

Abstract
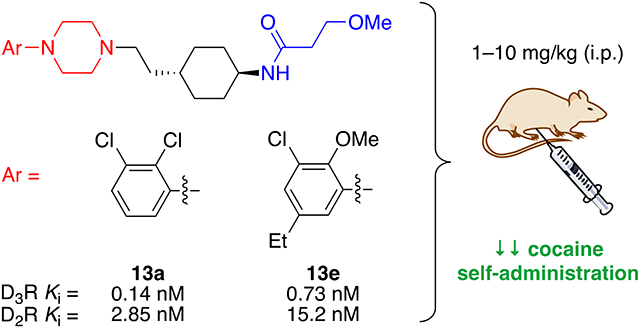
Highly selective dopamine D3 receptor (D3R) partial agonists/antagonists have been developed for the treatment of psychostimulant use disorders (PSUD). However, none have reached the clinic due to insufficient potency/efficacy or potential cardiotoxicity. Cariprazine, an FDA approved drug for the treatment of schizophrenia and bipolar disorder, is a high affinity D3R partial agonist (Ki = 0.22 nM) with 3.6-fold selectivity over the homologous dopamine D2 receptor (D2R). We hypothesized that compounds that are moderately D3R/D2R selective partial agonists/antagonists, may be effective for treatment of PSUD. By systematically modifying the parent molecule, we discovered partial agonists/antagonists, as measured in BRET-based assays, with high D3R affinities (Ki = 0.14–50 nM) and moderate selectivity (<100-fold) over D2R. Cariprazine and two lead analogues, 13a and 13e, decreased cocaine self-administration (FR2; 1–10 mg/kg, i.p.) in rats, suggesting that partial agonists/antagonists with modest D3R/D2R-selectivity may be effective in treating PSUD and potentially comorbidities with other affective disorders.
Keywords: Dopamine D3 receptors, Dopamine D2 receptors, bitopic ligands, substance use disorders, psychostimulant use disorder, cocaine, affective disorders, schizophrenia, bipolar disorder, cariprazine, self-administration, reward
Graphical Abstarct
INTRODUCTION
In the midst of the COVID-19 pandemic, drug overdose fatalities soared to >100,000 in 2021.1 While opioids are involved with a majority of these deaths, polysubstance use is another contributing factor, leading to increases in morbidity that poses further challenges for prevention and treatment.2 To further complicate matters, substance use disorders (SUDs) frequently occur with affective disorders, including anxiety, major depressive disorder, and bipolar disorder. Such dual disorders have also increased during the COVID-19 pandemic3–5 and are particularly difficult to treat when comorbid with psychostimulant use disorder (PSUD),6, 7 for which there is currently no FDA-approved pharmacotherapeutic.
Development of highly selective dopamine D3 receptor (D3R) partial agonists/antagonists for the treatment of SUD has been of great interest due to their ability to attenuate drug reinforcement as well as inhibit cue- and stress-induced reinstatement for psychostimulants and opioids in animal models.8–11 D3R-selective antagonists, specifically, have been shown to block the expression of cocaine- or heroin-induced conditioned place preference and inhibit the rewarding effects of these drugs.12–14 In addition to promising preclinical work, gaining D3R selectivity over the dopamine D2 receptor (D2R) may avoid the extrapyramidal side effects, weight gain, metabolic disorders, and motor coordination deficits associated with D2R antagonism.15–20 To this end, highly selective compounds (Figure 1), such as SB277011A (>100-fold),21 PG01037 (>100-fold),22 (±)-VK4–116 (1,700-fold),23, 24 (±)-ABS-1–113 (>300-fold),25 and BP1.4979 (~200-fold),26 have been discovered in spite of the ~80% homology between the D2R and D3R subtypes.27–29
Figure 1.
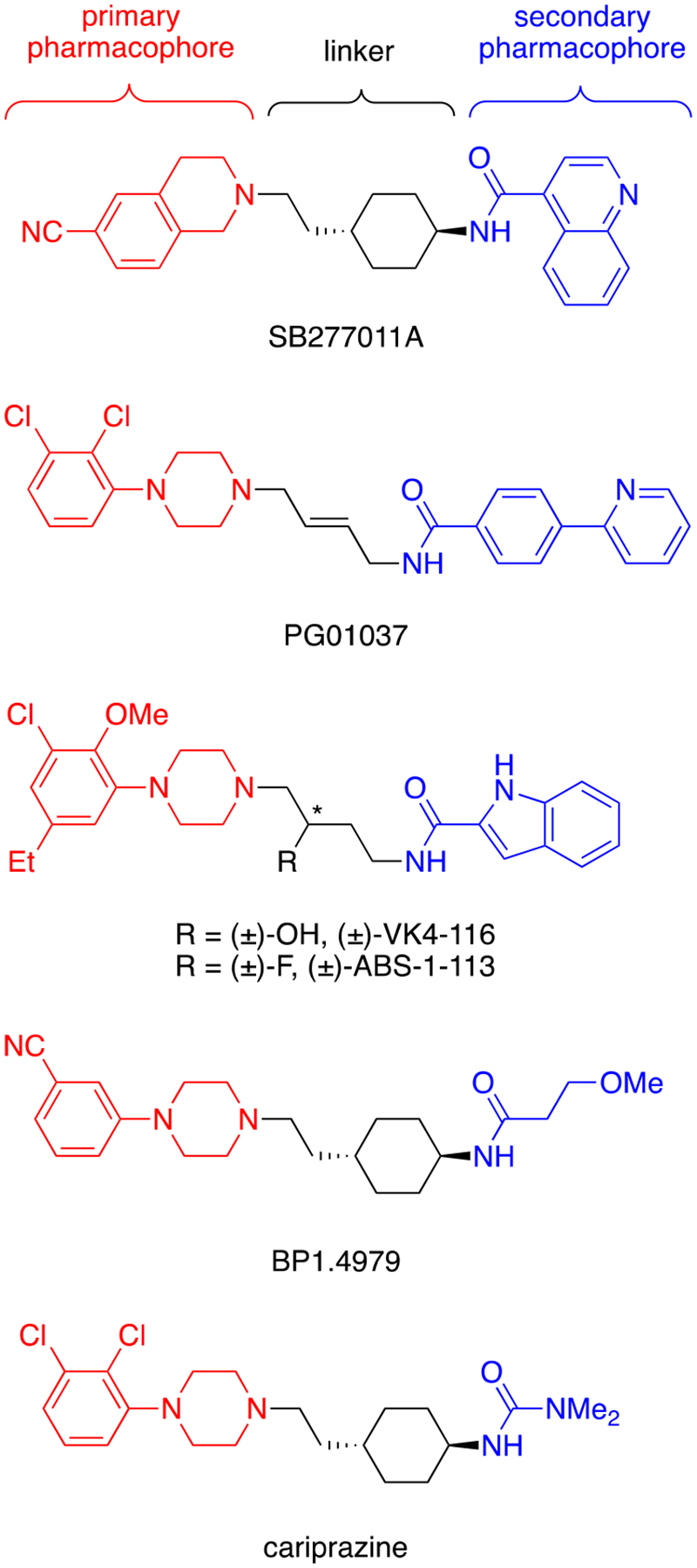
Structures of representative D3R-selective partial agonists/antagonists.
While highly D3R-preferential antagonists and low-efficacy partial agonists demonstrate potential as treatments for SUD,29 there are two challenges that still need to be addressed. The first is that these compounds generally show favorable results in animal models of opioid use disorder but are often not as effective in mitigating cocaine or methamphetamine self-administration, especially at low fixed-ratio schedules (e.g., FR1 or FR2) of reinforcement.18, 29 Secondly, relatively high doses (e.g., 10–56 mg/kg, i.p.) are required to observe reductions in drug-seeking behaviors despite having very high affinities (Ki values in the low nM to pM range) for the desired target, D3R. These challenges have led us to reconsider whether some D2R agonist activity would improve both the efficacy and potency of our compounds. Bearing in mind that high affinity D2R antagonists would likely cause negative side effects, including neuroleptic dysphoria30 (especially in a patient population with SUD), we sought to design molecules with D3R and D2R agonist activities and binding selectivities in the 10- to 100-fold range. Ultimately, the goal of this study was to identify a pharmacological profile that would not only have promise for treating patients with PSUD but might also be efficacious in patients with dual diagnoses.
Of note, there is one D3R-preferential agonist that is in clinical use for the treatment of schizophrenia and more recently, bipolar disorder. Cariprazine (see Figure 1), formerly described in the literature as RGH188, is a high affinity D2R/D3R agonist with preference for the D3R (3- to 10-fold).31–34 Most D2-like antipsychotics on the market generally reduce positive symptoms (e.g., delusions, disorganized speech, and hallucinations) of schizophrenia and manic episodes of bipolar disorder but are not effective at ameliorating the spectrum of other debilitating symptoms.34 However, cariprazine appears to be unique in that it also improves the negative (e.g., anhedonia, social and emotional withdrawal) and cognitive symptoms (e.g., attention deficit and executive function impairment) while significantly decreasing global illness severity of schizophrenia.34 Cariprazine can effectively treat the manic and mixed episodes without induction of depression in bipolar 1 disorder (BD-I).35, 36 Most patients with BD-I take atypical antipsychotics in combination with an antidepressant to treat both poles of the disorder.36 It is believed that these therapeutic benefits are associated with cariprazine’s profile as a relatively modest D3R preferential agonist.34, 37, 38
Herein, we report the design of cariprazine-based analogues resulting in a wide range of D3R/D2R selectivities while maintaining high (low nM to pM) binding affinities at D3R. These ligands are considered bitopic, meaning they include a primary pharmacophore (PP), which binds to the orthosteric binding site, that is then linked to a secondary pharmacophore (SP), which binds to D3R-unique secondary binding pocket (see Figure 1).39 By changing the PP, SP, and linker portions of various bitopic ligands, we have previously altered their D3R/D2R selectivities and functional efficacies.17, 40–42 Recent work by Keserű and coworkers, in particular, suggests that adjustments to the aliphatic SP of cariprazine have the potential to “tune” efficacy by repositioning the molecule in the orthosteric binding site through its interactions with the secondary binding pocket.43, 44 Further investigation of this hypothesis with additional aliphatic SPs will be of interest to support these findings. Hence, using the bitopic ligand design, we explored different modifications to the 2,3-dichlorophenylpiperazine (PP), trans-1,4-cyclohexane ring system (linker), and N,N-dimethylurea (SP) of cariprazine. Analogues were tested for D2R and D3R binding affinities in transfected HEK293 cells, and a subset was selected for functional bioluminescence resonance energy transfer (BRET) assays to assess their intrinsic efficacy. Based on the results of these experiments and additional off-target studies, two analogues (13a and 13e) along with cariprazine were evaluated for effectiveness in reducing cocaine self-administration in rats.
RESULTS AND DISCUSSION
Chemistry
We modified the PP and SP of cariprazine following the synthetic routes outlined in Scheme 1. Initially, 2-(trans-4-((tert-butoxycarbonyl)amino)cyclohexyl)acetic acid (1) was reduced with borane-dimethyl sulfide (BMS) complex, and the resulting alcohol (2) was converted to aldehyde 3 via Swern oxidation. In the next step, primary pharmacophores were installed by reductive amination using various aryl piperazines (4a-e).42 Deprotection of these intermediates with trifluoroacetic acid (TFA) gave amines 5a-e, which were then functionalized with different SPs. To that end, cariprazine and substituted ureas 6b-7e were prepared from either N,N-dimethyl- or N-methylcarbamyl chloride. Other urea-containing analogues were accessed by treating a selection of the aforementioned amines (5a, 5b, and 5e) with potassium cyanate under acidic conditions (8a and 8e) or phosgene followed by 5,6,7,8-tetrahydro-1,6-naphthyridine (9a and 9b).45 Lastly, amides 10a-13e were synthesized from a variety of carboxylic acids with either 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide•HCl (EDCI) or O-(1H-6-chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU) serving as the coupling reagent.
Scheme 1.
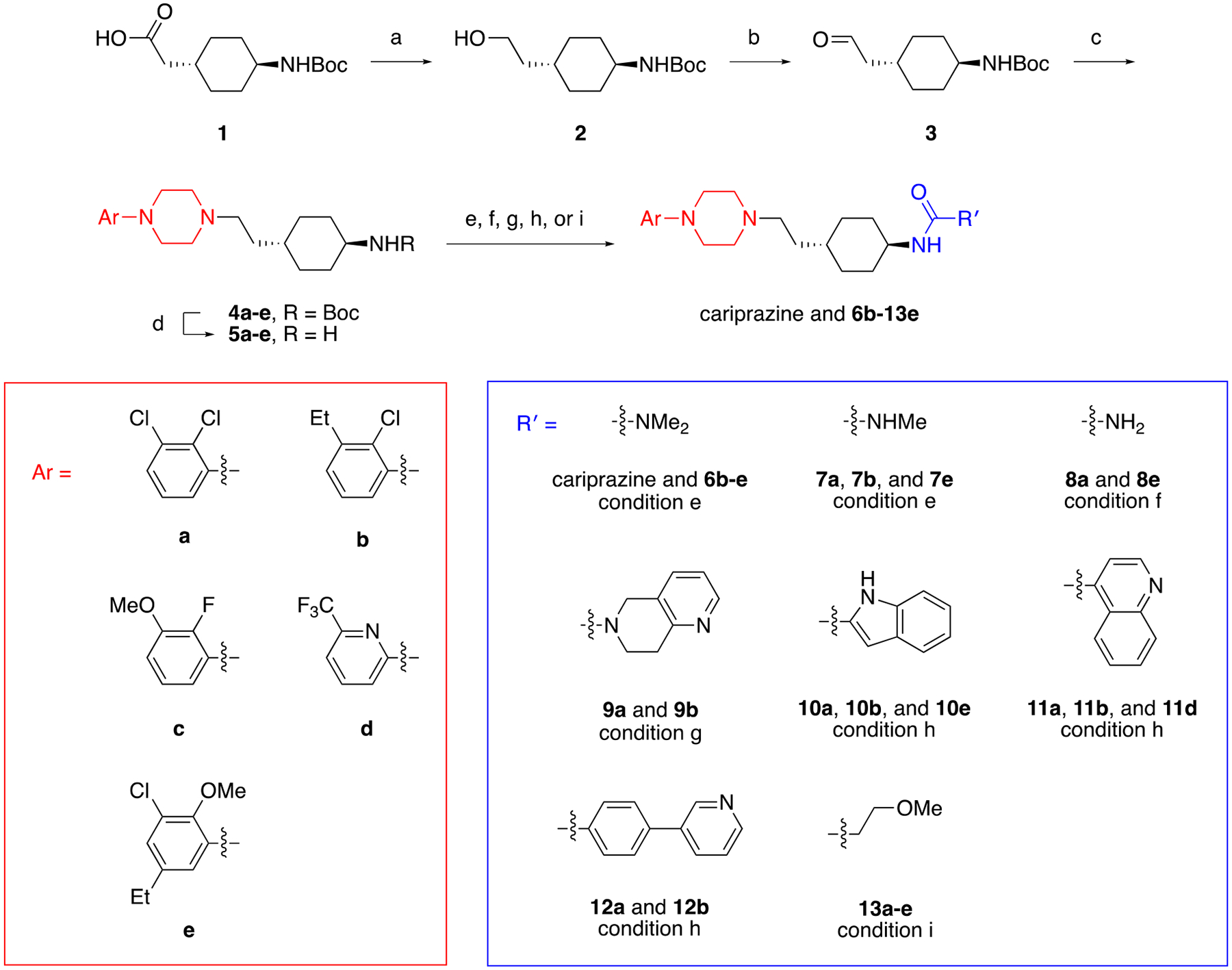
Synthesis of cariprazine and its derivativesa
a Reagents and Conditions: (a) BMS, THF, 0 °C to rt; (b) DMSO, (COCl)2, NEt3, DCM, −78 °C to rt; (c) aryl piperazine, NaBH(OAc)3, DCE, rt; (d) TFA, DCM, rt; (e) N,N-dimethyl- or N-methylcarbamyl chloride, DIPEA, DCM, rt; (f) KOCN, HCl, H2O, THF, rt; (g) COCl2, NEt3, toluene, THF, 0 °C, then 5,6,7,8-tetrahydro-1,6-naphthyridine, THF, 0 °C to rt; (h) carboxylic acid, EDCI, HOBt, DIPEA, CHCl3, rt; (i) 3-methoxyproponic acid, HCTU, DIPEA, DCM, rt.
Manipulations to the linker between the primary and secondary pharmacophores of cariprazine were additionally explored. As shown in Scheme 2, the cyclohexylamine was replaced with a piperidine ring using one of two aldehydes. The first (17a) retained the two-carbon chain found in cariprazine and was prepared from tert-butyl 4-oxopiperidine-1-carboxylate (14). Briefly, a Horner–Wadsworth–Emmons reaction furnished α,β-unsaturated ester 15, which was sequentially reduced by hydrogenation over palladium on carbon (16) and DIBALH to afford 17a. The second aldehyde (17b), possessing an extended carbon chain, was obtained using a Swern oxidation on commercially available tert-butyl 4-(3-hydroxypropyl)piperidine-1-carboxylate (18). With these materials in hand, we followed the same steps described in Scheme 1 to prepare analogues 21a-22b (i.e., reductive amination, Boc-deprotection of 19a and 19b, followed by functionalization of amines 20a and 20b).
Scheme 2.
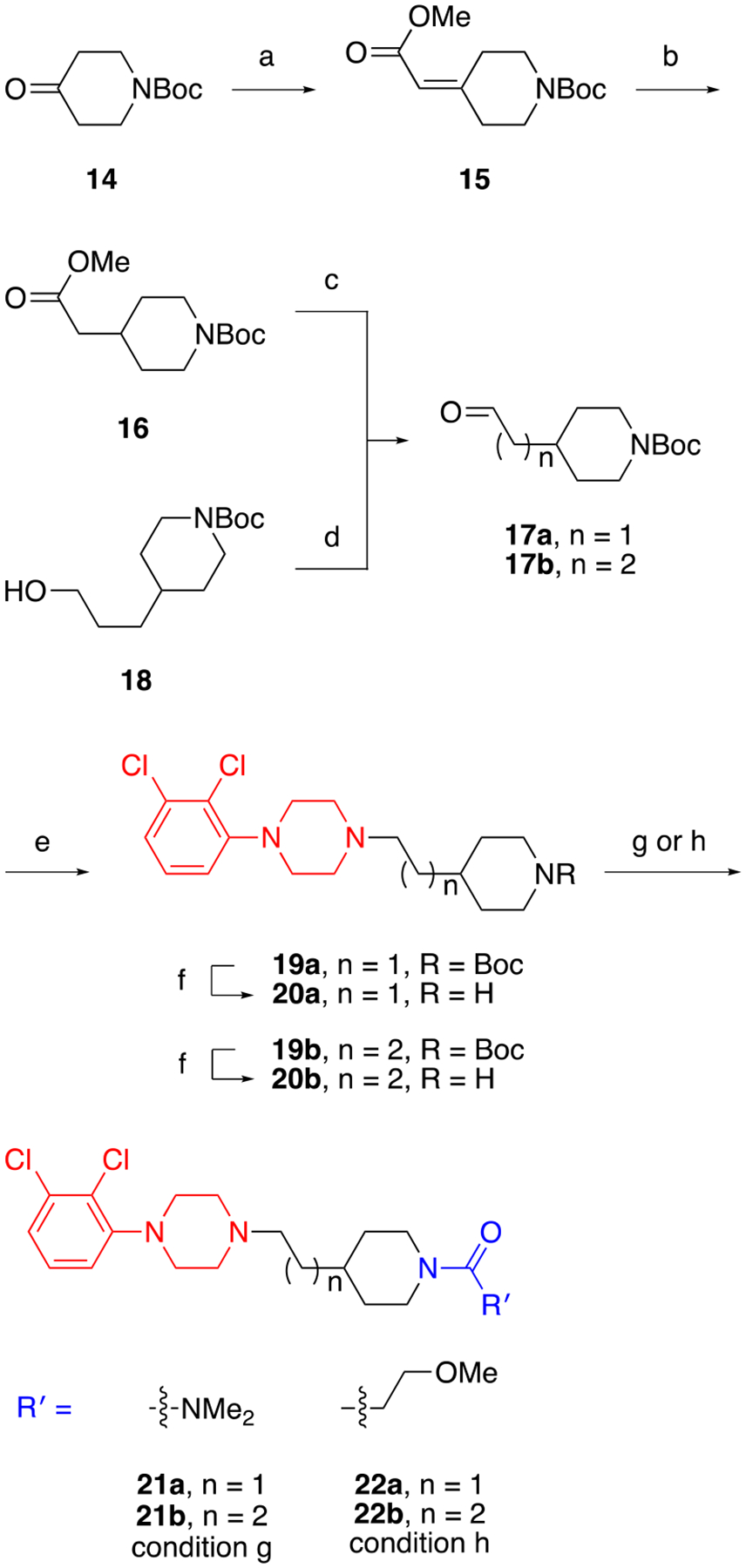
Routes to linker modified cariprazine analoguesa
a Reagents and Conditions: (a) NaH, methyl 2-(dimethoxyphosphoryl)acetate, THF, 0 °C to rt; (b) Pd/C, H2 (50 psi), MeOH, rt; (c) DIBALH, toluene, DCM, −78 °C to rt; (d) DMSO, (COCl)2, NEt3, DCM, −78 °C to rt; (e) 1-(2,3-dichlorophenyl)piperazine•HCl, NaBH(OAc)3, DCE, rt; (f) TFA, DCM, rt; (g) N,N-dimethylcarbamyl chloride, DIPEA, DCM, rt; (h) 3-methoxyproponic acid, HCTU, DIPEA, DCM, rt.
Expanding on the previous set of compounds, we also introduced a hydroxyl group onto the linker, while simultaneously adjusting the ring size (Scheme 3). In this case, cariprazine analogues were constructed from building blocks 26a and 26b, which were accessed using a parallel sequence of transformations. Specifically, a three-carbon chain was introduced onto tert-butyl 3-oxopyrrolidine-1-carboxylate (23a) and tert-butyl 4-oxopiperidine-1-carboxylate (23b) through separate Grignard reactions with allyl magnesium bromide. The terminal alkenes of 24a and 24b were then subjected to hydroboration–oxidation reactions, and the corresponding alcohols (25a and 25b) were converted to hemiacetals 26a and 26b. Like before (see Schemes 1 and 2), the synthesis of analogues 29a-30b proceeded through intermediates 27a-28b.
Scheme 3.
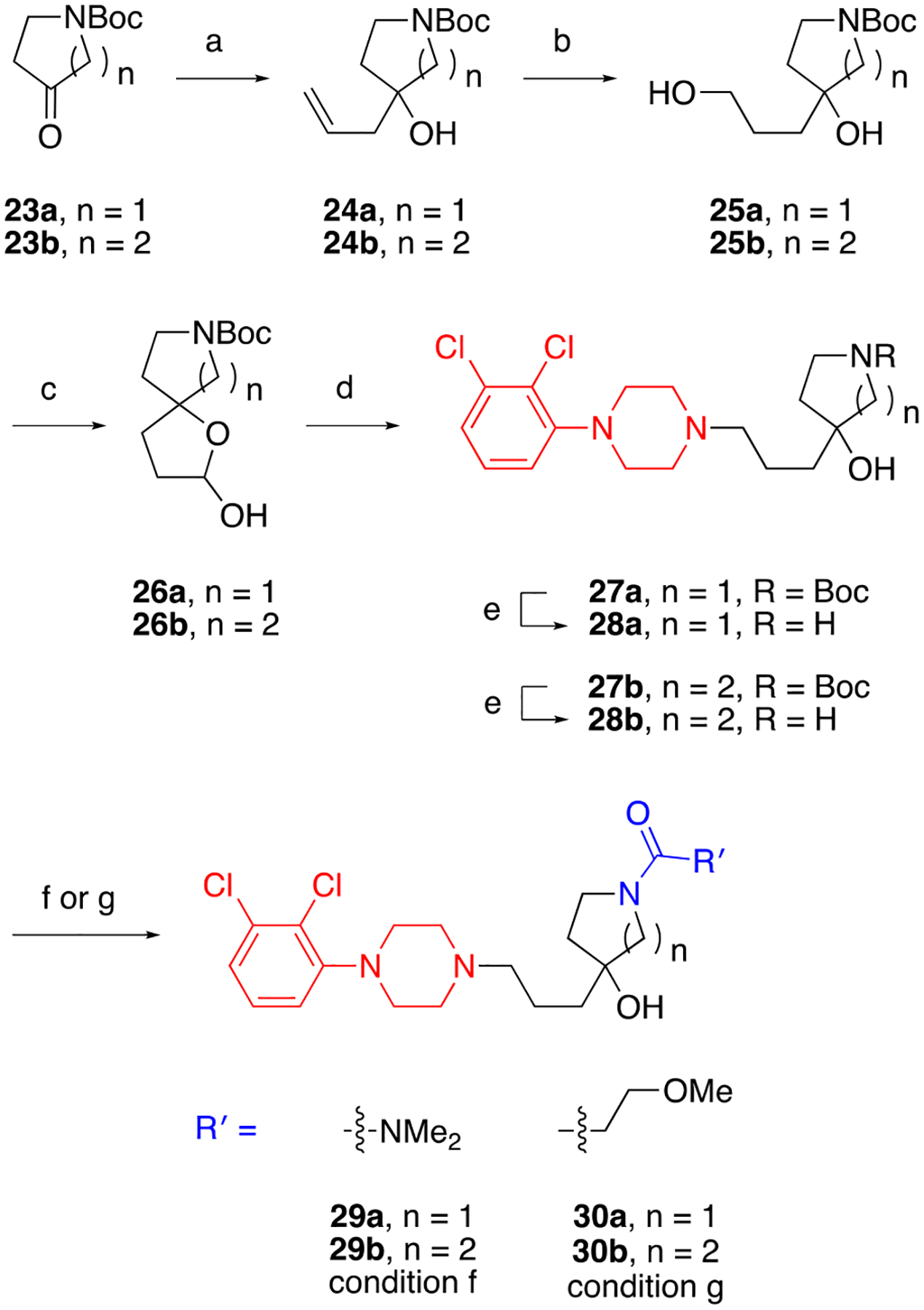
Preparation of compounds bearing a hydroxyl group within the linkera
a Reagents and Conditions: (a) allyl magnesium bromide, Et2O, 0 °C to rt; (b) BH3•THF, THF, 0 °C to rt, then NaOH, H2O2, H2O, 0 °C to rt; (c) DMSO, (COCl)2, NEt3, DCM, −78 °C to rt; (d) 1-(2,3-dichlorophenyl)piperazine•HCl, NaBH(OAc)3, DCE, rt; (e) TFA, DCM, rt; (f) N,N-dimethylcarbamyl chloride, DIPEA, DCM, rt; (g) 3-methoxyproponic acid, HCTU, DIPEA, DCM, rt.
Binding affinities of cariprazine and its analogues at D2R and D3R
To investigate the structure-activity relationships (SAR) of our cariprazine analogues, we determined their affinities at D2R and D3R. Competitive binding experiments using membrane preparations from stably transfected HEK293 cells expressing human D2L and D3 receptors were performed with [3H]N-methylspiperone serving as the radioligand.46 The Ki value for each compound is presented in Tables 1 and 2 along with the corresponding multiparameter optimization (MPO) scores, which are a prediction of brain penetrability.47, 48 For our purposes, derivatives with MPO scores >3 were considered as potentially drug-like.
Table 1.
D3R and D2R binding affinities of cariprazine and its analogues.a
| ||||||
|---|---|---|---|---|---|---|
| compound | Ar | R | MPO | Ki ± SEM (nM) | D2R Ki / D3R Ki | |
| D2R | D3R | |||||
| 4a |
|
|
2.8 | 8.2 ± 2.6 | 2.13 ± 0.85 | 3.8 |
| cariprazine |
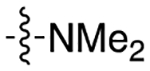
|
3.6 | 0.78 ± 0.17 | 0.22 ± 0.06 | 3.6 | |
| 7a |

|
3.5 | 1.21 ± 0.06 | 0.21 ± 0.02 | 5.7 | |
| 8a |
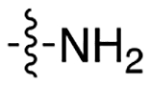
|
3.8 | 1.49 ± 0.03 | 0.26 ± 0.10 | 5.7 | |
| 9a |
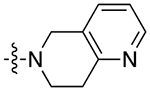
|
2.8 | 1.86 ± 0.06 | 0.36 ± 0.05 | 5.1 | |
| 10a |
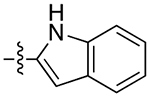
|
2.5 | 6.5 ± 1.9 | 0.31 ± 0.10 | 21 | |
| 11a |
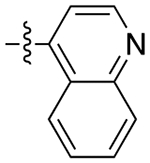
|
2.8 | 29 ± 13 | 0.66 ± 0.28 | 44 | |
| 12a |
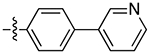
|
2.8 | 3.75 ± 0.65 | 0.59 ± 0.13 | 6.4 | |
| 13a |
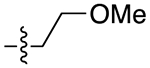
|
3.6 | 2.85 ± 0.63 | 0.14 ± 0.05 | 20 | |
| 6b |
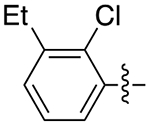
|

|
3.5 | 0.75 ± 0.07 | 0.26 ± 0.03 | 2.9 |
| 7b |

|
3.4 | 0.48 ± 0.11 | 0.18 ± 0.01 | 2.6 | |
| 9b |
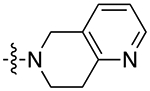
|
2.8 | 0.87 ± 0.08 | 0.29 ± 0.08 | 3.0 | |
| 10b |
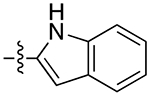
|
2.5 | 7.8 ± 1.9 | 1.31 ± 0.46 | 6.0 | |
| 11b |
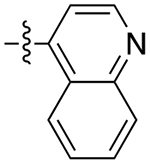
|
2.8 | 5.76 ± 0.93 | 0.34 ± 0.06 | 17 | |
| 12b |
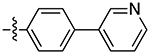
|
2.8 | 1.80 ± 0.34 | 0.58 ± 0.07 | 3.1 | |
| 13b |
|
3.5 | 1.78 ± 0.18 | 0.25 ± 0.04 | 7.1 | |
| 6c |
|

|
5.2 | 134 ± 17 | 9.4 ± 1.2 | 14 |
| 13c |
|
5.1 | 479 ± 26 | 1.31 ± 0.02 | 370 | |
| 6d |
|

|
4.7 | 26.7 ± 5.4 | 2.38 ± 0.54 | 11 |
| 11d |
|
2.6 | 162 ± 48 | 1.00 ± 0.08 | 160 | |
| 13d |
|
4.6 | 127 ± 14 | 1.06 ± 0.08 | 120 | |
| 4e |
|
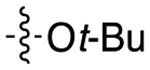
|
3.1 | 68.1 ± 4.3 | 38.8 ± 3.0 | 1.8 |
| 6e |

|
3.4 | 4.12 ± 0.17 | 1.76 ± 0.11 | 2.3 | |
| 7e |

|
3.3 | 5.8 ± 1.3 | 0.64 ± 0.10 | 9.1 | |
| 8e |
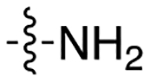
|
3.5 | 5.3 ± 1.8 | 0.78 ± 0.28 | 6.8 | |
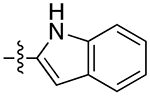
| 10e |
|
2.3 | 114 ± 36 | 1.03 ± 0.26 | 110 | |
| 13e |
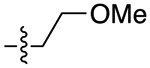
|
3.3 | 15.2 ± 2.0 | 0.73 ± 0.16 | 21 | |
Ki values are derived from IC50 values using the Cheng-Prusoff equation,50 and calculated as the mean of at least three independent experiments. The radioligand used in these assays was [3H]-N-methylspiperone.
Table 2.
D3R and D2R binding affinities of linker modified compounds.a
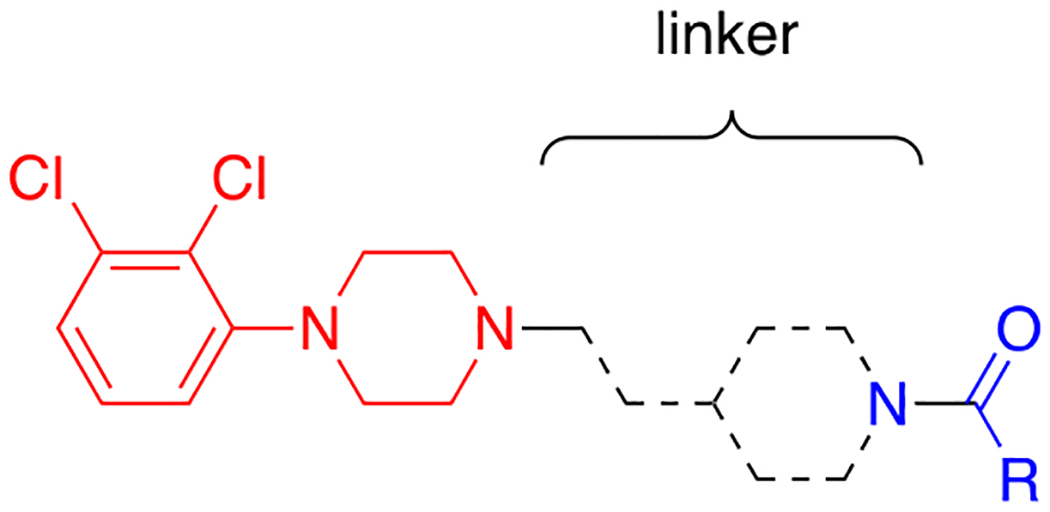
| ||||||
|---|---|---|---|---|---|---|
| compound | linker | R | MPO | Ki ± SEM (nM) | D2R Ki / D3R Ki | |
| D2R | D3R | |||||
| 21a |
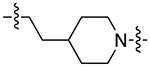
|

|
3.9 | 20.5 ± 2.1 | 1.02 ± 0.10 | 20 |
| 22a |
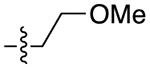
|
4.4 | 86 ± 28 | 3.5 ± 1.3 | 25 | |
| 21b |
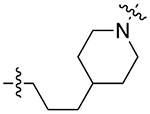
|

|
3.4 | 7.2 ± 1.1 | 1.86 ± 0.10 | 3.9 |
| 22b |
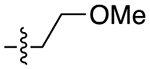
|
3.7 | 19.4 ± 2.6 | 0.96 ± 0.05 | 20 | |
| 29a |
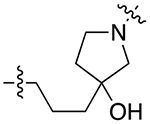
|

|
5.3 | 89.8 ± 5.9 | 29.5 ± 3.7 | 3.0 |
| 30a |
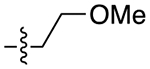
|
5.1 | 175 ± 19 | 18.7 ± 2.5 | 9.4 | |
| 29b |
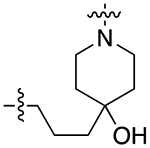
|

|
5.0 | 38.4 ± 5.2 | 50.7 ± 5.7 | 0.76 |
| 30b |
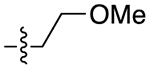
|
5.1 | 125 ± 27 | 27.5 ± 3.3 | 4.5 | |
Ki values are derived from IC50 values using the Cheng-Prusoff equation,50 and calculated as the mean of at least three independent experiments. The radioligand used in these assays was [3H]-N-methylspiperone.
In the first series of compounds, we focused on modifying the SP (blue) of cariprazine (Table 1, 4a, cariprazine, and 7a-13a). Compared to the parent compound, similar D3R binding affinities and selectivities were observed for the primary metabolites, 7a and 8a.49 When the N,N-dimethylamino group of the SP was replaced with different heterocycles (9a-12a), D3R affinity was retained (Ki = 0.31–0.66 nM), while selectivity over D2R increased from 5- to 44-fold. Importantly, the analogue inspired by BP1.4979 (13a) exhibited similar affinity (Ki = 0.14 nM) but better selectivity (20-fold) for D3R than cariprazine, while simultaneously maintaining a desirable MPO score (3.6).
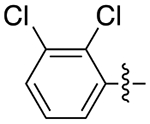
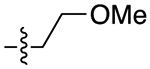
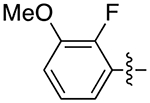
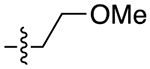
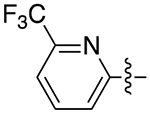
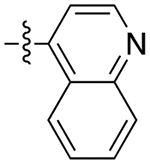
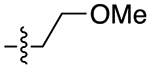
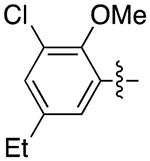
The next several series of compounds explored different PPs (red) in varying combinations with the aforementioned SPs. As shown in Table 1, the 2,3-dichlorophenyl was substituted with a 2-chloro-3-ethylphenyl (6b, 7b, and 9b-13b) derived from our previously reported D3R agonists.23, 25 Although high D3R affinities were achieved (Ki = 0.18–1.31 nM), none of these analogues were substantially more selective than cariprazine, and most had poor MPO scores (<3). Conversely, the 2-fluoro-3-methoxyphenyl analogue (6c) noticeably decreased D3R and D2R affinities (Ki = 9.4 and 134 nM, respectively). Of note, compound 13c showed much greater D3R/D2R selectivity than any of the analogues in this series, which was a bit surprising. We have previously reported that D2R agonists are sensitive to the radioligand they are displacing and typically show much lower affinities when tested against a D2-like antagonist (e.g., [3H]-N-methylspiperone).51, 52 We considered that the 2-fluoro-3-methoxy-substituted phenyl piperazine PP might be conferring agonist actions and thus binding affinities using the D2-like agonist [3H]7-OH-DPAT were tested (Supporting Information, Table S1). Indeed, we discovered that D3R selectivities were much lower in this binding assay (D2R Ki/D3RKi = 0.9 and 15 for 6c and 13c, respectively), suggesting these analogues were agonists at D2R and possibly D3R. The 2-fluoro-3-methoxy-containing PP also caused a loss in affinity when the N,N-dimethylurea was replaced with the 3-methoxypropanamide (13c), and further analogues were not pursued. The 2-trifluoromethyl substituted pyridine analogues (6d, 11d, and 13d) showed promising increases in D3R selectivity. Specifically, SPs with the 4-quinoline (11d) and 3-methoxypropanamide (13d) showed a 160- and 120-fold D3R/D2R selectivity, respectively. However, replacing the 2,3-dichlorophenyl substituent with the 3-chloro-5-ethyl-2-methoxyphenyl group (4e, 6e-8e, 10e, and 13e) discovered in the highly D3R-selective VK4–11623 resulted in D3R affinities and selectivities over D2R that were only modestly different from the parent compound, unless the SP contained an indole (10e). Nevertheless, the 3-methoxypropanamide SP in this series gave compound 13e with a 21-fold D3R/D2R selectivity, subnanomolar affinity for D3R (Ki = 0.73 nM), and potentially drug-like MPO score (3.3).
For the last series of analogues, the terminal urea of cariprazine was incorporated into the trans-1,4-cyclohexane ring system of the linker. As shown in Table 2, analogues 21a and 22a retained the two-carbon chain between the 6-membered ring and the PP, which resulted in ~20-fold D3R selectivity over D2R regardless of the SP. When the linking chain was increased by an additional methylene group, the D3R binding affinity for 21b, which is a constitutional isomer of cariprazine, was reduced by ~9-fold compared to the parent drug. Interestingly, only the 3-methoxypropanamide containing analogue (22b) retained the ~20-fold D3R selectivity. If a hydroxyl group was appended to the linking chain between the PP and SP (29a, 30a, 29b, and 30b), the MPO scores noticeably improved to ≥5, but the D3R affinities were substantially reduced (Ki = 18.7–50.7 nM). The D3R/D2R selectivities for these analogues were also undesirable (<10-fold), so the synthesis of additional analogues containing this hydroxyl group was not pursued further.
Characterization of selected compounds in assays of D2R and D3R activation
With the binding studies and SAR established, we then characterized the abilities of 14 selected ligands to signal through both D2R and D3R along with quinpirole and dopamine as reference agonists and haloperidol as a reference antagonist.
To measure D2R activation, we used two BRET assays. The first consisted of a relatively amplified and sensitive G protein activation (GPA) assay that measures Gβγ release from GαoA, which allows detection of signals from less efficacious agonists (Table 3, Figure 2A, C and E). The second measured recruitment of a truncated, soluble ‘mini’ Gi-protein fused with a Venus fluorescent protein (mGsi-Venus) to a D2LR fused with a nanoluciferase (D2LR-Nluc). This less amplified, proximal assay allows us to distinguish between full agonists and more efficacious agonists (miniG assay, Table 3, Figure 2B, D and F) that gave a maximal response relative to the reference agonist quinpirole in the GPA assay.53–55
Table 3.
The maximal effects (Emax) and potencies (EC50) of compounds determined in two assays of D2R activation.a
| compound | D2R GPA GαoA | D2R mini Gsi recruitment | ||||
|---|---|---|---|---|---|---|
| pEC50 ± SEM | EC50 (nM) | Emax ± SEM (%) | pEC50 ± SEM | EC50 (nM) | Emax ± SEM (%) | |
| cariprazine | 8.87 ± 0.08 | 1.35 | 94.2 ± 1.7 | 8.87 ± 0.28 | 1.34 | 22.5 ± 1.7 |
| 13a | 8.77 ± 0.10 | 1.70 | 94.7 ± 2.2 | 8.65 ± 0.27 | 2.26 | 18.4 ± 1.4 |
| 11b | 7.37 ± 0.19 | 42.60 | 41.4 ± 3.0 | ND | ND | ND |
| 13b | 8.22 ± 0.10 | 5.98 | 75.0 ± 2.3 | ND | ND | ND |
| 6c | 9.57 ± 0.15 | 0.27 | 102.3 ± 3.6 | 8.39 ± 0.06 | 4.10 | 92.4 ± 1.6 |
| 13c | 9.13 ± 0.13 | 0.75 | 96.4 ± 3.3 | 7.96 ± 0.06 | 11.02 | 90.6 ± 2.0 |
| 11d | <7 | >100 | <60% | ND | ND | ND |
| 13d | 7.57 ± 0.15 | 27.12 | 64.3 ± 3.2 | ND | ND | ND |
| 6e | 8.14 ± 0.14 | 7.18 | 74.3 ± 3.1 | ND | ND | ND |
| 7e | 8.30 ± 0.11 | 5.00 | 95.9 ± 2.9 | 8.15 ± 0.43 | 7.08 | 11.1 ± 1.9 |
| 13e | 8.28 ± 0.11 | 5.22 | 93.0 ± 2.9 | 7.58 ± 0.21 | 26.21 | 12.5 ± 0.9 |
| 21a | 8.40 ± 0.10 | 3.97 | 95.9 ± 2.8 | 7.93 ± 0.12 | 11.86 | 36.7 ± 1.5 |
| 22a | 8.20 ± 0.06 | 6.28 | 94.5 ± 1.9 | 7.66 ± 0.11 | 21.85 | 25.1 ± 0.9 |
| 22b | 7.96 ± 0.08 | 10.89 | 86.7 ± 2.4 | 8.00 ± 0.18 | 9.92 | 15.4 ± 1.0 |
| Quinpirole | 9.03 ± 0.05 | 0.93 | 100.1 ± 1.3 | 7.40 ± 0.08 | 39.78 | 100.0 ± 2.7 |
| Dopamine | - | - | - | 7.18 ± 0.03 | 66.25 | 99.9 ± 1.1 |
Compounds were tested in an amplified GαoA G protein (GPA GαoA) activation assay, shown on the left columns, and a less amplified miniG recruitment assay, shown on the right columns. ND indicates no agonist response detected, ‘-’ indicates compound not tested. Data represent the mean ± SEM of three independent experiments performed in duplicate.
Figure 2. Agonism profiles at D2R.
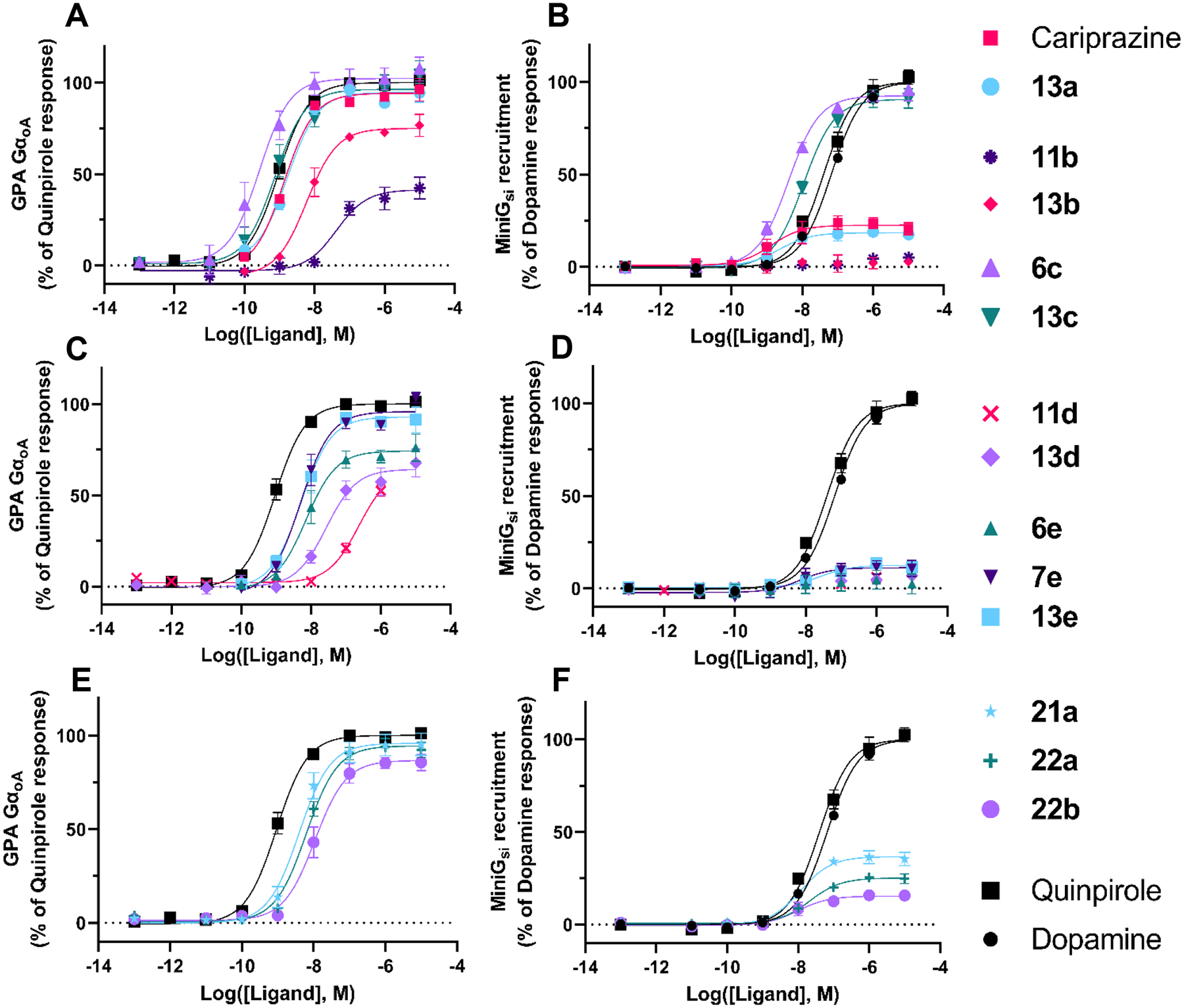
Two differents BRET assays were used to assess D2R activation: an amplified GαoA G protein (GPA GαoA) activation assay, shown on the left panels, and a less amplified miniG recruitment assay shown on the right panels. Each panel shows concentration response curves for a subset of the 14 selected ligands: 2,3-dichlorophenyl, 2-chloro-3-ethylphenyl, and 2-fluoro-3-methoxyphenyl compounds (A) GPA GαoA and (B) miniGsi; 2-trifluoromethyl substituted pyridine and 3-chloro-5-ethyl-2-methoxyphenyl compounds (C) GPA GαoA and (D) miniGsi; linker modified compounds (E) GPA GαoA and (F) miniGsi. Data points represent the mean ± SEM of three independent experiments performed in duplicate.
As expected, the reference agonist quinpirole displayed 43-fold lower potency in the miniG assay as compared to the GPA assay consistent with a lower degree of signal amplification. Quinpirole and dopamine displayed equivalent maximal responses in the miniG assay (Table 3). All ligands tested retained D2R agonism to various levels of intrinsic efficacy. The 2,3-dichlorophenyl compounds (cariprazine and 13a) are D2R agonists (Figure 2B, Emax value of 22.5% and 18.4% of the dopamine maximal response respectively in miniGsi recruitment assay) but displayed a maximal effect similar to quinpirole in the GPA assay. The 2-chloro-3-ethylphenyl compounds (11b and 13b) are weak agonists at D2R displaying lower efficacy than cariprazine with submaximal responses in the GPA assay (41% and 75% of quinpirole response, respectively) and no detectable effect in the miniG assay.40, 42 The 2-fluoro-3-methoxyphenyl compounds (6c and 13c) displayed a maximal effect similar to that of dopamine and quinpirole in both assays indicating they are full D2R agonists. Indeed, these data corroborated the binding data obtained using [3H]7-OH-DPAT as the radioligand, as described above (see Table S1). The 2-trifluoromethyl substituted pyridine compounds (11d and 13d) were both weak agonists displaying lower efficacy than cariprazine in both assays. The 3-chloro-5-ethyl-2-methoxyphenyl compounds (6e, 7e, and 13e) were also weak agonists displaying lower maximal effects than both quinpirole and cariprazine, but with higher potency than the 2-trifluoromethyl substituted pyridine compounds. Finally the modified linker compounds (21a, 22a, and 22b) all behaved as weak agonists in the miniG assay, displaying slightly higher (21a, 38% of dopamine Emax), similar (22a, 25%), and lower (22b, 15%) efficacy as compared to cariprazine (23%). Thus, apart from the compounds with the 2-fluoro-3-methoxyphenyl PP, all compounds exhibited weak agonism at the D2R like cariprazine but with subtle differences in maximal effects across the compound set. As we have previously suggested,41 the efficacies of D2R and D3R compounds can be modified through modifications to PP, linker, and SP. As there is a range of potencies and efficacies in this series of analogues, we now have the tools necessary to determine the optimal pharmacological profile, including binding affinities, selectivities and functional efficacies at both D2R and D3R.
All 14 selected compounds were then tested for D3R signaling profiles with the aim of selecting agonists or antagonists. The same GPA assay overexpressing GαoA was used to investigate D3R agonism as the D3R has been shown to selectively couple to this G protein α subunit (Table 4, Figure 3A, C and E).56 The 2,3-dichlorophenyl compounds (cariprazine and 13a) both behaved as D3R agonists in this assay with similar maximal effects (45% and 49% of quinpirole maximal effect), suggesting that this GPA assay would be suitable to detect D3R agonism with a similar or lower level than that of cariprazine. The 2-fluoro-3-methoxyphenyl compounds did not display the desired weak agonist/antagonist properties at the D3R but instead behaved as full agonists at D3R in this assay, consistent with their action at the D2R. The modified linkers compounds were all D3R agonists with 21a exhibiting the strongest response with a higher efficacy than cariprazine (Figure 3E, Emax value of 73.2 %). Finally, no agonist response was detected for the 2-chloro-3-ethylphenyl (11b and 13b), the 2-trifluoromethyl substituted pyridine (11d and 13d), and the 3-chloro-5-ethyl-2-methoxyphenyl compounds (6e, 7e, and 13e).
Table 4.
The maximal effects (Emax) and potencies (EC50 and IC50) of compounds determined in an assay of D3R activation.a
| compound | D3R GPA GαoA agonism | D3R GPA GαoA antagonism | |||
|---|---|---|---|---|---|
| pEC50 ± SEM | EC50 (nM) | Emax ± SEM (%) | pIC50 ± SEM | IC50 (nM) | |
| cariprazine | 8.72 ± 0.14 | 1.89 | 45.1 ± 1.6 | 8.70 ± 0.32 | 2.02 |
| 13a | 8.80 ± 0.20 | 1.60 | 49.3 ± 2.3 | 8.34 ± 0.23 | 4.62 |
| 11b | ND | ND | ND | 7.44 ± 0.25 | 36.00 |
| 13b | ND | ND | ND | 8.73 ± 0.16 | 1.87 |
| 6c | 8.79 ± 0.14 | 1.64 | 95.4 ± 3.8 | - | - |
| 13c | 8.54 ± 0.16 | 2.87 | 104.8 ± 4.8 | - | - |
| 11d | ND | ND | ND | 7.47 ± 0.15 | 33.63 |
| 13d | ND | ND | ND | 8.49 ± 0.24 | 3.21 |
| 6e | ND | ND | ND | 7.87 ± 0.13 | 13.41 |
| 7e | ND | ND | ND | 7.70 ± 0.19 | 19.85 |
| 13e | ND | ND | ND | 7.76 ± 0.13 | 17.45 |
| 21a | 8.22 ± 0.20 | 6.07 | 73.2 ± 4.9 | - | - |
| 22a | 7.70 ± 0.13 | 19.91 | 41.5 ± 1.9 | 7.92 ± 0.35 | 12.12 |
| 22b | 7.80 ± 0.28 | 16.02 | 33.0 ± 2.9 | 7.89 ± 0.37 | 12.93 |
| Quinpirole | 8.59 ± 0.06 | 2.59 | 100.1 ± 1.7 | - | - |
| Haloperidol | - | - | - | 8.60 ± 0.08 | 2.54 |
Compounds were tested in an amplified GαoA G protein (GPA GαoA) activation assay as agonists and antagonists. ND indicates no agonist response detected, ‘-’ indicates compound not tested. Data represent the mean ± SEM of three independent experiments performed in duplicate.
Figure 3. Functional profiles at the D3R.
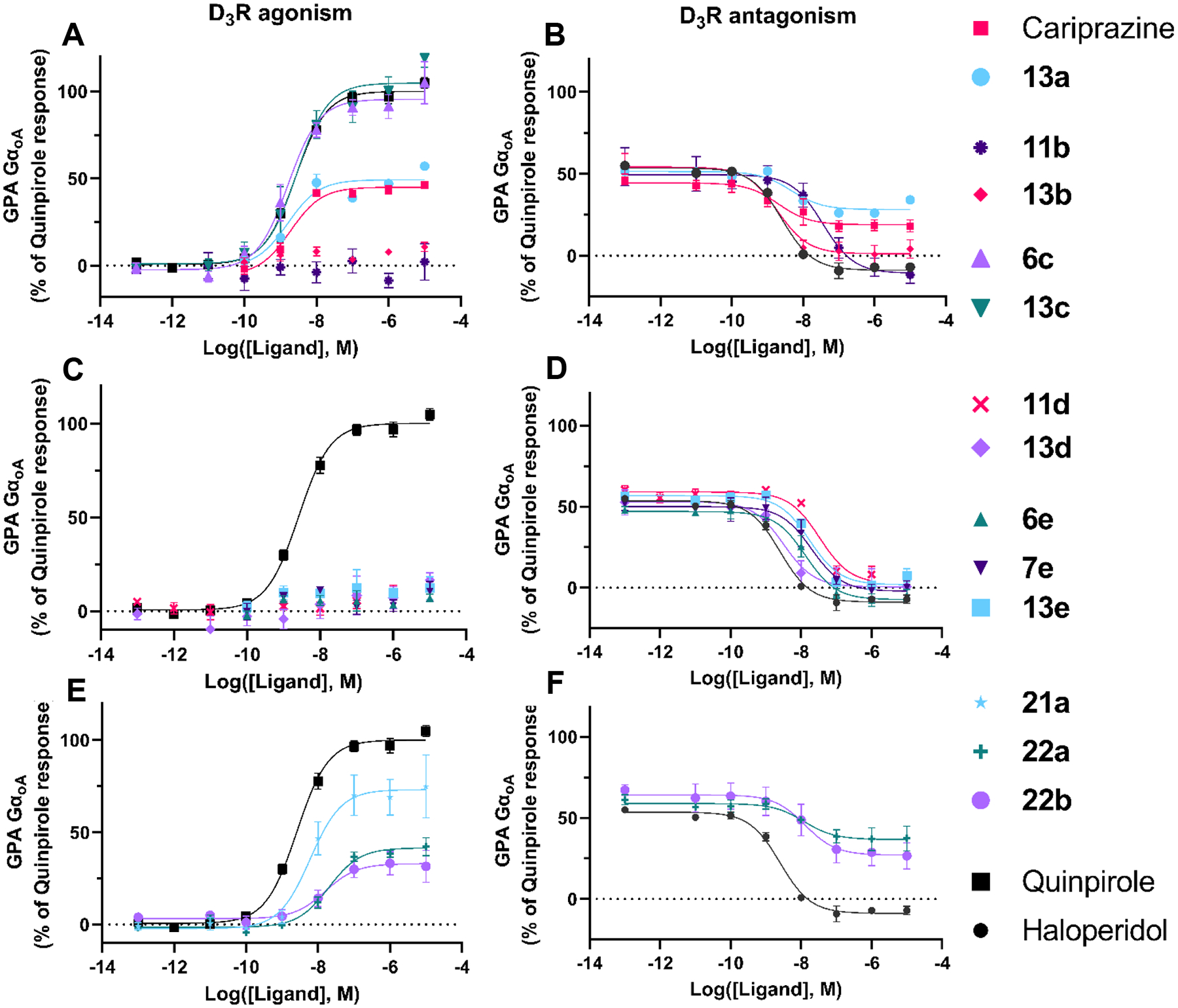
Using the GαoA assay, all ligands were tested for D3R agonism, shown on the left panels and 11 out of the 14 selected ligands were tested for D3R antagonism (compounds were added to cells together with and EC50 concentration (3 nM) of quinpirole), shown on the right panels. 6c, 13c, and 21a signaled as robust D3R agonists and were thus not tested as antagonists as they would not displace quinpirole signal. Each panel shows concentration response curves for a subset of the selected ligands: 2,3-dichlorophenyl, 2-chloro-3-ethylphenyl, and 2-fluoro-3-methoxyphenyl compounds (A) D3R agonism and (B) D3R antagonism; 2-trifluoromethyl substituted pyridine and 3-chloro-5-ethyl-2-methoxyphenyl compounds (C) D3R agonism and (D) D3R antagonism; linker modified compounds (E) D3R agonism and (F) D3R antagonism. Data points represent the mean ± SEM of three independent experiments performed in duplicate.
To quantify D3R antagonism, a competitive assay was used where test compounds were added together with an EC50 concentration of quinpirole to assess their ability to displace and antagonize the agonist response (Table 4, Figures 3B, 3D, and 3F). Haloperidol was used as a reference D3R antagonist. Compounds that signaled as robust D3R agonists (6c, 13c, and 21a) were not tested as antagonists. Out of the 11 remaining compounds, all showed some antagonist activity, with the agonists acting to reduce the quinpirole response down to their level of agonism (around 25% for cariprazine and 13a, around 30% for 22a and 22b). The most potent antagonists were 13b, 13d, and 13a, showing similar potencies to haloperidol. In addition, 13a showed a very similar signaling profile to cariprazine (weak agonist with similar efficacy at both the D2R and D3R) but with improved selectivity for D3R. Thus, this compound, like 13e, is a agonist at D2R (with 2-fold lower maximal effect than cariprazine) in the miniG assay but acted as a D3R antagonist.
Off-target data for cariprazine and related derivatives
Eleven compounds with a range of selectivities for D3R over D2R (between 7.1- to 370-fold) were then evaluated for off-target binding affinities (Table 5) and functional potencies/efficacies (Supporting Information, Tables S2–S5) against cariprazine. All tested analogues were selective for D3R over other dopamine receptors (i.e., D1R and D4R), exhibiting markedly lower affinities for these two subtypes (Ki ≥ 3 μM and 200 nM, respectively). Like cariprazine, several derivatives (e.g., 13a, 13b, and 13d) exhibited high binding affinities for the 5-HT1A receptor (Ki <10 nM), which is a key target in the treatment of schizophrenia.57, 58 Interestingly, 5-HT1A affinity decreased by 18-fold between 13a and 13e (Ki = 6.0 vs. 108 nM, respectively) when the classic 2,3-dichlorophenyl containing PP was replaced by a 3-chloro-5-ethyl-2-methoxyphenyl group. In general, compounds were far more selective for D3R over 5-HT2A and 5-HT2C (≥216- and ≥253-fold, respectively). The only exceptions to this trend were 11b and 13b, which were not pursued further. The full subset of cariprazine analogues also displayed high affinities for 5-HT2B (Ki ≤ 34.2 nM). Although activation of this serotonin receptor subtype has been associated with cardiotoxic effects,59 all tested compounds behaved as 5-HT2B antagonists in the functional assays, including cariprazine itself (see Table S5). Therefore, none of our analogues are expected to induce cardiotoxicity from 5-HT2B stimulation.
Table 5.
Off-target binding affinities of cariprazine-based analogues.a
| compound | Ki ± SEM (nM) | |||||
|---|---|---|---|---|---|---|
| D1Rb | D4Rc | 5-HT1Ad | 5-HT2Ae | 5-HT2Be | 5-HT2Ce | |
| cariprazine | 2920 ± 300 | 233 ± 20 | 1.81 ± 0.46 | 568 ± 72 | 0.151 ± 0.023 | 221.1 ± 8.0 |
| 11a | >3160 | 1390 ± 480 | 184 ± 25 | 1810 ± 640 | 1.99 ± 0.62 | 644 ± 58 |
| 13a | 4600 ± 400 | 756 ± 63 | 6.0 ± 1.8 | 54 ± 15 | 1.47 ± 0.36 | 252 ± 64 |
| 11b | 5390 ± 730 | 417 ± 40 | 75 ± 25 | 15.1 ± 4.8 | 1.27 ± 0.39 | 253 ± 86 |
| 13b | 6800 ± 480 | 342 ± 15 | 7.7 ± 1.8 | 20.8 ± 6.9 | 1.71 ± 0.43 | 34.0 ± 8.1 |
| 6c | >10000 | 1257 ± 23 | 33.8 ± 5.9 | >10000 | 34.2 ± 9.3 | >10000 |
| 13c | >10000 | 734 ± 16 | 69.9 ± 1.7 | >10000 | 29.6 ± 9.5 | >10000 |
| 11d | >3160 | 1790 ± 430 | 22.3 ± 6.4 | 1450 ± 340 | 18.8 ± 6.2 | 5800 ± 1300 |
| 13d | >10000 | 2520 ± 170 | 9.6 ± 2.6 | 4420 ± 340 | 11.3 ± 2.2 | 3170 ± 200 |
| 6e | >10,000 | 2434± 87 | 45.4 ± 9.0 | 680 ± 200 | 7.3 ± 1.9 | 1062 ± 86 |
| 7e | 8000 ± 1100 | 1346 ± 29 | 54.9 ± 4.2 | 207 ± 74 | 6.2 ± 1.5 | 162 ± 48 |
| 13e | >10,000 | 1870 ± 240 | 108 ± 11 | 158 ± 45 | 5.8 ± 1.7 | 800 ± 280 |
Data were obtained through the NIDA Addiction Treatment Discovery Program Contract (ADA12013) with Oregon Health & Science University. The radioligands used in these assays were
[3H]-SCH23390,
[3H]-spiperone,
[3H]-8-OH-DPAT, and
[3H]5-HT.
Effects of cariprazine and selected analogues on cocaine self-administration in rats
Cariprazine was tested in a rat model of cocaine self-administration under a fixed-ratio 2 (FR2, i.e., two-active lever presses lead to one cocaine infusion) schedule of reinforcement. Figure 4A shows dose-dependent biphasic effects of cariprazine on cocaine self-administration, specifically an increase at a very low dose (0.3 mg/kg) and a decrease at the higher doses (1, 3 mg/kg). One-way ANOVA with repeated measures (RM) over drug doses revealed a significant treatment main effect (Figure 4A, F(3, 24) = 9.616, p < 0.001). Dunnett’s post-hoc tests indicated a significant reduction in the number of cocaine infusions only after 3 mg/kg cariprazine administration compared to the vehicle control group (q’ = 3.497, p = 0.005). In contrast, cariprazine failed to alter the number of inactive lever presses (Figure 4B, F(3, 24) = 1.362, p > 0.05), suggesting that cariprazine did not produce sedation or locomotor impairment. Figure 4C shows the representative records of cocaine self-administration before and after the different doses of cariprazine administration, illustrating that at low doses (0.3mg/kg, i.p.), cariprazine produced an increase in drug taking, as previously reported,60 which is possibly a compensatory response to a reduction in cocaine reward. However, at high doses (1, 3 mg/kg), cariprazine caused a typical extinction-like pattern of cocaine self-administration (i.e., an initial burst-like increase in drug intake followed by cessation of cocaine intake), indicating a robust reduction in cocaine reward after high dose cariprazine administration. In total, this classical pattern of drug self-administration suggests that cariprazine pretreatment produces a dose-dependent reduction in cocaine’s reinforcing effects (efficacy).
Figure 4. Dose-dependent effects of cariprazine on cocaine self-administration in rats.
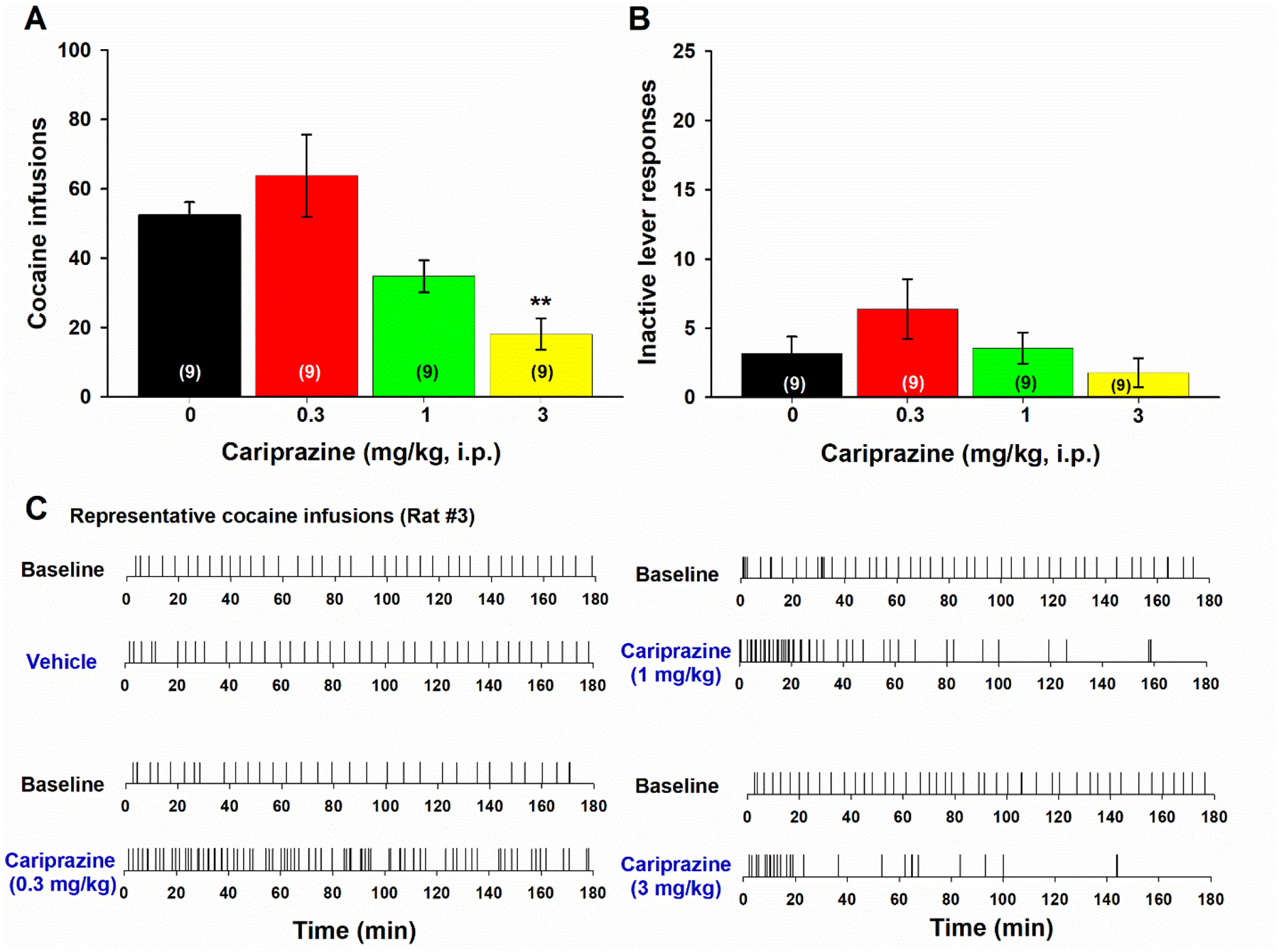
(A) The dose-dependent effects of cariprazine on cocaine self-administration (infusions). (B) Effects of cariprazine on inactive lever presses. (C) Representative cocaine self-administration (infusions) records, illustrating the typical extinction-like patterns of drug taking and drug seeking—lower doses of cariprazine (0.3 mg/kg) increased, while at higher doses it produced an initial burst-like drug infusions followed by cessation of drug self-administration. *p < 0.05, **p < 0.01, compared to vehicle (0 mg/kg).
Based on D3R binding affinities and selectivity profiles, as well as favorable MPO scores (>3) and agonist/antagonist profiles at both D3R and D2R, we selected 13a and 13e for behavioral testing in comparison to the parent compound. Similar to cariprazine (Figure 4), 13a also produced a dose-dependent biphasic effect on cocaine self-administration. That is, at 0.3 mg/kg, 13a produced a significant increase, while at higher doses (1, 3 mg/kg) it produced a dose-dependent reduction in the number of cocaine infusions (Figure 5A, F(3, 24)= 23.822, p < 0.001; Dunnett’s post-hoc, q’= 2.556, p = 0.045 at 0.3 mg/kg; q’= 2.873, p = 0.022, at 1 mg/kg; q’= 5.395, p < 0.001, at 3 mg/kg). Like cariprazine, 13a pretreatment also failed to affect the number of inactive lever presses (Figure 5B; F(3, 24)= 2.708, p > 0.05).
Figure 5. Dose-dependent effects of 13a on cocaine self-administration.
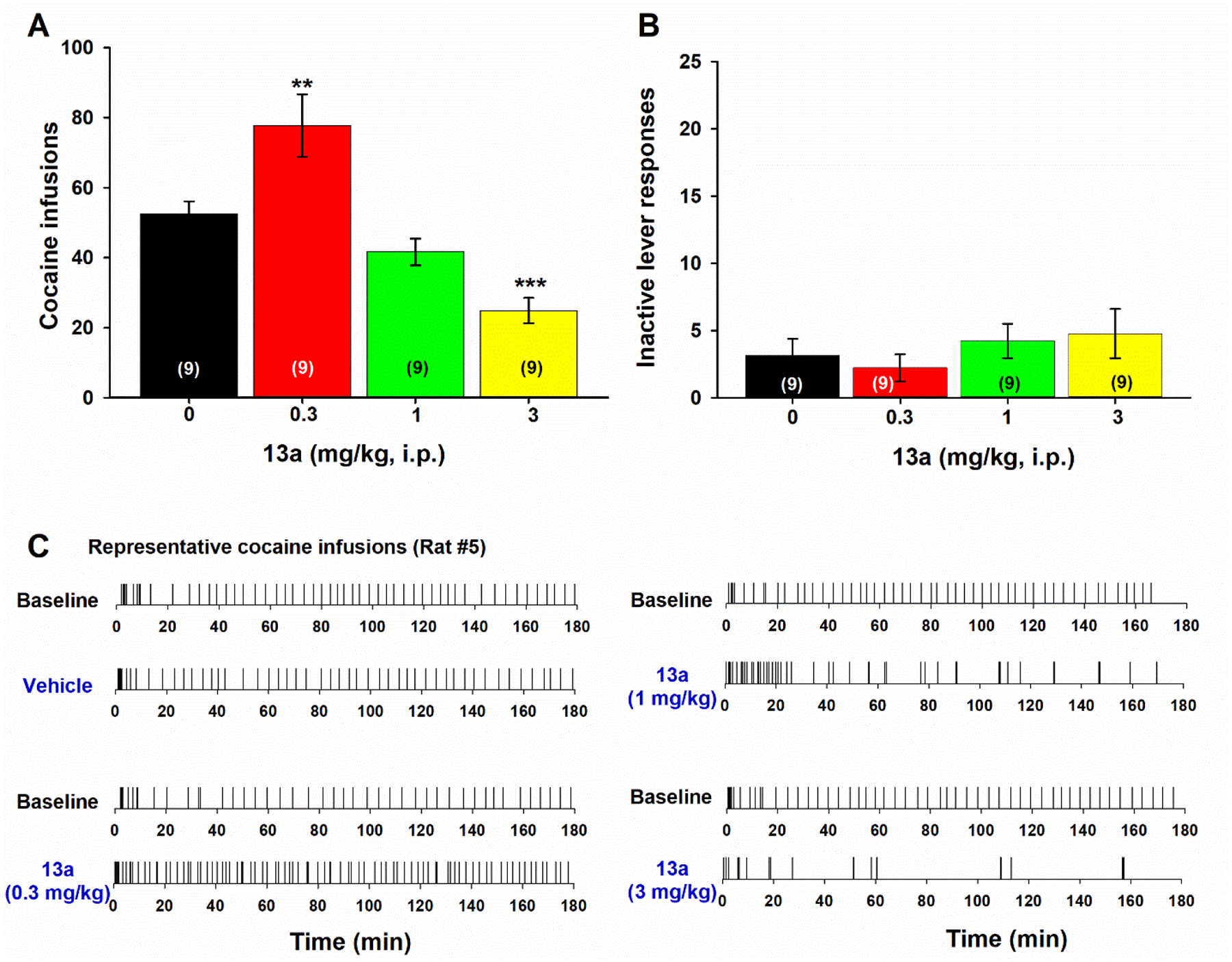
(A) Mean numbers after different doses of 13a administration. (B) Mean numbers of inactive lever presses after different doses of 13a pretreatments. (C) Representative cocaine infusions behavior indicating an extinction-like pattern of cocaine self-administration after 13a administration in a way similar to cariprazine. (*p < 0.05, **p < 0.01, ***p < 0.001, compared to the vehicle control group).
Next, we tested 13e and found that higher doses (10 mg/kg) of 13e are required to inhibit cocaine self-administration under FR2 reinforcement, in rats (Figure 6). One-way ANOVA revealed a significant treatment main effect on cocaine infusions (Figure 6A, F(4, 43) = 4.130, p = 0.006; Dunnett’s post-hoc, q’= 3.283, p = 0.007, at 10 mg/kg compared to vehicle). As observed with cariprazine and 13a, there was no effect on the number of inactive lever presses (Figure 6B; F(4, 43)= 0.736, p > 0.05).
Figure 6. Dose-dependent effects of 13e on cocaine self-administration.
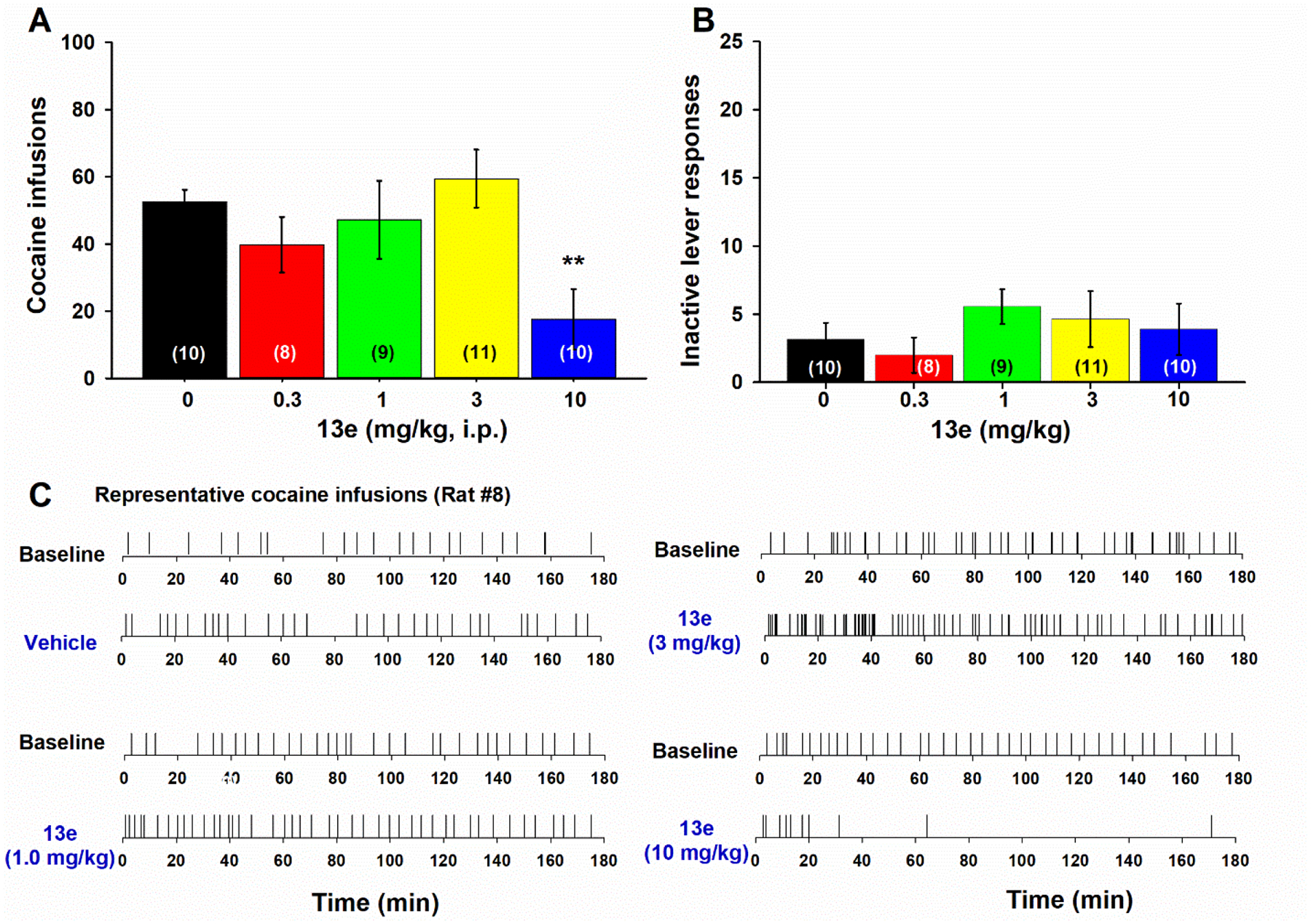
(A) Mean number of self-infusions after different doses of 13e. (B) Mean numbers of inactive lever presses 13e pretreatments. (C) Representative cocaine infusions illustrating the patterns of cocaine self-administration after vehicle or 13e administration. (*p < 0.05; **p < 0.01, compared to the vehicle control group).
There are three important findings in this behavioral test. First, cariprazine and its analogs appear to be more potent than highly selective D3R antagonists in reducing cocaine self-administration under low-cost high-payoff (FR2) reinforcement conditions. In particular, considerably lower doses (0.3, 1, 3 mg/kg) of cariprazine and 13a are required to alter cocaine self-administration. Second, cariprazine and 13a produced unique biphasic effects on cocaine self-administration—increasing the number of cocaine infusions at low doses and decreasing drug intake at higher doses. These biphasic effects are usually seen after pretreatment with non-selective dopamine receptor antagonists, such as pimozide and (+)-butaclamol.61 In contrast, highly selective D3R antagonists usually produced a monophasic reduction in cocaine or opioid self-administration, and higher doses (10–30 mg/kg) are required to inhibit cocaine or oxycodone self-administration.8, 24 The increase in cocaine self-administration after low doses of cariprazine and 13a is a compensatory response to reduced cocaine reward, as the pattern of cocaine self-administration is comparable to the responses produced by lower doses of cocaine - evenly distributed drug intake with shorter infusion intervals.61, 62 Thus, the increased behavioral performance reveals a reduction in cocaine’s rewarding effects. Congruently, at higher doses, cariprazine, 13a and 13e all produced a classical extinction-like pattern of cocaine self-administration—initial burst-like increase in drug infusions followed by cessation of drug taking.
Taken together, these data show that cariprazine and its two analogs, 13a and 13e, produce a dose-dependent reduction in cocaine reward, which is consistent with their pharmacological profiles as D3R agonists/antagonists with moderate D3R/D2R selectivities (cariprazine, 3.6-fold; 13a/13e, ~20-fold). We note that 13a displays similar pharmacological potency to cariprazine in attenuating cocaine reward, while 13e displays slightly lower potency as a higher dose (10 mg/kg) of 13e is required to inhibit cocaine self-administration. This behavioral potency difference is in line with their receptor binding profiles. Whereas 13a has a similar binding affinity to D2R and D3R as cariprazine (Table 1), 13e displays ~5-fold lower affinities for D2R and D3R compared to 13a (Table 3). Similarly, in vitro functional assays indicate that the EC50 value of 13e in activating D2R is ~10-fold higher than that of 13a. This is consistent with the effective dose of 13e (10 mg/kg, Figure 6) in attenuating cocaine self-administration that is also ~3-fold higher than that of 13a (3 mg/kg, Figure 5).
CONCLUSIONS
We have hypothesized that compounds that are moderately D3R/D2R selective and are partial agonists, especially at D2R may be more effective than highly D3R selective antagonists for treatment of PSUD. Previously, highly selective D3R partial agonists/antagonists have been shown to block the reinforcing effects of opioids, such as oxycodone,24, 63, 64 but are often less effective for psychostimulants, such as cocaine or methamphetamine,18, 65 especially when the drug is readily available (low fixed ratio schedule of reinforcement e.g., FR1 or FR2). Nevertheless, compounds that are D2R antagonists are generally not well tolerated in this patient population.30 To test this hypothesis, we designed a series of compounds, based on cariprazine, with varying selectivities and functional efficacies at D2R and D3R, depending on their PP, SP, and/or linker. By systematically modifying each piece of the parent molecule, we have discovered compounds with high D3R affinities (Ki < 1 nM) and improved selectivities (≥20-fold) over D2R that have the desired D2R partial agonist functional efficacies and are either low efficacy partial agonists or antagonists at D3R, as measured in BRET-based assays for each receptor subtype. We have obtained off target activities (D1R, D4R, 5HT1A, 5HT2A, 5HT2B, and 5HT2C) on a subset of these compounds that had desirable D3R/D2R selectivity profiles as well as MPO scores (>3) that further demonstrated D3R-selectivity.
Based on all the in vitro data obtained, we selected 13a and 13e as our initial lead compounds, which were tested side-by-side with cariprazine in a rat model of cocaine self-administration. The two new compounds had higher but still moderate D3R/D2R selectivities (~20-fold) as compared to cariprazine (3.6-fold) and were effective in blocking cocaine self-administration in the rats (FR2; 1–10 mg/kg, i.p.). Overall, these convincing behavioral data support the notion that moderately D3R selective partial agonists/antagonists that are also partial agonists at D2R, may be more effective than highly selective (>200-fold) D3R antagonists in reducing drug-seeking behavior. Of course, additional testing will be needed to determine the suitability of 13a and 13e for further development. For example, the extrapyramidal side effects of our lead compounds will need to be examined as well as their efficacy in animal models of affective disorders, such as BD-I.66, 67 Nonetheless, the cariprazine analogs described herein demonstrate high potential for treating PSUD that may be dual diagnosed with affective disorders.
EXPERIMENTAL METHODS
Chemistry
General Information.
Chemicals and solvents were purchased from commercial suppliers and used as received. Unless stated otherwise, reactions were performed under ambient conditions and monitored by thin-layer chromatography using Analtech silica gel GHLF (250 microns) coated glass plates, which were visualized with either phosphomolybdic acid, potassium permanganate, or vanillin stain. Normal phase column chromatography was conducted with a Teledyne Isco Combiflash Rf or EZ-Prep purification system (ELS detector associated). All nuclear magnetic resonance (NMR) spectra (1H and 13C) were acquired in deuterated solvents (CDCl3 or CD3OD) on a Varian Mercury Plus 400 spectrometer. Chemical shifts are reported in parts per million (ppm) and were adjusted using the residual solvents (CDCl3: 7.26 ppm for 1H NMR, 77.2 ppm for 13C NMR; CD3OD: 3.31 ppm for 1H NMR, 49.0 ppm for 13C NMR) as an internal reference. Coupling constants are reported in Hertz (Hz) and peak multiplicities as either a singlet (s), doublet (d), triplet (t), quartet (q), or multiplet (m). Infrared (IR) spectra were acquired on a Perkin Elmer Spectrum Two FT-IR spectrometer. Melting points were determined on an Optimelt MPA100 instrument and are uncorrected. High-resolution mass spectrometry (HRMS) data were collected on a Thermo Scientific LTQ-Orbitrap Velos spectrometer using either electrospray ionization (ESI) or matrix-assisted laser desorption/ionization (MALDI). High-performance liquid chromatography (HPLC) was conducted on an Agilent Technologies 1260 Infinity system coupled to a diode-array detector. A Phenonmenex Gemini C18 100 Å LC column (50 × 4.6 mm, 3 μm particle size) was used as the stationary phase. For basic conditions, the mobile phase consisted of H2O with 0.1% diethylamine (solvent A) and MeCN with 0.1% diethylamine (solvent B). For acidic conditions, solvents contained 0.1% TFA instead of diethylamine. All samples were prepared at a concentration of ca. 1 mg/mL in MeOH, and 20 μL of each solution was injected onto the column, which was maintained at 40 °C. Using a flow rate of 1.0 mL/min, the solvent gradient was as follows: 10% B held for 10 min, 10–40% B ramped over 10 min, 40% B held for 10 min, 40–80% B over 10 min, 80% B held for 20 min. Compound purity was determined based on peak integration (area under the curve) of the absorbance signals at 254 and 214 nm. Elemental analysis was performed by Atlantic Microlab, Inc. (Norcross, GA). All tested compounds were >95% pure by either HPLC or elemental analysis.
General Procedure A.
Using oven-dried glassware under an Ar atmosphere, a 0.60 M solution of (COCl)2 in anhydrous DCM (1.5 equiv) was cooled to −78 °C. Then, anhydrous DMSO (3.0 equiv) was added over ca. 5 min, and the reaction was stirred for 30 min. Next, a 0.30 M solution of the alcohol in anhydrous DCM (1.0 equiv) was added over ca. 10 min, and the reaction was stirred for an additional 30 min. Finally, anhydrous NEt3 (6.0 equiv) was added over ca. 5 min, and the mixture was allowed to warm to rt. When TLC analysis suggested the complete disappearance of starting material (ca. 2 h), the reaction was quenched with a 1.0 M aq solution of HCl, and the aq layer was extracted with DCM (2 ×). The combined organic layers were dried over Na2SO4, filtered, and concentrated. The crude product was purified by chromatography as described.
General Procedure B.
To a 0.08–0.13 M solution of 3 in anhydrous DCE (1.0 equiv) was added AcOH (1.0 equiv) followed by the appropriate aryl piperazine (1.1 equiv). The mixture was stirred for 30 min, and NaBH(OAc)3 (1.5 equiv) was added in one portion. After stirring overnight, the reaction was quenched with a saturated aq solution of NaHCO3, and the aq layer was extracted with DCM (2 ×). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated. The crude product was purified by chromatography as described.
General Procedure C.
TFA (27–144 equiv) was added to a 0.03–0.15 M solution of the appropriate Boc-protected amine in DCM (1.0 equiv). The reaction mixture was stirred for ca. 4 h and then concentrated. The resulting residue was suspended in a 1.0 M aq solution of NaOH, and the aq layer was extracted with CHCl3 (3 ×). The combined organic layers were dried over Na2SO4, filtered, and concentrated to afford the corresponding amine.
General Procedure D.
To a 0.07–0.10 M solution of the amine in DCM (1.0 equiv) was added DIPEA (1.5 equiv) followed by the appropriate carbamyl chloride (1.25 equiv) over 5 min. After stirring overnight, the reaction was quenched with a saturated aq solution of NaHCO3, and the aq layer was extracted with CHCl3 (3 ×). The combined organic layers were dried over Na2SO4, filtered, and concentrated. The crude product was purified by chromatography as described.
General Procedure E.
A 0.03–0.05 M mixture of the appropriate carboxylic acid (1.1 equiv) in CHCl3 was cooled to 0 °C, and EDCI (1.3 equiv) followed by HOBt (1.2 equiv) were each added in one portion. After 30 min, the amine (1.0 equiv) was added followed by DIPEA (1.4 equiv). The mixture was allowed to warm to rt and stirred overnight. Then, a saturated aq solution of NaHCO3 was added, and the aq layer was extracted with CHCl3 (3 ×). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated. The crude product was purified by chromatography as described.
General Procedure F.
To a 0.10 M mixture of HCTU (1.2 equiv) in DCM was added 3-methoxypropanoic acid (1.1 equiv) in one portion. After stirring the mixture for 10 min, a solution of the amine (1.0 equiv) and DIPEA (1.2 equiv) in DCM (0.04 M with respect to the limiting reagent) was added over 5 min. The reaction was stirred overnight, quenched with a saturated aq solution of NaHCO3, and the aq layer was extracted with DCM (3 ×). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated. The crude product was purified by chromatography as described to afford the corresponding amide.
tert-Butyl (trans-4-(2-hydroxyethyl)cyclohexyl)carbamate (2).
Using oven-dried glassware under an Ar atmosphere, a solution of 2-(trans-4-((tert-butoxycarbonyl)amino)cyclohexyl)acetic acid (6.01 g, 23.4 mmol) in anhydrous THF (30 mL) was cooled to 0 °C. Then, borane dimethylsulfide complex (3.4 mL, 36 mmol) was added dropwise over 5 min. The solution was allowed to warm to rt and stirred for 15 h. Afterward, the reaction was cooled back to 0 °C, quenched with a saturated aq solution of NaHCO3 (60 mL), and the aq layer was extracted with EtOAc (2 × 60 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to afford 2 (5.18 g, 21.3 mmol, 91% yield) as a white solid. An analytical sample was prepared by recrystallization using Et2O—hexanes (slow evaporation of solvent mixture). Rf = 0.4 (50% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 4.44–4.27 (m, 1H), 3.71–3.64 (m, 2H), 3.43–3.28 (m, 1H), 2.04–1.96 (m, 2H), 1.81–1.73 (m, 2H), 1.51–1.26 (m, 4H), 1.43 (s, 9H), 1.15–0.96 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 155.4, 79.2, 60.9, 50.0, 39.8, 33.6 (3C), 32.0 (2C), 28.6 (3C); IR (neat) 3425, 3365, 1682, 1517 cm−1; mp 102–103 °C (Et2O—hexanes); HRMS (MALDI) m/z [M + Na]+ calcd for C13H25NNaO3 266.1727, found 266.1730.
tert-Butyl (trans-4-(2-oxoethyl)cyclohexyl)carbamate (3).
General Procedure A was followed using 2 (5.18 g, 21.3 mmol). After work-up, the crude product was purified by chromatography (120 g of silica gel, 0–40% EtOAc/hexanes) to afford 3 (3.04 g, 12.6 mmol, 59% yield) as a white solid. Rf = 0.5 (40% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 9.78–9.73 (m, 1H), 4.47–4.29 (m, 1H), 3.45–3.26 (m, 1H), 2.35–2.29 (m, 2H), 2.06–1.95 (m, 2H), 1.90–1.74 (m, 3H), 1.44 (s, 9H), 1.20–1.03 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 202.3, 155.3, 79.3, 50.8, 49.6, 33.3 (2C), 31.9 (2C), 31.8, 28.6 (3C); IR (neat) 3377, 1715, 1686 cm−1; mp 90–91 °C; HRMS (MALDI) m/z [M + Na]+ calcd for C13H23NNaO3 264.1570, found 264.1578.
tert-Butyl (trans-4-(2-(4-(2,3-dichlorophenyl)piperazin-1-yl)ethyl)cyclohexyl)carbamate (4a).
General procedure B was followed using 3 (2.00 g, 8.29 mmol) and 1-(2,3-dichlorophenyl)piperazine•HCl (2.44 g, 9.12 mmol) in DCE (56 mL). After work-up, the crude product was purified by chromatography (80 g of silica gel, 0–50% EtOAc/hexanes) to afford 4a (1.79 g, 3.92 mmol, 47% yield) as a white solid. Rf = 0.2 (50% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 7.16–7.08 (m, 2H), 6.98–6.91 (m, 1H), 4.44–4.24 (m, 1H), 3.44–3.27 (m, 1H), 3.14–2.97 (m, 4H), 2.71–2.52 (m, 4H), 2.46–2.36 (m, 2H), 2.05–1.92 (m, 2H), 1.81–1.71 (m, 2H), 1.49–1.37 (m, 2H), 1.44 (s, 9H), 1.29–1.15 (m, 1H), 1.14–0.95 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 155.7, 151.8, 135.5, 128.0, 127.9, 125.0, 119.0, 79.5, 57.1, 53.9, 51.8, 50.4, 35.9, 34.4, 33.9 (2C), 32.5 (2C), 28.9 (3C); IR (neat) 3366, 1677 cm−1; mp 147–148 °C; HRMS (MALDI) m/z [M + H]+ calcd for C23H36Cl2N3O2 456.2179, found 456.2174; tR = 39.4 min (HPLC, basic). The oxalate salt was precipitated from a 0.03 M solution of the free base in 50% CHCl3/acetone using oxalic acid (1.25 equiv). Dec >185 °C. Anal. calcd for C23H35Cl2N3O2•1.25C2H2O4: C, 53.83; H, 6.64; N, 7.39. Found: C, 54.06; H, 6.67; N, 7.48.
tert-Butyl (trans-4-(2-(4-(2-chloro-3-ethylphenyl)piperazin-1-yl)ethyl)cyclohexyl)carbamate (4b).
General procedure B was followed using 3 (0.710 g, 2.94 mmol) and 1-(2-chloro-3-ethylphenyl)piperazine (0.810 g, 3.60 mmol) in DCE (35 mL). After work-up, the crude product was purified by chromatography (silica gel, 0–70% EtOAc/hexanes) to afford 4b (0.990 g, 2.20 mmol, 75% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.15 (t, J = 7.7 Hz, 1H), 6.96–6.91 (m, 2H), 4.41–4.31 (m, 1H), 3.45–3.29 (m, 1H), 3.16–2.95 (m, 4H), 2.77 (q, J = 7.6 Hz, 2H), 2.72–2.48 (m, 4H), 2.46–2.38 (m, 2H), 2.02–1.95 (m, 2H), 1.82–1.73 (m, 2H), 1.48–1.40 (m, 2H), 1.44 (s, 9H), 1.27–1.19 (m, 1H), 1.22 (t, J = 7.5 Hz, 3H), 1.13–0.99 (m, 4H).
tert-Butyl (trans-4-(2-(4-(2-fluoro-3-methoxyphenyl)piperazin-1-yl)ethyl)cyclohexyl)carbamate (4c).
General procedure B was followed using 3 (0.725 g, 3.00 mmol) and 1-(2-fluoro-3-methoxyphenyl)piperazine•HCl (0.816 g, 3.31 mmol) in DCE (25 mL). After work-up, the crude product was purified by chromatography (40 g of silica gel, 0–80% EtOAc/hexanes) to afford 4c (0.695 g, 1.60 mmol, 53% yield) as a light orange solid. Rf = 0.4 (80% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 6.95 (dt, J = 8.3, 1.9 Hz, 1H), 6.66–6.53 (m, 2H), 4.43–4.26 (m, 1H), 3.86 (s, 3H), 3.43–3.25 (m, 1H), 3.19–2.98 (m, 4H), 2.68–2.47 (m, 4H), 2.43–2.35 (m, 2H), 2.03–1.92 (m, 2H), 1.82–1.69 (m, 2H), 1.48–1.36 (m, 2H), 1.43 (s, 9H), 1.29–1.15 (m, 1H), 1.12–0.95 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 155.4, 148.7 (d, J = 10 Hz), 145.8 (d, J = 245 Hz),141.2 (d, J = 6 Hz), 123.6 (d, J = 5 Hz), 111.2 (d, J = 2 Hz), 107.2, 79.2, 56.8, 56.6, 53.6 (2C), 50.8 (d, J = 3 Hz, 2C), 50.0, 35.6, 34.0, 33.6 (2C), 32.1 (2C), 28.6 (3C); IR (neat) 3370, 1685 cm−1; mp 118–119 °C; HRMS (ESI) m/z [M + H]+ calcd for C24H39FN3O3 436.2970, found 436.2964.
tert-Butyl (trans-4-(2-(4-(6-(trifluoromethyl)pyridin-2-yl)piperazin-1-yl)ethyl)cyclohexyl)carbamate (4d).
General procedure B was followed using 3 (1.00 g, 4.16 mmol) and 1-(6-(trifluoromethyl)pyridin-2-yl)piperazine (1.06 g, 4.58 mmol) in DCE (28 mL). Note: 3 was added to a solution of the aryl piperazine. After work-up, the crude product was purified by chromatography (40 g of silica gel, 0–80% EtOAc/hexanes) to afford 4d (1.23 g, 2.69 mmol, 65% yield) as a white solid. Rf = 0.3 (80% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 7.55 (t, J = 8.0 Hz, 1H), 6.92 (d, J = 7.3 Hz, 1H), 6.75 (d, J = 8.7 Hz, 1H), 4.44–4.28 (m, 1H), 3.60 (t, J = 5.1 Hz, 4H), 3.43–3.27 (m, 1H), 2.51 (t, J = 5.1 Hz, 4H), 2.42–2.35 (m, 2H), 2.03–1.93 (m, 2H), 1.81–1.68 (m, 2H), 1.50–1.38 (m, 2H), 1.43 (s, 9H), 1.30–1.16 (m, 1H), 1.14–0.97 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 159.0, 155.4, 146.6 (q, J = 34 Hz), 138.3, 121.8 (q, J = 274 Hz), 109.5, 108.9 (q, J = 3 Hz), 79.2, 56.8, 53.1 (2C), 50.0, 44.9 (2C), 35.6, 34.0 (2C), 33.6, 32.1 (2C), 28.6 (3C); IR (neat) 3369, 1678 cm−1; mp 131–132 °C; HRMS (ESI) m/z [M + H]+ calcd for C23H36F3N4O2 457.2785, found 457.2777.
tert-Butyl (trans-4-(2-(4-(3-chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)ethyl)cyclohexyl)carbamate (4e).
General procedure B was followed using 3 (1.00 g, 4.16 mmol) and 1-(3-chloro-5-ethyl-2-methoxyphenyl)piperazine•HCl (1.33 g, 4.57 mmol) in DCE (27 mL). After work-up, the crude product was purified by chromatography (40 g of silica gel, 0–50% EtOAc/hexanes) to afford 4e (1.10 g, 2.29 mmol, 55% yield) as a yellow solid. Rf = 0.5 (50% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 6.84 (s, 1H), 6.61 (s, 1H), 4.44–4.26 (m, 1H), 3.83 (s, 3H), 3.44–3.26 (m, 1H), 3.23–3.02 (m, 4H), 2.68–2.49 (m, 6H), 2.45–2.35 (m, 2H), 2.03–1.94 (m, 2H), 1.81–1.71 (m, 2H), 1.50–1.37 (m, 2H), 1.43 (s, 9H), 1.30–1.15 (m, 4H), 1.14–0.97 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 155.1, 146.2, 146.0, 140.6, 128.0, 122.0, 116.5, 78.90, 58.8, 56.5, 53.7 (2C), 50.1 (2C), 49.7, 35.3, 33.7, 33.3 (2C), 31.8 (2C), 28.30, 28.27 (3C), 15.2; IR (neat) 3364, 1681 cm−1; mp 86–88 °C; HRMS (MALDI) m/z [M + H]+ calcd for C26H43ClN3O3 480.2987, found 480.2985. The oxalate salt was precipitated from a 0.02 M solution of the free base in 50% CHCl3/acetone using oxalic acid (1.25 equiv). mp 132–135 °C. Anal. calcd for C26H42ClN3O3•C2H2O4: C, 58.99; H, 7.78; N, 7.37. Found: C, 58.70; H, 7.81; N, 7.30.
trans-4-(2-(4-(2,3-Dichlorophenyl)piperazin-1-yl)ethyl)cyclohexan-1-amine (5a).
General procedure C was followed using 4a (1.33 g, 2.91 mmol) and TFA (9.0 mL, 0.12 mol) in DCM (20 mL). After work-up, 5a (1.00 g, 2.81 mmol, 96% yield) was isolated as a white solid. Rf = 0.1 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 7.17–7.09 (m, 2H), 6.95 (dd, J = 6.4, 3.1 Hz, 1H), 3.15–2.96 (m, 4H), 2.72–2.51 (m, 5H), 2.47–2.37 (m, 2H), 1.84 (d, J = 12.2 Hz, 2H), 1.76 (d, J = 12.3 Hz, 2H), 1.48–1.32 (m, 4H), 1.32–1.17 (m, 1H), 1.14–0.92 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 151.5, 134.2, 127.7, 127.6, 124.6, 118.7, 56.9, 53.6 (2C), 51.5 (2C), 50.9, 36.9 (2C), 35.8, 34.2, 32.3 (2C); IR (neat) 1578 cm−1; mp 72–74 °C; HRMS (MALDI) m/z [M + H]+ calcd for C18H28Cl2N3 356.1655, found 356.1660.
trans-4-(2-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)ethyl)cyclohexan-1-amine (5b).
General procedure C was followed using 4b (0.990 g, 2.20 mmol) and TFA (5.0 mL, 60 mmol) in DCM (15 mL). After work-up, 5b (0.775 g, 2,20 mmol, 100% yield) was isolated as a beige solid. 1H NMR (400 MHz, CDCl3) δ 7.15 (t, J = 7.8 Hz, 1H), 6.97–6.91 (m, 2H), 3.13–2.98 (m, 4H), 2.77 (q, J = 7.5 Hz, 2H), 2.72–2.54 (m, 5H), 2.47–2.40 (m, 2H), 1.88–1.81 (m, 2H), 1.80–1,73 (m, 2H), 1.48–1.40 (m, 2H), 1.28–1.18 (m, 1H), 1.22 (t, J = 7.5 Hz, 3H), 1.14–0.94 (m, 4H).
trans-4-(2-(4-(2-Fluoro-3-methoxyphenyl)piperazin-1-yl)ethyl)cyclohexan-1-amine (5c).
General procedure C was followed using 4c (0.251 g, 0.577 mmol) and TFA (1.8 mL, 24 mmol) in DCM (4.0 mL). After work-up, 5c (0.191 g, 0.569 mmol, 99% yield) was isolated as a tan solid. Rf = 0.1 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 6.95 (dt, J = 8.3, 1.9 Hz, 1H), 6.66–6.53 (m, 2H), 3.85 (s, 3H), 3.18–3.01 (m, 4H), 2.69–2.52 (m, 5H), 2.44–2.37 (m, 2H), 1.88–1.80 (m, 2H), 1.79–1.70 (m, 2H), 1.48–1.16 (m, 5H), 1.12–0.91 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 148.7 (d, J = 10 Hz), 145.8 (d, J = 245 Hz), 141.2 (d, J = 6 Hz), 123.6 (d, J = 5 Hz), 111.2 (d, J = 2 Hz), 107.1, 56.9, 56.6, 50.9, 50.8 (d, J = 3 Hz, 2C), 36.9, 35.8, 34.2, 32.3; IR (film) 1611, 1576 cm−1; mp 59–60 °C; HRMS (ESI) m/z [M + H]+ calcd for C19H31FN3O 336.2446, found 336.2442.
trans-4-(2-(4-(6-(Trifluoromethyl)pyridin-2-yl)piperazin-1-yl)ethyl)cyclohexan-1-amine (5d).
General procedure C was followed using 4d (0.301 g, 0.659 mmol) and TFA (2.0 mL, 26 mmol) in DCM (4.0 mL). After work-up, 5d (0.234 g, 0.656 mmol, 100% yield) was isolated as a clear, colorless oil. Rf = 0.1 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 7.55 (t, J = 8.0 Hz, 1H), 6.92 (d, J = 7.3 Hz, 1H), 6.75 (d, J = 8.7 Hz, 1H), 3.60 (t, J = 5.1 Hz, 4H), 2.63–2.55 (m, 1H), 2.52 (t, J = 5.1 Hz, 4H), 2.42–2.35 (m, 2H), 1.88–1.81 (m, 2H), 1.79–1.71 (m, 2H), 1.47–1.38 (m, 2H), 1.34–1.16 (m, 3H), 1.13–0.93 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 159.0, 146.6 (q, J = 34 Hz), 138.3, 121.8 (q, J = 274 Hz), 117.7, 108.9 (q, J = 3 Hz), 56.9, 53.1 (2C), 50.9, 44.9 (2C), 36.5 (2C), 35.7, 34.1, 32.2 (2C); IR (neat) 1604 cm−1; HRMS (ESI) m/z [M + H]+ calcd for C18H28F3N4 357.2261, found 357.2255.
trans-4-(2-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)ethyl)cyclohexan-1-amine (5e).
General procedure C was followed using 4e (1.10 g, 2.29 mmol) and TFA (7.0 mL, 91 mmol) in DCM (16 mL). After work-up, 5e (0.87 g, 2.3 mmol, 100% yield) was isolated as a clear, orange oil. Rf = 0.1 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 6.83 (d, J = 1.9 Hz, 1H), 6.61 (d, J = 2.0 Hz, 1H), 3.83 (s, 3H), 3.20–3.04 (m, 4H), 2.65–2.49 (m, 6H), 2.44–2.37 (m, 2H), 1.84 (d, J = 11.9 Hz, 2H), 1.76 (d, J = 12.0 Hz, 2H), 1.47–1.33 (m, 4H), 1.30–1.15 (m, 5H), 1.13–0.93 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 146.5, 146.3, 140.9, 128.3, 122.3, 116.8, 59.1, 57.0, 54.0 (2C), 50.9, 50.4 (2C), 36.9 (2C), 35.8, 34.2, 32.3 (2C), 28.6, 15.6; IR (neat) 1593, 1565 cm−1; HRMS (MALDI) m/z [M + H]+ calcd for C21H35ClN3O 380.2463, found 380.2467.
3-(trans-4-(2-(4-(2,3-Dichlorophenyl)piperazin-1-yl)ethyl)cyclohexyl)-1,1-dimethylurea (cariprazine).
General procedure D was followed using 5a (1.00 g, 2.81 mmol) and N,N-dimethylcarbamyl chloride (0.33 mL, 3.6 mmol) in DCM (28 mL). After work-up, the crude product was purified by chromatography (40 g of silica gel, 0–60% EtOAc/hexanes) to afford cariprazine (1.02 g, 2.39 mmol, 85% yield) as a white solid. Rf = 0.5 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 7.17–7.10 (m, 2H), 6.95 (dd, J = 6.3, 3.2 Hz, 1H), 4.11 (d, J = 7.6 Hz, 1H), 3.64–3.52 (m, 1H), 3.15–2.98 (m, 4H), 2.88 (s, 6H), 2.71–2.53 (m, 4H), 2.47–2.38 (m, 2H), 2.07–1.96 (m, 2H), 1.83–1.70 (m, 2H), 1.49–1.39 (m, 2H), 1.30–1.18 (m, 1H), 1.16–1.01 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 158.0, 151.5, 134.2, 127.7, 127.6, 124.6, 118.7, 56.8, 53.6 (2C), 51.5 (2C), 50.0, 36.3 (2C), 35.8, 34.2 (2C), 34.1, 32.3 (2C); IR (neat) 3338, 1622 cm−1; mp 212–213 °C (dec); HRMS (MALDI) m/z [M + H]+ calcd for C21H33Cl2N4O 427.2026, found 427.2023; tR = 32.1 min (HPLC, basic). The HCl salt was precipitated from 0.03 M solution of the free base in 50% CHCl3/acetone using a 2.0 M solution of HCl in Et2O (5.0 equiv). Dec >200 °C. Anal. calcd for C21H32Cl2N4O•2HCl: C, 50.41; H, 6.85; N, 11.20. Found: C, 50.43; H, 6.84; N, 11.01.
3-(trans-4-(2-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)ethyl)cyclohexyl)-1,1-dimethylurea (6b).
General procedure D was followed using 5b (0.500 g, 1.43 mmol) and N,N-dimethylcarbamyl chloride (0.16 mL, 1.8 mmol) in DCM (20 mL). After work-up, the crude product was purified by chromatography (silica gel, 0–10% MeOH/CHCl3) to afford 6b (0.414 g, 0.983 mmol, 69% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.15 (t, J = 7.7 Hz, 1H), 6.97–6.89 (m, 2H), 4.11 (d, J = 7.6 Hz, 1H), 3.63–3.53 (m, 1H), 3.14–2.98 (m, 4H), 2.88 (s, 6H), 2.77 (q, J = 7.6 Hz, 2H), 2.72–2.54 (m, 4H), 2.46–2.38 (m, 2H), 2.07–1.97 (m, 2H), 1.83–1.72 (m, 2H), 1.48–1.40 (m, 2H), 1.30–1.18 (m, 1H), 1.22 (t, J = 7.5 Hz, 3H), 1.15–1.01 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 158.0, 149.8, 143.4, 128.8, 127.0, 124.1, 118.1, 56.9, 53.7 (2C), 51.7 (2C), 50.0, 36.3 (2C), 35.8, 34.2 (2C), 34.1, 32.3, 27.6, 14.2; tR = 20.3 min (HPLC, acidic). The HCl salt was precipitated from 50% acetone/CHCl3 using a 2.0 M solution of HCl in Et2O. Dec >200 °C. Anal. calcd for C23H37ClN4O•HCl•H2O: C, 58.10; H, 8.48; N, 11.78. Found: C, 58.32; H, 8.24; N, 11.56.
3-(trans-4-(2-(4-(2-Fluoro-3-methoxyphenyl)piperazin-1-yl)ethyl)cyclohexyl)-1,1-dimethylurea (6c).
General procedure D was followed using 5c (0.189 g, 0.565 mmol) and N,N-dimethylcarbamyl chloride (66 μL, 0.72 mmol) in DCM (5.6 mL). After work-up, the crude product was purified by chromatography (12 g of silica gel, 0–10% MeOH/DCM) to afford 6c (0.179 g, 0.440 mmol, 78% yield) as a white solid. Rf = 0.5 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 6.95 (dt, J = 8.3, 1.8 Hz, 1H), 6.66–6.52 (m, 2H), 4.12 (d, J = 7.6 Hz, 1H), 3.85 (s, 3H), 3.63–3.50 (m, 1H), 3.18–3.01 (m, 4H), 2.87 (s, 6H), 2.68–2.50 (m, 4H), 2.44–2.35 (m, 2H), 2.05–1.94 (m, 2H), 1.82–1.70 (m, 2H), 1.47–1.38 (m, 2H), 1.30–1.16 (m, 1H), 1.14–0.99 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 157.9, 148.7 (d, J = 10 Hz), 145.8 (d, J = 245 Hz), 141.2 (d, J = 6 Hz), 123.6 (d, J = 5 Hz), 111.2 (d, J = 2 Hz), 107.1, 56.8, 56.6, 53.6 (2C), 50.8 (d, J = 3 Hz, 2C), 50.0, 36.3 (2C), 35.8, 34.2 (2C), 34.1, 32.2 (2C); IR (film) 3342, 1626 cm−1; mp 159–160 °C (dec); HRMS (ESI) m/z [M + H]+ calcd for C22H36FN4O2 407.2817, found 407.2811; tR = 22.2 min (HPLC, basic). The HCl salt was precipitated from a 0.03 M solution of the free base in 50% CHCl3/acetone using a 2.0 M solution of HCl in Et2O (5.0 equiv). mp 175–177 °C (dec). Anal. calcd for C22H35FN4O2•2HCl•1.25H2O: C, 52.64; H, 7.93; N, 11.16. Found: C, 52.50; H, 7.79; N, 11.01.
1,1-Dimethyl-3-(trans-4-(2-(4-(6-(trifluoromethyl)pyridine-2-yl)piperazin-1-yl)ethyl)cyclohexyl)urea (6d).
General procedure D was followed using 5d (0.232 g, 0.651 mmol) and N,N-dimethylcarbamyl chloride (80 μL, 0.81 mmol) in DCM (9.5 mL). After work-up, the crude product was purified by chromatography (silica gel, 0–10% MeOH/CHCl3) to afford 6d (0.107 g, 0.249 mmol, 38% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.55 (t, J = 8.2 Hz, 1H), 6.92 (d, J = 7.7 Hz, 1H), 6.75 (d, J = 8.5 Hz, 1H), 4.11 (d, J = 7.7 Hz, 1H), 3.65–3.52 (m, 5H), 2.88 (s, 6H), 2.57–2.46 (m, 4H), 2.43–2.35 (m, 2H), 2.05–1.96 (m, 2H), 1.83–1.72 (m, 2H), 1.47–1.39 (m, 2H), 1.32–1.18 (m, 1H), 1.16–1.00 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 159.0, 158.0, 146.6 (q, J = 34 Hz), 138.3, 121.8 (q, J = 274 Hz), 109.5, 108.9 (q, J = 3 Hz), 56.8, 53.1 (2C), 50.0, 44.9 (2C), 36.3 (2C), 35.8, 34.2 (2C), 34.0, 32.2 (2C); HRMS (MALDI) m/z [M + H]+ calcd for C21H33F3N5O 428.2632, found 428.2629;; tR = 18.5 min (HPLC, acidic).
3-(trans-4-(2-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)ethyl)cyclohexyl)-1,1-dimethylurea (6e).
General procedure D was followed using 5e (0.275 g, 0.723 mmol) and N,N-dimethylcarbamyl chloride (82 μL, 0.90 mmol) in DCM (10 mL). After work-up, the crude product was purified by chromatography (silica gel, 0–10% MeOH/CHCl3) to afford 6e (0.215 g, 0.477 mmol, 66% yield) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 6.87 (d, J = 1.9 Hz, 1H), 6.63 (d, J = 2.0 Hz, 1H), 4.13 (d, J = 7.6 Hz, 1H), 3.82 (s, 3H), 3.64–3.49, (m, 1H), 3.46–3.21 (m, 4H), 3.06–2.77 (m, 4H), 2.87 (s, 6H), 2.76–2.60 (m, 2H), 2.54 (q, J = 7.6 Hz, 2H), 2.08–1.97 (m, 2H), 1.83–1.72 (m, 2H), 1.67–1.54 (m, 2H), 1.39–1.25 (m, 1H), 1.19 (t, J = 7.6 Hz, 3H), 1.16–1.02 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 158.0, 146.6, 146.4, 140.9, 128.4, 122.3, 116.8, 66.0, 59.1, 56.9, 54.0 (2C), 50.4 (2C), 50.0, 36.3 (2C), 35.8, 34.2 (2C), 32.2 (2C), 28.6, 15.4. The HCl salt was precipitated from 50% CHCl3/acetone using a 2.0 M solution of HCl in Et2O. Dec >189 °C. Anal. calcd for C24H39ClN4O2•2HCl: C, 55.02; H, 7.89; N, 10.69. Found: C, 54.96; H, 7.88; N, 10.54.
1-(trans-4-(2-(4-(2,3-Dichlorophenyl)piperazin-1-yl)ethyl)cyclohexyl)-3-methylurea (7a).
General procedure D was followed using 5a (0.250 g, 0.702 mmol) and N-methylcarbamyl chloride (83.0 mg, 0.456 mmol) in DCM (10 mL). After work-up, the crude product was purified by chromatography (silica gel, 0–10% MeOH/CHCl3) to afford 7a (0.141 g, 0.341 mmol, 49% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.19–7.13 (m, 2H), 6.97 (dd, J = 5.7, 3.9 Hz, 1H), 5.11–5.05 (m, 1H), 5.00 (d, J = 8.0 Hz, 1H), 3.53–3.40 (m, 1H), 3.18–2.94 (m, 4H), 2.72 (d, J = 4.7 Hz, 3H), 2.68–2.55 (m, 4H), 2.47–2.38 (m, 2H), 2.04–1.92 (m, 2H), 1.82–1.70 (m, 2H), 1.49–1.39 (m, 2H), 1.30–1.19 (m, 1H), 1.14–1.00 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 158.6, 151.1, 133.6, 127.3, 127.1, 124.3, 118.5, 56.4, 53.1 (2C), 51.1 (2C), 48.9, 35.3, 33.7 (3C), 31.9 (2C), 26.5. The HCl salt was precipitated from a 0.02 M solution of the free base in 50% CHCl3/acetone using a 2.0 M solution of HCl in Et2O (35 equiv). Dec >175 °C. Anal. calcd for C20H30Cl2N4O•HCl•1.25H2O: C, 50.85; H, 7.15; N, 11.86. Found: C, 50.86; H, 6.86; N, 11.60.
1-(trans-4-(2-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)ethyl)cyclohexyl)-3-methylurea (7b).
General procedure D was followed using 5b (0.172 g, 0.491 mmol) and N-methylcarbamyl chloride (0.139 g, 1.47 mmol) in DCM (15 mL). After work-up, the crude product was purified by chromatography (silica gel, 0–10% MeOH/CHCl3) to afford 7b (0.150 g, 0.368 mmol, 75% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.15 (t, J = 7.7 Hz, 1H), 6.96–6.90 (m, 2H), 4.28–4.21 (m, 1H), 4.15 (d, J = 8.0 Hz, 1H), 3.51–3.39 (m, 1H), 3.16–2.94 (m, 4H), 2.81–2.73 (m, 5H), 2.72–2.52 (m, 4H), 2.46–2.36 (m, 2H), 2.06–1.94 (m, 2H), 1.85–1.69 (m, 2H), 1.49–1.41 (m, 2H), 1.22 (t, J = 7.5 Hz, 3H), 1.31–1.17 (m, 1H), 1.15–1.01 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 158.3, 149.8, 143.4, 128.8, 127.0, 124.1, 118.1, 56.8, 53.7 (2C), 51.7 (2C), 49.8, 35.7, 34.09, 34.06 (2C), 32.2 (2C), 27.6, 27.4, 14.2. The HCl salt was precipitated from a 0.03 M solution of the free base in 50% CHCl3/acetone using a 2.0 M solution of HCl in Et2O (29 equiv). Dec >199 °C. Anal. calcd for C22H35ClN4O•HCl•1.25H2O: C, 56.71; H, 8.33; N, 12.02. Found: C, 56.54; H, 7.92; N, 11.94.
1-(trans-4-(2-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)ethyl)cyclohexyl)-3-methylurea (7e).
General procedure D was followed using 5e (0.420 g, 1.11 mmol) and N-methylcarbamyl chloride (0.130 g, 1.38 mmol) in DCM (15 mL). After work-up, the crude product was purified by chromatography (silica gel, 0–10% MeOH/CHCl3) to afford 7e (0.416 g, 0.952 mmol, 86% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 6.84 (d, J = 2.0 Hz, 1H), 6.61 (d, J = 2.0 Hz, 1H), 4.16 (d, J = 5.1 Hz, 1H), 4.08 (d, J = 8.0 Hz, 1H), 3.83 (s, 3H), 3.52–3.40 (m, 1H), 3.22–3.05 (m, 4H), 2.77 (d, J = 4.9 Hz, 3H), 2.66–2.49 (m, 6H), 2.44–2.36 (m, 2H), 2.06–1.95 (m, 2H), 1.84–1.73 (m, 2H), 1.48–1.40 (m, 2H), 1.31–1.23 (m, 1H), 1.20 (t, J = 7.6 Hz, 3H), 1.15–1.01 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 158.3, 146.5, 146.3, 141.0, 128.4, 122.3, 116.8, 59.2, 56.9, 54.0 (2C), 50.4 (2C), 49.9, 35.7, 34.1 (2C), 34.0, 32.2 (2C), 28.6, 27.4, 15.6. The HCl salt was precipitated from a 0.04 M solution of the free base in 50% CHCl3/acetone using a 2.0 M solution of HCl in Et2O (27 equiv). Dec >220 °C. Anal. calcd for C23H37ClN4O2•2HCl•1.25H2O: C, 51.88; H, 7.86; N, 10.52. Found: C, 51.89; H, 7.60; N, 10.36.
1-(trans-4-(2-(4-(2,3-Dichlorophenyl)piperazin-1-yl)ethyl)cyclohexyl)urea (8a).
To a solution of 5a (0.108 g, 0.303 mmol) and KOCN (0.256 g, 3.16 mmol) in H2O (1.5 mL) and THF (3.0 mL) was added a 1.0 M aq solution of HCl (1.5 mL, 1.5 mmol) over 3 min. After stirring overnight, the reaction was quenched with a saturated aq solution of NaHCO3 (20 mL), and the aq layer was extracted with EtOAc (2 × 20 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated. The crude product was purified by chromatography (12 g of silica gel, 0–10% MeOH/DCM) to afford 8a (71.3 mg, 0.179 mmol, 59% yield) as a white solid. Rf = 0.3 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 7.18–7.10 (m, 2H), 6.95 (dd, J = 6.5, 3.1 Hz, 1H), 4.38–4.23 (m, 3H), 3.50–3.37 (m, 1H), 3.16–2.97 (m, 4H), 2.72–2.52 (m, 4H), 2.42 (t, J = 8.0 Hz, 2H), 2.08–1.96 (m, 2H), 1.85–1.64 (m, 3H), 1.51–1.39 (m, 2H), 1.17–0.99 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 157.8, 151.5, 134.2, 127.7, 127.6, 124.7, 118.7, 56.7, 53.6 (2C), 51.5 (2C), 50.2, 35.6, 34.1, 33.8 (2C), 32.1 (2C); IR (neat) 3477, 3329, 1645 cm−1; mp 212–213 °C (dec); HRMS (MALDI) m/z [M + H]+ calcd for C19H29Cl2N4O 399.1713, found 399.1711; tR = 24.9 min (HPLC, basic).
1-(trans-4-(2-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)ethyl)cyclohexyl)urea (8e).
The same procedure as the one described for 8a was followed starting from 5e (0.203 g, 0.534 mmol). After work-up, the crude product was purified by chromatography (12 g of silica gel, 0–10% MeOH/DCM) to afford 8e (93.0 mg, 0.220 mmol, 41% yield) as a tan solid. Rf = 0.3 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 6.83 (d, J = 2.0 Hz, 1H), 6.61 (d, J = 2.0 Hz, 1H), 4.58–4.33 (m, 3H), 3.83 (s, 3H), 3.49–3.34 (m, 1H), 3.26–2.99 (m, 4H), 2.68–2.48 (m, 6H), 2.45–2.34 (m, 2H), 2.06–1.94 (m, 2H), 1.85–1.70 (m, 2H), 1.48–1.38 (m, 2H), 1.30–1.22 (m, 1H), 1.19 (t, J = 7.6 Hz, 3H), 1.16–1.00 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 158.2, 146.5, 146.3, 140.9, 128.3, 122.3, 116.8, 59.1, 56.8, 54.0 (2C), 50.4 (2C), 50.0, 35.6, 34.0, 33.8 (2C), 32.1 (2C), 28.6, 15.6; IR (neat) 3492, 3348, 1647 cm−1; mp 163–166 °C (dec); HRMS (MALDI) m/z [M + H]+ calcd for C22H36ClN4O2 423.2521, found 423.2518; tR = 32.0 min (HPLC, basic).
N-(trans-4-(2-(4-(2,3-Dichlorophenyl)piperazin-1-yl)ethyl)cyclohexyl)-7,8-dihydro-1,6-naphthyridine-6(5H)-carboxamide (9a).
Using oven-dried glassware under an Ar atmosphere, a solution of NEt3 (0.21 mL, 1.5 mmol) in anhydrous THF (3.5 mL) was cooled to 0 °C, and a 15 wt% solution of phosgene in toluene (0.40 mL, 0.56 mmol) was added over 3 min. Next, a solution of 5a (0.179 g, 0.502 mmol) in anhydrous THF (3.5 mL) was added over 3 min, and the reaction was stirred for 1 h. Then, a solution of 5,6,7,8-tetrahydro-1,6-naphthyridine (80.5 mg, 0.600 mmol) in anhydrous THF (3.5 mL) was added over 3 min, and the reaction was allowed to warm to rt. After stirring for 19 h, the reaction was quenched with a saturated aq solution of NaHCO3 (40 mL), and the aq layer was extracted with EtOAc (40 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated. The crude product was purified by chromatography (12 g of silica gel, 0–10% MeOH/DCM) to afford 9a (35.1 mg, 68.0 μmol, 14% yield) as a white solid. Rf = 0.4 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 8.42 (d, J = 4.2 Hz, 1H), 7.42 (d, J = 7.6 Hz, 1H), 7.17–7.10 (m, 3H), 6.95 (dd, J = 6.4, 3.2 Hz, 1H), 4.57 (s, 2H), 4.32 (d, J = 7.6 Hz, 1H), 3.71–3.57 (m, 3H), 3.14–2.96 (m, 6H), 2.76–2.52 (m, 4H), 2.47–2.39 (m, 2H), 2.08–2.00 (m, 2H), 1.83–1.75 (m, 2H), 1.48–1.39 (m, 2H), 1.30–1.20 (m, 1H), 1.18–1.03 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 156.9, 155.0, 151.4, 148.0, 134.2, 134.1, 129.1, 127.62, 127.56, 124.6, 121.7, 118.7, 56.8, 53.5 (2C), 51.5 (2C), 50.1, 45.0, 41.6, 35.8, 34.08 (2C), 34.05, 32.2 (3C); IR (neat) 3326, 1620 cm−1; mp 163–164 °C (dec); HRMS (MALDI) m/z [M + H]+ calcd for C27H36Cl2N5O 516.2291, found 516.2283; tR = 33.8 min (HPLC, basic).
N-(trans-4-(2-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)ethyl)cyclohexyl)-7,8-dihydro-1,6-naphthyridine-6(5H)-carboxamide (9b).
The same procedure as the one described for 9a was followed starting from 5b (0.205 g, 0.586 mmol). After work-up, the crude product was purified by chromatography (12 g of silica gel, 0–10% MeOH/DCM) to afford 9b (0.192 g, 0.376 mmol, 64% yield) as a white solid. Rf = 0.4 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 8.42 (d, J = 4.7 Hz, 1H), 7.43 (d, J = 7.7 Hz, 1H), 7.18–7.10 (m, 2H), 6.93 (t, J = 7.2 Hz, 2H), 4.57 (s, 2H), 4.32 (d, J = 7.5 Hz, 1H), 3.71–3.59 (m, 3H), 3.12–2.98 (m, 6H), 2.77 (q, J = 7.5 Hz, 2H), 2.71–2.54 (m, 4H), 2.47–2.39 (m, 2H), 2.10–2.01 (m, 2H), 1.84–1.74 (m, 2H), 1.50–1.40 (m, 2H), 1.30–1.18 (m, 1H), 1.22 (t, J = 7.5 Hz, 3H), 1.15–1.04 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 156.9, 155.1, 149.8, 148.0, 143.4, 134.2, 129.1, 128.8, 127.0, 124.1, 121.7, 118.1, 56.9, 53.7 (2C), 51.7 (2C), 50.2, 45.0, 41.6, 35.8, 34.1 (3C), 32.2 (3C), 27.6, 14.4; IR (neat) 3323, 1619 cm−1; mp 58–60 °C; HRMS (MALDI) m/z [M + H]+ calcd for C29H41ClN5O 510.2994, found 510.2988; tR = 35.2 min (HPLC, basic).
N-(trans-4-(2-(4-(2,3-Dichlorophenyl)piperazin-1-yl)ethyl)cyclohexyl)-1H-indole-2-carboxamide (10a).
General procedure E was followed using 5a (0.101 g, 0.283 mmol) and 1H-indole-2-carboxylic acid (62.0 mg, 0.385 mmol) in CHCl3 (8.0 mL). After work-up, the crude product was product was purified by chromatography (12 g of silica gel, 0–10% MeOH/DCM) to afford 10a (20.2 mg, 40.2 μmol, 14% yield) as a white solid. Rf = 0.5 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 9.22 (s, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.43 (d, J = 8.3 Hz, 1H), 7.32–7.28 (m, 1H), 7.19–7.10 (m, 3H), 6.97 (dd, J = 6.5, 3.1 Hz, 1H), 6.80 (s, 1H), 5.95 (d, J = 8.2 Hz, 1H), 4.03–3.89 (m, 1H), 3.17–2.00 (m, 4H), 2.76–2.55 (m, 4H), 2.47 (t, J = 7.9 Hz, 2H), 2.13 (d, J = 11.8 Hz, 2H), 1.86 (d, J = 12.4 Hz, 2H), 1.54–1.42 (m, 2H), 1.37–1.10 (m, 5H); 13C NMR (101 MHz, CDCl3) δ 160.8, 151.4, 136.2, 134.2, 131.2, 127.9, 127.7, 127.6, 124.7, 124.6, 122.0, 120.8, 118.7, 112.0, 101.6, 56.7, 53.6 (2C), 51.5 (2C), 49.1, 35.6, 34.0, 33.4 (2C), 32.1 (2C); IR (neat) 3616, 3268, 1624 cm−1; mp 240–241 °C (dec); HRMS (MALDI) m/z [M + H]+ calcd for C27H33Cl2N4O 499.2026, found 499.2025; tR = 37.8 min (HPLC, basic).
N-(trans-4-(2-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)ethyl)cyclohexyl)indole-2-carboxamide (10b).
General procedure E was followed using 5b (0.100 g, 0.286 mmol) and 1H-indole-2-carboxylic acid (58.0 mg, 0.360 mmol) in CHCl3 (10 mL). After work-up, the crude product was purified by chromatography (silica gel, 0–50% EtOAc/hexanes) to afford 10b (70 mg, 0.142 mmol, 50% yield) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 9.16 (s, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.31–7.26 (m, 1H), 7.18–7.11 (m, 2H), 6.97–6.92 (m, 2H), 6.80 (dd, J = 2.1, 0.9 Hz, 1H), 5.94 (d, J = 8.2 Hz, 1H), 4.00–3.89 (m, 1H), 3.16–2.98 (m, 4H), 2.78 (q, J = 7.5 Hz, 2H), 2.73–2.55 (m, 4H), 2.50–2.41 (m, 2H), 2.17–2.08 (m, 2H), 1.90–1.82 (m, 2H), 1.53–1.45 (m, 2H), 1.38–1.10 (m, 7H); 13C NMR (101 MHz, CDCl3) δ 160.8, 149.8, 143.4, 136.2, 131.2, 128.9, 127.9, 127.0, 124.6, 124.1, 122.0, 120.8, 118.2, 112.0, 101.6, 56.8, 53.8 (2C), 51.8 (2C), 49.1, 35.7, 34.1, 33.4 (2C), 32.1 (2C), 27.6, 14.3. The HCl salt was precipitated from 50% CHCl3/acetone using a 2.0 M solution of HCl in Et2O. Anal. calcd for C29H37ClN4O•HCl•0.75H2O: C, 64.14; H, 7.33; N, 10.32. Found: C, 64.11; H, 7.09; N, 10.04.
N-(trans-4-(2-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)ethyl)cyclohexyl)indole-2-carboxamide (10e).
General procedure E was followed using 5e (0.125 g, 0.329 mmol) and 1H-indole-2-carboxylic acid (58.3 mg, 0.362 mmol) in CHCl3 (8.0 mL). After work-up, the crude product was purified by chromatography (silica gel, 0–10% MeOH/CHCl3) to afford 10e (0.133 g, 0.254 mmol, 77% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 9.33 (s, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.44 (d, J = 8.3 Hz, 1H), 7.33–7.24 (m, 1H), 7.14 (t, J = 7.5 Hz, 1H), 6.85 (s, 1H), 6.81 (s, 1H), 6.62 (s, 1H), 5.97 (d, J = 8.2 Hz, 1H), 4.02–3.90 (m, 1H), 3.85 (s, 3H), 3.25–3.06 (m, 4H), 2.69–2.51 (m, 6H), 2.47–2.40 (m, 2H), 2.17–2.08 (m, 2H), 1.90–1.81 (m, 2H), 1.53–1.44 (m, 2H), 1.39–1.09 (m, 8H). The HCl salt was precipitated from a 0.06 M solution of the free base in 50% CHCl3/acetone using a 2.0 M solution of HCl in Et2O (20 equiv). Dec >220 °C. Anal. calcd for C30H39ClN4O2•2HCl•0.25H2O: C, 60.00; H, 6.97; N, 9.33. Found: C, 60.16; H, 7.10; N, 9.12.
N-(trans-4-(2-(4-(2,3-Dichlorophenyl)piperazin-1-yl)ethyl)cyclohexyl)quinoline-4-carboxamide (11a).
General procedure E was followed using 5a (0.221 g, 0.620 mmol) and quinoline-4-carboxylic acid (0.118 g, 0.680 mmol) in CHCl3 (15 mL). Note: The reaction mixture was washed with a 1.0 M aq solution of NaOH. After work-up, the crude product was product was purified by chromatography (24 g of silica gel, 0–10% MeOH/DCM) to afford 11a (0.231 g, 0.451 mmol, 73% yield) as a white solid. Rf = 0.5 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 8.89 (d, J = 4.3 Hz, 1H), 8.18 (d, J = 8.4 Hz, 1H), 8.12 (d, J = 8.4 Hz, 1H), 7.75 (ddd, J = 8.4, 6.8, 1.2 Hz, 1H), 7.60 (ddd, J = 8.3, 6.9, 1.2 Hz, 1H), 7.38 (d, J = 4.3 Hz, 1H), 7.17–7.11 (m, 2H), 6.96 (dd, J = 6.4, 3.1 Hz, 1H), 5.96 (d, J = 8.2 Hz, 1H), 4.10–3.98 (m, 1H), 3.18–2.94 (m, 4H), 2.75–2.51 (m, 4H), 2.50–2.38 (m, 2H), 2.24–2.12 (m, 2H), 1.92–1.81 (m, 2H), 1.54–1.43 (m, 2H), 1.37–1.11 (m, 5H); 13C NMR (101 MHz, CDCl3) δ 166.7, 151.4, 150.0, 148.8, 142.5, 134.2, 130.1, 130.0, 127.8, 127.64, 127.57, 125.3, 124.7, 124.6, 118.7, 118.4, 56.7, 53.6 (2C), 51.5 (2C), 49.6, 35.6, 34.0, 33.2 (2C), 32.0 (2C); IR (neat) 3274, 1633 cm−1; mp 222–223 °C (dec); HRMS (ESI) m/z [M + H]+ calcd for C28H33Cl2N4O 511.2026, found 511.2024; tR = 36.0 min (HPLC, basic). The HCl salt was precipitated from 1 0.03 M solution of the free base in 50% CHCl3/acetone using a 2.0 M solution of HCl in Et2O (14 equiv). Dec >270 °C. Anal. calcd for C28H32Cl2N4O•2HCl•0.5H2O: C, 56.67; H, 5.95; N, 9.44. Found: C, 56.68; H, 5.81; N, 9.34.
N-(trans-4-(2-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)ethyl)cyclohexyl)quinoline-4-carboxamide (11b).
General procedure E was followed using 5b (0.125 g, 0.358 mmol) and quinoline-4-carboxylic acid (68.0 mg, 0.393 mmol) in CHCl3 (9.0 mL). After work-up, the crude product was purified by chromatography (silica gel, 0–8% MeOH/CHCl3) to afford 11b (0.185 g, mmol, ~100% yield) as a white solid, which was purified further by HCl salt formation. 1H NMR (400 MHz, CDCl3) δ 8.90 (d, J = 4.3, 1H), 8.19 (d, J = 8.5 Hz, 1H), 8.13 (d, J = 8.5 Hz, 1H), 7.75 (t, J = 7.7 Hz, 1H), 7.60 (t, J = 7.7 Hz, 1H), 7.39 (d, J = 4.3, 1H), 7.18–1.7.12 (m, 1H), 6.94 (t, J = 7.6 Hz, 2H), 5.94 (d, J = 8.3 Hz, 1H), 4.11–3.98 (m, 1H), 3.19–2.95 (m, 4H), 2.77 (q, J = 7.3 Hz, 2H), 2.72–2.51 (m, 4H), 2.50–2.41 (m, 2H), 2.23–2.15 (m, 2H), 1.91–1.83 (m, 2H), 1.54–1.45 (m, 2H), 1.41–1.11 (m, 8H); 13C NMR (101 MHz, CDCl3) δ 166.7, 150.0, 149.8, 148.8, 143.4, 142.5, 130.2, 130.1, 128.8, 127.8, 127.0, 125.3, 124.6, 124.1, 118.5, 118.1, 56.8, 53.7 (2C), 51.7 (2C), 49.6, 35.6, 34.0, 33.2 (2C), 32.0 (2C), 27.6, 14.3. The HCl salt was precipitated from 50% CHCl3/acetone using a 2.0 M solution of HCl in Et2O. Dec >250 °C. Anal. calcd for C30H37ClN4O•2HCl•2H2O: C, 58.68; H, 7.06; N, 9.12. Found: C, 58.74; H, 6.97; N, 8.94.
N-(trans-4-(2-(4-(6-(Trifluoromethyl)pyridin-2-yl)piperazin-1-yl)ethyl)cyclohexyl)quinoline-4-carboxamide (11d).
General procedure E was followed using 5d (0.234 g, 0.656 mmol) and quinoline-4-carboxylic acid (0.125 g, 0.720 mmol) in CHCl3 (16 mL). Note: The reaction mixture was washed with a 1.0 M aq solution of NaOH. After work-up, the crude product was product was purified by chromatography (24 g of silica gel, 0–10% MeOH/DCM) to afford 11d (0.234 g, 0.458 mmol, 70% yield) as a white solid. Rf = 0.2 (5% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 8.88 (d, J = 4.3 Hz, 1H), 8.18 (d, J = 8.5 Hz, 1H), 8.11 (d, J = 8.4 Hz, 1H), 7.74 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 7.63–7.52 (m, 2H), 7.37 (d, J = 4.3 Hz, 1H), 6.93 (d, J = 7.3 Hz, 1H), 6.76 (d, J = 8.7 Hz, 1H), 5.98 (d, J = 8.2 Hz, 1H), 4.10–3.97 (m, 1H), 3.61–3.53 (m, 4H), 2.60–2.47 (m, 4H), 2.46–2.35 (m, 2H), 2.23–2.14 (m, 2H), 1.91–1.82 (m, 2H), 1.53–1.43 (m, 2H), 1.38–1.10 (m, 5H); 13C NMR (101 MHz, CDCl3) δ 166.7, 159.0, 150.0, 148.8, 146.5 (q, J = 34 Hz), 142.5, 138.4, 130.1, 130.0, 127.8, 125.3, 124.6, 121.8 (q, J = 274 Hz), 118.4, 109.5, 109.0 (q, J = 3 Hz), 56.7, 53.1 (2C), 49.6, 44.9 (2C), 35.5, 33.9, 33.2 (2C), 32.0 (2C); IR (neat) 3328, 1635 cm−1; mp 187–188 °C (dec); HRMS (ESI) m/z [M + H]+ calcd for C28H33F3N5O 512.2632, found 512.2627; tR = 34.3 min (HPLC, basic). The HCl salt was precipitated from a 0.03 M solution of the free base in 50% CHCl3/acetone using a 2.0 M solution of HCl in Et2O (14 equiv). mp 292–294 °C (dec). Anal. calcd for C28H32F3N5O•2HCl•0.25H2O: C, 57.10; H, 5.90; N, 11.89. Found: C, 57.06; H, 5.93; N, 11.78.
N-trans-4-(2-(4-(2,3-Dichlorophenyl)piperazin-1-yl)ethyl)cyclohexyl)-4-(pyridin-3-yl)benzamide (12a).
General procedure E was followed using 5a (0.101 g, 0.284 mmol) and 4-(pyridin-3-yl)benzoic acid•HCl (73.1 mg, 0.310 mmol) in CHCl3 (8.0 mL). After work-up, the crude product was product was purified by chromatography (12 g of silica gel, 0–10% MeOH/DCM) to afford 12a (0.120 g, 0.224 mmol, 79% yield) as a white solid. Rf = 0.5 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 8.88–8.84 (m, 1H), 8.63 (d, J = 4.9 Hz, 1H), 7.92–7.83 (m, 3H), 7.67–7.61 (m, 2H), 7.39 (dd, J = 7.9, 4.8 Hz, 1H), 7.18–7.10 (m, 1H), 6.96 (dd, J = 6.3, 3.2 Hz, 1H), 5.98 (d, J = 8.1 Hz, 1H), 4.02–3.90 (m, 1H), 3.16–2.97 (m, 4H), 2.74–2.53 (m, 4H), 2.49–2.40 (m, 2H), 4.02–3.90 (m, 2H), 2.18–2.09 (m, 2H), 1.89–1.80 (m, 2H), 1.54–1.42 (m, 2H), 1.37–1.00 (m, 5H); 13C NMR (101 MHz, CDCl3) δ 166.3, 151.5, 149.3, 148.5, 140.9, 135.7, 134.7, 134.5, 134.5, 134.2, 127.8 (2C), 127.7, 127.6, 127.4 (2C), 123.8, 118.7, 56.7, 53.6 (2C), 51.5 (2C), 49.4, 35.7, 34.1 (2C), 33.3, 32.1 (2C); IR (neat) 3269, 1629 cm−1; mp 257–258 °C (dec); HRMS (MALDI) m/z [M + H]+ calcd for C30H35Cl2N4O 537.2182, found 537.2185; tR = 36.9 min (HPLC, basic).
N-(trans-4-(2-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)ethyl)cyclohexyl)-4-(yridine-3-yl)benzamide (12b).
General procedure E was followed using 5b (101 mg, 0.289 mmol) and 4-(pyridine-3-yl)benzoic acid•HCl (74.8 mg, 0.317 mmol) in CHCl3 (8.0 mL). After work-up, the crude product was purified by chromatography (silica gel, 0–10% MeOH/CHCl3) to afford 12b (0.150 g, 0.282 mmol, 98% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.86 (s, 1H), 8.63 (s, 1H), 7.94–7.81 (m, 3H), 7.69–7.61 (m, 2H), 7.43–7.34 (m, 1H), 7.19–7.11 (m, 1H), 6.98–6.90 (m, 2H), 5.95 (d, J = 8.0 Hz, 1H), 4.03–3.90 (m, 1H), 3.19–2.96 (m, 4H), 2.78 (q, J = 7.6 Hz, 2H), 2.73–2.52 (m, 4H), 2.51–2.40 (m, 2H), 2.19–2.08 (m, 2H), 1.92–1.81 (m, 2H), 1.54–1.45 (m, 2H), 1.37–1.11 (m, 8H); 13C NMR (101 MHz, CDCl3) δ 166.3, 149.8, 149.3, 148.5, 143.4, 140.9, 135.8, 134.7, 134.6, 128.9, 127.8, 127.4, 127.0, 124.1, 123.8, 118.2, 56.8, 53.8 (2C), 51.7 (2C), 49.4, 35.7, 34.1, 33.4 (2C), 32.1 (2C), 27.6, 14.3. The HCl salt was precipitated from a 0.02 M solution of the free base in 50% CHCl3/acetone using a 2.0 M solution of HCl in Et2O (44 equiv). Dec >223 °C. Anal. calcd for C32H39ClN4O•2HCl•2.25H2O: C, 59.63; H, 7.12; N, 8.69. Found: C, 59.24; H, 6.82; N, 8.54.
N-(trans-4-(2-(4-(2,3-Dichlorophenyl)piperazin-1-yl)ethyl)cyclohexyl)-3-methoxypropanamide (13a).
General procedure F was followed using 5a (0.85 g, 2.4 mmol). After work-up, the crude product was purified by chromatography (40 g of silica gel, 0–5% MeOH/DCM) to afford 13a (1.04 g, 2.35 mmol, 99% yield) as a white solid. Rf = 0.5 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 7.17–7.11 (m, 2H), 6.95 (dd, J = 6.4, 3.2 Hz, 1H), 5.95 (d, J = 8.2 Hz, 1H), 3.77–3.66 (m, 1H), 3.62 (t, J = 5.8 Hz, 2H), 3.36 (s, 3H), 3.17–2.98 (m, 4H), 2.73–2.52 (m, 4H), 2.47–2.37 (m, 4H), 2.04–1.92 (m, 2H), 1.82–1.72 (m, 2H), 1.47–1.38 (m, 2H), 1.31–1.18 (m, 1H), 1.17–1.01 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 170.7, 151.4, 134.2, 127.65, 127.57, 124.7, 118.7, 69.0, 58.9, 56.7, 53.6 (2C), 51.5 (2C), 48.6, 37.4, 35.6, 34.0, 33.2 (2C), 32.0 (2C); IR (neat) 3279, 1635 cm−1; mp 180–181 °C (dec); HRMS (MALDI) m/z [M + H]+ calcd for C22H34Cl2N3O2 442.2023, found 442.2023; tR = 30.3 min (HPLC, basic). The HCl salt was precipitated from a 0.03 M solution of the free base in 50% CHCl3/acetone using a 2.0 M solution of HCl in Et2O (5.0 equiv). Dec >235 °C. Anal. calcd for C22H33Cl2N3O2•HCl•0.75H2O: C, 53.66; H, 7.27; N, 8.53. Found: C, 53.54; H, 6.95; N, 8.49.
N-(trans-4-(2-(4-(2-Chloro-3-ethylphenyl)piperazin-1-yl)ethyl)cyclohexyl)-3-methoxypropanamide (13b).
General procedure F was followed using 5b (0.197 g, 0.563 mmol). After work-up, the crude product was purified by chromatography (silica gel, 0–5% MeOH/CHCl3) to afford 13b (0.245 g, 0.562 mmol, 100% yield) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 7.15 (t, J = 7.8 Hz, 1H), 6.96–6.91 (m, 2H), 5.93 (d, J = 8.2 Hz, 1H), 3.77–3.66 (m, 1H), 3.63 (t, J = 5.8 Hz, 2H), 3.36 (s, 3H), 3.13–2.98 (m, 4H), 2.77 (q, J = 7.5 Hz, 2H), 2.71–2.53 (m, 4H), 2.46–2.39 (m, 4H), 2.03–1.95 (m, 2H), 1.82–1.73 (m, 2H), 1.48–1.41 (m, 2H), 1.32–1.25 (m, 1H), 1.22 (t, J = 7.5 Hz, 3H), 1.17–1.02 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 170.7, 149.8, 143.4, 128.9, 127.0, 124.1, 118.2, 69.0, 58.9, 56.8, 53.7 (2C), 51.8 (2C), 48.6, 37.4, 35.7, 34.1, 33.2 (2C), 32.0 (2C), 27.6, 14.3. The HCl salt was precipitated from a 0.04 M solution of the free base in 50% CHCl3/acetone using a 2.0 M solution of HCl in Et2O (22 equiv). Dec >240 °C. Anal. calcd for C28H38ClN3O2•HCl: C, 61.01; H, 8.32; N, 8.89. Found: C, 60.82; H, 8.21; N, 8.81.
N-(trans-4-(2-(4-(2-Fluoro-3-methoxyphenyl)piperazin-1-yl)ethyl)cyclohexyl)-3-methoxypropanamide (13c).
General procedure F was followed using 5c (0.243 g, 0.724 mmol). After work-up, the crude product was purified by chromatography (24 g of silica gel, 0–5% MeOH/DCM) to afford 13c (0.159 g, 0.377 mmol, 52% yield) as a tan solid. Rf = 0.2 (5% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 6.96 (td, J = 8.3, 1.8 Hz, 1H), 6.66–6.54 (m, 2H), 5.95 (d, J = 8.2 Hz, 1H),3.86 (s, 3H), 3.76–3.65 (m, 1H), 3.61 (t, J = 5.8 Hz, 2H), 3.35 (s, 3H), 3.15–3.03 (m, 4H), 2.66–2.53 (m, 4H), 2.45–2.35 (m, 4H), 2.03–1.92 (m, 2H), 1.81–1.72 (m, 2H), 1.48–1.38 (m, 2H), 1.30–1.18 (m, 1H), 1.16–1.00 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 170.6, 148.7 (d, J = 10 Hz), 145.8 (d, J = 245 Hz), 141.2 (d, J = 6 Hz), 123.6 (d, J = 5 Hz), 111.2 (d, J = 2 Hz), 107.2, 69.0, 58.9, 56.8, 56.6, 53.6 (2C), 50.8 (d, J = 3 Hz, 2C), 48.5, 37.4, 35.6, 34.0, 33.2 (2C), 32.0 (2C); IR (neat) 3275, 1635 cm−1; mp 156–157 °C (dec); HRMS (ESI) m/z [M + H]+ calcd for C23H37FN3O3 422.2813, found 422.2807; tR = 21.6 min (HPLC, basic). The HCl salt was precipitated from a 0.03 M solution of the free base in 50% CHCl3/acetone using a 2.0 M solution of HCl in Et2O (5.0 equiv). mp 211–213 °C (dec). Anal. calcd for C23H36FN3O3•2HCl•2H2O: C, 52.07; H, 7.98; N, 7.92. Found: C, 51.96; H, 7.64; N, 7.78.
3-Methoxy-N-(trans-4-(2-(4-(6-(trifluoromethyl)pyridin-2-yl)piperazin-1-yl)ethyl)cyclohexyl)propenamide (13d).
General procedure F was followed using 5d (0.424 g, 1.19 mmol). After work-up, the crude product was purified by chromatography (40 g of silica gel, 0–5% MeOH/CHCl3) to afford 13d (0.289 g, 0.654 mmol, 55% yield) as a white solid. Rf = 0.2 (5% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 7.56 (t, J = 8.1 Hz, 1H), 6.93 (d, J = 6.8 Hz, 1H), 6.76 (d, J = 8.6 Hz, 1H), 5.95 (d, J = 8.2 Hz, 1H), 3.77–3.66 (m, 1H), 3.65–3.54 (m, 6H), 3.36 (s, 3H), 2.58–2.47 (m, 4H), 2.44–2.33 (m, 4H), 2.04–1.92 (m, 2H), 1.83–1.73 (m, 2H), 1.48–1.39 (m, 2H), 1.32–1.21 (m, 1H), 1.17–1.01 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 170.6, 159.0, 146.6 (q, J = 35 Hz), 138.4, 121.8 (q, J = 274 Hz), 109.5, 108.9, 69.0, 58.9, 56.8, 53.1 (2C), 48.5, 44.9 (2C), 37.4, 35.6, 33.9, 33.2 (2C), 32.0 (2C); IR (film) 3277, 1634 cm−1; mp 167–168 °C (dec); HRMS (ESI) m/z [M + H]+ calcd for C22H34F3N4O2 443.2628, found 443.2625; tR = 14.5 min (HPLC, acidic). The HCl salt was precipitated from a 0.04 M solution of the free base in 50% CHCl3/acetone using a 2.0 M solution of HCl in Et2O (3.0 equiv). Note: Et2O was added to initiate precipitation. Dec >229 °C. Anal. calcd for C22H33F3N4O2•HCl•0.5H2O: C, 54.15; H, 7.23; N, 11.48. Found: C, 53.96; H, 6.99; N, 11.50.
N-(trans-4-(2-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)ethyl)cyclohexyl)-3-methoxypropanamide (13e).
General procedure F was followed using 5e (0.87 g, 2.3 mmol). After work-up, the crude product was purified by chromatography (40 g of silica gel, 0–5% MeOH/DCM) to afford 13e (0.89 g, 1.9 mmol, 83% yield) as a white solid. Rf = 0.2 (5% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 6.85 (d, J = 2.0 Hz, 1H), 6.61 (d, J = 2.0 Hz, 1H), 6.02 (d, J = 8.1 Hz, 1H), 3.83 (s, 3H), 3.75–3.65 (m, 1H), 3.62 (t, J = 5.8 Hz, 2H), 3.36 (s, 3H), 3.24–3.10 (m, 4H), 2.77–2.62 (m, 4H), 2.58–2.46 (m, 4H), 2.41 (t, J = 5.8 Hz, 2H), 1.97 (d, J = 9.7 Hz, 2H), 1.78 (d, J = 10.2 Hz, 2H), 1.15–1.42 (m, 2H), 1.33–1.22 (m, 1H), 1.19 (t, J = 7.6 Hz, 3H), 1.15–1.01 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 170.6, 146.4, 146.0, 140.8, 128.2, 122.3, 116.7, 68.8, 59.0, 58.7, 56.6, 53.8 (2C), 50.0 (2C), 48.4, 37.2, 35.4, 33.6, 33.0 (2C), 31.8 (2C), 28.4, 15.4; IR (neat) 3303, 1634 cm−1; mp 118–120 °C; HRMS (MALDI) m/z [M + H]+ calcd for C25H41ClN3O3 466.2831, found 466.2824; tR = 35.1 min (HPLC, basic). The HCl salt was precipitated from a 0.03 M solution of the free base in 50% CHCl3/acetone using a 2.0 M solution of HCl in Et2O (5.0 equiv). Dec >240 °C. Anal. calcd for C25H40ClN3O3•2HCl•0.25H2O: C, 55.25; H, 7.88; N, 7.73. Found: C, 55.06; H, 7.69; N, 7.62.
tert-Butyl 4-(2-methoxy-2-oxoethylidene)piperidine-1-carboxylate (15).
Using oven-dried glassware under an Ar atmosphere, a mixture of 90 wt% NaH (0.521 g, 19.5 mmol) in anhydrous THF (76 mL) was cooled to 0 °C, and methyl 2-(dimethoxyphosphoryl)acetate (3.2 mL, 20 mmol) was added over 10 min. After stirring the mixture vigorously for 30 min, a solution of tert-butyl 4-oxopiperidine-1-carboxylate (3.01 g, 15.1 mmol) in anhydrous THF (38 mL) was added over 10 min. The reaction was allowed to warm to rt and stirred for an additional 18 h. Then, H2O (50 mL) was added, and the aq layer was extracted with EtOAc (2 × 50 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous Na2SO4, filtered, and concentrated. The crude product was purified by chromatography (80 g of silica gel, 0–20% EtOAc/hexanes) to afford 15 (3.69 g, 14.5 mmol, 96% yield) as a white solid. Rf = 0.4 (20% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 5.71 (s, 1H), 3.69 (s, 3H), 3.54–3.43 (m, 4H), 2.97–2.89 (m, 2H), 2.31–2.23 (m, 2H), 1.47 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 166.9, 158.4, 154.7, 115.0, 80.0, 51.2, 44.7 (2C), 36.6, 29.7, 28.6 (3C); IR (neat) 1711, 1680, 1655 cm−1; mp 64–65 °C; HRMS (MALDI) m/z [M + Na]+ calcd for C13H21NNaO4 278.1363, found 278.1367.
tert-Butyl 4-(2-methoxy-2-oxoethyl)piperidine-1-carboxylate (16).
Using a Parr shaker, a mixture of 15 (2.00 g, 7.84 mmol), 10 wt% Pd/C (0.839 g, 0.787 mmol), and MeOH (60 mL) was agitated under H2 (50 psi) for 5 h. Afterward, the mixture was passed through a pad of Celite (ca. 30 g) using EtOAc (3 × 50 mL), and the filtrate was concentrated. The crude product was purified by chromatography (40 g of silica gel, 0–20% EtOAc/hexanes) to afford 16 (1.81 g, 7.03 mmol, 90% yield) as a clear, colorless oil. Rf = 0.3 (20% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 4.21–3.96 (m, 2H), 3.67 (s, 3H), 2.71 (t, J = 12.9 Hz, 2H), 2.24 (d, J = 7.1 Hz, 2H), 1.98–1.86 (m, 1H), 1.68 (d, J = 13.1 Hz, 2H), 1.44 (s, 9H), 1.22–1.07 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 173.0, 154.9, 79.5, 51.6, 43.8 (2C), 41.0, 33.2, 32.0 (2C), 28.6 (3C); IR (neat) 1737, 1688 cm−1; HRMS (MALDI) m/z [M + Na]+ calcd for C13H23NNaO4 280.1519, found 280.1525.
tert-Butyl 4-(2-oxoethyl)piperidine-1-carboxylate (17a).
Using oven-dried glassware under an Ar atmosphere, a solution of 16 (1.10 g, 4.27 mmol) in anhydrous DCM (33 mL) was cooled to −78 °C, and a 1.0 M solution of DIBALH in toluene (5.4 mL, 5.4 mmol) was added over 5 min. The reaction was stirred for 3 h and allowed to warm to rt. Then, a saturated aq solution of Rochelle salt (60 mL) was added, and the aq layer was extracted with DCM (3 × 30 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated. The crude product was purified by chromatography (80 g of silica gel, 0–40% EtOAc/hexanes) to afford 17a (0.492 g, 2.17 mmol, 51% yield) as a clear, colorless oil. Rf = 0.5 (40% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 9.80–9.76 (m, 1H), 4.21–3.96 (m, 2H), 2.73 (t, J = 12.8 Hz, 2H), 2.38 (d, J = 6.7 Hz, 2H), 2.11–1.98 (m, 1H), 1.68 (d, J = 13.2 Hz, 2H), 1.45 (s, 9H), 1.24–1.09 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 201.6, 154.9, 79.6, 50.3, 43.9 (2C), 32.1 (2C), 30.8, 28.6 (3C); IR (neat) 1722, 1686 cm−1; HRMS (MALDI) m/z [M + Na]+ calcd for C12H21NNaO3 250.1414, found 250.1416.
tert-Butyl 4-(3-oxopropyl)piperidine-1-carboxylate (17b).
General procedure A was followed using tert-butyl 4-(3-hydroxypropyl)piperidine-1-carboxylate (1.02 g, 4.18 mmol). After work-up, the crude product was purified by chromatography (40 g of silica gel, 0–30% EtOAc/hexanes) to afford 17b (0.769 g, 3.19 mmol, 76% yield) as a clear, colorless oil. Rf = 0.4 (30% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 9.77 (t, J = 1.3 Hz, 1H), 4.22–3.94 (m, 2H), 2.65 (t, J = 13.1 Hz, 2H), 2.46 (td, J = 7.5, 1.7 Hz, 2H), 1.68–1.54 (m, 4H), 1.44 (s, 9H), 1.42–1.33 (m, 1H), 1.15–1.02 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 202.4, 154.9, 79.4, 44.0 (2C), 41.3, 35.6, 32.0 (2C), 28.58 (3C), 28.55; IR (neat) 1724, 1686 cm−1; HRMS (MALDI) m/z [M + Na]+ calcd for C13H23NNaO3 264.1570, found 264.1575.
tert-Butyl 4-(2-(4-(2,3-dichlorophenyl)piperazin-1-yl)ethyl)piperidine-1-carboxylate (19a).
General procedure B was followed using 17a (0.486 g, 2.14 mmol) and 1-(2,3-dichlorophenyl)piperazine•HCl (0.688 g, 3.22 mmol) in DCE (14 mL). After work-up, the crude product was purified by chromatography (40 g of silica gel, 0–80% EtOAc/hexanes) to afford 19a (0.61 g, 1.4 mmol, 64% yield) as a white solid. Rf = 0.3 (80% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 7.18–7.10 (m, 2H), 6.95 (dd, J = 6.5, 3.1 Hz, 1H), 4.18–3.94 (m, 2H), 3.15–2.96 (m, 4H), 2.78–2.52 (m, 6H), 2.49–2.40 (m, 2H), 1.67 (d, J = 13.0 Hz, 2H), 1.52–1.39 (m, 3H), 1.45 (s, 9H), 1.20–1.05 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 155.0, 151.4, 134.2, 127.7, 127.6, 124.7, 118.7, 79.4, 56.3, 53.6 (2C), 51.5 (2C), 44.2 (2C), 34.7, 33.7, 32.4 (2C), 28.6 (3C); IR (neat) 1682 cm−1; mp 116–117 °C; HRMS (MALDI) m/z [M + H]+ calcd for C22H34Cl2N3O2 442.2023, found 442.2021.
tert-Butyl 4-(3-(4-(2,3-dichlorophenyl)piperazin-1-yl)propyl)piperidine-1-carboxylate (19b).
General procedure B was followed using 17b (0.764 g, 3.17 mmol) and 1-(2,3-dichlorophenyl)piperazine•HCl (0.933 g, 3.49 mmol) in DCE (31 mL). After work-up, the crude product was purified by chromatography (40 g of silica gel, 0–100% EtOAc/hexanes) to afford 19b (0.884 g, 1.94 mmol, 61% yield) as a clear, orange oil. Rf = 0.2 (EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.19–7.07 (m, 2H), 6.95 (dd, J = 6.4, 3.1 Hz, 1H), 4.19–3.95 (m, 2H), 3.17–2.96 (m, 4H), 2.78–2.48 (m, 6H), 2.43–2.35 (m, 2H), 1.71–1.62 (m, 2H), 1.61–1.49 (m, 2H), 1.45 (s, 9H), 1.42–1.33 (m, 1H), 1.30–1.22 (m, 2H), 1.15–1.01 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 155.0, 151.4, 134.2, 127.7, 127.6, 124.7, 118.7, 79.3, 59.0, 53.5 (2C), 51.5 (2C), 44.2 (2C), 36.2, 34.5, 32.4 (2C), 28.6 (3C), 24.2; IR (film) 1692 cm−1; HRMS (ESI) m/z [M + H]+ calcd for C23H36Cl2N3O2 456.2179, found 456.2175.
1-(2,3-Dichlorophenyl)-4-(2-(piperidin-4-yl)ethyl)piperazine (20a).
General procedure C was followed using 19a (0.250 g, 0.566 mmol) and TFA (7.0 mL, 91 mmol) in DCM (37 mL). After work-up, 20a (0.191 g, 0.558 mmol, 99% yield) was isolated as a tan solid. Rf = 0.1 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 7.16–7.10 (m, 2H), 6.95 (dd, J = 6.4, 3.2 Hz, 1H), 3.14–2.94 (m, 6H), 2.73–2.49 (m, 6H), 2.47–2.38 (m, 2H), 1.73–1.55 (m, 3H), 1.51–1.34 (m, 3H), 1.00–1.06 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 151.5, 134.1, 127.64, 127.56, 124.6, 118.7, 56.3, 53.6 (2C), 51.5 (2C), 47.0 (2C), 35.0, 34.5, 34.0 (2C); IR (neat) 1577 cm−1; mp 78–80 °C; HRMS (MALDI) m/z [M + H]+ calcd for C17H26Cl2N3 342.1498, found 342.1500.
1-(2,3-Dichlorophenyl)-4-(3-(piperidin-4-yl)propyl)piperazine (20b).
General procedure C was followed using 19b (0.244 g, 0.535 mmol) and TFA (1.6 mL, 21 mmol) in DCM (3.7 mL). After work-up, 20b (0.188 g, 0.527 mmol, 99% yield) was isolated as a clear, yellow oil. Rf = 0.1 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 7.16–7.08 (m, 2H), 6.98–6.90 (m, 1H), 2.17–2.92 (m, 6H), 2.74–2.49 (m, 6H), 2.42–2.31 (m, 2H), 1.95–1.77 (m, 1H), 1.72–1.60 (m, 2H), 1.59–1.43 (m, 2H), 1.42–1.16 (m, 3H), 1.15–0.98 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 151.5, 134.2, 127.7, 127.6, 124.7, 118.7, 59.0, 53.5 (2C), 51.5 (2C), 46.0 (2C), 35.8, 34.7, 32.2 (2C), 24.1; IR (neat) 1577 cm−1; HRMS (ESI) m/z [M + H]+ calcd for C18H28Cl2N3 356.1655, found 356.1652.
4-(2-(4-(2,3-Dichlorophenyl)piperazin-1-yl)ethyl)-N,N-dimethylpiperidine-1-carboxamide (21a).
General procedure D was followed using 20a (0.184 g, 0.538 mmol) and N,N-dimethylcarbamyl chloride (65 μL, 0.71 mmol) in DCM (5.4 mL). After work-up, the crude product was purified by chromatography (12 g of silica gel, 0–10% MeOH/DCM) to afford 21a (0.159 g, 0.385 mmol, 71% yield) as a tan solid. Rf = 0.5 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 7.17–7.10 (m, 2H), 6.95 (dd, J = 6.4, 3.1 Hz, 1H), 3.68–3.60 (m, 2H), 3.14–2.94 (m, 4H), 2.80 (s, 6H), 2.72 (td, J = 12.7, 2.3 Hz, 2H), 2.67–2.52 (m, 4H), 2.48–2.40 (m, 2H), 1.73–1.65 (m, 2H), 1.53–1.40 (m, 3H), 1.28–1.13 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 165.3, 151.4, 134.1, 127.64, 127.57, 124.7, 118.7, 56.3, 53.6 (2C), 51.5 (2C), 47.3 (2C), 38.7 (2C), 34.9, 33.8, 32.4 (2C); IR (neat) 1640 cm−1; mp 96–97 °C; HRMS (MALDI) m/z [M + H]+ calcd for C20H31Cl2N4O 413.1869, found 413.1870; tR = 32.6 min (HPLC, basic).
4-(3-(4-(2,3-Dichlorophenyl)piperazin-1-yl)propyl)-N,N-dimethylpiperidine-1-carboxamide (21b).
General procedure D was followed using 20b (0.188 g, 0.527 mmol) and N,N-dimethylcarbamyl chloride (65 μL, 0.71 mmol) in DCM (5.5 mL). After work-up, the crude product was purified by chromatography (12 g of silica gel, 0–10% MeOH/DCM) to afford 21b (0.176 g, 0.410 mmol, 78% yield) as a tan, amorphous solid. Rf = 0.5 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 7.17–7.10 (m, 2H), 6.95 (dd, J = 6.3, 3.3 Hz, 1H), 3.68–3.59 (m, 2H), 3.16–2.96 (m, 4H), 2.80 (s, 6H), 2.75–2.51 (m, 6H), 2.43–2.34 (m, 2H), 1.73–1.69 (m, 2H), 1.60–1.48 (m, 2H), 1.45–1.34 (m, 1H), 1.31–1.22 (m, 2H), 1.21–1.08 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 165.3, 151.4, 134.1, 127.63, 127.56, 124.7, 118.7, 59.0, 53.5 (2C), 51.5 (2C), 47.3 (2C), 38.7 (2C), 36.4, 34.6, 32.3 (2C), 24.2; IR (film) 1643 cm−1; mp 71–73 °C; HRMS (ESI) m/z [M + H]+ calcd for C21H33Cl2N4O 427.2026, found 427.2021; tR = 35.5 min (HPLC, basic).
1-(4-(2-(4-(2,3-Dichlorophenyl)piperazin-1-yl)ethyl)piperidin-1-yl)-3-methoxypropan-1-one (22a).
General procedure F was followed using 20a (0.192 g, 0.560 mmol). After work-up, the crude product was purified by chromatography (12 g of silica gel, 0–10% MeOH/DCM) to afford 22a (0.121 g, 0.283 mmol, 50% yield) as a tan solid. Rf = 0.5 (10% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 7.19–7.11 (m, 2H), 6.96 (dd, J = 7.0, 2.6 Hz, 1H), 4.61 (d, J = 13.3 Hz, 1H), 3.87 (d, J = 13.5 Hz, 1H), 3.70 (t, J = 6.7 Hz, 2H), 3.36 (s, 3H), 3.18–2.95 (m, 5H), 2.82–2.64 (m, 4H), 2.60 (t, J = 6.7 Hz, 2H), 2.57–2.44 (m, 3H), 1.74 (t, J = 12.1 Hz, 2H), 1.64–1.45 (m, 3H), 1.23–1.07 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 169.2, 151.3, 134.2, 127.7, 127.6, 124.8, 118.7, 69.0, 59.0, 56.2, 53.5 (2C), 51.3 (2C), 46.1, 42.0, 34.7, 33.7, 33.4, 33.0, 32.1; IR (neat) 1644 cm−1; mp 88–90 °C; HRMS (MALDI) m/z [M + H]+ calcd for C21H32Cl2N3O2 428.1866, found 428.1860; tR = 27.4 min (HPLC, basic).
1-(4-(3-(4-(2,3-Dichlorophenyl)piperazin-1-yl)propyl)piperidin-1-yl)-3-methoxypropan-1-one (22b).
General procedure F was followed using 20b (0.183 g, 0.513 mmol). After work-up, the crude product was purified by chromatography (24 g of silica gel, 0–5% MeOH/DCM) to afford 22b (0.175 g, 0.396 mmol, 77% yield) as a clear, orange oil. Rf = 0.2 (5% MeOH/DCM); 1H NMR (400 MHz, CDCl3) δ 7.21–7.11 (m, 2H), 7.01–6.94 (m, 1H), 4.69–4.55 (m, 1H), 3.95–3.83 (m, 1H), 3.76–3.64 (m, 2H), 3.36 (s, 3H), 3.21–3.04 (m, 4H), 3.03–2.93 (m, 1H), 2.85–2.66 (m, 4H), 2.65–2.42 (m, 5H), 1.82–1.68 (m, 2H), 1.66–1.42 (m, 3H), 1.36–1.21 (m, 2H), 1.18–1.01 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 169.2, 151.1, 134.2, 127.7, 127.6, 125.0, 118.8, 69.0, 59.0, 58.9, 53.5 (2C), 51.1 (2C), 46.1, 42.1, 36.2, 34.2, 33.7, 32.9, 32.0, 23.8; IR (neat) 1634 cm−1; HRMS (ESI) m/z [M + H]+ calcd for C22H34Cl2N3O2 442.2023, found 442.2019; tR = 17.8 min (HPLC, acidic). The HCl salt was prepared from a 0.03 M solution of the free base in 50% CHCl3/acetone using a 2.0 M solution of HCl in Et2O (15 equiv). The solution was concentrated, and the resulting residue was sonicated with hexanes to obtain a yellow solid, which was recrystallized from CHCl3—hexanes (slow evaporation of solvent mixture). Mp 193–194 °C (dec). Anal. calcd for C22H33Cl2N3O2•HCl•0.25H2O: C, 54.66; H, 7.19; N, 8.69. Found: C, 54.37; H, 7.04; N, 8.52.
tert-Butyl 3-allyl-3-hydroxypyrrolidine-1-carboxylate (24a).
Using oven-dried glassware under an Ar atmosphere, a solution of tert-butyl 3-oxopyrrolidine-1-carboxylate (1.52 g, 8.21 mmol) in anhydrous Et2O (50 mL) was cooled to 0 °C. Then, a 1.0 M solution of allyl magnesium bromide in Et2O (9.8 mL, 9.8 mmol) was added over 5 min, and the mixture was allowed to warm to rt. After stirring for 4 h, the reaction was quenched with a saturated aq solution of NH4Cl (10 mL), and the aq layer was extracted with Et2O (3 × 10 mL). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated. The crude product was purified by chromatography (40 g of silica gel, 0–30% EtOAc/hexanes) to afford 24a (1.11 g, 4.88 mmol, 60% yield) as a clear, yellow oil. Rf = 0.2 (30% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3, mixture of rotamers) δ 5.94–5.79 (m, 1H), 5.25–5.15 (m, 2H), 3.59–3.42 (m, 2H), 3.41–3.20 (m, 2H), 2.39 (d, J = 7.5 Hz, 2H), 1.90–1.80 (m, 3H), 1.46 (s, 9H); 13C NMR (101 MHz, CDCl3, mixture of rotamers) δ 154.8, 133.0, 119.9, 79.5, 79.0, 78.2, 57.7, 57.5, 44.9, 44.4, 43.5, 37.8, 37.3, 28.7 (3C); IR (film) 3412, 1697, 1670 cm−1; mp 110–112 °C; HRMS (ESI) m/z [M + Na]+ calcd for C12H21NO3Na 250.1414, found 250.1414.
tert-Butyl 4-allyl-4-hydroxypiperidine-1-carboxylate (24b).
The same procedure as the one described for 24a was followed starting from tert-butyl 4-oxopiperidine-1-carboxylate (2.00 g, 10.0 mmol). After work-up, the crude product was purified by chromatography (silica gel, 0–40% EtOA/hexanes) to afford 24b (1.44 g, 6.00 mmol, 60% yield) as a clear, colorless oil. 1H NMR (400 MHz, CDCl3) δ 5.93–5.79 (m, 1H), 5.25–5.10 (m, 2H), 4.00–3.60 (m, 2H), 3.28–3.04 (m, 2H), 2.23 (d, J = 7.6 Hz, 2H), 1.74–1.34 (m, 5H), 1.45 (s, 9H).
tert-Butyl 3-hydroxy-3-(3-hydroxypropyl)pyrrolidine-1-carboxylate (25a).
A 1.0 M solution of BH3•THF in THF (15 mL, 15 mmol) was added over 5 min to a solution of 24a (1.75 g, 7.72 mmol) in THF (15 mL) at 0 °C. The reaction mixture was stirred for 3 h, and a 2.0 M aq solution of NaOH (24 mL, 47 mmol) was added followed by 30 wt% H2O2 in H2O (24 mL, 0.23 mol). The resulting mixture was stirred for an additional 3 h and allowed to warm to rt. The mixture was diluted with H2O (30 mL) and extracted with EtOAc (3 × 150 mL). The combined organic extracts were dried over anhydrous Na2SO4, filtered, and concentrated. The crude product was purified by chromatography (120 g of silica gel, 0–100% hexanes/EtOAc) to afford 25a (1.34 g, 5.48 mmol, 71% yield) as a clear, colorless oil. Rf = 0.3 (EtOAc); 1H NMR (400 MHz, CDCl3, mixture of rotamers) δ 3.79–3.61 (m, 2H), 3.55–3.32 (m, 3H), 3.26–3.16 (m, 1H), 1.97–1.89 (m, 1H), 1.86–1.66 (m, 5H), 1.45 (s, 9H); 13C NMR (101 MHz, CDCl3, mixture of rotamers) δ 155.1, 154.9, 79.5, 79.4, 78.3, 63.1, 58.3, 57.9, 45.1, 44.5, 38.4, 37.7, 36.9, 36.6, 28.7 (3C), 28.1, 27.8; IR (film) 3374, 1668 cm−1; HRMS (ESI) m/z [M + Na]+ calcd for C12H23NO4Na 268.1519, found 268.1517.
tert-Butyl 4-hydroxy-4-(3-hydroxypropyl)piperidine-1-carboxylate (25b).
The same procedure as the one described for 25a was followed starting from 24b (2.60 g, 10.8 mmol). After work-up, the crude product was purified by chromatography (silica gel, 0–100% hexanes/EtOAc) to afford 25b (1.14 g, 4.40 mmol, 41% yield) of a clear, colorless oil. 1H NMR (400 MHz, CDCl3) δ 3.80 (d, J = 12.9 Hz, 2H), 3.70 (t, J = 6.1 Hz, 2H), 3.16 (t, J = 12.3 Hz, 2H), 2.06–1.80 (m, 2H), 1.73–1.65 (m, 2H), 1.63–1.48 (m, 6H), 1.45 (s, 9H).
tert-Butyl 2-hydroxy-1-oxa-7-azaspiro[4.4]nonane-7-carboxylate (26a).
General procedure A was followed using 25a (1.34 g, 5.46 mmol). After work-up, the crude product was purified by chromatography (80 g of silica gel, 0–50% EtOAc/hexanes) to afford 26a (0.958 g, 3.94 mmol, 72% yield) as a white solid. Rf = 0.3 (50% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3, mixture of rotamers and diastereomers) δ 5.53 (s, 1H), 3.67–2.99 (m, 5H), 2.27–1.67 (m, 6H), 1.44 (s, 9H); 13C NMR (101 MHz, CDCl3, mixture of rotamers and diastereomers) δ 154.7, 98.9, 89.2, 89.1, 88.4, 88.3, 79.4, 57.9, 57.5, 57.0, 56.4, 45.3, 44.8, 38.6, 38.1, 37.8, 37.1, 33.7, 33.6, 32.1, 32.0, 31.9, 31.4, 28.7 (3C); IR (film) 3406, 1695, 1675 cm−1; HRMS (ESI) m/z [M + Na]+ calcd for C12H21NO4Na 266.1363, found 266.1362.
tert-Butyl 2-hydroxy-1-oxa-8-azaspiro[4.5]decane-8-carboxylate (26b).
General procedure A was followed using 25b (1.14 g, 4.40 mmol). After work-up, the crude product was purified by chromatography (silica gel, 0–40% EtOAc/hexanes) to afford 26b (0.450 g, 1.75 mmol, 40% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 5.55–5.48 (m, 1H), 3.69–3.48 (m, 2H), 3.44–3.26 (m, 2H), 2.56–2.50 (m, 1H), 2.01–1.85 (m, 2H), 1.85–1.64 (m, 3H), 1.62–1.39 (m, 3H), 1.45 (s, 9H).
tert-Butyl 3-(3-(4-(2,3-dichlorophenyl)piperazin-1-yl)propyl)-3-hydroxypyrrolidine-1-carboxylate (27a).
General procedure B was followed using 26a (0.958 g, 3.94 mmol) and 1-(2,3-dichlorophenyl)piperazine•HCl (1.16 g, 4.33 mmol) in DCE (38 mL). After work-up, the crude product was purified by chromatography (silica gel, 0–10% MeOH/DCM) to afford 27a (0.754 g, 1.64 mmol, 42% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3, mixture of rotamers) δ 7.19–7.10 (m, 2H), 6.98–6.93 (m, 1H), 3.60–3.38 (m, 2H), 3.35–3.18 (m, 2H), 3.17–2.97 (m, 4H), 2.90–2.58 (m, 4H), 2.56–2.47 (m, 2H), 1.92–1.69 (m, 6H), 1.48–1.41 (m, 9H).
tert-Butyl 4-(3-(4-(2,3-Dichlorophenyl)piperazin-1-yl)propyl)-4-hydroxypiperidine-1-carboxylate (27b).
General procedure B was followed using 26b (0.445 g, 1.73 mmol) and 1-(2,3-dichlorophenyl)piperazine•HCl (0.509 g, 1.90 mmol) in DCE (18 mL). After work-up, the crude product was purified by chromatography (silica gel, 0–10% MeOH/DCM) to afford 27b (0.810 g, 1.71 mmol, 99% yield) as a clear, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.19–7.12 (m, 2H), 6.96 (dd, J = 7.1, 2.5 Hz, 1H), 3.89–3.64 (m, 2H), 3.24 (t, J = 11.8 Hz, 2H), 3.16–2.96 (m, 4H), 2.83–2.57 (m, 4H), 2.53–2.46 (m, 2H), 1.74–1.63 (m, 4H), 1.56–1.38 (m, 5H), 1.45 (s, 9H).
3-(3-(4-(2,3-Dichlorophenyl)piperazin-1-yl)propyl)pyrrolidin-3-ol (28a).
General procedure C was followed using 27a (0.754 g, 1.64 mmol) and TFA (2.5 mL, 33 mmol) in DCM (12 mL). After work-up, 28a (0.422 g, 1.18 mmol, 72% yield) was isolated and used immediately.
4-(3-(4-(2,3-Dichlorophenyl)piperazin-1-yl)propyl)piperidin-4-ol (28b).
General procedure C was followed using 27b (0.814 g, 1.72 mmol) and TFA (2.6 mL, 34 mmol) in DCM (12 mL). After work-up, 28b (0.500 g, 1.34 mmol, 78% yield) was isolated and used immediately.
3-(3-(4-(2,3-Dichlorophenyl)piperazin-1-yl)propyl)-3-hydroxy-N,N-dimethylpyrrolidine-1-carboxamide (29a).
General procedure D was followed using 28a (0.150 g, 0.419 mmol) and N,N-dimethylcarbamyl chloride (48 μL, 0.52 mmol) in DCM (6.0 mL). After work-up, the crude product was purified by chromatography (silica gel, 0–5% MeOH/DCM) to afford 29a (68.0 mg, 0.158 mmol, 38% yield) as a clear, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.16–7.08 (m, 2H), 6.97–6.91 (m, 1H), 3.70–3.60 (m, 1H), 3.45–3.35 (m, 2H), 3.24–3.18 (m, 1H), 3.15–2.95 (m, 4H), 2.82–2.58 (m, 4H), 2.81 (s, 6H), 2.53–2.46 (m, 2H), 1.87–1.67 (m, 6H); HRMS (MALDI) m/z [M + H]+ calcd for C20H31Cl2N4O2 429.1819, found 429.1802. The HCl salt was precipitated from a 0.06 M solution of the free base in 50% acetone/CHCl3 using a 2.0 M solution of HCl in Et2O (4.0 equiv). Mp 184–186 °C. Anal. calcd for C20H30Cl2N4O2•1.25HCl: C, 50.58; H, 6.63; N, 11.80. Found: C, 50.64; H, 6.39; N, 11.60.
4-(3-(4-(2,3-Dichlorophenyl)piperazin-1-yl)propyl)-4-hydroxy-N,N-dimethylpiperidine-1-carboxamide (29b).
General procedure D was followed using 28b (0.804 g, 2.16 mmol) and N,N-dimethylcarbamyl chloride (0.25 mL, 2.7 mmol) in DCM (42 mL). After work-up, the crude product was purified by chromatography (silica gel, 0–5% MeOH/DCM) to afford 29b (0.272 g, 0.613 mmol, 28% yield) as a clear, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.18–7.11 (m, 2H), 6.99–6.92 (m, 1H), 3.45–3.37 (m, 2H), 3.31–3.19 (m, 2H), 3.18–2.96 (m, 4H), 2.87–2.57 (m, 4H), 2.80 (s, 6H), 2.53–2.44 (m, 2H), 1.74–1.63 (m, 4H), 1.58–1.48 (m, 4H); HRMS (MALDI) m/z [M + H]+ calcd for C21H33Cl2N4O2 443.1975, found 443.1971. The HCl salt was precipitated from a 0.06 M solution of the free base in 50% acetone/CHCl3 using a 2.0 M solution of HCl in Et2O (2.5 equiv). mp 189–192 °C. Anal. calcd for C21H32Cl2N4O2•HCl•0.25H2O: C, 52.07; H, 6.97; N, 11.57. Found: C, 51.98; H, 7.01; N, 11.22.
1-(3-(3-(4-(2,3-Dichlorophenyl)piperazin-1-yl)propyl)-3-hydroxypyrrolidin-1-yl)-3-methoxypropan-1-one (30a).
General procedure F was followed using 28a (0.422 g, 1.18 mmol). After work-up, the crude product was purified by chromatography (silica gel, 0–5% MeOH/DCM) to afford 30a (0.137 g, 0.309 mmol, 26% yield) as a clear, yellow oil. 1H NMR (400 MHz, CD3OD) δ 7.31–7.24 (m, 2H), 7.16–7.10 (m, 1H), 3.76–3.60 (m, 4H), 3.59–3.45 (m, 2H), 3.35–3.32 (m, 3H), 3.28–3.15 (m, 4H), 3.14–2.96 (m, 4H), 2.94–2.82 (m, 2H), 2.68–2.51 (m, 2H), 2.04–1.70 (m, 6H); HRMS (MALDI) m/z [M + H]+ calcd for C21H32Cl2N3O3 444.1815, found 444.1814; tR = 16.9 min (HPLC, acidic).
1-(4-(3-(4-(2,3-Dichlorophenyl)piperazin-1-yl)propyl)-4-hydroxypiperidin-1-yl)-3-methoxypropan-1-one (30b).
General procedure F was followed using 28b (0.500 g, 1.34 mmol). After work-up, the crude product was purified by chromatography (silica gel, 0–5% MeOH/CHCl3) to afford 30b (0.313 g, 0.309 mmol, 50% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.22–7.11 (m, 2H), 7.00–6.94 (m, 1H), 4.38–4.29 (m, 1H), 3.74–3.67 (m, 2H), 3.66–3.58 (m, 1H), 3.43 (t, J = 12.6 Hz, 1H), 3.35 (s, 3H), 3.27–3.03 (m, 5H), 2.95–2.69 (m, 4H), 2.61 (t, J = 6.9 Hz, 4H), 1.84–1.53 (m, 6H), 1.50–1.36 (m, 2H); HRMS (MALDI) m/z [M + H]+ calcd for C22H34Cl2N3O3 458.1972, found 458.1962. The HCl salt was precipitated from a 0.08 M solution of the free base in 50% acetone/CHCl3 using a 2.0 M solution of HCl in Et2O (10 equiv). Dec >160 °C. Anal. calcd for C22H33Cl2N3O3•HCl•H2O: C, 51.52; H, 7.08; N, 8.19. Found: C, 51.62; H, 6.81; N, 8.14.
hD2R and hD3R Radioligand Binding Studies
Radioligand binding assays were conducted similarly as previously described.46 HEK293 cells stably expressing human D2LR or D3R were grown in a 50:50 mix of DMEM and Ham’s F12 culture media, supplemented with 20 mM HEPES, 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1X antibiotic/antimycotic, 10% heat-inactivated fetal bovine serum, and 200 μg/mL hygromycin (Life Technologies, Grand Island, NY) and kept in an incubator at 37 °C and 5% CO2. Upon reaching 80−90% confluence, cells were harvested using premixed Earle’s balanced salt solution with 5 mM EDTA (Life Technologies) and centrifuged at 3000 rpm for 10 min at 21 °C. The supernatant was removed, and the pellet was resuspended in 10 mL hypotonic lysis buffer (5 mM MgCl2, 5 mM Tris, pH 7.4 at 4 °C) and centrifuged at 14500 rpm (~25000 g) for 30 min at 4 °C. The pellet was then resuspended in binding buffer. Bradford protein assay (Bio-Rad, Hercules, CA) was used to determine the protein concentration. For [3H]-N-methylspiperone binding studies membranes were diluted to 500 μg/mL, in fresh EBSS binding buffer made from 8.7 g/L Earle’s Balanced Salts without phenol red (US Biological, Salem, MA), 2.2 g/L sodium bicarbonate, pH to 7.4, and stored in a −80 °C freezer for later use. For [3H]-(R)-(+)-7- OH-DPAT binding studies,51 membranes were harvested and used fresh; the binding buffer was made from 50 mM Tris, 10 mM MgCl2, 1 mM EDTA, pH 7.4. On the test day, each test compound was diluted into half-log serial dilutions using the 30% dimethyl sulfoxide (DMSO) vehicle. When it was necessary to assist solubilization of the drugs at the highest tested concentration, 0.1% AcOH (final concentration v/v) was added alongside the vehicle. Membranes were diluted in fresh binding buffer. Radioligand competition experiments were conducted in 96-well plates containing 300 μL fresh binding buffer, 50 μL of the diluted test compound, 100 μL of membranes (for [3H]-N-methylspiperone assays: 10−20 μg/well total protein for hD2LR and hD3R; for [3H]-(R)-(+)-7- OH-DPAT assays: 40−80, and 20−40 μg/well total protein for hD2LR, and hD3R, respectively), and 50 μL of radioligand diluted in binding buffer ([3H]-N-methylspiperone: 0.4 nM final concentration for all the hD2-like receptor subtypes; [3H]-(R)-(+)-7-OH-DPAT: 1.5 nM final concentration for hD2L, and 0.5 nM final concentration for hD3; Perkin Elmer). Aliquots of radioligands solution were also quantified accurately in each experiment replicate, to determine how much radioactivity was added, taking in account the experimentally determined counter efficiency. Nonspecific binding was determined using 10 μM (+)-butaclamol (Sigma-Aldrich, St. Louis, MO), and total binding was determined with the 30% DMSO vehicle. All compound dilutions were tested in triplicate, and the reaction incubated for 60 min ([3H]-N-methylspiperone assays) or 90 min ([3H]-(R)-(+)-7-OH-DPAT assays) at RT. The reaction was terminated by filtration through PerkinElmer Uni-Filter-96 GF/B, presoaked for the incubation time in 0.5% polyethylenimine, using a Brandel 96-Well Plates Harvester Manifold (Brandel Instruments, Gaithersburg, MD). The filters were washed thrice with 3 mL (3 X ~1 mL/well) of ice-cold binding buffer. PerkinElmer MicroScint 20 Scintillation Cocktail (65 μL) was added to each well, and filters were counted using a PerkinElmer MicroBeta Microplate Counter. IC50 values for each compound were determined from dose−response curves, and Ki values were calculated using the Cheng−Prusoff equation.50 Kd values were determined via separate homologous competitive binding experiments. When a complete inhibition could not be achieved at the highest tested concentrations, Ki values have been extrapolated by constraining the bottom of the dose−response curves (=0% residual specific binding) in the nonlinear regression analysis. These analyses were performed using GraphPad Prism version 8 for Macintosh (GraphPad Software, San Diego, CA). All results were rounded to the third significant figure. Ki values were determined from at least three independent experiments, each performed in triplicate, and are reported as the mean ± standard error of the mean (SEM).
BRET Studies of D2R and D3R Signaling
All reagents were purchased from Sigma Aldrich-Merck unless otherwise stated. BRET experiments were performed in transiently transfected human embryonic kidney 293 T (HEK 293T) cells, as described previously.53, 68 Briefly, cells were grown and maintained at 37 °C in 5% CO2 in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS). Cells were seeded in 10 cm Petri dishes (2.5 × 106 cells per dish) and allowed to grow overnight in media at 37 °C, 5% CO2. The following day, cells were transiently transfected in full media supplemented with antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin, Gibco) using a 1:6 total DNA to PEI (PolySciences Inc) ratio. BRET constructs were as follows: 2 μg of WT-GαoA, 1 μg of Gβ1-Venus(156–239), 1 μg of Gγ2-Venus(1–155), 1 μg of masGRK3ct-Rluc8 and 1 μg of receptor (D2LR or D3R) for GPA assays and 4 μg of Venus-mGsi and 1 μg of D2LR-Rluc8 for mini G recruitment assays. Cells were then allowed to grow overnight at 37 °C, 5% CO2. The next day, cells were plated in Greiner poly-D-lysine-coated 96-well plates (SLS) in media and allowed to grow overnight. On the day of the assay (48h post-transfection), cells were washed once with D-PBS (Lonza, SLS) and incubated in D-PBS for 30 min at 37 °C, 5% CO2. The Rluc substrate coelenterazine h (NanoLight) was added to each well (final concentration of 5 μM) and cells were incubated for 5 minutes at 37 °C. After 5 minutes, ligands (final concentration from 10 μM to 0.01 nM in D-PBS) were added to the plate and cells were incubated for a further 10 minutes at 37 °C. For the antagonist-mode assays that require co-addition, quinpirole was added together with the ligands (final concentration of 3 nM) to generate 50–70% of the signal that can then be displaced by an antagonist. The plate was then read in a PHERAstar FSX microplate reader (Venus and Rluc emission signals at 535 and 475 nm respectively, BMG Labtech). The ratio of Venus:Rluc counts was used to quantify the BRET signal in each well. Data were normalized to the maximal response of dopamine/quinpirole or no drug for the 100% or 0% response, respectively and as indicated in the figure axes titles. All experiments were performed in duplicate and at least three times independently. All data points represent the mean value and error bars represent the standard error of the mean (SEM). Data were fitted using the built-in log(agonist) vs. response (three parameters) model in Prism 9.0 (GraphPad software Inc., San Diego, CA). For agonist-mode assays, data were fitted to a three-parameter concentration-response model where EC50 is the concentration of the agonist needed to elicit half the maximal response of the particular agonist, defined as Emax. For the antagonist-mode assays, data points were fitted using a three-parameter concentration-response model where IC50 is the concentration required to inhibit half the maximum response of the agonist used at a particular concentration. Values of pEC50 or pIC50 plus/minus error are given as the error has a gaussian distribution whereas the error associated with the antilog value does not.
Cocaine Self Administration
Animals.
Male Long–Evans rats 275–325 g (Charles River Laboratories) were used in the experiments. All animals were housed individually in a climate-controlled animal room on a reversed light–dark cycle with free access to food and water. All experimental procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the United States National Research Council and were approved by the National Institute on Drug Abuse Animal Care and Use Committee.
Surgery.
Rats used in cocaine self-administration experiments were implanted with a microrenathane intravenous (i.v.) catheter (Braintree Scientific Inc., Braintree, MA, USA). Each rat was first anaesthetized with ketamine (100 mg/kg i.p.) and xylazine (10 mg/kg, i.p.) and then a small incision was made to the right of the midline of the neck to expose the external jugular vein. One end of the i.v. catheter was next inserted into the vein with the catheter tip reaching the right atrium. The catheter was then secured to the vein with silk suture and the other end fed subcutaneously around the back of the neck to exit near the back of the skull, connected to a bent 24-gauge stainless steel cannula (Plastics One Inc., Roanoke, VA, USA) with a threaded head used to secure a dummy cannula and, during experimentation, an infusion line. The catheter and the guide cannula were secured to the skull with four stainless steel screws threaded into the skull and dental cement. After surgery, the catheters were flushed daily with gentamicin– and heparin–saline solutions (0.1 mg/ml gentamicin and 30 IU/ml heparin; ICN Biochemicals, Cleveland, OH, USA) as precaution against catheter clogging and infection. The animals were allowed to recover for at least 7 days before behavioral training started.
Apparatus.
Standard MED associates operant test chambers were used for self-administration experiments. Each box was equipped with two levers (active and inactive) and a house light set to on at the beginning of each session. Presses on the active lever triggered a compound light and tone cue. Data were collected and analyzed using MED PC software.
Cocaine self-administration under Fixed Ratio (FR) reinforcement.
A total of 48 animals underwent intravenous catheterization surgery. Following recovery, subjects were trained to lever press for cocaine (1 mg/kg/infusion) under an FR1 reinforcement schedule for five sessions. Then cocaine self-administration continued under FR2 reinforcement at 0.5 mg/kg per infusion for approximately two weeks until stable responding was observed (more than 20 infusions, <20% variability in responding across three consecutive sessions and a ratio of at least 2:1 active to inactive lever presses). Each session was 3 h long with a cap of 50 infusions at a dose of 1 mg/kg/infusion to prevent overdose and a cap of 100 infusions at a dose of 0.5 mg/kg/infusion. Rats were randomly assigned to receive intraperitoneal injections of vehicle (25% 2-hydroxypropyl-beta-cyclodextrin; Onbio Inc., Ontario, Canada) or cariprazine (0.3, 1, and 3 mg/kg), 13a (0.3, 1, and 3 mg/kg), 13e (0.3, 1, 3, and 10 mg/kg), 30 min before cocaine self-administration testing. Each animal received 4 or 5 injections for the different drug doses. The sequence of the drug doses was counterbalanced. Between tests, subjects underwent an additional three to five sessions of cocaine self-administration until the baseline response was re-established.
Data analysis.
Behavioral experiments were analyzed with SigmaPlot 12.5 Software (Systat, Palo Alto, CA). All dose treatments groups were compared to vehicle group using One-way Repeated Measures ANOVA or One-way ANOVA tests followed by Dunnet’s post-hoc test.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by the National Institute on Drug Abuse – Intramural Research Program (ZIADA000424) and the NIDA-IRP Medications Development Program. The authors thank Dr. Ning Sheng Cai from the Integrative Neurobiology Section at NIDA-IRP for assistance with cell lines and tissue culture protocols. We also acknowledge Dr. Shelley Jackson (Translational Analytical Core, NIDA-IRP) for providing all HRMS data. The ACUC approved animal protocol number is 22-BNRB-48.
ABBREVIATIONS
- BD-I
bipolar 1 disorder
- BMS
borane-dimethyl sulfide
- BRET
bioluminescence resonance energy transfer
- D2R
dopamine D2 receptor
- D3R
dopamine D3 receptor
- DIPEA
N,N-diisopropylethylamine
- EDCI
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide•HCl
- GPA
G-protein Activation
- HCTU
O-(1H-6-chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HEK 293 cells
human embryonic kidney 293 cells
- HOBt
hydroxybenzotriazole
- MPO
multiparameter optimization
- PP
primary pharmacophore
- PSUD
psychostimulant use disorders
- SP
secondary pharmacophore
- SUD
substance use disorders
Footnotes
The Supporting Information is available free of charge on the ACS Publications website.
Supplementary Tables and representative HPLC traces (pdf)
SMILES strings (excel)
The authors declare no competing financial interest.
REFERENCES
- 1.Ahmad FB; Cisewski JA; Rossen LM; Sutton P Provisional drug overdose death counts. National Center for Health Statistics: 2022. [Google Scholar]
- 2.Ciccarone D The rise of illicit fentanyls, stimulants and the fourth wave of the opioid overdose crisis. Curr. Opin. Psychiatry 2021, 34 (4), 344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Angarita GA; Hadizadeh H; Cerdena I; Potenza MN Can pharmacotherapy improve treatment outcomes in people with co-occurring major depressive and cocaine use disorders? Expert Opin. Pharmacother 2021, 22 (13), 1669–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hellem TL; Lundberg KJ; Renshaw PF A Review of Treatment Options for Co-Occurring Methamphetamine Use Disorders and Depression. J. Addict. Nurs 2015, 26 (1), 14–23; quiz E11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murthy P; Mahadevan J; Chand PK Treatment of substance use disorders with co-occurring severe mental health disorders. Curr. Opin. Psychiatry 2019, 32 (4), 293–299. [DOI] [PubMed] [Google Scholar]
- 6.Coles AS; Sasiadek J; George TP Pharmacotherapies for co-occurring substance use and bipolar disorders: A systematic review. Bipolar Disord. 2019, 21 (7), 595–610. [DOI] [PubMed] [Google Scholar]
- 7.Peris L; Szerman N Partial Agonists and Dual Disorders: Focus on Dual Schizophrenia. Front. Psychiatry 2021, 12, 769623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jordan CJ; Humburg B; Rice M; Bi GH; You ZB; Shaik AB; Cao J; Bonifazi A; Gadiano A; Rais R; Slusher B; Newman AH; Xi ZX The highly selective dopamine D3R antagonist, R-VK4–40 attenuates oxycodone reward and augments analgesia in rodents. Neuropharmacology 2019, 158, 107597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prieto GA Abnormalities of Dopamine D3 Receptor Signaling in the Diseased Brain. J. Cent. Nerv. Syst. Dis 2017, 9, 1179573517726335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xi ZX; Gilbert J; Campos AC; Kline N; Ashby CR Jr.; Hagan JJ; Heidbreder CA; Gardner EL Blockade of mesolimbic dopamine D3 receptors inhibits stress-induced reinstatement of cocaine-seeking in rats. Psychopharmacology 2004, 176 (1), 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xi ZX; Gilbert JG; Pak AC; Ashby CR Jr.; Heidbreder CA; Gardner EL Selective dopamine D3 receptor antagonism by SB-277011A attenuates cocaine reinforcement as assessed by progressive-ratio and variable-cost-variable-payoff fixed-ratio cocaine self-administration in rats. Eur. J. Neurosci 2005, 21 (12), 3427–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ashby CR Jr.; Paul M; Gardner EL; Heidbreder CA; Hagan JJ Acute Administration of the Selective D3 Receptor Antagonist SB-277011A Blocks the Acquisition and Expression of the Conditioned Place Preference Response to Heroin in Male Rats. Synapse 2003, 48 (3), 154–156. [DOI] [PubMed] [Google Scholar]
- 13.Heidbreder CA; Newman AH Current perspectives on selective dopamine D3 receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann. N. Y. Acad. Sci 2010, 1187 (1), 4–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Payer D; Balasubramaniam G; Boileau I What is the role of the D3 receptor in addiction? A mini review of PET studies with [11C]-(+)-PHNO. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 52, 4–8. [DOI] [PubMed] [Google Scholar]
- 15.Ballon JS; Pajvani U; Freyberg Z; Leibel RL; Lieberman JA Molecular pathophysiology of metabolic effects of antipsychotic medications. Trends Endocrinol. Metab 2014, 25 (11), 593–600. [DOI] [PubMed] [Google Scholar]
- 16.Farino ZJ; Morgenstern TJ; Maffei A; Quick M; De Solis AJ; Wiriyasermkul P; Freyberg RJ; Aslanoglou D; Sorisio D; Inbar BP; Free RB; Donthamsetti P; Mosharov EV; Kellendonk C; Schwartz GJ; Sibley DR; Schmauss C; Zeltser LM; Moore H; Harris PE; Javitch JA; Freyberg Z New roles for dopamine D2 and D3 receptors in pancreatic beta cell insulin secretion. Mol. Psychiatry 2020, 25 (9), 2070–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fyfe TJ; Kellam B; Sykes DA; Capuano B; Scammells PJ; Lane JR; Charlton SJ; Mistry SN Structure-Kinetic Profiling of Haloperidol Analogues at the Human Dopamine D2 Receptor. J. Med. Chem 2019, 62 (21), 9488–9520. [DOI] [PubMed] [Google Scholar]
- 18.Keck TM; John WS; Czoty PW; Nader MA; Newman AH Identifying Medication Targets for Psychostimulant Addiction: Unraveling the Dopamine D3 Receptor Hypothesis. J. Med. Chem 2015, 58 (14), 5361–5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meltzer HY Update on typical and atypical antipsychotic drugs. Annu. Rev. Med 2013, 64 (1), 393–406. [DOI] [PubMed] [Google Scholar]
- 20.Miyamoto S; Miyake N; Jarskog LF; Fleischhacker WW; Lieberman JA Pharmacological treatment of schizophrenia: a critical review of the pharmacology and clinical effects of current and future therapeutic agents. Mol. Psychiatry 2012, 17 (12), 1206–1227. [DOI] [PubMed] [Google Scholar]
- 21.Reavill C; Taylor SG; Wood MD; Ashmeade T; Austin NE; Avenell KY; Boyfield I; Branch CL; Cilia J; Coldwell MC; Hadley MS; Hunter AJ; Jeffrey P; Jewitt F; Johnson CN; Jones DN; Medhurst AD; Middlemiss DN; Nash DJ; Riley GJ; Routledge C; Stemp G; Thewlis KM; Trail B; Vong AK; Hagan JJ Pharmacological Actions of a Novel, High-Affinity, and Selective Human Dopamine D3 Receptor Antagonist, SB-277011-A. J. Pharmacol. Exp. Ther 2000, 294 (3), 1154–1165. [PubMed] [Google Scholar]
- 22.Grundt P; Carlson EE; Cao J; Bennett CJ; McElveen E; Taylor M; Luedtke RR; Newman AH Novel Heterocyclic Trans Olefin Analogues of N-{4-[4-(2,3-Dichlorophenyl)piperazin-1-yl]butyl}arylcarboxamides as Selective Probes with High Affinity for the Dopamine D3 Receptor. J. Med. Chem 2005, 48 (3), 839–848. [DOI] [PubMed] [Google Scholar]
- 23.Kumar V; Bonifazi A; Ellenberger MP; Keck TM; Pommier E; Rais R; Slusher BS; Gardner E; You ZB; Xi ZX; Newman AH Highly Selective Dopamine D3 Receptor (D3R) Antagonists and Partial Agonists Based on Eticlopride and the D3R Crystal Structure: New Leads for Opioid Dependence Treatment. J. Med. Chem 2016, 59 (16), 7634–7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.You ZB; Bi GH; Galaj E; Kumar V; Cao J; Gadiano A; Rais R; Slusher BS; Gardner EL; Xi ZX; Newman AH Dopamine D3R antagonist VK4–116 attenuates oxycodone self-administration and reinstatement without compromising its antinociceptive effects. Neuropsychopharmacology 2019, 44 (8), 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shaik AB; Kumar V; Bonifazi A; Guerrero AM; Cemaj SL; Gadiano A; Lam J; Xi ZX; Rais R; Slusher BS; Newman AH Investigation of Novel Primary and Secondary Pharmacophores and 3-Substitution in the Linking Chain of a Series of Highly Selective and Bitopic Dopamine D3 Receptor Antagonists and Partial Agonists. J. Med. Chem 2019, 62 (20), 9061–9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Ciano P; Mansouri E; Tong J; Wilson AA; Houle S; Boileau I; Duvauchelle T; Robert P; Schwartz JC; Le Foll B Occupancy of dopamine D2 and D3 receptors by a novel D3 partial agonist BP1.4979: a [11C]-(+)-PHNO PET study in humans. Neuropsychopharmacology 2019, 44 (7), 1284–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chien EYT; Liu W; Zhao Q; Katritch V; Won Han G; Hanson MA; Shi L; Newman AH; Javitch JA; Cherezov V; Stevens RC Structure of the Human Dopamine D3 Receptor in Complex with a D2/D3 Selective Antagonist. Science 2010, 330 (6007), 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keck TM; Burzynski C; Shi L; Newman AH Chapter Seven - Beyond Small-Molecule SAR: Using the Dopamine D3 Receptor Crystal Structure to Guide Drug Design. In Advances in Pharmacology, Dwoskin LP Ed.; Vol. 69; Academic Press, 2014; pp 267–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newman AH; Xi Z-X; Heidbreder C Current Perspectives on Selective Dopamine D3 Receptor Antagonists/Partial Agonists as Pharmacotherapeutics for Opioid and Psychostimulant Use Disorders. In Current Topics in Behavioral Neurosciences; Springer; Berlin Heidelberg, 2022; pp 1–45. DOI: 10.1007/7854_2022_347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Awad AG; Voruganti LN Neuroleptic dysphoria: revisiting the concept 50 years later. Acta Psychiatr. Scand., Suppl 2005, 111 (427), 6–13. [DOI] [PubMed] [Google Scholar]
- 31.Agai-Csongor E; Domany G; Nogradi K; Galambos J; Vago I; Keseru GM; Greiner I; Laszlovszky I; Gere A; Schmidt E; Kiss B; Vastag M; Tihanyi K; Saghy K; Laszy J; Gyertyan I; Zajer-Balazs M; Gemesi L; Kapas M; Szombathelyi Z Discovery of cariprazine (RGH-188): a novel antipsychotic acting on dopamine D3/D2 receptors. Bioorg. Med. Chem. Lett 2012, 22 (10), 3437–3440. [DOI] [PubMed] [Google Scholar]
- 32.Calabrese F; Tarazi FI; Racagni G; Riva MA The role of dopamine D3 receptors in the mechanism of action of cariprazine. CNS Spectr. 2020, 25 (3), 343–351. [DOI] [PubMed] [Google Scholar]
- 33.Kiss B; Laszlovszky I; Kramos B; Visegrady A; Bobok A; Levay G; Lendvai B; Roman V Neuronal Dopamine D3 Receptors: Translational Implications for Preclinical Research and CNS Disorders. Biomolecules 2021, 11 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laszlovszky I; Barabassy A; Nemeth G Cariprazine, A Broad-Spectrum Antipsychotic for the Treatment of Schizophrenia: Pharmacology, Efficacy, and Safety. Adv. Ther 2021, 38 (7), 3652–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Earley W; Burgess MV; Rekeda L; Dickinson R; Szatmári B; Németh G; McIntyre RS; Sachs GS; Yatham LN Cariprazine Treatment of Bipolar Depression: A Randomized Double-Blind Placebo-Controlled Phase 3 Study. Am. J. Psychiatry 2019, 176 (6), 439–448. [DOI] [PubMed] [Google Scholar]
- 36.Earley WR; Burgess MV; Khan B; Rekeda L; Suppes T; Tohen M; Calabrese JR Efficacy and safety of cariprazine in bipolar I depression: A double-blind, placebo-controlled phase 3 study. Bipolar Disord. 2020, 22 (4), 372–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guma E; Rocchetti J; Devenyi GA; Tanti A; Mathieu AP; Lerch JP; Elgbeili G; Courcot B; Mechawar N; Chakravarty MM; Giros B Role of D3 dopamine receptors in modulating neuroanatomical changes in response to antipsychotic administration. Sci. Rep 2019, 9 (1), 7850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang M; He W; Kiss B; Farkas B; Adham N; Meltzer HY The Role of Dopamine D3 Receptor Partial Agonism in Cariprazine-Induced Neurotransmitter Efflux in Rat Hippocampus and Nucleus Accumbens. J. Pharmacol. Exp. Ther 2019, 371 (2), 517–525. [DOI] [PubMed] [Google Scholar]
- 39.Michino M; Donthamsetti P; Beuming T; Banala A; Duan L; Roux T; Han Y; Trinquet E; Newman AH; Javitch JA; Shi L A Single Glycine in Extracellular Loop 1 Is the Critical Determinant for Pharmacological Specificity of Dopamine D2 and D3 Receptors. Mol. Pharmacol 2013, 84 (6), 854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herenbrink CK; Verma R; Lim HD; Kopinathan A; Keen A; Shonberg J; Draper-Joyce CJ; Scammells PJ; Christopoulos A; Javitch JA; Capuano B; Shi L; Lane JR Molecular Determinants of the Intrinsic Efficacy of the Antipsychotic Aripiprazole. ACS Chem. Biol 2019, 14 (8), 1780–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Newman AH; Battiti FO; Bonifazi A 2016 Philip S. Portoghese Medicinal Chemistry Lectureship: Designing Bivalent or Bitopic Molecules for G-Protein Coupled Receptors. The Whole Is Greater Than the Sum of Its Parts. J. Med. Chem 2020, 63 (5), 1779–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shonberg J; Herenbrink CK; Lopez L; Christopoulos A; Scammells PJ; Capuano B; Lane JR A Structure-Activity Analysis of Biased Agonism at the Dopamine D2 Receptor. J. Med. Chem 2013, 56 (22), 9199–9221. [DOI] [PubMed] [Google Scholar]
- 43.Egyed A; Domany-Kovacs K; Kovanyi B; Horti F; Kurko D; Kiss DJ; Pandy-Szekeres G; Greiner I; Keseru GM Controlling receptor function from the extracellular vestibule of G-protein coupled receptors. Chem. Commun 2020, 56 (91), 14167–14170. [DOI] [PubMed] [Google Scholar]
- 44.Egyed A; Kiss DJ; Keseru GM The Impact of the Secondary Binding Pocket on the Pharmacology of Class A GPCRs. Front. Pharmacol 2022, 13, 847788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lim J; Kelley EH; Methot JL; Zhou H; Petrocchi A; Chen H; Hill SE; Hinton MC; Hruza A; Jung JO; Maclean JK; Mansueto M; Naumov GN; Philippar U; Raut S; Spacciapoli P; Sun D; Siliphaivanh P Discovery of 1-(1H-Pyrazolo[4,3-c]pyridin-6-yl)urea Inhibitors of Extracellular Signal-Regulated Kinase (ERK) for the Treatment of Cancers. J. Med. Chem 2016, 59 (13), 6501–6511. [DOI] [PubMed] [Google Scholar]
- 46.Bonifazi A; Battiti FO; Sanchez J; Zaidi SA; Bow E; Makarova M; Cao J; Shaik AB; Sulima A; Rice KC; Katritch V; Canals M; Lane JR; Newman AH Novel Dual-Target μ-Opioid Receptor and Dopamine D3 Receptor Ligands as Potential Nonaddictive Pharmacotherapeutics for Pain Management. J. Med. Chem 2021, 64 (11), 7778–7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wager TT; Hou X; Verhoest PR; Villalobos A Moving beyond Rules: The Development of a Central Nervous System Multiparameter Optimization (CNS MPO) Approach To Enable Alignment of Druglike Properties. ACS Chem. Neurosci 2010, 1 (6), 435–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wager TT; Hou X; Verhoest PR; Villalobos A Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem. Neurosci 2016, 7 (6), 767–775. [DOI] [PubMed] [Google Scholar]
- 49.Kiss B; Nemethy Z; Fazekas K; Kurko D; Gyertyan I; Saghy K; Laszlovszky I; Farkas B; Kirschner N; Bolf-Terjeki E; Balazs O; Lendvai B Preclinical pharmacodynamic and pharmacokinetic characterization of the major metabolites of cariprazine. Drug Des. Devel. Ther 2019, 13, 3229–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yung-Chi C; Prusoff WH Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol 1973, 22 (23), 3099–3108. [DOI] [PubMed] [Google Scholar]
- 51.Bonifazi A; Yano H; Ellenberger MP; Muller L; Kumar V; Zou M-F; Cai NS; Guerrero AM; Woods AS; Shi L; Newman AH Novel Bivalent Ligands Based on the Sumanirole Pharmacophore Reveal Dopamine D2 Receptor (D2R) Biased Agonism. J. Med. Chem 2017, 60 (7), 2890–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zou M-F; Keck TM; Kumar V; Donthamsetti P; Michino M; Burzynski C; Schweppe C; Bonifazi A; Free RB; Sibley DR; Janowsky A; Shi L; Javitch JA; Newman AH Novel Analogues of (R)-5-(Methylamino)-5,6-dihydro-4H-imidazo[4,5,1-ij]quinolin-2(1H)-one (Sumanirole) Provide Clues to Dopamine D2/D3 Receptor Agonist Selectivity. J. Med. Chem 2016, 59 (7), 2973–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mann A; Keen AC; Mark H; Dasgupta P; Javitch JA; Canals M; Schulz S; Robert Lane J New phosphosite-specific antibodies to unravel the role of GRK phosphorylation in dopamine D2 receptor regulation and signaling. Sci. Rep 2021, 11 (1), 8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Masuho I; Martemyanov KA; Lambert NA Monitoring G Protein Activation in Cells with BRET. In G Protein-Coupled Receptors in Drug Discovery: Methods and Protocols, Filizola M Ed.; Springer; New York, 2015; pp 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nehme R; Carpenter B; Singhal A; Strege A; Edwards PC; White CF; Du H; Grisshammer R; Tate CG Mini-G proteins: Novel tools for studying GPCRs in their active conformation. PLoS One 2017, 12 (4), e0175642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lane JR; Powney B; Wise A; Rees S; Milligan G G Protein Coupling and Ligand Selectivity of the D2L and D3 Dopamine Receptors. J. Pharmacol. Exp. Ther 2008, 325 (1), 319–330. [DOI] [PubMed] [Google Scholar]
- 57.Newman-Tancredi A The importance of 5-HT1A receptor agonism in antipsychotic drug action: rationale and perspectives. Curr. Opin. Investig. Drugs 2010, 11 (7), 802–812. [PubMed] [Google Scholar]
- 58.Newman-Tancredi A; Kleven MS Comparative pharmacology of antipsychotics possessing combined dopamine D2 and serotonin 5-HT1A receptor properties. Psychopharmacology 2011, 216 (4), 451–473. [DOI] [PubMed] [Google Scholar]
- 59.Rothman RB; Baumann MH; Savage JE; Rauser L; McBride A; Hufeisen SJ; Roth BL Evidence for Possible Involvement of 5-HT2B Receptors in the Cardiac Valvulopathy Associated With Fenfluramine and Other Serotonergic Medications. Circulation 2000, 102 (23), 2836–2841. [DOI] [PubMed] [Google Scholar]
- 60.Roman V; Gyertyan I; Saghy K; Kiss B; Szombathelyi Z Cariprazine (RGH-188), a D3-preferring dopamine D3/D2 receptor partial agonist antipsychotic candidate demonstrates anti-abuse potential in rats. Psychopharmacology 2013, 226 (2), 285–293. [DOI] [PubMed] [Google Scholar]
- 61.Yokel RA; Wise RA Attenuation of Intravenous Amphetamine Reinforcement by Central Dopamine Blockade in Rats. Psychopharmacology 1976, 48 (3), 311–318. [DOI] [PubMed] [Google Scholar]
- 62.Galaj E; Newman AH; Xi ZX Dopamine D3 receptor-based medication development for the treatment of opioid use disorder: Rationale, progress, and challenges. Neurosci. Biobehav. Rev 2020, 114, 38–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.de Guglielmo G; Kallupi M; Sedighim S; Newman AH; George O Dopamine D3 Receptor Antagonism Reverses the Escalation of Oxycodone Self-administration and Decreases Withdrawal-Induced Hyperalgesia and Irritability-Like Behavior in Oxycodone-Dependent Heterogeneous Stock Rats. Front. Behav. Neurosci 2019, 13, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Galaj E; Bi G-H; Klein B; Hempel B; Shaik AB; Gogarnoiu ES; Friedman J; Lam J; Rais R; Reed JF; Bloom SH; Swanson TL; Schmachtenberg JL; Eshleman AJ; Janowsky A; Xi Z-X; Newman AH A highly D3R-selective and efficacious partial agonist (S)-ABS01–113 compared to its D3R-selective antagonist enantiomer (R)-ABS01–113 as potential treatments for opioid use disorder. Neuropsychopharmacology 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Newman AH; Ku T; Jordan CJ; Bonifazi A; Xi ZX New Drugs, Old Targets: Tweaking the Dopamine System to Treat Psychostimulant Use Disorders. Annu. Rev. Pharmacol. Toxicol 2021, 61 (1), 609–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ozburn AR; Larson EB; Self DW; McClung CA Cocaine self-administration behaviors in ClockΔ19 mice. Psychopharmacology 2012, 223 (2), 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ozburn AR; Falcon E; Twaddle A; Nugent AL; Gillman AG; Spencer SM; Arey RN; Mukherjee S; Lyons-Weiler J; Self DW; McClung CA Direct Regulation of Diurnal Drd3 Expression and Cocaine Reward by NPAS2. Biol. Psychiatry 2015, 77 (5), 425–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gillis A; Gondin AB; Kliewer A; Sanchez J; Lim HD; Alamein C; Manandhar P; Santiago M; Fritzwanker S; Schmiedel F; Katte TA; Reekie T; Grimsey NL; Kassiou M; Kellam B; Krasel C; Halls ML; Connor M; Lane JR; Schulz S; Christie MJ; Canals M Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signal 2020, 13 (625), eaaz3140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.