

Abstract
The isoguanosine self-assembled pentamer (isoG-star) has exhibited remarkable selectivity for Cs+ binding over competing alkali and alkali earth metal cation, rendering it a promising extractor for radioactive waste 137Cs separation. However, to make isoG-star a pracrtical material for Cs+ isolation, the development of recyclable isoG-star material is required. In this study, a systematic screening of functional isoG derivatives was performed. By employing well-defined complex formation and post-assembly modification, a covalently tethered isoG5-star was prepared through olefin metathesis, utilizing a designed isoG monomer. The application of this newly developed covalently linked isoG-star enabled selective Cs+ extraction, followed by controled solvent-induced H-bond dessociation. This resulted in the creation of a recyclable Cs+ extractor, demonstrating excellent cation selectivity and good reusability (over seven cycles) the first time. Consequently, this new supramolecular macrocycle offers a practical new platform for the treatment of radiocesium (134Cs and 137Cs) in an environmentally friendly and highly effective manner.
Graphica Abstract:
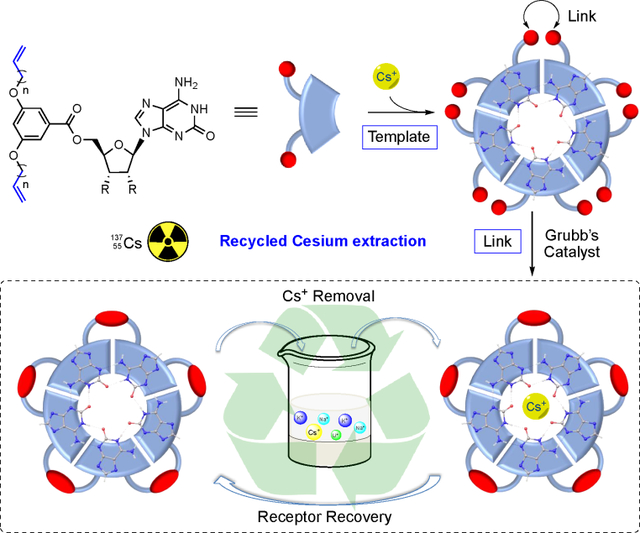
By employing well-defined isoguanosine self-assembled pentamer formation and post-assembly modification, covalently tethered isoG-star through olefin metathesis was prepared as recyclable receptor for selective Cesium separation.
Introduction
Radiocesium ions represent significant constituents of radioactive wastewaters in nuclear power plant, necessitating efficient treatment and isolation methods for Caesium (Cs) ions.1–3 In 2011, the Fukushima Daiichi Nuclear Power Plant (FDNPP) accident resulted in the release of substantial amounts of radiocesium (134Cs and 137Cs with half-lives 2.07 and 30.1 years, respectively) into the environment, further highlighted the requirement for novel systems to effectively extract Cs cation from aqueous solutions with a focus on ecological security and sustainable development.4–7 Considering the abundant existence of Sodium (Na) and Potassium (K) cations in aqueous solutions, along with their chemical similarities, achieving selective separation of Cs+ from such solutions can pose significant challenges.8–11 Crown ethers and calixarenes derivatives are typical receptor for Cs+ separation. Reinhoud and Ungaro made fundamental contribution to the hybrids of calix[4]arene and crown ethers as excellent Cs+ selective ionophores.12–15 Selective binding is the first part of recognition, subsequent controlled release is critical for practical application. Although certain crown ether and calixarene derivatives can selectively coordinate with Cs+ through ion-dipole interaction or cation-π interaction, making them promising Cs+ extractants, concerns arise regarding their limited operating pH range and poor recyclability for practical applications. Sessler and co-workers developed calix[4]pyrrole which could extract Cs+ from aqueous phase while allowing its subsequent release by addition of K+. Calix[4]pyrrole-containing diblock copolymers synthesis demonstrated more effective as an extractant than the corresponding free ion pair receptor.16–21 The development of new systems exhibiting high Cs+ binding selectivity and practical operational conditions is thus of paramount importance for environmental protection and energy sustainability.
Molecular self-assembly is a prevalent process in nature that gives rise to various receptors, providing a powerful approach for designing selective ionophores.22–24 Guanosine (G) rich nucleic acids are recognized for their ability to coordinate alkali and alkaline earth cations through the formation of Hydrogen-bonded G4-quartet motif.25–33 This rigid macrocyclic structure exhibits a high binding affinity towards K+, Sr2+, and Ba2+.34–39 However, despite its structural reversibility, the H-bonding supramolecular architecture lacks flexibility and tends to maintain a fixed size to maximize H-bonding interactions. Consequently, the G4-quartet demonstrates no binding affinity towards Cs+ due to a size mismatch. Interestingly, the structure isomer of G, known as isoguanosine (isoG, also referred to as 2-hydroxy-adenosine) features a larger H-bond donors and acceptors angle (108° for isoG compare to 90° in G-quartet), leading to the formation of a pentameric assembly, isoG5, in contrast to the tetrameric G4-quartet.40–46 With the significantly larger central core, the isoG5 exhibits strong binding towards Cs+ (radii=174 pm) with high affinity and excellent selectivity over Na+ and K+ (Scheme 1A).47–50 Although self-assembly is a powerful tool for creating supramolecular scaffold, it faces challenges as a stable molecular host due to non-covalent bond dissociation, limiting its practical applications. To address these limitations and improve the binding affinity (minimizing entropy penalty associated with self-assembly) and practical usability (recyclability), we have directed our efforts towards the development of the first covalently tethered isoG5-star through post-assembly supramolecular modification (Scheme 1B).
Scheme 1.
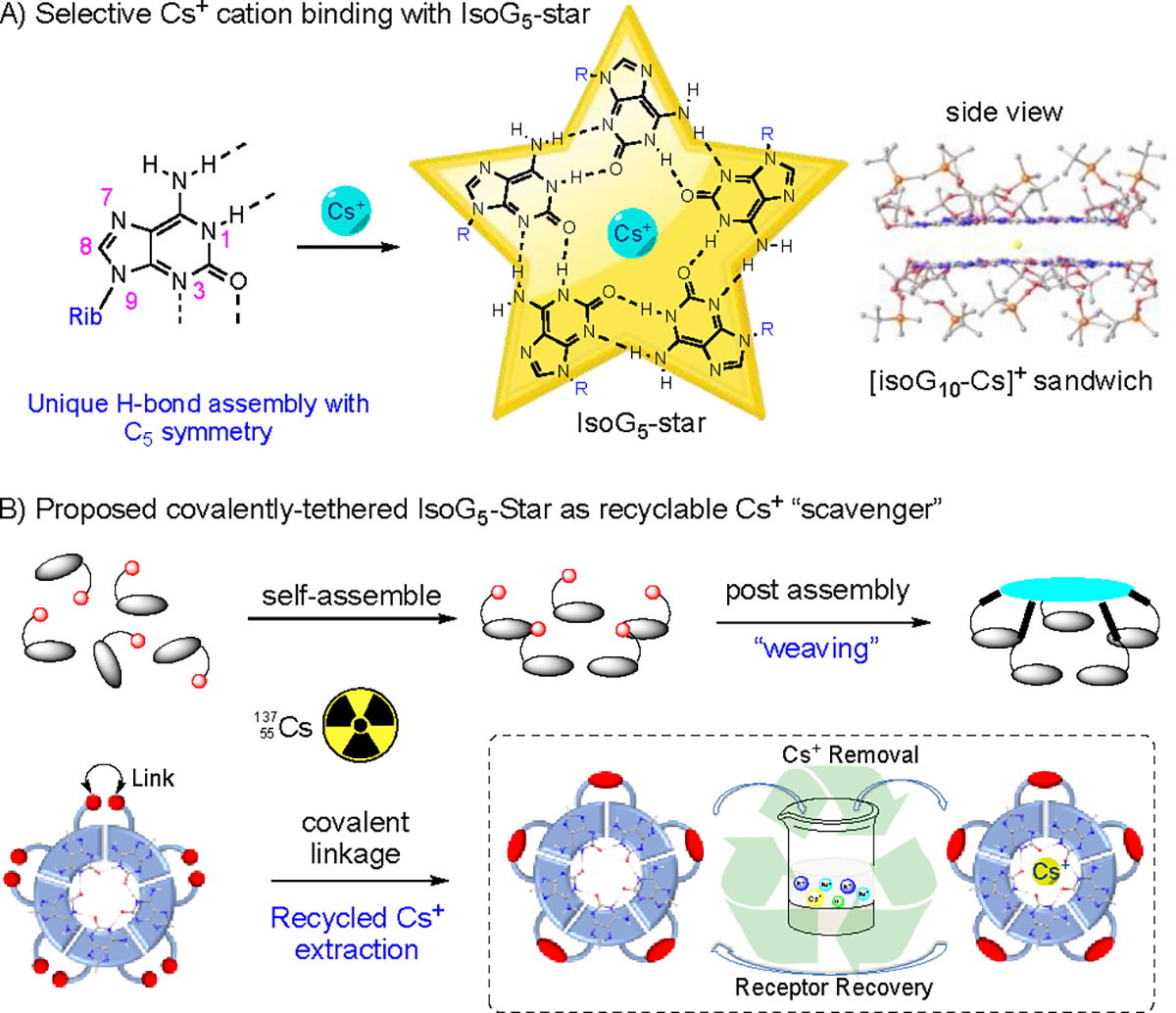
A) Cs+ templated isoG5-star pentaplex formation; B) Proposed covalently tethered isoG5-star as recyclable Cs+ extractor.
In theory, covalently tethered supramolecular scaffold can be achieved through two approaches: A) covalent linkage of a monomer to a template or B) post-assembly modification. The first approach offers greater synthetically accessibility, allowing for easy functional group modification.51–54 However, it requires precise template design to ensure the formation of self-assembly with optimal non-covalent binding, which can be challenging. On the other hand, the post-assembly modification approach ensures the formation of a supramolecular assembly with strong non-covalent interaction, similar to the self-assembled process. However, it requires compatible synthetic methods and appropriate linkage design to ensure the proper weaving of supramolecular scaffold without disrupting the non-covalent binding. Nonetheless, in either approach, the availability of good synthetic handles and the establishment of effective self-assembly are critical factors.
To evaluate different building blocks in the formation of isoG5-star, various isoG derivatives were synthesized and applied in the Cs+ binding experiments (Figure 1A). Typically, the purine C-8 and N-9 positions provide potential synthetic sites without disrupting H-bond formation. Non-sugar isoG derivatives 1a-1d were synthesized and subjected to evaluate Cs+ binding.55 While these compounds were able to effectively extract from aqueous solution, analysis using NMR and MS revealed the presence of complex reaction mixtures with no clear detection of isoG-star formation (Figure S1, S2). This outcome is attributed to the formation of various stacking isomers. Further investigation revealed that the structure of isopropylidene modified isoG 2a could form a stable decamer [(2a)10Cs]+. However, the introduction of a benzyl group at the 5’-position, serving as a potential synthetic handle, significantly reduced solubility in organic solvents. Modification of the C-8 position with a phenyl group gave compounds 2c and 2d, both of which exhibited good solubility in organic solvents. However, despite their favourable solubility properties, neither of these compounds was able to form isoG-star. The X-ray crystal structure of 2c and 2d revealed the configuration is energetically favored by avoiding repulsion.49 As a result, the O5’ was placed close to N3, blocking the critical H-bonding in isoG5 formation. Armed with this mechanistic understanding, our research focused on exploring the isopropylidene modified isoG derivatives 3a-b, and the 2’-deoxy isoG derivatives 4a-4c with the intention of striking a balance between solubility and H-bonding formation (Figure 1B).
Figure 1.
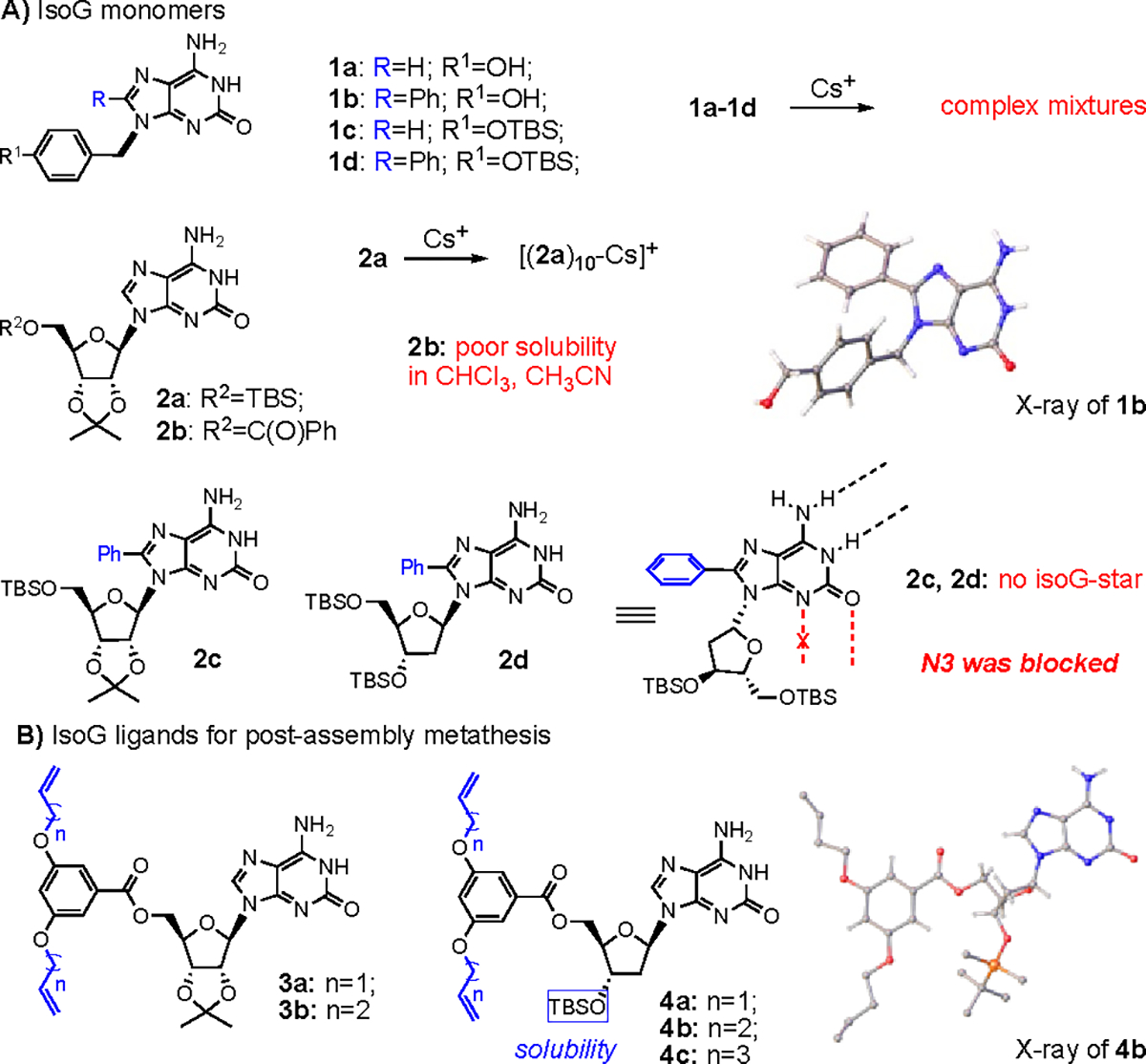
(A) Non-sugar IsoG derivatives 1 and 8-phenyl-isoG derivatives 2; (B) IsoG ligand 3 and 4 for post-assembly weaving.
With the incorporation of longer ester chain, both compounds 3a and 3b exhibited improved solubility compared to 2b. Treatment of isoG 3a in CDCl3 solution with CsCl and NaBPh4 (in an aqueous solution) resulted in one sets of signals in 1H NMR. The proton integration between isoG ligand and anion was 10:1. Both N1-H (at 13.60 ppm) and N6-HA (at 11.02 ppm) protons displayed clear downfiled shifts, indicating the foramtion [(3a)10Cs]+(BPh4−) and successful extraction of Cs+ into the organic layer. Interestingly, BARF− anion led to the formation of a different Cs+ complex, [(3a)5Cs]+(BARF−), with 5:1 integration ratio, suggesting a specificl role of the BARF anion in this assembly. Additionally, isoG 3b, with one extra carbon on the side chain, could also form similar Cs+ complexes, [(3b)10Cs]+(BPh4−) and [(3b)5Cs]+(BARF−). The detailed NMR spectra are provided in Figure S3, S4.
Following isoG-star formation, we proceeded with the supramolecular weaving process in an attempt to covalently link the isoG-star through olefin metathesis. Both the decamer and pentamer from 3a and 3b were treated with Hoveyda-Grubbs-II (HG-II) catalyst, and the reaction process was monitored by MALDI-TOF. Subsequent analysis through NMR and MS confirmed the occurrence of metathesis when treating [(3a)10Cs]+(BPh4−) and [(3a)5Cs]+(BARF−) with HG-II. However, a mixture of oligomers was observed, and the desired formation of pentameric isoG5-star did not materialize. Our hypothesis is that the ester linker might not be sufficiently long to establish a connection with neighboring isoG units. As expected, treating [(3b)10Cs]+ with HG-II led to the detection of the alkene-linked cyclic isoG5 by MALDI-TOF with MW of 2829.15. However, the resulting cyclic structure showed very poor solubility in CHCl3, pacipitating from the reaction mixture, which makes it unsuitable for the proposed Cs+ extraction application.
To address the solubility issue, we investigated the metathesis reaction with 2’-deoxy isoG derivatives 4a-4c. Figure 2A illustrates the treatment of deoxy isoG 4a and 4b in CDCl3 solution with CsCl and NaBARF aqueous solution, leading to the formation of simple complexes exhibiting a single set of signals in 1H NMR. Diffusion NMR experiments deoxy isoG derivatives confirmed the presence of C5-symmetric decamer with the formula [(4a)10Cs2]2+(BARF−)2 and [(4b)10Cs2]2+(BARF−)2. This finding establishes that the enhanced CDCl3 solubility of these deoxy-isoG derivatives enables effective Cs+ extraction into the organic layer through straightforward procedures. Moreover, it is notaworthy that these deoxy-isoG derivatives can also form isoG-star structures with both Na+ and K+. To assess the binding selectivity of these alkene-modified isoG compounds, extrations of Cs+ in the presence of competing Na+ and K+ were performed. The summarized results are presented in Figure 2B and 2C.
Figure 2.
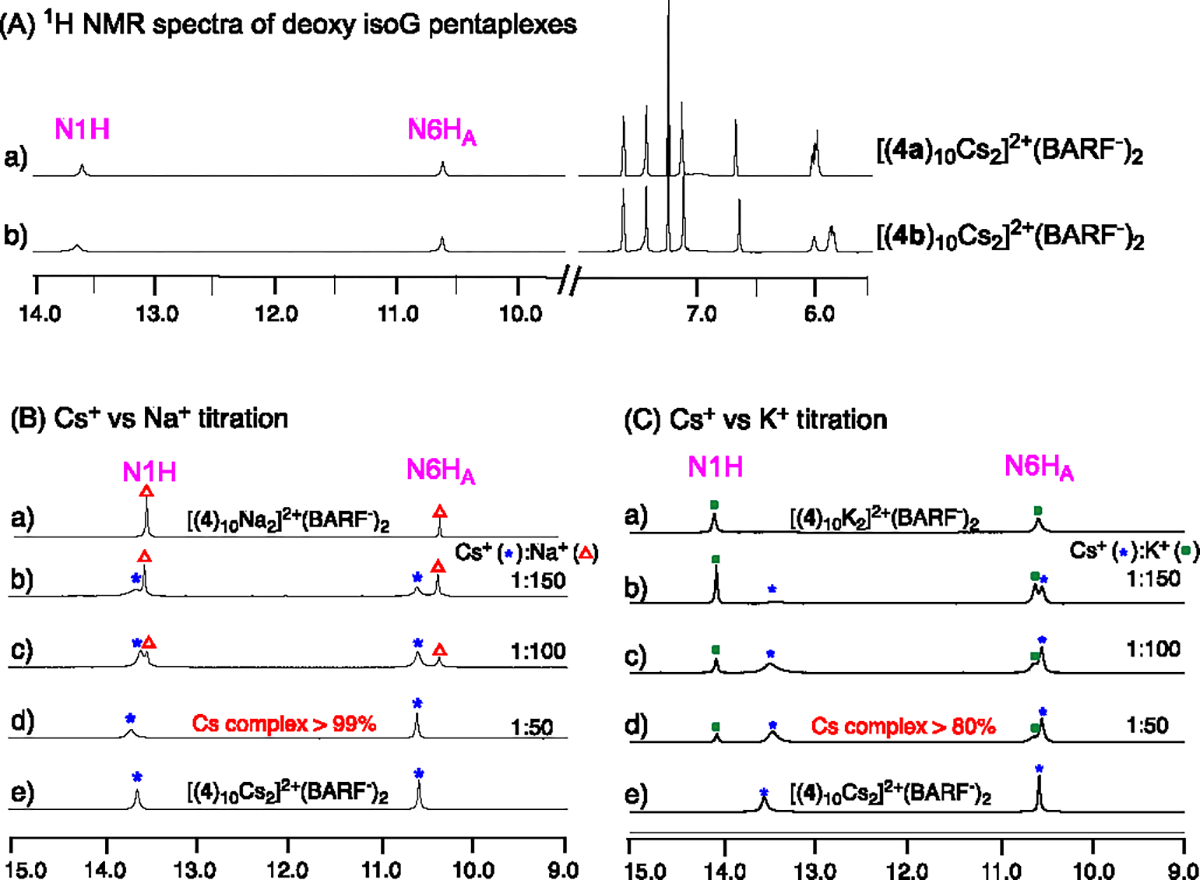
(A) Self-assembly experiments of 4a and 4b: a) 1H NMR spectrum of [(4a)10Cs2]2+(BARF−)2 in CDCl3, b) 1H NMR spectrum of [(4b)10Cs2]2+(BARF−)2 in CDCl3. B) 1H NMR experiments of 4b with Cs+ and Na+ mixture at 25°C in CDCl3, (a) [(4)10Na2]2+(BARF−)2, Cs+ : Na+ molar ratio of : (b) 1:150, (c) 1:100, (d) 1:50, (e) [(4)10Cs2]2+(BARF−)2. The concentrations of Cs+ in aqueous solution are the same in all cases. (C) 1H NMR experiments of 4b with Cs+ and K+ mixture at 25°C in CDCl3, (a) [(4b)10K2]2+(BARF−)2, Cs+ : K+ molar ratio of (b) 1:150, (c) 1:100, (d) 1:50, (e) [(4b)10Cs2]2+(BARF−)2. The concentrations of Cs+ in aqueous solution are the same in all cases. The portion of the spectra shows the region of the N1H and N6HA peaks.
As anticipated, 1H NMR spectra demonstrated excellent Cs+ cation binding selectivity over Na+ and K+ cations. At a Cs+/Na+ ratio of 1:50, deoxy isoG 4b formed over 99% Cs+ complex with minimal Na+ complex formation observed. Slightly reduced Cs+ selectivity was observed when treating 4b with a large excess of K+, resulting in 80% Cs+ complex formation with a 1:50 Cs+/K+ mixture. Other alkaline earth metal cations (Mg2+, Ca2+, and Ba2+) were applied to investigate their influence on the selectivity of Cs+. Nearly zero amount of Mg2+, Ca2+, Ba2+ was extracted in the competition experiments with a 1:100 Cs+/alkaline earth metal ions mixture separately. Remarkably, at a Cs+/ Mg2+ ratio of 1:150, Cs+ complex is still dominant in solution. Slightly reduced Cs+ selectivity was observed when treating 4b with a large excess of Ca2+, resulting in 90% Cs+ complex formation with a 1:150 Cs+/ Ca2+ mixture (Figure S9–S11).
Remarkably, the resulting Cs+ complexes derived from isoG 4b remain stable even at very low concentration (0.0001 M) with no significant dissociation. Variable-temperature NMR (VT-NMR) experiments confirmed that these Cs+ complexes maintained stability even at elevated temperature (55 °C, see dilution and VT NMR in Figure S8). These results strongly support the potential of using deoxy isoG 4 as a new host for Cs+ extraction even in the presence of excess of Na+ and K+ cations.
Following the confirmaiton of Cs+ binding selectivity, excellent solubility, and good complex stability, we investigated the possiblity of forming covalently-linked isoG-star using these deoxy-isoG derivatives 4a - 4c. Complexes derived from these compounds were treated with HG-II catalyst, and the metathesis reactions were monitored using NMR and MALDI-TOF. As depicted in Figure 3A, both isoG ligand and the Cs+ complex [(4)10Cs2]2+(BARF−)2) were treated with HG-II catalyst (5 mol%). Interestingly, no olefin metathesis products were observed while reacting with monomer 4, likely due to the catalyst being quenched by the purine moiety. In the presence of the complex [(4)10Cs2]2+(BARF−)2, the metathesis reaction was observed with complete consumption of the terminal olefin within 8 h in CHCl3 at 55 °C. Monitoring the process with MALDI-TOF sugested the formaiton of isoG trimer and tetramer with allyl ether 4a, indicating the chain length was not sufficient enough to accommodate the isoG5-pemtermer cyclization, leading to compelx dissociation after pentamer formation. With a longer linker, the desired cyclic pentamer was successfully observed with m/z = 3120.9732, consistent with convalently linked cyclic isoG5 containing one Cs cation (C145H195N25O35Si5Cs+, Figure 3D). The overall yield was over 90%, giving the cyclic deoxy isoG pentamer 5a as the dominant product. Compound 5a could be readily purified through column chromatography, with no terminal olefin signals observed in 1H NMR. Similar cyclic isoG-pentamer structures were observed with 4c, the ligand contianing even longer chain. However, complex NMR was received, likely due to the overly flexible side arms.
Figure 3.
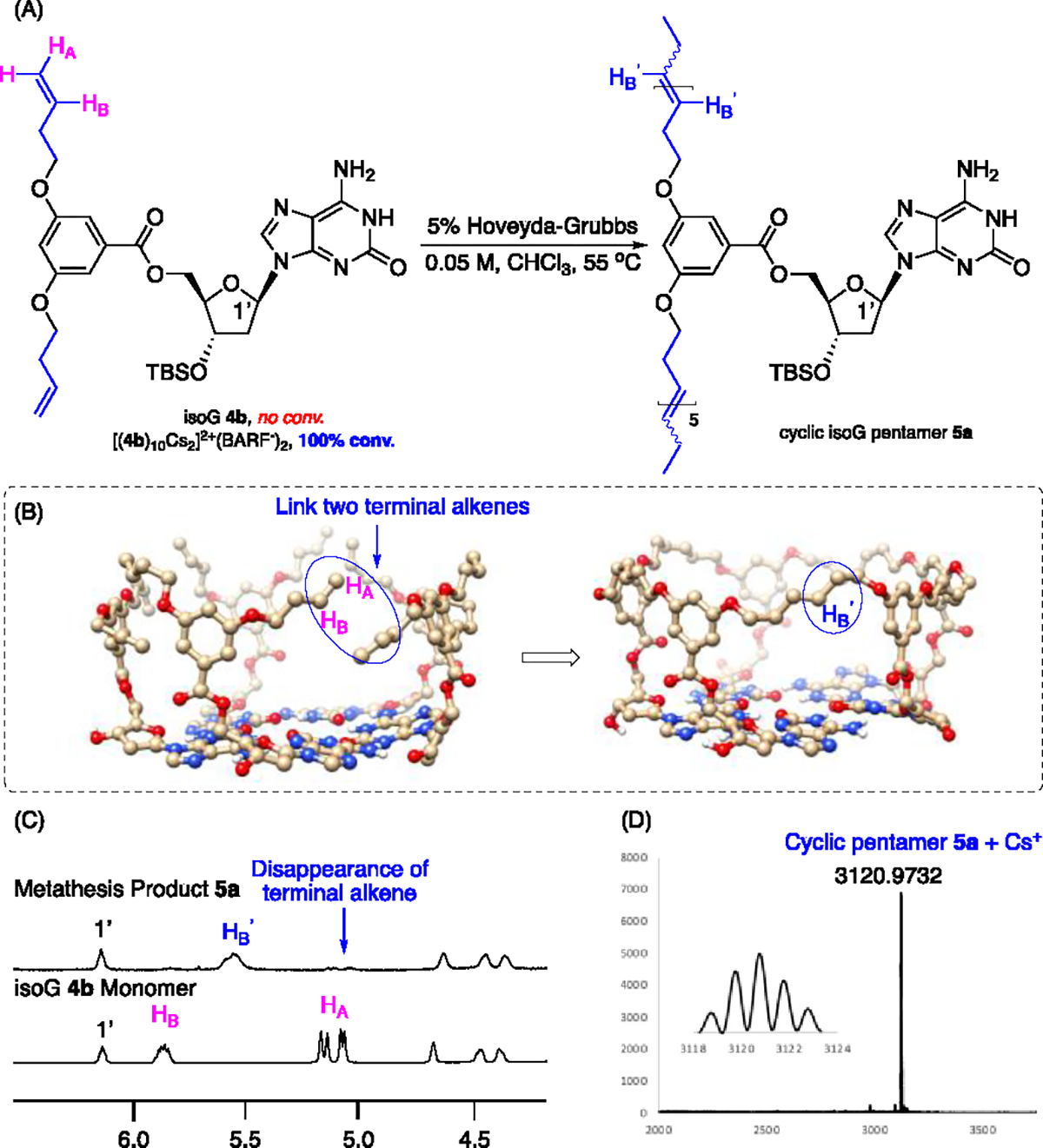
Chemical structure, model and characterizations of cyclic doxy isoG pentamer 5a. (A) Macrocyclization process through olefin metathesis; (B) Computational model of cyclic pentamer; (C) 1H NMR spectra of deoxy isoG 4b monomer and metathesis product cyclic doxy isoG pentamer 5a in DMSO; (D) MALDI-TOF spectrum showing the effective extraction of Cs+ from aqueous phase.
Having successfully prepared cyclic-isoG 5a, aqueous extraction experiments to evaluate its Cs+ extraction capabilities were performed. As expected, these covalently linked isoG-star effectively extract Cs+ from the aqueous solution, forming Cs+ complexes in CHCl3. The formation of new complexes was confirmed by 133Cs NMR that 25 ppm and 53 ppm signals clearly proved the coordination between Cs+ and cyclic pentamer product. In recyclebility experiment, protic solvent MeOH can disrupt the H-bond in deoxy isoG pentamer and cause the decomposition of isoG complex. Upon treating the complex MeOH, complex dissociation occurred, resulting in the release of Cs+. The fact that cyclic pentamer 5a is not soluble in MeOH led it starts to precipitate from solution. The receptor itself can be regenerated after filtration. In this way, Cs+ can be released or storage in another container by adding MeOH solvent, giving C145H195N25O35Si5Na+ and C145H195N25O35Si5K+ on MALDI-TOF. The resulting recycled cyclic isoG 5a could be reapplied for Cs+ extraction by dissolving it in CHCl3 and reacting it with Cs+ containing aqueous solution. Remarkably, this process was performed multiple cycles (7 times) without losing Cs+ selectivity and binding affinity (Figrue 4). The detailed results for each cycle were provided in Figure S14. Furthermore, the pH influence on Cs+ selective extraction was investigated, showing effective extraction of Cs+ between pH=1 and pH=14. The effective pH range covers a broad region, making cyclic deoxy isoG pentamer widely applicable ionophores. To the best of klnowledge, this is the first covalently-linked isoG-star that has been synthesized, and its ease of operation for effective Cs+ extraction demonstrates its potential as a promising solution for the treatment of radioactive Cs+ waste.
Figure 4.
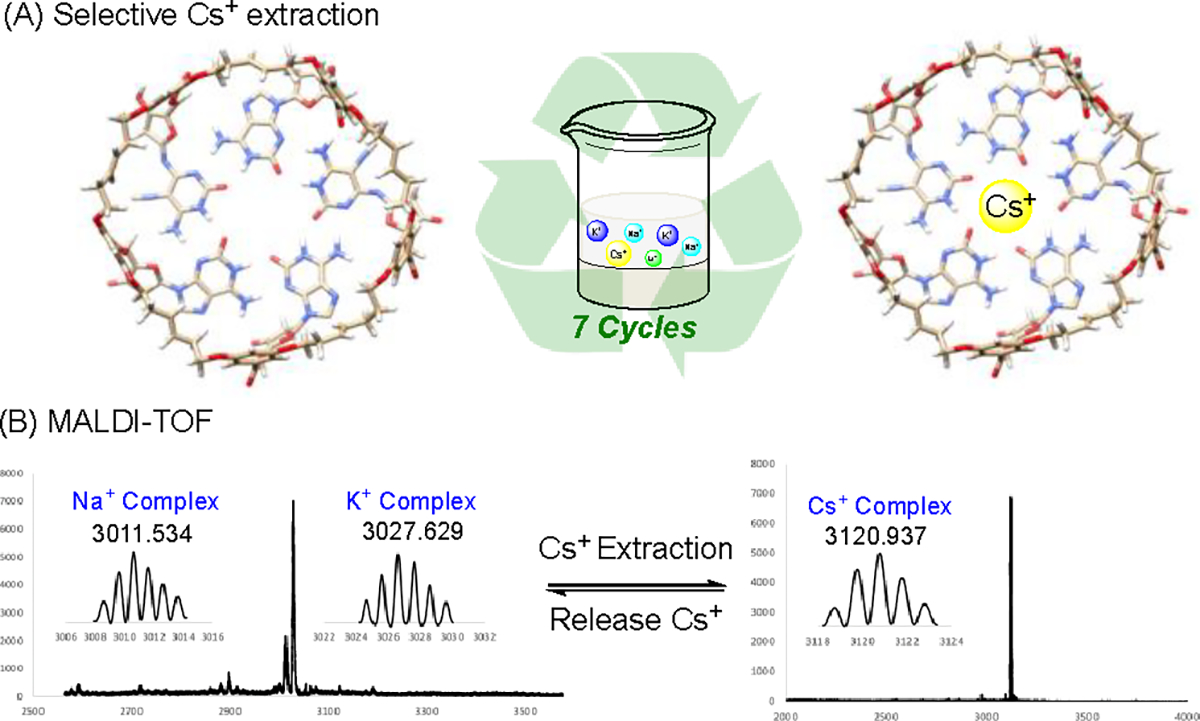
Selective Cs+ extraction by cyclic pentamer 5. (A) 7 cycles extraction without significant efficiency decrease; (B) MALDI-TOF spectra of Cs+ extraction and release.
Conclusion
In summary, we have successfully developed the first covalently linked isoG-star through olefin metathesis with selected deoxy-isoG derivatives. The resulting cyclic isoG-star has demonstrated exceptional performance in Cs+ extraction and separation with ease of operation and excellent recyclability. This breakthrough not only offers a novel material for potential Cs+-containing radioactive nuclear waste treatment, but also introduces a new strategy for the preparation of extra-large macrocycles through post-assembly modification. We anticipate that this strategy will be applicable in the development of new supramolecular hosts, and our ongoing investigations in the laboratory are currently exploring its potential applications.
Supplementary Material
Acknowledgements
Financial support for this work was provided by NSF (CHE-1665122) and NIH (1R01GM120240–01). This work has been supported in part by University of South Florida Interdisciplinary NMR Facility and the Chemical Purification, Analysis, and Screening (CPAS) Core Facility.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic supplementary information (ESI) available: Experimental section, NMR spectra, MALDI-TOF spectra and crystallographic data. CCDC 2287051 and 2287052. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/x0xx00000x
References
- 1.Koo YH, Yang YS and Song KW, Prog. Nucl. Energy, 2014, 74, 61–70. [Google Scholar]
- 2.Wang JL, Zhuang ST and Liu Y, Coord. Chem. Rev, 2018, 374, 430–438. [Google Scholar]
- 3.Wang JL and Zhuang ST, Reviews in Environmental Science and Bio-Technology, 2019, 18, 231–269. [Google Scholar]
- 4.Katata G, Chino M, Kobayashi T, Terada H, Ota M, Nagai H, Kajino M, Draxler R, Hort MC, Malo A, Torii T and Sanada Y, Atmospheric Chemistry and Physics, 2015, 15, 1029–1070. [Google Scholar]
- 5.Saito K, Tanihata I, Fujiwara M, Saito T, Shimoura S, Otsuka T, Onda Y, Hoshi M, Ikeuchi Y, Takahashi F, Kinouchi N, Saegusa J, Seki A, Takemiya H and Shibata T, J. Environ. Radioact, 2015, 139, 308–319. [DOI] [PubMed] [Google Scholar]
- 6.Andoh M, Nakahara Y, Tsuda S, Yoshida T, Matsuda N, Takahashi F, Mikami S, Kinouchi N, Sato T, Tanigaki M, Takamiya K, Sato N, Okumura R, Uchihori Y and Saito K, J. Environ. Radioact, 2015, 139, 266–280. [DOI] [PubMed] [Google Scholar]
- 7.Russell BC, Croudace IW and Warwick PE, Anal. Chim. Acta, 2015, 890, 7–20. [DOI] [PubMed] [Google Scholar]
- 8.Shannon RD, Acta Crystallographica Section A, 1976, 32, 751–767. [Google Scholar]
- 9.Chen YW and Wang JL, Nucl. Sci. Tech, 2016, 27. [Google Scholar]
- 10.Yin YA, Hu J and Wang JL, Environmental Progress & Sustainable Energy, 2017, 36, 989–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ellis RJ, Reinhart B, Williams NJ, Moyer BA and Bryantsev VS, Chem. Commun, 2017, 53, 5610–5613. [DOI] [PubMed] [Google Scholar]
- 12.Casnati A, Sansone F, Dozol J-F, Rouquette H, Arnaud-Neu F. o, Byrne D, Fuangswasdi S, Schwing-Weill M-J and Ungaro R, J. Inclusion Phenom.Macrocyclic Chem., 2001, 41, 193–200. [Google Scholar]
- 13.Casnati A, Pochini A, Ungaro R, Ugozzoli F, Arnaud F, Fanni S, Schwing M-J, Egberink RJM, de Jong F and Reinhoudt DN, J. Am. Chem. Soc, 1995, 117, 2767–2777. [Google Scholar]
- 14.Chrisstoffels LAJ, de Jong F, Reinhoudt DN, Sivelli S, Gazzola L, Casnati A and Ungaro R, J. Am. Chem. Soc, 1999, 121, 10142–10151. [Google Scholar]
- 15.Ungaro R, Casnati A, Ugozzoli F, Pochini A, Dozol J-F, Hill C and Rouquette H, Angew. Chem. Int. Ed, 1994, 33, 1506–1509. [Google Scholar]
- 16.Hanna TA, Liu LH, Angeles-Boza AM, Kou XD, Gutsche CD, Ejsmont K, Watson WH, Zakharov LN, Incarvito CD and Rheingold AL, J. Am. Chem. Soc, 2003, 125, 6228–6238. [DOI] [PubMed] [Google Scholar]
- 17.Levitskaia TG, Maya L, Van Berkel GJ and Moyer BA, Inorg. Chem, 2007, 46, 261–272. [DOI] [PubMed] [Google Scholar]
- 18.Kolesnichenko IV and Anslyn EV, Chem. Soc. Rev, 2017, 46, 2385–2390. [DOI] [PubMed] [Google Scholar]
- 19.Kim SK, Lee HG, Vargas-Zuniga GI, Lynch VM, Kim C and Sessler JL, Chem. Eur. J, 2014, 20, 11750–11759. [DOI] [PubMed] [Google Scholar]
- 20.Kim SK, Vargas-Zuniga GI, Hay BP, Young NJ, Delmau LH, Masselin C, Lee CH, Kim JS, Lynch VM, Moyer BA and Sessler JL, J. Am. Chem. Soc, 2012, 134, 1782–1792. [DOI] [PubMed] [Google Scholar]
- 21.Chi X, Peters GM, Brockman C, Lynch VM and Sessler JL, J. Am. Chem. Soc, 2018, 140, 13219–13222. [DOI] [PubMed] [Google Scholar]
- 22.Davis JT, Tirumala S, Jenssen JR, Radler E and Fabris D, J. Org. Chem, 1995, 60, 4167–4176. [Google Scholar]
- 23.Piotrowski H, Polborn K, Hilt G and Severin K, J. Am. Chem. Soc, 2001, 123, 2699–2700. [DOI] [PubMed] [Google Scholar]
- 24.Debnath M, Chakraborty S, Kumar YP, Chaudhuri R, Jana B and Dash J, Nat. Coummun, 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuhrman F, Fuhrman G, Nachman R and Mosher H, Science, 1981, 212, 557–558. [DOI] [PubMed] [Google Scholar]
- 26.Davis JT and Spada GP, Chem. Soc. Rev, 2007, 36, 296–313. [DOI] [PubMed] [Google Scholar]
- 27.Jiang D and Seela F, J. Am. Chem. Soc, 2010, 132, 4016–4024. [DOI] [PubMed] [Google Scholar]
- 28.Cheng Q, Gu J, Compaan KR and Schaefer III HF, Chem. Eur. J, 2012, 18, 4877–4886. [DOI] [PubMed] [Google Scholar]
- 29.Ingale SA, Leonard P, Tran QN and Seela F, J. Org. Chem, 2015, 80, 3124–3138. [DOI] [PubMed] [Google Scholar]
- 30.Zhao H, Feng H, Liu J, Tang F, Du Y, Ji N, Xie L, Zhao X, Wang Z and Chen Q, Biomaterials, 2020, 230, 119598. [DOI] [PubMed] [Google Scholar]
- 31.Gubala V, Betancourt JE and Rivera JM, Org. Lett, 2004, 6, 4735–4738. [DOI] [PubMed] [Google Scholar]
- 32.Betancourt JE, Martín-Hidalgo M, Gubala V and Rivera JM, J. Am. Chem. Soc, 2009, 131, 3186–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia-Arriaga M, Hobley G and Rivera JM, J. Org. Chem, 2016, 81, 6026–6035. [DOI] [PubMed] [Google Scholar]
- 34.van Leeuwen FWB, Verboom W, Shi X, Davis JT and Reinhoudt DN, J. Am. Chem. Soc, 2004, 126, 16575–16581. [DOI] [PubMed] [Google Scholar]
- 35.He Y, Zhang Y, Liu M, Zhao K, Shan C, Wojtas L, Guo H, Ding A and Shi X, Cell Rep. Phys. Sci, 2021, 2, 100519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He Y, Zhang Y, Wojtas L, Akhmedov NG, Thai D, Wang H, Li X, Guo H and Shi X, Chem. Sci, 2019, 10, 4192–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.González-Rodríguez D, van Dongen JLJ, Lutz M, Spek AL, Schenning APHJ and Meijer EW, Nat. Chem, 2009, 1, 151–155. [DOI] [PubMed] [Google Scholar]
- 38.He Y, Liu MJ, Teng S, Wojtas L, Gu GX and Shi XD, Chin. Chem. Lett, 2022, 33, 4203–4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi X, Fettinger JC and Davis JT, Angew. Chem. Int. Ed, 2001, 40, 2827–2831. [PubMed] [Google Scholar]
- 40.Cai M, Shi X, Sidorov V, Fabris D, Y.-f. Lam and J. T. Davis, Tetrahedron, 2002, 58, 661–671. [Google Scholar]
- 41.Davis JT, Angew. Chem. Int. Ed, 2004, 43, 668–698. [DOI] [PubMed] [Google Scholar]
- 42.Ding T, Tang F, Ni G, Liu J, Zhao H and Chen Q, RSC Advances, 2020, 10, 6223–6248. [Google Scholar]
- 43.Meyer M and Sühnel J, J. Phys. Chem. A, 2003, 107, 1025–1031. [Google Scholar]
- 44.Gu J, Wang J and Leszczynski J, J. Comput. Chem, 2007, 28, 1790–1795. [DOI] [PubMed] [Google Scholar]
- 45.Gu J, Wang J and Leszczynski J, Chem. Phys. Lett, 2007, 445, 243–245. [Google Scholar]
- 46.Zhao H, Schäfer AH and Seela F, ChemPlusChem, 2017, 82, 826–833. [DOI] [PubMed] [Google Scholar]
- 47.Shi X, Fettinger JC, Cai M and Davis JT, Angew. Chem. Int. Ed, 2000, 39, 3124–3127. [DOI] [PubMed] [Google Scholar]
- 48.Cai M, Marlow AL, Fettinger JC, Fabris D, Haverlock TJ, Moyer BA and Davis JT, Angew. Chem. Int. Ed, 2000, 112, 1339–1341. [DOI] [PubMed] [Google Scholar]
- 49.Liu M, He Y, Shan C, Wojtas L, Ghiviriga I, Fathalla O, Yan Y, Li X and Shi X, Chem. Sci, 2021, 12, 7569–7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evan‐Salem T, Frish L, van Leeuwen FW, Reinhoudt DN, Verboom W, Kaucher MS, Davis JT and Cohen Y, Chem. Eur. J, 2007, 13, 1969–1977. [DOI] [PubMed] [Google Scholar]
- 51.Ashcraft A, Liu KX, Mukhopadhyay A, Paulino V, Liu C, Bernard B, Husainy D, Phan T and Olivier JH, Angew. Chem. Int. Ed, 2020, 59, 7487–7493. [DOI] [PubMed] [Google Scholar]
- 52.Paulino V, Liu KX, Cesiliano V, Tsironi I, Mukhopadhyay A, Kaufman M and Olivier JH, Nanoscale, 2023, 15, 4448–4456. [DOI] [PubMed] [Google Scholar]
- 53.Metze FK, Filipucci I and Klok HA, Angew. Chem. Int. Ed, 2023, DOI: 10.1002/anie.202305930. [DOI] [PubMed] [Google Scholar]
- 54.Kaucher MS, Harrell WA and Davis JT, J. Am. Chem. Soc, 2006, 128, 38–39. [DOI] [PubMed] [Google Scholar]
- 55.Lu WB, Sengupta S, Petersen JL, Akhmedov NG and Shi XD, J. Org. Chem, 2007, 72, 5012–5015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.