

Summary
CBASS is a common anti-phage immune system that uses cyclic oligonucleotide signals to limit phage replication. In turn, phages encode anti-CBASS (Acb) proteins like Acb2, which can sequester the cyclic dinucleotide (CDN) cGAMP. Here, we identified that Acb2 sequesters many CBASS and cGAS-produced CDNs and inhibits cGAMP-mediated STING activity in human cells. Surprisingly, the Acb2 hexamer also binds with high affinity to CBASS cyclic trinucleotides (CTNs) 3’3’3’-cyclic AMP-AMP-AMP and 3’3’3’-cAAG at a distinct site from CDNs. One Acb2 hexamer can simultaneously bind two CTNs and three CDNs. Phage-encoded Acb2 provides protection from type III-C CBASS that uses cA3 signaling molecules. Moreover, phylogenetic analysis of >2,000 Acb2 homologs encoded by diverse phages and prophages revealed that most are expected to bind both CTNs and CDNs. Altogether, Acb2 sequesters nearly all known CBASS signaling molecules through two distinct binding pockets and therefore serves as a broad-spectrum inhibitor of cGAS-based immunity.
Introduction
Anti-viral immune pathways across all kingdoms of life sense and respond to viral infection. Cyclic GMP-AMP synthase (cGAS) is an evolutionarily conserved enzyme that performs a pivotal role in innate immunity against viruses. 1 In mammalian cells, cGAS binds viral DNA and is activated to produce 2’,3’-cyclic GMP-AMP (2’,3’-cGAMP) dinucleotides, which activate the STING (stimulator of interferon genes) effector protein to initiate a potent interferon response. 2, 3 In bacteria, cGAS-like enzymes named cGAS/DncV-like nucleotidyltransferases (CD-NTases) have been identified and enzymatically characterized. 4–6 CD-NTases have been classified into 8 enzymatic clades and at least 12 cyclic di- and trinucleotide products have been identified. 4–9 During phage infection, these enzymes are activated and produce cyclic oligonucleotides that bind to and activate a downstream effector protein. The activated effector proteins are proposed to induce premature cell death through various mechanisms, including membrane impairment, 8, 10 DNA degradation, 9, 11, 12 NAD+ depletion, 13, 14 amongst others. This anti-phage strategy was named cyclic-oligonucleotide-based anti-phage signaling system (CBASS).
As a countermeasure to CBASS immunity, phages encode anti-CBASS (Acb) proteins. Acb1 degrades the cyclic nucleotide messengers to inhibit CBASS 15 and Acb2 is a cyclic dinucleotide (CDN) sponge. 16, 17 Interestingly, Acb2 also binds to a variety of other CDNs: 2’,3’-cGAMP and 3’,3’-cUU/UA/UG/AA with varying affinities. Structures of Acb2 from P. aeruginosa phage PaMx33 and from E. coli phage T4 in complex with 3’,3’-cGAMP showed that Acb2 forms an interlocked hexamer and binds to three cyclic dinucleotides (CDNs), each with a binding pocket located in one Acb2 dimer within the hexamer. 16, 17 However, it remains unknown whether Acb2 binds to cyclic nucleotides that are utilized in Pycsar, CBASS, and Type III CRISPR-Cas signaling systems. 18–20 Here, we find that Acb2 not only binds to and sequesters a broad spectrum of CDNs, but it also binds to cyclic trinucleotides (CTNs) 3’3’3’-cyclic AMP-AMP-AMP (cA3 hereafter) and 3’3’3’-cAAG (cAAG) with an order of magnitude higher affinity. CBASS systems commonly use these CTN signals, 7 as do some Type III CRISPR-Cas systems. 21 Structural characterization identified that one Acb2 hexamer binds two CTNs within binding pockets that are different from those binding CDNs. A co-structure of Acb2 bound to cA3 and 3’,3’-cGAMP at the same time is presented, as are mutants that independently disrupt the different binding sites. Plasmid and phage-encoded Acb2 effectively protects phage when infecting cells that independently and simultaneously express cA3 and cGAMP CBASS systems. Together, this work identifies two distinct cyclic oligonucleotide binding sites on Acb2 and demonstrates that Acb2 is an effective, broad-spectrum inhibitor of CBASS and cGAS-STING signaling pathways.
Results
Acb2 sequesters diverse cyclic dinucleotides and is active in human cells
To understand the selectivity of the newly identified Acb2 protein fold, we comprehensively tested an array of cyclic oligonucleotides that Acb2 may bind to. Previous work revealed that Acb2 binds to 3’,3’-cGAMP, 2’,3’-cGAMP and 3’,3’-cUU/UA/UG/AA, but not 3’,3’-cGG 16 (Figure S1A–B). In this current study, we first tested Acb2 binding of 3’,2’-cGAMP, which was recently identified as a signaling molecule for both CBASS and cGAS-like enzymes in eukaryotes. 9, 22 A native gel assay showed a significant shift of the Acb2 protein upon adding 3’,2’-cGAMP (Figure S1A), and isothermal calorimetry (ITC) experiments verified that Acb2 binds to 3’,2’-cGAMP with a KD of ~297.7 nM (Figures 1A and S2). Next, we solved the structure of Acb2 complexed with 3’,2’-cGAMP (2.33 Å, Figure 1B), in which 3’,2’-cGAMP binds in the same binding pocket and shows a similar binding mode as 3’,3’-cGAMP and c-di-AMP (Figures 1C–E, S3A, and Table 1). Specifically, these CDN molecules are bound by the N-terminal domains of the two interacting Acb2 protomers, each from one Acb2 dimer (Figures 1D). The π-π stacking from Y11 residue and salt bridges from K26 residue of both protomers further stabilize this interaction (Figures 1C–D). The structure of Acb2 complexed with another cGAMP isomer, 2’,3’-cGAMP, solved at 2.24 Å resolution further confirmed this mode of binding (Figures 1D–E, S3B, and Table 1). Interestingly, multiple CDNs are tolerated in the binding pocket. Their base groups are mainly stabilized by the π-π stacking from the Y11 residue (Figures 1C–D). Therefore, all the three tested base groups (adenine, guanine, and uridine) are tolerated, albeit with different affinities. For the phosphate-ribose backbone, interestingly, 3’,3’-, 2’,3’- and 3’,2’-cGAMP linkages are all tolerated by Acb2. Detailed analysis further shows that for each linkage, its phosphate group can be stabilized by polar interactions from both K26 and Y11 (Figure 1D), while the other interacting Acb2 residues might vary a little. However, the cavity in Acb2 is large enough for all the three linkages (Figure 1E). Since 3’,2’-cGAMP and 2’,3’-cGAMP are ligands used in eukaryotic cGAS-STING immunity, we also tested whether Acb2 can antagonize cGAS-STING signaling pathway in human cells. The results showed that upon expression of WT Acb2, interferon (IFN) signaling mediated by 2’,3’-cGAMP is significantly reduced while Y11A and K26A Acb2 mutants were less active (Figure 1F). Consistent with these data, native gel assays showed that Y11A and K26A mutations abrogated 2’,3’-cGAMP binding (Figure S1C). Taken together, our data demonstrates that Acb2 harbors a binding pocket that is well suited for many CDNs.
Figure 1. Acb2 from phage PaMx33 binds cyclic trinucleotides and 3’, 2’-cGAMP.
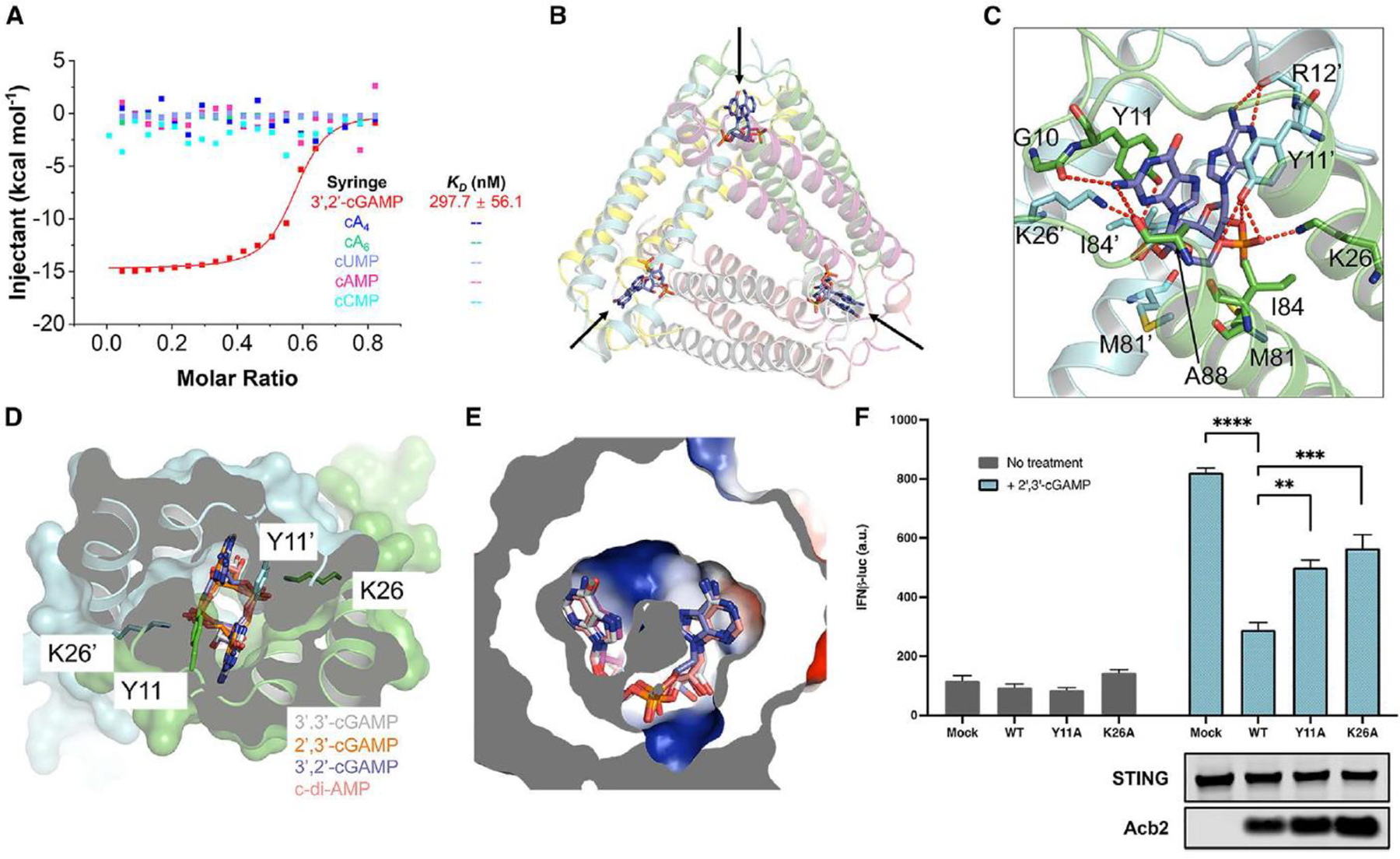
(A) ITC assays to test binding of cyclic nucleotides to PaMx33-Acb2. Representative binding curves and binding affinities are shown. The KD values are mean ± s.d. (n=3). Raw data for these curves are shown in Figure S2.
(B) Overall structure of Acb2 complexed with 3’,2’-cGAMP, which are indicated by arrows.
(C) Detailed binding between Acb2 and 3’,2’-cGAMP. Residues involved in 3’,2’-cGAMP binding are shown as sticks. Red dashed lines represent polar interactions.
(D) Structural alignment among 3’,2’-cGAMP, 2’,3’-cGAMP, 3’,3’-cGAMP and c-di-AMP bound Acb2. Surface representation overlaid to cartoon representation, highlighting the binding pocket of CDNs.
(E) Electrostatic surface model showing the binding pocket of CDNs. The CDNs are colored as in D.
(F) 293T-Dual cells were transfected with hSTING and Acb2 or its mutants, then treated with 2’,3’-cGAMP. STING activation was read as luciferase signal controlled by an interferon promoter. A western blot is shown probing the expression of STING and Acb2.
Table 1.
Data collection and refinement statistics
| Data set | Acb2–2’,3’-cGAMP | Acb2–3’,2’-cGAMP | Acb2-cA3 | Acb2-cAAG | Acb2–3’,3’-cGAMP-cA3 |
|---|---|---|---|---|---|
|
| |||||
| PDB code | 8J8O | 8IXZ | 8IY0 | 8IY1 | 8IY2 |
| Data collection | |||||
|
| |||||
| Space group | P321 | P321 | P321 | P321 | P321 |
| Cell dimensions | |||||
| a, b, c (Å) | 106.0 106.0 101.6 | 104.0 104.0 101.1 | 103.9 103.9 101.9 | 103.6 103.6 101.4 | 103.4 103.4 101.5 |
| α, β, γ (°) | 90 90 120 | 90 90 120 | 90 90 120 | 90 90 120 | 90 90 120 |
| Resolution (Å) | 50–2.24 (2.28–2.24) |
50–2.32 (2.36–2.32) |
50–2.26 (2.30–2.26) |
50–2.10 (2.14–2.10) |
50–2.76 (2.81–2.76) |
| Unique reflections | 31815 (1484) | 27614 (1352) | 29547 (1474) | 36272 (1648) | 16565 (824) |
| Completeness (%) | 99.5 (94.3) | 100.0 (100.0) | 98.7 (99.1) | 98.1 (92.8) | 99.7 (100.0) |
| Rmeas (%) | 10.6 (96.7) | 8.4 (60.8) | 12.3 (103.7) | 9.9 (87.9) | 13.3 (101.2) |
| Redundancy | 4.8 (3.9) | 17.6 (13.1) | 4.8 (4.8) | 17.6 (13.6) | 9.8 (8.9) |
| I/σ(I) | 18.2 (1.4) | 50.0 (4.8) | 18.3 (2.2) | 47.9 (4.1) | 20.8 (2.5) |
| Statistics for Refinement | |||||
|
| |||||
| Rwork (%) | 23.8 (32.4) | 23.0 (27.3) | 21.5 (25.2) | 22.1 (24.6) | 24.0 (34.8) |
| Rfree (%) | 27.3 (34.6) | 27.4 (32.6) | 25.8 (35.0) | 24.9 (28.8) | 28.9 (40.1) |
| No. atoms | 4915 | 4741 | 5019 | 5072 | 5051 |
| Protein | 4416 | 4396 | 4440 | 4440 | 4396 |
| Ligand/ion | 270 | 270 | 396 | 402 | 628 |
| Solvent | 229 | 75 | 183 | 230 | 27 |
| B factors | 51.90 | 55.65 | 48.17 | 45.56 | 57.43 |
| Protein | 52.14 | 56.07 | 49.77 | 47.10 | 58.87 |
| Ligand/ion | 50.60 | 51.32 | 33.26 | 31.55 | 47.68 |
| Solvent | 47.16 | 47.03 | 41.65 | 40.42 | 49.83 |
| R.m.s.d. | |||||
| Bond angles (°) | 1.49 | 2.49 | 2.37 | 2.90 | 2.42 |
| Bond length (Å) | 0.013 | 0.016 | 0.014 | 0.029 | 0.016 |
| Ramachandran Plot (%) | |||||
| Favored region | 99.07 | 96.26 | 97.59 | 96.67 | 97.01 |
| Allowed region | 0.93 | 3.74 | 2.41 | 3.33 | 2.99 |
| Outliers | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
Acb2 sequesters cyclic trinucleotides with higher affinity than cyclic dinucleotides
Based on the binding pocket of Acb2, we hypothesized that Acb2 may not bind cyclic mononucleotides or oligonucleotides, such as cA3, cA4 or cA6, due to potential steric clash caused by the nucleotides. Of note, cA3 and cAAG are major products of the CD-NTase enzymes involved in CBASS whereas cA4 is only a minor product of a single CD-NTase. 11 cA6 has not been identified as a product of any known CD-NTases. However, all three cyclic oligoadenylates are known products involved in Type III CRISPR-Cas anti-phage immunity. 23 A native gel assay showed that the Acb2 protein does not shift upon adding cA4 or cA6 molecules (Figures S1A–B). Furthermore, both native gel and ITC assays showed that Acb2 does not bind to cUMP, cCMP or cAMP (Figures S1–2 and 1A). However, the native gel assay revealed a significant shift of the Acb2 protein upon adding cA3 or cAAG (Figure S1B). ITC experiments revealed that Acb2 binds to cA3 and cAAG with a KD of ~1.5 and ~2.7 nM (Figures 2A and S2), respectively, which is more than an order of magnitude stronger than Acb2 binding to 3’,3’-cGAMP (KD of ~87 nM). 16 To determine whether Acb2 sequesters or cleaves the CTN molecules, high-performance liquid chromatography (HPLC) revealed that incubating Acb2 with cA3 depletes any detectable molecules, and following proteolysis of Acb2, cA3 is released back into the buffer unmodified (Figure 2B). Collectively, these results demonstrate that Acb2 binds to and sequesters CTNs commonly used in CBASS immunity with a significantly higher affinity than CDNs.
Figure 2. Acb2 binds to cyclic trinucleotides with binding sites different from those of cyclic dinucleotides.
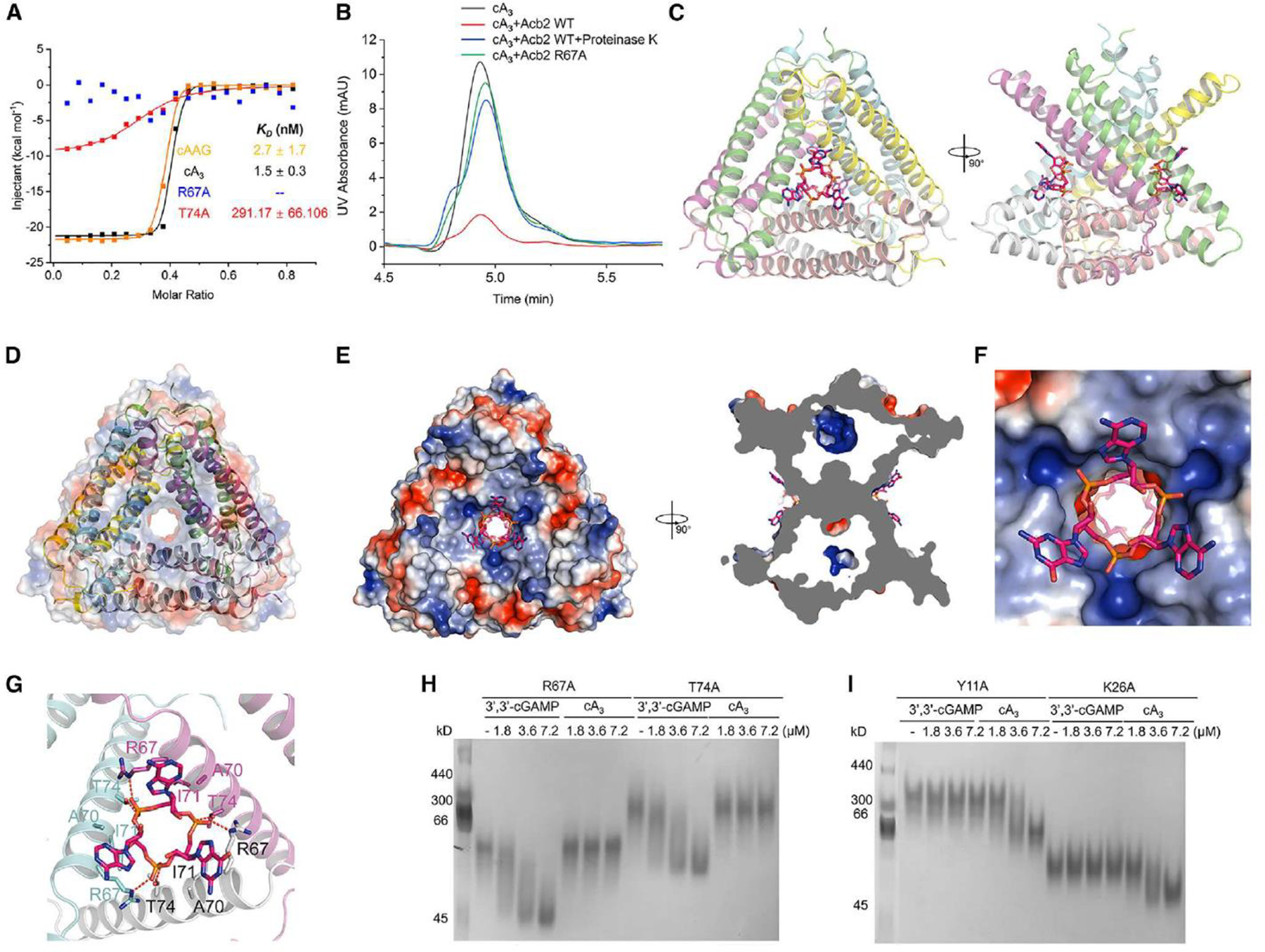
(A) ITC assays to test the binding of cAAG and cA3 to PaMx33-Acb2, and binding of cA3 to PaMx33-Acb2 mutants. Representative binding curves and binding affinities are shown. The KD values are mean ± s.d (n = 3). Raw data for these curves are shown in Figure S2. The two mutants R67A and T74A in the panel represent their binding to cA3.
(B) The ability of PaMx33-Acb2 to bind and release cA3 when treated with proteinase K was analyzed by HPLC. cA3 standard was used as a control. The remaining cA3 after incubation with PaMx33-Acb2 was tested.
I Overall structure of Acb2 complexed with cAAG, which are shown as sticks. Two views are shown.
(D) Electrostatic surface of Acb2 overlaid on the cartoon model shows the channel in the center of the Acb2 hexamer.
I Electrostatic surface of Acb2 bound with cAAG. Two views are shown.
(F) A closer view of the binding pocket shown in the left panel of D.
(G) Detailed binding between Acb2 and cAAG. Residues involved in cAAG binding are shown as sticks. Red dashed lines represent polar interactions.
(H-I) Native PAGE showed the binding of PaMx33 Acb2 mutants to cyclic oligonucleotides.
Acb2 binds cyclic trinucleotides and dinucleotides with different binding sites
The binding of CTNs was unexpected because the Acb2 binding pocket appears well suited for only CDNs. To understand how Acb2 interacts with CTNs, we determined the crystal structures of Acb2 in complex with cA3 (2.26 Å, Figure S3C) or cAAG (2.10 Å, Figure S3D) (Table 1). Surprisingly, the structures showed that one Acb2 hexamer binds two CTNs with two distinct binding pockets that are far from the three pockets for the CDN binding (Figures 2C and S4A–B). An Acb2 hexamer can be viewed as a trimer of dimers, which forms a channel in the center of the hexamer (Figure 2D). Interestingly, each binding pocket of the CTNs is formed by three Acb2 protomers, each from one different Acb2 dimer, in a three-fold symmetry (Figure S4B). The two CTN molecules bind at the two ends of the channel, blocking the channel from two opposite sides (Figure 2E). The binding modes of CDNs and CTNs within Acb2 can be described as follows: Each of the two protomers that together bind a CDN is involved in binding to one out of the two CTNs, respectively (Figure S4A). Correspondingly, each of the three protomers that together bind a CTN is involved in binding to one out of the three CDNs, respectively (Figure S4B).
The CTN is bound mainly through its three phosphate groups, each of which is coordinated by R67 of one protomer and T74 of another protomer through hydrogen bonds (Figures 2F–G). Moreover, the CTN is also stabilized by hydrophobic interactions from R67, A70, and I71 from each of the three protomers (Figure 2G). Consistent with this analysis, the Acb2 T74A mutant displayed a significantly decreased binding affinity to cA3 (KD of ~291 nM), and the Acb2 R67A mutant abolished Acb2 binding of cA3 in vitro (Figures 2A and S2). To confirm that the binding sites of the CTNs and CDNs in Acb2 are independent of each other, we tested the binding of 3’,3’-cGAMP with the T74A or R67A Acb2 mutant proteins. A native gel assay showed similar shifts of the two Acb2 mutants as WT Acb2 upon adding 3’,3’-cGAMP (Figure 2H), suggesting that the binding to 3’,3’-cGAMP is not affected by the two mutations. In turn, we tested the binding of cA3 with Y11A and K26A Acb2 mutants, which lose their binding to 3’,3’-cGAMP. 16 The native gel results showed a significant shift of Y11A and K26A mutant proteins upon adding cA3 (Figure 2I). Taken together, these data collectively show that one Acb2 hexamer binds two CTNs through two pockets independent of those that bind CDNs.
Structural alignment between apo Acb2 and its complexes with CTNs showed that the binding of CTNs does not induce a conformational change of Acb2, with a root mean square deviation (RMSD) of 0.224 and 0.261 Å (Cα atoms) for Acb2-cA3 and Acb2-cAAG compared to the apo Acb2, respectively (Figure S4C). Therefore, we co-crystallized Acb2 with both cA3 and 3’,3’-cGAMP and then solved its crystal structure at a resolution of 2.76 Å (Table 1). The structure clearly showed that Acb2 binds to two cA3 and three 3’,3’-cGAMP molecules simultaneously (Figures 3A–C, Movie S1). Structural alignment between Acb2-cA3-3’,3’-cGAMP and apo Acb2 also showed little conformational changes with an RMSD of 0.298 Å for Cα atoms (Figure S4D).
Figure 3. Acb2 binds to cyclic trinucleotides and dinucleotides simultaneously.
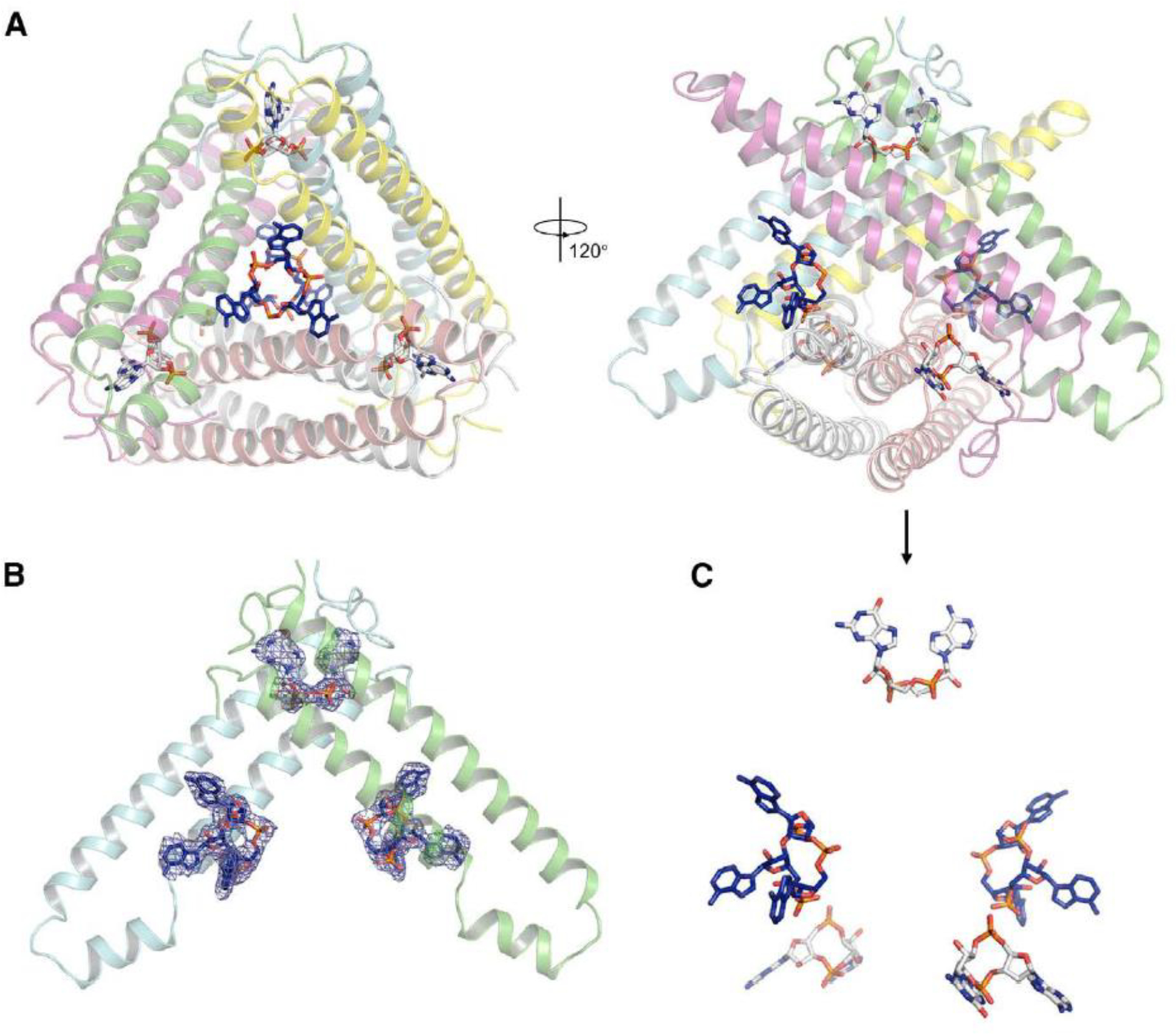
(A) Overall structure of Acb2 complexed with cA3 and 3’,3’-cGAMP. cA3 and 3’,3’-cGAMP are shown as blue and light gray sticks. Two views are shown.
(B) 2Fo-Fc electron density of cA3 and 3’,3’-cGAMP within an Acb2 dimer contoured at 1 σ.
(C) Distribution of the small molecules within the Acb2 hexamer. The nucleotides are shown as they are in the right panel of (A).
Acb2 binds to cA3 with a novel fold
Dali search did not return entries of experimentally determined proteins with the same fold as Acb2 nor did Foldseek searches of computationally predicted proteins, 24 suggesting that both the CDN and CTN-binding folds are novel. Foldseek also did not reveal any similar structures encoded by viruses that infect eukaryotes. Several experimentally determined proteins have been reported to bind CTNs, including the CBASS effector proteins NucC 12 and Cap4 11 that directly bind cA3, as well as the human CDN sensor RECON that directly binds cAAG. 4 Compared to the cA3 binding pocket in Acb2, those in NucC, Cap4, and RECON are significantly different. In NucC, one cA3 molecule is bound in a three-fold symmetric allosteric pocket at the “bottom” of the protein trimer, mainly formed by an extended hairpin loop from each protomer. Additionally, each adenine base is stabilized by hydrogen bonds and π stacking interactions in NucC (Figure S5A, PDB code: 6Q1H). In Cap4, cA3 is bound within its SAVED (SMODS-associated) domain, which is a fusion of two CARF (CRISPR-associated Rossman fold) domains derived from Type III CRISPR-Cas system (Figure S5B, PDB code: 6WAN). RECON adopts a TIM barrel fold with eight parallel β strands surrounded by eight crossover α-helixes and cAAG is bound in a deep crevice at the top of the β barrel (Figure S5C, PDB code: 6M7K). Moreover, the conformation of cA3 within Acb2 is also different from those within NucC, Cap4, and RECON complex structures (Figure S5D). Specifically, cA3 in both NucC and Cap4 are almost in an overall planar conformation, and two adenine bases of cAAG within RECON are nearly in the same plane as the phosphodiester ring and the third guanine base is extended out. However, each base of cA3 forms a ~46.8 degree angle with the phosphate plane in Acb2. Together, the structure of Acb2 complexed with cA3 reveals a novel CTN-binding fold.
Cyclic nucleotide binding spectra are different among Acb2 homologs
To determine the conservation of each binding site across the Acb2 family, PSI-BLAST was used to identify 2,242 total homologs. From these, clustering of the Acb2 homologs revealed 878 unique and non-redundant proteins (see Methods for details; Table S1). Multi-sequence alignments and phylogenetic analyses of these unique Acb2 homologs revealed that both binding sites are predicted to be intact in most homologs (78%). However, some homologs have a mutation in a residue homologous to R67 or T74 that are essential for CTN binding (19%), and very few proteins had mutated Y11 or K26 sites that are essential for CDN binding (3%; Figure 4A and S6). We therefore assessed the binding spectrum of representative Acb2 homologs to determine their cyclic oligonucleotide binding preferences. We chose Acb2 homologs from P. aeruginosa phage JBD67 (44.4% a.a. identity), in which both R67 and T74 residues are conserved, alongside Serratia phage CHI14 (23.5% a.a. identity) and Escherichia phage T4 (24.2% a.a. identity), in which only the R67 (Serratia phage) or T74 (Escherichia phage) residue is conserved. ITC analyses showed that JBD67-Acb2 directly binds to 3’,3’-cGAMP with a KD of ~99 nM and cA3 with a KD of ~3.5 nM (Figure 4B and S7A–B), both of which are comparable to those of PaMx33-Acb2. Native gel assays also suggest that JBD67-Acb2 binds to the same spectrum of cyclic nucleotides as PaMx33-Acb2 (Figure S8A). ITC analyses showed that T4-Acb2 directly binds to 3’,3’-cGAMP with a KD of ~84.4 nM, consistent with previous work, 17 but does not bind to cA3 (Figure 4C and S7C–D). Native gel assays also suggest that T4-Acb2 binds the same spectrum of CDNs as PaMx33-Acb2, but not to the CTNs cA3 and cAAG (Figure S8B). Next, we mutated D61 of T4-Acb2 to Arginine to see whether it can endow T4-Acb2 with the binding activity of cA3 because T4-Acb2 already has T68 residue in the place of T74 of PaMx33-Acb2 that is essential for cA3 binding. However, based on ITC assays, we observed that the D61R mutant of T4-Acb2 was still unable to bind cA3 (Figures 4C and S7E). Interestingly, structural alignment between T4-Acb2 17 and PaMx33-Acb2 complexed with cA3 showed that the helix lining the cA3 binding pocket of PaMx33-Acb2 has a kink at E64, which enlarges the pocket to accommodate the base groups of cA3 (Figure 4D). However, the corresponding helix of T4-Acb2 does not kink here, so the Y37, A57, L60, D61, and T64 residues of T4-Acb2 may undergo steric clashing and prevent cA3 binding (Figure 4D). More importantly, the relative angles among the three helices lining the binding pocket are also different between PaMx33-Acb2 and T4-Acb2, resulting in a smaller binding pocket in T4-Acb2 (Figure 4E). Together, these observations may explain the inability of T4-Acb2 binding to CTNs. Lastly, ITC analyses showed that CHI14-Acb2 directly binds to 3’,3’-cGAMP with a KD of ~62.4 nM, but also does not bind to cA3 (Figure 4F). Of note, native gel assays showed almost no shift of the CHI14-Acb2 protein upon adding any cyclic oligonucleotides, including 3’,3’-cGAMP (Figure S8C), suggesting that native gel assay is not suitable for studying the binding spectrum of CHI14-Acb2. Using ITC, CHI14-Acb2 displayed the same binding spectrum to all cyclic oligonucleotides as T4-Acb2 (Figures 4F–G and S9). The outcomes of binding experiments are summarized, along with a comparison to the enzyme Acb1 (Figure 4H). In summary, Acb2 homologs bind to many CTNs and CDNs used in cGAS-based immunity with certain homologs having a more limited spectrum.
Figure 4. The binding spectra are different among Acb2 homologs.
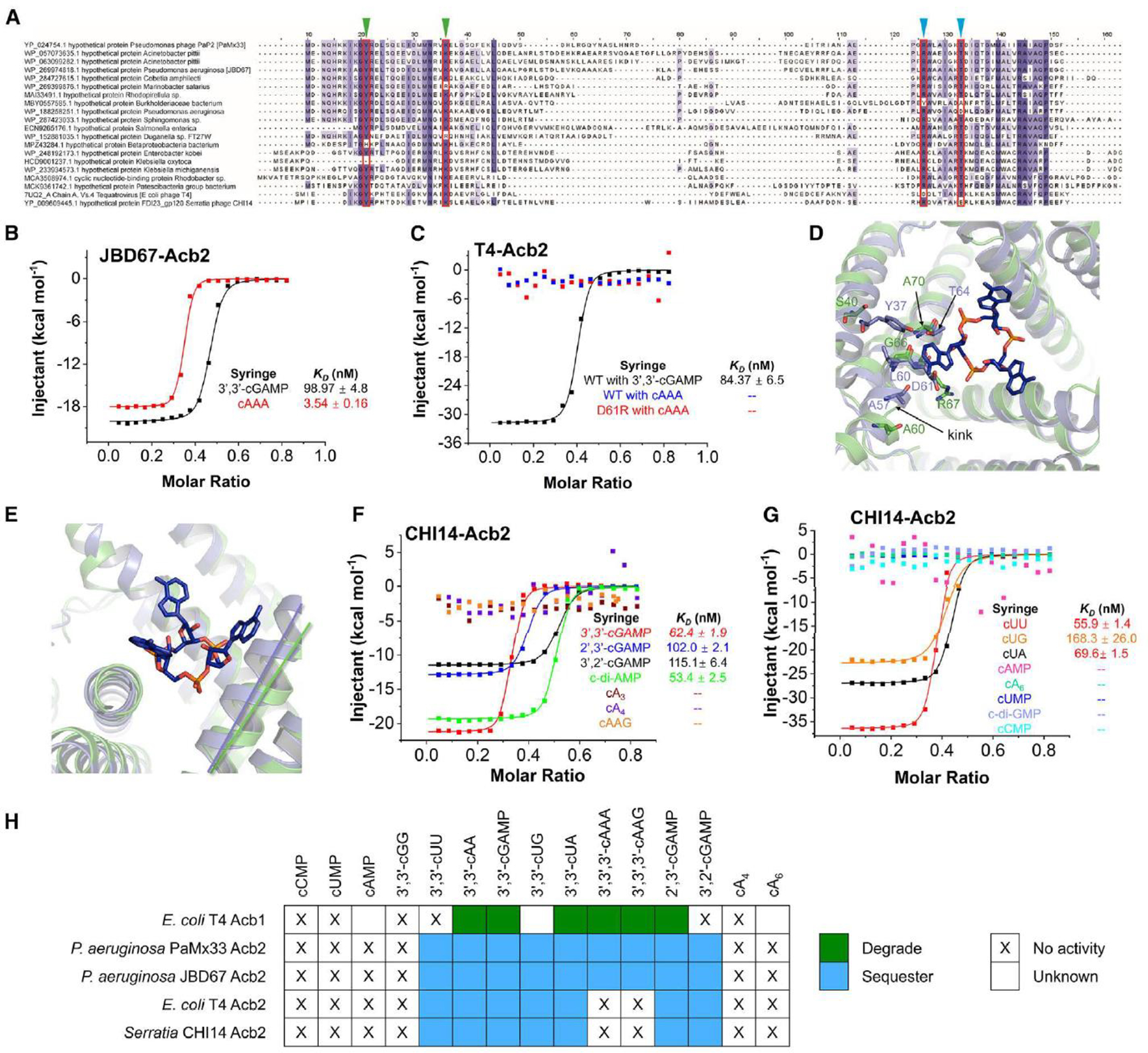
(A) Sequence alignment among Acb2 homologs. Residues that are >80 % conserved, >60 % conserved and >40% conserved are shaded in dark purple, light purple, and light grey, respectively. Residues involved in binding of cyclic CDNs and CTNs are marked with green and blue triangles, respectively.
(B) ITC assays to test binding of cyclic oligonucleotides to JBD67-Acb2. Representative binding curves and binding affinities are shown. The KD values are mean ± s.d. (n=3). Raw data for these curves are shown in Figure S7.
(C) ITC assays to test binding of cyclic oligonucleotides to T4-Acb2. Representative binding curves and binding affinities are shown. The KD values are mean ± s.d. (n=3). Raw data for these curves are shown in Figure S7.
(D) Structural alignment between PaMx33-Acb2 and T4-Acb2 at one monomer. Residues with potential steric clash with cA3 in T4-Acb2 and the corresponding residues in PaMx33-Acb2 are shown in sticks.
(E) The same alignment shown in (D), highlighting the different relative angels formed by the three helices lining the binding pocket of cA3.
(F-G) ITC assays to test binding of cyclic nucleotides to CHI14-Acb2. Representative binding curves and binding affinities are shown. The KD values are mean ± s.d. (n=3). Raw data for these curves are shown in Figure S9.
(H) Summary of the binding results of Acb2 homologs and Acb1. 15
Acb2 antagonizes Type III-C CBASS immunity
Since Acb2 displays high affinity binding to CTNs, we tested whether phage-encoded Acb2 can antagonize Type III-C CBASS immunity that uses a cA3 signaling molecule to activate the endonuclease (NucC) effector protein. NucC is a cyclic nucleotide-activated effector in both CBASS and Type III CRISPR-Cas systems, which non-specifically degrades DNA and limits phage replication. 12, 21 First, we established an in vitro NucC activity assay using purified NucC from the P. aeruginosa strain ATCC 27853 (Pa278) and Acb2 from P. aeruginosa phage PaMx33 (Figure 5A–B). While cA3 activates the DNA cleavage activity of NucC, WT Acb2 significantly decreased NucC activity (Figure 5C). Moreover, following proteolysis of WT Acb2, the released cA3 molecule again activated NucC activity (Figure 5C; last two lanes). The R67A and T74A Acb2 mutant proteins, which lost or exhibited decreased cA3 binding, displayed minimal inhibition of NucC activity. However, the Y11A and K26A Acb2 mutants, whose CDN binding pockets are disrupted, inhibited NucC activity similarly to WT Acb2 (Figure 5C). These results demonstrate that Acb2 antagonizes Type III-C CBASS immunity in vitro through sequestering the cA3 molecule.
Figure 5. Acb2 antagonizes tri- and di-nucleotide based CBASS immunity.
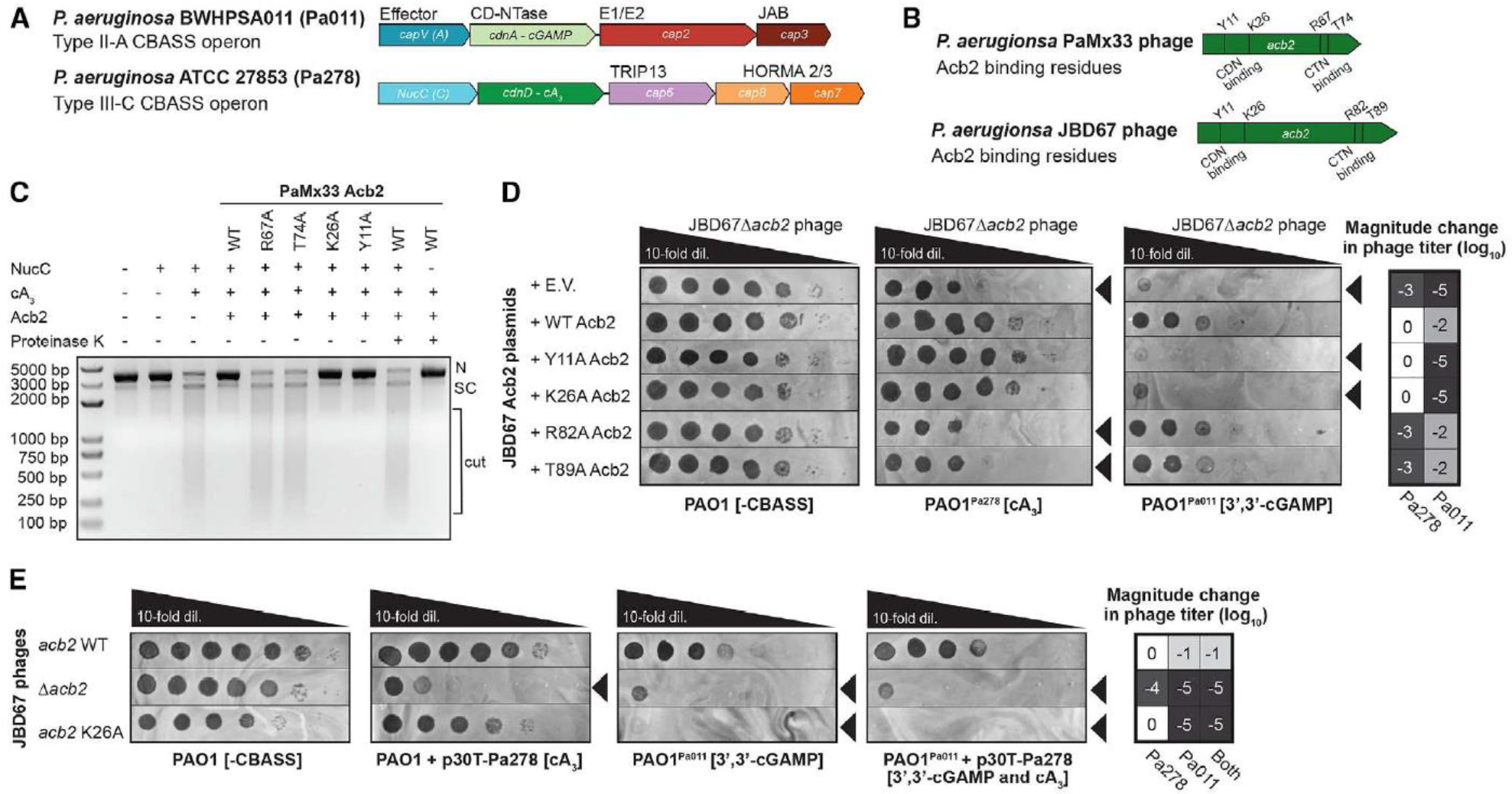
(A) Pseudomonas aeruginosa BWHPSA011 (Pa011) Type II-A CBASS and ATCC 27853 (Pa278) Type III-C CBASS operons.
(B) Pseudomonas aeruginosa PaMx33 and JBD67 phages acb2 gene annotated with residues essential for CDN (3’,3’-cGAMP) binding and CTN (cA3) binding.
(C) Effect of PaMx33 Acb2 or its mutants on cA3-activated NucC effector protein function. After treatment with proteinase K, the released cA3 also showed the ability to activate the nuclease activity of NucC. The concentration of NucC, cA3, Acb2 and proteinase K is 10 nM, 5 nM, 50 nM and 1 μM, respectively. N denotes nicked plasmid, SC denotes closed-circular supercoiled plasmid, and cut denotes fully digested DNA.
(D) Plaque assays with JBD67Δacb2 phage spotted in 10-fold serial dilutions on PAO1 strains harboring an empty vector (E.V.) plasmid or JBD67 Acb2 variants. The PAO1 strains either contain no CBASS operon (-CBASS), a chromosomally integrated Pa011 CBASS operon (PAO1Pa011), or a chromosomally integrated Pa278 CBASS operon (PAO1Pa278). These plaque assays were used to quantify the order of magnitude change in phage titer by comparing the number of spots (with plaques, or clearings if plaques were not visible) on the PAO1Pa011 or PAO1Pa278 CBASS-expressing strains divided by the PAO1 (-CBASS) strain (n=3). Basal expression of the Pa011 CBASS operon and 0.3mM IPTG-inducible expression of the Pa278 CBASS operon is sufficient for phage targeting. Black arrowheads highlight significant CBASS-dependent reductions in phage titer.
(E) Plaque assays with JBD67 phages spotted in 10-fold serial dilutions on PAO1 strains with and without CBASS. Pa278 CBASS was expressed from the pHERD30T (p30T) plasmid, while Pa011 CBASS was expressed from the chromosome. These plaque assays were used to quantify the order of magnitude change in phage titer (n=3). Basal expression (i.e. no arabinose added) of the Pa278 CBASS operon is sufficient for phage targeting. Black arrowheads highlight significant CBASS-dependent reductions in phage titer.
To determine whether Acb2 can inhibit this same cA3-based CBASS system in vivo, we performed phage infection assays with plasmid or phage-encoded Acb2 and the Pa278 Type III-C CBASS operon (Figure 5B). Phages naturally expressing acb2 were unable to replicate on the native P. aeruginosa ATCC 27853 strain, so the Pa278 system was chromosomally integrated into PAO1 that is a phage sensitive strain and naturally lacks CBASS. In the presence Pa278 Type III-C CBASS, the titer of JBD67 phage lacking acb2 (JBD67Δacb2) was reduced by 3 orders of magnitude compared to its replication in the absence of CBASS (Figure 5D–E). Plasmid-based expression of WT Acb2 or Y11A and K26A Acb2 (CDN binding mutants) fully rescued phage titer, while R82A and T89A (CTN binding mutants) did not (Figure 5D). In the presence of the Pa011 Type II-A CBASS (cGAMP producing), the titer of JBD67Δacb2 phage was reduced by 5 orders of magnitude (Figure 5D–E), whereas JBD67 WT phage was reduced by 1–2 orders of magnitude (Figure S10). Plasmid-based expression of WT Acb2 or R82A and T89A Acb2 (CTN binding mutants) partially rescued phage titer, while Y11A and K26A Acb2 (CDN binding mutants) did not (Figure 5D). The partial targeting of JBD67 WT phage (i.e. naturally encoding Acb2) was fully reversed by plasmid-based expression of WT Acb2 or the CTN binding mutants, but not the CDN binding mutants (Figure S10A). We additionally introduced a K26A mutation into acb2 within the genome of JBD67 WT phage. This rendered mutant phage completely sensitive to cells expressing the cGAMP-based system but maintained resistance against the cA3 system (Figures 5E and S10B). These findings demonstrate Acb2 protects phage from Type III-C (cA3-producing) and Type II-A (3’,3’-cGAMP-producing) CBASS using different interfaces, further highlighting the versatility of the Acb2 protein.
Discussion
Following phage infection, CBASS immunity functions via the activation of a cGAS-like enzyme to catalyze the synthesis of a cyclic oligonucleotide signaling molecule. To date, two phage proteins have been discovered to antagonize the CBASS immunity: Acb1 and Acb2. Acb1 uses an inhibitory mechanism common to the eukaryotic cGAS-STING signaling system, 25 that is, enzymatically cleaving and depleting an array of CDNs and CTNs. 15 In contrast, we, alongside another independent group, reported that Acb2 acts as a “sponge” and sequesters 3’,3’-cGAMP 16, 17 as well as a variety of other CBASS CDN signaling molecules. 16 A sponging mechanism was also reported for inhibitors of the anti-phage system Thoeris, including Tad1 26 and Tad2, 27 that sequester gcADPR signaling molecules. Here, we extend the CDN binding spectrum of Acb2 to include 3’,2’-cGAMP, a signaling molecule not cleaved by Acb1, but recently implicated in both CBASS and cGAS like signaling in eukaryotes. 9, 22, 28 The ability of Acb2 to function in human cells against 2’,3’-cGAMP reinforces the flexibility of the “sponging” mechanism (i.e. no need to bind to cGAS, STING, or host proteins), and the remarkable cross-kingdom conservation of this cyclic-oligonucleotide-based immune system. The activity of Acb2 in mammalian cells also implies the possibility that pathogenic bacteria with prophage-encoded Acb1, Acb2, or other undiscovered cGAMP interactors could use them to dampen the human immune response during intracellular infection. Similar cross-kingdom interactions have been previously reported with bacterial c-di-GMP 3 and c-di-AMP produced by Listeria monocytogenes, 29 serving as ligands for human STING. While we could not identify proteins with similar structures to Acb2 in eukaryotic viruses, we speculate that novel cGAMP sponges await discovery in eukaryotes and their viruses. Taken together, our work suggests that discoveries with human health implications remain to be made at the interface of cyclic oligonucleotide inhibitors and cGAS-STING immunity.
Acb2 binds to a wider spectrum of cyclic oligonucleotides compared to the enzyme Acb1, including 3’,3’-cUU, 3’,3’-cUG, and 3’,2’-cGAMP, while also sequestering CTNs with a stronger binding affinity. In some Acb2 homologs, however, we observed that the CTN binding site is mutated and non-functional. Although it is unclear if there is a cost to the CTN binding site that would lead to its loss, in the case of phage T4, its genome encodes both Acb1 and Acb2 and therefore suggests that this phage is broadly evasive of most CBASS types. To our knowledge, Acb2 represents the first phage anti-immune protein, as well as the first protein more broadly, that can bind two types of cyclic oligonucleotides simultaneously with different binding pockets. This highlights Acb2 as a nearly universal inhibitor of CBASS, which could enhance phage therapeutics by enabling the evasion of many common CBASS types and subtypes encoded by human bacterial pathogens. Moreover, with the recent unveiling of counter-defense “sponge” mechanisms (i.e. Tad1 and Tad2 for Thoeris), our paper sets precedent to consider other small, versatile proteins that can soak up multiple immune signaling molecules.
Limitations of the Study
The difference in affinity that Acb2 displays for CDNs and CTNs raises a very interesting hypothesis about whether this correlates with the affinity of the CBASS effectors to their respective activating ligands. The affinity of cA3 and cGAMP for their respective effectors, the concentrations of these nucleotides over the course of phage infection, and the amount of Acb2 synthesized during phage infection are all unknown and limit a complete quantitative understanding of the CBASS-Acb2 interaction. Moreover, the binding mode of cyclic trinucleotides is a novel one which depends on trimerization of Acb2 dimers. Therefore, an interesting question is whether some of the Acb2 homologs may undergo tetramerization or hexamerization of the Acb2 dimers. If so, they might be able to bind to cA4 or cA6, which are commonly used by Type III CRISPR-Cas systems. This can be investigated through biochemical characterization combined with structure-based predictions in future studies. Moreover, since Acb2 has two different pockets for different types of cyclic oligonucleotides, it will be interesting to test whether any other phage “sponge” proteins may also have two or more unique binding pockets for different types of nucleotides.
STAR Methods:
RESOURCE AVAILABILITY
Lead contact
Further Information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yue Feng (fengyue@mail.buct.edu.cn).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The accession numbers for the coordinate and structure factors reported in this paper are PDB: 8IXZ (Acb2–3’,2’-cGAMP), 8J8O (Acb2–2’,3’-cGAMP), 8IY0 (Acb2-cA3), 8IY1 (Acb2-cAAG) and 8IY2 (Acb2–3’,3’-cGAMP-cA3). This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains and phages
The bacterial strains and phages used in this study are listed in the Key Resources Table. The P. aeruginosa strains (BWHPSA011, ATCC 27853, PAO1) and E. coli strains (DH5ɑ) were grown in Lysogeny broth (LB) medium at 37°C both with aeration at 225 r.p.m. Plating was performed on LB solid agar with 10 mM MgSO4 when performing phage infections, and when indicated, gentamicin (50 μg ml−1 for P. aeruginosa and 15 μg ml−1 for E. coli) was used to maintain the pHERD30T plasmid. Gene expression was induced by the addition of L-arabinose (0.3%) unless stated otherwise. The E. coli BL21 (DE3) strain was used for recombinant protein overexpression and grown in Lysogeny broth (LB) medium. The cells were grown at 37°C until OD600nm reached 0.8 and then induced at 18°C for 12 h.
Key Resources Table.
| Resource or Reagent | Source | Identifier |
|---|---|---|
| Bacterial and virus strains | ||
| P. aeruginosa BWHPSA011 (Pa011) WT | Deborah Hung Lab | NCBI: NZ_AXQR01000000.1 |
| P. aeruginosa ATCC 27853 (Pa278) WT | ATCC | NCBI: CP015117.1 |
| P. aeruginosa PAO1 WT | Joe Bondy-Denomy Lab | NCBI: NC_002516.2 |
| PAO1Pa011 (PAO1 attTn7::Pa011 Type II-A CBASS) | Joe Bondy-Denomy Lab 16 | N/A |
| PAO1Pa278 (PAO1 attTn7::Pa278 Type III-C CBASS) | This study | N/A |
| JBD67 | Alan Davidson Lab 30 | NCBI: NC_042135.1 |
| JBD67Δacb2 (Removal of 23005–23232 bp orf24 (acb2)) |
Joe Bondy-Denomy Lab 16 | N/A |
| JBD67 acb2K26A | This study | N/A |
| JBD67 acb2R82A | This study | N/A |
| JBD67 acb2R82A/T89A | This study | N/A |
| JBD67 acb2K26A | This study | N/A |
| Mammalian cell lines | ||
| HEK293T Dual-Null | Invivogen | Cat#: 293d-null |
| Recombinant DNA | ||
| pET28a-His6-SUMO- PaMx33-Acb2 | This study | N/A |
| pET28a-His6-SUMO- PaMx33-Acb2 Y11A | This study | N/A |
| pET28a-His6-SUMO- PaMx33-Acb2 K26A | This study | N/A |
| pET28a-His6-SUMO- PaMx33-Acb2 T74A | This study | N/A |
| pET28a-His6-SUMO- PaMx33-Acb2 R67A | This study | N/A |
| pET28a-His6-SUMO- JBD67-Acb2 | This study | N/A |
| pET28a-His6-CHI14-Acb2 | This study | N/A |
| pET28a-His6-CHI14-Acb2 E76T | This study | N/A |
| pET28a-His6-CHI14- Acb2 E76T/R67P | This study | N/A |
| pET28a-His6-SUMO-T4-Acb2 | This study | N/A |
| pET28a-His6-SUMO-T4-Acb2 D61R | This study | N/A |
| pUC18-mini-Tn7-LAC (pTn7) | 31 | N/A |
| pTn7-Pa278 Type III-C CBASS (6283357–6286926) | This study | N/A |
| pNTS3 | 32 | N/A |
| pMQ30 | 33 | N/A |
| pMQ30-HDR-JBD67 Acb2 WT | This study | N/A |
| pMQ30-HDR-JBD67 Acb2 K26A | This study | N/A |
| pMQ30-HDR-JBD67 Acb2 R82A | This study | N/A |
| pMQ30-HDR-JBD67 Acb2 T89A | This study | N/A |
| pHERD30T (p30T) | 34 | N/A |
| p30T-JBD67 WT Acb2 | This study | N/A |
| p30T-JBD67 Y11A Acb2 | This study | N/A |
| p30T-JBD67 K26A Acb2 | This study | N/A |
| p30T-JBD67 R82A Acb2 | This study | N/A |
| p30T-JBD67 T89A Acb2 | This study | N/A |
| p30T-Pa27853 Type III-C CBASS (6283357–6286926) | This study | N/A |
| pcDNA3 | Lingyin Li Lab | N/A |
| pcDNA3-hSTING-232R | Lingyin Li Lab | N/A |
| pcDNA3-Acb2 WT | This paper | N/A |
| pcDNA3-Acb2 Y11A | This paper | N/A |
| pcDNA3-Acb2 K26A | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Phusion High-Fidelity DNA polymerase | NEB | Cat #M0530S |
| dNTPs | NEB | Cat #N0447S |
| Phusion GC Buffer | NEB | Cat #B0519 |
| Gibson Assembly HiFi DNA Master Mix | NEB | Cat #E2621 |
| SacI | NEB | Cat #R3156S |
| PstI | NEB | Cat #R3140S |
| HEPES sodium salt | Sigma-Aldrich | CAS: 7365–45-9 Cat #: V900477–500G |
| Tris base | Sigma-Aldrich | CAS: 77–86-1 Cat #: RDD008–2.5KG |
| Sodium dihydrogen phosphate dihydrate | Sigma-Aldrich | CAS: 13472–35-0 Cat #: 1063420250 |
| Disodium hydrogen phosphate dodecahydrate | Sigma-Aldrich | CAS: 10039–32-4 Cat #: 1065790500 |
| Bis-Tris propane | Sigma-Aldrich | CAS: 64431–96-5 Cat #: B6755–25G |
| Sodium bromide | Sigma-Aldrich | CAS: 7647–15-6 Cat #: 310506–100G |
| PEG 3350 | Biorigin | CAS: 25322–68-3 Cat #: BN33640 |
| Glycerol | Sigma-Aldrich | CAS: 56–81-5 Ca t#: V900122–500ML |
| Imidazole | CAS: 288–32-4 Cat #: V900153–500G |
|
| Sigma-Aldrich | Vazyme | Cat #: P515–03 |
| 2× Rapid Taq Master Mix | Vazyme | Cat #: P222-AA |
| KOD-Plus-Neo | TOYOBO | Cat #: KOD-401 |
| DpnI | NEB | Cat #: R0176S |
| Proteinase K | NEB | Cat #: P8107S |
| High Affinity Ni-NTA Resin | GenScript | Cat #: L00250–100 |
| α-tubulin (DM1A), Mouse mAb | Sigma-Aldrich | Cat# T6199; RRID: AB_477583 |
| STING (D2P2F) Rabbit mAb | Cell Signaling Technologies | Cat# 13647; RRID: AB_2732796 |
| Acb2 custom polyclonal antibody | GenScript | N/A |
| Goat Anti-Mouse IgG Antibody, IRDye® 680RD Conjugated | Li-COR Biosciences | Cat# 926–68070; RRID: AB_10956588 |
| IRDye 800CW Goat anti-Rabbit IgG (H+L) | Li-COR Biosciences | Cat# 925–32211; RRID: AB_2651167 |
| PaMx33-Acb2 WT recombinant protein | This study | N/A |
| PaMx33-Acb2Y11A recombinant protein | This study | N/A |
| PaMx33-Acb2R67A recombinant protein | This study | N/A |
| PaMx33-Acb2T74A recombinant protein | This study | N/A |
| PaMx33-Acb2K26A recombinant protein | This study | N/A |
| CHI14-Acb2 WT recombinant protein | This study | N/A |
| JBD67-Acb2 WT recombinant protein | This study | N/A |
| T4-Acb2 WT recombinant protein | This study | N/A |
| T4-Acb2 D61R recombinant protein | This study | N/A |
| Critical Commerical Assays | ||
| Plasmid Miniprep Kit | Vazyme | Cat #: DC201–01 |
| Gel DNA Extraction Mini Kit | Vazyme | Cat #: DC301–01 |
| QUANTI-Luc | Invivogen | Cat #: rep-qlc2 |
| Deposited Data | ||
| Structure of Acb2–2’,3’-cGAMP | This study | PDB: 8J8O |
| Structure of Acb2–3’,2’-cGAMP | This study | PDB: 8IXZ |
| Structure of Acb2-cA3 | This study | PDB: 8IY0 |
| Structure of Acb2-cAAG | This study | PDB: 8IY1 |
| Structure of Acb2–3’,3’-cGAMP-cA3 | This study | PDB: 8IY2 |
| Oligonucleotides | ||
| 3’,3’-cGAMP | Sigma-Aldrich | CAS: 849214–04-6 Cat #: SML1232-.5UMO |
| 2’,3’-cGAMP | Sigma-Aldrich | CAS:1441190–66-4 Cat #SML1229-.5UMO |
| c-di-AMP | Sigma-Aldrich | CAS: 54447–84-6 Cat #SML1231–1UMO |
| c-di-GMP | Sigma-Aldrich | CAS: 61093–23-0 Cat #SML1228–1UMO |
| 3’,3’-c-UMP-GMP | Biolog Life Science Institute | CAS: 232933–52-7 Cat #C371 |
| 3’,3’-c-di-UMP | Biolog Life Science Institute | CAS: 73120–97-5 Cat #C256 |
| 3’,3’-c-UMP-AMP | Biolog Life Science Institute | CAS: 83799–66-0 Cat #C357 |
| 3’,2’-cGAMP | Biolog Life Science Institute | CAS: 1615704–64-7 Cat #C238 |
| 3’,3’,3’-cAAG | Biolog Life Science Institute | CAS: 2365165–54-2 Cat #C361 |
| 3’,3’,3’-cAAA (cA3) | Biolog Life Science Institute | CAS: 54447–85-7 Cat #C362 |
| cA4 | Biolog Life Science Institute | CAS: 118004–81-2 Cat #C335 |
| cA6 | Biolog Life Science Institute | CAS: 232933–63-0 Cat #C332 |
| cAMP | MedChemExprss | CAS: 60–92-4 Cat #HY-B1511S |
| cUMP | Sigma-Aldrich | CAS: 56632–58-7 Cat #SML3286–50UMOL |
| Y11A_F 5’–gcacaagaaaatcaagggcgccagagatctgtctcagga–3’ |
This study | N/A |
| Y11A_R 5’–ctcctgagacagatctctggcgcccttgattttcttgtgc–3’ |
This study | N/A |
| K26A_F 5’–gacatgatgaacagagtggcggaactgggcagccagtt–3’ |
This study | N/A |
| K26A_R 5’–aactggctgcccagttccgccactctgttcatcatgtc–3’ |
This study | N/A |
| Software and algorithms | ||
| National Center for Biotechnology Information (NCBI) database | 35 | https://blast.ncbi.nlm.nih.gov/ |
| HKL2000 | 36 | http://www.hkl-xray.com/ |
| PHENIX | 37 | http://www.phenix-online.org |
| COOT | 38 | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| PyMOL | The PyMOL Molecular Graphics System, Version 2.5.2., Schrodinger, LLC | https://pymol.org/2/ |
| DALI | 39 | http://ekhidna2.biocenter.helsinki.fi/dali/ |
| Foldseek | 24 | https://search.foldseek.com/search |
| MAFFT | 40 | N/A |
| MMSeq2 | 41 | N/A |
| FastTree | 42 | N/A |
| iTOL | 43 | https://itol.embl.de/ |
| OriginPro 8 | OriginPro Software | N/A |
| GraphPad Prism 9 | GraphPad Software | https://www.graphpad.com/ |
| Other | ||
| Amicon Ultra-0.5 centrifugal filter unit | Merck | Cat #: UFC500396 |
| Amicon concentrators (3 K) | Millipore | Cat #: UFC800308 |
| Amicon concentrators (10 K) | Millipore | Cat #: UFC901096 |
| Amicon concentrators (30 K) | Millipore | Cat #: UFC903024 |
| HisTrap FF (5 mL) | GE Healthcare | Cat #: 17–5255-01 |
| HiTrap Heparin HP (5 mL) | GE Healthcare | Cat #: 17–0407-03 |
| HiTrap Q Sepharose FF (5 mL) | GE Healthcare | Cat #: 17–5156-01 |
| Superdex 200 increase 10/300 GL | GE Healthcare | Cat #: 17517501 |
METHOD DETAILS
Protein expression and purification
The PaMx33-Acb2, JBD67-Acb2, T4-Acb2, and CHI14-Acb2 genes were synthesized by GenScript. The full-length Acb2 gene was amplified by PCR and cloned into a modified pET28a vector in which the expressed Acb2 protein contains a His6-SUMO tag or His6 tag. The Acb2 mutants were generated by two-step PCR and were subcloned, overexpressed and purified in the same way as wild-type protein. The proteins were expressed in E. coli strain BL21 (DE3) and induced by 0.2 mM isopropyl-β-D-thiogalactopyranoside (IPTG) when the cell density reached an OD600nm of 0.8. After growth at 18°C for 12 h, the cells were harvested, re-suspended in lysis buffer (50 mM Tris–HCl pH 8.0, 300 mM NaCl, 10 mM imidazole and 1 mM PMSF) and lysed by sonication. The cell lysate was centrifuged at 20,000 g for 50 min at 4°C to remove cell debris. The supernatant was applied onto a self-packaged Ni-affinity column (2 mL Ni-NTA, Genscript) and contaminant proteins were removed with wash buffer (50 mM Tris pH 8.0, 300 mM NaCl, 30 mM imidazole). The fusion protein of Acb2 with His6-SUMO tag was then digested with Ulp1 at 18°C for 2 h, and then the Acb2 protein was eluted with wash buffer. The eluant of Acb2 was concentrated and further purified using a Superdex-200 increase 10/300 GL (GE Healthcare) column equilibrated with a buffer containing 10 mM Tris-HCl pH 8.0, 500 mM NaCl and 5 mM DTT. The purified protein was analyzed by SDS-PAGE. The fractions containing the target protein were pooled and concentrated.
The His6-Acb2 proteins bound to Ni-NTA beads were washed with wash buffer (50 mM Tris pH 8.0, 300 mM NaCl, 30 mM imidazole) and then eluted with the 50 mM Tris pH 8.0, 300 mM NaCl, 300 mM imidazole. The eluant of His6-Acb2 was concentrated, then further purified and analyzed as described above.
Crystallization, data collection and structural determination
The crystals of Acb2 were grown with reservoir solution containing 0.2 M Sodium bromide, 0.1 M Bis-Tris propane pH 6.5, 10% Ethylene glycol and 20% v/v PEG 3350 at 18°C. Prior to crystallization, cA3, cAAG, cA3+3’,3’-cGAMP or 3’,2’-cGAMP were mixed with the protein at a molar ratio of 2:1, respectively, and the concentration of Acb2 was 24 mg/mL. The crystals appeared overnight and grew to full size in about two to three days. The crystals were cryoprotected in the reservoir solution containing 20% glycerol before its transferring to liquid nitrogen.
All the data were collected at SSRF beamlines BL02U1 and BL19U1, integrated and scaled using the HKL2000 package. 36 The initial model of Acb2 was used from PDB: 8H2X. The structures of Acb2 and its complex with cyclic oligonucleotides were solved through molecular replacement and refined manually using COOT. 38 The structure was further refined with PHENIX 37 using non-crystallographic symmetry and stereochemistry information as restraints. The final structure was obtained through several rounds of refinement. Data collection and structure refinement statistics are summarized in Table 1.
Isothermal titration calorimetry binding assay
The dissociation constants of binding reactions of Acb2 or Acb2 mutants with the cA3/cAAG/3’,2’-cGAMP/3’,3’-cGAMP/2’,3’-cGAMP/c-di-AMP/c-di-GMP/3’,3’-cUU/3’,3’-cUA/3’,3’-cUG/cUMP/cCMP/cAMP/cA6/cA4 were determined by isothermal titration calorimetry (ITC) using a MicroCal ITC200 calorimeter. Both proteins and cyclic-oligonucleotides were desalted into the working buffer containing 20 mM HEPES pH 7.5 and 200 mM NaCl. The titration was carried out with 19 successive injections of 2 μL cA3/cAAG/cA6/cA4 at the 0.04 mM concentration, spaced 120 s apart, into the sample cell containing the Acb2 or Acb2 mutants with a concentration of 0.01 mM by 700 rpm at 25°C. Correspondingly, the 3’,2’-cGAMP/3’,3’-cGAMP/2’,3’-cGAMP/c-di-AMP/c-di-GMP/3’,3’-cUU/3’,3’-cUA/3’,3’-cUG/cUMP/cAMP at the 0.4 mM concentration was titrated into 0.1 mM Acb2 or Acb2 mutants at the same experimental conditions. The Origin software was used for baseline correction, integration, and curve fitting to a single site binding model.
Native-PAGE assay
Acb2 or Acb2 mutants was pre-incubated with CDNs for 10 min at 18°C, where Acb2 or Acb2 mutants was 14.3 μM and the concentrations of CDNs ranged from 1.8 to 7.2 μM (1.8, 3.6, 7.2 μM). Products of the reaction were analyzed using 5% native polyacrylamide gels and visualized by Coomassie blue staining.
High-performance liquid chromatography (HPLC)
40 μM Acb2 or Acb2 mutants was pre-incubated with 4 μM cA3 for 10 min at 18°C. Proteinase K was subsequently added to the reaction system at a final concentration of 0.5 μM and the reaction was performed at 58°C for 1 h. Reaction products were transferred to Amicon Ultra-15 Centrifugal Filter Unit 3 kDa and centrifuged at 4°C, 4,000 g. The products obtained by filtration were further filtered with a 0.22 μm filter and subsequently used for HPLC experiments. The HPLC analysis was performed on an Agilent 1200 system with a ZORBAX Bonus-RP column (4.6 × 150 mm). A mixture of acetonitrile (2%) and 0.1% trifluoroacetic acid solution in water (98%) were used as mobile phase with 0.8 mL/min. The compounds were detected at 254 nm.
In vitro NucC activity assay
For nuclease activity assay, a pUC19 plasmid was used as substrate. Pa-NucC (10 nM) and cA3 molecules (5 nM) were mixed with 0.5 μg DNA in a buffer containing 25 mM Tris-HCl pH 8.0, 10 mM NaCl, 10 mM MgCl2, and 2 mM DTT (20 μL reaction volume), incubated 10 min at 37°C, then separated on a 1% agarose gel. Gels were stained with Goldview and imaged by UV illumination.
To determine the function of Acb2, 50 nM Acb2 or its mutants were pre-incubated with the system at 18°C for 15 min, and the subsequent reaction and detection method was as described above. To examine whether the released molecule from Acb2 is able to activate NucC, 5 nM cA3 was incubated with 50 nM Acb2 for 15 min at 18°C. Proteinase K was subsequently added to the reaction system at a final concentration of 1 μM and the reaction was performed at 58°C for 1 h, then the proteinase K-treated samples were heated with 100°C for 10 min to extinguish proteinase K and the subsequent detection method was as described above.
Episomal gene expression
The shuttle vector that replicates in P. aeruginosa and E. coli, pHERD30T 34 was used for cloning and episomal expression of P. aeruginosa ATCC 27853 Type III-C CBASS operons into the PAO1 WT strain. This vector has an arabinose-inducible promoter and a selectable gentamicin marker. Vector was digested with SacI and PstI restriction enzymes and then purified. Inserts were amplified by PCR using bacterial overnight culture or phage lysate as the DNA template, and joined into the pHERD30T vector at the SacI-PstI restriction enzyme cut sites by Hi-Fi DNA Gibson Assembly (NEB) following the manufacturer’s protocol. The resulting plasmids were transformed into E. coli DH5ɑ. All plasmid constructs were verified by sequencing using primers that annealed to sites outside the multiple cloning site. P. aeruginosa cells were electroporated with the pHERD30T constructs and selected on gentamicin.
Chromosomal CBASS integration
For chromosomal insertion of the Pa011 CBASS operon, the integrating vector pUC18-mini-Tn7T-LAC 31 and the transposase expressing helper plasmid pTNS3 32 were used to insert the Pa278 Type III-C CBASS operon at the Tn7 locus in P. aeruginosa PAO1 strain (PaPa011), or an pUC18-mini-Tn7T-LAC empty vector (E.V.) control strain (PAO1). The vector was linearized using around-the-world PCR, treated with DpnI, and then purified. Two overlapping inserts encompassing the CBASS operon were amplified by PCR using Pa011 overnight culture as the DNA template, and joined into the pUC18-mini-Tn7T-LAC vector a the SacI-PstI restriction enzyme cut sites by Hi-Fi DNA Gibson Assembly (NEB) following the manufacturer’s protocol. The resulting plasmids were used to transform E. coli DH5ɑ. All plasmid constructs were verified by sequencing using primers that annealed to sites outside the multiple cloning site. P. aeruginosa PAO1 cells were electroporated with pUC18-mini-Tn7T-LAC and pTNS3 and selected for on gentamicin. Potential integrants were screened by colony PCR with primers PTn7R and PglmS-down, and then verified by sequencing using primers that anneal to sites outside the attTn7 site. Electrocompetent cell preparations, transformations, integrations, selections, plasmid curing, and FLP-recombinase-mediated marker excision with pFLP were performed as described previously. 31
Phage growth
All phages were grown at 37°C with solid LB agar plates containing 20 ml of bottom agar containing 10 mM MgSO4 and any necessary inducers or antibiotics. Phages were grown on the permissible host P. aeruginosa PAO1, which naturally lacks CBASS. 150 μl of overnight cultures of PAO1 were infected with 10 μl of low titer phage lysate (>104–7 pfu/ml) and then mixed with 3 ml of 0.7% top agar 10 mM MgSO4 for plating on the LB solid agar. After incubating at 37°C overnight, individual phage plaques were picked from top agar and resuspended in 200 μl SM phage buffer. For high titer lysates, the purified phage was further amplified on LB solid agar plates with PAO1 WT. After incubating 37°C overnight, 3 ml SM phage buffer was added until the solid agar lawn was completely covered and then incubated for 5–10 minutes at room temperature. The whole cell lysate was collected and a 1% volume of chloroform was added, and then left to shake gently on an orbital shaker at room temperature for 15 min followed by centrifugation at maximum g for 3 min to remove cell debris. The supernatant phage lysate was stored at 4°C for downstream assays.
Plaque assays
Plaque assays were conducted at 37°C with solid LB agar plates. 150 μL of overnight bacterial culture was mixed with top agar and plated. Phage lysates were diluted 10-fold then 2 μL spots were applied to the top agar after it had been poured and solidified.
Homologous recombination-mediated mutation of phage gene
Construction of template plasmids for homologous recombination consisted of homology arms >500bp up- and downstream of the mutation of interest encoded in JBD67 acb2. The homology arms were amplified by PCR using JBD67 WT phage genomic DNA as the template. Template primers were designed to symmetrically flank the JBD67 acb2 CDN or CTN binding sites. PCR products were purified and assembled as a recombineering substrate and then inserted into the SacI-PstI site of the pMQ30 vector. The resulting plasmids were electroporated into P. aeruginosa PAO1-JBD67 WT lysogen strain as previously described. 16 PAO1 lysogen strains carrying the recombination plasmid were grown in LB media supplemented with gentamicin. 150 μl of overnight cultures were infected with 10 μl of high titer phage lysate (>109 pfu/ml; JBD67 WT) and then plated on LB solid agar. After incubating at 37°C overnight, SM phage buffer was added to the entire lawn and whole cell lysate collected. The resulting phage lysate containing both WT and recombinant phages and were screen by colony PCR with the appropriate pairs of primers amplifying the region outside of the homology arms and subject to Sanger Sequencing. Once confirmed, the PAO1-JBD67 lysogens were grown in liquid culture, and the presence of spontaneously produced phage in the supernatant that could plaque on the PAO1 wildtype strain confirmed lysogeny.
Interferon reporter assay in human cell line
The PaMx33-Acb2 gene was codon-optimized for human expression and synthesized by GenScript with overhangs that enabled insertion into the XhoI–BamHI sites of pcDNA3 via Gibson assembly. The wild-type plasmid was modified by site-directed mutagenesis using the QuikChange protocol with the indicated primers, and Dpn1-digested to obtain all point mutants. All recombinant plasmids were transformed into XL1-Blue competent cells (Agilent) and sequenced for verification.
293T-Dual Null cells were cultured in DMEM (Gibco) supplemented with 10% FBS (Atlanta Biologics) (v/v) and 100 U/mL penicillin-streptomycin (Gibco) and maintained in 37°C incubators with 5% CO2. Four days prior to measurement, 293T-Dual cells were passaged and plated in 12-well tissue cultured-treated plates at 100000 cells/well. After 20 hours they were transfected with 100 ng of pcDNA3-hSTING and 100 ng of pcDNA3 empty vector or containing Acb2 using Fugene6 transfection reagent (Promega) according to its associated protocol. After 20 hours, the growth media was replaced with fresh growth media containing 50 μM 2’,3’-cGAMP or regular growth media as negative controls. The cells were further incubated for 18 h, and the media was harvested to measure luciferase activity using the QuantiLuc system (Invivogen). The cells were directly lysed in 1 × LSB and western blot was performed on cell lysates to verify expression of STING and Acb2.
Western blot
A rabbit Acb2 polyclonal antibody was generated by a commercial vender (GenScript) using a synthetic peptide from Acb2 (CHNRDEITRIANAEP). The polyclonal Acb2 antibody was further purified by antigen affinity (GenScript).
After harvesting conditioned media, cells were directly lysed on the plate using 1× LSB. Lysates were separated on a SurePage Bis-Tris polyacrylamide gel (GenScript) and transferred to a nitrocellulose membrane using the semi-dry iBlot2 system (Invitrogen). The membrane was blocked for 1 h at room temperature (Intercept blocking buffer, Li-COR Biosciences), and incubated with primary antibodies (rabbit anti-STING (Cell Signaling Technologies), mouse anti-alpha-tubulin (Sigma-Aldrich), rabbit anti-Acb2 (GenScript)) overnight at 4°C. Following three washes in 1×TBS-0.1% tween, secondary antibody (Anti-rabbit or anti-mouse (Li-Cor Biosciences)) was added for 1 hour at room temperature, followed by three additional washes in TBS-T. Blots were imaged in IR using a Li-Cor Odyssey Blot Imager.
Bioinformatics and phylogenetic tree analysis
Acb2 homologs were identified using PaMx33 Acb2 (NCBI: ANA48877.1) as a query protein to seed a position-specific iterative blast (PSI-BLAST) search of the NCBI non-redundant protein database. Three rounds of PSI-BLAST searches were performed with a max target sequence of 5,000 and E value cut-off of 0.005 for inclusion in the next search round, BLOSUM62 scoring matrix, gap costs settings existence 11 and extension 1, and using conditional compositional score matrix adjustment. Hits from the third search round of PSI-BLAST with >70% coverage and an E value of < 0.0005 were aligned using MAFFT (FFT-NS-I iterative refinement method). 40 Manual analysis of the MAFFT protein alignment was performed to ensure the presence of at least one of the cyclic oligonucleotide binding site motifs (CDN site <YX(14)K and CTN site RX(6)T>) and to remove large gaps, resulting in 2,242 total sequences. MMSeq2 41 was used to remove protein redundancies (minimum sequence identity=0.95, minimum alignment coverage=1), which resulted in 878 representative or unique Acb2 homolog sequences. The final aligned 878 sequences were used to construct a phylogenetic tree using FastTree 42 and then visualized and annotated in iTOL. 43 The full list Acb2 homologs, as well as the representative or unique Acb2 homologs included in the phylogenetic tree and MSA figures, are included in Table S1.
Supplementary Material
Acknowledgments:
We thank the staff at beamlines BL02U1 and BL19U1 of the Shanghai Synchrotron Radiation Facility for their assistance with data collection. We thank the Tsinghua University Branch of China National Center for Protein Sciences Beijing and Dr. Shilong Fan for providing facility support for X-ray diffraction of the crystal samples. We thank Drs. Yuanyuan Chen, Zhenwei Yang, Bingxue Zhou at the Institute of Biophysics, Chinese Academy of Sciences for technical help with ITC experiments. Y. F. is supported by National key research and development program of China (2022YFC3401500 and 2022YFC2104800), the National Natural Science Foundation of China (32171274), Beijing Nova Program (20220484160) and the Fundamental Research Funds for the Central Universities (QNTD2023-01). E.H. is supported by the National Science Foundation Graduate Research Fellowship Program [Grant No. 2038436]. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. J.B.-D. is supported by the National Institutes of Health [R21AI168811, R01GM127489], the Vallee Foundation, and the Searle Scholarship.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Declaration of interests: J.B.-D. is a scientific advisory board member of SNIPR Biome and Excision Biotherapeutics, a consultant to LeapFrog Bio, and a scientific advisory board member and co-founder of Acrigen Biosciences. The Bondy-Denomy lab received research support from Felix Biotechnology. UCSF has filed a patent application related to this work with J.B.-D. and E.H. listed as inventors.
References
- 1.Li XD, Wu J, Gao D, Wang H, Sun L, and Chen ZJ (2013). Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341, 1390–1394. 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishikawa H, and Barber GN (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 456, 274–274. 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, and Vance RE (2011). STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–U111. 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whiteley AT, Eaglesham JB, de Oliveira Mann CC, Morehouse BR, Lowey B, Nieminen EA, Danilchanka O, King DS, Lee ASY, Mekalanos JJ, et al. (2019). Bacterial cGAS-like enzymes synthesize diverse nucleotide signals. Nature 567, 194–199. 10.1038/s41586-019-0953-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies BW, Bogard RW, Young TS, and Mekalanos JJ (2012). Coordinated regulation of accessory genetic elements produces cyclic di-nucleotides for V. cholerae virulence. Cell 149, 358–370. 10.1016/j.cell.2012.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burroughs AM, Zhang D, Schäffer DE, Iyer LM, and Aravind L (2015). Comparative genomic analyses reveal a vast, novel network of nucleotide-centric systems in biological conflicts, immunity and signaling. Nucleic acids research 43, 10633–10654. 10.1093/nar/gkv1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Millman A, Melamed S, Amitai G, and Sorek R (2020). Diversity and classification of cyclic-oligonucleotide-based anti-phage signalling systems. Nature microbiology 5, 1608–1615. 10.1038/s41564-020-0777-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duncan-Lowey B, McNamara-Bordewick NK, Tal N, Sorek R, and Kranzusch PJ (2021). Effector-mediated membrane disruption controls cell death in CBASS antiphage defense. Mol Cell 81, 5039–5051.e5035. 10.1016/j.molcel.2021.10.020. [DOI] [PubMed] [Google Scholar]
- 9.Fatma S, Chakravarti A, Zeng X, and Huang RH (2021). Molecular mechanisms of the CdnG-Cap5 antiphage defense system employing 3’,2’-cGAMP as the second messenger. Nat Commun 12, 6381. 10.1038/s41467-021-26738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen D, Melamed S, Millman A, Shulman G, Oppenheimer-Shaanan Y, Kacen A, Doron S, Amitai G, and Sorek R (2019). Cyclic GMP-AMP signalling protects bacteria against viral infection. Nature 574, 691–695. 10.1038/s41586-019-1605-5. [DOI] [PubMed] [Google Scholar]
- 11.Lowey B, Whiteley AT, Keszei AFA, Morehouse BR, Mathews IT, Antine SP, Cabrera VJ, Kashin D, Niemann P, Jain M, et al. (2020). CBASS Immunity Uses CARF-Related Effectors to Sense 3’–5’- and 2’–5’-Linked Cyclic Oligonucleotide Signals and Protect Bacteria from Phage Infection. Cell 182, 38–49.e17. 10.1016/j.cell.2020.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lau RK, Ye Q, Birkholz EA, Berg KR, Patel L, Mathews IT, Watrous JD, Ego K, Whiteley AT, Lowey B, et al. (2020). Structure and Mechanism of a Cyclic Trinucleotide-Activated Bacterial Endonuclease Mediating Bacteriophage Immunity. Mol Cell 77, 723–733 e726. 10.1016/j.molcel.2019.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ko TP, Wang YC, Yang CS, Hou MH, Chen CJ, Chiu YF, and Chen Y (2022). Crystal structure and functional implication of bacterial STING. Nat Commun 13, 26. 10.1038/s41467-021-26583-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morehouse BR, Yip MCJ, Keszei AFA, McNamara-Bordewick NK, Shao S, and Kranzusch PJ (2022). Cryo-EM structure of an active bacterial TIR-STING filament complex. Nature 608, 803–807. 10.1038/s41586-022-04999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hobbs SJ, Wein T, Lu A, Morehouse BR, Schnabel J, Leavitt A, Yirmiya E, Sorek R, and Kranzusch PJ (2022). Phage anti-CBASS and anti-Pycsar nucleases subvert bacterial immunity. Nature 605, 522–526. 10.1038/s41586-022-04716-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huiting E, Cao X, Ren J, Athukoralage JS, Luo Z, Silas S, An N, Carion H, Zhou Y, Fraser JS, et al. (2023). Bacteriophages inhibit and evade cGAS-like immune function in bacteria. Cell 186, 864–876 e821. 10.1016/j.cell.2022.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jenson JM, Li T, Du F, Ea CK, and Chen ZJ (2023). Ubiquitin-like Conjugation by Bacterial cGAS Enhances Anti-phage Defence. Nature 616, 326–331. 10.1038/s41586-023-05862-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Molina R, Sofos N, and Montoya G (2020). Structural basis of CRISPR-Cas Type III prokaryotic defence systems. Curr Opin Struct Biol 65, 119–129. 10.1016/j.sbi.2020.06.010. [DOI] [PubMed] [Google Scholar]
- 19.van Beljouw SPB, Sanders J, Rodriguez-Molina A, and Brouns SJJ (2022). RNA-targeting CRISPR-Cas systems. Nat Rev Microbiol 21, 21–34. 10.1038/s41579-022-00793-y. [DOI] [PubMed] [Google Scholar]
- 20.Tal N, Morehouse BR, Millman A, Stokar-Avihail A, Avraham C, Fedorenko T, Yirmiya E, Herbst E, Brandis A, Mehlman T, et al. (2021). Cyclic CMP and cyclic UMP mediate bacterial immunity against phages. Cell 184, 5728–5739 e5716. 10.1016/j.cell.2021.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayo-Muñoz D, Smith LM, Garcia-Doval C, Malone LM, Harding KR, Jackson SA, Hampton HG, Fagerlund RD, Gumy LF, and Fineran PC (2022). Type III CRISPR-Cas provides resistance against nucleus-forming jumbo phages via abortive infection. Mol Cell 82, 4471–4486.e4479. 10.1016/j.molcel.2022.10.028. [DOI] [PubMed] [Google Scholar]
- 22.Slavik KM, Morehouse BR, Ragucci AE, Zhou W, Ai X, Chen Y, Li L, Wei Z, Bähre H, König M, et al. (2021). cGAS-like receptors sense RNA and control 3’2’-cGAMP signalling in Drosophila. Nature 597, 109–113. 10.1038/s41586-021-03743-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Athukoralage JS, and White MF (2022). Cyclic Nucleotide Signaling in Phage Defense and Counter-Defense. Annual review of virology 9, 451–468. 10.1146/annurev-virology-100120-010228. [DOI] [PubMed] [Google Scholar]
- 24.van Kempen M, Kim SS, Tumescheit C, Mirdita M, Lee J, Gilchrist CLM, Söding J, and Steinegger M (2023). Fast and accurate protein structure search with Foldseek. Nature biotechnology. 10.1038/s41587-023-01773-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eaglesham JB, and Kranzusch PJ (2020). Conserved strategies for pathogen evasion of cGAS-STING immunity. Current opinion in immunology 66, 27–34. 10.1016/j.coi.2020.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leavitt A, Yirmiya E, Amitai G, Lu A, Garb J, Herbst E, Morehouse BR, Hobbs SJ, Antine SP, Sun Z-YJ, et al. (2022). Viruses inhibit TIR gcADPR signaling to overcome bacterial defense. Nature 611, 326–331. 10.1016/j.coi.2020.04.002. [DOI] [PubMed] [Google Scholar]
- 27.Yirmiya E, Leavitt A, Lu A, Avraham C, Osterman I, Garb J, Antine SP, Mooney SE, Hobbs SJ, Kranzusch PJ, et al. (2023). Phages overcome bacterial immunity via diverse anti-defense proteins. bioRxiv, 2023.2005.2001.538930. 10.1101/2023.05.01.538930. [DOI] [PubMed] [Google Scholar]
- 28.Cai H, Li L, Slavik K, Huang J, Yin T, Hédelin L, Xiang Z, Yang Y, Li X, Chen Y, et al. (2023). A novel virus-induced cyclic dinucleotide, 2′3′-c-di-GMP, mediates STING-dependent antiviral immunity in Drosophila. Immunity 56, 1991–2005. 10.1101/2023.05.08.539652. [DOI] [PubMed] [Google Scholar]
- 29.Woodward JJ, Iavarone AT, and Portnoy DA (2010). C-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328, 1703–1705. 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cady KC, Bondy-Denomy J, Heussler GE, Davidson AR, and O’Toole GA (2012). The CRISPR/Cas adaptive immune system of Pseudomonas aeruginosa mediates resistance to naturally occurring and engineered phages. J Bacteriol 194, 5728–5738. 10.1128/JB.01184-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi K-H, and Schweizer HP (2006). Mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nature Protocols 1, 153–161. 10.1038/nprot.2006.24. [DOI] [PubMed] [Google Scholar]
- 32.Choi KH, Mima T, Casart Y, Rholl D, Kumar A, Beacham IR, and Schweizer HP (2008). Genetic tools for select-agent-compliant manipulation of Burkholderia pseudomallei. Appl Environ Microbiol 74, 1064–1075. 10.1128/AEM.02430-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shanks RM, Caiazza NC, Hinsa SM, Toutain CM, and O’Toole GA (2006). Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from gram-negative bacteria. Appl Environ Microbiol 72, 5027–5036. 10.1128/AEM.00682-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qiu D, Damron FH, Mima T, Schweizer HP, and Yu HD (2008). PBAD-based shuttle vectors for functional analysis of toxic and highly regulated genes in Pseudomonas and Burkholderia spp. And other bacteria. Appl Environ Microbiol 74, 7422–7426. 10.1128/AEM.01369-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sayers EW, Bolton EE, Brister JR, Canese K, Chan J, Comeau DC, Connor R, Funk K, Kelly C, Kim S, et al. (2022). Database resources of the national center for biotechnology information. Nucleic Acids Res. 50, D20–D26. 10.1093/nar/gkab1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otwinowski Z, and Minor W (1997). Processing of X-ray diffraction data collected in oscillation mode – ScienceDirect. Methods in Enzymology 276, 307–326. 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 37.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, and Terwilliger TC (2002). PHENIX: building new software for automated crystallographic structure determination. Acta Crystallographica Section D Biological Crystallography 58, 1948–1954. 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 38.Emsley P, Lohkamp B, Scott W, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D Biol Crystallogr 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holm L (2022). Dali server: structural unification of protein families. Nucleic acids research 50, W210–w215. 10.1093/nar/gkac387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katoh K, Rozewicki J, and Yamada KD (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Briefings in bioinformatics 20, 1160–1166. 10.1093/bib/bbx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steinegger M, and Söding J (2017). MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nature biotechnology 35, 1026–1028. 10.1038/nbt.3988. [DOI] [PubMed] [Google Scholar]
- 42.Price MN, Dehal PS, and Arkin AP (2010). FastTree 2--approximately maximum-likelihood trees for large alignments. PloS one 5, e9490. 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Letunic I, and Bork P (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic acids research 49, W293–w296. 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the coordinate and structure factors reported in this paper are PDB: 8IXZ (Acb2–3’,2’-cGAMP), 8J8O (Acb2–2’,3’-cGAMP), 8IY0 (Acb2-cA3), 8IY1 (Acb2-cAAG) and 8IY2 (Acb2–3’,3’-cGAMP-cA3). This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.