

Abstract
This first‐in‐human study evaluated the safety, tolerability, single‐ and multiple‐dose pharmacokinetic profiles with dietary influence, and pharmacodynamics (PD) of DFV890, an oral NLRP3 inhibitor, in healthy participants. In total, 122 participants were enrolled into a three‐part trial including single and 2‐week multiple ascending oral doses (SAD and MAD, respectively) of DFV890, and were randomized (3:1) to DFV890 or placebo (SAD [3–600 mg] and MAD [fasted: 10–200 mg, once‐daily or fed: 25 and 50 mg, twice‐daily]). DFV890 was generally well‐tolerated. Neither deaths nor serious adverse events were reported. A less than dose‐proportional increase in exposure was observed with the initially used crystalline suspension (3–300 mg); however, an adjusted suspension formulation using spray‐dried dispersion (SDD; 100–600 mg) confirmed dose‐proportional increase in exposure. Relative bioavailability between crystalline suspension and tablets, and food effect were evaluated at 100 mg. Under fasting conditions, C max of the tablet yielded 78% compared with the crystalline suspension, and both formulations showed comparable AUC. The fed condition led to a 2.05‐ and 1.49‐fold increase in C max and AUC0–last compared with the fasting condition. The median IC50 and IC90 for ex‐vivo lipopolysaccharide‐stimulated interleukin IL‐1β release inhibition (PD) were 61 (90% CI: 50, 70) and 1340 ng/mL (90% CI: 1190, 1490). Crystalline tablets of 100 mg once‐daily or 25 mg twice‐daily were sufficient to maintain ~90% of the IL‐1β release inhibition over 24 h at steady state. Data support dose and formulation selection for further development in diseases, in which an overactivated NLRP3 represents the underlying pathophysiology.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
DFV890 is a low‐molecular‐weight compound that blocks the inflammasome assembly by inhibiting the activation of the NLRP3. DFV890 is designed to prevent caspase‐1 and subsequent activation of the inflammatory interleukins IL‐1β and IL‐19 in response to a broad range of NLRP3‐dependent activators. Non‐clinical studies suggest that DFV890 was well‐tolerated and has a favorable pharmacokinetic (PK) profile.
WHAT QUESTION DID THIS STUDY ADDRESS?
In this first‐in‐human study, DFV890 was tested in healthy adult subjects to determine the safety, tolerability, pharmacodynamic (PD), and PK parameters prior to subsequent therapeutic investigations in patients with NLRP3 related inflammatory conditions.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Increasing oral doses of DFV890 were well tolerated for up to 14 days by healthy subjects, with no safety or tolerability concerns. To achieve and maintain a stable steady state concentrations, DFV890 is compatible with a twice‐daily dosing regimen.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
The safety, tolerability, PK, and PD results suggest that DFV890 has the potential to become an effective oral first‐in‐class innate immune modulator warranting further clinical evaluation
INTRODUCTION
Chronic inflammation and dysregulated immune activity contribute to various human diseases. The nucleotide oligomerization domain (NOD), leucine‐rich repeats (LRR), and pyrin domain‐containing 3 protein (NLRP3) inflammasome represent an intracellular multiprotein complex of the innate immune system associated with the onset and progression of metabolic disorders, neurologic diseases such as multiple sclerosis, inflammatory bowel disease, and genetically caused fever syndromes, as well as other autoimmune and auto‐inflammatory diseases. 1 , 2 The NLRP3 inflammasome regulates immune responses by activation of caspase‐1, and maturation and secretion of pro‐inflammatory cytokines such as interleukin 1β (IL‐1β) and IL‐18. 3 , 4 IL‐1β neutralizing drugs such as anakinra and the anti‐IL 1β antibody canakinumab are approved for the treatment of a range of auto‐inflammatory diseases, including the cryopyrin‐associated periodic syndrome (CAPS), familial Mediterranean fever (FMF), TNF receptor‐associated periodic fever syndrome (TRAPS), and hyperimmunoglobulin D syndrome (HIDS). In addition, canakinumab demonstrated a reduced cardiovascular risk in patients postmyocardial infarction with persistent inflammation (CANTOS study; Ridker et al. 5 ). Further analysis of this study revealed that canakinumab‐treated subjects showed a reduced lung cancer incidence (Ridker et al. 6 ), less osteoarthritis requiring knee and hip replacements (Schieker et al. 7 ), and incident anemia (Vallurupalli et al. 8 ), suggesting that IL‐1β blockade might be beneficial in a broad range of inflammatory conditions.
Other than targeted IL‐1 receptor blockage (anakinra) or cytokine neutralization (canakinumab), direct NLRP3 inhibition blocks not only IL‐1β but also IL‐18 production and pyroptosis, theoretically offering therapeutic benefits beyond IL‐1β (Ridker et al. 9 ) including diseases such as rheumatoid arthritis, 10 gout, 11 atherosclerosis, 12 Alzheimer disease, 13 Parkinson's disease, 14 and non‐alcoholic steatohepatitis. 15
DFV890 (also known as IFM‐2427) is an orally administered low‐molecular‐weight compound designed to inhibit NLRP3 activation (chemical structure depicted in Figure 1). DFV890 is a highly permeable compound (apparent permeability coefficient of 24.7 × 10−6 cm/s in the human Caco‐2 cells) with limited solubility of a crystalline material. It is metabolically stable in rat, monkey, and human hepatocytes and is characterized by excellent oral bioavailability (80%–100%) in animals (data on file). DFV890 directly binds to the NLRP3 protein, thereby blocking its activation and preventing caspase‐1‐dependent maturation of pro‐inflammatory cytokines, such as IL‐1β, IL‐18 along with pyroptosis, an inflammatory form of cell death. 2 , 16 , 17 DFV890 demonstrated inhibition of lipopolysaccharide (LPS)‐induced IL‐1β release with similar potency in a range of human myeloid cell populations including peripheral blood mononuclear cells (PBMC), monocytes, and monocyte‐derived macrophages (free half‐maximal inhibitory concentration [IC50] range: 1.0–2.9 nM). Using the LPS‐induced assay in human whole blood in vitro, the respective IC50 and IC90 in plasma were 0.33 and 2.14 μM (data on file).
FIGURE 1.
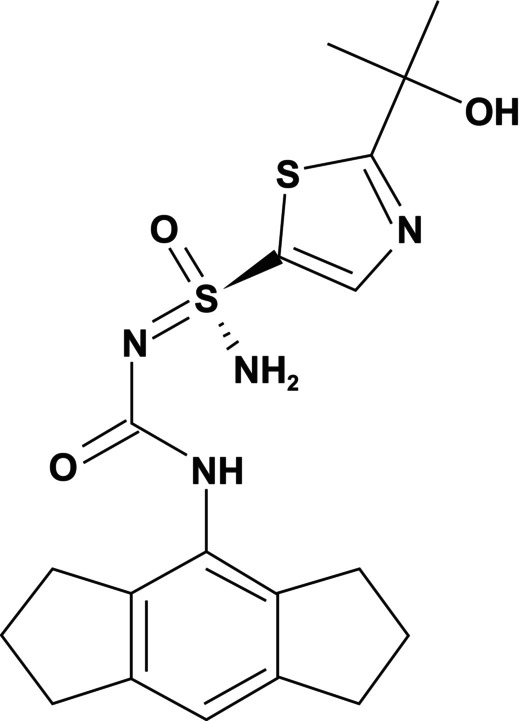
Chemical structure of DFV890.
Based on supportive nonclinical safety and pharmacology data translating into predicted human efficacious doses that can maintain 90% inhibition of IL‐1β release at trough plasma concentrations, the clinical development of DFV890 was initiated.
Here, we report the results of a first‐in‐human (FIH) study of DFV890 including safety, tolerability, pharmacokinetics (PK), pharmacodynamics (PD), and the effect of food on PK of DFV890, following administration of single‐ and multiple ascending oral doses (SAD and MAD, respectively) in healthy subjects. Due to limited drug solubility, two DFV890 suspensions (i.e., crystalline form, and spray‐dried dispersion [SDD]) were tested alongside of tablets containing a crystalline form of DFV890. To note that the SDD suspension was subsequently included due to a better DFV890 solubility of the amorphous form to improve dissolution and bioavailability at higher doses, enabling a broader exposure range, which is potentially required to support further clinical development in various indications. Finally, PK and PD data were supportive to select the clinical service form that was applied in subsequent development studies.
METHODS
This was a FIH, Phase I, randomized, single‐site, SAD, MAD, and food effect study of DFV890 in healthy subjects conducted at PRA Health Sciences in Groningen, the Netherlands, between February 2019 and April 2021 (EudraCT number: 2018‐004402‐26).
Objectives
The primary objective was to evaluate the safety and tolerability of SAD and MAD of DFV890 in healthy subjects. Key secondary objectives were to characterize the PK profile of DFV890 formulated as either suspensions or tablet, and to evaluate the effect of food on the DFV890 PK profile in its tablet formulation. In addition, the inhibitory PD effect of DFV890 on IL‐1β release from ex vivo stimulated whole‐blood samples was assessed as an exploratory measure.
Formulations
The drug product was tested in four different oral forms:
Crystalline suspension (CS) in water, methylcellulose, and polysorbate—different concentrations were tested in Parts A, C, and D
Spray‐dried dispersion (SDD) —suspension in water, methylcellulose, and polysorbate at 10 mg/mL with variable volumes were tested in Parts A and C
Crystalline tablets (CT)—25 mg tablets made with crystalline form were tested in Part D
Encapsulated crystalline tablets (ECT). CT tablets placed in gelatin capsule were tested in Part C.
Study design
Originally, the trial comprised four parts: (i) SAD (Part A), (ii) relative bioavailability of different tablet formulations (Part B, which was finally not conducted), (iii) MAD (Part C), and (iv) relative bioavailability and food effect (Part D) (Figure S1).
Each group in Part A (SAD) and Part C (MAD) enrolled eight subjects randomized (3:1) to receive either DFV890 (6 subjects) or matching placebo (2 subjects).
Part A included eight SAD groups (A1–8). Participants received a single oral dose of DFV890 (3, 10, 30, 100, 300 mg CS, or 100, 300, 600 mg SDD under fasting conditions). In Group A1 (3 mg), two sentinel subjects received treatment at least 24 h before the rest of the group to ensure uneventful safety. A starting dose of 3 mg was selected based on the minimum anticipated biological activity at C max targeting ≤75% of IL‐1β release inhibition following ex vivo LPS‐induced stimulation of human whole blood and large safety margins to the no‐observed‐adverse‐effect level (NOAEL) concluded from the 4‐week GLP toxicology animal studies (data on file) and based on the human PK prediction. Exposure from the highest 600 mg dose was predicted to be below the NOAEL.
Part B was skipped because data collected in Part A provided an adequate comparison between crystalline and SDD formulations.
Subjects in Part C only received study medication following a review of available safety, tolerability, and PK data from all preceding groups in Part A. Group C1 was initiated once group A4 was completed.
Part C included six MAD groups (C1–6) in which subjects received multiple doses of DFV890 or placebo. Groups C1/2 received CS (10 and 30 mg), C3/4 SDD (100 and 200 mg) for 14 days once‐daily (q.d.) in fasting condition. Groups C5/6 received over‐encapsulated crystalline tablets (ECT; 25 and 50 mg) for 13 days twice‐daily (b.i.d.) and a single dose on Day 14 under fed conditions. Prior to administration, tablets were placed in a gelatin capsule to maintain blinding.
Consenting participants were admitted to the clinical research center the day before dosing and were discharged on Day 3 (Part A) or Day 16 (Part C). In Parts A and C, subjects fasted overnight for at least 8 h before dosing, wherein no fluids were allowed except water until 2 h pre‐dose and 240 mL of water taken with the study medication. In Part A, fasting continued for 4 h after study drug administration. In Part C, fasting continued for at least 2 h after the q.d. drug administration. For b.i.d. dosing, doses were taken shortly after completion of the meals and subjects fasted for at least 4 h (Days 1 and 14) or 2 h (Days 2–13) after drug administration.
Part D recruited six subjects into a single group following an open‐label, randomized, three‐period crossover design. The DFV890 CT PK profile was compared between fed and fasting conditions, and against the DFV890 CS PK profile under fasting conditions. Subjects received three doses of DFV890 with a washout period of 7–14 days between each dose (Dose 1: 100 mg crystalline suspension, fasting condition; Dose 2: 100 mg tablet, fasting condition; Dose 3: 100 mg tablet, fed condition). Based on these doses, subjects were randomly assigned to 1 of 6 treatment sequences (1 subject per sequence) prepared using a Williams design. 18 For each period, subjects were admitted to the clinic on the day prior to dosing and discharged on Day 3. To study fed conditions, the trial provided the FDA recommended high‐fat breakfast with calculated caloric content consumed within 30 min before study drug administration. In the other two periods, subjects fasted overnight for at least 8 h prior to and for 4 h after administration of the study medication.
Subjects
Eligible subjects were healthy, aged between 18 and 64 years, with a body mass index (BMI) ≥18.5 and ≤30.0 kg/m2, and allowed to only participate in one group. Written informed consent was obtained prior to any study procedure. Subjects participating in Part D had to be willing and able to consume the entire high‐fat breakfast meal in the designated time frame. Subjects with a history of major psychiatric disorders, diagnosis of intellectual disability, clinically significant vital sign abnormality, or use of tobacco products within 90 days prior to (the first) drug administration were excluded. Detailed inclusion/exclusion criteria are provided in Table S1.
Blinding
In Parts A and C, active and placebo treatments could not be distinguished based on labeling, had identical appearance, and were similar in taste and smell. To maintain blinding, the same number of tablets or suspension was given to each subject in respective groups. The investigator and subjects remained blinded throughout the relevant parts of the study, and the blinding was maintained throughout. The Sponsor (IFM Management, Inc.) was given unblinded access to all study data and was provided with a copy of the randomization codes to support decision making concerning the study. Part D remained open‐label as only DFV890 was administered.
Assessments
Safety assessments
Safety assessments in all parts of the study included adverse event (AE) reporting using the Medical Dictionary for Regulatory Activities (version 22.1), clinical laboratory tests (biochemistry, hematology, and urinalysis), vital signs, electrocardiograms (ECG), physical examination, and skin biopsy (as applicable).
Pharmacokinetic assessments
To determine the concentrations of DFV890 in plasma, during the SAD part (Part A) and Part D, blood samples were collected at the following timepoints relative to dosing on Day 1: pre‐dose and 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, and 48 h post‐dose, and at the follow‐up visit (7–10 days after dosing). During the MAD part, relative to dosing on Days 1 and 14, blood samples were collected at pre‐dose; at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, and 12 h post‐dose; and on Days 2, 4, 7, 9, and 11; blood samples were taken pre‐dose; after the last dose on Day 14, at 24 and 36 h (Day 15), and 48 h (Day 16) post‐dose; and at the follow‐up visit (7–10 days after last dose). In addition, urine samples were collected over 24 h for q.d. doses (0–12 h and 12–24 h) and over 12 h for b.i.d. doses on Day 1 and Day 14 in the MAD part.
Plasma and urine samples were analyzed by a validated liquid chromatography–tandem mass spectrometry method (LC–MS/MS). DFV890 concentrations >1 ng/mL (the lower limit of quantification, LLOQ) were determined with a precision of ≤4.4% and an accuracy of −2.0% to 1.5% relative error. Concentrations below LLOQ were set to 0. A summary of the PK methodology is given in Supplementary Information S1.
The following plasma PK parameters were estimated using non‐compartmental analysis and software Phoenix Version 8.1: maximum plasma concentration (C max); time to maximum plasma concentration (t max); concentration at 24 h post‐dose (C24h) (Part A); lag time, time of observation prior to the first quantifiable concentration (t lag); time of the last quantifiable concentration (t last); area under the concentration‐time curve (AUC) from time 0 to the last quantifiable concentration (AUC0–last); AUC from time 0 to infinity (AUC0–inf); AUC from time 0 to 24 h post‐dose (AUC0–24); terminal phase half‐life (t 1/2). For Part C only, AUC over the dosing interval from time 0 to 12 h of post‐dose (AUC0–tau) and accumulation ratio based on AUC0–tau (Rac,AUC) and C max (Rac, C max) were estimated.
The following urine PK parameters were estimated: amount of drug excreted unchanged (Ae0–tau); fraction (%) of the dose excreted unchanged; and renal clearance calculated as the ratio between Ae0–tau and plasma AUC0–tau.
Pharmacodynamic assessments
To determine dose dependency of the PD response to NLRP3 inhibition, whole blood samples were collected for exploratory analyses (Groups A1–8, and Groups C1–3). In the Part A, samples were collected at pre‐dose and 1, 3, and 6 h of post‐dose; on Day 2 at 24, 27, and 30 h of post‐dose; and on Day 3 at 48 h of post‐dose. For multiple‐dose groups, blood samples were collected at the following timepoints: Days 1 and 14 at pre‐dose and 1, 3, and 6 h of post‐dose; on Days 2 and 7 at pre‐dose; and Day 15 at 24, 27, and 30 h post last dose.
Within 1 h after collection, whole blood samples were stimulated ex vivo with LPS (1 μg/mL) for 24 h at 37°C. After incubation, samples were centrifuged, plasma was separated and stored at −70°C until plasma IL‐1β release was measured using Proinflammatory Panel 1 (Human) V‐Plex from MSD (quantification range of IL‐1β in 100 times diluted samples: ULOQ: 375 pg/mL, LLOQ: 0.646 pg/mL).
Statistical analysis
All data were summarized using descriptive statistics and are listed and summarized in tabular and/or graphical form. Descriptive statistics for all relevant PK parameters included: n, arithmetic mean, standard deviation (SD), coefficient of variation (CV%), minimum, median, maximum, geometric mean, and geometric CV%. For t max only median, minimum, and maximum are presented. Concentrations below the LLOQ were treated as zero in summary statistics for concentration data alone. The linear trapezoidal rule was used for AUC calculation. Regression analysis of the terminal plasma elimination phase for the determination of t 1/2 included at least three data points after C max. The AUC0–inf with an %AUCextra above 20% were excluded from the descriptive statistics.
In Part A, dose proportionality was explored using a regression power model relating logarithmically (log)‐transformed C max, AUC0–last, and AUC0–inf to the log‐transformed dose level. Point estimates for the intercept and the slope and corresponding to 90% confidence intervals (CIs) for the slope were calculated. In Part C, dose proportionality was not explored. In Part D, the relative bioavailability of the crystalline tablet versus crystalline suspension, as well as the effect of food, was explored using an analysis of variance (ANOVA) model on the PK data. The ANOVA model included fixed effects for treatment, period, and sequence, and a random effect for subject within sequence.
For the following treatments, the least‐squares geometric mean ratios were presented together with 90% CI: DFV890 100 mg tablet fasted over 100 mg suspension fasted, and DFV890 100 mg tablet fed over 100 mg tablet fasted.
Combined individual and mean plots of IL‐1β concentrations versus time are presented by treatment.
To characterize the inhibitory potency of DFV890 on stimulated IL‐1β release, a Bayesian E max PK/PD model was developed using the rstan library in the free software environment R (version 4.0.5). Two results are presented: one with estimation of the maximal inhibition and the other one assuming the ability of full inhibition (E max = 1). Ex vivo LPS‐stimulated results were corrected for unstimulated results for the same sample. Concentrations below LLOQ were set to half of the LLOQ (IL‐1β) and to 0 (DFV890). As sensitivity analysis, the PK/PD model was re‐run with concentrations below LLOQ set to 0 for both IL‐1β and DFV890.
Ethics statement
The study was conducted in accordance with the principles of the Declaration of Helsinki, the International Council for Harmonization (ICH) E6(R2) Guideline for Good Clinical Practice (European Medicines Agency/Committee for Medicinal Products for Human Use/ICH/135/1995), and in compliance with the EU Clinical Trial Directive: Directive 2001/20/EC. All subjects provided written informed consent. The Independent Ethics Committee of the foundation (Evaluation of Ethics in Biomedical Research; The Netherlands) approved the protocol.
RESULTS
Subject disposition and demographics
A total of 122 subjects were enrolled and included in the safety and PD analysis sets, whereas the 94 subjects receiving active treatment (DFV890) were included in the PK analysis set. Overall, 64 (52%) female and 58 (48%) male subjects aged 18–64 years, and with a BMI between 18.9 and 29.4 kg/m2 participated in the study. The majority (105 [86%]) of the subjects (Part A, n = 57; Part C, n = 42; Part D, n = 6) were Caucasian (Table 1).
TABLE 1.
Subject disposition and demographics.
| Part A | Placebo | DFV890 | Total active | Total | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 mg CS | 10 mg CS | 30 mg CS | 100 mg CS | 300 mg CS | 100 mg SDD | 300 mg SDD | 600 mg SDD | ||||
| (N = 16) | (N = 6) | (N = 6) | (N = 6) | (N = 6) | (N = 6) | (N = 6) | (N = 6) | (N = 6) | (N = 48) | (N = 64) | |
| Age, years | 33 (14) | 22 (2) | 30 (10) | 37 (18) | 28 (15) | 28 (11) | 27 (14) | 21 (3) | 22 (2) | 27 (11) | 28 (12) |
| BMI, kg/m2 | 24.2 (2.6) | 24.3 (1.2) | 24.0 (2.9) | 24.9 (2.1) | 23.1 (1.9) | 22.9 (2.6) | 22.3 (2.2) | 22.6 (1.3) | 22.1 (2.2) | 23.3 (2.2) | 23.5 (2.3) |
| Male, n (%) | 8 (50%) | 2 (33%) | 3 (50%) | 3 (50%) | 1 (17%) | 4 (67%) | 4 (67%) | 2 (33%) | 2 (33%) | 21 (44%) | 29 (45%) |
| Ethnicity, n (%) | |||||||||||
| Not Hispanic or Latino | 14 (88%) | 6 (100%) | 6 (100%) | 5 (83%) | 6 (100%) | 6 (100%) | 6 (100%) | 6 (100%) | 6 (100%) | 47 (98%) | 61 (95%) |
| Hispanic or Latino | 2 (13%) | – | – | 1 (17%) | – | – | – | – | – | 1 (2%) | 3 (5%) |
| Race, n (%) | |||||||||||
| White | 15 (94%) | 2 (33%) | 5 (83%) | 6 (100%) | 6 (100%) | 6 (100%) | 6 (100%) | 5 (83%) | 6 (100%) | 42 (88%) | 57 (89%) |
| Multiple | – | 1 (17%) | 1 (17%) | – | – | – | – | – | – | 2 (4%) | 2 (3%) |
|
Black or African American |
– | 2 (33%) | – | – | – | – | – | – | – | 2 (4%) | 2 (3%) |
| Asian | – | 1 (17%) | – | – | – | – | – | 1 (17%) | – | 2 (4%) | 2 (3%) |
| American Indian or Alaska Native | 1 (6%) | – | 1 (2%) | ||||||||
| Part C | Placebo | DFV890 | Total | Part D | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| q.d./b.i.d. | 10 mg q.d. CS | 30 mg q.d. CS | 100 mg q.d. SDD | 200 mg q.d. SDD | 25 mg b.i.d. ECT | 50 mg b.i.d. ECT | Total active | DFV890 | ||
| All sequences | ||||||||||
| (N = 12) | (N = 6) | (N = 6) | (N = 6) | (N = 7) | (N = 8) | (N = 7) | (N = 40) | (N = 52) | (N = 6) | |
| Age, years | 33 (14) | 28 (4) | 26 (5) | 23 (3) | 42 (16) | 37 (16) | 23 (5) | 30 (12) | 31 (13) | 24 (2) |
| BMI, kg/m2 | 23.7 (1.9) | 24.4 (2.6) | 23.4 (2.9) | 23.1 (2.6) | 25.0 (3.3) | 24.3 (1.9) | 24.4 (2.0) | 24.1 (2.5) | 24.0 (2.4) | 21.1 (1.4) |
| Male, n (%) | 10 (83%) | 5 (83%) | 1 (17%) | 1 (17%) | 6 (86%) | 2 (25%) | 2 (29%) | 17 (43%) | 27 (52%) | 2 (33%) |
| Ethnicity, n (%) | ||||||||||
| Not Hispanic or Latino | 12 (100%) | 6 (100%) | 6 (100%) | 6 (100%) | 7 (100%) | 8 (100%) | 6 (86%) | 39 (98%) | 51 (98%) | 6 (100%) |
| Hispanic or Latino | – | – | – | – | – | – | 1 (14%) | 1 (3%) | 1 (2%) | – |
| Race, n (%) | ||||||||||
| White | 11 (92%) | 4 (67%) | 5 (83%) | 4 (67%) | 6 (86%) | 6 (75%) | 6 (86%) | 31 (78%) | 42 (81%) | 6 (100%) |
| Multiple | 1 (8%) | 1 (17%) | – | 1 (17%) | 1 (14%) | 1 (13%) | – | 4 (10%) | 5 (10%) | – |
| Asian | – | 1 (17%) | – | 1 (17%) | 1 (13%) | – | 3 (8%) | 3 (6%) | – | |
|
Black or African American |
– | – | 1 (17%) | – | – | – | 1 (14%) | 2 (5%) | 2 (4%) | – |
Note: Age and BMI are presented as mean (SD). % = (n/N) × 100%.
Abbreviations: b.i.d., twice‐daily; BMI, body mass index; CS, crystalline suspension; ECT, encapsulated crystalline tablet; n, number of subjects in each category; N, number of subjects randomized to treatment; q.d., once‐daily; SD, standard deviation; SDD, spray‐dried dispersion suspension.
Of the enrolled subjects, 107 (88%) completed the study per protocol and 15 (12%) discontinued early. The early discontinuations were in 1 of 64 (2%) subjects in Part A, 13 of 52 (25%) subjects in Part C, and 1 of 6 (17%) subjects in Part D. Reasons for discontinuation included withdrawal due to AE in 12 (10%) subjects, and 1 (1%) subject each discontinuing the study either due to withdrawal of consent, loss to follow‐up, or the study being on temporary hold due to the COVID‐19 pandemic (preventing visits; unrelated to the safety of DFV890) (Figure 2). A total of 4 discontinued subjects were replaced in Part C.
FIGURE 2.
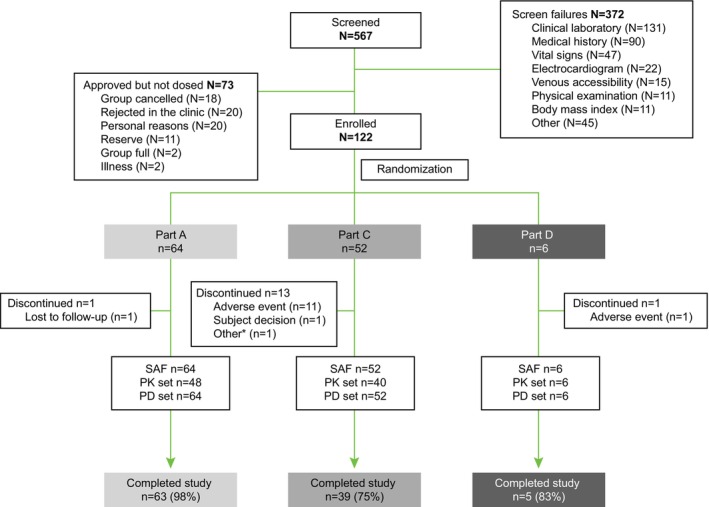
Disposition of subjects and analysis sets. *Subject had discontinued as the study was put on hold due to coronavirus disease 2019 pandemic. Part B was skipped because the data collected in Part A provided an adequate comparison of crystalline and SDD formulations. N, number of subjects randomized to treatment; n, number of subjects in each category; PD, pharmacodynamics; PK, pharmacokinetics; SAF, safety analysis set.
Safety
Single and multiple doses of DFV890 were generally well‐tolerated. Neither deaths nor serious AEs (SAEs) were reported. A summary of AEs is presented in Table 2. Overall, 87 of 122 subjects (71%) reported treatment‐emergent AE (TEAE): 66 of 94 subjects (70%) in the DFV890 arm and 21 of 28 (75%) subjects in the placebo arm. The majority of TEAEs reported by 84 (69%) subjects were of mild intensity, while 15 subjects (12%) reported moderate TEAEs. Overall, the frequently‐reported system organ class (SOC) events in >20% of subjects were nervous system disorders (DFV890 30% vs. placebo 46%), general disorders and administration site conditions (DFV890 27% vs. placebo 36%), and gastrointestinal disorders (DFV890 28% vs. placebo 25%) (Table S2). TEAE by treatment and SOC are shown in Figure 3 and Table S3.
TABLE 2.
Summary of TEAEs.
| Study part treatment | All TEAEs | Related TEAEs | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All severities | Mild | Moderate | All severities | Mild | Moderate | |||||||
| E | n (%) | E | n (%) | E | n (%) | E | n (%) | E | n (%) | E | n (%) | |
| All study parts | ||||||||||||
| Total active (N = 94) | 206 | 66 (70) | 186 | 63 (67) | 20 | 13 (14) | 40 | 21 (22) | 22 | 15 (16) | 18 | 11 (12) |
| Placebo (N = 28) | 82 | 21 (75) | 80 | 21 (75) | 2 | 2 (7) | 6 | 3 (11) | 5 | 2 (7) | 1 | 1 (4) |
| Total (N = 122) | 288 | 87 (71) | 266 | 84 (69) | 22 | 15 (12) | 46 | 24 (20) | 27 | 17 (14) | 19 | 12 (10) |
| Part A | ||||||||||||
| Total active (N = 48) | 64 | 30 (63) | 56 | 29 (60) | 8 | 5 (10) | 12 | 6 (13) | 5 | 5 (10) | 7 | 4 (8) |
| Placebo (N = 16) | 31 | 11 (69) | 31 | 11 (69) | 2 | 1 (6) | 2 | 1 (6) | ||||
| Part C | ||||||||||||
| Total active (N = 40) | 119 | 31 (78) | 109 | 29 (73) | 10 | 7 (18) | 26 | 14 (35) | 17 | 10 (25) | 9 | 6 (15) |
| Placebo (N = 12) | 51 | 10 (83) | 49 | 10 (83) | 2 | 2 (17) | 4 | 2 (17) | 3 | 1 (8) | 1 | 1 (8) |
| Part D | ||||||||||||
| Total active (N = 6) | 23 | 5 (83) | 21 | 5 (83) | 2 | 1 (17) | 2 | 1 (17) | 2 | 1 (17) | ||
Note: % = (n/N) × 100%.
Abbreviations: E, number of adverse events; N, number of subjects exposed; n, number of subjects that experienced the adverse events; TEAE, treatment‐emergent adverse event.
FIGURE 3.
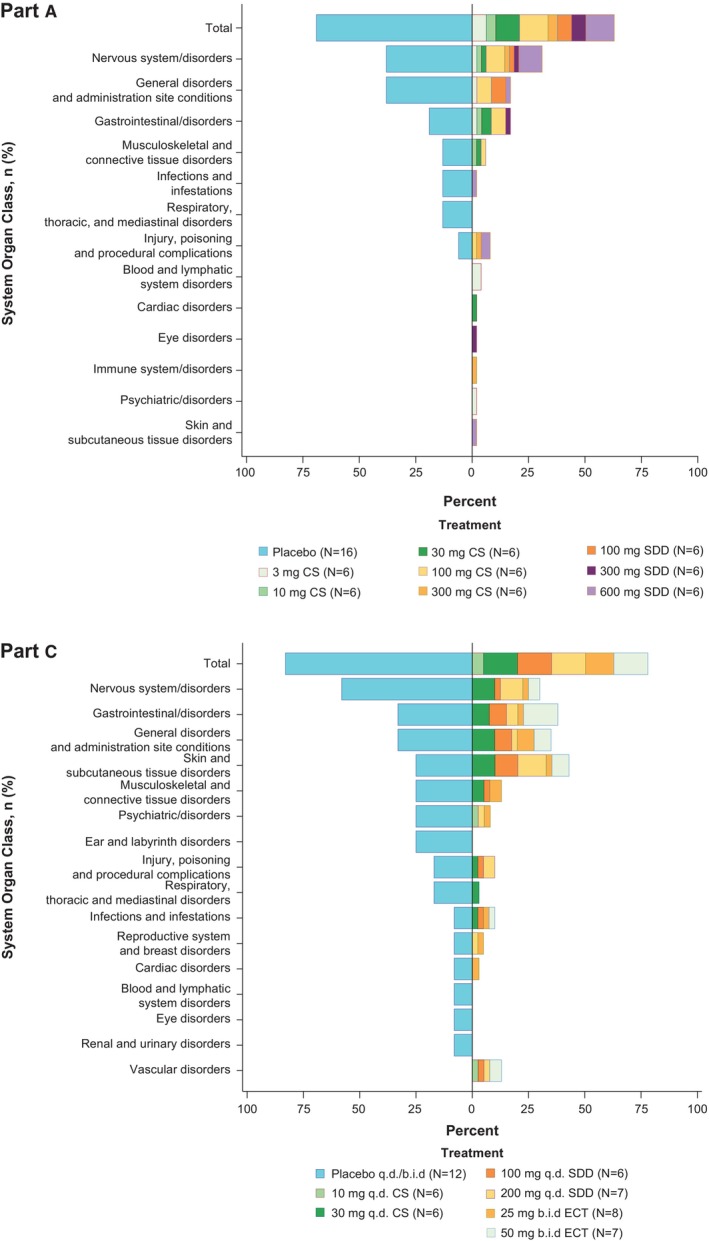
Summary of TEAE (Tornedo plot). The tornado plot for adverse events gives a visual interpretation of the scarcity of the events. The left side is maintained for the pooled placebo group with descending percentage of AE. The right side is the percentage of AE for the total active treatment groups in respective part with stacks inside to indicate contribution from different dose groups. The overall bar of active treatment groups represents the AE incidence percentage, but the stacks inside represent the portion of overall AE percentage coming from the particular dose group. b.i.d., twice‐daily; CS, crystalline suspension; CT, crystalline tablet; q.d., once‐daily; sd, single dose; ECT, encapsulated crystalline tablet; SDD, spray‐dried dispersion suspension; TEAE, treatment emergent adverse event; % = (n/N) × 100%.
Collectively, 46 TEAEs reported by 24 of 122 subjects (20%) were considered study drug‐related, including 21 of 94 (22%) receiving DFV890 and 3 of 28 (11%) receiving placebo. For 12 of 122 subjects (10%), 20 TEAEs of maculopapular skin rash and/or pruritus were considered AEs of special interest as they were considered to be related to the study drug. All 12 subjects received DFV890, either as single (100 mg CS [n = 1] or 600 mg SDD [n = 1]) or multiple doses (30 mg q.d. CS [n = 2], 100 mg q.d. SDD [n = 3], 200 mg q.d. SDD [n = 2], or 50 mg b.i.d. ECT [n = 3]). These TEAEs were of mild‐to‐moderate intensity; all cases resolved without concomitant treatment. For 10 subjects, TEAEs led to treatment discontinuation. Two other subjects discontinued early due to TEAE being unrelated to the study drug.
Leukocyte and neutrophil counts resulting from treatments at different time intervals are presented in Figure 4. Mild decreases in neutrophil and leukocyte counts were considered to be non‐clinically significant and only noted occasionally, which could be consistent with a PD effect of DFV890 resulting from inhibition of IL‐1β signaling downstream of NLRP3. One subject had a second‐degree atrioventricular block that was not considered related to the study drug. No other clinically relevant findings were reported in vital signs, 12‐lead ECG, 24‐h Holter monitoring, or physical examination. A histopathological evaluation of skin samples was performed in five subjects with skin rashes in Groups 4 and 6 of Part C. The skin rashes were generally diagnosed as superficial lymphohistiocytic infiltrates without epidermal involvement.
FIGURE 4.
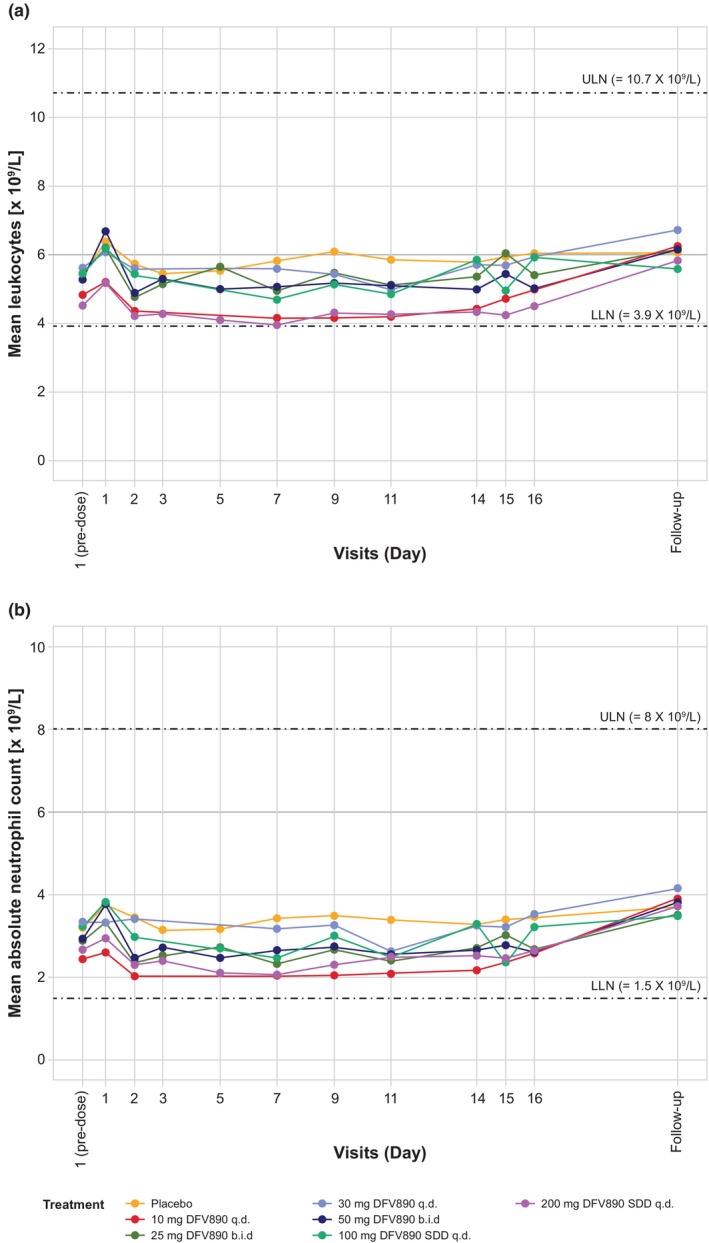
Leukocyte and neutrophil counts after multiple ascending dosing (Part C). Longitudinal trajectory of mean leukocytes (a) and absolute neutrophils counts (b) by treatment and visit days in MAD cohorts (Part C). Participants received once or twice‐daily DFV890 or placebo for 13 days and last treatment in the morning on Day 14. Blood samples were taken at predose (baseline) and 8 h post (morning) dose on Day 1; at predose on Days 2–14; at 24 h (Day 15) and 48 h (Day 16) post last (morning)dose; and at the follow‐up visit (7–10 days after last dose). Data from placebo treatment was pooled from all cohorts. Dashed lines represent lower and upper limit of normal range (LLN and ULN, respectively). b.i.d., twice‐daily; q.d., once‐daily; SDD, spray‐dried dispersion.
Pharmacokinetics
As indicated by dose‐normalized C max and AUC (Table 3), plasma exposure of single doses of DFV890 (Figure 5a) increased in a less than dose‐proportional manner when DFV890 was administered as a crystalline suspension (3–300 mg, slopes [90% CI]: 0.518 [0.460; 0.577] for C max and 0.701 [0.614; 0.788] for AUC0–last). Although the mean AUC0–last appeared to increase dose proportionally at the lower dose levels (3–30 mg), the geomean AUC0–last values were similar at the 100 and 300 mg dose levels (72,200 [CV 47.7%] vs. 63,000 ng h/mL [CV 22.6%], respectively). DFV890 administered as suspensions was rapidly absorbed with a median t max ranging from 0.76 to 3.00 h across dose levels. At higher doses 30–600 mg, the median t max was slightly delayed (1.5–3.0 h) compared with lower doses (≤10 mg: 0.76–1.00 h).
TABLE 3.
Pharmacokinetic parameters for DFV890 in plasma.
| Parameter, geomean (CV%) | DFV890 | |||||||
|---|---|---|---|---|---|---|---|---|
| Part A | 3 mg CS | 10 mg CS | 30 mg CS | 100 mg CS | 300 mg CS | 100 mg SDD | 300 mg SDD | 600 mg SDD |
| (N = 6) | (N = 6) | (N = 6) | (N = 6) | (N = 6) | (N = 6) | (N = 6) | (N = 6) | |
| C max (ng/mL) | 451 (18.8) | 1230 (15.1) | 1750 (27.2) | 4670 (25.3) | 4520 (19.0) | 11,200 (9.6) | 25,600 (21.9) | 59,100 (20.6) |
| C max/Dose (ng/mL mg) | 150 | 123 | 58.3 | 46.7 | 15.1 | 112 | 85.3 | 98.5 |
| t max, h (min–max) | 0.76 (0.75–1.0) | 1.00 (1.00–1.5) | 3.00 (1.5–6.0) | 2.00 (1.0–4.0) | 1.50 (1.0–2.0) | 3.0 (2.0–4.0) | 2.53 (1.5–12.0) | 2.5 (2.0–4.0) |
| AUC0–last (ng h/mL) | 3110 (19.1) | 9440 (18.7) | 27,700 (39.8) | 72,200 (47.7) | 63,000 (22.6) | 98,500 (29.6) | 327,000 (40.0) | 792,000 (41.9) |
| AUC0–last/Dose (ng h/mL mg) | 1037 | 944 | 923 | 722 | 210 | 985 | 1090 | 1320 |
| AUC0–inf (ng h/mL) | 3170 (19.9) | 9560 (19.0) | 23,800 (43.4) (N = 3) | 78,300 (48.6) (N = 5) | 63,700 (19.3) (N = 4) | 99,400 (29.1) | 329,000 (39.5) | 839,000 (46.6) |
| t 1/2 (h) | 8.62 (9.8) | 7.80 (9.9) | 16.4 (50.2) | 16.2 (74.3) | 19.9 (66.3) | 9.90 (38.4) | 10.9 (39.5) | 11.4 (21.3) |
| Part C | 10 mg q.d. CS | 30 mg q.d. CS | 100 mg q.d. SDD | 200 mg q.d. SDD | 25 mg b.i.d. ECT fed | 50 mg b.i.d. ECT fed |
|---|---|---|---|---|---|---|
| (N = 6) | (N = 6) | (N = 6) | (N = 7) | (N = 8) | (N = 7) | |
| Day 1 | ||||||
| C max (ng/mL) | 992 (11.1) | 2640 (10.0) | 10,700 (26.6) | 22,100 (19.8) | 2030 (13.7) | 3940 (14.0) |
| t max, h (min–max) | 1.0 (0.77–1.50) | 1.27 (0.87–2.0) | 3.0 (1.5–4.0) | 2.0 (1.0–3.02) | 4.0 (3.0–8.0) | 4.0 (3.0–8.0) |
| AUC0–tau (ng h/mL) | 8630 (20.6) | 24,800 (21.4) | 81,900 (13.6) | 163,000 (13.2) | 14,100 (17.3) | 27,000 (18.6) |
| Day 14 | ||||||
| C max (ng/mL) | 1040 (20.9) | 2770 (4.4) | 12,700 (25.2) | 26,300 (10.2) | 3470 (13.5) | 8520 (27.3) |
| t max, h (min–max) | 2.50 (1.50–3.00) | 1.50 (0.75–3.00) | 3.00 (3.00–3.00) | 2.00 (1.00–3.00) | 4.00 (3.00–4.03) | 2.25 (0.25–4.00) |
| AUC0–tau (ng h/mL) | 11,200 (28.1) | 29,400 (15.4) | 96,700 (32.2) | 181,000 (9.3) | 27,100 (17.5) | 61,200 (20.3) |
| AUC0–tau/Dose (ng h/mL mg) | 1120 | 980 | 967 | 905 | 1084 | 1224 |
| t 1/2 (h) | 9.58 (24.5) | 15.9 (22.1) | 9.58 (45.6) | 12.5 (47.6) | 10.4 (22.1) | 16.2 (4.8) |
| Rac (AUC) | 1.30 (11.1) | 1.28 (7.5) | 1.17 (11.0) | 1.20 (7.5) | 1.90 (14.4) | 2.24 (16.3) |
| Rac (C max) | 1.06 (14.9) | 1.08 (14.2) | 1.12 (10.7) | 1.15 (11.9) | 1.78 (21.5) | 2.20 (30.2) |
| Part D | 100 mg | 100 mg | 100 mg |
|---|---|---|---|
| CS fasted (N = 5) | CT fasted (N = 6) | CT fed (N = 6) | |
| C max (ng/mL) | 4820 (20.0) | 3750 (15.9) | 7680 (15.0) |
| t max, h (min–max) | 2.0 (1.0–3.0) | 5.0 (4.0–8.0) | 5.0 (3.0–8.0) |
| AUC0–last (ng h/mL) | 63,700 (25.7) | 74,900 (42.2) | 112,000 (25.3) |
| AUC0–inf (ng h/mL) | 77,100 (20.9) | 92,000 (36.0) | 117,000 (29.3) |
| t 1/2 (h) | 16.8 (38.7) | 18.4 (14.1) | 9.76 (21.7) |
Note: Values are represented as geometric mean (CV%) for each parameter, except for t max, for which the values are the median and (minimum–maximum) and except for Rac (mean (CV%)).
Abbreviations: b.i.d., twice‐daily; CS, crystalline suspension; CT, crystalline tablet; CV%, coefficient of variation; ECT, encapsulated crystalline tablet; N, number of subjects; NC, not calculated; q.d., once‐daily; SDD, spray‐dried dispersion suspension.
FIGURE 5.
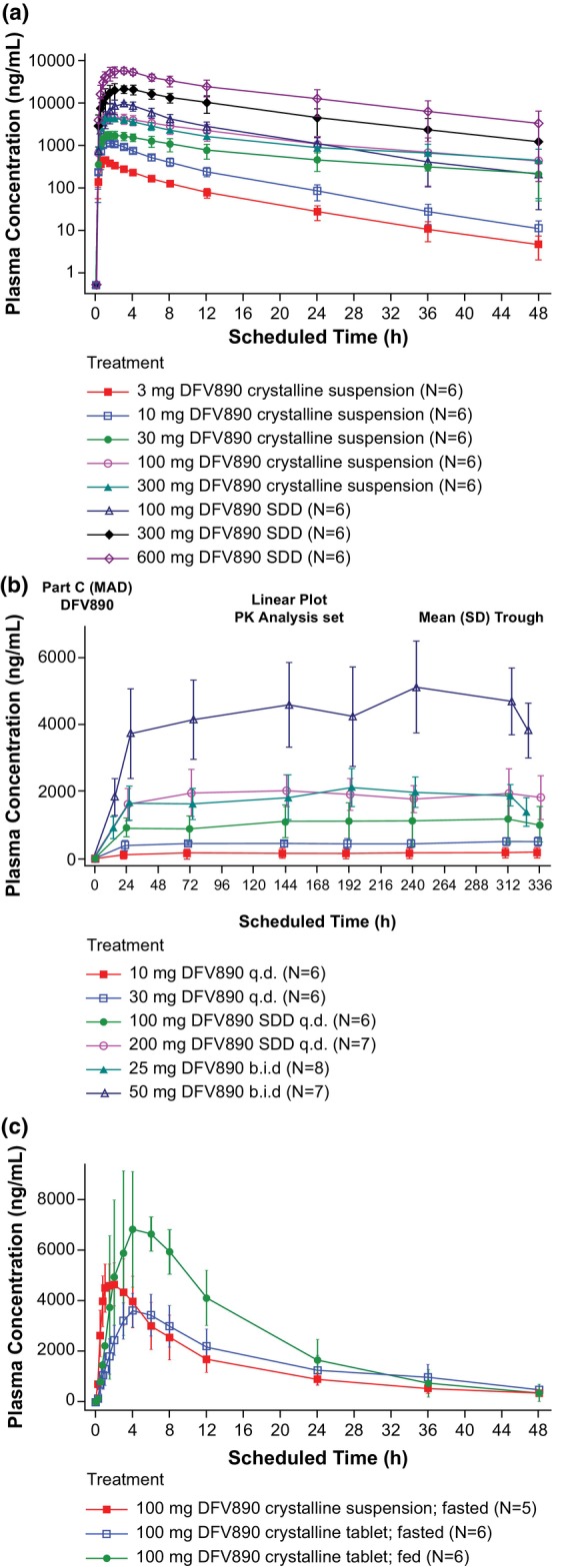
Plasma concentration‐time profiles of DFV890 (a) SAD, (b) MAD from Day 1 to Day 14, (c) Relative bioavailability and food effect. b.i.d., twice‐daily; MAD, multiple ascending dose; N, number of subjects randomized to treatment; n, number of subjects in each category; q.d., once‐daily; SAD, single ascending dose; SDD, spray‐dried dispersion.
Exposure increased dose‐proportionally when administered as SDD suspension (100–600 mg, slopes [90% CI]: 0.913 [0.802; 0.1.024] for C max and 1.121 [0.977; 0.1.265] for AUC0–last).
After q.d. administration of DFV890 dose range 30–200 mg for 2 weeks (Figure 5b), only limited drug accumulation of ~1.1 to 1.3‐fold was observed in reaching steady state. This is consistent with a geomean t 1/2 ranging from 9.58–16.2 h across q.d. and b.i.d. dose levels.
Total cumulative amount of DFV890 excreted in urine increased in linear manner with increasing multiple dose levels. DFV890 was mostly excreted within 12 h. On Days 1 and 14, the mean fraction of dose excreted was in the range 0.3%–0.4% and 0.7%–1.1% and renal clearance was in the range 3.6–5.0 mL/h and 6.8–9.1 mL/h, respectively.
Administration of a single 100 mg DFV890 dose as a crystalline suspension under fed conditions led to a 2.05‐fold increase in C max (90% CI: 1.78; 2.35) and 1.49‐fold increase in AUC0–last (90% CI: 1.21; 1.83) compared with fasting conditions (Figure 5b). For the crystalline tablet (100 mg DFV890 under fasting conditions), the median t max of DFV890 was delayed from 2 to 5 h, the C max was 78% of CS (geometric mean ratio [90% CI]: 0.78 [0.67; 0.90]), the AUC was similar (geometric mean ratio [90% CI]: 1.05 [0.84; 1.31]) and the t 1/2 was comparable between the crystalline tablet and suspension. The encapsulated crystalline tablets (25 and 50 mg b.i.d. under fed conditions) were characterized by a median lag time of 0.75 and 0.25 h, respectively, and a median t max of 4 h on Day 1. The geomean t 1/2 of DFV890 was comparable between the tablet (18.4 h) and suspension (16.8 h) formulations (Table 3).
Pharmacodynamics
Dose‐dependent decreases of IL‐1β concentrations in whole blood samples after ex vivo LPS stimulation (with mean nadir concentrations of ~5%–20% of the baseline value) were observed with increasing single and multiple oral DFV890 doses (Figure 6). At most dose levels, the inhibition of IL‐1β release was observed from 1 h after dosing until the last sampling timepoint for single (Day 3 or up to 6 h for the lowest ≤10 mg dose levels) and multiple (Day 15) oral doses of DFV890.
FIGURE 6.
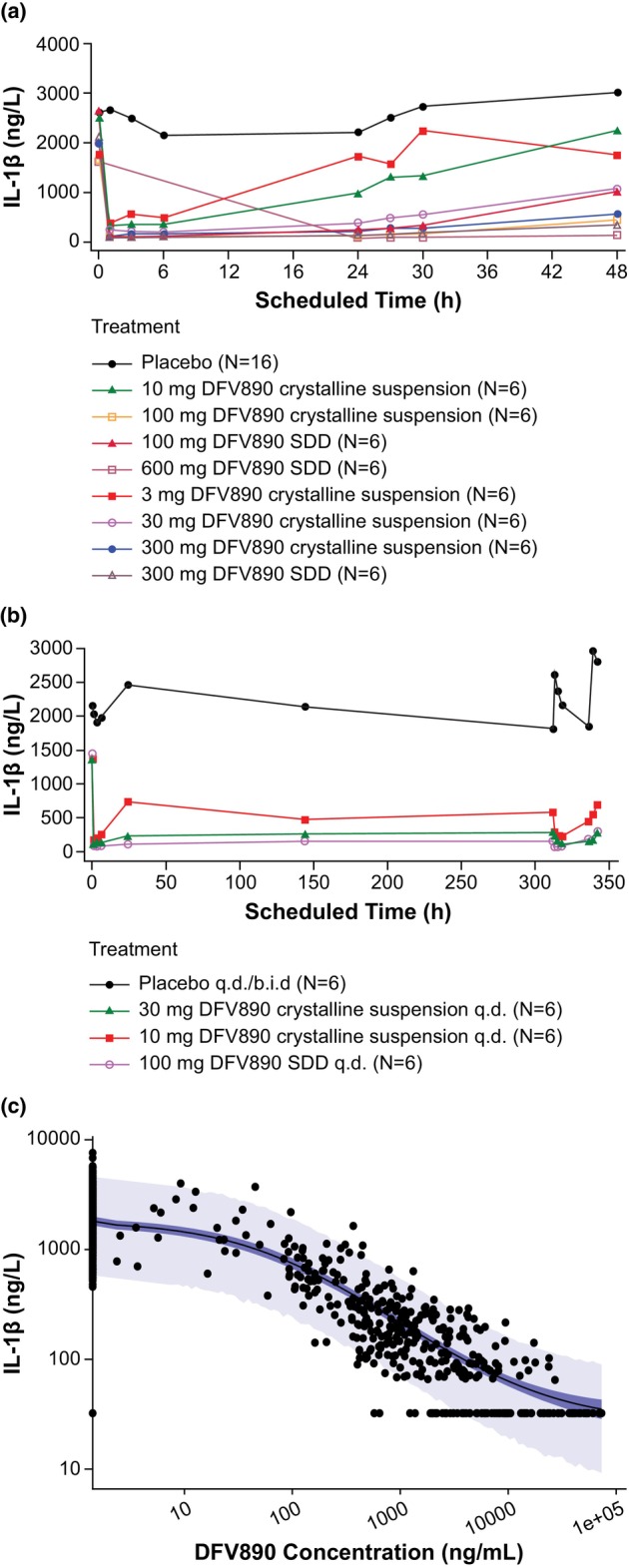
Geometric mean IL‐1β concentrations plots (a) in SAD, (b) in MAD, (c) IL‐1β versus DFV890 concentrations. Panel (c): Model‐estimated 90% credible interval (dark blue region), 90% prediction interval (light blue region), median estimate (black line), and observed IL‐1β concentration (points) versus DFV890 concentration, log–log scale. b.i.d., twice‐daily; MAD, multiple ascending dose; q.d., once‐daily; SAD, single ascending dose; SDD, spray‐dried dispersion.
Based on the fractional maximum stimulation effect (E max), a model tested using a Hill coefficient, the typical baseline (E 0) (±SD) of the observed stimulation effect of IL‐1β was 1820 (±102) ng/L; the E max of IL‐1β was −0.985 (±0.00277) and the Hill coefficient was 0.758 (±0.0351). The effective concentrations relative to the estimated maximum effect of DFV890 resulting from ex vivo stimulated IL‐1β release were EC50: 59 ng/mL (90% CI: 48, 72), and EC90: 1080 ng/mL (90% CI: 942, 1240). When considering full inhibition (fixing E max = 1) then the plasma concentrations of DFV890 inhibiting 50% and 90% of the LPS ex vivo stimulated IL‐1β release (IC50 and IC90) were 61 ng/mL (90% CI: 50; 70) and 1340 ng/mL (90% CI: 1190; 1490), respectively, and the Hill coefficient was 0.715 (±0.0333). A sensitivity analysis setting values below LLOQ to 0 for both PK and PD was performed. PK/PD model parameters were re‐estimated and the new estimates were in agreement with those reported (within approximately 3%–15% of the original estimates).
DISCUSSION
Although antibodies against inflammatory cytokines (i.e., IL‐1β and IL‐6) have been approved for clinical use, there is significant medical need to widen therapeutic modalities for patients suffering from NLRP3‐mediated inflammatory, metabolic, and neurodegenerative diseases that offer an effective, predictable, and convenient treatment option without increased risk of AEs. 19 In this study, a low‐molecular‐weight NLRP3 antagonist DFV890 was administered orally for the first time to human subjects exploring safety, tolerability, PK, and PD properties. We selected initial doses based on predicted human PK and anticipated efficacious doses, as well non‐clinical safety from animal and in vitro data. DFV890 is characterized by a good permeability and a low solubility of crystalline form at physiological pH. The real‐time manufacturing of drug product was introduced at the clinical site (so called Translational Pharmaceutics approach) for flexible dose and formulation preparation to support objective of the study. Initially, in the SAD part, we used crystalline suspension at doses of 3–300 mg. However, 300 mg dose provided similar exposure to 100 mg. The 100 mg dose was repeated with the SDD suspension containing amorphous form and this formulation was continued up to 600 mg to evaluate supra‐therapeutic DFV890 exposure in preparation for future clinical development. Based on initial PK and PD data from the SAD part, tablets made of 25 mg crystalline DFV890 were produced to evaluate the relative bioavailability/food effect at 100 mg. Tablets we also dosed in the two last MAD cohorts twice‐daily under fed conditions and were over‐encapsulated to maintain blinding.
Single and multiple doses of DFV890 or placebo were generally well‐tolerated, and neither deaths nor SAE were reported. Similar rates of TEAEs were observed between subjects receiving DFV890 (70%) and placebo (75%). The majority of TEAEs reported by subjects were mild (69%) or moderate (12%) in severity. Maculopapular and/or pruritic skin rashes of mild‐to‐moderate intensity were reported in 10% of subjects receiving higher DFV890 doses. Skin rashes were self‐limiting after treatment discontinuation and resolved without sequalae, but considered to be DFV890 related, independent of the formulation used. Skin rashes started to appear mainly at 100 mg q.d. after multiple dosing. To reach trough plasma levels ≥IC90 at steady‐state and to reduce C max (in case rash was C max‐driven) twice‐daily dose regimen was included. Skin reactions were seen in some participants dosed with 50 mg b.i.d., but not in 25 mg b.i.d. Tolerability data from other NLRP3 inhibitors like ZYIL1 or dapasuntrile 20 , 21 , 22 currently being tested in early clinical trials did not yet report any drug‐related skin reactions. In contrary, GDC‐2394, another NLRP3 inhibitor, caused rash in 22% healthy subjects at 900 mg, and thus it is unknown if skin reactions are structure related or whether it is an NLRP3 inhibitory on‐target effect.
Plasma PK data confirmed solubility/dissolution‐limited absorption of the crystalline suspension at dose levels ≥100 mg. Doses, formulations, and conditions selected for the MAD part showed no deviations from dose proportionality in drug‐exposure after 2 weeks indicated by similar dose‐normalized AUC and C max, signifying that multiple‐dose PK parameters were linear.
Slight drug accumulation of ~1.2‐fold after q.d. dosing and twofold after b.i.d. dosing was observed in reaching steady state, consistent with an effective t 1/2 of ~10 h, as it was determined for crystalline tablets when given with food. Doses ≥30 mg given as crystalline form under fasted conditions had longer terminal half‐lives. It is likely due to a flip‐flop mechanism, in which the terminal half‐life was partially influenced by the absorption rate. DFV890 had a very low apparent oral clearance, which relates to ≤2% of human liver blood flow and a low apparent volume of distribution indicating limited tissue distribution in line with preclinical data. Only <0.8% of an oral dose was excreted in urine as DFV890. This shows that renal clearance is not a major elimination route for this drug in humans.
Nonclinical studies have suggested that DFV890 blocks the maturation and release of IL‐1β related to a range of NLRP3‐dependent activators. This has also been observed with MCC950, which selectively inhibits NLRP3 activation, 23 GDC‐2394 24 and ZYIL1, which demonstrated >90% IL‐1β release inhibition in healthy subjects. 20 In contrast, dapansutrile (OLT1177), another NLRP3 inhibitor, only partially reduced IL‐1β release in healthy subjects and patients with gout flare. 21 , 22
In this study, dose‐dependent decreases in stimulated IL‐1β concentrations were observed with increasing single and multiple oral doses of DFV890. IL‐1 β production can be mediated by other inflammasomes or by inflammasome‐independent pathways; 25 thus, inhibitors aimed at IL‐1β can result in unintentional immunosuppressive effects. Therefore, pharmacological inhibitors that specifically target the NLRP3 inflammasome alone could be a better option for treatment of NLRP3‐associated diseases. 26 Safety laboratory findings, if at all, were mild; a clinically non‐relevant decrease in neutrophil and leukocyte counts was observed in 27 subjects, which appears to be consistent with a PD effect resulting from inhibition of signaling downstream of NLRP3, similar to the known effects of the anti‐IL‐1β monoclonal antibody canakinumab. 27
DFV890 exhibited rapid onset of action on IL‐1β inhibition with clear dose response over the entire dose range investigated, both after single and multiple doses (Figure 6a,b), suggesting a direct PK/PD relationship. To maintain ~90% of IL‐1β inhibition over 24 h, a dose of 25 mg twice‐daily administered as crystalline tablet was chosen for the ongoing Phase II a study in patients with inflammatory knee osteoarthritis.
A recent report from an early Phase IIa clinical trial of DFV890 in SARS‐CoV‐2 infected patients with COVID‐19‐associated pneumonia and impaired respiratory function in presence of standard of care (SoC) versus SoC alone, also showed that DFV890 tablets at a dose level of 50 mg b.i.d. were well‐tolerated, and no new safety signals were identified. 28 To date, several Phase II studies are ongoing to evaluate DFV890 in multiple indications including familial cold auto inflammatory syndrome (FCAS), knee osteoarthritis, myelodysplastic syndromes, chronic myelomonocytic leukemia, and coronary heart disease.
In summary, single and multiple doses of DFV890 were well‐tolerated for up to 14 days in healthy subjects, with no safety or tolerability concerns. The PK profile of DFV890 is compatible with a b.i.d. dosing regimen and PK/PD data supported dose and formulation selection for further development. The safety and tolerability, PK, and PD results suggest that DFV890 has the potential to be a clinically effective and orally available direct NLRP3 inhibitor warranting further clinical evaluation.
AUTHOR CONTRIBUTIONS
G.J., E.G., B.M., P.P., Y.F., U.S., E.V., D.D., and A.O. wrote the manuscript. D.D., A.O., G.J., E.T., and E.G. designed the research. M.V. performed the research; Y.F., U.S., G.L., P.P., W.S.D., and X.L. analyzed the data.
FUNDING INFORMATION
The study was funded by IFM Therapeutics in accordance with Good Publication Practice guidelines.
CONFLICT OF INTEREST STATEMENT
E.G., B.M., E.V., U.S., E.T., G.L., P.P., Y.F., and G.J. are employees of Novartis with Novartis stock. X.L. was an employee of Novartis when the study was conducted. A.O. and D.D. are employees of IFM Therapeutics with stock. M.V. is an employee of ICON and W.S.D. is a consultant for IFM Therapeutics.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
We thank the healthy participants who joined this study. The authors thank Md Zuber Birajdar, Rajeeb Ghosh and Shravanti Mukherjee, Novartis Healthcare Pvt. Ltd., for medical writing support and editorial guidance, Martin Fink, Novartis Pharma AG Switzerland, for scientific contribution to the PK/PD model and Raphael Sommer, Novartis Pharma AG, Basel, Switzerland, for operational support during execution and completion of the study.
Gatlik E, Mehes B, Voltz E, et al. First‐in‐human safety, tolerability, and pharmacokinetic results of DFV890, an oral low‐molecular‐weight NLRP3 inhibitor. Clin Transl Sci. 2024;17:e13789. doi: 10.1111/cts.13789
Clinical trials registration: EudraCT number: 2018‐004402‐26
DATA AVAILABILITY STATEMENT
The datasets generated and analyzed during the current study are not publicly available. Novartis is committed to sharing access to patient‐level data and supporting clinical documents from eligible studies with qualified external researchers. These requests are reviewed and approved based on scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the trial, in line with applicable laws and regulations. The data can be requested from the corresponding author of the manuscript. The protocol can be made available on request by contacting the corresponding author.
REFERENCES
- 1. Jiang N, Li Z, Luo Y, et al. Emodin ameliorates acute pancreatitis‐induced lung injury by suppressing NLRP3 inflammasome‐mediated neutrophil recruitment. Exp Ther Med. 2021;22:857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. 2015;6:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Franchi L, Eigenbrod T, Munoz‐Planillo R, Nunez G. The inflammasome: a caspase‐1‐activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210‐215. [DOI] [PubMed] [Google Scholar]
- 5. Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119‐1131. [DOI] [PubMed] [Google Scholar]
- 6. Ridker PM, MacFadyen JG, Thuren T, et al. Effect of interleukin‐1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double‐blind, placebo‐controlled trial. Lancet. 2017;390:1833‐1842. [DOI] [PubMed] [Google Scholar]
- 7. Schieker M, Conaghan PG, Mindeholm L, et al. Effects of interleukin‐1β inhibition on incident hip and knee replacement: exploratory analyses from a randomized, double‐blind, placebo‐controlled trial. Ann Intern Med. 2020;173:509‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vallurupalli M, MacFadyen JG, Glynn RJ, et al. Effects of interleukin‐1β inhibition on incident anemia: exploratory analyses from a randomized trial. Ann Intern Med. 2020;172:523‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ridker PM, Libby P, MacFadyen JG, et al. Modulation of the interleukin‐6 signalling pathway and incidence rates of atherosclerotic events and all‐cause mortality: analyses from the Canakinumab Anti‐Inflammatory Thrombosis Outcomes Study (CANTOS). Eur Heart J. 2018;39:3499‐3507. [DOI] [PubMed] [Google Scholar]
- 10. Guo C, Fu R, Wang S, et al. NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin Exp Immunol. 2018;194:231‐243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kingsbury SR, Conaghan PG, McDermott MF. The role of the NLRP3 inflammasome in gout. J Inflamm Res. 2011;4:39‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baldrighi M, Mallat Z, Li X. NLRP3 inflammasome pathways in atherosclerosis. Atherosclerosis. 2017;267:127‐138. [DOI] [PubMed] [Google Scholar]
- 13. Zhang Y, Dong Z, Song W. NLRP3 inflammasome as a novel therapeutic target for Alzheimer's disease. Signal Transduct Target Ther. 2020;5:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haque ME, Akther M, Jakaria M, Kim IS, Azam S, Choi DK. Targeting the microglial NLRP3 inflammasome and its role in Parkinson's disease. Mov Disord. 2020;35:20‐33. [DOI] [PubMed] [Google Scholar]
- 15. Knorr J, Wree A, Tacke F, Feldstein AE. The NLRP3 inflammasome in alcoholic and nonalcoholic steatohepatitis. Semin Liver Dis. 2020;40:298‐306. [DOI] [PubMed] [Google Scholar]
- 16. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20:3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xuan TQ, Gong G, du H, et al. Protective effect of pteryxin on LPS‐induced acute lung injury via modulating MAPK/NF‐kappaB pathway and NLRP3 inflammasome activation. J Ethnopharmacol. 2022;286:114924. [DOI] [PubMed] [Google Scholar]
- 18. Wang B‐S, Wang X‐J, Gong L‐K. The construction of a Williams design and randomization in cross‐over clinical trials using SAS. J Stat Softw. 2009;29:1‐10. [Google Scholar]
- 19. Chauhan D, Vande Walle L, Lamkanfi M. Therapeutic modulation of inflammasome pathways. Immunol Rev. 2020;297:123‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parmar DV, Kansagra KA, Momin T, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of the oral NLRP3 Inflammasome inhibitor ZYIL1: first‐in‐human phase 1 studies (single ascending dose and multiple ascending dose). Clin Pharmacol Drug Dev. 2023;12:202‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marchetti C, Swartzwelter B, Gamboni F, et al. OLT1177, a β‐sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci USA. 2018;115:E1530‐E1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klück V, Jansen TLTA, Janssen M, et al. Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: an open‐label, dose‐adaptive, proof‐of‐concept, phase 2a trial. Lancet Rheumatol. 2020;2:e270‐e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tapia‐Abellán A, Angosto‐Bazarra D, Martínez‐Banaclocha H, et al. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat Chem Biol. 2019;15:560‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tang F, Kunder R, Chu T, et al. First‐in‐human phase 1 trial evaluating safety, pharmacokinetics, and pharmacodynamics of NLRP3 inflammasome inhibitor, GDC‐2394, in healthy volunteers. Clin Transl Sci. 2023;16:1653‐1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gaidt MM, Hornung V. Alternative inflammasome activation enables IL‐1β release from living cells. Curr Opin Immunol. 2017;44:7‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zahid A, Li B, Kombe AJK, Jin T, Tao J. Pharmacological inhibitors of the NLRP3 inflammasome. Front Immunol. 2019;10:2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dhimolea E. Canakinumab. MAbs. 2010;2:3‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Madurka I, Vishnevsky A, Soriano JB, et al. DFV890: a new oral NLRP3 inhibitor‐tested in an early phase 2a randomised clinical trial in patients with COVID‐19 pneumonia and impaired respiratory function. Infection. 2023;51:641‐654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
The datasets generated and analyzed during the current study are not publicly available. Novartis is committed to sharing access to patient‐level data and supporting clinical documents from eligible studies with qualified external researchers. These requests are reviewed and approved based on scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the trial, in line with applicable laws and regulations. The data can be requested from the corresponding author of the manuscript. The protocol can be made available on request by contacting the corresponding author.