NIH Care and Use Guidelines and were approved by UCLA’s Animal Care and Use Committee (protocol number ARC-2018-026).
Behavioral tests
All tests were completed at the Behavioral Testing Core at UCLA. Animals were handled for 5 days prior to experiments. The order of experiments for genetic mapping was EPM -> FC cue/context. EPM -> 7 days-> FC training -> 24 h -> FC context -> 24 h -> FC tone.
Elevated plus maze
Animals were placed into an elevated plus maze apparatus (arms 5 cm × 30 cm) for 5 min with 15 lux at the center. We measured time spent in open/closed arms, and total entries into open/closed.
Fear-conditioning
Mice were placed in the conditioning chamber (context A) for 3 min before the onset of the discrete conditioned stimulus. The parameters were as follows:
Tone: pure tone, 2700Hz, 80dB, rise/fall 50, duration 30s.
Shock: 0.75 mAmps, duration 1s, inter-trial interval 180s.
Animals were exposed to three pairings of the CS and US. After the CS-US pairings, the mice were left in the conditioning chamber for another 60 s and then placed back in their home cages. FC boxes and software to control the boxes were purchased from MedAssociates.
Fear-conditioning context test: 24 h after conditioning, mice were returned to the same context (context A) with no shock or tone and freezing was measured for 8 min.
Fear-conditioning cued test: 24 h after the context test, mice were placed in a novel context (context B) where, they are exposed to the same procedure as the training but without any shocks (3 min exposure followed by 3 CS-US pairings, with parameters equal to fear acquisition). Freezing was measured during the initial 3 min exposure, during tone, and during the inter-trial interval.
Behavioral analysis
Behavior was recorded digitally from a camera mounted above each test chamber at 30 FPS. Following the completion of experiments, each recorded EPM video was analyzed using ANY-maze for positional tracking of the animal along the apparatus. For EPM mapping we measured open arm entries, time spent in open arms, and closed arm. VideoFreeze (MedAssociates) was used to measure animal freezing in QC testing from recorded videos. Freezing was determined as an absence of all visible movement except that required for respiration; in VideoFreeze, we set the motion threshold to 18 au with a minimum freeze duration of 1 s (30 frames). Contextual fear was assessed by total freezing time over the first 3 min of the test. Cued fear was assessed by total freezing time in response to the presentation of the first, second, and third CS during ITI. One exception was with freezing measurements during genetic mapping with BXD lines. Due to the variable coat color across lines, we used ANY-maze for animal tracking and freezing measurements as it performed better than VideoFreeze.
Genotyping
Our starting point for genotyping each mouse strain was variant calls from sequencing reads for those strains that have been sequenced,46,105 and genotypes from high density arrays for those that have not106 (https://www.informatics.jax.org/). We confirmed genotypes in the BxD recombinant inbred lines by randomly choosing 24 lines and subjecting to genotyping them on the GigaMUGA mouse array (140 thousand SNPs).107 This genotyping was done commercially (Neogen GeneSeek Operations, 4131 N 48th St. Lincoln, NE 68504). All lines were of the expected genotype.
Imputation of genotypes
We used imputation to generate a near complete set of genotyped markers for all strains. The highest density set of genotyped markers is that derived from 16 strains for which we have complete sequence data46 but this does not include any recombinant inbreds and lacks some of the strains used for mapping. Since the missing strains are descendants of a small number of founder haplotypes,106 we can impute the genotypes in the non-sequenced strains. Using genotypes from arrays as a scaffold56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,104,105,106,107,108 we imputed missing genotypes using data from sequenced animals. Most imputation algorithms are designed for fully outbred populations, where a key problem is correctly phasing haplotypes (e.g.,109). Alternative methods are needed for efficient imputation in mouse strains and so we used the software tool EMINIM,48 which requires a reference file, containing strains with known haplotypes, and a target file for imputation, containing strains genotyped at some SNPs. We ran this with the recommended parameter settings for inbred mice given according to the software documentation.
We prepared these two files by placing strains that were genotyped at all SNPs in the reference file, while strains with one or more missing SNPs were placed in the target file. For the BXD mice, SNPs between genotype markers from the same founder haplotype were pre-filled due to the negligible likelihood of multiple recombination events between consecutive genotyped SNPs. This resulted in 27 reference strains and 227 target strains. We then ran EMINIM imputation on each chromosome separately and obtained calls on 16,767,664 SNPs (triallelic SNPs, constituting 1% of the total, were discarded). Since EMINIM outputs high confidence values for one of the alleles, we translated a 90% or higher confidence for an allele into a hard call for that allele, with lower confidence calls being called as missing. We combined these imputation results with the genotyped data. The rate of missing calls was less than 0.8% for 18 of 19 autosomal chromosomes (chromosome 19 had a missing call rate of 1.03%).
We validated the imputation results by comparing imputed calls to genotypes from sequenced BXD strains obtained from.110 The sequence-derived set contained 4,325,552 SNPs, of which 3,812,095 variant sites overlapped with our imputation. Across all chromosomes, each BXD strain was imputed with between 93% and 99% accuracy. Imputation accuracy for all strains combined varied between 89% and 99% per chromosome, with 96.29% overall. Due to the pre-filling procedure described previously, this accuracy reflects only the most challenging SNPs to impute: those between genotyped SNPs from different founder haplotypes. When including the genotyped and pre-filled SNPs, the accuracy for each chromosome was greater than 99.64%.
We performed a second assessment of imputation accuracy by removing the C57BL/6J and DBA/2J strains from our reference panel, randomly masking 50% of the bases of those strains, and using the remaining inbred strains in the reference panel to impute the masked bases. Once again, we used EMINIM with its default inbred imputation parameter settings and translated a 90% or higher confidence call for an allele into a hard genotype call. Calls with less than 90% confidence were translated to a missing call, leading to a missing genotype rate of 0.09%–0.59% per chromosome. The accuracy of imputation on the masked bases was 95.6–99.7% per chromosome, and 97.9% overall.
Genetic mapping
All phenotypes were mapped in a two-stage approach. First, we used a mixed model, implemented in GEMMA51,52 (obtained from https://github.com/genetics-statistics/GEMMA), to map each study separately. Input files for GEMMA were generated using PLINK,103 obtained from https://zzz.bwh.harvard.edu/plink. We used the PLINK binary format of the required files, including all alleles with a frequency greater than 10%. A genetic relationship matrix was generated using the command:
gemma -bfile input.file -gk -o kinship.file.
Mapping was carried out using the command:
gemma -bfile pl input.file -k kinship.file -lmm -o output.file.
After mapping with GEMMA, the results files were combined with purpose written perl scripts into a format suitable for meta-analysis with METASOFT.53,54 METASOFT (downloaded from http://genetics.cs.ucla.edu/meta_jemdoc/) was invoked with the following command:
java -jar Metasoft.jar -input input.file -mvalue -output output.file.
GEMMA assigns effects to the minor allele, as determined by the.bim files from PLINK which means there is considerable variation between studies as to which allele gets an effect (due to allele frequency variation). After running each study though GEMMA we swapped alleles to the most common arrangement, before proceeding to the meta-analysis.
METASOFT calculates a genomic-control inflation factor for the mean effect (lambda_Mean Effect) and a genomic-control inflation factor for heterogeneity (lambda_heterogeneity). These values were calculated for each run, and the software was run a second time, to include the effect of these variables. For example:
java -jar Metasoft.jar -input input.file -mvalue -output output.file -lambda_hetero 2.738124 -lambda_mean 2.430179.
Significance threshold for the genome-wide association studies
Simulations to estimate the appropriate significance threshold were carried out using GCTA,55 obtained from https://yanglab.westlake.edu.cn/software/gcta. Phenotypes were simulated using real genotype data under a simple additive genetic model. A set SNPs was chosen as the causal variants that were seen to have effects in the mapping studies. We obtained estimated effect sizes for these SNPs from GEMMA output files and converted the beta estimate from the linear model to the values required by GCTA using
(where p is the allele frequency from the GEMMA output file)
Input SNP files were those used for the mapping with GEMMA. 1000 simulations were carried out using the command:
gcta64 --bfile input.SNP.file --simu-qt --simu-causal-loci causal.snplist --simu-hsq 0.2 --simu-rep 3 --out.
After simulating data for each component study, files were processed through GEMMA and METASOFT exactly as for the real data.
To investigate the contribution of genotype structure to the inflation seen in quantile-quantile plots we simulated data in the same way, but with no causal SNPs.
Generating knockout animals for QC testing
We identified genes lying within QTLs, and determine whether they are expressed in brain tissue from in situ hybridization data (Allen Brain Atlas,60 the mouse ENCODE transcription dataset61), and RNA-seq data obtained from the following publications: 62,63,64,65,66,67 For ISH data from the Allen dataset, we looked for presence of expression. For ENCODE transcriptome data we set a cutoff for an RPKM >5 in at least one brain region category, and for RNA-seq studies we ranked expression of each gene and averaged across studies; any gene in the top 25% of all genes (or >rank 6000) was considered expressed. For the Allen institute scRNA-seq dataset, we only included sequencing data from cell type clusters from CA1-CA3 of the hippocampus. Genes that passed expression thresholds for all three datasets were included for further consideration.
Where an existing knockout had been successfully created for the gene, usually on a C57BL/6N background, we adopted the same design, targeting the same part of the gene but in the C57BL/6J strain. To generate each KO, we used a CRISPR-Cas9 genome editing approach. For each gene, two single guide RNA (sgRNA) were generated by Synthego Corp. (Redwood City, California, USA) for targeting, except for 4933413L06Rik where we carried out a knock-in with a single-stranded DNA synthesized by IDT, Inc (Coralville, Iowa, USA). Guide RNAs (gRNAs) were designed to maximize both efficiency and specificity scores.111 The sgRNA and/or ssDNA, and Cas9 protein (Alt-R SpCas9 Nuclease V3 from IDT) were co-injected into zygotes of C57BL/6J mice (Jackson Laboratory, Stock#009086). After microinjection, zygotes were transferred into the oviduct of pseudo-pregnant Swiss Outbred mice (Jackson Laboratory, Stock#034608) to generate founder mice. Founder mice were obtained and confirmed by PCR genotyping and Sanger sequencing. The correctly targeted founders were bred with C57BL/6J mice, and PCR genotyping and Sanger sequencing were again performed to confirm germline transmission. We used quantitative PCR to confirm that the deletion affected abundance of RNA transcript in hippocampal tissue from F1 animals. Full descriptions of each knockout design, and sgRNA sequences are given at dx.doi.org/10.6084/m9.figshare.25521283. Genotyping primers are listed in Table S4.
Quantitative polymerase chain reactions
To confirm the effect of the deletion on RNA production at each locus we ran quantitative polymerase chain reactions using a standard RT–qPCR assay (Taqpath COVID-19 Combo Kit). We analyzed RNA extracted from the hippocampus of heterozygous knockouts on C57BL6/J animals. Primer sequences can be found in Table S5.
Quantitative complementation testing
We generated offspring from four crosses.4 C57BL/6J animals were mated to DBA/2J, and to heterozygote C57BL/6J animals where one allele was the knockout. DBA/2J animals were also mated to the heterozygote knockout mice. This generated four groups: wildtype C57BL/6J, F1 DBA/2J/C57BL/6J (wildtype), heterozygote C57BL/6J knockouts, and F1 DBA/2J/C57BL/6J (knockout). All four groups were assayed for the relevant phenotypes associated with the QTL, using the phenotyping protocols described above.
Evidence that the gene is the QTL gene was then detected as a statistical ‘cross’ (mutant or wildtype) by ‘strain’ (DBA/2J or C57BL/6J) interaction in a linear model, using the R statistical programming language.112 Batch and sex were included as covariates, using the R command
Ventral hippocampus microdissections
Adult male animals (Jackson Laboratories) were euthanized at 10–16 weeks old in an isoflurane chamber and decapitated. The brain was removed and the ventral region of the hippocampus was microdissected, snap frozen in dry ice, and stored at −80°C until processing. Tissue from ∼2 animals were combined into a single tube and considered a replicate, with 2 replicates per strain for snmC-seq2, snRNA-seq, and snATAC-seq experiments.
Amygdala microdissections
Adult male animals (Jackson Laboratories) were euthanized at 10–16 weeks old in an isoflurane chamber and decapitated. The brain was removed and coronal brain slices containing amygdala tissue were generated on a 1mm brain matrix (World Precision Instruments). Amygdala tissue was micro-dissected from these slices under a dissecting scope in cold PBS, snap frozen in dry ice, and stored at −80 until processing. Tissue from ∼2 to 3 animals were combined into a single tube and considered a replicate, with 2 replicates per strain for snmC-seq2, snRNA-seq, and snATAC-seq experiments.
Generating snmC-seq2 libraries
We carried out snmC-seq2 on microdissected tissue as previously described.70 Briefly, frozen tissue was homogenized into single nuclei suspensions with Dounce homogenization, then immediately sorted on into a 384-well plate with a FACSAria sorter (BD Biosciences) at the UCLA Flow Cytometry Core. We selected for a 75-25 enrichment of neuronal vs. non-neuronal nuclei during FACS sorting using NeuN-488/DAPI counterstains (Millipore Sigma MAB377X). Bisulfite conversion and single-cell methylome libraries were generated following this step.
Generating snRNA-seq libraries
Single nuclei suspension and library generation were completed at the Cedars Sinai Applied Genomics, Computation and Translational Core and followed the 10X protocol for the Chromium Next GEM Automated Single Cell 3′ Library and Gel Bead Kit v3.1 (cat# PN-100014) as described except for the following modifications:
Suspensions from cell nuclei were generated using the recommended method from the 10X scMultiome protocol (CG000375 Rev C) to lyse cells and obtain nuclei. Following single nuclei suspension generation, nuclei were counterstained for 7-AAD and NeuN-405 antibody (Novus Biologicals, 1:200) and sorted on a MACSQuant Tyto (Miltenyi Biotech) prior to GEM generation. We selected for a 75-25 split of NeuN+/7-AAD+ nuclei for neurons and NeuN-/7-AAD+ for non-neuronal nuclei respectively.
We captured ∼10,000 nuclei per genotype per region per replicate on a single 10X GEM chip. All downstream library preparation was done according to the 10X Genomics protocol (CG000286) and sequenced on a Novaseq 6000 with a target of ∼40-50k reads per nucleus.
Generating snATAC-seq libraries
Single nuclei suspension and library generation were completed at the Cedars Sinai AGCT core and followed the 10X protocol for Next GEM scATAC-Seq v1.1 (PN-1000175) as described except for the following modifications:
Nuclei suspensions were generated using the recommended method from the 10X scMultiome protocol (CG000375 Rev C) to lyse cells and obtain nuclei.
Following single nuclei suspension generation, nuclei were counterstained for 7-AAD and sorted on a MACSQuant Tyto prior to GEM generation. NeuN was not used for neuronal enrichment due to dye incompatibility between our NeuN antibody and a nuclear counterstain. After the sort, we carried out permeabilization of nuclei as per the protocol. We aimed to capture 10,000 nuclei per well x 8 wells, for a total of 80,000 nuclei over 8 total samples (∼10,000 nuclei per genotype per region per replicate). All downstream library preparation was done according to the 10X Genomics protocol (CG000209) and sequenced on a Novaseq 6000 with a target of >35k reads per nucleus.
Quantification and statistical analysis
Single-nucleus RNA sequencing data quality control and pre-processing
All quality control and pre-processing were done under the Seurat package framework.96 Per biological sample, we filtered out cells that (1) fall below the 5th percentile of the total UMI counts (nCount_RNA) or the 5th percentile total number of unique genes expressed (nFeature_RNA) or 700 unique genes expressed, whichever was more stringent; (2) are over the 95th percentile quantile in terms of either the total UMI counts or the total number of unique genes expressed; (3) have larger than 5% mitochondria fraction (percent.mt).
Global coverage normalization: counts per million (CPM) was applied to each cell followed by log transformation (“LogNormalize”). We then projected cells from each biological sample to low dimensional space using principal components analysis (PCA) on highly variable features selected by Seurat. Potential doublets were identified and subsequently removed from the downstream analysis by DoubletFinder,99 ran in the top 15 principal components space with the expected doublet rate set to the recommended amount from 10X genomics based on high quality yield volume.
Single-nucleus ATAC sequencing data quality control and pre-processing
All quality control and pre-processing were done under the Seurat and Signac package framework.97 Per sample, we first used Model-based Analysis for ChIP-Seq (MACS) to call sample specific de-novo peaks from its fragments file.98 We then merged sample-specific sets of peaks to a unified peaks set while removing peaks with length larger than 10000bp or smaller than 20bp. A unified peaks by cells count matrix was constructed from the fragments file while removing cells with lower than 200 peaks detected and peaks only present in less than 10 cells. Cells were filtered based on the following criteria: (1) appropriate number of non-duplicate, usable read-pairs (passed_filters from Cell Ranger’s output singlecell.csv). Specifically, we set it to larger than 3000, 4000, 2500, and 5000 for the 2 BL6 and 2 DBA samples collected in the hippocampus region. Similarly, we set it to be larger than 7500, 5000, 7500, and 7500 for their amygdala counterpart. (2) number of fragments overlapping peaks (peak_region_fragments from Cell Ranger’s output singlecell.csv) falls within the 5th percentile and the 95th percentile. (3) ratio of fragments overlapping peaks over the total number of non-duplicate, usable read-pairs falls within the 5th percentile and the 95th percentile. (4) nucleosome_signal: the ratio of fragments between 147 bp and 294 bp (mononucleosome) to fragments <147 bp (nucleosome-free) is smaller than 4 (5) TSS enrichment score is larger than 2. We did not include a filter for ratio of peaks in black list regions over the total number of non-duplicate, usable read-pairs as this was removed during the construction of the DBA SNP-swapped reference genome. We normalized the count data with Text Frequency Inverse Document Frequency (RunTFIDF) and performed Singular Value decomposition (RunSVD) on top 90% informative features selected by Signac (FindTopFeatures). The first low dimensional embedding was excluded from downstream doublet detection and clustering analysis due to high correlation with sequencing depth. Potential doublets were identified and subsequently removed from downstream analysis by DoubletFinder,99 ran on the 2nd - 11th low dimensional embedding with the expected doublet rate set to the recommended amount from 10X genomics based on high quality yield volume. Finally, we built a gene-by-cell transcriptional activity matrix that counts per cell, at the gene body and 2000bp upstream to capture the promoter region, the total number of ATAC-seq counts.
Single-nucleus methylation data quality control and pre-processing
Cells were filtered on the basis of several metadata metrics: (1) mCCC level <0.03; (2) global mCG level >0.5; (3) global mCH level <0.2; (4) total mapped reads >100,000; (5) Bismarck mapping rate >0.5; and 6 (percent genome covered >2). Methylation features were calculated as fractions of methylcytosine over total cytosine across gene bodies ± 2kb flanking regions and 100kb bins spanning the entire genome. Methylation features were further split into CG and CH methylation types. Features overlapping our methylation mm10 blacklist were removed. 100kb bin features were then filtered on minimum mean coverage >500 and maximum mean coverage <3000. Gene body features were filtered on minimum coverage >5 and all remaining features were normalized per cell using the beta binomial normalization technique in allcools.72 Individual CpG sites were only counted when they had ≥5 reads covering the site.
Single-nucleus RNA sequencing data integration, clustering and annotation
Gene counts were normalized using SCTransform,102 and regressed out percentage of reads from mitochondrial genes. We then integrated cells from all samples using reciprocal principal components analysis (rPCA) implemented in Seurat 4.0.596 on the top 5000 integration genes and in the top 30 reciprocal principal components space. For clustering, we standardized the integrated data, performed PCA on all integrated genes and ran de-novo Louvain clustering algorithm in the top 15 principal components space with resolution set to 0.1. Cluster markers that are conserved between the strains were called using non-parametric Wilcoxon rank-sum test and subsequently used for annotation. We annotated hippocampals clusters by manually checking conserved markers against the ALLEN BRAIN MAP’s Mouse Whole Cortex and Hippocampus dataset. For cells collected in the amygdala, we performed a second round of analysis restricted to those identified to be neuronal in the initial annotation to increase our annotation resolution. We used the same integration pipeline and parameters described above except when clustering we ran de-novo Louvain clustering algorithm in the top 25 principal components space with resolution set to 0.75, due to the increased expected complexity of cell types in this brain region. All clusters were first grouped into broad types of inhibitory or excitatory neurons except for one cluster that simultaneously expressed Gad2 and Slc17a7. We thus simply annotated it as “Neuronal” followed by its by cluster marker gene names. We also manually compared our clusters to those that were previously reported in amygdala,113 and adapted their annotation for clusters with high concordance in top marker genes.
Single-nucleus ATAC sequencing data integration, clustering and annotation
We first jointly projected all cells’ ATAC peak profile to uncorrected Latent Semantic Indexing (LSI) embeddings with TFIDF transformation followed by calculating SVD on the top 90% most informative peaks. Peak profile embeddings were then integrated in shared low dimensional space via integration anchors identified in the 2nd to 30th reciprocal LSI space as implemented in Signac 1.5.0. We then integrated cell transcriptional activity profiles by performing SCTransform after regressing out percentage of activity from mitochondrial genes and carried out rPCA integration on integration genes identified from the single-nucleus RNA experiment and in the top 10 reciprocal principal components. We transferred the single-nucleus RNA annotation onto the single-nucleus ATAC cells by linking the RNA’s expression profile with ATAC’s transcriptional activity profile through canonical correlation analysis (CCA) described in Seurat. Pairs of cells from each modality that are mutual nearest neighbors in the top 15 canonical components space were identified as “transfer anchors”. “Anchors” were further filtered and weighted by distances in the integrated peak embeddings prior to imputing ATAC cells’ annotation of cell type. For amygdala cells, after the initial assignment to cell type, we repeated the above-mentioned pipeline only on those cells predicted to be neuronal to obtain a set of higher resolution annotation. The second round of analysis was performed on integration genes identified from the subset of single-nucleus RNA data also categorized as neuronal.
Single-cell methylation data integration, clustering and annotation
We used the negative of the average mCH fraction of the gene body ± 2kb as the proxy of methylation cells’ transcriptional activity as described previously.72 We first integrated, across strains, single-nucleus methylation gene-body mCH profiles on RNA integration genes via “integration anchors” identified in the top 30 reciprocal PCA space. Gene body mCH profiles were then linked to single-nucleus RNA expression profiles via “transfer anchors” identified in the top 15 canonical components space. “Transfer anchors” were further filtered and weighted by distances in the integrated mCH embeddings prior to imputing methylation cells’ annotation using their RNA counterpart as reference. For amygdala, we directly linked to the higher resolution annotation used on the neuronal cells in RNA. These served as our final annotation for all the neuronal cells in both hippocampus and amygdala. Since mCH methylation, used to construct the “transfer anchors”, is largely not present in the non-neuronal population,114 we further performed de-novo annotation on methylation cells using both the genome wide mCG and mCH fraction at 100kb as described previously.115 Non-neuronal clusters were annotated by their canonical marker genes (gene-body hypo-methylation). These served as our final annotation for all the non-neuronal cells in both hippocampus and amygdala.
Co-embedding of single-nucleus RNA, single-nucleus ATAC, and single-nucleus methylation sequencing data
Both single-nucleus methylation and single-nucleus ATAC cells’ expression profiles were imputed with previously computed “transfer anchors”. All three modalities were merged on their integrated or imputed expression profiles, projected to low dimensional space via PCA, and visualized by UMAP (performed on the top 15 principal components).
Cell-type specific differential test for single-nucleus ATAC and single-nucleus RNA
We used DESeq273 for cell-type specific pseudobulk level differential expression analysis and differential accessibility analysis. Per cell type, raw counts were aggregated to replicate level and DESeq2 was run under default parameters to detect statistical evidence of strain differences. Any genes with no coverage in either or both strains were excluded.
Testing class and cell type effects on strain differences
We used the brms package in R to test for the effect of class analysis.116 We defined a Bayesian model using brms with a negative binomial distribution for the number of significant differences (for RNA and ATAC data) or the number of mutations for methylation. We used up to eight chains and visually inspected the output to ensure chains were mixed. We used the following code to define the model:
Acknowledgments
This work was funded in part through NIH grants R01MH115979, R01MH125252, U01MH130995, and DP1DA044371, the UCLA Jonsson Comprehensive Cancer Center, the Eli and Edythe Broad Center of Regenerative Medicine, and the Stem Cell Research Ablon Scholars Program. We would like to gratefully acknowledge the UCLA Behavioral Testing Core for assisting with the behavioral experiments, the Cedars-Sinai Applied Genomics, Computation & Translational Core for assisting with the sequencing experiments, and the Genome Editing Core at Augusta University for assisting with the generation of the mouse models necessary to support this research.
Author contributions
R.C. performed the mouse breeding and behavioral experiments. C.L. performed the single-cell methylation analyses, data from which were processed by M.G.H. and Z.C. P.B.C. performed the snRNA and snATAC analyses, with data processing by Z.C. J.F., P.B.C., and D.C.S. analyzed data from the single-cell experiments. J.F. and N.L. analyzed the mouse mapping data. J.M. provided statistical support. J.F. and P.B.C. designed the study. P.B.C., J.F., and E.M. co-wrote the manuscript, which was subsequently reviewed and edited by the rest of the authors.
Declaration of interests
The authors declare no competing interests.
Published: May 1, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2024.100545.
Supplemental information
References
- 1.Abdellaoui A., Yengo L., Verweij K.J.H., Visscher P.M. 15 years of GWAS discovery: Realizing the promise. Am. J. Hum. Genet. 2023;110:179–194. doi: 10.1016/j.ajhg.2022.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flint J., Eskin E. Genome-wide association studies in mice. Nat. Rev. Genet. 2012;13:807–817. doi: 10.1038/nrg3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mackay T.F. Quantitative trait loci in Drosophila. Nat. Rev. Genet. 2001;2:11–20. doi: 10.1038/35047544. [DOI] [PubMed] [Google Scholar]
- 4.Yalcin B., Willis-Owen S.A.G., Fullerton J., Meesaq A., Deacon R.M., Rawlins J.N.P., Copley R.R., Morris A.P., Flint J., Mott R. Genetic dissection of a behavioral quantitative trait locus shows that Rgs2 modulates anxiety in mice. Nat. Genet. 2004;36:1197–1202. doi: 10.1038/ng1450. [DOI] [PubMed] [Google Scholar]
- 5.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang H., Yang H., Shivalila C.S., Dawlaty M.M., Cheng A.W., Zhang F., Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bannerman D.M., Rawlins J.N.P., McHugh S.B., Deacon R.M.J., Yee B.K., Bast T., Zhang W.N., Pothuizen H.H.J., Feldon J. Regional dissociations within the hippocampus--memory and anxiety. Neurosci. Biobehav. Rev. 2004;28:273–283. doi: 10.1016/j.neubiorev.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 8.Tovote P., Fadok J.P., Lüthi A. Neuronal circuits for fear and anxiety. Nat. Rev. Neurosci. 2015;16:317–331. doi: 10.1038/nrn3945. [DOI] [PubMed] [Google Scholar]
- 9.Xu C., Krabbe S., Gründemann J., Botta P., Fadok J.P., Osakada F., Saur D., Grewe B.F., Schnitzer M.J., Callaway E.M., Lüthi A. Distinct Hippocampal Pathways Mediate Dissociable Roles of Context in Memory Retrieval. Cell. 2016;167:961–972.e16. doi: 10.1016/j.cell.2016.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolff S.B.E., Gründemann J., Tovote P., Krabbe S., Jacobson G.A., Müller C., Herry C., Ehrlich I., Friedrich R.W., Letzkus J.J., Lüthi A. Amygdala interneuron subtypes control fear learning through disinhibition. Nature. 2014;509:453–458. doi: 10.1038/nature13258. [DOI] [PubMed] [Google Scholar]
- 11.Felix-Ortiz A.C., Beyeler A., Seo C., Leppla C.A., Wildes C.P., Tye K.M. BLA to vHPC inputs modulate anxiety-related behaviors. Neuron. 2013;79:658–664. doi: 10.1016/j.neuron.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tye K.M., Prakash R., Kim S.Y., Fenno L.E., Grosenick L., Zarabi H., Thompson K.R., Gradinaru V., Ramakrishnan C., Deisseroth K. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature. 2011;471:358–362. doi: 10.1038/nature09820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis M. The role of the amygdala in fear and anxiety. Annu. Rev. Neurosci. 1992;15:353–375. doi: 10.1146/annurev.ne.15.030192.002033. [DOI] [PubMed] [Google Scholar]
- 14.LeDoux J.E. Emotion circuits in the brain. Annu. Rev. Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- 15.Maren S. Neurobiology of Pavlovian fear conditioning. Annu. Rev. Neurosci. 2001;24:897–931. doi: 10.1146/annurev.neuro.24.1.897. [DOI] [PubMed] [Google Scholar]
- 16.Bailey J.S., Grabowski-Boase L., Steffy B.M., Wiltshire T., Churchill G.A., Tarantino L.M. Identification of quantitative trait loci for locomotor activation and anxiety using closely related inbred strains. Genes Brain Behav. 2008;7:761–769. doi: 10.1111/j.1601-183x.2008.00415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolivar V.J., Caldarone B.J., Reilly A.A., Flaherty L. Habituation of activity in an open field: A survey of inbred strains and F1 hybrids. Behav. Genet. 2000;30:285–293. doi: 10.1023/a:1026545316455. [DOI] [PubMed] [Google Scholar]
- 18.Bolivar V.J., Pooler O., Flaherty L. Inbred strain variation in contextual and cued fear conditioning behavior. Mamm. Genome. 2001;12:651–656. doi: 10.1007/s003350020039. [DOI] [PubMed] [Google Scholar]
- 19.Bothe G.W.M., Bolivar V.J., Vedder M.J., Geistfeld J.G. Behavioral differences among fourteen inbred mouse strains commonly used as disease models. Comp. Med. 2005;55:326–334. [PubMed] [Google Scholar]
- 20.Brigman J.L., Mathur P., Lu L., Williams R.W., Holmes A. Genetic relationship between anxiety-related and fear-related behaviors in BXD recombinant inbred mice. Behav. Pharmacol. 2009;20:204–209. doi: 10.1097/FBP.0b013e32830c368c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carhuatanta K.A.K., Shea C.J.A., Herman J.P., Jankord R. Unique genetic loci identified for emotional behavior in control and chronic stress conditions. Front. Behav. Neurosci. 2014;8:341. doi: 10.3389/fnbeh.2014.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cook M.N., Williams R.W., Flaherty L. Anxiety-related behaviors in the elevated zero-maze are affected by genetic factors and retinal degeneration. Behav. Neurosci. 2001;115:468–476. [PubMed] [Google Scholar]
- 23.Delprato A., Bonheur B., Algéo M.P., Rosay P., Lu L., Williams R.W., Crusio W.E. Systems genetic analysis of hippocampal neuroanatomy and spatial learning in mice. Genes Brain Behav. 2015;14:591–606. doi: 10.1111/gbb.12259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geuther B.Q., Deats S.P., Fox K.J., Murray S.A., Braun R.E., White J.K., Chesler E.J., Lutz C.M., Kumar V. Robust mouse tracking in complex environments using neural networks. Commun. Biol. 2019;2:124. doi: 10.1038/s42003-019-0362-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gubner N.R., Wilhelm C.J., Phillips T.J., Mitchell S.H. Strain differences in behavioral inhibition in a Go/No-go task demonstrated using 15 inbred mouse strains. Alcohol Clin. Exp. Res. 2010;34:1353–1362. doi: 10.1111/j.1530-0277.2010.01219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keum S., Park J., Kim A., Park J., Kim K.K., Jeong J., Shin H.S. Variability in empathic fear response among 11 inbred strains of mice. Genes Brain Behav. 2016;15:231–242. doi: 10.1111/gbb.12278. [DOI] [PubMed] [Google Scholar]
- 27.Knoll A.T., Halladay L.R., Holmes A.J., Levitt P. Quantitative Trait Loci and a Novel Genetic Candidate for Fear Learning. J. Neurosci. 2016;36:6258–6268. doi: 10.1523/JNEUROSCI.0177-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koide T., Moriwaki K., Ikeda K., Niki H., Shiroishi T. Multi-phenotype behavioral characterization of inbred strains derived from wild stocks of Mus musculus. Mamm. Genome. 2000;11:664–670. doi: 10.1007/s003350010129. [DOI] [PubMed] [Google Scholar]
- 29.Lad H.V., Liu L., Paya-Cano J.L., Parsons M.J., Kember R., Fernandes C., Schalkwyk L.C. Behavioural battery testing: evaluation and behavioural outcomes in 8 inbred mouse strains. Physiol. Behav. 2010;99:301–316. doi: 10.1016/j.physbeh.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 30.Loos M., van der Sluis S., Bochdanovits Z., van Zutphen I.J., Pattij T., Stiedl O., Neuro-BSIK Mouse Phenomics consortium. Smit A.B., Spijker S. Activity and impulsive action are controlled by different genetic and environmental factors. Genes Brain Behav. 2009;8:817–828. doi: 10.1111/j.1601-183X.2009.00528.x. [DOI] [PubMed] [Google Scholar]
- 31.Miller B.H., Schultz L.E., Gulati A., Su A.I., Pletcher M.T. Phenotypic characterization of a genetically diverse panel of mice for behavioral despair and anxiety. PLoS One. 2010;5:e14458. doi: 10.1371/journal.pone.0014458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milner L.C., Crabbe J.C. Three murine anxiety models: results from multiple inbred strain comparisons. Genes Brain Behav. 2008;7:496–505. doi: 10.1111/j.1601-183X.2007.00385.x. [DOI] [PubMed] [Google Scholar]
- 33.O'Leary T.P., Gunn R.K., Brown R.E. What are we measuring when we test strain differences in anxiety in mice? Behav. Genet. 2013;43:34–50. doi: 10.1007/s10519-012-9572-8. [DOI] [PubMed] [Google Scholar]
- 34.Owen E.H., Christensen S.C., Paylor R., Wehner J.M. Identification of quantitative trait loci involved in contextual and auditory-cued fear conditioning in BXD recombinant inbred strains. Behav. Neurosci. 1997;111:292–300. doi: 10.1037//0735-7044.111.2.292. [DOI] [PubMed] [Google Scholar]
- 35.Park C.C., Gale G.D., de Jong S., Ghazalpour A., Bennett B.J., Farber C.R., Langfelder P., Lin A., Khan A.H., Eskin E., et al. Gene networks associated with conditional fear in mice identified using a systems genetics approach. BMC Syst. Biol. 2011;5:43. doi: 10.1186/1752-0509-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Philip V.M., Duvvuru S., Gomero B., Ansah T.A., Blaha C.D., Cook M.N., Hamre K.M., Lariviere W.R., Matthews D.B., Mittleman G., et al. High-throughput behavioral phenotyping in the expanded panel of BXD recombinant inbred strains. Genes Brain Behav. 2010;9:129–159. doi: 10.1111/j.1601-183X.2009.00540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Philip V.M., Sokoloff G., Ackert-Bicknell C.L., Striz M., Branstetter L., Beckmann M.A., Spence J.S., Jackson B.L., Galloway L.D., Barker P., et al. Genetic analysis in the Collaborative Cross breeding population. Genome Res. 2011;21:1223–1238. doi: 10.1101/gr.113886.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ponder C.A., Munoz M., Gilliam T.C., Palmer A.A. Genetic architecture of fear conditioning in chromosome substitution strains: relationship to measures of innate (unlearned) anxiety-like behavior. Mamm. Genome. 2007;18:221–228. doi: 10.1007/s00335-007-9013-9. [DOI] [PubMed] [Google Scholar]
- 39.Sittig L.J., Carbonetto P., Engel K.A., Krauss K.S., Barrios-Camacho C.M., Palmer A.A. Genetic Background Limits Generalizability of Genotype-Phenotype Relationships. Neuron. 2016;91:1253–1259. doi: 10.1016/j.neuron.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takahashi A., Kato K., Makino J., Shiroishi T., Koide T. Multivariate analysis of temporal descriptions of open-field behavior in wild-derived mouse strains. Behav. Genet. 2006;36:763–774. doi: 10.1007/s10519-005-9038-3. [DOI] [PubMed] [Google Scholar]
- 41.Trullas R., Skolnick P. Differences In Fear Motivated Behaviors Among Inbred Mouse Strains. Psychopharmacology. 1993;111:323–331. doi: 10.1007/BF02244948. [DOI] [PubMed] [Google Scholar]
- 42.Wahlsten D., Metten P., Crabbe J.C. Survey of 21 inbred mouse strains in two laboratories reveals that BTBR T/+ tf/tf has severely reduced hippocampal commissure and absent corpus callosum. Brain Res. 2003;971:47–54. doi: 10.1016/s0006-8993(03)02354-0. [DOI] [PubMed] [Google Scholar]
- 43.Wiltshire T., Ervin R.B., Duan H., Bogue M.A., Zamboni W.C., Cook S., Chung W., Zou F., Tarantino L.M. Initial locomotor sensitivity to cocaine varies widely among inbred mouse strains. Genes Brain Behav. 2015;14:271–280. doi: 10.1111/gbb.12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang R.J., Mozhui K., Karlsson R.M., Cameron H.A., Williams R.W., Holmes A. Variation in mouse basolateral amygdala volume is associated with differences in stress reactivity and fear learning. Neuropsychopharmacology. 2008;33:2595–2604. doi: 10.1038/sj.npp.1301665. [DOI] [PubMed] [Google Scholar]
- 45.de Mooij-van Malsen J.G., Yu K.L., Veldman H., Oppelaar H., van den Berg L.H., Olivier B., Kas M.J.H. Variations in ventral root axon morphology and locomotor behavior components across different inbred strains of mice. Neuroscience. 2009;164:1477–1483. doi: 10.1016/j.neuroscience.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 46.Lilue J., Doran A.G., Fiddes I.T., Abrudan M., Armstrong J., Bennett R., Chow W., Collins J., Collins S., Czechanski A., et al. Sixteen diverse laboratory mouse reference genomes define strain-specific haplotypes and novel functional loci. Nat. Genet. 2018;50:1574–1583. doi: 10.1038/s41588-018-0223-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marchini J., Howie B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 48.Kang H.M., Zaitlen N.A., Eskin E. EMINIM: an adaptive and memory-efficient algorithm for genotype imputation. J. Comput. Biol. 2010;17:547–560. doi: 10.1089/cmb.2009.0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yalcin B., Flint J., Mott R. Using progenitor strain information to identify quantitative trait nucleotides in outbred mice. Genetics. 2005;171:673–681. doi: 10.1534/genetics.104.028902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keane T.M., Goodstadt L., Danecek P., White M.A., Wong K., Yalcin B., Heger A., Agam A., Slater G., Goodson M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477:289–294. doi: 10.1038/nature10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou X., Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012;44:821–824. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou X., Stephens M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat. Methods. 2014;11:407–409. doi: 10.1038/nmeth.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Han B., Eskin E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet. 2011;88:586–598. doi: 10.1016/j.ajhg.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han B., Eskin E. Interpreting meta-analyses of genome-wide association studies. PLoS Genet. 2012;8:e1002555. doi: 10.1371/journal.pgen.1002555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang J., Lee S.H., Goddard M.E., Visscher P.M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bennett B.J., Farber C.R., Orozco L., Kang H.M., Ghazalpour A., Siemers N., Neubauer M., Neuhaus I., Yordanova R., Guan B., et al. A high-resolution association mapping panel for the dissection of complex traits in mice. Genome Res. 2010;20:281–290. doi: 10.1101/gr.099234.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zou J., Gopalakrishnan S., Parker C.C., Nicod J., Mott R., Cai N., Lionikas A., Davies R.W., Palmer A.A., Flint J. Analysis of Independent Cohorts of Outbred CFW Mice Reveals Novel Loci for Behavioral and Physiological Traits and Identifies Factors Determining Reproducibility. G3 (Bethesda) 2022;12:jkab394. doi: 10.1093/g3journal/jkab394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nolan M.F., Malleret G., Dudman J.T., Buhl D.L., Santoro B., Gibbs E., Vronskaya S., Buzsáki G., Siegelbaum S.A., Kandel E.R., Morozov A. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell. 2004;119:719–732. doi: 10.1016/j.cell.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 59.Pruim R.J., Welch R.P., Sanna S., Teslovich T.M., Chines P.S., Gliedt T.P., Boehnke M., Abecasis G.R., Willer C.J. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ng L., Bernard A., Lau C., Overly C.C., Dong H.W., Kuan C., Pathak S., Sunkin S.M., Dang C., Bohland J.W., et al. An anatomic gene expression atlas of the adult mouse brain. Nat. Neurosci. 2009;12:356–362. doi: 10.1038/nn.2281. [DOI] [PubMed] [Google Scholar]
- 61.Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C., Pope B.D., et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515:355–364. doi: 10.1038/nature13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ahrens S., Wu M.V., Furlan A., Hwang G.R., Paik R., Li H., Penzo M.A., Tollkuhn J., Li B. A Central Extended Amygdala Circuit That Modulates Anxiety. J. Neurosci. 2018;38:5567–5583. doi: 10.1523/JNEUROSCI.0705-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Levitan D., Liu C., Yang T., Shima Y., Lin J.Y., Wachutka J., Marrero Y., Ali Marandi Ghoddousi R., Veiga Beltrame E.D., Richter T.A., et al. Deletion of Stk11 and Fos in mouse BLA projection neurons alters intrinsic excitability and impairs formation of long-term aversive memory. Elife. 2020;9:e61036. doi: 10.7554/eLife.61036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McCullough K.M., Daskalakis N.P., Gafford G., Morrison F.G., Ressler K.J. Cell-type-specific interrogation of CeA Drd2 neurons to identify targets for pharmacological modulation of fear extinction. Transl. Psychiatry. 2018;8:164. doi: 10.1038/s41398-018-0190-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lori A., Maddox S.A., Sharma S., Andero R., Ressler K.J., Smith A.K. Dynamic Patterns of Threat-Associated Gene Expression in the Amygdala and Blood. Front. Psychiatry. 2018;9:778. doi: 10.3389/fpsyt.2018.00778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen P.B., Kawaguchi R., Blum C., Achiro J.M., Coppola G., O'Dell T.J., Martin K.C. Mapping Gene Expression in Excitatory Neurons during Hippocampal Late-Phase Long-Term Potentiation. Front. Mol. Neurosci. 2017;10:39. doi: 10.3389/fnmol.2017.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yao Z., van Velthoven C.T.J., Nguyen T.N., Goldy J., Sedeno-Cortes A.E., Baftizadeh F., Bertagnolli D., Casper T., Chiang M., Crichton K., et al. A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell. 2021;184:3222–3241.e26. doi: 10.1016/j.cell.2021.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Catania E.H., Pimenta A., Levitt P. Genetic deletion of Lsamp causes exaggerated behavioral activation in novel environments. Behav. Brain Res. 2008;188:380–390. doi: 10.1016/j.bbr.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Qiu S., Champagne D.L., Peters M., Catania E.H., Weeber E.J., Levitt P., Pimenta A.F. Loss of limbic system-associated membrane protein leads to reduced hippocampal mineralocorticoid receptor expression, impaired synaptic plasticity, and spatial memory deficit. Biol. Psychiatry. 2010;68:197–204. doi: 10.1016/j.biopsych.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Luo C., Rivkin A., Zhou J., Sandoval J.P., Kurihara L., Lucero J., Castanon R., Nery J.R., Pinto-Duarte A., Bui B., et al. Robust single-cell DNA methylome profiling with snmC-seq2. Nat. Commun. 2018;9:3824. doi: 10.1038/s41467-018-06355-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Preissl S., Fang R., Huang H., Zhao Y., Raviram R., Gorkin D.U., Zhang Y., Sos B.C., Afzal V., Dickel D.E., et al. Single-nucleus analysis of accessible chromatin in developing mouse forebrain reveals cell-type-specific transcriptional regulation. Nat. Neurosci. 2018;21:432–439. doi: 10.1038/s41593-018-0079-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu H., Zhou J., Tian W., Luo C., Bartlett A., Aldridge A., Lucero J., Osteen J.K., Nery J.R., Chen H., et al. DNA methylation atlas of the mouse brain at single-cell resolution. Nature. 2021;598:120–128. doi: 10.1038/s41586-020-03182-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guo Y., Zhang P., Sheng Q., Zhao S., Hackett T.A. lncRNA expression in the auditory forebrain during postnatal development. Gene. 2016;593:201–216. doi: 10.1016/j.gene.2016.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pimenta A.F., Zhukareva V., Barbe M.F., Reinoso B.S., Grimley C., Henzel W., Fischer I., Levitt P. The limbic system-associated membrane protein is an Ig superfamily member that mediates selective neuronal growth and axon targeting. Neuron. 1995;15:287–297. doi: 10.1016/0896-6273(95)90034-9. [DOI] [PubMed] [Google Scholar]
- 76.Tomita H., Cornejo F., Aranda-Pino B., Woodard C.L., Rioseco C.C., Neel B.G., Alvarez A.R., Kaplan D.R., Miller F.D., Cancino G.I. The Protein Tyrosine Phosphatase Receptor Delta Regulates Developmental Neurogenesis. Cell Rep. 2020;30:215–228.e5. doi: 10.1016/j.celrep.2019.11.033. [DOI] [PubMed] [Google Scholar]
- 77.Pelkey K.A., Barksdale E., Craig M.T., Yuan X., Sukumaran M., Vargish G.A., Mitchell R.M., Wyeth M.S., Petralia R.S., Chittajallu R., et al. Pentraxins coordinate excitatory synapse maturation and circuit integration of parvalbumin interneurons. Neuron. 2015;85:1257–1272. doi: 10.1016/j.neuron.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Irie F., Okuno M., Pasquale E.B., Yamaguchi Y. EphrinB-EphB signalling regulates clathrin-mediated endocytosis through tyrosine phosphorylation of synaptojanin 1. Nat. Cell Biol. 2005;7:501–509. doi: 10.1038/ncb1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schuske K.R., Richmond J.E., Matthies D.S., Davis W.S., Runz S., Rube D.A., van der Bliek A.M., Jorgensen E.M. Endophilin is required for synaptic vesicle endocytosis by localizing synaptojanin. Neuron. 2003;40:749–762. doi: 10.1016/s0896-6273(03)00667-6. [DOI] [PubMed] [Google Scholar]
- 80.Bregin A., Kaare M., Jagomäe T., Karis K., Singh K., Laugus K., Innos J., Leidmaa E., Heinla I., Visnapuu T., et al. Expression and impact of Lsamp neural adhesion molecule in the serotonergic neurotransmission system. Pharmacol. Biochem. Behav. 2020;198:173017. doi: 10.1016/j.pbb.2020.173017. [DOI] [PubMed] [Google Scholar]
- 81.Mann F., Zhukareva V., Pimenta A., Levitt P., Bolz J. Membrane-associated molecules guide limbic and nonlimbic thalamocortical projections. J. Neurosci. 1998;18:9409–9419. doi: 10.1523/JNEUROSCI.18-22-09409.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chapman G., Shanmugalingam U., Smith P.D. The Role of Neuronal Pentraxin 2 (NP2) in Regulating Glutamatergic Signaling and Neuropathology. Front. Cell. Neurosci. 2019;13:575. doi: 10.3389/fncel.2019.00575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chang S., Bok P., Tsai C.Y., Sun C.P., Liu H., Deussing J.M., Huang G.J. NPTX2 is a key component in the regulation of anxiety. Neuropsychopharmacology. 2018;43:1943–1953. doi: 10.1038/s41386-018-0091-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Reutens A.T., Begley C.G. Endophilin-1: a multifunctional protein. Int. J. Biochem. Cell Biol. 2002;34:1173–1177. doi: 10.1016/s1357-2725(02)00063-8. [DOI] [PubMed] [Google Scholar]
- 85.Pechstein A., Gerth F., Milosevic I., Jäpel M., Eichhorn-Grünig M., Vorontsova O., Bacetic J., Maritzen T., Shupliakov O., Freund C., Haucke V. Vesicle uncoating regulated by SH3-SH3 domain-mediated complex formation between endophilin and intersectin at synapses. EMBO Rep. 2015;16:232–239. doi: 10.15252/embr.201439260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gowrisankaran S., Houy S., Del Castillo J.G.P., Steubler V., Gelker M., Kroll J., Pinheiro P.S., Schwitters D., Halbsgut N., Pechstein A., et al. Endophilin-A coordinates priming and fusion of neurosecretory vesicles via intersectin. Nat. Commun. 2020;11:1266. doi: 10.1038/s41467-020-14993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Flint J., Valdar W., Shifman S., Mott R. Strategies for mapping and cloning quantitative trait genes in rodents. Nat. Rev. Genet. 2005;6:271–286. doi: 10.1038/nrg1576. [DOI] [PubMed] [Google Scholar]
- 88.Makino K., Ishikawa A. Genetic identification of Ly75 as a novel quantitative trait gene for resistance to obesity in mice. Sci. Rep. 2018;8:17658. doi: 10.1038/s41598-018-36073-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stringer C., Pachitariu M., Steinmetz N., Carandini M., Harris K.D. High-dimensional geometry of population responses in visual cortex. Nature. 2019;571:361–365. doi: 10.1038/s41586-019-1346-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.DiCarlo J.J., Zoccolan D., Rust N.C. How does the brain solve visual object recognition? Neuron. 2012;73:415–434. doi: 10.1016/j.neuron.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bugeon S., Duffield J., Dipoppa M., Ritoux A., Prankerd I., Nicoloutsopoulos D., Orme D., Shinn M., Peng H., Forrest H., et al. A transcriptomic axis predicts state modulation of cortical interneurons. Nature. 2022;607:330–338. doi: 10.1038/s41586-022-04915-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lovett-Barron M., Andalman A.S., Allen W.E., Vesuna S., Kauvar I., Burns V.M., Deisseroth K. Ancestral Circuits for the Coordinated Modulation of Brain State. Cell. 2017;171:1411–1423.e17. doi: 10.1016/j.cell.2017.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schneider-Mizell C.M., Bodor A.L., Collman F., Brittain D., Bleckert A., Dorkenwald S., Turner N.L., Macrina T., Lee K., Lu R., et al. Structure and function of axo-axonic inhibition. Elife. 2021;10:e73783. doi: 10.7554/eLife.73783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Flint J., Heffel M.G., Chen Z., Mefford J., Marcus E., Chen P.B., Ernst J., Luo C. Single-cell methylation analysis of brain tissue prioritizes mutations that alter transcription. Cell Genom. 2023;3:100454. doi: 10.1016/j.xgen.2023.100454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Krueger F., Andrews S.R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stuart T., Srivastava A., Madad S., Lareau C.A., Satija R. Single-cell chromatin state analysis with Signac. Nat. Methods. 2021;18:1333–1341. doi: 10.1038/s41592-021-01282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.McGinnis C.S., Murrow L.M., Gartner Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019;8:329–337.e4. doi: 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Valdar W., Solberg L.C., Gauguier D., Cookson W.O., Rawlins J.N.P., Mott R., Flint J. Genetic and environmental effects on complex traits in mice. Genetics. 2006;174:959–984. doi: 10.1534/genetics.106.060004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Srivastava A., Morgan A.P., Najarian M.L., Sarsani V.K., Sigmon J.S., Shorter J.R., Kashfeen A., McMullan R.C., Williams L.H., Giusti-Rodríguez P., et al. Genomes of the Mouse Collaborative Cross. Genetics. 2017;206:537–556. doi: 10.1534/genetics.116.198838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shifman S., Bell J.T., Copley R.R., Taylor M.S., Williams R.W., Mott R., Flint J. A High-Resolution Single Nucleotide Polymorphism Genetic Map of the Mouse Genome. PLoS Biol. 2006;4:e395. doi: 10.1371/journal.pbio.0040395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Morgan A.P., Fu C.P., Kao C.Y., Welsh C.E., Didion J.P., Yadgary L., Hyacinth L., Ferris M.T., Bell T.A., Miller D.R., et al. The Mouse Universal Genotyping Array: From Substrains to Subspecies. G3 (Bethesda) 2015;6:263–279. doi: 10.1534/g3.115.022087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kirby A., Kang H.M., Wade C.M., Cotsapas C., Kostem E., Han B., Furlotte N., Kang E.Y., Rivas M., Bogue M.A., et al. Fine mapping in 94 inbred mouse strains using a high-density haplotype resource. Genetics. 2010;185:1081–1095. doi: 10.1534/genetics.110.115014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Howie B.N., Donnelly P., Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ashbrook D.G., Sasani T., Maksimov M., Gunturkun M.H., Ma N., Villani F., Ren Y., Rothschild D., Chen H., Lu L., et al. Private and sub-family specific mutations of founder haplotypes in the BXD family reveal phenotypic consequences relevant to health and disease. bioRxiv. 2022 doi: 10.1101/2022.04.21.489063. Preprint at. [DOI] [Google Scholar]
- 111.Doench J.G., Hartenian E., Graham D.B., Tothova Z., Hegde M., Smith I., Sullender M., Ebert B.L., Xavier R.J., Root D.E. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 2014;32:1262–1267. doi: 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.R Development Core Team . R Foundation for Statistical Computing; 2010. R: A Language and Environment for Statistical Computing. [Google Scholar]
- 113.Yu B., Zhang Q., Lin L., Zhou X., Ma W., Wen S., Li C., Wang W., Wu Q., Wang X., Li X.M. Molecular and cellular evolution of the amygdala across species analyzed by single-nucleus transcriptome profiling. Cell Discov. 2023;9:19. doi: 10.1038/s41421-022-00506-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jeong H., Mendizabal I., Berto S., Chatterjee P., Layman T., Usui N., Toriumi K., Douglas C., Singh D., Huh I., et al. Evolution of DNA methylation in the human brain. Nat. Commun. 2021;12:2021. doi: 10.1038/s41467-021-21917-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu H., Zeng Q., Zhou J., Bartlett A., Wang B.A., Berube P., Tian W., Kenworthy M., Altshul J., Nery J.R., et al. Single-cell DNA Methylome and 3D Multi-omic Atlas of the Adult Mouse Brain. bioRxiv. 2023 doi: 10.1038/s41586-023-06805-y. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bürkner P.C. brms: An R Package for Bayesian Multilevel Models Using Stan. J. Stat. Software. 2017;80:1–28. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Raw and processed sequencing data generated for this study were deposited to NCBI GEO/SRA with accession number GSE245367 and GSE262259 and are publicly available at the time of publication.
-
•
Mapping data (including imputed genotypes of all mouse inbred strains and F1s, together with results from the meta-analysis of the three phenotypes) https://figshare.com/s/7fcb1eebfd9c607afd12.
-
•
Detailed CRISPR KO mouse generation for each gene, including exon targeted, sgRNA sequences, genotyping primers, and DNA gel images https://doi.org/10.6084/m9.figshare.25521283.
-
•
Knockout interaction data, including phenotypes and genotypes for all quantitative complementation tests https://figshare.com/s/cc86ed5777cb95b99cec.
-
•
Processed single nucleus epigenetic and gene expression data https://figshare.com/s/39e1c671ec0dc09d5b27.
-
•
All original code has been deposited at https://doi.org/10.5281/zenodo.10892164.