

Abstract
Immune thrombocytopenia (ITP) is an autoimmune bleeding disorder affecting approximately 1 in 20,000 people. While most patients with ITP are successfully managed with the current set of standard and approved therapeutics, patients who cannot be adequately managed with these therapies, considered to have refractory ITP, are not uncommon. Therefore, there remains an ongoing need for novel therapeutics and drug development in ITP. Several agents exploiting novel targets and mechanisms in ITP are presently under clinical development, with trials primarily recruiting heavily pre-treated patients and those with otherwise refractory disease. Such agents include the neonatal Fc receptor antagonist efgartigimod, the Bruton tyrosine kinase inhibitor rilzabrutinib, the complement inhibitors sutimlimab and iptacopan, and anti-CD38 monoclonal antibodies such as daratumumab and mezagitamab, among others. Each of these agents exploits therapeutic targets or other aspects of ITP pathophysiology currently not targeted by the existing approved agents (thrombopoietin receptor agonists and fostamatinib). This manuscript offers an in-depth review of the current available data for novel therapeutics in ITP presently undergoing phase 2 or 3 studies in patients with heavily pretreated or refractory ITP. It additionally highlights the future directions for drug development in refractory ITP, including discussion of innovative clinical trial designs, health-related quality of life as an indispensable clinical trial endpoint and balancing potential toxicities of drugs with their potential benefits in a bleeding disorder in which few patients suffer life-threatening bleeding.
Keywords: Platelets, immune thrombocytopenia, ITP, refractory, efgartigimod, rilzabrutinib, rozanolixizumab, daratumumab, iptacopan, mezagitamab, sutimlimab, umbrella trials
INTRODUCTION
Immune thrombocytopenia (ITP), an autoimmune acquired bleeding disorder, results from a combination of increased platelet destruction in the reticuloendothelial system as well as inadequate compensatory platelet production. Multiple pathophysiologic mechanisms have been demonstrated in ITP, including glycoprotein-specific platelet autoantibodies responsible for platelet destruction and megakaryocyte apoptosis, complement-mediated platelet destruction, and the direct action of cytotoxic T-cells on platelets (1–3) Patients with ITP are currently managed primarily with corticosteroids and intravenous immunoglobulin (IVIG) in the newly-diagnosed and rescue settings, with thrombopoietin receptor agonists, rituximab, fostamatinib and splenectomy forming the therapeutic armamentarium used to treat patients with longer lasting disease (4, 5).
Despite the FDA approval of four drugs for ITP (romiplostim, eltrombopag, avatrombopag and fostamatinib) and development of a strong body of evidence for a fifth (rituximab), a small but significant minority of patients have disease inadequately responsive to these therapies. These patients are variably managed with experimental combination approaches, toxic use of chronic corticosteroids, regular IVIG infusions, or a whole host of salvage therapies, off-label immunosuppressants with generally only limited observational retrospective data to guide their use in ITP (4, 6). While the ideal definition of “refractory ITP” in the modern day remains unclear and controversial, for the purposes of this review, we will consider patients with inadequate responses to multiple or all approved therapies to have refractory ITP. Many such patients ultimately seek treatment in the setting of a clinical trial of a novel agent. Thankfully, many such trials are currently ongoing, evaluating agents with completely distinct targets and mechanisms of action than currently approved drugs. In this review, we describe agents being evaluated in patients with refractory ITP that have reached the phase 2 or phase 3 stage in ongoing or completed clinical trials. We additionally highlight the necessary future directions for drug development in refractory ITP, including emphasis on health-related quality of life as an indispensable clinical trial endpoint and balance of potential toxicities of drugs with their potential benefits in a bleeding disorder in which few patients suffer life-threatening bleeding.
NEONATAL FC RECEPTOR ANTAGONISM
Principles and Rationale
The neonatal Fc receptor (FcRn), named as a result of its initial discovery in the neonatal rodent gut, is critical in IgG and albumin recycling (thus enabling the normal half-life of circulating IgG of 21 days) as well as in the passive antibody transfer from mother to fetus (7, 8). Under normal circumstances, IgG bound to the FcRn in cellular endosomes is rescued from degradation in lysosomes (9, 10). This significantly prolongs the half-life of, and thereby increases the concentration of, circulating IgG, Figure 1A. Therefore, antagonists of the FcRn have potential therapeutic value in the treatment of humoral autoimmune diseases. In antagonizing the FcRn, the half-life of circulating IgG and therefore its plasma concentration is reduced significantly. This results in increased degradation of both desirable protective antibodies as well as pathologic autoantibodies (Figure 1B), with the therapeutic aim being to reduce autoantibody titers sufficiently to reduce or eliminate manifestations of the autoimmune disorder while not excessively reducing overall IgG levels such that the individual is at a significantly increased risk of infection. These agents have no impact on levels of other immunoglobulin isotypes. Because the FcRn also recycles albumin at a distinct binding site from IgG (11), the impact of FcRn antagonists on albumin levels has been an important consideration during drug development.
Figure 1. FcRn mode of action in protection of IgG from degradation and how FcRn inhibitors disrupt IgG recycling.
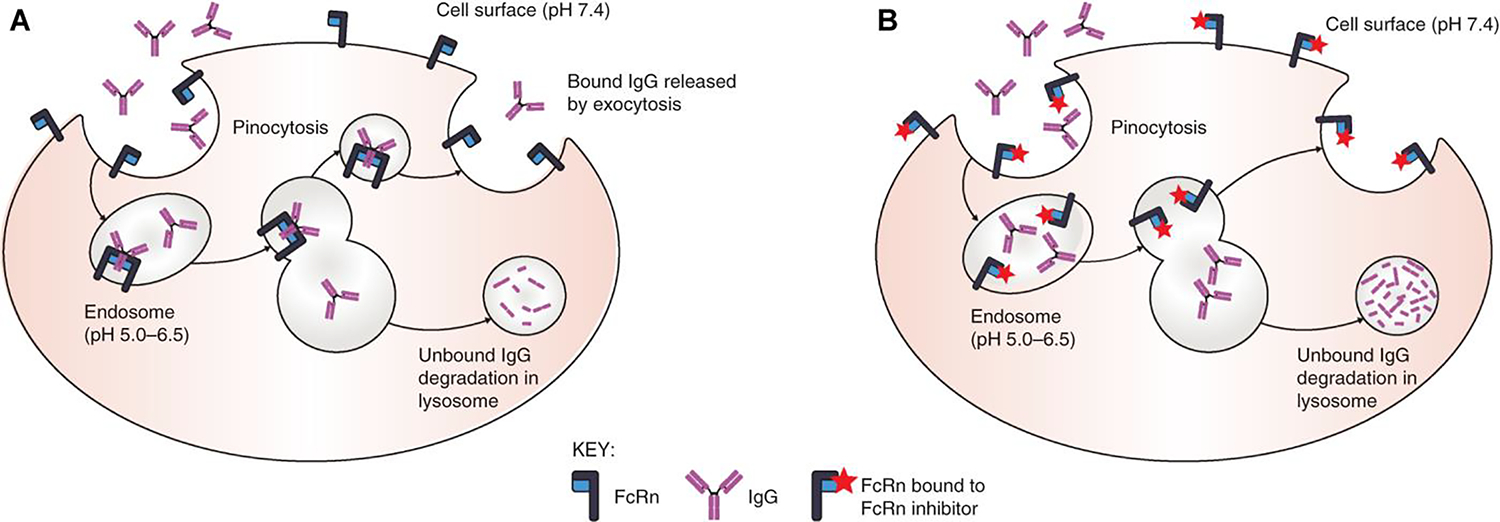
(A) IgG is ingested by pinocytosis. Pinocytotic vesicles fuse with acidic endosomes in which FcRn can bind IgG. Excess unbound IgG and other proteins enter the lysosome and are degraded. IgG bound to FcRn is retained and released by exocytosis. (B) FcRn inhibitors bind to FcRn in both neutral and acidic environments; in the presence of FcRn inhibitors, ingested IgG is unable to bind to FcRn; the unbound IgG enters the lysosome and is degraded. For illustrative purposes, albumin binding is not shown. Reproduced with permission from Patel and Bussel (56).
While of obvious therapeutic potential in ITP given the well-documented role of pathologic glycoprotein-specific autoantibodies in this disease (12, 13), FcRn antagonists have been or are presently being evaluated in a wide spectrum of autoimmune disorders (14). For example, the first FDA and EMA approvals of an FcRn antagonist (efgartigimod) were for the treatment of generalized myasthenia gravis (15).
Efgartigimod alfa
Efgartigimod alfa (Vyvgart, argenx SE, the Netherlands) is a first-in-class FcRn antagonist currently approved for the treatment of adults with myasthenia gravis in the US and EU. Efgartigimod is a human IgG1 antibody Fc fragment, engineered via a five amino acid substitution to have increased affinity for the FcRn at both neutral and acidic pH (16). By outcompeting IgG for the FcRn, greater quantities of IgG are susceptible to, and therefore undergo, lysosomal degradation. Efgartigimod does not reduce serum albumin levels (16, 17). It may be administered either as an intravenous infusion or a subcutaneous injection. In a phase 1 study in healthy volunteers, efgartigimod safely and sustainably reduced total IgG levels (16).
In a published Phase 2 study from Newland and colleagues (NCT03102593), 38 adults with ITP, mostly patients refractory to many lines of therapy, were randomized 1:1:1 to receive four weekly intravenous infusions of either placebo or efgartigimod at a dose of 5 mg/kg or 10 mg/kg (18). More patients receiving efgartigimod had a clinically relevant improvement in platelet count: a count of >50 × 109/L on 2 or more measures was achieved by 46% of patients receiving efgartigimod versus 25% of patients receiving placebo, and a count of >50 × 109/L on 10 or more consecutive days was achieved by 38% of patients receiving efgartigimod versus 0% of patients receiving placebo. Concurrent with the rapid platelet count improvement was a rapid reduction in total plasma IgG, with maximum mean reductions of 60.4% in patients receiving efgartigimod 5 mg/kg and 63.7% in those receiving efgartigimod 10 mg/kg; IgG levels in the placebo group remained essentially unchanged. Consistent with this, a reduction in measured platelet autoantibody signal declined by over 40% in 66.7% of patients with glycoprotein-specific platelet autoantibodies measured prior to treatment receiving efgartigimod 5 mg/kg and in 70% of such patients receiving efgartigimod 10 mg/kg. The proportion of patients with bleeding events decreased in both efgartigimod groups (46.2% in 5 mg/kg group and 38.5% in 10 mg/kg group to 7.7% in both groups after 4 weeks) to a much greater degree than the placebo group (33.3% to 25.0%). Platelet counts, IgG levels, and bleeding events for the three groups over the course of the study are illustrated in Figure 2. While approximately one-fifth of subjects receiving efgartigimod developed anti-drug antibodies after treatment, these did not impact pharmacokinetic or pharmacodynamic parameters. This short treatment cycle of efgartigimod was well tolerated and had a favorable safety profile, with headache as the most common adverse event.
Figure 2. Course of platelet counts, IgG levels, and bleeding scores over the course of a phase 2 study of efgartigimod to treat ITP.
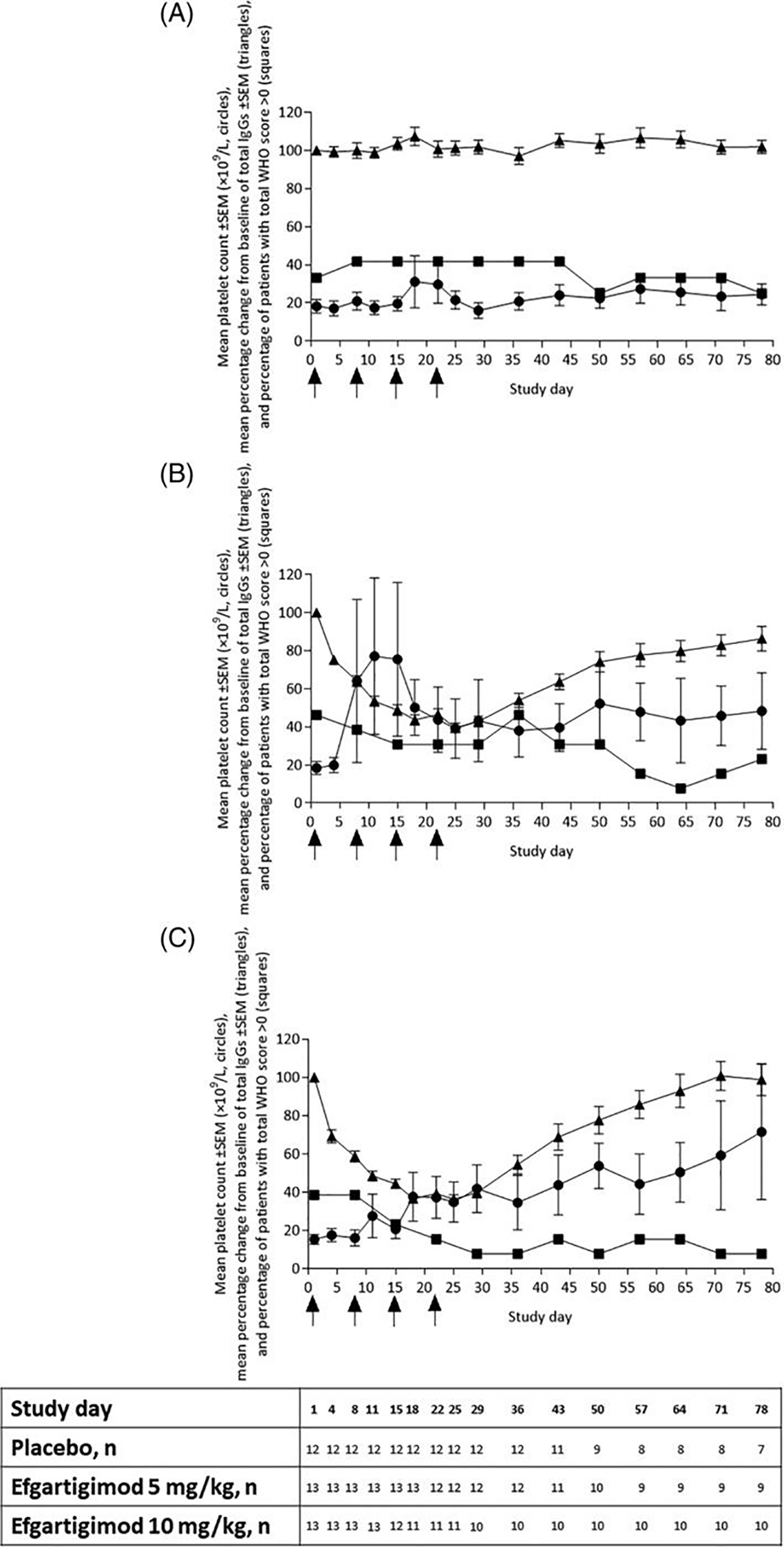
Mean platelet count ±SEM (×109/L, circles), mean percentage change from baseline of total IgGs ±SEM (triangles), and percentage of patients with total WHO score >0 (squares) assessed per treatment group. (A) Placebo, (B) efgartigimod 5 mg/kg, and (C) efgartigimod 10 mg/kg. Patients receiving rescue medication were excluded from the analysis from the day of rescue (as indicated in the table below the figure). Arrows on the X-axis indicate time points of treatment administration. Reproduced with permission from Newland et al (18).
There are two pivotal global phase 3 randomized, double-blind, placebo-controlled trials of efgartigimod in adults with persistent or chronic primary ITP: ADVANCE IV (NCT04188379), which evaluated efgartigimod administered via intravenous infusion, and ADVANCE SC (NCT04687072), which is evaluating efgartigimod administered via subcutaneous injection. ADVANCE IV has been completed, and while the full results have not yet been published other than in abstract form, the main results of the trial were presented at the American Society of Hematology 2022 Annual Meeting’s Plenary Session by Broome and colleagues (19). ADVANCE IV randomized 131 patients with platelet counts <30 × 109/L 2:1 to receive efgartigimod 10 mg/kg weekly or placebo. Patients were allowed to receive certain concurrent ITP therapies at a stable dose during the study. Nearly 70% of enrolled patients had received ≥3 prior ITP therapies and the mean time since diagnosis was over 10 years in both study groups, indicating likely enrichment of the study population with refractory ITP patients. 21.8% of patients in the efgartigimod group versus 5% of patients in the placebo group achieved the primary endpoint of a sustained platelet count response (platelet count ≥50 × 109/L for at least 4 or the 6 final 2-weekly visits in the main study period without bleeding events), a significant difference. International Working Group Response rates (which incorporate both clinically meaningful platelet count improvements and the absence of bleeding events), a prespecified secondary endpoint, were more impressive: 51.2% in the efgartigimod group versus 20.0% in the placebo group. The efgartigimod group additionally outperformed the placebo group in terms of total duration of disease control and number of patients achieving a more durable sustained platelet count response, but the number of bleeding events (which were rare overall) were similar between the two groups. As in the phase 2 trial, platelet count improvements were relatively rapid, occurring after 1 week in many patients, and 10 patients were able to transition to every-other-week infusions after 4 weeks owing to achievement of a platelet count of ≥100 × 109/L for 3 out of the 4 initial platelet count measurements. The magnitude of total IgG reduction (>60%) was nearly identical to the phase 2 study. Treatment-emergent adverse event rates were similar in the efgartigimod and placebo arms, with the most frequent treatment-emergent adverse events (TEAEs) reported being bruising, headache, hematuria, and petechiae. Serious TEAEs were reported in approximately double the patients in the placebo group (15.6%) than the efgartigimod group (8.1%), and none were considered treatment related. The findings from the companion ADVANCE SC trial are awaited.
Given the accumulated efficacy data, novel mechanism of action, and demonstrated safety, efgartigimod represents a promising treatment modality for patients with refractory ITP. While we await key findings from the phase 3 trial program, most notably results of health-related quality of life measurements and the performance of the drug when administered as a subcutaneous injection, efgartigimod has progressed further than any other current agent under development in ITP and is already FDA and EMA-approved for another indication. A home-administered once-weekly efgartigimod subcutaneous injection could be an ideal means of chronic ITP management for many patients, refractory and otherwise.
Rozanolixizumab
Rozanolixizumab is a subcutaneously infused humanized monoclonal antibody targeted against the IgG binding region of the FcRn. Similar to the effect of efgartigimod, this increases lysosomal degradation and reduces the half-life of circulating IgG (20). Also, like efgartigimod, rozanolixizumab does not affect levels of albumin or other immunoglobulin isotypes (21).
A phase 2 open-label study evaluating rozanolixizumab in 65 adults with persistent or chronic primary ITP (NCT02718716) was completed and published by Robak and colleagues (22). Enrolled patients had a median ITP duration of 5.8 years and a median of 4 prior ITP therapies, once again a heavily pre-treated population likely enriched with refractory ITP. Patients received 1 to 5 once-weekly subcutaneous infusions of rozanolixizumab, for a total cumulative dose of 15–21 mg/kg. The percentage of patients achieving a platelet response (defined as a platelet count improvement to ≥50 × 109/L or more at least one time) ranged between 35 and 66%, depending on the dose cohort, with most responses occurring in the first week (Figure 3). Major decreases in IgG levels were observed across all dose groups, and rozanolixizumab was well-tolerated across all dose groups, with headache, diarrhea and vomiting as the most commonly reported adverse events. With the success of this phase 2 trial, two phase 3 trials evaluating rozanolixizumab in persistent or chronic primary ITP was launched and began enrolling patients. However, both trials were terminated in 2022 by UCB, their sponsor, due to “a strategic business decision, not a safety decision” (NCT04200456, NCT04224688). Like efgartigimod, a phase 3 trial of rozanolixizumab in generalized myasthenia gravis (NCT03971422) was successful. It is not clear at present whether development of rozanolixizumab in ITP will resume at some point in the future.
Figure 3. Clinical efficacy of rozanolixizumab in a phase 2 trial in ITP.
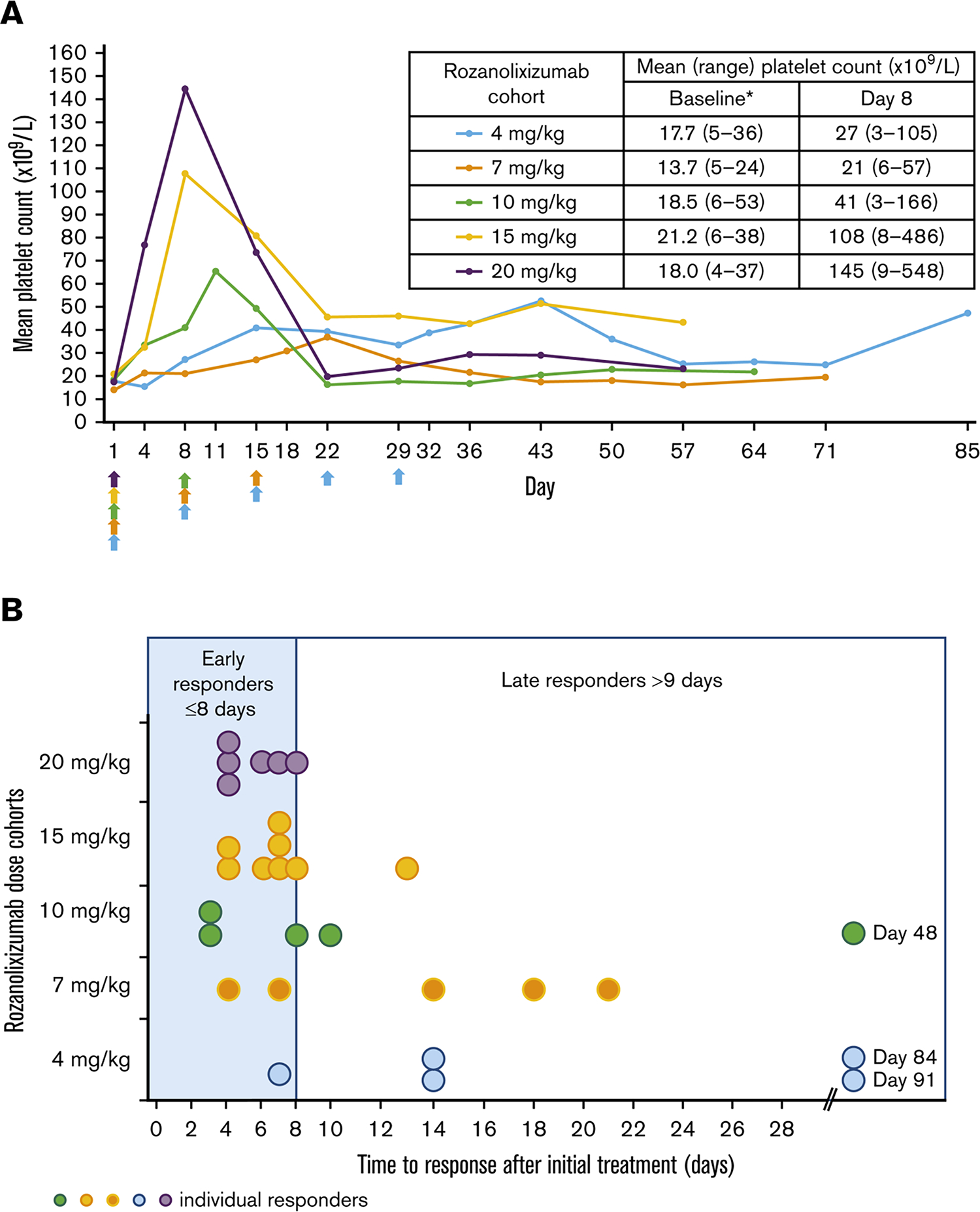
(A) Mean platelet count over time after rozanolixizumab subcutaneous infusion (per protocol set). Arrows indicate time of rozanolixizumab subcutaneous infusion. *Baseline platelet counts were derived from central laboratory data. (B) Time to first clinically relevant response (platelet count ≥50 × 109/L) in the patients classified as responders (per protocol set). Reproduced with permission from Robak et al (22).
BRUTON’S TYROSINE KINASE INHIBITION
Principles and Rationale
Bruton’s tyrosine kinase (BTK) is critical in macrophage Fcγ receptor-mediated signaling pathways as well as B-cell maturation and antibody production (23). Accordingly, BTK inhibitors have become a mainstay of treatment for B-cell malignancies. The potential of BTK inhibitors in the management of non-malignant immune disorders, however, is becoming clear. Ibrutinib, for example, is FDA-approved for chronic graft versus host disease (24), and other BTK inhibitors are currently under investigation for various autoimmune disorders including rheumatoid arthritis, Sjogren syndrome and pemphigus (25–27). Given the potential impact of BTK inhibition on autoantibody production and phagocyte-mediated platelet destruction, as well as a convenient oral route of administration, this is a potentially promising new target in the treatment of ITP. Early studies of ibrutinib in patients with B-cell malignancy complicated by ITP demonstrated high rates of ITP remission after initiation of ibrutinib treatment for the malignancy (28).
The main concern regarding use of ibrutinib and other currently approved BTK inhibitors for the treatment of ITP is inhibition of platelet function. Ibrutinib and other currently approved BTK inhibitors exert platelet inhibitory effects via inhibition of platelet aggregation and adhesion mechanisms downstream of the collagen receptor GPVI, GPIb and integrin αIIbβ3 (29). This appears to occur due to broad inhibition of many tyrosine kinases by insufficiently selective small molecule tyrosine inhibitors and does not occur due to inhibition of BTK alone, as platelets have alternative signaling pathways that can bypass selective BTK inhibition and allow for normal function (30). The latter fact has been recognized for some time as patients with X-linked agammaglobulinemia (Bruton’s agammaglobulinemia, caused by a congenital isolated BTK defect) do not have a bleeding phenotype, albeit they do have abnormal platelet function when tested (31). So, without exquisite selectivity, BTK inhibitors may inhibit platelet function and cause increased bleeding risk, an obvious problem in their use to treat ITP.
Rilzabrutinib
Rilzabrutinib (PRN1008, Principia, United States) is an oral, reversible, potent BTK inhibitor designed to treat immunologic disorders rather than malignancies (30). The molecule covalently binds to and inhibits BTK after only a short period of drug exposure and is then rapidly cleared, which theoretically reduces off-target toxicity potential (27). Unlike other existing BTK inhibitors, rilzabrutinib is highly selective. In an enzymatic inhibition panel including 251 kinases, rilzabrutinib demonstrated exquisite kinase selectivity with >90% inhibition of just 6 kinases (BTK, RLK, TEC, BMX, BLK, and ERBB4) (30). This compares to 21 kinases inhibited >90% by ibrutinib. This selectivity results in preservation of platelet function following exposure to rilzabrutinib in both healthy subjects and patients with ITP, where it would otherwise be reduced following exposure to ibrutinib (Figure 4). Additionally, rilzabrutinib’s selectivity in avoiding significant inhibition of the phosphatidylinositol 3-kinase (PI3K)-AKT signaling pathway should decrease risk of some other typical BTK inhibitor toxicities, notably atrial fibrillation (30).
Figure 4. Platelet aggregation and function in healthy volunteers and ITP patients treated with rilzabrutinib or ibrutinib.
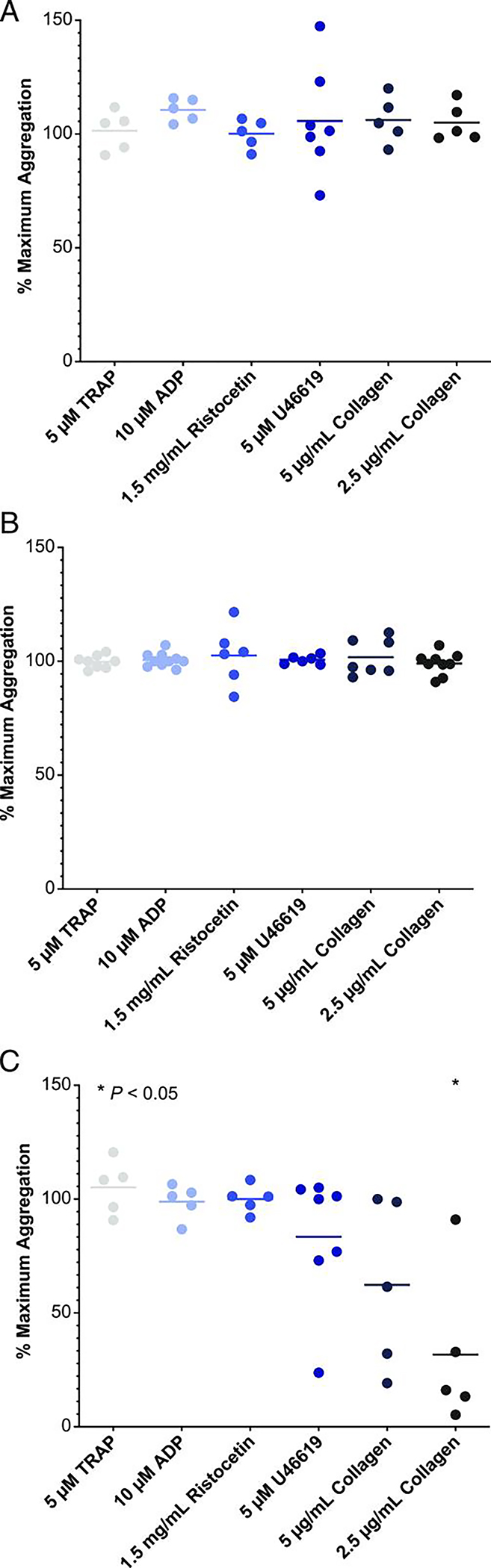
Plasma from human healthy volunteers [HVs; n = 5 (A)] or ITP patients treated with rilzabrutinib 1 μM [n = 7 (B)] or with ibrutinib 1 μM in HVs [n = 5 (C)] were studied to evaluate their impact on platelet aggregation. Plotted is the percent of maximum platelet aggregation of compound-treated samples normalized to that of untreated samples for each of the indicated platelet agonists and compared using a two-tailed t-test versus DMSO control. Only the ibrutinib-treated 2.5 μg/ml collagen group (in C) was statistically significant at *p < 0.05. Reproduced with permission from Langrish et al (30). Copyright 2021. The American Association of Immunologists, Inc.
Results of an adaptive, open-label, dose-finding phase 1/2 trial of oral rilzabrutinib in 60 adults with chronic ITP (NCT03395210) have been published by Kuter and colleagues (32). At baseline, the median platelet count was 15 × 109/L, the median duration of disease was 6.3 years, and patients had received a median of four different immune thrombocytopenia therapies previously. All enrolled patients could be reasonably judged to have refractory ITP, as one of the trial’s eligibility criteria was “refractory or relapsed patients with no available and approved therapeutic options.” Patients were allowed to enroll on a stable, low dose of a corticosteroid or a stable dose of a chronic TPO-RA as concomitant therapy, which was kept constant during the duration of the trial. Despite enrolling a population of chronic ITP patients with refractory disease, 24 of 60 patients (40%) overall and 18 of 45 patients (40%) who had started rilzabrutinib treatment at the highest dose (400 mg twice daily) met the primary endpoint of platelet response (defined in the study as a platelet count of at least 50 × 109/L plus an increase from baseline of at least 20 × 109/L). The median time to the first platelet count of at least 50 × 109/L was 11.5 days, and those patients who responded had impressive durability of response with continued treatment (Figure 5) with a mean percentage of weeks with a platelet count of ≥50 × 109/L of 65%. Response rates were similar in all relevant subgroup analyses, including 36% in patients previously receiving ≥4 therapies, 45% in patients receiving no concurrent ITP treatment, and 33% in patients who were previously splenectomized. Rilzabrutinib was well tolerated: all treatment-related adverse events were of grade 1 or 2 and transient. The most common adverse events were gastrointestinal in nature (diarrhea in 32% and nausea in 30%), and no patients had treatment-related bleeding or thrombotic events of grade 2 or higher. There was additionally no evidence of infections, liver toxicity, or cardiac arrhythmias, adverse events well-documented in patients receiving other BTK inhibitors. Based on these very promising findings, a phase 3 randomized trial of rilzabrutinib in adults and adolescents (age ≥12 years) with persistent or chronic ITP has now begun (LUNA3, NCT04562766) and rilzabrutinib is now also undergoing development to treat autoimmune hemolytic anemia.
Figure 5. Platelet counts over time in a phase 1/2 study of rilzabrutinib in ITP.
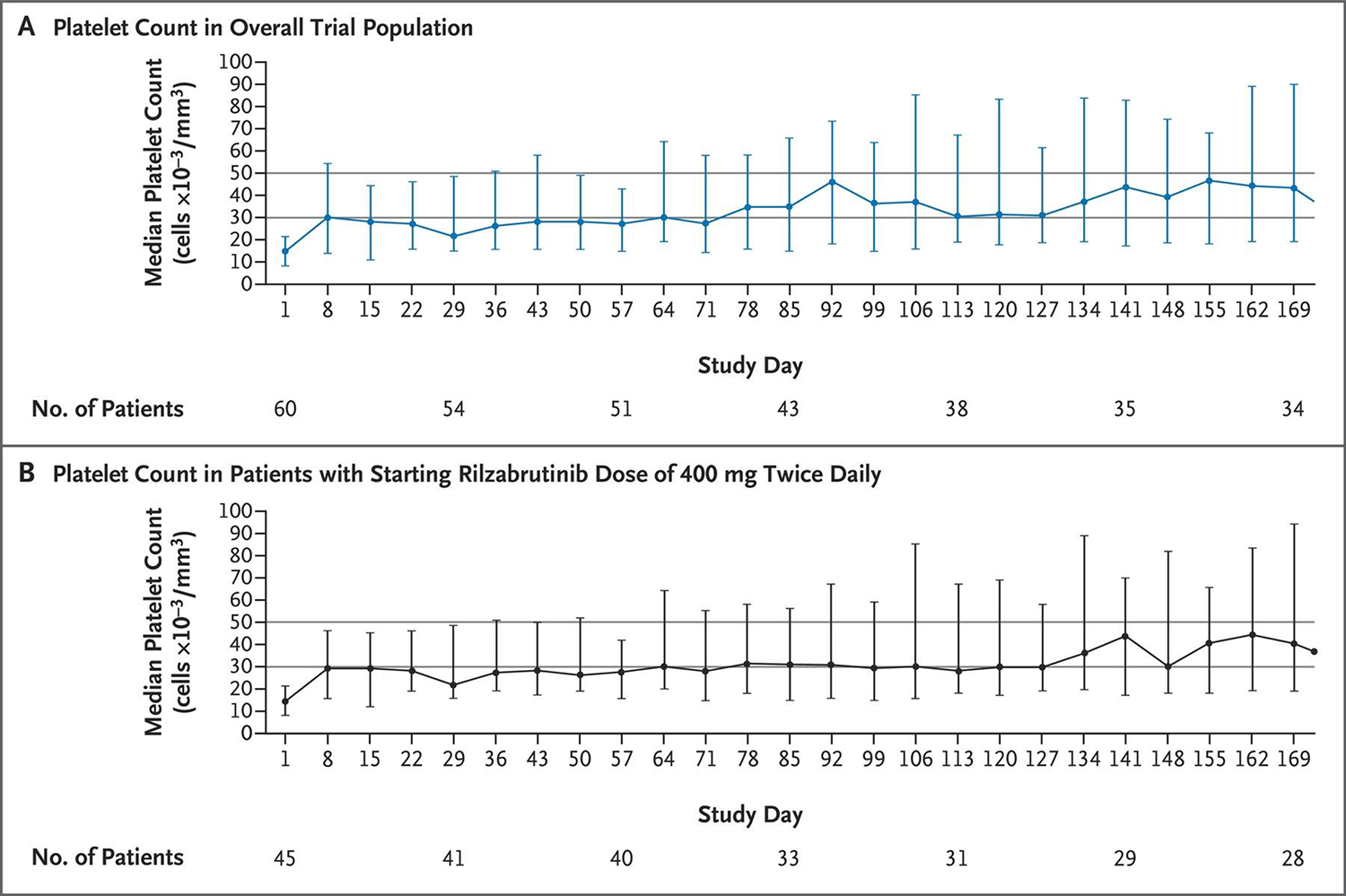
The median platelet counts from the initiation of treatment through the 24-week treatment period are shown for all 60 patients (Panel A) and for the 45 patients with a starting rilzabrutinib dose of 400 mg twice daily (Panel B). 𝙸 bars indicate the interquartile range. The first platelet count was obtained on day 8. Horizontal lines at platelet counts of 30 × 109/L and 50 × 109/L represent clinically significant thresholds for platelet response. The primary end point of platelet response was defined as at least two consecutive platelet counts, separated by at least 5 days, of at least 50 × 109/L and an increase from baseline of at least 20 × 109/L without the use of rescue medication in the 4 weeks before the latest elevated platelet count. Reproduced with permission from Kuter et al (32).
PLASMA CELL DEPLETION VIA TARGETING CD38
Principles and Rationale
It has been long-recognized that B-cell targeted therapies such as rituximab do not target the nondividing, long-lived plasma cells in the bone marrow and spleen that chronically produce platelet autoantibodies in ITP. Awareness of these cells has increased substantially following the advent of rituximab treatment in ITP. Therefore, agents depleting these cells are of substantive interest and represent yet another novel target not exploited by any existing approved therapies. CD38 (cyclic ADP ribose hydrolase), an enzymatic glycoprotein found on the surface of many immune cells including T-cells, B-cells and NK cells, is highly expressed on plasma cells (33). Originally developed for the treatment of multiple myeloma, anti-CD38 monoclonal antibodies are now under investigation for the treatment of autoimmune disorders, including ITP (34). Depletion of long-lived platelet autoantibody-producing plasma cells may allow for treatment responses in patients with otherwise refractory ITP.
Daratumumab
Daratumumab (Darzalex, Janssen, Belgium) is a first-in-class human anti-CD38 monoclonal antibody currently approved for the treatment of multiple myeloma (35). Daratumumab targets CD38-rich plasma cells via multiple mechanisms, including antibody-dependent cellular toxicity, antibody-dependent cellular phagocytosis, complement-dependent cytotoxicity, and direct apoptosis (33), Figure 6. Published cases describe successful use of daratumumab to treat refractory autoimmune cytopenias, in the post-hematopoietic stem cell transplant setting, and in patients with refractory systemic lupus erythematosus (36–39). Against this background, there is an ongoing multicenter, open-label, phase 2 dose-escalation study (the DART study, NCT04703621) evaluating the safety and efficacy of daratumumab to treat primary ITP in adults (40). Patients enrolled in this study have progressed beyond second-line therapy and failed a second-line therapy including either rituximab or TPO-RAs. The results of treatment of the first 3 patients (safety run-in phase) have been published in abstract form (40). These patients received 4 weekly subcutaneous daratumumab injections followed by a four-week observational period. All 3 patients had very low platelet counts at baseline and all responded well to daratumumab while it was being administered. One patient maintained a durable response after the four weekly injections and the other two ultimately lost their responses after daratumumab was discontinued. No serious or grade 3 adverse events occurred during this safety run-in. At the time of writing, the study is ongoing.
Figure 6.
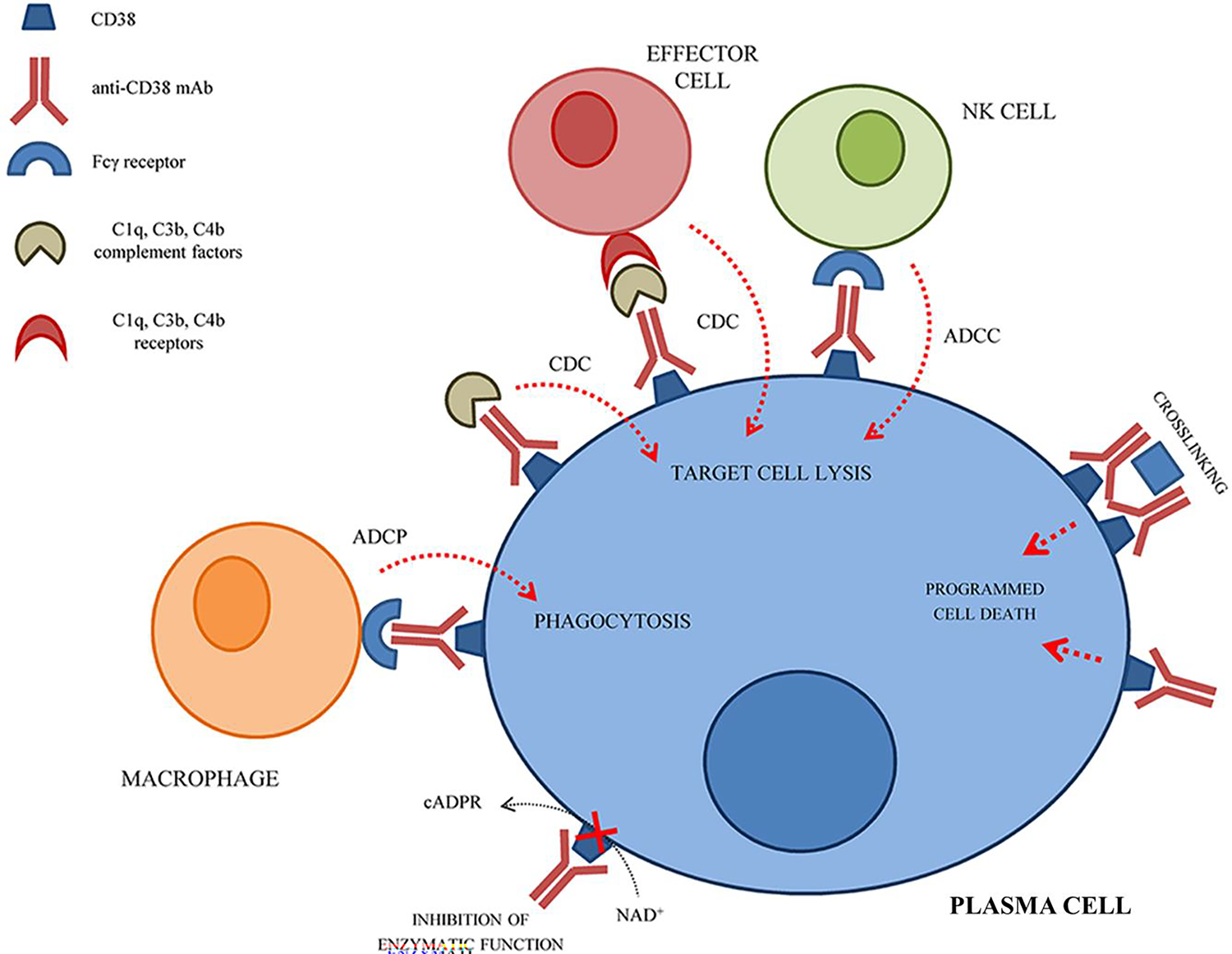
Schematic representation of the mechanism(s) of action of anti-CD38 monoclonal antibodies on plasma cells. NK cell, natural killer cell; ADCC, antibody-dependent cell-medicated cytotoxicity; CDC, complement-mediated cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; cADPR, cyclic adenosine diphosphate ribose; NAD+, nicotinamide adenine dinucleotide. Adapted from Morandi et al (33)., originally published in Frontiers in Immunology (copyright owner Frontiers Media S.A.) as per the Creative Commons Attribution License (CC BY).
Mezagitamab
Mezagitamab (TAK-079, Takeda, Japan) is a fully humanized anti-CD38 monoclonal antibody currently under investigation in a randomized phase 2 study (NCT04278924) to treat ITP. Mezagitamab destroys both plasma cells and plasmablasts via antibody-dependent cell-mediated cytotoxicity, complement-dependent cytotoxicity, antibody-dependent cellular phagocytosis, and direct apoptosis (41). Like daratumumab in the DART study, mezagitamab is administered as a weekly subcutaneous injection in the ongoing phase 2 trial. This study will enroll up to 54 participants with persistent or chronic primary ITP.
COMPLEMENT INHIBITION
Principles and Rationale
The complement system, a complex cascade of proteins produced in the liver involved in innate immune defense, is composed of three pathways: the classical, alternative, and lectin pathways (42). The complement system serves three main functions: the formation of the membrane attack complex (formed by C5b, C6, C7, C8, and C9), a powerful innate immune weapon against bacteria; inflammation, as a result of the anaphylatoxins C3a and C5a; and opsonization, primarily by C3b, to promote phagocytosis of foreign organisms (42). The classical pathway activates when C1q, a portion of the C1 complex, binds IgM or IgG complexed with an antigen. The alternative pathway, which serves as an internal amplification loop, is continuously activated at a low level due to spontaneous C3 hydrolysis; when C3b attaches to a pathogen, the alternative pathway proceeds. The lectin pathway is homologous to the classical pathway, with the exception that it is initiated by an opsonin, mannose-binding lectin (42).
Glycoprotein-specific platelet autoantibodies bound to the platelet membrane in patients with ITP retain complement fixing capability, which results in local deposition of C3b on the platelet membrane and subsequent phagocytosis and destruction of these opsonized platelets ensues (43). Additionally, direct assault by the membrane attack complex (C5b-9) may additionally contribute to platelet destruction. As no current therapeutics approved for ITP target the complement system, this is another potential novel target to reduce platelet destruction in patients with refractory ITP.
Sutimlimab
Sutimlimab (Enjayvo, Sanofi, France), is a humanized monoclonal anti-C1s antibody that selectively inhibits the C1 complex of complement, preventing complement activation, while leaving the lectin and alternative pathways intact (44). Sutimlimab prevents antibody-mediated, complement-enhanced activation of autoimmune human B cells. Under typical circumstances, the C1 complex would bind to the autoantibody-opsonized autoantigen and activate the classical complement pathway. This would result in deposition of C3 split products on the autoantigen surface, which then results in complement-enhanced activation of autoreactive B cells. However, the presence of sutimilimab inhibits classical complement activation and therefore the deposition of C3 on the autoantigen surface is reduced, thereby blunting the activation of autoreactive B cells or resulting in their anergy (45). In addition to the reduction of autoreactive B cell activation, direct deposition of C3b on the surface of platelets is lessened, thereby further reducing immune-mediated platelet destruction. Sutimlimab is presently FDA-approved for cold agglutinin disease (44). Because only the classical pathway of complement is inhibited by this agent and the alternative and lectin pathways are left intact, the encapsulated organism infectious risk of sutimlimab may be limited relative to more drastic complement inhibition.
In a phase 1 study of sutimlimab in ITP (NCT03275454), 12 adults with longstanding and generally refractory ITP were treated with sutimlimab infusions at a dose of 6.5 g if weight <75 kg or 7.5 g if weight ≥75 kg every 2 weeks (46). As demonstrated in Figure 7, platelet counts rapidly improved and complement functional activity plummeted quickly after the first infusion. Remarkably, meaningful platelet count improvements were observed in some patients within just hours of their first sutimlimab infusion suggesting the opsonization effect is important in these patients. During the study’s planned washout period and withholding of sutimlimab, the platelet count dropped steeply and complement activity recovers, both to their pretreatment baselines; once sutimlimab is restarted, the treatment effect is restored. In this small phase 1 study, 42% of patients achieved an overall response, which was durable with continued sutimlimab treatment. The drug was well-tolerated overall; one patient experienced a serious adverse event of migraine (thought to be possibly related to sutimlimab), and no patient discontinued drug due to an AE.
Figure 7. Changes in CH50 versus platelet count over the course of the study in a phase 1 trial of sutimlimab in ITP.
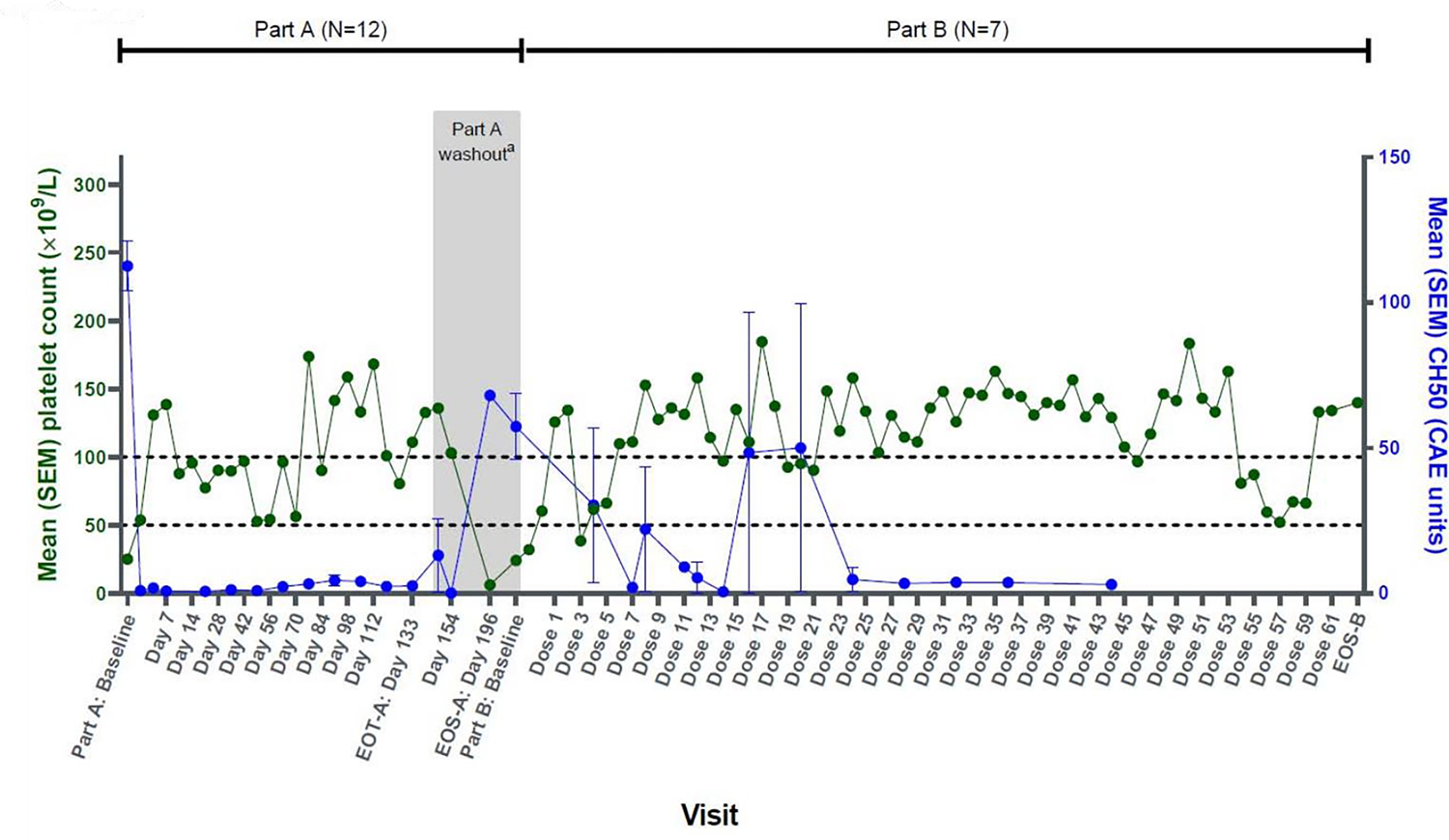
CAE, complement activity enzyme; EOS, end of study; EOT, end of treatment; SEM, standard error of the mean.
aFor patients enrolled in protocol version 3 or higher, washout period starts at Day 147 and ends at Day 196.
The value at Part A baseline is the average of all platelet counts during the screening period, including Day 0 predose. The value at Part B baseline is the average of all platelet counts during the screening period in Part B. Reproduced with permission from Broome et al (46).
Iptacopan
Iptacopan (LNP023, Novartis, Switzerland) is an oral, first-in-class, potent and selective inhibitor of factor B, a component of the alternative pathway C3 convertase (47). In reducing generation of the alternative pathway C3 convertase, iptacopan reduces downstream production of the C3a and C5a anaphylatoxins as well as the membrane attack complex. The classical and lectin pathways are left alone, which may reduce the overall risk of infection with encapsulated bacteria relative to more complete complement blockers. Data for iptacopan in other autoimmune conditions, such as PNH and IgA nephropathy, has been made available and is promising both in terms of safety and efficacy (47, 48). The most common side-effects in a phase 2 study in patients with PNH included headache and abdominal discomfort (47). Iptacopan is being investigated in ITP currently in an ongoing phase 2 basket study in autoimmune hematologic disorders (NCT05086744), currently enrolling patients with ITP and CAD.
FUTURE DIRECTIONS FOR RESEARCH IN REFRACTORY ITP
The principles discussed at length below are summarized in Table 1 and Table 2.
Table 1.
Research considerations for future studies in refractory ITP.
| Challenges Based on Ultra-Rare Status of Refractory ITP | Logistical Study Considerations Not Yet Resolved | Key Principles that Must Underlie Research in Refractory ITP Studies |
|---|---|---|
| Low N of research subjects: limited power to detect treatment and toxicity effects | Refractory ITP difficult to distinguish from some non-ITP conditions (such patients not expected to respond to ITP therapies in trials) | Must include health-related quality of life and patient-reported outcome data |
| Limited ability to stratify subjects of define sub-groups | Could non-refractory patients who have contraindications to current treatment be included? | Keep in mind risk of over-treatment or overly immunosuppressive therapy in non-bleeding yet refractory patients |
| Inefficient trials because number of sites must be large; affects trial budgets, experience, enrolment timelines | Which and how many therapies would be required to “fail” to enroll in a refractory ITP trial? | Consider that cost, patient preference, tolerability, convenience, and shared decision making all affect what treatment will be chosen |
| Biological heterogeneity means a given subgroup is even smaller population | Would “rescue” therapy be allowed for bleeding? What would this mean in refractory ITP? | |
| Pharma unlikely to be interested in refractory ITP alone as a primary study target | Include or exclude Evans syndrome/multiple cytopenia patients with active yet refractory thrombocytopenia? |
Table 2.
Potential research strategies for future studies in refractory ITP.
| Interventional (Clinical Trial) Methods | Rationale | Logistic Advantage |
|---|---|---|
| Umbrella Trials | Open a new arm of the master trial for a novel therapy, rather than one new trial for each new treatment | Much faster to open/close arms than individual trials; common control arm of best current treatment; speeds every stage of discovery |
| Adding “refractory” and “other” patient group arms to single drug trials | Depends on advocacy from investigators and patient support groups; assess outcomes separately from responsive ITP | This arm can be underpowered compared to phase 2/3 primary outcome; learn potential without massive investment |
| Non-Interventional Strategies | Rationale | Pros and Cons |
| Comparative effectiveness studies | Lower cost, opportunistic studies that require investigators to agree about treatment guidelines, choices for study; easier to accrue sites without interventional trial burden | Observational in nature, allow comparison of use of approved therapies, as single agents or combinations; allows rapid assessment of adding a treatment to standard care |
| Prospective registries | Paired with biobanking and biology ancillary studies, good way to assess who was refractory, what treatments they received, and whether they respond to anything | May take many years to bear fruit, but possible to include virtually every patient in a country or region; large advantage in ultra-rare disorders |
| Ancillary biology studies to sort ITP heterogeneous etiologies | Biological heterogeneity is a likely cause of ‘refractoriness’ in many cases; biological assessment/genetics can point the way for ultra-rare subgroups | Some immunogenetic outcomes now routinely point the way to immunomodulators that would otherwise not have been studied in immune cytopenia |
Underlying Challenges
Regardless of a particular definition of “refractory” ITP, research into this ultra-rare subset of a rare disease poses some fundamental limitations. First, due to its rarity alone, the number of patients able to be studied in any interventional trial, no matter how ambitious, will be limited, and therefore the power of a trial to detect a meaningful treatment effect (or a toxicity of therapy) would be limited unless the magnitude expected of an effect size is very large. Second, small study populations limit ability to randomize or stratify trials. Third, research in very rare disorders is inefficient for site enrollments (by way of example, the initial pediatric rituximab trial, open to refractory or chronic ITP patients, required ten sites to enroll 36 subjects (49)). Another significant limitation is the fact that refractory ITP is almost certainly biologically heterogeneous. Taken together, these limitations, which are not lost on the pharmaceutical industry, make it unlikely that a manufacturer of a promising therapy would choose “refractory ITP” as a specific indication to target, despite the unmet need, because the broader ITP population is the majority of the potential market.
Unresolved Logistical Questions
Designing trials in refractory ITP demands addressing some vexing questions besides patient numbers. First and foremost, the question, “are we certain this patient has ITP?” This question is especially important when all trial sites are not ITP referral centers. Entering “non-ITP” in a refractory ITP study would act to dilute treatment effects and potentially expose more subjects to unnecessary side effects. Another key question is whether patients who do not have truly refractory disease, but a combination of failed treatments and contraindications to others, might be included. Examples include steroid-responsive patients with morbid obesity, or eltrombopag-responsive patients with overt hepatotoxicity. Including these patients might be a reasonable “positive control” for response in some trial situations. What therapies must be “failed” by a patient to be considered refractory for trial eligibility? It is no longer feasible, with a menagerie of a dozen available therapies, to need to fail them all. In a well-reasoned article discussing refractory ITP management (50), the authors propose a framework of “Tier 1 agents” including low dose prednisone, rituximab, and approved TPO agents, versus “Tier 2 agents”, 6-mercaptopurine/azathioprine, cyclosporin A, danazol, dapsone, mycophenylate mofetil, and vincristine. Newer therapies (51) might fall into either tier because some (e.g. fostamatinib) have entered common practice. What rescue therapies can be allowed in a refractory ITP trial? Are patients with Evans syndrome included or excluded, and why? There are potential advantages or disadvantages to either approach (including or excluding patients with Evans syndrome), both in terms of expected treatment biology and in terms of “success” definitions.
Basic Principles
With these limitations in mind, investigators, clinicians and patient advocacy groups can best approach the challenge by keeping some basic principles in mind. First, it will be vital to include patient-reported outcomes and health-related quality of life measures (ideally those with salience to ITP patients) in every study. A second basic principle is that all parties need to be keenly aware of a general risk of overtreatment in ITP patients without bleeding (52). A vital correlate of this principle is that the risks of very strong immunosuppression may be greater than the potential benefits in patients with ITP who are not experiencing life-threatening bleeding episodes (even if the alternative is to have a profoundly low platelet count without bleeding). An example of this phenomenon was discovered in a different disease population yet leaves an important lesson: Early in the development of rituximab for chronic lymphocytic leukemia, it was noted that rituximab and high dose steroids in combination used in elderly patients led to high risk of fungal pneumonias and other complications (53). Finally, investigators need to keep in mind that QoL considerations, cost, and patient preferences often play key roles in ITP decision making, and how best to keep in mind these decision points.
Armed with understanding of these basic principles and fundamental limitations, there is nevertheless reason for hope in the field, and several potential strategies to move forward.
Master Trial Strategies
In a 2019 draft guidance document, US FDA presented information for industry trials in rare cancers that laid out conditions for master trial protocol designs (54). They distinguished “basket trials” (one drug for several disorders based on mechanism of action), from “umbrella trials” (one disorder for several available treatment strategies). Refractory ITP would fit into the latter, umbrella, category. Advantages of an umbrella design include markedly faster regulatory approvals to add a new therapy as a new arm, compared to launching an entirely new trial. Further, common control groups could be used, treated with “best available therapy.” Biobanking, longitudinal follow up, genetics, immunological and QoL studies could be shared as ancillary measures across arms. Umbrella trials may be especially helpful in the rare setting of refractory ITP, where at any given time, the number of patients available for study might be less than the required number for several discrete trials. As was previously mentioned in the section of this article discussing iptacopan, basket trials are actually already underway including ITP (the ongoing trial of iptacopan in autoimmune hematologic disorders includes immune thrombocytopenia and cold agglutinin disease, with the potential to open in other autoimmune hematologic disorders).
Additional Non-interventional Opportunities
Several opportunities exist in the ITP research community to improve available research in the rare subset of refractory patients. Three examples are proposed here. (1) There exists an advocacy opportunity among ITP investigators and thought leaders who carry out interventional trials: to insist that in phase 2 and 3 trials of novel ITP agents, an arm might be added for both ‘refractory ITP’ and ‘other’ considerations. This group might be assessed separately from responsive ITP. Trials in this group might include those adding a novel agent to standard care, or trials of biologics which target specific immune effectors, especially in Evans syndrome (aka immune multilineage cytopenias). (2) Use of ancillary studies of ITP biology in the setting of trials of biologics with target specificity (e.g. T- or B-cell directed, FcRn inhibition, etc.) to identify patients who might benefit from one approach or another. (3) Use of prospective registries may capture refractory patients without bias. Such registries ideally would capture agents used (whether or not approved for other indications), and biobanking should be used to the greatest extent possible to allow retrieval of biological samples. An example of such a registry is the French registry, CEREVANCE, which is a model to be emulated in this regard (55).
Acknowledgements:
Information contained within this manuscript was presented at the 7th ICIS Expert Meeting, 2022, in Lenzerheide, Switzerland (www.itpbasel.ch). H.A. is the recipient of the American Society of Hematology Scholar Award. H.A. is funded by the National Heart, Lung, and Blood Institute (1K23HL159313).
Footnotes
Conflict of Interest Statement/Disclosures: Dr. Al-Samkari reports research funding to his institution (Agios, Sobi, Novartis, Vaderis, Amgen) and consultancy (Agios, Sobi, Moderna, Novartis, Rigel, argenx, Forma, Pharmacosmos). Dr. Neufeld reports consultancy (Genentech, Pfizer), payment for honoraria or lectures (Octapharma, Takeda), participation in Data Safety Monitoring Board or Advisory Board (Acceleron/Merck, Sobi/Dova, Agios, Takeda, Genentech, Novo Nordisk), and stock or stock options (Saliogen).
REFERENCES
- 1.Jiang D, Al-Samkari H, Panch SR. Changing Paradigms in ITP Management: Newer Tools for an Old Disease. Transfus Med Rev. 2022;36(4):188–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al-Samkari H, Kuter DJ. Immune Thrombocytopenia in Adults: Modern Approaches to Diagnosis and Treatment. Semin Thromb Hemost. 2020;46(3):275–88. [DOI] [PubMed] [Google Scholar]
- 3.Cheloff AZ, Kuter DJ, Al-Samkari H. Serum complement levels in immune thrombocytopenia: Characterization and relation to clinical features. Res Pract Thromb Haemost. 2020;4(5):807–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neunert C, Terrell DR, Arnold DM, Buchanan G, Cines DB, Cooper N, et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood advances. 2019;3(23):3829–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Provan D, Arnold DM, Bussel JB, Chong BH, Cooper N, Gernsheimer T, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood advances. 2019;3(22):3780–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song F, Al-Samkari H. Management of Adult Patients with Immune Thrombocytopenia (ITP): A Review on Current Guidance and Experience from Clinical Practice. J Blood Med. 2021;12:653–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akilesh S, Christianson GJ, Roopenian DC, Shaw AS. Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J Immunol. 2007;179(7):4580–8. [DOI] [PubMed] [Google Scholar]
- 8.Latvala S, Jacobsen B, Otteneder MB, Herrmann A, Kronenberg S. Distribution of FcRn Across Species and Tissues. J Histochem Cytochem. 2017;65(6):321–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ober RJ, Martinez C, Lai X, Zhou J, Ward ES. Exocytosis of IgG as mediated by the receptor, FcRn: an analysis at the single-molecule level. Proc Natl Acad Sci U S A. 2004;101(30):11076–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7(9):715–25. [DOI] [PubMed] [Google Scholar]
- 11.Sand KM, Bern M, Nilsen J, Noordzij HT, Sandlie I, Andersen JT. Unraveling the Interaction between FcRn and Albumin: Opportunities for Design of Albumin-Based Therapeutics. Front Immunol. 2014;5:682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nugent D, McMillan R, Nichol JL, Slichter SJ. Pathogenesis of chronic immune thrombocytopenia: increased platelet destruction and/or decreased platelet production. Br J Haematol. 2009;146(6):585–96. [DOI] [PubMed] [Google Scholar]
- 13.Al-Samkari H, Rosovsky RP, Karp Leaf RS, Smith DB, Goodarzi K, Fogerty AE, et al. A modern reassessment of glycoprotein-specific direct platelet autoantibody testing in immune thrombocytopenia. Blood advances. 2020;4(1):9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ward ES, Ober RJ. Targeting FcRn to Generate Antibody-Based Therapeutics. Trends Pharmacol Sci. 2018;39(10):892–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howard JF Jr., Bril V, Burns TM, Mantegazza R, Bilinska M, Szczudlik A, et al. Randomized phase 2 study of FcRn antagonist efgartigimod in generalized myasthenia gravis. Neurology. 2019;92(23):e2661–e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ulrichts P, Guglietta A, Dreier T, van Bragt T, Hanssens V, Hofman E, et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J Clin Invest. 2018;128(10):4372–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goebeler M, Bata-Csorgo Z, De Simone C, Didona B, Remenyik E, Reznichenko N, et al. Treatment of pemphigus vulgaris and foliaceus with efgartigimod, a neonatal Fc receptor inhibitor: a phase II multicentre, open-label feasibility trial. Br J Dermatol. 2022;186(3):429–39. [DOI] [PubMed] [Google Scholar]
- 18.Newland AC, Sanchez-Gonzalez B, Rejto L, Egyed M, Romanyuk N, Godar M, et al. Phase 2 study of efgartigimod, a novel FcRn antagonist, in adult patients with primary immune thrombocytopenia. Am J Hematol. 2020;95(2):178–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Broome CM, McDonald V, Miyakawa Y, Carpenedo M, Kuter DJ, Al-Samkari H, et al. Efficacy and Safety of Intravenous Efgartigimod in Adults with Primary Immune Thrombocytopenia: Results of a Phase 3, Multicenter, Double-Blinded, Placebo-Controlled, Randomized Clinical Trial (ADVANCE IV). Blood. 2022;140(Supplement 1):6–8.35797019 [Google Scholar]
- 20.Smith B, Kiessling A, Lledo-Garcia R, Dixon KL, Christodoulou L, Catley MC, et al. Generation and characterization of a high affinity anti-human FcRn antibody, rozanolixizumab, and the effects of different molecular formats on the reduction of plasma IgG concentration. MAbs. 2018;10(7):1111–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiessling P, Lledo-Garcia R, Watanabe S, Langdon G, Tran D, Bari M, et al. The FcRn inhibitor rozanolixizumab reduces human serum IgG concentration: A randomized phase 1 study. Sci Transl Med. 2017;9(414). [DOI] [PubMed] [Google Scholar]
- 22.Robak T, Kazmierczak M, Jarque I, Musteata V, Trelinski J, Cooper N, et al. Phase 2 multiple-dose study of an FcRn inhibitor, rozanolixizumab, in patients with primary immune thrombocytopenia. Blood advances. 2020;4(17):4136–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez-Herrera G, Vargas-Hernandez A, Gonzalez-Serrano ME, Berron-Ruiz L, Rodriguez-Alba JC, Espinosa-Rosales F, et al. Bruton’s tyrosine kinase--an integral protein of B cell development that also has an essential role in the innate immune system. J Leukoc Biol. 2014;95(2):243–50. [DOI] [PubMed] [Google Scholar]
- 24.Miklos D, Cutler CS, Arora M, Waller EK, Jagasia M, Pusic I, et al. Ibrutinib for chronic graft-versus-host disease after failure of prior therapy. Blood. 2017;130(21):2243–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crofford LJ, Nyhoff LE, Sheehan JH, Kendall PL. The role of Bruton’s tyrosine kinase in autoimmunity and implications for therapy. Expert Rev Clin Immunol. 2016;12(7):763–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nyhoff LE, Barron BL, Johnson EM, Bonami RH, Maseda D, Fensterheim BA, et al. Bruton’s Tyrosine Kinase Deficiency Inhibits Autoimmune Arthritis in Mice but Fails to Block Immune Complex-Mediated Inflammatory Arthritis. Arthritis Rheumatol. 2016;68(8):1856–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Owens TD, Brameld KA, Verner EJ, Ton T, Li X, Zhu J, et al. Discovery of Reversible Covalent Bruton’s Tyrosine Kinase Inhibitors PRN473 and PRN1008 (Rilzabrutinib). J Med Chem. 2022;65(7):5300–16. [DOI] [PubMed] [Google Scholar]
- 28.Rogers KA, Ruppert AS, Bingman A, Andritsos LA, Awan FT, Blum KA, et al. Incidence and description of autoimmune cytopenias during treatment with ibrutinib for chronic lymphocytic leukemia. Leukemia. 2016;30(2):346–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng TJ, Lofurno ER, Melrose AR, Lakshmanan HHS, Pang J, Phillips KG, et al. Assessment of the effects of Syk and BTK inhibitors on GPVI-mediated platelet signaling and function. Am J Physiol Cell Physiol. 2021;320(5):C902–C15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langrish CL, Bradshaw JM, Francesco MR, Owens TD, Xing Y, Shu J, et al. Preclinical Efficacy and Anti-Inflammatory Mechanisms of Action of the Bruton Tyrosine Kinase Inhibitor Rilzabrutinib for Immune-Mediated Disease. J Immunol. 2021;206(7):1454–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quek LS, Bolen J, Watson SP. A role for Bruton’s tyrosine kinase (Btk) in platelet activation by collagen. Curr Biol. 1998;8(20):1137–40. [DOI] [PubMed] [Google Scholar]
- 32.Kuter DJ, Efraim M, Mayer J, Trneny M, McDonald V, Bird R, et al. Rilzabrutinib, an Oral BTK Inhibitor, in Immune Thrombocytopenia. N Engl J Med. 2022;386(15):1421–31. [DOI] [PubMed] [Google Scholar]
- 33.Morandi F, Horenstein AL, Costa F, Giuliani N, Pistoia V, Malavasi F. CD38: A Target for Immunotherapeutic Approaches in Multiple Myeloma. Front Immunol. 2018;9:2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ye X, Zhao Y, Ma W, Ares I, Martinez M, Lopez-Torres B, et al. The potential of CD38 protein as a target for autoimmune diseases. Autoimmunity reviews. 2023;22(4):103289. [DOI] [PubMed] [Google Scholar]
- 35.McKeage K Daratumumab: First Global Approval. Drugs. 2016;76(2):275–81. [DOI] [PubMed] [Google Scholar]
- 36.Strussmann T, Jung J, Heinz J, Duque Afonso J, Wasch R, Engelhardt M, et al. Long-term complete remission of refractory severe idiopathic immune thrombocytopenia (ITP) treated with daratumumab. Ann Hematol. 2023;102(1):245–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rieger MJ, Stolz SM, Ludwig S, Benoit TM, Bissig M, Widmer CC, et al. Daratumumab in rituximab-refractory autoimmune haemolytic anaemia. Br J Haematol. 2021;194(5):931–4. [DOI] [PubMed] [Google Scholar]
- 38.Schuetz C, Hoenig M, Moshous D, Weinstock C, Castelle M, Bendavid M, et al. Daratumumab in life-threatening autoimmune hemolytic anemia following hematopoietic stem cell transplantation. Blood advances. 2018;2(19):2550–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khandelwal P, Teusink-Cross A, Kumar AR, Bleesing JJ, Mehta PA, Jordan MB, et al. Daratumumab for the management of autoimmune cytopenias in children and young adults: a case series. Br J Haematol. 2021;194(5):e84–e9. [DOI] [PubMed] [Google Scholar]
- 40.Tsykunova G, Holme PA, Tran H, Tvedt THA, Munthe LA, Michel M, et al. Daratumumab As a Treatment for Adult Immune Thrombocytopenia: A Phase II Study with Safety Run-in (the DART Study). Blood. 2021;138(Supplement 1):2088. [Google Scholar]
- 41.Fedyk ER, Zhao L, Koch A, Smithson G, Estevam J, Chen G, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of the anti-CD38 cytolytic antibody TAK-079 in healthy subjects. Br J Clin Pharmacol. 2020;86(7):1314–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reddy YN, Siedlecki AM, Francis JM. Breaking down the complement system: a review and update on novel therapies. Curr Opin Nephrol Hypertens. 2017;26(2):123–8. [DOI] [PubMed] [Google Scholar]
- 43.Najaoui A, Bakchoul T, Stoy J, Bein G, Rummel MJ, Santoso S, et al. Autoantibody-mediated complement activation on platelets is a common finding in patients with immune thrombocytopenic purpura (ITP). Eur J Haematol. 2012;88(2):167–74. [DOI] [PubMed] [Google Scholar]
- 44.Dhillon S Sutimlimab: First Approval. Drugs. 2022;82(7):817–23. [DOI] [PubMed] [Google Scholar]
- 45.Nikitin PA, Rose EL, Byun TS, Parry GC, Panicker S. C1s Inhibition by BIVV009 (Sutimlimab) Prevents Complement-Enhanced Activation of Autoimmune Human B Cells In Vitro. J Immunol. 2019;202(4):1200–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Broome CM, Roth A, Kuter DJ, Scully M, Smith R, Wang J, et al. Safety and Efficacy of Classical Complement Pathway Inhibition with Sutimlimab in Chronic Immune Thrombocytopenia. Blood advances. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jang JH, Wong L, Ko BS, Yoon SS, Li K, Baltcheva I, et al. Iptacopan monotherapy in patients with paroxysmal nocturnal hemoglobinuria: a 2-cohort open-label proof-of-concept study. Blood advances. 2022;6(15):4450–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bomback AS, Kavanagh D, Vivarelli M, Meier M, Wang Y, Webb NJA, et al. Alternative Complement Pathway Inhibition With Iptacopan for the Treatment of C3 Glomerulopathy-Study Design of the APPEAR-C3G Trial. Kidney Int Rep. 2022;7(10):2150–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bennett CM, Rogers ZR, Kinnamon DD, Bussel JB, Mahoney DH, Abshire TC, et al. Prospective phase 1/2 study of rituximab in childhood and adolescent chronic immune thrombocytopenic purpura. Blood. 2006;107(7):2639–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cuker A, Neunert CE. How I treat refractory immune thrombocytopenia. Blood. 2016;128(12):1547–54. [DOI] [PubMed] [Google Scholar]
- 51.Provan D, Semple JW. Recent advances in the mechanisms and treatment of immune thrombocytopenia. EBioMedicine. 2022;76:103820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chaturvedi S, McCrae KR. Blind men and the refractory ITP elephant. Blood. 2016;128(12):1537–8. [DOI] [PubMed] [Google Scholar]
- 53.Smolej L Therapy of elderly/comorbid patients with chronic lymphocytic leukemia. Curr Pharm Des. 2012;18(23):3399–405. [DOI] [PubMed] [Google Scholar]
- 54.FDA US. FDA Modernizes Clinical Trials with Master Protocols. CDER Small Business and Industry Assistance (SBIA) Chronicles 2019. [Google Scholar]
- 55.Ducassou S, Gourdonneau A, Fernandes H, Leverger G, Pasquet M, Fouyssac F, et al. Second-line treatment trends and long-term outcomes of 392 children with chronic immune thrombocytopenic purpura: the French experience over the past 25 years. Br J Haematol. 2020;189(5):931–42. [DOI] [PubMed] [Google Scholar]
- 56.Patel DD, Bussel JB. Neonatal Fc receptor in human immunity: Function and role in therapeutic intervention. J Allergy Clin Immunol. 2020;146(3):467–78. [DOI] [PubMed] [Google Scholar]