

Abstract
Schistosomiasis caused by Schistosoma spp. is a disease that causes a considerable health burden to millions of people worldwide. The limited availability of effective drugs on the market and the increased risk of resistance development due to extensive usage, highlight the urgent need for new antischistosomal drugs. Recent studies have shown that robenidine derivatives, containing an aminoguanidine core, exhibit promising activities against Plasmodium falciparum, motivating further investigation into their efficacy against Schistosoma mansoni, due to their similar habitat and the resulting related cellular mechanisms like the heme detoxification pathway. The conducted phenotypic screening of robenidine and 80 derivatives against newly transformed schistosomula and adult Schistosoma mansoni yielded 11 candidates with low EC50 values for newly transformed schistosomula (1.12–4.63 μM) and adults (2.78–9.47 μM). The structure-activity relationship revealed that electron-withdrawing groups at the phenyl moiety, as well as the presence of methyl groups adjacent to the guanidine moiety, enhanced the activity of derivatives against both stages of Schistosoma mansoni. The two compounds 2,2′-Bis[(3-cyano-4-fluorophenyl)methylene] carbonimidic Dihydrazide Hydrochloride (1) and 2,2′-Bis[(4-difluoromethoxyphenyl) ethylidene] carbonimidic Dihydrazide Hydrochloride (19), were selected for an in vivo study in Schistosoma mansoni-infected mice based on their potency, cytotoxicity, pharmacokinetic-, and physicochemical properties, but failed to reduce the worm burden significantly (worm burden reduction <20%). Thus, robenidine derivatives require further refinements to obtain higher antischistosomal specificity and in vivo activity.
Keywords: Robenidine derivative, Aminoguanidine, Schistosoma mansoni, Drug discovery, Structure-activity relationship
Graphical abstract
1. Introduction
Schistosomiasis, also known as bilharzia, is an acute and chronic parasitic disease caused by blood-dwelling flukes (trematodes) of the genus Schistosoma. Over 240 million people are infected, leading to 11792 deaths annually (WHO, 2023). Acute symptoms, known as the Katayama fever, occur in individuals from non-endemic areas upon initial exposure to schistosome antigens and include fever, fatigue, myalgia, and abdominal pain (McManus et al., 2018). Intestinal chronic schistosomiasis caused by Schistosoma mansoni (S. mansoni) and Schistosoma japonicum leads to fibrotic tissue, hepatosplenomegaly, and rectal bleeding, while Schistosoma haematobium triggers the urogenital form, causing hematuria, renal dysfunction, or even a carcinoma of the bladder (Colley et al., 2014).
Preventive chemotherapy using praziquantel (PZQ) is the core strategy in the fight against schistosomiasis (Cioli et al., 1995; Panic et al., 2014). Since 1973, PZQ has been the only drug against schistosomiasis on the market, and concerns about drug resistance, its inactivity against juvenile parasites, the poor pharmacokinetics, and the bitter taste highlighting the urgent need to develop new antischistosomal drugs (Fallon and Doenhoff, 1994; Doenhoff et al., 2002; Wang et al., 2012; Bergquist et al., 2017; Dziwornu et al., 2020).
Antimalarials often serve as a starting point for developing new antischistosomal lead compounds (Keiser et al., 2009; Ezra and Stefan, 2022; Bottieau et al., 2024). One underlying reason is that the parasites causing malaria or schistosomiasis both reside in the blood, using it as the source of nutrients, and have to cope with the generated by-products like heme (Keiser and Utzinger, 2012; Fong and Wright, 2013; Xiao and Sun, 2017).
A recent study demonstrated that the anti-coccidial drug robenidine, a symmetrical aminoguanidine, and its derivatives exhibited promising antimalarial activity against Plasmodium falciparum (Anadón, 2014; Krollenbrock et al., 2021). Subsequently, additional analogs were prepared for a total library of 80 antimalarial aminoguanidines. It is noteworthy that robenidine and related derivatives also have effects on other organisms like fungi, or bacteria (Abraham et al., 2016; Khazandi et al., 2019; Mei et al., 2020). Furthermore, robenidine does not demonstrate genotoxic, mutagenic, or teratogenic effects, and poses no risk to terrestrial or aquatic systems (Bampidis et al., 2019).
The present work aimed to assess this antimalarial aminoguanidine library and to identify robenidine derivatives that serve as potential antischistosomal drugs. First, these structurally diverse robenidine derivatives were tested on newly transformed schistosomula (NTS). Compounds exhibiting an activity of ≥60% were tested in adult S. mansoni. In parallel rat myoblast (L6) cell and Human hepatoma (HepG2) cell cytotoxicity assays were taken into consideration for further compound selection. Additionally, two in silico prediction tools were used, one identifying possible toxicity-related side targets (VirtualToxLab™) and one looking at violations of the Lipinski's and Veber's rules (SwissADME) (Vedani et al., 2014; Daina et al., 2017). Furthermore, we considered microsomal stability data, if available, when selecting the compounds to be tested in vivo. Finally, an in vivo evaluation of the two robenidine derivatives 1 and 19 was conducted.
2. Materials and methods
2.1. General synthesis of aminoguanidines
Aminoguanidine compounds were synthesized as previously described (Krollenbrock et al., 2021). A solution of starting benzaldehyde or acetophenone (2.2 eq.) and 1,3-diaminoguanidine hydrochloride (1 eq.) in ethanol (10 ml) was refluxed for 16 h. Diethyl ether (10 ml) was added and the product carbonimidic dihydrazide crashed out of solution as a white solid. The product was filtered, washed with diethyl ether, and recrystallized from methanol as a hydrochloride salt. All aminoguanidine compounds were characterized by 1H NMR, 13C NMR, and mass spectrometry and confirmed to be >95% pure by HPLC. 1H and 13C NMR Spectra were taken on a Bruker 400 MHz instrument and chemical shifts are reported relative to TMS (0.0 ppm). Fluorescence measurements were recorded using a Molecular Devices Spectramax iD3 equipped with Softmax Pro 7 software. Final compounds were measured to be >95% pure by high-performance liquid chromatography (HPLC) using an Agilent Technologies 1260 Infinity II system (unless otherwise noted). Gas Chromatography Mass Spectrometry (GCMS) was performed on an Agilent GCMS system including a GC System (7890B), MSD (5977A), and autosampler (7693), and data was visualized and processed using Agilent MassHunter software. High-resolution mass spectrometry (HRMS) using electrospray ionization was performed by the Portland State University BioAnalytical Mass Spectrometry Facility. Melting points were measured using a Stanford Research Systems OptiMelt Automated Melting Point System (model MPA100). For individual syntheses and characterization, see supplemental file 1.
2.2. Compounds and media
For in vitro assays with NTS and adult S. mansoni, stock solutions (10 mM) of robenidine and its synthesized derivatives were prepared in dimethyl sulfoxide (DMSO) (Thermofisher, USA). All solvents, starting materials, and reagents for synthesis were acquired from commercial sources (Sigma-Aldrich, Combi-Blocks, and Ambeed, USA). Robenidine was obtained from Santa Cruz Biotechnology (Santa Cruz, California, USA). All materials were used without further purification. For in vivo efficacy studies, the stock solutions of selected robenidine derivatives were suspended in Tween 80/ethanol (10% of final volume, 70:30) (Sigma-Aldrich, Switzerland/Merck, Germany). Tap water (90% of the final volume) was added stepwise while sonicating and heating the formulation in a water bath (35 °C).
Hank's balanced salt solution (HBSS, 1x), medium 199 (M199), and Roswell Park Memorial Institute Medium (RPMI) 1640 were commercially obtained from Gibco (USA). Fetal calf serum (FCS) was purchased from Bioconcept AG (Allschwil, Switzerland). Penicillin/streptomycin (pen./strep.) 10,000 U/ml, L-glutamine, amphotericin B, resazurin, and podophyllotoxin were purchased from Sigma-Aldrich (Switzerland). Stock solutions of podophyllotoxin (5 mg/ml) were prepared in RPMI 1640 supplemented with L-glutamine and FCS. For the L6 cytotoxicity assays, RPMI 1640 was supplemented with FCS and L-glutamine reaching a final concentration of 10% and 1.7 μM. The Mastermix consists of penicillin G (6 mg/ml), kanamycin (10 mg/ml), 5-fluorocysteine (5 mg/ml), chloramphenicol (1 mg/ml) (all Sigma-Aldrich, Switzerland) and 1% ethanol dissolved in MilliQ water (Mäser et al., 2002).
2.3. Ethics
Adult S. mansoni, Trichuris muris (T. muris), and Heligmosomoides polygyrus (H. polygyrus) collection, as well as in vivo efficacy studies were performed following the Swiss national and cantonal regulations on animal welfare at the Swiss Tropical and Public Health Institute (Swiss TPH) (Basel-Landschaft, Switzerland) under permission number 520.
2.4. In vitro screening of NTS and adult worms
The S. mansoni life cycle is maintained at the Swiss TPH, as described by Lombardo et al. (2019). Infected Biomphalaria glabrata snails were placed for 4 h under a light source to initiate the shedding of cercariae, which were collected, mechanically transformed into NTS, and kept in M199 medium supplemented with 5% FCS, 1% Mastermix, and 1% pen./strep. at 37 °C and 5% CO2.
To collect adult S. mansoni, mesenteric veins of infected mice were dissected on day 49 post-infection. The worms were placed in supplemented RPMI 1640 medium (containing 5% FCS, 1% pen., and 100 μg/ml strep.) and incubated at 37 °C with 5% CO2 until further usage.
For the drug screening in NTS, the stock solutions of robenidine and the synthesized derivatives (10 mM) were diluted with M199 medium (supplemented with 1% pen./strep., 1% Mastermix, 5% FCS) and NTS solution (0.7 NTS/μl) in a transparent flat-bottom 96-well plate (Sarstedt, Switzerland), resulting in a final volume of 200 μl, 40–50 NTS, and drug concentrations of 10, 7.5, 5, 3.75, 2.5, 1, or 0.25 μM. For adults, the derivatives were diluted to the respective concentrations (40, 30, 20, 10, 7.5, 5, 2.5, or 1 μM) in 2 ml with RPMI 1640 medium (supplemented with 1% pen./strep. and 5% FCS) in a 24-well plate (Sarstedt, Switzerland) and two adult pairs were added. In both approaches, DMSO served as a negative control (<1% of the final volume). The plates were incubated at 37 °C and 5% CO2, and the viability was scored after 24 and 72 h through visual inspection. The viability scores were assigned based on the protocol of Lombardo et al. (2019). It was scored in steps of 0.5 in the range from 0 to 3. All compounds were tested in triplicates for NTS or duplicates for adults and the whole assays were repeated once.
2.5. Analysis of structural, physicochemical, and biochemical properties
The website SwissADME was used for analyzing structural- and physicochemical properties of the remaining 11 robenidine derivatives (Daina et al., 2017). VirtualToxLab™ was used for an in silico prediction to evaluate possible binding of the 11 remaining robenidine derivatives to toxicity-related proteins (Vedani et al., 2014).
2.6. Rat skeletal myoblast L6 cytotoxicity assay
At the Swiss TPH 2 × 10000 cells/ml L6 cells were seeded in a transparent flat bottom 96-well plate. During the experiment, cells were kept at 37 °C and 5% CO2. After adherence of the cells overnight, selected robenidine derivatives were added to the cells with three-fold serial drug dilution steps (concentration range: 100 − 0.0017 μM). Podophyllotoxin was used as a positive reference substance, ranging from 0.05 μM to 1.3 pM. Four wells were left untreated to serve as a cell viability control. 10 μl resazurin was added to the wells after 69 h of incubation, and after another 1.5 h of incubation, the fluorescence was read using a SpectraMax M2 (SoftMax Pro Software, version 5.4.6.005) at an excitation wavelength of 536 nm and an emission wavelength of 588 nm. Compounds were tested as singletons and the whole assay was repeated three times.
2.7. HepG2 cytotoxicity assay
At the Oregon Health & Science University, Oregon, USA derivatives were prepared as 10 mM stock solutions in DMSO. HepG2 cells were maintained in culture at 37 °C in a humidified 5% CO2 atmosphere in RPMI 1640 medium containing 10% fetal bovine serum. HepG2 cells were added to each well of flat bottom 96-well tissue culture plates at an initial density of 2 × 104 cells per well, and an initial volume of 160 μl complete medium per well. After adherence overnight incubation at 37 °C, 40 μl drug solutions in complete medium were applied to each well at a final concentration range of 0–200 μM across each plate. Drugs were tested in triplicate or quadruplicate. The cells were incubated for 24 h at 37 °C and 5% CO2 with the drug solutions, which were then aspirated and replaced with 200 μl per well of complete medium for an additional 24 h incubation under the same conditions. To each well 20 μl of resazurin (Alamar Blue) in PBS buffer was added reaching a final concentration of 10 μM, followed by 3 h of incubation. Fluorescence was measured at 560 nm excitation and 590 nm emission bands using a Spectramax iD3 plate reader. Fluorescence readings were normalized with respect to the untreated control wells and plotted against the logarithm of drug concentration.
2.8. Metabolic stability assay
Metabolic stability studies of selected aminoguanidines were performed at ChemPartner, Shanghai, China. Compounds were incubated at 37 °C and 1 μM concentration in murine liver microsomes (Corning, China) for 1 h at a protein concentration of 0.5 mg/ml in potassium phosphate buffer at pH 7.4 containing 1.0 mM EDTA. The metabolic reaction was initiated by NADPH and quenched with ice-cold acetonitrile at 15 min increments for up to 1 h. The progress of compound metabolism was followed by LC-MS/MS (ESI positive ion, LC-MS/MS-034(API-6500+)) using a C18 stationary phase (ACQUITY UPLC BEH C18 (2.1 × 50 mm, 1.7 μm)) and a MeOH/water mobile phase containing 0.25% FA and 1 mM NH4OAc. Imipramine or Osalmid were used as internal standards, and ketanserin was used as an internal standard for a drug with intermediate stability. Concentration versus time data for each compound was fitted to an exponential decay function to determine the first-order rate constant for substrate depletion, which was then used to calculate the degradation half-life (t1/2) and predicted intrinsic clearance value (Clint) from an assumed murine hepatic blood flow of 90 ml/min/kg.
2.9. In vivo efficacy study in S. mansoni-infected mice
For an in vivo efficacy study, female, three week-old NMRI mice were purchased from Charles River (Germany). For acclimatization, the mice were kept under environmentally controlled conditions in individually ventilated cages (temperature: 21.5 °C ± 0.5 °C; relative humidity: 55% ± 15%; artificial lighting with a circadian cycle of 12 h of light) with access to water and rodent diet ad libitum for one week. For infecting mice, S. mansoni cercariae were gained as described earlier, the cercarial concentration was adjusted to one cercaria/μl, and 100 μl of the cercarial suspension was injected subcutaneously into the nuchal fold of the mice. After 49 days post-infection, two groups of four mice each received one oral gavage of either 200 mg/kg of compound 1 or 100 mg/kg of compound 19, while four mice served as untreated controls. 21 days post-dosing the mice were euthanized using CO2, the intestine was dissected, worms were pulled out of the mesenteric veins, and the liver was pressed between two transparent plastic layers. The worms were sexed, counted based on their localization (mesenteric veins or liver), and categorized into dead or alive (Lombardo et al., 2019).
2.10. Data analysis
Drug activities of the robenidine derivatives were calculated by normalizing the obtained viability scores of the visual scoring to the vehicle-control scores and averaging across replicates, as previously described (Lombardo et al., 2019). The determination of EC50 values in NTS and adult S. mansoni involved a nonlinear least-squares analysis using a four-parameter sigmoid function. In cases where the four-parameter function was not applicable, a two-parameter function was used instead. To assess statistical significance (p < 0.05), a Kruskal-Wallis test was performed. The above-mentioned calculations were performed in R (version 4.1.3). The SoftMaxPro Software (version 5.4.6.005) calculated the IC50 values of the cytotoxicity assay in L6 cells. The IC50 values for the HepG2 cells were generated using a nonlinear regression with GraphPad Prism software (v.8). Selectivity indices (SI) were calculated by dividing the L6 or HepG2 cytotoxicity IC50 values by the respective EC50 values in adult S. mansoni. The in vivo worm burden reductions were determined by calculating the mean values of living worms of each treatment group relative to the untreated mice. Results were given in percentage (±SD). OriginPro 2022b was used for graph generation. All plots show the mean ± standard deviation. Molecular structures were created with MarvinSketch (version 20.20).
3. Results and discussion
3.1. Chemistry
Robenidine derivatives as depicted in Fig. 1 were synthesized as described by Krollenbrock et al. (2021). Briefly, two equivalents of appropriate benzaldehyde or acetophenone were combined with one equivalent of 1,3-diaminoguanidine and refluxed in ethanol for 16 h. The resulting aminoguanidine hydrochloride product crashed out of solution and was recrystallized from methanol (please refer to Supplemental file 1 for the synthesis pathways and further experimental details).
Fig. 1.

Chemical structure of the symmetrical aminoguanidine robenidine, including possible modification sites.
3.2. In vitro antischistosomal evaluation
The 80 synthesized robenidine derivatives and robenidine were first tested for their activity against NTS (Supplemental Table 1). Among the 80 tested derivatives, 78 compounds were symmetrical molecules, whereas only two compounds showed asymmetry. To conclude the structure-activity relationship (SAR) on NTS summarized below, the data obtained from incubation at 10 μM for 72 h were used (Table 1).
Table 1.
Added molecular groups and activities of 29 robenidine derivatives tested. Activities against NTS and adult S. mansoni are presented after 72 h. EC50 values are shown after 72 h.
| Compound | Added molecular groups |
NTS |
Adult S. mansoni |
||||||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | X | % Activity 10 μM, (SD) |
EC50 [μM] | % Activity 10 μM, (SD) |
EC50 [μM] | |
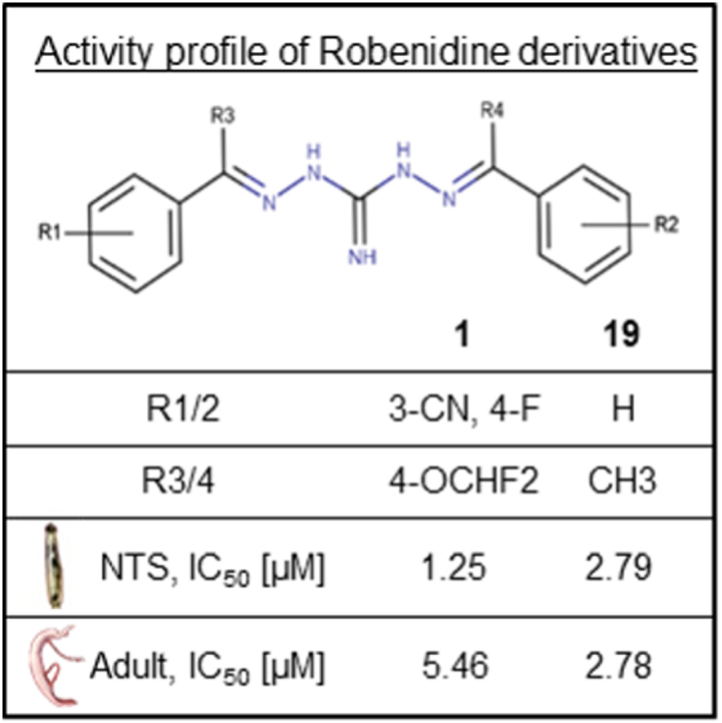
| 1 | 3-CN, 4-F | 3-CN, 4-F | H | H | NH | 100 (0) | 1.25a | 72 (19) | 5.46b |
| 12 | 4-CF3 | 4-CF3 | H | H | NH | 68 (35) | 6.23b | 25 (11) | |
| 13 | 4-CF3 | 4-CF3 | CH3 | CH3 | NH | 96 (9) | 1.67b | 45 (14) | |
| 16 | 4-OCF3 | 4-OCF3 | H | H | NH | 96 (9) | 3.31a | 59 (22) | 8.06a |
| 17 | 4-OCF3 | 4-OCF3 | CH3 | CH3 | NH | 100 (0) | 2.01b | 64 (19) | 4.91a |
| 18 | 4-OCHF2 | 4-OCHF2 | H | H | NH | 90 (12) | 4.63b | 64 (19) | 5.12a |
| 19 | 4-OCHF2 | 4-OCHF2 | CH3 | CH3 | NH | 96 (11) | 2.79a | 99 (3) | 2.78b |
| 20ç | 2-CN | 2-CN | H | H | NH | 22 (8) | |||
| 21 | 3-CN | 3-CN | H | H | NH | 98 (7) | 1.66a | 79 (14) | 4.04a |
| 23 | 3-CN | 3-CN | CH3 | CH3 | NH | 100 (0) | 1.12b | 91 (7) | 2.69b |
| 24ç | No Substitution | No Substitution | H | H | NH | 22 (18) | |||
| 26 | 4-Phenyl | 4-Phenyl | H | H | NH | 76 (8) | 4.43a | 9 (10) | |
| 27 | 4-NO2 | 4-NO2 | H | H | NH | 63 (13) | 5.71a | 32 (13) | |
| 32 | 4-Propynoxy | 4-Propynoxy | H | H | NH | 62 (15) | 5.12a | 49 (10) | |
| 33 | 4-Morpholino | 4-Morpholino | H | H | NH | 83 (20) | 2.44b | 60 (15) | 6.45a |
| 37 | 2, 4-OCH3, 5-Cl | 2, 4-OCH3, 5-Cl | CH3 | CH3 | NH | 100 (0) | 2.7a | 49 (14) | |
| 39ç | 4-OCF3 | 4-OCF3 | H | H | O | 19 (15) | |||
| 49 | 3-NO2 | 3-NO2 | H | H | NH | 100 (0) | 3.3a | 76 (14) | 9.47b |
| 52 | 3-Phenyl | 3-Phenyl | H | H | NH | 75 (21) | 3.09b | 52 (22) | 7.57b |
| 53 | 4-Phenyl, 4′-F | 4-Phenyl, 4′-F | H | H | NH | 90 (11) | 3.37a | 22 (16) | |
| 57 | 4-Ophenyl | 4-Ophenyl | H | H | NH | 100 (0) | 3.84b | 66 (15) | 7.07a |
| 58 | 4-Ophenyl, 4′-F | 4-Ophenyl, 4′-F | H | H | NH | 100 (0) | 4.51b | 56 (17) | 8.59a |
| 59 | 4-Ophenyl, 4′-Cl | 4-Ophenyl, 4′-Cl | H | H | NH | 100 (0) | 2.7a | 75 (6) | 5.52a |
| 62 | 2-Quinoline | 2-Quinoline | H | H | NH | 90 (26) | 2.5b | 54 (17) | 7.49b |
| 63 | 3-Quinoline | 3-Quinoline | H | H | NH | 96 (9) | 0.65a | 39 (11) | |
| 64 | 4-Quinoline | 4-Quinoline | H | H | NH | 93 (11) | 1.57b | 34 (11) | |
| 71 | 3-CN, 4-Cl | 3-CN, 4-Cl | H | H | NH | 100 (0) | 1.24a | 47 (15) | |
| 77 | 4-OCF3 | 3-CN, 4-F | H | H | NH | 63 (16) | 4.02a | 68 (18) | 6.76a |
| 86 | 4-BnCOBn | 4-Propynoxy | H | H | NH | 78 (14) | 4.44a | 45 (15) | |
Compounds marked with ç did not reached significant activity in NTS but give insights regarding the SAR. Blank cells = data not available. All concentrations were tested in duplicates and the corresponding experiments were repeated once.
EC50 values obtained by applying a nonlinear least-squares analysis using a four-parameter sigmoid function.
EC50 values obtained by applying a nonlinear least-squares analysis using a two-parameter sigmoid function.
It was seen that substitution with electron-withdrawing groups (e.g. –CN, –NO2, –OCF3) on the phenyl rings (Fig. 1, R1 and R2) potentiated the activity up to fourfold (e.g. 24 with 22% activity versus 16 with 96% activity and 1 with 100% activity). Furthermore, the meta or para position at the phenyl moiety enhanced this effect up to fourfold compared to ortho-substituted derivatives (e.g. 20 with 22% activity versus 21 with 96% activity). Additionally, phenoxy substitutions were more active compared to biphenyl substitutions (e.g. 57 with 100% activity versus 26 with 76% activity). Furthermore, the addition of a methyl group next to the guanidine moiety (Fig. 1, R3 and R4), improved the activity of the respective derivatives (e.g. 12 with 68% activity versus 13 with 96% activity). Modification of the imine nitrogen of the aminoguanidine core to oxygen or sulfur significantly reduced activity (e.g. 39 with 15% activity versus 16 with 96% activity).
Only the 26 compounds with activities ≥60% after 24 and 72 h in the 10 μM drug screening assay against NTS were selected for determining EC 50 values against NTS and for testing against adult S. mansoni (Fig. 2 and Supplemental Table 1).
Fig. 2.

Activity of the 26 hit derivatives against NTS (24 h and 72 h at 10 μM). All compounds were tested in duplicates and the whole experiment was repeated once. All depicted compounds showed an activity higher than 60% at both time points and were subsequently considered hits.
The SAR against adult S. mansoni slightly differed from what was seen against NTS (Table 1). The following activity-increasing trend could be observed with fluorinated substituents at the para position of the phenyl rings (Fig. 1, R1 and R2): OCHF2 > OCF3 > CF3 (e.g. 19 with 99% activity versus 17 with 64% activity versus 13 with 45% activity). The activity of meta cyano-derivatives could be enhanced by the addition of a para-position halogen. In these compounds, fluorine was preferred over chlorine (e.g. 1 with 72% activity versus 71 with 47% activity). The preference for meta over para was more pronounced with a nitro substitution (e.g. 27 with 32% activity versus 49 with 76% activity). The addition of methyl groups next to the guanidine moiety (R3 and R4) improved the activity of the compounds as seen for NTS, but was more pronounced against adult S. mansoni (e.g. 18 with 64% activity versus 19 with 99% activity). It is worth highlighting that the 26 compounds were additionally screened against the non-blood-feeding adult helminths T. muris and H. polygyrus (Supplemental file 1 and Supplemental Table 1) where only compound 18 showed significant activity against H. polygyrus. Furthermore, for the 11 compounds with the highest activity against adult S. mansoni, nine showed low IC50 values (>100 nM) against the P. falciparum D6 strain (Supplemental Table 1), with compound 1 having the most potent antimalarial activity in vitro and in vivo. Despite the vitality and overall appearance no significant abnormalities were observed in any compound tested in NTS, as well as in adult schistosomes. These correlations further point in the direction of shared drug targets. For the 11 robenidine derivatives that had an activity ≥60% after 24 and 72 h against adult S. mansoni a drug likeness evaluation was done.
3.3. Drug likeness evaluation
The 11 compounds showing promising activity against NTS and adult S. mansoni were further analyzed for their drug-likeness (Table 2). The toxicity potential was evaluated using cytotoxicity assays in two mammalian cell lines, HepG2 cells and L6 cells, as well as the in silico toxicity prediction tool VirtualToxLab™. Overall, the cytotoxicity of the derivatives was more pronounced in L6 cells compared to HepG2 cells. The indices for HepG2 cells ranged from 1.13 to >34.30, whereas for L6 cells the indices were between 0.59 and 2.89. The fact that the tested compounds were less cytotoxic against HepG2 cells than L6 cells, is not uncommon and underlines the challenges associated with accurately predicting the cytotoxic profile of a compound (Probst et al., 2021). This strongly emphasizes the need to evaluate the cytotoxicity of these compounds across multiple mammalian cell lines to obtain a comprehensive understanding of their cytotoxic behavior (Probst et al., 2021). Furthermore, the VirtualToxLab™ analysis revealed that six out of the 11 selected derivatives were predicted to bind to the estrogen receptor alpha, estrogen receptor beta, or the mineralocorticoid receptor with binding affinities in the low micromolar range (Supplemental Table 1). However, it is important to note that compounds with elevated toxic potential may not necessarily be inherently toxic, as the assessment of ADME properties as well as the kinetic stability of the protein-ligand interaction is not taken into account (Vedani et al., 2014).
Table 2.
Structure and drug likeness evaluation of 11 robenidine derivatives showing promising activity in NTS and adult S. mansoni. Including cytotoxicity in HepG2 and L6 cells with the corresponding SI, the in silico toxicity potential generated through the VirtualToxLab™, the SwissADME predicted solubility, and if available their microsomal stability.
| Compound | Structure | HepG2 IC50 [μM] | SIa | L6 IC50 [μM] (SD) | SIa | Toxic potential | Predicted solubility logS [mg/ml] | Microscomal stability t1/2 [min] |
|---|---|---|---|---|---|---|---|---|
| 1 |  |
8 | 1.49 | 5.95 (0.35) | 1.09 | 0.084 | 1.05e-02 | 186 |
| 17 | 9 | 1.58 | 4.02 (0.63) | 0.68 | 0.079 | 2.89e-05 | – | |
| 18 | 19 | 4.07 | 13.40 (2.94) | 2.89 | 0.359 | 2.47e-04 | – | |
| 19 | 3 | 1.13 | 2.03 (0.14) | 0.73 | 0.277 | 9.24e-05 | – | |
| 21 |  |
7 | 1.87 | 6.65 (2.79) | 1.71 | 0.300 | 1.52e-02 | 58 |
| 23 |  |
9 | 3.18 | 2.63 (1.02) | 0.98 | 0.099 | 5.78e-03 | – |
| 33 | 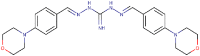 |
>200 | >26.33 | 2.30 (1.35) | 0.30 | 0.128 | 5.43e-02 | – |
| 49 | 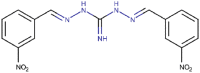 |
44 | 4.64 | 17.67 (6.36) | 1.87 | 0.348 | 4.07e-03 | – |
| 57 | 65 | 8.67 | 4.44 (1.13) | 0.59 | 0.357 | 1.55e-05 | – | |
| 59 | 43 | 8.47 | 3.18 (1.72) | 0.63 | 0.300 | 8.81e-07 | – | |
| 77 | 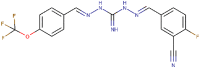 |
>200 | >34.30 | 7.05 (0.17) | 1.21 | 0.183 | 9.19e-04 | 279.57 |
SI is calculated by dividing the corresponding IC50 by EC50 adult S. mansoni. Blank cells = data not available.
SwissADME was used for an in silico prediction of the compound's solubility. Moderate solubility was predicted for three compounds: 1, 21, and 33 as summarized in Table 2 (Ali et al., 2012; Daina et al., 2017).
The microsomal stability suggests a sufficient half-life (ranging from 58 to 279.57 min) for five of the ten compounds for which the assays were performed. For comparison, previous studies showed that the standard drug PZQ is quickly metabolized (46 min for R-PZQ, 29.50 min for S-PZQ) (Kapungu et al., 2020). Furthermore, a study by Abla et al. demonstrated that the efficacy of PZQ is driven by the concentration of the drug reaching the mesenteric veins before undergoing first-pass metabolism in mice (Abla et al., 2017). However, as not all antischistosomal drugs act as rapidly as PZQ, a longer half-life is preferred (e.g. 1, 186 min versus 21, 58 min), because it allows for extended exposure (Probst et al., 2022).
For a final decision on compound progression into in vivo, Lipinski's- and Veber's rules were taken into consideration, but besides compounds 49 (11 hydrogen bond acceptors and a polar surface area >140 Å2), 57, and 59 (both a consensus logP >5, and 59 a molecular weight >500 Da), no compound violated any of the rules (Supplemental Table 1).
3.4. In vivo oral efficacy evaluation
Two compounds (1 and 19) with high in vitro activity against NTS and adult S. mansoni, and an overall acceptable drug candidate profile, were selected for an in vivo efficacy study in mice with an established patent S. mansoni infection (Table 3).
Table 3.
In vivo oral efficacy of selected robenidine derivatives. Four mice received either 200 mg/kg of 1 or 100 mg/kg of 19 through an oral gavage.
| Compound | Dose [mg/kg] | Number of mice cured/treated | Mean number of worms (SD) | Worm burden reduction [%] (SD) | p-value |
|---|---|---|---|---|---|
| 1 | 200 | 0/4 | 24.3 (4.3) | 14 (15) | 0.15 |
| 19 | 100 | 0/4 | 28.3 (10.6) | 0 (38) | 0.88 |
| Control | – | – | 28.0 (4.3) | – | – |
The worm burden reduction was calculated by comparing the number of worms found in the control mice versus treated mice.
The doses of 200 mg/kg for 1 and 100 mg/kg for 19 were selected based on PZQ as reference and the predicted solubility profile. The lower dosage of compound 19 was chosen based on lower selectivity indices and a higher predicted toxic potential. Both compounds were well tolerated by the mice, but no significant worm burden reduction was detected as presented in Table 3.
A possible explanation for the in vitro/in vivo discrepancy could be insufficient solubility, despite a predicted moderate solubility for 1 and a low to moderate solubility for 19 (Table 2), resulting in poor absorption of the compounds (Kerns and Di, 2008). This potential issue may not have arisen in the context of malaria due to the administration of lower doses (Delaney, 2004; Krollenbrock et al., 2021).
Not only low solubility could be the reason for the in vitro/in vivo discrepancy, but also poor bioavailability. The most relevant uptake mechanism is passive diffusion, which is strongly influenced by various factors such as lipophilicity (e.g. expressed as logP), size, polarity, and ionizable groups (pKa values) (Kerns and Di, 2008). Here, the functional groups of the derivatives have only slight effects on the pKa leading to an uncharged appearance in the small intestine, the main site for absorption. Yet, it is difficult to predict what impact these parameters had on the corresponding permeability as well as on the effect on the parasite. Moreover, the serum components in mice, such as those binding to albumin or inducing enzymatic inactivation, can significantly influence the availability of the unbound, hence active, drug in vivo (Beckmann et al., 2014; Tayyab and Feroz, 2021).
Lastly as mentioned before, the microsomal stability is an important factor, but for the selected derivatives these values should be sufficient since the superiority of 1 compared to the standard drug PZQ. We assume compound 19 to be better protected against microsomal degradation due to the para substitution at the phenyl moiety, as seen with compound 21 with a t1/2 of 50 min versus 1 with a t1/2 of 186 min.
4. Conclusion
In summary, we examined antimalarial robenidine derivatives hypothesizing that they would be efficacious against schistosomes. Our research revealed that derivatives of robenidine, particularly those with electron-withdrawing groups positioned at the para or meta locations on the phenyl ring, exhibited significant in vitro activity against both NTS and adult S. mansoni. Notably, these compounds demonstrated a stronger effect on NTS compared to adult forms. Based on the SAR, it would be intriguing to investigate whether the addition of methyl groups next to the guanidine moiety could further improve the activity of selected derivatives, such as 1. The most potent derivative we identified against adult schistosomes in vitro (19) has a difluoromethoxy group at the para position. Thus, synthesizing a new compound that combines a difluoromethoxy group at the para position with a cyanide group at the meta position mirroring the structure of the other lead compound (1), might yield a derivative with improved efficacy and specificity against schistosomiasis. Despite promising in vitro results, our lead compounds, selected based on their performance against NTS and adult stages and their promising drug-like properties, did not exhibit satisfactory activity in vivo. Nevertheless, the easy and versatile synthesis of robenidine derivatives and their in vitro activity underscore their potential as promising agents in the fight against schistosomiasis.
Ethics approval
This study was conducted in accordance with the Swiss national and cantonal regulations on animal welfare at the Swiss Tropical and Public Health Institute (Basel-Landschaft, Switzerland) under permission number 520.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
CRediT authorship contribution statement
Christian N. Lotz: Writing – original draft, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Alina Krollenbrock: Writing – review & editing, Methodology, Investigation. Lea Imhof: Writing – original draft, Methodology, Investigation, Formal analysis. Michael Riscoe: Writing – review & editing, Supervision, Project administration, Conceptualization. Jennifer Keiser: Writing – review & editing, Supervision, Project administration, Methodology, Funding acquisition, Conceptualization.
Declaration of generative AI and AI-assisted technologies in the writing process
During the preparation of this work the authors used ChatGPT 3.5 in order to improve readability. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Declaration of competing interest
None.
Acknowledgements
J.K. and C.L. are grateful to the European Research Council for financial support (No. 101019223).
The authors wish to thank Stefan Biendl for the provision of an R-Script for the in vitro data analysis, Cécile Häberli for maintaining the schistosome life cycle as well as taking care of the laboratory at the Swiss Tropical and Public Health Institute. We are grateful to Monica Cal for her support with the cytotoxicity assay in L6 cells. Furthermore, we want to thank Prof. Dr. Martin Smiesko from the University of Basel for providing and helping with the VirtualToxLab™ analysis.
A.K. and M.R. conceived the idea. A.K. synthesized and chemically characterized the compounds.
C.L., L.I., and J.K. designed the in vitro and in vivo experiments. C.L. and L.I. carried out the aforementioned experiments. C.L., A.K., L.I., M.R., and J.K. contributed to the conceptualization of the study, obtaining and analyzing the data, writing, and approving the final version of the manuscript.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ijpddr.2024.100546.
Contributor Information
Christian N. Lotz, Email: christian.lotz@swisstph.ch.
Alina Krollenbrock, Email: alinakrollenbrock@gmail.com.
Lea Imhof, Email: lea.imhof28@gmail.com.
Michael Riscoe, Email: riscoem@ohsu.edu.
Jennifer Keiser, Email: jennifer.keiser@swisstph.ch.
Appendix A. Supplementary data
The following are the Supplementary data to this article.
References
- Abla N., Keiser J., Vargas M., Reimers N., Haas H., Spangenberg T. Evaluation of the pharmacokinetic-pharmacodynamic relationship of praziquantel in the Schistosoma mansoni mouse model. PLoS Negl. Trop. Dis. 2017;11(9) doi: 10.1371/journal.pntd.0005942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham R.J., Stevens A.J., Young K.A., Russell C., Qvist A., Khazandi M., Wong H.S., Abraham S., Ogunniyi A.D., Page S.W., O’Handley R., McCluskey A., Trott D.J. Robenidine analogues as gram-positive antibacterial agents. J. Med. Chem. 2016;59(5):2126–2138. doi: 10.1021/acs.jmedchem.5b01797. [DOI] [PubMed] [Google Scholar]
- Ali J., Camilleri P., Brown M.B., Hutt A.J., Kirton S.B. Revisiting the General solubility equation: in Silico prediction of aqueous solubility incorporating the effect of topographical polar surface area. JCIM. 2012;52(2):420–428. doi: 10.1021/ci200387c. [DOI] [PubMed] [Google Scholar]
- Anadón A.M.M.-L. Veterinary drugs Residues: Coccidiostats. Encycl. Food Saf. 2014;3:63–75. [Google Scholar]
- Bampidis V., Azimonti G., Bastos M.D.L., Christensen H., Dusemund B., Kouba M., Kos Durjava M., López‐Alonso M., López Puente S., Marcon F., Mayo B., Pechová A., Petkova M., Ramos F., Sanz Y., Villa R.E., Woutersen R., Aquilina G., Bories G., Brantom P., Cocconcelli P.S., Halle I., Kolar B., Wester P., Van Beelen P., Holczknecht O., Vettori M.V., Gropp J. Safety and efficacy of Robenz® 66G (robenidine hydrochloride) for chickens for fattening and turkeys for fattening. EFSA J. 2019;17(3) doi: 10.2903/j.efsa.2019.5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann S., Long T., Scheld C., Geyer R., Caffrey C.R., Grevelding C.G. Serum albumin and α-1 acid glycoprotein impede the killing of Schistosoma mansoni by the tyrosine kinase inhibitor Imatinib. Int. J. Parasitol. Drugs Drug Resist. 2014;4(3):287–295. doi: 10.1016/j.ijpddr.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergquist R., Utzinger J., Keiser J. Controlling schistosomiasis with praziquantel: how much longer without a viable alternative? Infect. Dis. Poverty. 2017;6(1):74. doi: 10.1186/s40249-017-0286-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottieau E., Mbow M., Brosius I., Roucher C., Gueye C.T., Mbodj O.T., Faye B.T., De Hondt A., Smekens B., Arango D., Burm C., Tsoumanis A., Paredis L., Van Herrewege Y., Potters I., Richter J., Rosanas-Urgell A., Cissé B., Mboup S., Polman K. Antimalarial artesunate–mefloquine versus praziquantel in African children with schistosomiasis: an open-label, randomized controlled trial. Nat. Med. 2024;30(1):130–137. doi: 10.1038/s41591-023-02719-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioli D., Pica-Mattoccia L., Archer S. Antischistosomal drugs: past, present and future? Pharmacol. Ther. 1995;68(1):35–85. doi: 10.1016/0163-7258(95)00026-7. [DOI] [PubMed] [Google Scholar]
- Colley D.G., Bustinduy A.L., Secor W.E., King C.H. Human schistosomiasis. Lancet. 2014;383(9936):2253–2264. doi: 10.1016/s0140-6736(13)61949-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7(1) doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney J.S. ESOL: estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. 2004;44(3):1000–1005. doi: 10.1021/ci034243x. [DOI] [PubMed] [Google Scholar]
- Doenhoff M.J., Kusel J.R., Coles G.C., Cioli D. Resistance of Schistosoma mansoni to praziquantel: is there a problem? Trans. R. Soc. Trop. Med. Hyg. 2002;96(5):465–469. doi: 10.1016/s0035-9203(02)90405-0. [DOI] [PubMed] [Google Scholar]
- Dziwornu G.A., Attram H.D., Gachuhi S., Chibale K. Chemotherapy for human schistosomiasis: how far have we come? What's new? Where do we go from here? RSC Med. Chem. 2020;11(4):455–490. doi: 10.1039/d0md00062k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezra J.M., Stefan L.D. Parasitic Helminths and Zoonoses-From Basic to Applied Research. IntechOpen; 2022. Recent advances in anti-schistosomiasis drug discovery. Chapter 3. [DOI] [Google Scholar]
- Fallon P.G., Doenhoff M.J. Drug-resistant schistosomiasis: resistance to praziquantel and oxamniquine induced in Schistosoma mansoni in mice is drug specific. ASTMH. 1994;51(1):83–88. doi: 10.4269/ajtmh.1994.51.83. [DOI] [PubMed] [Google Scholar]
- Fong K.Y., Wright D.W. Hemozoin and antimalarial drug discovery. Future Med. Chem. 2013;5(12):1437–1450. doi: 10.4155/fmc.13.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapungu N.N., Li X., Nhachi C., Masimirembwa C., Thelingwani R.S. In vitro and in vivo human metabolism and pharmacokinetics of S‐ and R‐praziquantel. Pharmacol. Res. Perspect. 2020;8(4) doi: 10.1002/prp2.618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiser J., Chollet J., Xiao S.-H., Mei J.-Y., Jiao P.-Y., Utzinger J., Tanner M. Mefloquine—an aminoalcohol with promising antischistosomal properties in mice. PLoS Negl. Trop. Dis. 2009;3(1) doi: 10.1371/journal.pntd.0000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiser J., Utzinger J. Antimalarials in the treatment of schistosomiasis. Curr. Pharm. Des. 2012;18(24):3531–3538. doi: 10.2174/138161212801327293. [DOI] [PubMed] [Google Scholar]
- Kerns E.H., Di L. First edition. 2008. Drug-like Properties: Concepts, Structure Design, and Methods; pp. 3–415. [Google Scholar]
- Khazandi M., Pi H., Chan W.Y., Ogunniyi A.D., Sim J.X.F., Venter H., Garg S., Page S.W., Hill P.B., McCluskey A., Trott D.J. In vitro antimicrobial activity of robenidine, ethylenediaminetetraacetic acid and polymyxin B nonapeptide against important human and veterinary pathogens. Front. Microbiol. 2019;10:837. doi: 10.3389/fmicb.2019.00837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krollenbrock A., Li Y., Kelly J.X., Riscoe M.K. Robenidine analogues are potent antimalarials in drug-resistant Plasmodium falciparum. ACS Infect. Dis. 2021;7(7):1956–1968. doi: 10.1021/acsinfecdis.1c00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo F.C., Pasche V., Panic G., Endriss Y., Keiser J. Life cycle maintenance and drug-sensitivity assays for early drug discovery in Schistosoma mansoni. Nat. Protoc. 2019;14(2):461–481. doi: 10.1038/s41596-018-0101-y. [DOI] [PubMed] [Google Scholar]
- Mäser P., Grether-Bühler Y., Kaminsky R., Brun R. An anti-contamination cocktail for the in vitro isolation and cultivation of parasitic protozoa. Parasitol. Res. 2002;88(2):172–174. doi: 10.1007/s00436-001-0511-5. [DOI] [PubMed] [Google Scholar]
- McManus D.P., Dunne D.W., Sacko M., Utzinger J., Vennervald B.J., Zhou X.-N. Schistosomiasis. Nat. Rev. Dis. Primers. 2018;4(1) doi: 10.1038/s41572-018-0013-8. [DOI] [PubMed] [Google Scholar]
- Mei Y., Jiang T., Zou Y., Wang Y., Zhou J., Li J., Liu L., Tan J., Wei L., Li J., Dai H., Peng Y., Zhang L., Lopez-Ribot J.L., Shapiro R.S., Chen C., Liu N.N., Wang H. FDA approved drug library screening identifies robenidine as a repositionable antifungal. Front. Microbiol. 2020;11:996. doi: 10.3389/fmicb.2020.00996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panic G., Duthaler U., Speich B., Keiser J. Repurposing drugs for the treatment and control of helminth infections. Int. J. Parasitol. Drugs Drug Resist. 2014;4(3):185–200. doi: 10.1016/j.ijpddr.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst A., Biendl S., Keiser J. Improving translational power in antischistosomal drug discovery. Adv. Parasitol. 2022;117:47–73. doi: 10.1016/bs.apar.2022.05.002. [DOI] [PubMed] [Google Scholar]
- Probst A., Chisanga K., Dziwornu G.A., Haeberli C., Keiser J., Chibale K. Expanding the activity profile of pyrido[1,2-a]benzimidazoles: synthesis and evaluation of novel N1-1-phenylethanamine derivatives against Schistosoma mansoni. ACS Infect. Dis. 2021;7(5):1032–1043. doi: 10.1021/acsinfecdis.0c00278. [DOI] [PubMed] [Google Scholar]
- Tayyab S., Feroz S.R. Serum albumin: clinical significance of drug binding and development as drug delivery vehicle. Adv. Protein Chem. Strct. 2021;123:193–218. doi: 10.1016/bs.apcsb.2020.08.003. [DOI] [PubMed] [Google Scholar]
- Vedani A., D M, H Z., S M. OpenVirtualToxLab—a platform for generating and exchanging in silico toxicity data. Toxicol. Lett. 2014;232 doi: 10.1016/j.toxlet.2014.09.004. [DOI] [PubMed] [Google Scholar]
- Wang W., Wang L., Liang Y.-S. Susceptibility or resistance of praziquantel in human schistosomiasis: a review. Parasitol. Res. 2012;111(5):1871–1877. doi: 10.1007/s00436-012-3151-z. [DOI] [PubMed] [Google Scholar]
- WHO Schistosomiasis fact-sheet. 2023. https://www.who.int/news-room/fact-sheets/detail/schistosomiasis
- Xiao S.-h., Sun J. Schistosoma hemozoin and its possible roles. Int. J. Parasitol. 2017;47(4):171–183. doi: 10.1016/j.ijpara.2016.10.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.