

Abstract
Adeno-associated virus (AAV) is a relatively safe and efficient vector for gene therapy. However, due to its 4.7-kb limit of cargo, SpCas9-mediated base editors cannot be packaged into a single AAV vector, which hinders their clinical application. The development of efficient miniature base editors becomes an urgent need. Un1Cas12f1 is a class II V-F-type CRISPR-Cas protein with only 529 amino acids. Although Un1Cas12f1 has been engineered to be a base editor in mammalian cells, the base-editing efficiency is less than 10%, which limits its therapeutic applications. Here, we developed hypercompact and high-efficiency base editors by engineering Un1Cas12f1, fusing non-specific DNA binding protein Sso7d, and truncating single guide RNA (sgRNA), termed STUminiBEs. We demonstrated robust A-to-G conversion (54% on average) by STUminiABEs or C-to-T conversion (45% on average) by STUminiCBEs. We packaged STUminiCBEs into AAVs and successfully introduced a premature stop codon on the PCSK9 gene in mammalian cells. In sum, STUminiBEs are efficient miniature base editors and could readily be packaged into AAVs for biological research or biomedical applications.
Keywords: MT: RNA/DNA Editing, miniature CRISPR, Un1Cas12f1, base editing, protein engineering, Sso7d, AAV
Graphical abstract
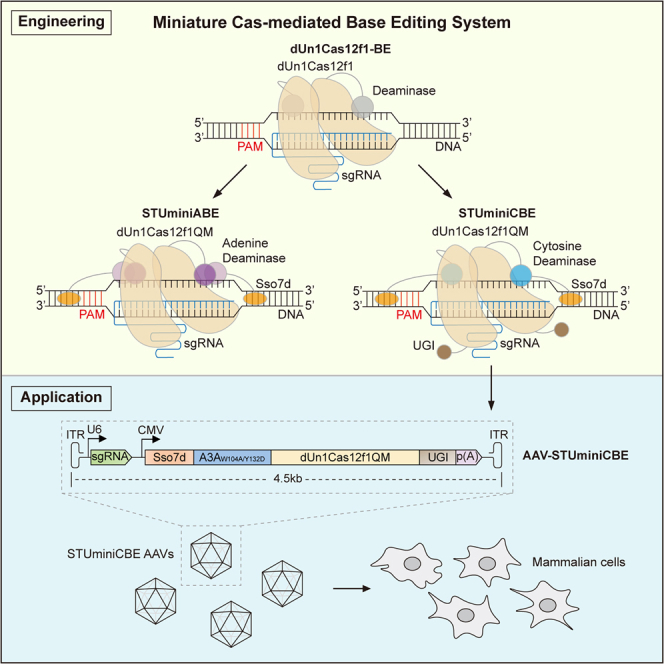
Hu and colleagues developed robust miniature Cas-mediated base-editing systems, STUminiBEs, by engineering Un1Cas12f1 protein, fusing non-specific DNA-binding protein Sso7d, and truncating sgRNA. STUminiBEs are efficient and can readily be packaged into AAVs for biological research or biomedical applications.
Introduction
Approximately 60% of currently known human genetic diseases are caused by base mutations.1,2 CRISPR-mediated base editors allow the conversion of a single base with high efficiency at target sites in the human genome without double-strand DNA breaks,3 which have the potential for gene therapy to cure single-base-mutation diseases. The most widely used base editors at present are cytosine base editors (CBEs) for C•G-to-T•A mutations and adenine base editors (ABEs) for A•T-to-G•C mutations.3,4 It is critical to deliver base editors into specific tissues safely and efficiently. Due to its high efficiency and tissue specificity, the adeno-associated virus (AAV) has become one of the major in vivo delivery tools for gene therapy.5,6 Nevertheless, SpCas9 nickase (D10A)-mediated base editors failed to be delivered by a single AAV, owing to their sizes being larger than the AAV packaging limit of ∼4.7 kb.7,8 Thus, the development of efficient miniature base editors is an urgent need.
Recently, a series of compact RNA-guided nucleases, including Cas12f, Cas12m, Cas12n, TnpB, IscB, Fanzor, etc., were discovered and engineered for genome editing.9,10,11,12,13,14,15,16,17,18,19,20,21 Un1Cas12f1 is a type V-F nuclease consisting of 529 amino acids, less than 39% of SpCas9 (1,368 aa) in size, which cleaves double-stranded DNA (dsDNA) targets with TTTR (where R is A or G) protospacer adjacent motifs.22,23 Un1Cas12f1-derived base editor dCasMINI (D143R/T147R/K330R/E528R) -ABE in mammalian cells with an efficiency of less than 10% in the genomic sites examined,10 which is not sufficient for its application in gene therapy. The high-resolution crystal structure of Un1Cas12f1-single guide RNA (sgRNA)-dsDNA allows us to engineer Un1Cas12f1 to be an efficient base editor.24,25
In this study, we engineered Un1Cas12f1, resulting in high efficient base editors, UminiBEs (UminiABEs for A-to-G transition or UminiCBEs for C-to-T transition). We fused a non-specific dsDNA-binding protein, Sso7d, to the N terminus of the deaminase in UminiABEs or UminiCBEs, which further increased base editing (BE) efficiency, termed SUminiABEs or SUminiCBEs. In addition, we optimized the sgRNA scaffold of Un1Cas12f1 to make it smaller in size while maintaining the BE efficiency, resulting in SUminiBEs with truncated sgRNAs, named STUminiBEs. We packaged STUminiCBEs into a single AAV vector and efficiently installed a premature stop codon on the PCSK9 gene in mammalian cells.
Results
Engineering Un1Cas12f1 to be an efficient miniature ABE, STUminiABE
Un1Cas12f1 has been repurposed for BE in mammalian cells, but the current efficiency was less than 10% in the genomic sites tested.10 Improvement of the Un1Cas12f1-mediated BE efficiency becomes essential for its applications. Un1Cas12f1 has only one nuclease domain, the RuvC domain.22 Nuclease-dead mutations D326A and D510A were introduced into Un1Cas12f1,22,23 resulting in dUn1Cas12f1. To test the efficiency of dUn1Cas12f1-mediated BE, we fused adenine deaminase TadA and adenine deaminase mutant TadA-8e to its N terminus4,10,26 along with the sgRNA, resulting in dUn1Cas12f1-ABEs (Figure 1A).
Figure 1.

Engineering dUn1Cas12f1-mediated miniature adenine base editors with high efficiency
(A) Schematic of nuclease-dead Un1Cas12f1-derived miniature adenine base editor dUn1Cas12f1-ABE. dUn1Cas12f1-ABE consists of wild-type dUn1Cas12f1 (light orange) and adenine deaminases (TadA is in pink, TadA-8e is in purple) at the N terminus, along with original sgRNA (blue) for DNA targeting and adenine base editing. (B) Sixteen point mutations of Un1Cas12f1 residues to enhance the interaction between Un1Cas12f1-sgRNA and target dsDNA (Un1Cas12f1, PDB: 7C7L). Un1Cas12f1 is in gray, target strand (TS) DNA is in purple, non-target strand (NTS) DNA is in orange, and sgRNA is in white. (C) Comparison of A-to-G editing efficiencies mediated by dUn1Cas12f1 variants at DNMT3B-2 site in HEK293FT cells. The most efficient position A3 of the DNMT3B-2 site is chosen for the comparison. WT is the wild-type dUn1Cas12f1. (D) Comparison of A-to-G editing efficiencies mediated by the combinations of dUn1Cas12f1 variants D143R, T147R, T203R, or E206R at DNMT3B-2 site. dCasMINI is the dUn1Cas12f1 variant with D143R/T147R/K330R/E528R mutations. The orange arrow refers to the variant dUn1Cas12f1QM with D143R/T147R/T203R/E206R mutations. (E) Diagram of dUn1Cas12f1-ABE, UminiABE, and the orientations of DNA-binding domain (DBD) fused to UminiABE. All ABEs consist of the adenine deaminase heterodimer TadA-TadA-8e (TadA is in pink, TadA-8e is in purple) and dUn1Cas12f1 or dUn1Cas12f1QM (light orange) with the original sgRNA that is not shown in the diagram. DBD (orange) is fused to the N terminus, middle, or C terminus of TadA-TadA-8e-dUn1Cas12f1QM, resulting in DBD-UminiABE, UminiABE-DBD-I, or UminiABE-DBD-C, respectively. (F) Comparison of A-to-G editing efficiencies induced by dUn1Cas12f1-ABE, dCasMINI-ABE, UminiABE, and DBD-UminiABEs at DNMT3B-2 site. DBDs contain Sso7d, HMGN1 (HN1), HMGB1 box A (HB1), and histone H1 central globular domain (H1G). (G) Comparison of A-to-G editing efficiencies induced by dUn1Cas12f1-ABE, dCasMINI-ABE, UminiABE, Sso7d-UminiABE, UminiABE-Sso7d-I, and UminiABE-Sso7d-C at DNMT3B-2 site. The orange arrow refers to SUminiABE, which is an Sso7d-UminiABE containing the original sgRNA. (H) The secondary structure of original sgRNA for Un1Cas12f1 interacting with target dsDNA. The sequence of the protospacer adjacent motif (PAM) is in red. D1 (marked in orange), D2 (light yellow), and D3 (light blue) are the three regions of sgRNA marked for deletion. (I) Comparison of A-to-G editing efficiencies at DNMT3B-2 site induced by Sso7d-UminiABEs containing original sgRNA (NT, natural tracrRNA, spacer length = 20 nt) or truncated sgRNA variants. (J) Comparison of A-to-G editing efficiencies at DNMT3B-2 site induced by Sso7d-UminiABEs containing original sgRNA (NT, spacer length = 20 nt) or the combinations of truncated sgRNA variants D1, D2, or D3. The orange arrow refers to STUminiABE, which is an Sso7d-UminiABE containing truncated sgRNA-D1/D2/D3. (K) Comparison of A-to-G editing efficiencies mediated by dUn1Cas12f1-ABE, UminiABE, SUminiABE, and STUminiABE at DNMT1-2, HEK3-1, HEK3-3, or KLF4-2 site. All values and error bars represent means ± SD, n = 3 independent biological replicates.
We speculated that dUn1Cas12f1-ABE activity could be enhanced by increasing the binding affinity of dUn1Cas12f1 to the target DNA. Based on the cryoelectron microscopy (cryo-EM) structure of the Un1Cas12f1-sgRNA-dsDNA complex,24 we selected 14 different amino acid residues that are adjacent to the target strand (TS) DNA or non-target strand (NTS) and mutated them into basic amino acid arginine (R), histidine (H), or lysine (K). After fitting these mutations in the Un1Cas12f1-sgRNA-dsDNA structure, we designed 16 different single point mutants: D143R, Y146R, Y146H, T147R, R163H, K167R, V195K, Q197R, Q197H, G199R, G200R, T203R, E206R, Q242R, E393R, and E425R (Figure 1B). Among these point mutations, D143R and T147R are also the mutations in dCasMINI (D143R/T147R/K330R/E528R).10
As shown in Figures 1C and S1A, a few variants (D143R, T147R, T203R, E206R, or E393R) enhanced ABE efficiency at the DNMT3B-2 or KLF4-1 site in HEK293FT cells, while most variants showed no improvement of ABE efficiency over the wild-type dUn1Cas12f1. Subsequently, we chose the top 4 high-efficiency variants (D143R, T147R, T203R, and E206R) to obtain dUn1Cas12f1 variants with double, triple, or quadruple mutations. The dUn1Cas12f1-D143R/T147R/T203R/E206R variant enhanced A-to-G editing efficiency most significantly at both the DNMT3B-2 and KLF4-1 sites (Figures 1D and S1B). We named this quadruple-mutation variant dUn1Cas12f1QM and the ABE with the combination of dUn1Cas12f1QM and original sgRNA UminiABEs (Figure 1E). Compared to the wild-type dUn1Cas12f1, dUn1Cas12f1QM increased A-to-G editing efficiency from 1.84% to 30.99% at the DNMT3B-2 site and from 0.31% to 23.12% at the KLF4-1 site. Compared to dCasMINI,10 dUn1Cas12f1QM enhanced A-to-G editing efficiency by 3.78-fold at the DNMT3B-2 site and 7.25-fold at the KLF4-1 site.
dUn1Cas12f1QM-mediated A-to-G editing efficiency was increased by 15.50-fold (above 30%) if compared to the wild-type dUn1Cas12f1 at the DNMT3B-2 site in HEK293FT cells. However, 30% of the ABE efficiency might still not be enough for it to be utilized in clinical applications. Therefore, alternative strategies are required to further improve ABE efficiency. It has been reported that the fusion of a DNA-binding domain (DBD) can improve genome editing efficiency.27,28,29 Sso7d, a small protein with 64 amino acids and 7.1 kDa in size from Sulfolobus solfataricus, is a non-specific dsDNA-binding protein.30,31,32 This property has been repurposed to improve the processivity of DNA polymerase.33 There are three other non-specific DBDs, high-mobility group nucleosome-binding domain 1 (HMGN1), HMG box 1 (HMGB1) box A, and histone H1 central globular domain (abbreviated as H1G), that have been reported to improve the genome editing activity of Cas proteins.27,28,29 HMGN1 (abbreviated as HN1), an HMG protein with 100 amino acids and 11.1 kDa in size from Homo sapiens, can decompact chromatin structure locally.34,35,36 HMGB1 box A (abbreviated as HB1), which has 84 amino acids and is 9.3 kDa from Homo sapiens, has the activity of bending DNA and competes with histone H1 for chromatin binding.37,38,39 The human H1G, with 81 amino acids and 9 kDa in size, binds near the entry/exit site of linker DNA on the nucleosome.27,39,40
To test whether Sso7d, HN1, HB1, or H1G is able to enhance the BE efficiencies of UminiBEs, we fused Sso7d, HN1, HB1, or H1G to the N terminus of UminiABEs, respectively (Figure 1E). As shown in Figures 1F and S2A, all DBD-UminiABEs enhanced ABE efficiencies if compared to UminiABEs at both the DNMT3B-2 and KLF4-1 sites. The increase of A-to-G editing efficiency by Sso7d-UminiABEs was most prominent at the DNMT3B-2 site, which was 1.45-fold that of UminiABEs (Figure 1F). The increase by Sso7d-UminiABEs at the KLF4-1 site was 1.39-fold that of UminiABEs (Figure S2A). Due to the small size of Sso7d and a slightly higher increase of A-to-G editing efficiency by Sso7d-UminiABEs than other DBD-UminiABEs, Sso7d was selected in subsequent studies. To test whether different fusion positions of Sso7d would affect the BE efficiency, we fused Sso7d to the middle or C terminus of UminiABEs, resulting in UminiABE-Sso7d-I or UminiABE-Sso7d-C (Figure 1E). As shown in Figures 1G and S2B, regardless of the fusion positions of Sso7d to the UminiABE, A-to-G editing efficiencies can be increased by 1.1- to 1.5-fold if compared to UminiABEs at the DNMT3B-2 or KLF4-1 site. The increase of ABE efficiency by Sso7d-UminiABEs was more prominent at the DNMT3B-2 site (Figure 1G). We named Sso7d-UminiBEs containing original sgRNAs SUminiBEs.
In addition, we also optimized the sgRNA scaffold of Un1Cas12f1 by deleting non-essential regions with a minimal effect on the BE efficiency. First, we optimized the spacer length of sgRNA. The spacer length we used in Figures 1C–1G, S1, and S2 was 23 nucleotides (nt), which was the same as the sgRNA spacer length used in dCasMINI-ABEs.10 Previous in vitro assays showed that sgRNA spacers of 17–21 nt supported robust dsDNA cleavage activity.23 We compared the A-to-G editing efficiency of SUminiABEs containing 23-, 20-, or 18-nt spacer sgRNA and investigated which spacer length results in higher editing efficiency. As shown in Figure S3, SUminiABEs containing the 20-nt spacer sgRNA showed slightly better ABE efficiency at the DNMT3B-2 or KLF4-1 site. Thus, we used the 20-nt spacer length sgRNA for further study. Based on the cryo-EM structure of Un1Cas12f1-sgRNA-target dsDNA and the previous sgRNA engineering strategy,9,24 we deleted three regions of the sgRNA, which do not interact with Un1Cas12f1 or DNA (D1, the deletion of stem-loop 5 disordered region; D2, the deletion of stem-loop 1 disordered region; D3, the deletion of stem-loop 2 disordered region, Figure 1H). As shown in Figures 1I and S4A, A-to-G editing efficiency slightly increased when either sgRNA-D1 or -D3 was used if compared to the original sgRNA at both the DNMT3B-2 and KLF4-1 sites, while ABE efficiency slightly decreased when sgRNA-D2 was used. Subsequently, we combined different deletions of disordered regions, resulting in the following sgRNA variants: sgRNA-D1/D2, sgRNA-D1/D3, sgRNA-D2/D3, and sgRNA-D1/D2/D3. sgRNA-D1/D2/D3 was truncated by almost half in size (96 nt in length) if compared to the original sgRNA (160 nt in length) and also slightly increased the ABE efficiency at the DNMT3B-2 or KLF4-1 site (Figures 1J and S4B). We named Sso7d-UminiBEs containing truncated sgRNA-D1/D2/D3 STUminiBEs.
To confirm the enhancement of A-to-G editing efficiency by STUminiABEs, we compared the BE efficiency of dUn1Cas12f1-ABEs, UminiABEs, SUminiABEs, or STUminiABEs at the DNMT1-2, HEK3-1, HEK3-3, or KLF4-2 site (Figure 1K). The ABE efficiency by STUminiABEs ranged from 35.76% to 62.24% in the most efficient position A3 or A4 with a mean of 53.68%, which was significantly higher than that by dUn1Cas12f1-ABEs (range: 1.54%–23.66%, mean: 12.15%), slightly higher than that by UminiABEs (range: 29.28%–51.53%, mean: 45.43%), and comparable to that by SUminiABEs (range: 34.76%–58.83%, mean: 52.23%). The proportion of A-to-G base conversion induced by UminiABEs, SUminiABEs, or STUminiABEs was close to 100%, slightly higher than that induced by dUn1Cas12f1-ABEs (range: 93.99%–99.86%, mean: 98.29%) (Figure S5).
Generation of Un1Cas12f1-mediated CBE STUminiCBE
To test whether the STUminiABE (combination of dUn1Cas12f1QM, Sso7d, and sgRNA-D1/D2/D3) can be adapted to CBEs for C-to-T transition, we replaced the adenine deaminase by the cytosine deaminase APOBEC3A (abbreviated as A3A) mutant A3AW104A/Y132D, and uracil glycosylase inhibitor (UGI) was added to the C terminus of dUn1Cas12f1QM,3,41,42 along with truncated sgRNA-D1/D2/D3, resulting in STUminiCBEs (Figure 2A). As controls, we fused A3AW104A/Y132D to the N terminus of wild-type dUn1Cas12f1 or dUn1Cas12f1QM and UGI to the C terminus, along with the original sgRNA, resulting in dUn1Cas12f1-CBEs or UminiCBEs, respectively (Figure 2B). We also fused Sso7d to the N terminus of UminiABEs, resulting in SUminiCBEs (Figure 2B).
Figure 2.

Development of dUn1Cas12f-mediated cytosine base editors
(A) Schematic of nuclease-dead Un1Cas12f1-derived miniature cytosine base editor (STUminiCBE). STUminiCBE consists of dUn1Cas12f1QM (light orange) fused with cytosine deaminase (light blue) and Sso7d (orange) at the N terminus and UGI (brown) at the C terminus, along with truncated sgRNA (blue) for DNA targeting and cytosine base editing. (B) Diagram of dUn1Cas12f1-CBE, UminiCBE, SUminiCBE, and STUminiCBE. All CBEs consist of the cytosine deaminase A3AW104A/Y132D (light blue) at the N terminus, dUn1Cas12f1 or dUn1Cas12f1QM (light orange) in the middle, and UGI (brown) at the C terminus. SUminiCBE or STUminiCBE contains Sso7d (orange) at the N terminus of A3AW104A/Y132D-dUn1Cas12f1QM-UGI. Original sgRNA and truncated sgRNA-D1/D2/D3 are not shown in the diagram. (C) Comparison of C-to-T editing efficiencies mediated by dUn1Cas12f1-CBE, UminiCBE, SUminiCBE, and STUminiCBE at EMX1-5, KLF4-5, DNMT3B-4, DNMT3B-5, DNMT3B-6, RUNX1-4, or RUNX1-6 site. All values and error bars represent means ± SD, n = 3 independent biological replicates.
We compared C-to-T editing efficiencies by dUn1Cas12f1-CBEs, UminiCBEs, SUminiCBEs, and STUminiCBEs at the EMX1-5, DNMT3B-4, DNMT3B-5, DNMT3B-6, KLF4-5, RUNX1-4, or RUNX1-6 site (Figure 2C). Consistent with engineered dUn1Cas12f1-mediated ABEs, the CBE efficiency by STUminiCBEs ranged from 25.33% to 64.80% with a mean of 45.22%, which was substantially higher than that by dUn1Cas12f1-CBEs (range: 0.32%–12.46%, mean: 6.54%), 1.11- to 1.97-fold higher than that by UminiCBEs (range: 13.17%–52.81%, mean: 31.76%), and comparable to SUminiCBEs (range: 23.65%–66.70%, mean: 44.43%). The proportion of C-to-T base conversion induced by UminiCBEs, SUminiCBEs, or STUminiCBEs was close to 100%, slightly higher than that induced by dUn1Cas12f1-CBEs (range: 88.60%–99.83%, mean: 97.68%) (Figure S6).
To further evaluate the editing capabilities of STUminiBEs, we compared the A-to-G editing efficiency of STUminiABEs with Sso7d-dCas9-ABEs, TaRGET-ABE-C3.0, and TaRGET-ABE-C3.1 at the spacer-sequence-overlapped target sites (Figure 3).12 We also compared the C-to-T editing efficiency of STUminiCBEs and Sso7d-dCas9-CBEs at the same sites as in ABEs (Figure 3). The ABE efficiency of STUminiABEs (range: 41.20%–53.36%, mean: 45.30%) was comparable to that of Sso7d-dCas9-ABEs (range: 26.53%–56.40%, mean: 46.34%) and substantially higher than that of TaRGET-ABE-C3.0 (range: 0.64%–5.15%, mean: 2.45%) and TaRGET-ABE-C3.1 (range: 12.88%–30.49%, mean: 21.98%) at A3 or A4. The CBE efficiency of STUminiCBEs ranged from 14.50% to 52.42% with a mean of 35.08% at C8, which was comparable to that of Sso7d-dCas9-CBEs (range: 29.82%–48.85%, mean: 36.41%) at C8. The C-to-T editing efficiency of STUminiCBEs was higher than that of Sso7d-dCas9-CBEs when the edited cytosine was in C10-C19. The proportion of base substitution (A to G or C to T) by STUminiBEs was close to 100%, similar to that of Sso7d-dCas9-BEs (Figure S7).
Figure 3.

Comparison of the base-editing efficiencies induced by STUminiBEs, dCas9-derived BEs, or TaRGET-ABEs
(A) Diagram of Sso7d-dCas9-ABE, Sso7d-dCas9-CBE, TaRGET-ABE-C3.0, and TaRGET-ABE-C3.1. Sso7d-dCas9-ABE consists of Sso7d (orange) at the N terminus, adenine deaminases (TadA is in pink, TadA-8e is in purple) in the middle, and dead SpCas9 (purple brown) at the C terminus. Sso7d-dCas9-CBE contains Sso7d and the cytosine deaminase A3AW104A/Y132D (light blue) at the N terminus, dCas9 in the middle, and UGI (brown) at the C terminus. TaRGET-ABE-C3.0 and TaRGET-ABE-C3.1 both consist of dCWCas12f (D354A) (dark orange) at the N terminus and adenine deaminases at the C terminus. TaRGET-ABE-C3.0 contains TadA∗∗ that denotes the engineered form with V106W and N108Q mutations in Tad∗(7.10), while TaRGET-ABE-C3.1 contains TadA-8e∗ that denotes the engineered form with V106W and N108Q mutations in TadA-8e. sgRNA is not shown in the diagram. (B) Comparison of A-to-G editing efficiencies mediated by STUminiABE, Sso7d-dCas9-ABE, TaRGET-ABE-C3.0, and TaRGET-ABE-C3.1 and C-to-T editing efficiencies mediated by STUminiCBE and Sso7d-dCas9-CBE at PWAR5, NEK10-1, INIP, or KLHL29 site. dUn1Cas12f1 or dCWCas12f PAM sequences are in light purple, and dCas9 PAM sequences are in brown. All values and error bars represent means ± SD, n = 3 independent biological replicates.
CGBEs (C-to-G base editors) are CBE derivatives that remove the UGI domain and often add uracil DNA N-glycosylase (UNG) or other DNA repair proteins to improve the efficiency and purity of C-to-G conversion.43,44,45,46,47 AYBE (adenine transversion base editor) is a derivative of ABEs by fusing with an N-methylpurine DNA glycosylase (MPG).48,49 To investigate whether our engineered dUn1Cas12f1 and truncated sgRNA could be applied to achieve other base substitutions, we tested dUn1Cas12f1-mediated CGBEs or AYBEs (Figure S8). To test whether dUn1Cas12f1-mediated CBEs could be transformed to be dUn1Cas12f1-mediated CGBEs, we fused three different cytosine deaminase variants to the N terminus of dUn1Cas12f1 or dUn1Cas12f1QM and different UNG orthologs to the C terminus, resulting in dUn1Cas12f1-CGBEs or STUminiCGBEs (Figure S8A). As shown in Figure S8B, the C-to-G editing efficiency was lower than 5%. We fused an MPG mutant (MPGm) to the C terminus of dUn1Cas12f1-ABEs or STUminiABEs,48 resulting in dUn1Cas12f1-AYBEs or STUminiAYBEs (Figure S8C). As shown in Figure S8D, the A-to-C/T editing efficiency was also lower than 5%. Thus, more optimization is required to generate robust dUn1Cas12f1-mediated CGBEs or AYBEs.
Evaluating the specificity of UminiBEs, SUminiBEs, and STUminiBEs
To evaluate the specificity of UminiBEs, SUminiBEs, and STUminiBEs, we screened 1- or 2-nt mismatch with 0-, 1-, or 2-bulge or 1- to 3-nt mismatch with no bulge off-target sites for DNMT1-2, HEK3-3, KLF4-1, RUNX1-4, EMX1-5, and RUNX1-6 in the human genome using Cas-OFFinder.50 There were four potential off-target loci for the DNMT1-2 site, three for the HEK3-3 site, seven for the KLF4-1 site, four for the RUNX1-4 site, seven for the EMX1-5 site, and seven for the RUNX1-6 site. As shown in Figure S9A, the A-to-G on-target editing efficiency of UminiABEs, SUminiABEs, or STUminiABEs reached 53.21%, 60.61%, or 60.99% in A3 at the DNMT1-2 site; 54.00%, 62.30%, or 67.14% in A4 at the HEK3-3 site, and 35.77%, 43.97%, or 44.83% in A4 at the KLF4-1 site, respectively. The A-to-G off-target editing efficiency of UminiABEs, SUminiABEs, or STUminiABEs corresponding to the DNMT1-2 site were 20.79%, 31.31%, or 35.36% at OT1 (2-nt mismatches, 1 bulge); 10.78%, 21.07%, or 24.67% at OT2 (2-nt mismatches, 1 bulge); 1.59%, 1.69%, or 4.27% at OT3 (2-nt mismatches, 2 bulges); and barely detectable at OT4 (3-nt mismatches, no bulge). The A-to-G off-target editing efficiency of UminiABEs, SUminiABEs or STUminiABEs corresponding to the HEK3-3 site were 34.93%, 48.84%, or 57.20% at OT1 (2-nt mismatches, 1 bulge); 1.91%, 6.39%, or 17.17% at OT2 (2-nt mismatches, 1 bulge), and barely detectable at OT3 (2-nt mismatches, 1 bulge). The A-to-G off-target editing efficiency of UminiABEs, SUminiABEs, or STUminiABEs corresponding to the KLF4-1 site were 19.55%, 32.39%, or 42.84% at OT1 (2-nt mismatches, 2 bulges); 7.70%, 17.09%, or 29.52% at OT5 (3-nt mismatches, no bulge); and barely detectable at other off-target loci (OT2, OT3, OT4, OT6, and OT7). As shown in Figure S9B, the C-to-T on-target editing efficiency of UminiCBEs, SUminiCBEs, or STUminiCBEs reached 35.38%, 46.62%, or 46.36% in C11 at the RUNX1-4 site; 33.87%, 46.69%, or 57.92% in C13 at the EMX1-5 site; and 47.49%, 59.95%, or 58.37% in C13 at the RUNX1-6 site. The C-to-T off-target editing efficiency of UminiCBEs, SUminiCBEs, or STUminiCBEs corresponding to the RUNX1-4 site were 1.80%, 3.27%, or 2.90% at OT4 (3-nt mismatches, no bulge) and barely detectable at other off-target loci (OT1, OT2, and OT3). The C-to-T off-target editing efficiency of UminiCBEs, SUminiCBEs, or STUminiCBEs corresponding to the EMX1-5 site were 1.53%, 2.60%, or 4.78% at OT1 (2-nt mismatches, 2 bulges); 16.04%, 22.23%, or 33.30% at OT5 (3-nt mismatches, no bulge); 2.67%, 3.33%, or 9.72% at OT7 (3-nt mismatches, no bulge); and barely detectable at other off-target loci (OT2, OT3, OT4, and OT6). The C-to-T off-target editing efficiency of UminiCBEs, SUminiCBEs, or STUminiCBEs corresponding to the RUNX1-6 site were 19.79%, 33.48%, or 43.2% at OT5 (3-nt mismatches, no bulge); 1.52%, 2.41%, or 12.09% at OT6 (3-nt mismatches, no bulge); 10.44%, 13.35%, or 36.47% at OT7 (3-nt mismatches, no bulge), and barely detectable at other off-target loci (OT1, OT2, OT3, and OT4). These data indicated that UminiCBEs, SUminiCBEs, or STUminiCBEs showed more specificity than UminiABEs, SUminiABEs, or STUminiABEs from the sites we examined. The off-target activity of UminiABEs, SUminiABEs, or STUminiABEs was high overall, which is a drawback for downstream applications. The further reduction of off-target activity for UminiBEs, SUminiBEs, and STUminiBEs will be required for their application in basic research or potential therapeutic applications.
Installation of a premature stop codon on the PCSK9 gene by STUminiCBE AAVs
SpCas9 nickase (D10A)-mediated base editors need to be packaged in dual-AAV vectors for their application in animal models or clinic trials because of their large size.7,8 However, the STUminiBEs can be packed in a single AAV vector owing to their smaller size. To demonstrate the potential applications of STUminiCBEs in vivo, we applied them to edit the therapeutic target gene PCSK9 by introducing a premature stop codon to repress the expression of PCSK9 protein.51 We first examined the efficiency of STUminiCBEs at 5 different sites to introduce a stop codon on the PCSK9 gene in HEK293FT cells (Figure 4A). The C-to-T editing efficiency in C12 of the PCSK9-2 site reached 63.52%, which was the most efficient CBE among the target cytosines for stop codon installation at five PCSK9 sites by STUminiCBEs. Thus, we selected the PCSK9-2 site for further study. We packaged STUminiCBEs (a sgRNA-PCSK9-2 expression cassette and a Sso7d-A3AW104A/Y132D-dUn1Cas12f1QM-UGI expression cassette) into a single AAV vector with a total size of 4.5 kb (Figure 4B) and generated STUminiCBE AAVs. As shown in Figure 4C, STUminiCBE AAVs successfully infected HEK293FT cells and transduced STUminiCBEs into cells, inducing the C-to-T transition with an editing efficiency of 34.02% in C12 when the MOI was 1 × 107 vector genomes (vg) per cell.
Figure 4.

Premature stop codons on the PCSK9 gene introduced by STUminiCBE AAVs in HEK293FT cells
(A) Comparison of C-to-T editing efficiencies mediated by STUminiCBE at different PCSK9 sites. The target cytosines (red) are used to be mutated and generate premature stop codons. PAM sequences are in light purple. Sequences in an open reading frame are translated into amino acids, which are shown in light blue. The red arrow refers to the target cytosines with the highest editing efficiency. (B) Schematic of STUminiCBE AAV delivered into HEK293FT cells. STUminiCBE is packaged into a single AAV vector within a cargo size of 4.5 kb, including the inverted terminal repeats. Cells are lysed to analyze the editing efficiencies 5 days after infection. (C) The C-to-T editing efficiency at PCSK9-2 site induced by STUminiCBE AAVs when MOI is 1 × 106 or 1 × 107 vg/cell. The red arrow refers to the CBE efficiency in C12 when the MOI is 1 × 107 vg/cell. All values and error bars represent means ± SD, n = 3 independent biological replicates.
Discussion
In summary, we obtained UminiBEs by engineering dUn1Cas12f1 and greatly improved their editing efficiency in mammalian cells if compared to previously engineered dCasMINI.10 By fusing the non-specific dsDNA-binding protein Sso7d to the N terminus of deaminases in UminiBEs, which barely affected cell viability (Figure S10), we obtained SUminiBEs with further enhanced editing efficiency. Moreover, by truncating the sgRNA of Un1Cas12f1, we obtained an sgRNA variant, which had almost half the length of the original sgRNA. Base editors containing truncated sgRNA variants were termed STUminiBEs, which maintained high editing efficiency.
dUn1Cas12f1QM variant (D143R/T147R/T203R/E206R)-mediated UminiBEs showed significantly increased BE activity over dUn1Cas12f1-BEs or dCasMINI-ABEs, which suggested that enhancing the interactions between dUn1Cas12f1 and the target dsDNA was critical. The mutations of dCasMINI were based on the alignment of the protein sequences between Un1Cas12f1 and Cas12a proteins.10 However, the high-resolution crystal structure of Un1Cas12f1-sgRNA-dsDNA showed that Un1Cas12f1, as an asymmetric protein dimer configuration, bound one sgRNA and target dsDNA,24,25 which was structurally distinct from the Cas12a protein.52,53 Thus, we rationally designed Un1Cas12f1 mutations based on the structure of the Un1Cas12f1-sgRNA-dsDNA complex. The point mutations of dUn1Cas12f1QM (D143R/T147R/T203R/E206R) were different from dCasMINI (D143R/T147R/K330R/E528R). The comparison of these mutations in the Un1Cas12f1-sgRNA-dsDNA structures is shown in Figure S11. D143R and T147R, the common mutations in dCasMINI, increased the dUn1Cas12f1 binding to the NTS of the target DNA. T203R and E206R could increase the dUn1Cas12f1 binding to either the TS or NTS of the target DNA on the opposite side of D143R and T147R.
SpCas9 nickase (D10A)-mediated base editors need to be packaged in dual-AAV vectors for their application in animal models of human diseases due to their large size.54,55,56,57,58 However, the dual-AAV system was constrained by recombination efficiency and dose-limiting toxicity.59 Thus, the development of a single-AAV BE system was urgent, which could decrease the required dose of AAV and was more suitable for use in clinical applications. Although single-AAV delivery of Cas9-based ABEs has been reported,60 the size is close or slightly over the 4.7-kb capacity. In this study, a STUminiCBE has been successfully packaged into a single AAV vector due to its more compact size compared to SpCas9-mediated CBEs. The size of the AAV-STUminiCBE was about 4.5 kb, which was less than the 4.7-kb packaging limit of AAV. We delivered STUminiCBEs into HEK293FT cells by AAVs to introduce premature stop codons at the PCSK9 site. We used the AAV8 vector, which targets the liver, pancreas, skeletal muscle, and other tissues.61 In addition to introducing premature stop codons into genomic target sites, STUminiBEs can be used in other applications in vivo, such as exon skipping and lineage tracing. Although the size of STUminiBEs is smaller than the packaging limit of AAV, it is still close to the 4.7-kb capacity, which affects the yield. In the future, other strategies need to be combined and further reduce the size of the STUminiBEs to increase the AAV yield.
STUminiBEs showed high off-target editing at some sgRNA-dependent off-target sites (Figure S9) when compared to Sso7d-dCas9-BEs (Figures S12 and S13). Off-target editing may cause safety issues and hinder the therapeutic applications of miniature base editors. The transformer BE (tBE) system constructed by using a cleavable deoxycytidine deaminase inhibitor domain could eliminate sgRNA-dependent off-target editing.58 It might be able to adopt the tBE system to STUminiBEs to reduce off-target activity in future studies.
The dUn1Cas12f1-derived base editors UminiBEs, SUminiBEs, and STUminiBEs introduced base conversions while generating very few indels (insertions and deletions) (<0.15%), although it was relatively higher than non-transfected control (mean: 0.03%) (Figure S14), which were potentially safer than nuclease or nickase-based genome editing. The detected indels of non-transfected control might be caused by amplification, deep sequencing, or alignment artifacts. The reason that the indels were higher in cells transfected with dUn1Cas12f1-derived BEs than in the control cells was not clear. It could be related to DNA damage induced by dUn1Cas12f1-mediated R-loop formation. The C-to-T editing efficiency by STUminiCBEs ranged from 25.33% to 64.80% with a mean of 45.22%, which was comparable to the C-to-T editing efficiency by hA3AW104A/Y132D-dCas12a-BE-op (BEACON1) ranging from ∼38% to ∼68%.41 Overall, the development of STUminiBEs enhances the potential of miniature CRISPR-Cas systems for biological research or therapeutical applications.
Materials and methods
Plasmid construction
Human codon-optimized Un1Cas12f1 gene fragments were synthesized by Azenta. The vector of miniABEmax (Addgene no. 131311) was used to generate dUn1Cas12f1-ABE protein expression plasmids. Firstly, P2A-EGFP in miniABEmax was replaced by P2A-sfGFP to get pCMV-miniABEmax-P2A-sfGFP. Secondly, TadA7.10-nCas9 (D10A) was replaced with TadA-TadA8e-dUn1Cas12f1 (D326A/D510A), resulting in pCMV-TadA-TadA8e-dUn1Cas12f1-P2A-sfGFP. Thirdly, dUn1Cas12f1 mutants were constructed through a series of site-specific mutations, and dUn1Cas12f1QM was the Un1Cas12f1 mutant resulting in the highest editing efficiency. DBD proteins were used as follows: Sso7d, HN1, HB1, and H1G were synthesized and cloned to pCMV-TadA-TadA8e-dUn1Cas12f1QM-P2A-sfGFP, resulting in pCMV-DBD-TadA-TadA8e-dUn1Cas12f1QM-P2A-sfGFP, pCMV-TadA-TadA8e-Sso7d-dUn1Cas12f1QM-P2A-sfGFP, and pCMV-TadA-TadA8e-dUn1Cas12f1QM-Sso7d-P2A-sfGFP, respectively. pCMV-BEACON1 (Addgene no. 171697) was used to generate dUn1Cas12f1-CBE protein expression plasmids. Firstly, P2A-sfGFP was inserted downstream of pCMV-BEACON1, resulting in pCMV-BEACON1-P2A-sfGFP. Secondly, dCas12a was replaced by dUn1Cas12f1QM, resulting in pCMV-A3AW104A/Y132D-dUn1Cas12f1QM-UGI-P2A-sfGFP. Finally, Sso7d was cloned into the pCMV-A3AW104A/Y132D-dUn1Cas12f1QM-UGI-P2A-sfGFP, resulting in pCMV-Sso7d-A3AW104A/Y132D-dUn1Cas12f1QM-UGI-P2A-sfGFP. dUn1Cas12f1-CGBEs and STUminiCGBEs protein expression plasmids were constructed based on pCMV_BE4max (Addgene no. 112093). Firstly, P2A-sfGFP was cloned downstream of UGI to obtain pCMV_BE4max-P2A-sfGFP. Subsequently, A1-nCas9-UGI-UGI in pCMV_BE4max-P2A-sfGFP was replaced by A1R33A-dUn1Cas12f1-UNG1, A1-dUn1Cas12f1-UdgX, eA3A-dUn1Cas12f1, Sso7d-A1R33A-dUn1Cas12f1-UNG1, Sso7d-A1-dUn1Cas12f1-UdgX, or Sso7d-eA3A-dUn1Cas12f1, respectively, resulting in dUn1Cas12f1-CGBE1, dUn1Cas12f1-CGBE2, dUn1Cas12f1-CGBE3, STUminiCGBE1, STUminiCGBE2, or STUminiCGBE3 protein expression plasmid. To construct the dUn1Cas12f1-AYBE or STUminiAYBE expression plasmid, MPGm was synthesized and cloned into pCMV-TadA-TadA-8e-dUn1Cas12f1-P2A-sfGFP or pCMV-Sso7d-TadA-TadA-8e-dUn1Cas12f1QM-P2A-sfGFP, resulting in pCMV-TadA-TadA-8e-dUn1Cas12f1-MPGm-P2A-sfGFP or pCMV-Sso7d-TadA-TadA-8e-dUn1Cas12f1QM-MPGm-P2A-sfGFP. To construct sgRNA expression plasmids for dUn1Cas12f1-BEs, the sgRNA scaffold with the U6 sequence was cloned into pDONOR5.1-CcdB-hPGK-puromycin-P2A-tagBFPnls, resulting in pDONOR5.1-U6-sgRNA scaffold-CcdB-hPGK-puromycin-P2A-tagBFPnls. Then, the engineered sgRNA scaffold was cloned into pDONOR5.1-U6-sgRNA scaffold-CcdB-hPGK-puromycin-P2A-tagBFPnls, resulting in pDONOR5.1-U6-engineered sgRNA scaffold-CcdB-hPGK-puromycin-P2A-tagBFPnls, respectively. Finally, the oligos for sgRNA spacers of target sites synthesized by Azenta were annealed and cloned into empty plasmids, resulting in target sgRNA expression plasmids. The sgRNA spacer sequences are listed in Tables S1 and S2. The AAV-STUminiCBE plasmid was constructed based on the AAV-CMV-EGFP plasmid. CMV-EGFP in AAV-CMV-EGFP was replaced by U6-sgRNA-D1/D2/D3-PCSK9-2 and CMV-Sso7d-A3AW104A/Y132D-dUn1Cas12f1QM-UGI to obtain the AAV-STUminiCBE plasmid.
Cell culture and transfection and genomic DNA extraction
HEK293FT cells were cultured in DMEM with high glucose supplemented with 10% fetal bovine serum (Gibco, 16000-044) and 1% Pen-Strep (100×) (Gibco, 15140-163) at 37°C with 5% CO2 in a humidified incubator. Cells were seeded in 12-well plates with 60%–80% confluency for transient transfection by Lipofectamine 2000 (Invitrogen, 11668019) following the manufacturer’s protocols. Cells were co-transfected with 1.5 μg of dUn1Cas12f1-BE protein expression plasmid and 1.0 μg of dUn1Cas12f1-BE sgRNA expression plasmid. The medium was replaced 12–24 h after transfection. The GFP (dUn1Cas12f1-BE protein) and BFP (dUn1Cas12f1-BE sgRNA) double-positive cells were sorted by a BD FACS Aria III flow cytometer 60–72 h after transfection. The genomic DNAs of cells were extracted using 20 μL of QuickExtract DNA Extraction Solution 1.0 (Lucigen, QE0905T) following the manufacturer’s protocol. The resulting genomic DNA mixture was stored at 4°C for the BE efficiency test.
Detection of base-editing efficiency
For deep sequencing analysis of BE efficiency, the samples were prepared by two rounds of polymerase chain reaction (PCR). For the primary PCR, 1 μL genomic DNA mixture was amplified by target-specific primers with adapters in 20 μL total volume to produce 200- to 250-bp-long amplicons. The target-specific primers to amplify the genome sequence regions of interests were designed by using the online Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast) and synthesized by Azenta. For the second PCR, 1 μL primary PCR products were amplified by primers with TruSeq indexes in 20 μL total volume. All PCR reactions were performed using Phanta Flash Master Mix (Vazyme, P510). PCR products with different indexes were pooled together in equal amounts, and the gel purification of the PCR product mixture was performed with the FastPure Gel DNA Extraction Mini Kit (Vazyme, DC301). The purified samples were sequenced by the 150-bp paired-end reads Illumina NovaSeq 6000 platform (LianChuan). Primers used in amplifying target genomic DNAs are provided in Table S3.
For analyzing the deep sequencing data, the quantification of editing efficiency, including indels and A-to-G or C-to-T conversions, was calculated by a homemade script to execute CRISPResso2 with the quality control parameter --min_average_read_quality 10.62 The sequenced reads were locally aligned with the unedited original reference sequences. Specifically, after alignment, the target positions corresponding to sgRNA were examined. If the input sequences matched to the reference sequences of sgRNA, then they would be classified as unedited reads. Otherwise, it would be checked whether the alignment lengths were consistent. Inconsistent lengths indicated that the input sequences had insertion or deletion mutations, labeled as indels. If the alignment lengths were consistent, each base would be evaluated individually. Bases consistent with the expected conversions induced by specific base editors were marked as effective edits (A-to-G conversions induced by ABEs, C-to-T conversions induced by CBEs). The efficiencies of conversions were defined as reads of effective edits/total reads. Script is available at https://github.com/Hanhui-Ma-Lab/NGS_Tools.
AAV production and cell transduction
The product of STUminiCBE AAVs was provided by Belief BioMed (Shanghai) with a final physical titration of 7.88 × 1012 vg/mL. The specific AAV serotype we used was AAV8. 1 mg/mL AAV-STUminiCBE plasmid, 1 mg/mL Rep/Cap plasmid, and 2 mg/mL AdHelper plasmid were co-transfected into HEK293 cells when the cell density reached 5 × 106 cells/mL. After transfection for 72 h, the pellet and supernatant were collected, and the AAV viral particles were purified using iodixanol gradient ultracentrifugation. The purified viral particles were digested by DNase I (Roche, 10104159001) for 30 min at 37°C. The amount of VP3 capsid protein was determined by Pierce Silver Stain Kit (Thermo Scientific, 24612) based on the standard curve prepared from serial dilutions of standard BSA protein.
HEK293FT cells were seeded at 5 × 104 per well in 24-well plates. Cells were infected by STUminiCBE AAVs at an MOI of 106 or 107 vg per cell, respectively. Cells were collected 5 days after AAV infection, and the editing efficiencies were evaluated by deep sequencing.
CellTiter-Lumi assay
HEK293FT cells were seeded in 12-well plates with 60%–80% confluency for transient transfection with 1.5 μg of UminiBE protein expression plasmid, 1.5 μg of UminiBE protein expression plasmid, and 1.0 μg of UminiBE sgRNA expression plasmid; 1.5 μg of SUminiBE protein expression plasmid, 1.5 μg of SUminiBE protein expression plasmid, and 1.0 μg of SUminiBE sgRNA expression plasmid; or Lipofectamine 2000 control. 5×103 cells of each well were subcultured into black 96-well plates 24 h after transfection. After transfection for 72 h, the same volume of CellTiter-Lumi Luminescent Cell Viability Assay Kit (Beyotime, C0065S) as the culture medium (100 μL) was added to each well and incubated at room temperature for 10–15 min in the dark, and luminescence parameters were detected on a microplate reader to evaluate the activity of cells.
Data and code availability
The next-generation sequencing data have been deposited in the NCBI Sequence Read Archive database under the BioProject accession code PRJNA1022844. All other relevant data are available from the corresponding authors upon reasonable request.
Acknowledgments
We thank the Wei Qi Lab for the gift of HEK293FT cells. We thank Pengwei Zhang and Lishuang Zhang for their help with cell sorting. Fluorescence-activated cell sorting was provided by the Shanghai Institute for Advanced Immunochemical Studies (SIAIS) at ShanghaiTech University. We are grateful to the Jia Chen Lab for the gifts of pCMV-BEACON1, miniABEmax, and pCMV_BE4max plasmids. We are grateful to the Guisheng Zhong Lab for the gift of the AAV-CMV-EGFP plasmid. This work was funded by the National Natural Science Foundation of China (no. 31970591 to H.M.) and the Shanghai Science and Technology Innovation Action Plan (21JC1404800 to H.M.).
Author contributions
H.M. conceived the project. Y.H., L.H., and H.M. designed the experiments. Y.S. performed the structural analysis for Un1Cas12f1. Y.H. and L.H. performed the experiments for Un1Cas12f1 and sgRNA engineering and base-editing assays. Z.D., W.J., and X.W. performed the experiments for AAV packaging. Q.M., Y.H., and L.H. analyzed the next-generation sequencing data. Y.H., X.X., J.Z., and H.M. interpreted the data. Y.H., L.H., and H.M. wrote the paper with input from all authors.
Declaration of interests
The authors have filed patent applications on UminiBEs, SUminiBEs, and STUminiBEs through ShanghaiTech University.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2024.102201.
Supplemental information
References
- 1.Landrum M.J., Lee J.M., Riley G.R., Jang W., Rubinstein W.S., Church D.M., Maglott D.R. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:980–985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rees H.A., Liu D.R. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018;19:770–788. doi: 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tong S., Moyo B., Lee C.M., Leong K., Bao G. Engineered materials for in vivo delivery of genome-editing machinery. Nat. Rev. Mater. 2019;4:726–737. doi: 10.1038/s41578-019-0145-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zincarelli C., Soltys S., Rengo G., Rabinowitz J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 7.Dong J.Y., Fan P.D., Frizzell R.A. Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum. Gene Ther. 1996;7:2101–2112. doi: 10.1089/hum.1996.7.17-2101. [DOI] [PubMed] [Google Scholar]
- 8.Wu Z., Yang H., Colosi P. Effect of genome size on AAV vector packaging. Mol. Ther. 2010;18:80–86. doi: 10.1038/mt.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim D.Y., Lee J.M., Moon S.B., Chin H.J., Park S., Lim Y., Kim D., Koo T., Ko J.H., Kim Y.S. Efficient CRISPR editing with a hypercompact Cas12f1 and engineered guide RNAs delivered by adeno-associated virus. Nat. Biotechnol. 2022;40:94–102. doi: 10.1038/s41587-021-01009-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu X., Chemparathy A., Zeng L., Kempton H.R., Shang S., Nakamura M., Qi L.S. Engineered miniature CRISPR-Cas system for mammalian genome regulation and editing. Mol. Cell. 2021;81:4333–4345.e4. doi: 10.1016/j.molcel.2021.08.008. [DOI] [PubMed] [Google Scholar]
- 11.Wu Z., Zhang Y., Yu H., Pan D., Wang Y., Wang Y., Li F., Liu C., Nan H., Chen W., Ji Q. Programmed genome editing by a miniature CRISPR-Cas12f nuclease. Nat. Chem. Biol. 2021;17:1132–1138. doi: 10.1038/s41589-021-00868-6. [DOI] [PubMed] [Google Scholar]
- 12.Kim D.Y., Chung Y., Lee Y., Jeong D., Park K.H., Chin H.J., Lee J.M., Park S., Ko S., Ko J.H., Kim Y.S. Hypercompact adenine base editors based on a Cas12f variant guided by engineered RNA. Nat. Chem. Biol. 2022;18:1005–1013. doi: 10.1038/s41589-022-01077-5. [DOI] [PubMed] [Google Scholar]
- 13.Zhang S., Song L., Yuan B., Zhang C., Cao J., Chen J., Qiu J., Tai Y., Chen J., Qiu Z., et al. TadA reprogramming to generate potent miniature base editors with high precision. Nat. Commun. 2023;14:413. doi: 10.1038/s41467-023-36004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu T., Liu C., Zou S., Lyu R., Yang B., Yan H., Zhao M., Tang W. An engineered hypercompact CRISPR-Cas12f system with boosted gene-editing activity. Nat. Chem. Biol. 2023;19:1384–1393. doi: 10.1038/s41589-023-01380-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu W.Y., Mohanraju P., Liao C., Adiego-Pérez B., Creutzburg S.C.A., Makarova K.S., Keessen K., Lindeboom T.A., Khan T.S., Prinsen S., et al. The miniature CRISPR-Cas12m effector binds DNA to block transcription. Mol. Cell. 2022;82:4487–4502.e7. doi: 10.1016/j.molcel.2022.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Chen W., Ma J., Wu Z., Wang Z., Zhang H., Fu W., Pan D., Shi J., Ji Q. Cas12n nucleases, early evolutionary intermediates of type V CRISPR, comprise a distinct family of miniature genome editors. Mol. Cell. 2023;83:2768–2780.e6. doi: 10.1016/j.molcel.2023.06.014. [DOI] [PubMed] [Google Scholar]
- 17.Karvelis T., Druteika G., Bigelyte G., Budre K., Zedaveinyte R., Silanskas A., Kazlauskas D., Venclovas Č., Siksnys V. Transposon-associated TnpB is a programmable RNA-guided DNA endonuclease. Nature. 2021;599:692–696. doi: 10.1038/s41586-021-04058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Altae-Tran H., Kannan S., Demircioglu F.E., Oshiro R., Nety S.P., McKay L.J., Dlakić M., Inskeep W.P., Makarova K.S., Macrae R.K., et al. The widespread IS200/IS605 transposon family encodes diverse programmable RNA-guided endonucleases. Science. 2021;374:57–65. doi: 10.1126/science.abj6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han D., Xiao Q., Wang Y., Zhang H., Dong X., Li G., Kong X., Wang S., Song J., Zhang W., et al. Development of miniature base editors using engineered IscB nickase. Nat. Methods. 2023;20:1029–1036. doi: 10.1038/s41592-023-01898-9. [DOI] [PubMed] [Google Scholar]
- 20.Saito M., Xu P., Faure G., Maguire S., Kannan S., Altae-Tran H., Vo S., Desimone A., Macrae R.K., Zhang F. Fanzor is a eukaryotic programmable RNA-guided endonuclease. Nature. 2023;620:660–668. doi: 10.1038/s41586-023-06356-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang K., Lim J., Sgrizzi S., Trinh M., Kayabolen A., Yutin N., Bao W., Kato K., Koonin E.V., Gootenberg J.S., Abudayyeh O.O. Programmable RNA-guided DNA endonucleases are widespread in eukaryotes and their viruses. Sci. Adv. 2023;9 doi: 10.1126/sciadv.adk0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harrington L.B., Burstein D., Chen J.S., Paez-Espino D., Ma E., Witte I.P., Cofsky J.C., Kyrpides N.C., Banfield J.F., Doudna J.A. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science. 2018;362:839–842. doi: 10.1126/science.aav4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karvelis T., Bigelyte G., Young J.K., Hou Z., Zedaveinyte R., Budre K., Paulraj S., Djukanovic V., Gasior S., Silanskas A., et al. PAM recognition by miniature CRISPR-Cas12f nucleases triggers programmable double-stranded DNA target cleavage. Nucleic Acids Res. 2020;48:5016–5023. doi: 10.1093/nar/gkaa208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takeda S.N., Nakagawa R., Okazaki S., Hirano H., Kobayashi K., Kusakizako T., Nishizawa T., Yamashita K., Nishimasu H., Nureki O. Structure of the miniature type V-F CRISPR-Cas effector enzyme. Mol. Cell. 2021;81:558–570.e3. doi: 10.1016/j.molcel.2020.11.035. [DOI] [PubMed] [Google Scholar]
- 25.Xiao R., Li Z., Wang S., Han R., Chang L. Structural basis for substrate recognition and cleavage by the dimerization-dependent CRISPR–Cas12f nuclease. Nucleic Acids Res. 2021;49:4120–4128. doi: 10.1093/nar/gkab179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richter M.F., Zhao K.T., Eton E., Lapinaite A., Newby G.A., Thuronyi B.W., Wilson C., Koblan L.W., Zeng J., Bauer D.E., et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020;38:883–891. doi: 10.1038/s41587-020-0453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding X., Seebeck T., Feng Y., Jiang Y., Davis G.D., Chen F. Improving CRISPR-Cas9 genome editing efficiency by fusion with chromatin-modulating peptides. CRISPR J. 2019;2:51–63. doi: 10.1089/crispr.2018.0036. [DOI] [PubMed] [Google Scholar]
- 28.Yang C., Ma Z., Wang K., Dong X., Huang M., Li Y., Zhu X., Li J., Cheng Z., Bi C., Zhang X. HMGN1 enhances CRISPR-directed dual-function A-to-G and C-to-G base editing. Nat. Commun. 2023;14:2430. doi: 10.1038/s41467-023-38193-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin S., Zhang M., Liu Y., Sun X., Guan Y., Chen X., Yang L., Huo Y., Yang J., Zhang X., et al. Engineering of efficiency-enhanced Cas9 and base editors with improved gene therapy efficacies. Mol. Ther. 2023;31:744–759. doi: 10.1016/j.ymthe.2022.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choli T., Henning P., Wittmann-Liebold B., Reinhardt R. Isolation, characterization and microsequence analysis of a small basic methylated DNA-binding protein from the Archaebacterium, Sulfolobus solfataricus. Biochim. Biophys. Acta. 1988;950:193–203. doi: 10.1016/0167-4781(88)90011-5. [DOI] [PubMed] [Google Scholar]
- 31.Agback P., Baumann H., Knapp S., Ladenstein R., Härd T. Architecture of nonspecific protein–DNA interactions in the Sso7d–DNA complex. Nat. Struct. Biol. 1998;5:579–584. doi: 10.1038/836. [DOI] [PubMed] [Google Scholar]
- 32.Kalichuk V., Béhar G., Renodon-Cornière A., Danovski G., Obal G., Barbet J., Mouratou B., Pecorari F. The archaeal “7 kDa DNA-binding” proteins: extended characterization of an old gifted family. Sci. Rep. 2016;6 doi: 10.1038/srep37274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y., Prosen D.E., Mei L., Sullivan J.C., Finney M., Vander Horn P.B. A novel strategy to engineer DNA polymerases for enhanced processivity and improved performance in vitro. Nucleic Acids Res. 2004;32:1197–1207. doi: 10.1093/nar/gkh271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bustin M. Chromatin unfolding and activation by HMGN∗ chromosomal proteins. Trends Biochem. Sci. 2001;26:431–437. doi: 10.1016/s0968-0004(01)01855-2. [DOI] [PubMed] [Google Scholar]
- 35.Kugler J.E., Deng T., Bustin M. The HMGN family of chromatin-binding proteins: dynamic modulators of epigenetic processes. Biochim. Biophys. Acta. 2012;1819:652–656. doi: 10.1016/j.bbagrm.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.González-Romero R., Eirín-López J.M., Ausió J. Evolution of high mobility group nucleosome-binding proteins and its implications for vertebrate chromatin specialization. Mol. Biol. Evol. 2015;32:121–131. doi: 10.1093/molbev/msu280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klass J., Murphy F.V., 4th, Fouts S., Serenil M., Changela A., Siple J., Churchill M.E.A. The role of intercalating residues in chromosomal high-mobility-group protein DNA binding, bending and specificity. Nucleic Acids Res. 2003;31:2852–2864. doi: 10.1093/nar/gkg389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomas J.O., Stott K. H1 and HMGB1: modulators of chromatin structure. Biochem. Soc. Trans. 2012;40:341–346. doi: 10.1042/BST20120014. [DOI] [PubMed] [Google Scholar]
- 39.Harshman S.W., Young N.L., Parthun M.R., Freitas M.A. H1 histones: current perspectives and challenges. Nucleic Acids Res. 2013;41:9593–9609. doi: 10.1093/nar/gkt700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hergeth S.P., Schneider R. The H1 linker histones: multifunctional proteins beyond the nucleosomal core particle. EMBO Rep. 2015;16:1439–1453. doi: 10.15252/embr.201540749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X., Ding C., Yu W., Wang Y., He S., Yang B., Xiong Y.C., Wei J., Li J., Liang J., et al. Cas12a base editors induce efficient and specific editing with low DNA damage response. Cell Rep. 2020;31 doi: 10.1016/j.celrep.2020.107723. [DOI] [PubMed] [Google Scholar]
- 42.Mol C.D., Arvai A.S., Sanderson R.J., Slupphaug G., Kavli B., Krokan H.E., Mosbaugh D.W., Tainer J.A. Crystal structure of human uracil–DNA glycosylase in complex with a protein inhibitor: protein mimicry of DNA. Cell. 1995;82:701–708. doi: 10.1016/0092-8674(95)90467-0. [DOI] [PubMed] [Google Scholar]
- 43.Zhao D., Li J., Li S., Xin X., Hu M., Price M.A., Rosser S.J., Bi C., Zhang X. Glycosylase base editors enable C-to-A and C-to-G base changes. Nat. Biotechnol. 2021;39:35–40. doi: 10.1038/s41587-020-0592-2. [DOI] [PubMed] [Google Scholar]
- 44.Kurt I.C., Zhou R., Iyer S., Garcia S.P., Miller B.R., Langner L.M., Grünewald J., Joung J.K. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol. 2021;39:41–46. doi: 10.1038/s41587-020-0609-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen L., Park J.E., Paa P., Rajakumar P.D., Prekop H.T., Chew Y.T., Manivannan S.N., Chew W.L. Programmable C:G to G:C genome editing with CRISPR-Cas9-directed base excision repair proteins. Nat. Commun. 2021;12:1384. doi: 10.1038/s41467-021-21559-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koblan L.W., Arbab M., Shen M.W., Hussmann J.A., Anzalone A.V., Doman J.L., Newby G.A., Yang D., Mok B., Replogle J.M., et al. Efficient C•G-to-G•C base editors developed using CRISPRi screens, target-library analysis, and machine learning. Nat. Biotechnol. 2021;39:1414–1425. doi: 10.1038/s41587-021-00938-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun N., Zhao D., Li S., Zhang Z., Bi C., Zhang X. Reconstructed glycosylase base editors GBE2.0 with enhanced C-to-G base editing efficiency and purity. Mol. Ther. 2022;30:2452–2463. doi: 10.1016/j.ymthe.2022.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tong H., Wang X., Liu Y., Liu N., Li Y., Luo J., Ma Q., Wu D., Li J., Xu C., Yang H. Programmable A-to-Y base editing by fusing an adenine base editor with an N-methylpurine DNA glycosylase. Nat. Biotechnol. 2023;41:1080–1084. doi: 10.1038/s41587-022-01595-6. [DOI] [PubMed] [Google Scholar]
- 49.Chen L., Hong M., Luan C., Gao H., Ru G., Guo X., Zhang D., Zhang S., Li C., Wu J., et al. Adenine transversion editors enable precise, efficient A•T-to-C•G base editing in mammalian cells and embryos. Nat. Biotechnol. 2024;42:638–650. doi: 10.1038/s41587-023-01821-9. [DOI] [PubMed] [Google Scholar]
- 50.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ding Q., Strong A., Patel K.M., Ng S.L., Gosis B.S., Regan S.N., Cowan C.A., Rader D.J., Musunuru K. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ. Res. 2014;115:488–492. doi: 10.1161/CIRCRESAHA.115.304351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamano T., Nishimasu H., Zetsche B., Hirano H., Slaymaker I.M., Li Y., Fedorova I., Nakane T., Makarova K.S., Koonin E.V., et al. Crystal structure of Cpf1 in complex with guide RNA and target DNA. Cell. 2016;165:949–962. doi: 10.1016/j.cell.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gao P., Yang H., Rajashankar K.R., Huang Z., Patel D.J. Type V CRISPR-Cas Cpf1 endonuclease employs a unique mechanism for crRNA-mediated target DNA recognition. Cell Res. 2016;26:901–913. doi: 10.1038/cr.2016.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Villiger L., Grisch-Chan H.M., Lindsay H., Ringnalda F., Pogliano C.B., Allegri G., Fingerhut R., Häberle J., Matos J., Robinson M.D., et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 2018;24:1519–1525. doi: 10.1038/s41591-018-0209-1. [DOI] [PubMed] [Google Scholar]
- 55.Yeh W.H., Shubina-Oleinik O., Levy J.M., Pan B., Newby G.A., Wornow M., Burt R., Chen J.C., Holt J.R., Liu D.R. In vivo base editing restores sensory transduction and transiently improves auditory function in a mouse model of recessive deafness. Sci. Transl. Med. 2020;12 doi: 10.1126/scitranslmed.aay9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koblan L.W., Erdos M.R., Wilson C., Cabral W.A., Levy J.M., Xiong Z.M., Tavarez U.L., Davison L.M., Gete Y.G., Mao X., et al. In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice. Nature. 2021;589:608–614. doi: 10.1038/s41586-020-03086-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chemello F., Chai A.C., Li H., Rodriguez-Caycedo C., Sanchez-Ortiz E., Atmanli A., Mireault A.A., Liu N., Bassel-Duby R., Olson E.N. Precise correction of Duchenne muscular dystrophy exon deletion mutations by base and prime editing. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abg4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang L., Xue W., Zhang H., Gao R., Qiu H., Wei J., Zhou L., Lei Y.N., Wu X., Li X., et al. Eliminating base-editor-induced genome-wide and transcriptome-wide off-target mutations. Nat. Cell Biol. 2021;23:552–563. doi: 10.1038/s41556-021-00671-4. [DOI] [PubMed] [Google Scholar]
- 59.Kuzmin D.A., Shutova M.V., Johnston N.R., Smith O.P., Fedorin V.V., Kukushkin Y.S., van der Loo J.C.M., Johnstone E.C. The clinical landscape for AAV gene therapies. Nat. Rev. Drug Discov. 2021;20:173–174. doi: 10.1038/d41573-021-00017-7. [DOI] [PubMed] [Google Scholar]
- 60.Davis J.R., Wang X., Witte I.P., Huang T.P., Levy J.M., Raguram A., Banskota S., Seidah N.G., Musunuru K., Liu D.R. Efficient in vivo base editing via single adeno-associated viruses with size-optimized genomes encoding compact adenine base editors. Nat. Biomed. Eng. 2022;6:1272–1283. doi: 10.1038/s41551-022-00911-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Issa S.S., Shaimardanova A.A., Solovyeva V.V., Rizvanov A.A. Various AAV Serotypes and Their Applications in Gene Therapy: An Overview. Cells. 2023;12:785. doi: 10.3390/cells12050785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R., Hsu J.Y., Cole M.A., Liu D.R., Joung J.K., Bauer D.E., Pinello L. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019;37:224–226. doi: 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The next-generation sequencing data have been deposited in the NCBI Sequence Read Archive database under the BioProject accession code PRJNA1022844. All other relevant data are available from the corresponding authors upon reasonable request.