

Abstract
Background:
Basal zone hyperplasia (BZH) and dilated intercellular spaces (DIS) are thought to contribute to the clinical manifestations of Eosinophilic esophagitis (EoE); however, the molecular pathways that drive BZH remain largely unexplored.
Objective:
To define the role of IL-13-induced transcriptional programs in esophageal epithelial proliferation in EoE.
Methods:
We performed RNA sequencing, bioinformatics, western blot, RT-qPCR, and histological analyses on esophageal biopsies from healthy control and EoE patients, primary esophageal cells derived from patients with EoE and IL-13-stimulated esophageal epithelial keratinocytes (EPC2-ALI). Genetic (shRNA) and pharmacologic (PROTAC degrader) approaches and in vivo model of IL-13-induced esophageal epithelial remodeling (Krt5-rtTA x tetO-IL-13Tg) were used to define the role of STAT3 and STAT6 and SFRP1 in esophageal epithelial proliferation.
Results:
RNAseq analysis of esophageal biopsies (healthy control vs. EoE) and EPC2-ALI revealed 82 common differentially expressed genes (DEGs) that were enriched for putative STAT3 target genes. In vitro and in vivo analyses revealed a link between IL-13-induced STAT3 and STAT6 phosphorylation, secreted frizzled-related protein 1 (SFRP1) mRNA expression and esophageal epithelial proliferation. In vitro studies showed that IL-13-induced esophageal epithelial proliferation was STAT3-dependent and regulated by the STAT3 target SFRP1. SFRP1 mRNA is increased in esophageal biopsies from patients with active EoE compared to healthy controls or patients in remission and identifies an esophageal suprabasal epithelial cell subpopulation that uniquely expressed the core EoE pro-inflammatory transcriptome genes (CCL26, ALOX15, CAPN14, ANO1, TNFAIP6).
Conclusions:
These studies identify SFRP1 as a key regulator of IL-13-induced and STAT3-dependent esophageal proliferation and BZH in EoE, and links SFRP1+ esophageal epithelial cells with the proinflammatory and epithelial remodeling response in EoE.
INTRODUCTION
Eosinophilic esophagitis (EoE) is a food antigen-mediated chronic inflammatory disease of the esophagus clinically characterized by upper gastrointestinal (GI) symptoms including, dysphagia, vomiting, and food impaction 1–6. The histopathology of EoE is defined by eosinophil-rich inflammation (≥15 eosinophils per high-power field [eos/hpf]) and esophageal remodeling, consisting of basal zone hyperplasia (BZH) and dilated intercellular spaces (DIS) 1, 2, 4, 5, 7–9. BZH and DIS are thought to contribute to esophageal barrier dysfunction, luminal narrowing, and chronic inflammation and the fibrostenotic phenotype in EoE 1, 6, 10–13. Currently, there is an urgent and unmet need for a better understanding of the biological processes that regulate esophageal epithelial remodeling to identify new therapeutic targets for the treatment of EoE.
Corroborative clinical and experimental studies support the concept that an underlying allergic sensitization to dietary food antigens and development of a CD4+ Th2 and ILC2 inflammatory response in the esophageal mucosa drive the eosinophilic inflammation and esophageal remodeling in EoE2, 14–16. The frequency of CD4+ Th2 cells in the peripheral blood and esophageal biopsy samples from EoE individuals17–19 are increased and the percentage of CD4+ Th2 cells correlated with esophageal tissue eosinophilia19. ScRNAseq analysis of T cells derived from esophageal biopsies of patients with EoE revealed enrichment of GATA3+ effector Th2 cells (CRTH2+ IL-17RB+ FFAR3+ CD4+) which expressed prodigious levels of IL-5 and IL-1319.
IL-13 is thought to be the central cytokine that drives the allergic inflammatory response and histopathological features of EoE. Esophageal epithelial transcriptional changes observed in the esophagus of mice engineered to overexpress IL-13 significantly overlap with that of esophageal biopsy samples of EoE patients20, 21. Treatment of adult EoE patients with RPC4046, a recombinant humanized monoclonal antibody against IL-13, improves histologic and endoscopic outcomes after 16 weeks. Patients that completed a 52-week open-label, long-term extension (LTE) study receiving open-label RPC4046 360 mg/week demonstrated sustained endoscopic, histologic, and clinical improvement22, 23. Furthermore, a phase 2 multi-center study of adults with active EoE who received weekly subcutaneous injections of dupilumab, a fully human IgG4 monoclonal antibody that binds the IL4Rα chain and blocks both IL-4 and IL-13 signaling, demonstrated reduced peak esophageal eosinophil count, decreased histologic and endoscopic severity scores, and increased esophageal distensibility24, 25. IL-13 is thought to dysregulate the expression of several key epithelial barrier regulatory genes including desmosomal cadherin desmoglein-1 (DSG1), leucine-rich repeat-containing protein 31 (LRRC31), kallikrein (KLK) serine proteases and calpain-14 (CAPN14) and promote esophageal epithelial proliferation altering esophageal epithelial barrier function and inducing esophageal remodeling7, 20, 26.
IL-13 can signal through the Type II IL-4 receptor that is composed of an IL-4Rα1 and IL-13Rα1 chain and the IL-13 receptor which consists of the IL-13Rα1 and IL-13Rα2 chains27–32. IL-13 binding to the Type II IL-4 receptor leads to activation of downstream JAK1- or JAK2/TYK2 and subsequent phosphorylation of STAT6 and STAT3. IL-13-induced phosphorylation of STAT6 induces expression of key EoE inflammatory genes including CCL26 and CAPN1433. Intratracheal IL-13 administration to wild-type mice resulted in dose-dependent esophageal eosinophilia and EoE-like pathophysiology, which was ablated in STAT6-deficient mice34. On the contrary, the contribution of IL-13-induced STAT3 signaling to the inflammatory and esophageal epithelial remodeling response is not well defined. STAT3 is phosphorylated at Y705 and S727 residues. Phosphorylation at the Y705 residue is required for STAT3 dimerization and nuclear translocation, and phosphorylation at the S727 residue promotes STAT3 mitochondrial translocation and enhances gene transcription27–32, 35–38. Importantly, STAT3 phosphorylation and activation has been implicated in many cellular functions including cell growth, apoptosis, cell migration and proliferation.
Herein, we identified 82 genes that were differentially expressed in EoE patient biopsies and IL-13-stimulated EPC2-ALI. Computational analysis revealed that these differentially expressed genes (DEGs) were enriched for putative STAT3 targets. Employing esophageal epithelial keratinocytes, a primary esophageal culture system derived from patient biopsies, and an in vivo model of esophageal IL-13-overexpression, we show that IL-13 induces esophageal epithelial STAT3 phosphorylation and activation (pSTAT3-Y705 and pSTAT3-S727) and esophageal epithelial proliferation. Importantly, STAT3 was required for IL-13-induced esophageal epithelial proliferation and expression of EoE proliferative genes. In silico analyses identified the putative STAT3 target secreted frizzled-related protein 1 (SFRP1) as a DEG associated with the IL-13-induced esophageal epithelial proliferative network. IL-13 induced SFRP1 mRNA expression in esophageal epithelial cells via a STAT3-dependent mechanism. Agonism and antagonism of SFRP1 revealed that SFRP1 plays a key regulatory role in IL-13-induced STAT3-dependent esophageal proliferation. Strikingly, we showed that SFRP1 was expressed predominantly in esophageal epithelial cells in active EoE compared to EoE in remission or normal control samples and that SFRP1+ esophageal epithelial cells were enriched for the core EoE pro-inflammatory transcriptome (CCL26, ALOX15, TNFAIP6, POSTN, ANO1)7. These studies identify SFRP1 as a key regulator of IL-13-induced and STAT3-dependent esophageal proliferation and BZH in EoE and identify an SFRP1+ esophageal epithelial cell population in esophageal inflammation and remodeling in EoE.
MATERIALS AND METHODS:
Bulk RNAseq pre-processing and quality control of human subjects –
Healthy control patients (NL) were defined as having no history of EoE diagnosis, 0 esophageal eos/hpf and no evidence of esophagitis within distal esophageal biopsies obtained during the same endoscopy procedure as the analyzed samples. EoE was defined as described in the recent consensus guidelines39, 40. Specifically, patients needed to have ≥15 eos/hpf in at least 1 high-power field in an esophageal biopsy without other causes of esophageal eosinophilia. RNAseq files were downloaded from NCBI GEO (GSE58640) and the FASTQC program and Trimmomatic tools were used to examine the quality of raw reads and filtering poor quality reads, respectively. Genome indexing was performed using Bowtie2 and the reads were aligned to the human reference genome (GRCh38) using HiSAT2 program with the default options. Read counts were generated using the feature-counts function from the subRead package. Downstream analysis was performed using DEseq2 and EdgeR in R (R Core Team, Vienna, Austria). DEGs were filtered using FDR: adjusted p-value < 0.05 and absolute log2-fold-change (absLog2FC) > 1.
EPC2-ALI culture –
hTERT immortalized human esophageal epithelial keratinocytes (hTERT-EPC2) were cultured under air-liquid-interface conditions, as previously described 9, 41. In brief, EPC2-hTERT cells were seeded onto permeable (0.4 μm) transwell support (Corning Incorporated, Corning, NY, USA) and grown to confluence while fully submerged in low-calcium ([Ca2+] = 0.09 mM) keratinocyte serum-free media (K-SFM) (Life Technologies; Carlsbad, CA). Epithelial differentiation was induced by culturing submerged cell monolayers in high-calcium K-SFM ([Ca2+] = 1.8 mM) for 4 days (day 3 to 7). Culture medium was removed from the apical chamber for 3 days (day 7 to 10) to induce stratification. For IL-13 time-course studies, cells were stimulated with IL-13 for 0–120 minutes. For STAT3 degradation studies using SD-36, EPC2-ALI were stimulated with Vehicle and IL-13 in the presence and absence of SD-36 for 48 hours. For STAT3-knockdown studies, EPC2-ALICTRL and EPC2-ALIΔSTAT3 were stimulated with IL-13 for 48 hours. For studies with agonism and antagonism of SFRP1, EPC2-ALI were stimulated with Vehicle or IL-13 in the presence and absence of recombinant SFRP1 (rSFRP1) or SFRP1 pharmacologic inhibitor (SFRP1inh, WAY316606) for 48 hours.
Primary esophageal cells cultured from biopsies –
Primary esophageal cell cultures were generated as previously 42. In brief, esophageal biopsies were collected in HYENAC media from males and females who underwent endoscopy. Biopsies were finely minced and seeded onto irradiated NIH 3T3-J2 mouse embryonic fibroblast (MEF) cells (7.5 × 104 cells per well; Kerafast; Catalog: EF3003) in a 6 well plate and were maintained and split upon reaching confluency for 8 passages. For western blot and RT-qPCR studies, primary esophageal cells were stimulated with IL-13 (100ng/ml) for 0 – 120 minutes and 48 hours and were collected and lysed for protein (NP40 lysis buffer with protease and phosphatase inhibitor) and mRNA (Tripure with Phenol chloroform) as described below. For immunofluorescence studies, primary esophageal cells were grown on MEFs which were cultured on top of 12mm siliconized glass coverslides in 12-well tissue culture plates. Esophageal cells were seeded at high-density (500,000 cells) and were cultured for 14 days before fixation, primary and secondary antibody staining, and fluorescent protein detection using confocal microscopy as described below. For access to detailed protocols see https://www.umichttml.org/protocols. The study was approved by the Institutional review boards at the Icahn School of Medicine at Mount Sinai and at the University of Michigan. All patients provided consent and assent when applicable to participate in the study.
Bulk RNAseq pre-processing and quality control of esophageal epithelial keratinocytes –
For bulk RNAseq, EPC2-ALI were cultured and stimulated with vehicle or IL-13 (100ng/mL) for 4 hours or 48 hours. RNA was isolated from cells with the RNeasy Kit (Qiagen, Germantown, Md), according to the manufacturer’s protocol. RNA quality was assessed by using the Agilent 2100 Expert Bioanalyzer (Agilent Technologies, Santa Clara, Calif), and only the samples with RNA integrity numbers of greater than 8 were processed for sequencing. RNA samples were subjected to RNA-seq at the Cincinnati Children’s Hospital Medical Center (CCHMC) Gene Discovery and Genetic Variation Core, as previously described 43. We performed the same pre-processing steps as described above until read counts were generated using subRead. The IDEP 9.1 webtool and rStudio DeSeq2 was used to analyze read counts and identify the differentially expressed genes (DEGs). DEGs were filtered using FDR: adjusted p-value < 0.05 and absLog2FC > 1.
RNAseq analysis of esophageal biopsies and EPC2-ALI –
GO biological processes were identified using DAVID Bioinformatics Resources 6.8. Common and unique DEGs were identified via DeSeq2 and represented as Venn diagrams (https://bioinformatics.psb.ugent.be/webtools/Venn/). Putative transcription factor targets analysis was performed using the ChIP-X Enrichment Analysis (ChEA3), which is a gene-set enrichment analysis tool to probe whether DEG lists contain putative targets of transcription factors. For our ChEA assessments, we employed the ENCODE TF libraries derived from ChIP-seq experiments (n = 7 experiments of STAT3 and n = 3 experiments for STAT6). The STAT3 TF ChIP-seq experiments were performed in HM12878 (lymphoblastoid cell line), Hela S3, MCF10A (epithelial cell line- mammary gland) cells. N = 5 STAT3 TF ChIP-seq experiments were public and passed specific quality control threshold including inclusion of isogenic replicates, utilization of well characterized antibody (clone sc-482; rabbit polyclonal IgG antibody with an epitope mapping at the C-terminus of STAT3)44, 45, possessed a read depth (> 10 million and < 20 million) for isogenic replicates and read length ~ 36bp. N = 2 experiments did not reach ENCODE standard (low read depth, low read length) quality control threshold and were not used in our analyses. The n = 3 STAT6 TF ChIP-seq experiments were performed in K562 (lymphoblast cells isolated from bone marrow), WTC11 (iPSC cell line) and HEPG2 (epithelial like morphology isolated from hepatocellular carcinoma) cells. All experiments passed the ENCODE quality control threshold with n = 2 experiments with > 20 million reads and n = 1 experiment (> 10 million, < 20 million). All three experiments had isogenic replicates and read length of > 50bp. These experiments were conducted using STAT6 protein conjugated with FLAG peptide sequence to permit detection of STAT6 using a validated monoclonal mouse anti-FLAG M2 antibody that recognizes the FLAG “tag” sequence “DYKDDDDK” 46, 47. All three experiments were used in our ChEA assessment analyses. The TF target libraries for STAT1, STAT3, STAT4, STAT5a, and STAT6 were used and mapped onto the 82 common DEGs identified through the RNAseq analysis. 82 DEGs were also mapped onto the “cell population proliferation” gene ontology term (GO:0008283) to identify DEGs which were putative STAT3 targets and associated with “cell population proliferation.”
Single-cell RNAseq analysis of esophageal epithelium in homeostasis and disease –
Data (UMI) count matrices, feature counts, and barcodes for each sample were acquired from NCBI GEO (GSE201153) and uploaded to Seurat v4 for downstream analysis. In brief, for each sample, the standard Seurat “Introduction to scRNA-seq Integration” vignette was used to perform downstream analysis. The data was processed through the standard pre-processing workflow including quality control (QC) metrics, such as removal of low-quality cells, empty droplets, cell doublets and cell multiplets. Cells with unique feature counts greater than 2500 or less than 200, and cells with >5% mitochondrial counts were filtered out. The UMI count matrix was then normalized using LogNormalize and scaled using a scale factor of 10,000, according to the standard Seurat pipeline. Samples were then integrated by all 10 samples using the IntegrateData function, to compare the cellular populations in an unbiased manner. Initial clustering was performed using the FindClusters function with a resolution of 0.5, to identify the top 20 principal components and detect predominant cell types, including epithelial cells, mast cells, fibroblasts, lymphocytes, myeloid cells, and endothelial cells. FindConservedMarkers was employed to identify the canonical cell type-specific markers that are conserved across samples. Epithelial cells were isolated for the secondary clustering to identify 12 epithelial subpopulations (resolution: 0.5), broadly categorized into quiescent, proliferating, transdifferentiated 1, transdifferentiated 2, differentiated HI, and differentiated LO clusters; cell type-specific marker gene identification was performed the same way as the initial clustering. A seventh disease-associated epithelial subpopulation was mapped onto “Basal” and “Suprabasal” epithelial markers to determine the phenotype of this cluster. Further, differential gene expression analysis was performed using DeSeq2 to identify differentially expressed genes in the new epithelial subcluster. StringDB and Cytoscape were used to represent the gene networks present in the novel epithelial subcluster.
Data visualization –
DEG comparisons were visualized using the Set Comparison Appyter from the Ma’ayan Lab (https://appyters.maayanlab.cloud/#/CompareSets). Network analysis was performed in Cytoscape3.7.1. Graphs and quantifications were performed on GraphPad Prism. Figures were constructed using Inkscape 1.1.1 and Adobe Illustrator 26.0.2.
Lentiviral transduction of EPC2 cells –
EPC2 cells were transduced at 60–70% confluency with lentiviral particles containing Mission® STAT3 shRNA (TRCN0000329887, Sigma; St. Louis, MO, USA), Mission® STAT6 shRNA TRC 0000019409 shRNA, or Mission® non-target control shRNA (Sigma; St. Louis, MO, USA). Lentiviral particles were incubated with EPC2 cells for 6 hours. All the viral particles were added in the presence of 5 μg/mL Hexadimethrine Bromide (Polybrene®) (Sigma; St. Louis, MO, USA). During the first hour of incubation, cells were spun down at 1,000*g for 1 hour at room temperature. 6 hours following transduction cells were put in fresh KSFM media, and 24 hours later media containing 1 μg/mL of Puromycin (Thermo Fisher Scientific Incorporated; Rockford, IL, USA) was used for selection. Cells were grown under selective pressure and cultured as regular EPC2 cells. Stable knockdown for STAT3 was demonstrated by western blot and RNAseq analyses.
Krt5-rtTA x tetO-IL-13 mouse model of EoE-like epithelial remodeling –
Krt5-rtTA mice (C57BL6; provided by Professor Andrzej Dlugoz MD)48 were backcrossed on the tetO-IL-13 (BALB/c; provided by Professor Marc E. Rothenberg MD PhD)49 In this system, the Krt5 promoter directs the expression of rtTA to the esophagus basal progenitors cells50. In the presence of doxycycline (Dox+), rtTA binds in trans to the tet-O and the VP-16 transactivator activates IL-13 gene transcription49. 8–10-week-old mice received doxycycline chow (625 ppm, Envigo) daily for two weeks to activate esophageal IL-13 expression and histological and molecular analyses were performed as described below. A total of n = 12 Krt5-rtTA x WT mice and n = 15 Krt5-rtTA x tetO-IL-13Tg mice were used in three independent experiments.
Quantitative PCR –
RNA was extracted from EPC2-ALI cells by using the Quick-RNA Microprep Kit (Zymo Research Corporation) according to the manufacturer’s protocol. Purified RNA (500 ng) was DNase treated and reverse transcribed to cDNA by using Superscript II RNase H Reverse Transcriptase (Thermo Fisher Scientific, Rockford, Ill), according to the manufacturer’s instructions. cDNA for ANO1, CAPN14, CCL26, TP63, SFRP1, STAT3 and hypoxanthine phosphoribosyltransferase (HPRT) was quantified by using real-time PCR with the IQ SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA) with the CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories). Quantitative PCR (qPCR) analyses were performed by using Bio-Rad CFX Manager Software (version 3.1; Bio-Rad Laboratories), and the results were normalized with HPRT amplified from the same cDNA mix and expressed as fold induction (2^-(ΔΔCT)). Primers used for amplification are reported in Supplementary Table 1.
Western blot –
EPC2 ALI cells and primary esophageal cells were lysed following stimulation, using a protein extraction reagent (10% Glycerol, 20 mM Tris HCl pH7, 137 mM NaCl, 2 mM EDTA, 1% NP-40 in H2O) with Halt™ protease inhibitor cocktail (Thermo Fisher Scientific Incorporated; Rockford, IL, USA) and PhosSTOP phosphatase inhibitor cocktail tablet (Roche Diagnostics; Mannheim, Germany). 25 μg of protein extract were separated on a 4%–12% Bis-Tris gel and transferred to a nitrocellulose membrane (Life Technologies; Carlsbad, CA). The following primary antibodies were used: anti-Phospho-STAT3 (Tyr705) (1:500, #9145, Cell Signaling Technology; Danvers, MA), anti-Phospho-STAT3 (Ser727) (1:500, #, Cell Signaling Technology; Danvers, MA), anti-STAT3 (1:1000, #4094, Cell Signaling Technology; Danvers, MA), anti-Phospho-STAT6 (Y641) (1:1000, #9361, Cell Signaling Technology; Danvers, MA), anti-STAT6 (1:1000, #9362, Cell Signaling Technology; Danvers, MA), anti-SFRP1 (1:500, #3534, Cell Signaling Technology; Danvers, MA), anti-Phospho-Jak2(Tyr1007/1008) (1:100, #3771, Cell Signaling Technologies, Danvers, MA), anti-Jak2 (1:250, #3230, Cell Signaling Technology; Danvers, MA), and anti- β-Actin (1:5000, #4970, Cell Signaling Technology; Danvers, MA). Anti-rabbit IgG HRP-linked antibody (1:10,000, #7074, Cell Signaling Technology; Danvers, MA) was used as the secondary antibody. For chemiluminescent detection of proteins, SuperSignal™ West Femto Maximum Sensitivity Substrate was diluted 1:5 in SuperSignal™ West Pico PLUS Chemiluminescence Substrate detection reagent (34094 and 34579, Thermo Scientific™, Life Technologies; Carlsbad, CA).
Histopathological analysis –
EPC2 cells were cultured in ALI conditions for 5 days and treated for 48 hours with their respective stimulation conditions. EPC2-ALI cells were then fixed on Transwell (Corning Incorporated, Corning, NY, USA) support with 4% paraformaldehyde (PFA) for 1 hour at room-temperature and underwent a series of ethanol (EtOH) washes (30% EtOH → 50% EtOH → 70% EtOH) for seven minutes per wash. Esophageal tissues from mice were fixed in 4% PFA overnight at 4°C and were then submerged in 70% EtOH. Formalin-fixed Transwell membranes with EPC2 cells and esophageal tissues were embedded in paraffin and sectioned at 5μm using a microtome. Sections were mounted on slides, deparaffinized and rehydrated using standard histological techniques. Hematoxylin & eosin (H&E) staining was used to study esophageal eosinophilia and esophageal remodeling (DIS, BZH, epithelial thickness). Stained slides were imaged with the Olympus DP-72 microscope and CellSens standard software (Olympus, Semrock, New York, NY). FIJI ImageJ was used for histologic and morphometric analysis. A photomicrograph (10x magnification) of a transverse section of the entire esophagus was obtained per mouse. Morphometric parameters (DIS, total epithelial thickness, BZH) were quantified from ten randomly selected regions from the entire circumference of the esophageal epithelium. DIS was measured in nanometers (nM) and total epithelial thickness was measured in micrometers (μM). For BZH quantification (% basal zone to total epithelial thickness), the basal zone measurement was divided by the total epithelial thickness from ten randomly selected regions from the entire circumference of the esophageal epithelium.
Immunofluorescence Staining –
Formalin-fixed and paraffin-embedded (FFPE) esophageal tissues on glass slides were deparaffinized and rehydrated using standard histological techniques. Slides were then permeabilized in Tris-EDTA (1 mM, pH 9.0) with 0.1% Tween-20, and antigen exposure performed at 125°C for 30 seconds in a decloaking chamber. Slides were washed in 1X PBS twice for 5 minutes each wash and then incubated blocked in 4% Normal donkey serum (Jackson ImmunoResearch; Catalog: 017–000-121), diluted in 1X PBS, for 1 hour to reduce non-specific hydrophobic interaction between primary antibody and the tissue. This was followed by overnight incubation of primary antibodies diluted in 4% normal donkey serum: anti-Krt15 (Cytokeratin 15 monoclonal antibody (LHK15); Thermo Fisher Scientific; Catalog: MA5–11344), anti-KRT4 (Cytokeratin 4 polyclonal antibody; Thermo Fisher Scientific; Catalog: 16572–1-AP), anti-Ki67 (Ki67 monoclonal antibody, Thermo Fisher Scientific; Catalog: 14–5698-82). Slides were then washed and incubated with secondary antibody at RT for 1 hour. Slides were mounted with DAPI Fluoromount-G (SouthernBiotech, Birmingham, Ala) mounting solution. Fluorescence imaging was performed with the Zeiss Apotome fluorescent microscope (Carl Zeiss Meditec, Dublin, California) with Nikon Elements software and ImageJ software (National Institutes of Health, Bethesda, Md).
Immunohistochemistry Staining –
Formalin-fixed and paraffin-embedded (FFPE) esophageal tissues on glass slides were deparaffinized and rehydrated using standard histological techniques. Slides were then permeabilized in Tris-EDTA (1 mM, pH 9.0) with 0.1% Tween-20, and antigen exposure performed at 125°C for 30 seconds in a decloaking chamber. Slides were washed in 1X PBS twice for 5 minutes each wash and then incubated in 3% hydrogen peroxide (H2O2) for 30 minutes. Sides were washed again in 1X PBS twice for 5 minutes and blocked in 4% normal donkey serum (Jackson ImmunoResearch; Catalog: 017–000-121), diluted in 1X PBS, for 1 hour to reduce non-specific hydrophobic interaction between primary antibody and the tissue. To block endogenous biotin binding, samples were incubated in 3 drops of avidin blocking reagent for 15 minutes. To block subsequent binding to avidin, samples were incubated with 3 drops of biotin blocking reagent for 15 minutes, followed by overnight incubation of primary antibody anti-STAT3 (1:1000, #30835, Cell Signaling Technology, Danvers, MA), anti-pSTAT3-Y705 (1:500, #9145, Cell Signaling Technology, Danvers, MA), anti-STAT6 (1:500, #5397, Cell Signaling Technology, Danvers, MA), anti-SFRP1 (1:500, #PA5–119647, ThermoFisher Scientific, Waltham, MA), and anti-B-actin (1:2000, #8457, Cell Signaling Technology, Danvers, MA) at 4°C. The next day, samples were washed three times with 1X PBS for 5 minutes each and incubated with biotinylated anti-rabbit IgG secondary antibody for 60 minutes. Slides were rinsed in 1X PBS for 3 times and incubated with ABC reagents (Vectastain PK-6100, Vector Labs, Burlingame, CA) for 30 minutes at room temperature. DAB peroxidase substrate was prepared immediately before use (Vectastain, SK-4100, Vector Labs), applied to the slides and observed for brown staining. Slides were added to tap water to stop the reaction and counterstained with Harris Modified Hematoxylin Solution for 4 minutes (#HHS32, Sigma Aldrich, Saint Louis, MO). Slides were then dehydrated using standard histological techniques and mounted with Cytoseal mounting media (C860G35, ThermoFisher Scientific, Kalamazoo, MI). Stained slides were imaged with the Olympus DP-72 microscope and processed by the CellSens standard software (Olympus, Semrock, New York, NY) and FIJI ImageJ software (National Institutes of Health, Bethesda, Md).
Masson’s Trichrome histopathological staining –
PFA-fixed and paraffin-embedded (FFPE) mouse esophageal tissue sections were deparaffinized according to standard histological procedures. The Masson’s Trichrome staining was performed exactly according to manufacturer’s protocol and instructions (Trichrome Stain Kit, Connective Tissue Stain, ab150686; Abcam).
Quantification of 5-bromo-2’-deoxyuridine-positive EPC2-ALI cells –
5-Bromo-2′-deoxyuridine (BrdU) was obtained from Sigma-Aldrich. EPC2-ALICTRL and EPC2-ALIΔSTAT3 cells were cultured as described above, treated for 48 hours with vehicle or IL-13 (100 ng/mL), and then exposed to BrdU (10 μmol/L) in dimethyl sulfoxide for 2 hours at 37°C. Cells were then fixed on support for 2 hours with 4% PFA at RT. Fixed membranes were then processed, embedded, and sectioned as described above. Immunofluorescence staining was performed using methods described in the previous section. Samples were stained for detection of BrdU+ cells by using 2.5 μg/μL G3G4 anti-BrdU antibody (Developmental Studies Hybridoma Bank, Iowa City, IA) and 4′−6-diamidino-2-phenylindole dihydrochloride (DAPI). The number of BrdU+-labeled cells were quantitated under a 20 × objective by using the Zeiss Apotome fluorescent microscope (Carl Zeiss Meditec). The BrdU+-labeled cells were quantified as the number of BrdU+-labeled cells per total linear length of filter-attached EPC2-ALI cells using FIJI ImageJ and calculated based on the ratio of BrdU+ cells per millimeter of membrane. Three filters per condition from three individual experiments were quantitated.
Quantification of Ki67-positive EPC2-ALI cells –
EPC2-ALI cells were cultured in the presence or absence of their respective stimulations for 48 hours. For immunofluorescence (IF) staining, formalin- or paraformaldehyde-fixed, paraffin-embedded transwell sections were sectioned, mounted on slides, and deparaffinized by using standard histologic procedures. Slides were then permeabilized in Tris-EDTA (1 mmol/L, pH 9.0) with 0.1% Tween-20, and antigen exposure was performed at 125°C for 30 seconds in a decloaking chamber by using a pressure cooker. Slides were then blocked by 10% normal donkey serum for 1 hour, followed by overnight incubation of primary antibodies diluted in 10% normal donkey serum at a final concentration of 1 μg/mL anti-Ki67 (1:1000; clone: SolA15; Invitrogen), slides were then washed and incubated with secondary antibody at RT for 1 hour. Slides were mounted with DAPI Fluoromount-G (SouthernBiotech, Birmingham, AL) mounting solution. Fluorescent imaging was performed with the Zeiss Apotome fluorescent microscope (Carl Zeiss Meditec, Dublin, CA) using Nikon Elements software. Ki67+ cells were quantified as the number of Ki67+-labeled cells per total linear length of filter-attached EPC2-ALI cells using FIJI ImageJ. Three filters per condition from three individual experiments were quantitated.
Statistical analysis –
Statistical significance of EPC2-ALI samples were established using unpaired t-test (two-tailed), or two-way ANOVA when there was more than one variable. Repeated measures ANOVA was performed on IL-13 time-course experimentation in the EPC2-ALI and primary esophageal cells. In the in vivo model of EoE-like epithelial remodeling, Welch’s t tests were performed when comparing epithelial thickness, percent basal zone to total epithelial thickness, Ki67+ cells, and DIS between Krt5-rtTA x WT to Krt5-rtTA x tetO-IL-13Tg. Simple linear regression was performed to assess the correlation between epithelial thickness and Ki67+ cells/mm epithelium. For non-normally distributed data Spearman correlation and Mann-Whitney test were used. Graphs and statistical analyses were performed using GraphPad Prism 9.1 (GraphPad Software Incorporated, La Jolla, CA).
RESULTS
Enrichment of STAT3-regulated genes in EoE and IL-13-stimulated EPC2 ALI.
To identify the key genes involved in IL-13-induced esophageal proliferation, we mapped the DEGs identified from RNAseq analysis of IL-13-stimulated EPC2-ALI cells onto the DEGs biopsy samples from normal healthy controls and pediatric EoE patients (GSE58640; n = 6 controls and n = 9 EoE patients). Analyses of differential gene expression in biopsy samples from normal healthy controls and pediatric EoE patients identified 1470 dysregulated genes (absolute LogFC > 1; FDR: adjusted p-value < 0.05; Supplementary Table 2). Consistent with previous analyses, GO gene ontology analyses identified the significant enrichment of genes that are associated with immune and inflammatory regulation (CCL26, CDH26), epithelial cell proliferation (ANO1, SLC26A4-AS1), and epithelial barrier regulation (SLC9A3, ALOX15, POSTN)26. RNAseq analysis of EPC2-ALI stimulated with vehicle or IL-13 for 4-hours and 48-hours identified 1563 DEGs (absolute LogFC > 1; Supplementary Table 3) and 1425 DEGs respectively (absolute LogFC > 1; Supplementary Table 4; Fig 1A). Notably, when we mapped the 1470 DEGs identified in pediatric EoE onto 1425 IL-13-induced DEGs in epithelial cells we revealed 82 common DEGs referred to as esophageal epithelial-specific EoE genes (Fig 1B). The 82 esophageal epithelial-specific EoE DEGs revealed significant enrichment of pathways involving “cytokine-cytokine receptor interaction” and “Interleukin-4 and Interleukin-13 signaling” (Supplementary Table 5). Key genes associated with “cytokine-cytokine receptor interaction” and “Interleukin-4 and Interleukin-13 signaling” pathways included C-C chemokine CCL24 (eotaxin 2), CXCL14, and SOCS1 (suppressor of cytokine signaling-1). Other highly enriched pathways in the 82 DEGs included “Wnt signaling pathway” (fold enrichment: 4.30), “Signaling by WNT” (fold enrichment: 2.05), and “Pathways in cancer” (fold enrichment: 1.35). To gain insight into the transcriptional regulation of the 82 esophageal epithelial-specific EoE DEGs we utilized ChEA TF gene sets and revealed that the 82 common DEGs were enriched for putative STAT protein targets including STAT1, STAT3, STAT4, STAT5a and STAT6 (Fig 1C). Of the 82 common DEGs, n = 32 (39%) were putative STAT3 targets and n = 8 (9.8%) were putative STAT6 targets (Fig 1C–D; Supplementary Table 6). Collectively, these studies show that esophageal epithelial-specific EoE DEGs are enriched for pathways associated with allergic inflammation and Wnt signaling pathways and are enriched for predominantly putative STAT3 targets.
Figure 1: Enrichment of STAT3-regulated genes in EoE and IL-13-stimulated EPC2-ALI.
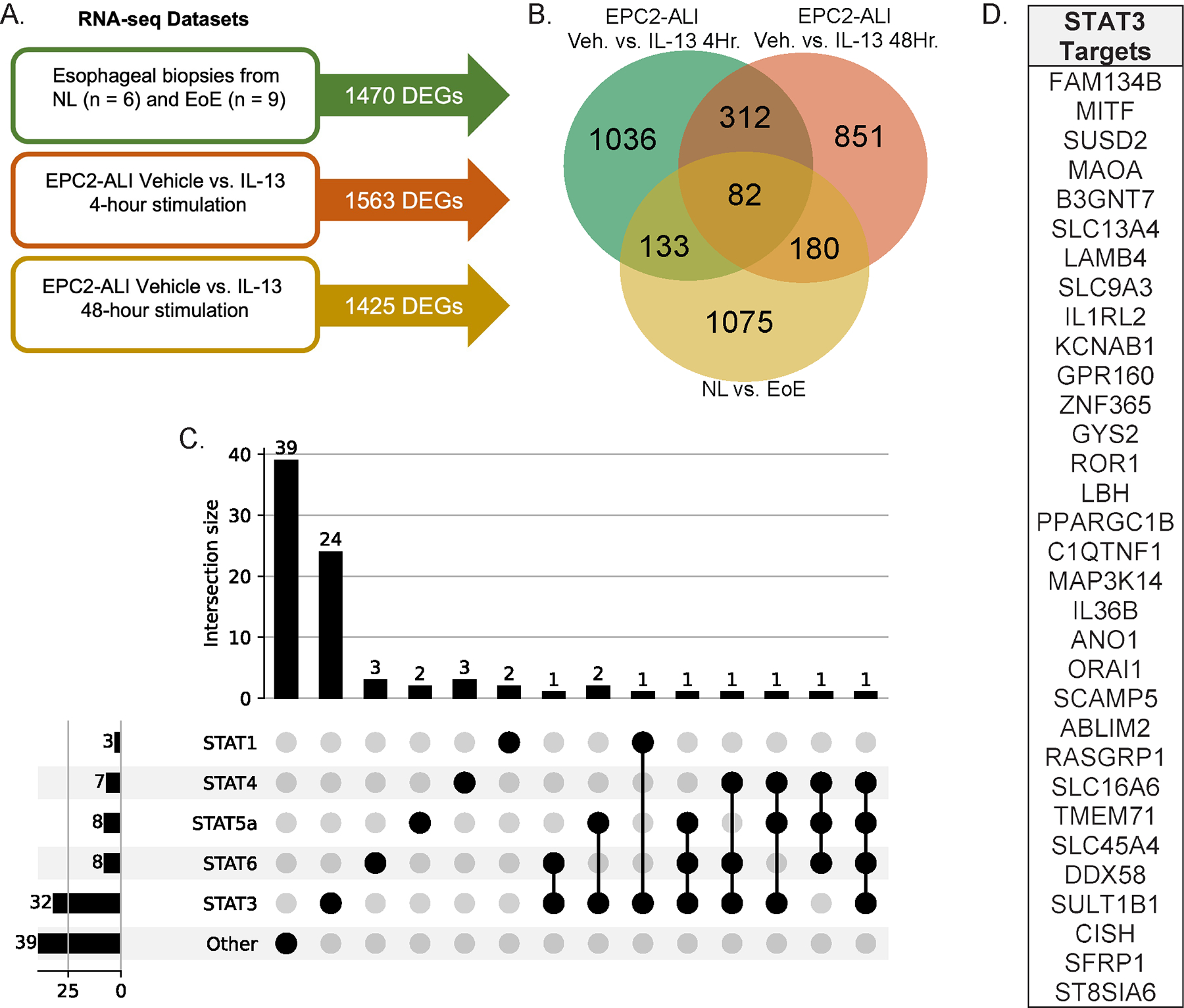
A. RNA sequencing (RNAseq) datasets and associated differentially expressed gene (DEG) counts B. Venn diagram of common and unique DEGs from three independent RNAseq datasets C. UpSet plot of putative STAT protein targets from the 82 common esophageal epithelial-specific EoE genes D. List of putative STAT3 targets from the 82 esophageal epithelial-specific EoE genes.
IL-13 activates STAT3 in EPC2-ALI and primary esophageal cells derived from patient biopsies.
To gain insight into the involvement of STAT3 in IL-13-induced esophageal epithelial signaling and proliferation, we utilized two in vitro culture systems, the EPC2-ALI and primary esophageal cell culture derived from patient biopsies9, 41, 42. Western blot analyses of IL-13-stimulated EPC2-ALI cells revealed a time-dependent phosphorylation of STAT6 with pSTAT6 (Y641) observed at 5 minutes post-stimulation and peaking by 60 minutes (Fig 2A–C, Supplementary Fig 2A). IL-13 also induced a time-dependent increase in phosphorylation of STAT3 at tyrosine 705 (Y705) and serine 727 (S727) (Fig 2A–C, Supplementary Fig 2B). Consistent with this, we observed phosphorylation of the primary STAT3-activating kinase, Jak2 in the EPC2-ALI cells following IL-13 stimulation (Supplementary Figure 2C). Notably, the kinetics of STAT3 phosphorylation differed from that of STAT6, in that STAT3 underwent a gradual and constitutive phosphorylation reaching maximal at 60- and 120-minutes post-stimulation (Fig 2B, Supplementary Fig 2A–B). Next, we examined IL-13- induced STAT3 and STAT6 phosphorylation and activation in an invitro complex 3D primary esophageal cell culture derived from human esophageal biopsy42. Immunofluorescence analyses reveals that the complex 3D esophageal cell culture consists of immature basal cells (Fig 2D–F; Krt15+) at the base of the culture and a suprabasal (Fig 2D–E, G; Krt4+) zone consisting of maturing cells apically, representing increasing maturation of esophageal cells from the basolateral to apical surface. Notably, colocalization of Krt15 and Ki67, indicates the presence of proliferating basal cells within the 3D primary esophageal cell culture (Fig 2H; Krt15+, Ki67+). IL-13 stimulation of 3D primary esophageal cell culture led to phosphorylation and activation of STAT3 and STAT6 (Fig 2I). IL-13-induction of STAT6 (Y641) peaked at 30 minutes and plateaued by 60 minutes (Supplementary Fig 2D). IL-13-induced rapid phosphorylation of STAT3 at Y705 residue reaching maximal by 30 minutes and declining over the 120-minute time course (Supplementary Fig 2D). IL-13 phosphorylation of S727 residue was delayed and gradually increased over the 120-minute time course (Fig 2I, Supplementary Fig 2E). Consistent with previous studies, IL-13 stimulation of primary esophageal epithelial cells at different passages induced increased mRNA expression of canonical EoE markers, ANO1 and CCL26 (Fig 2J–K)9, 26. Together, these data show that IL-13-induces phosphorylation and activation of STAT6 (Y641) and STAT3 (Y705 and S727) in esophageal epithelial cells.
Figure 2:
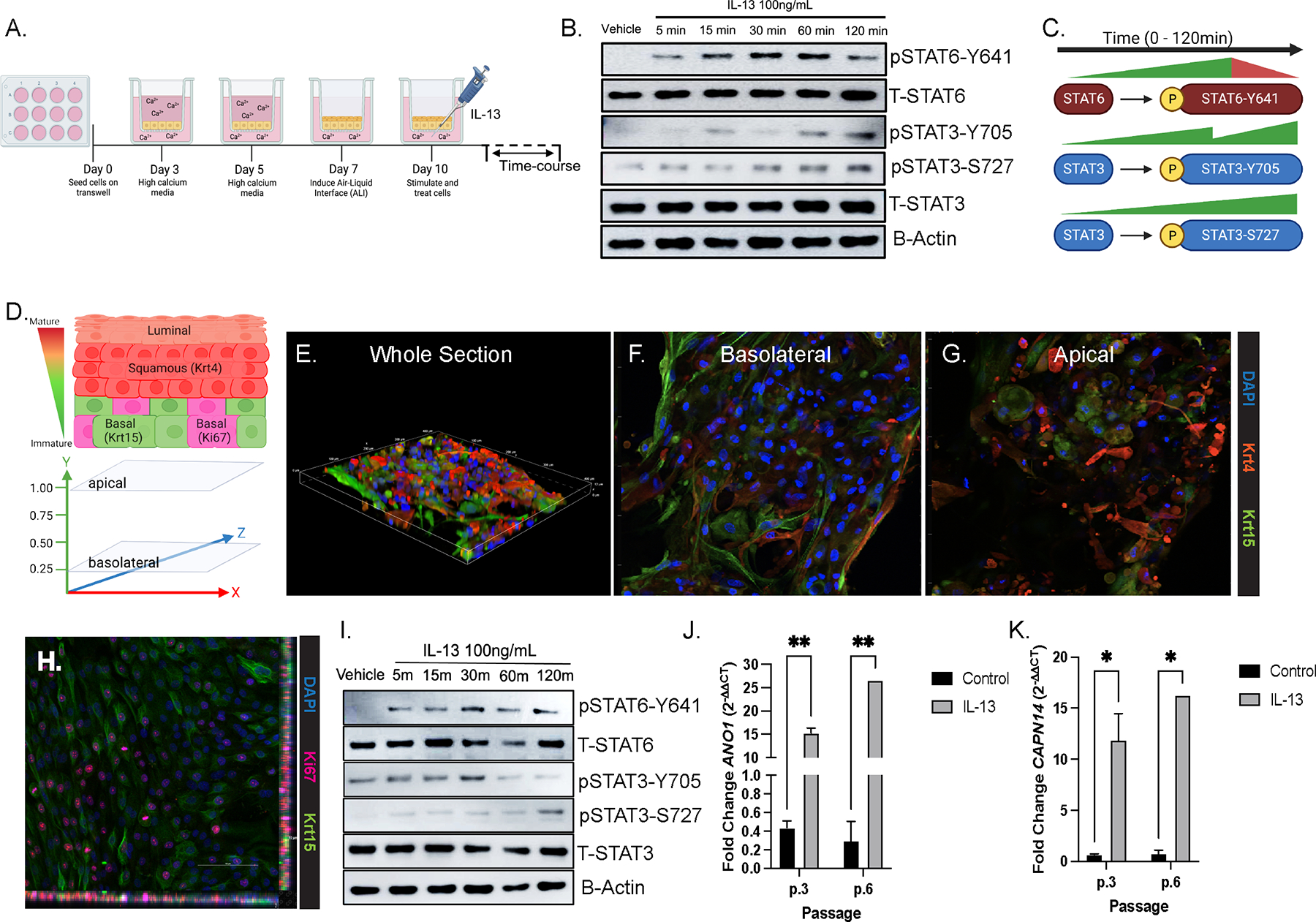
IL-13 activates STAT3 in EPC2-ALI and primary esophageal cells derived from patient biopsies. A. Schematic of EPC2-ALI culture system B. Western blot of EPC2-ALI under Vehicle and IL-13-stimulated conditions with probing for phosphorylation of STAT3 (Y705, S727) and STAT6 (Y641) C. Schematic of STAT6 and STAT3 activation kinetics from EPC2-ALI stimulated with Vehicle or IL-13 time-course D. Representative diagram of 3D esophageal epithelial cell organization and marker expression with Krt15- expressing basal cells at the basolateral surface and Krt4-expressing squamous cells towards the apical surface E. Immunofluorescence (IF) staining of 3D primary esophageal epithelium stained for Krt15 (green), Krt4 (red), and DAPI (blue) F. IF image from the basolateral surface of the 3D stack with Krt15+ basal epithelial cells G. IF image from the apical surface of the 3D stack with Krt4+ squamous epithelial cells H. IF staining of basal cells (Krt15; green) and proliferative cells (Ki67; pink). I. Western blot of IL-13-stimulated primary esophageal cells for STAT3 and STAT6 phosphorylation J-K. qPCR mRNA fold-change of ANO1 and CCL26 in IL-13-stimulated primary esophageal cells with representative passages 3 and 6. Repeated measures ANOVA were performed on IL-13-time course western blot densitometry analyses to determine the statistical significance of IL-13 stimulation on expression of proteins over time. Ordinary one-way ANOVA with Tukey’s multiple comparisons test were performed on qPCR analyses of the mean fold-change expression values across groups. * p < .05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Mice with inducible overexpression of IL-13 in esophageal epithelial cells exhibit epithelial remodeling and increased STAT3 expression.
To validate our in vitro approaches and to define the role of IL-13 induction of STAT3 in esophageal epithelial cells and remodeling, we employed a mouse model using Krt5-rtTA tetO-IL-13Tg mice where IL-13 is induced in esophageal epithelial cells upon receiving Dox chow ([Dox+], 625 ppm)50 (Fig 3A). Dox treatment of Krt5-rtTA x tetO-IL-13Tg mice, and not Krt5-rtTA x WT counterparts, induced esophageal epithelial remodeling including increased total esophageal epithelial thickness, percentage basal zone to total epithelial thickness, and DIS (Fig 3B–C, 3H–J). Krt5-rtTA x tetO-IL-13Tg mice which did not receive Dox (Krt5-rtTA x tetO-IL-13Tg, Dox-) resembled the histological characteristics like that observed in Dox-treated Krt5-rtTA x WT mice (results not shown). Immunohistochemistry analysis probing for Ki67 revealed that Dox+ Krt5-rtTA x tetO-IL-13Tg mice had increased esophageal epithelial proliferation (Ki67+ cells/mm epithelium) compared to their WT counterparts (Fig 3D–E, 3K), and the number of Ki67+ cells/mm epithelium positively correlated with total epithelial thickness (r = 0.7926, p < 0.0001; Fig 3L). Masson’s trichrome staining revealed a dense band of outer-epithelial collagen in WT mice (Fig 3F–G). In contrast, in Dox+ Krt5-rtTA x tetO-IL-13Tg mice, collagen was more disperse and was observed within esophageal epithelial invaginations that resembled papillary structures (Fig 3F–G). Immunohistochemistry analyses of the esophagus revealed low basal expression of STAT3 and STAT6 protein within the esophageal epithelial compartment at steady state (Fig 4B and Supplementary Figure S1A). Overexpression of IL-13 in the esophageal epithelium led to increased total levels of STAT3 and STAT6 in the esophageal epithelium (Fig 4C and Supplementary Figure S1A). Notably, increased esophageal epithelial proliferation was associated with increased phosphorylated STAT3 (pSTAT3-Y705) in esophageal epithelial cells in Dox+ Krt5-rtTA x tetO-IL-13Tg compared to WT counterparts (Fig 4D–E). Collectively, these studies show that esophageal IL-13 expression is associated with esophageal epithelial remodeling (BZH, DIS) and STAT3 protein expression and activation, and that esophageal epithelial proliferation is associated with increased STAT3 activation.
Figure 3:
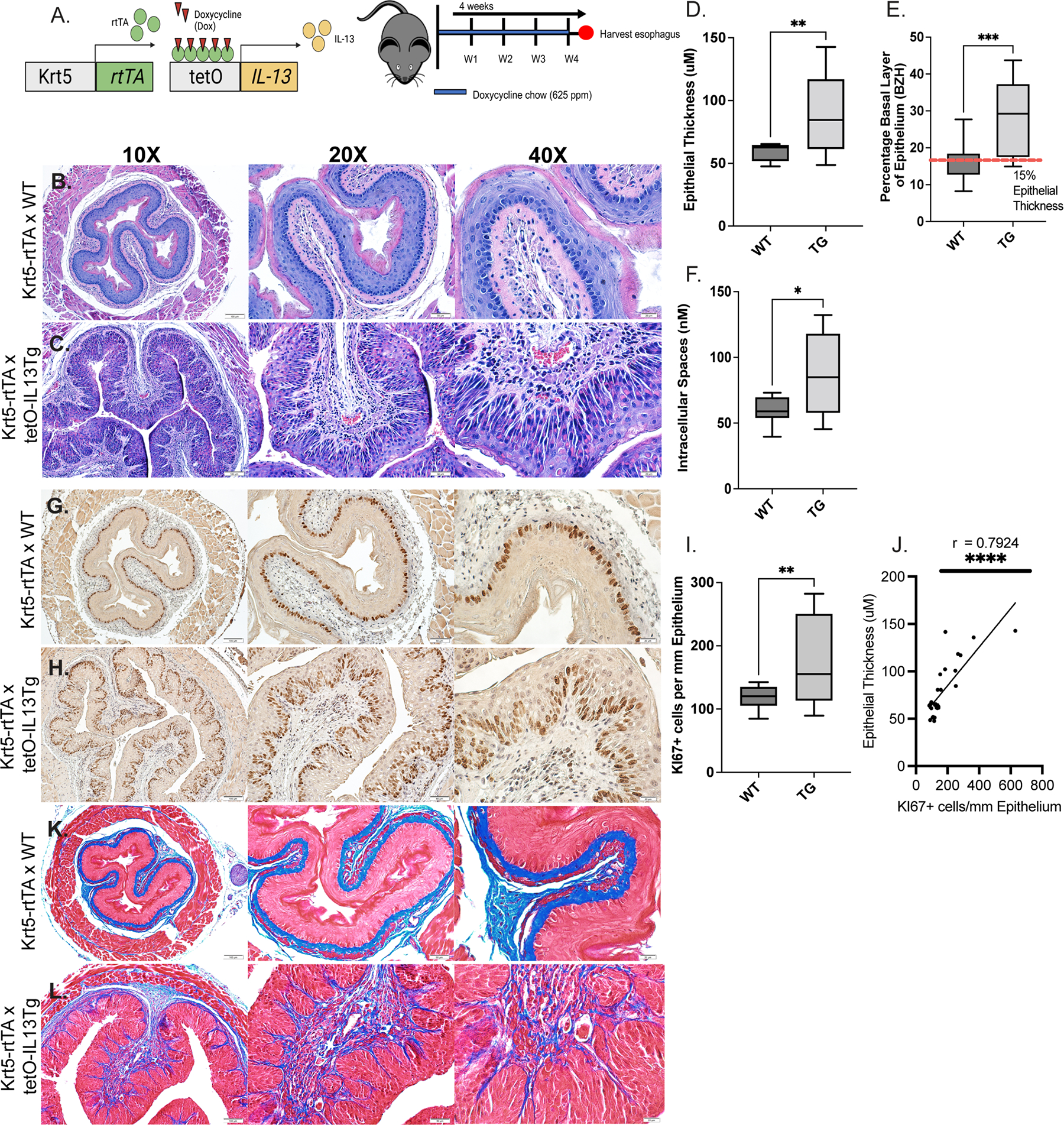
Mice with inducible overexpression of IL-13 in esophageal epithelial cells exhibit epithelial remodeling and increased STAT3 expression. A. Schematic of in vivo model of IL-13-induced esophageal epithelial remodeling B-C. H&E staining of Dox+ Krt5-rtTA x WT (WT) and Dox+ Krt5-rtTA xTetO-IL-13Tg (Tg) mouse esophagus; yellow arrows indicate DIS, green bars indicate BZH and red bars indicate total epithelial thickness D-E. Immunohistochemistry (IHC) staining for Ki67 for esophageal basal proliferation of WT and Tg esophagus; green arrows indicate Ki67+ cells F-G. Masson’s Trichrome staining for collagen deposition in WT and Tg mice H. Measurement of total epithelial thickness I. Percentage of basal zone to total epithelial thickness J. Measurement of dilated intercellular spaces (nM) K. Quantification of Ki67+ cells/mm epithelium L. Spearman correlation analysis between Ki67+ cells/mm epithelium and esophageal epithelial thickness in WT and Tg mice. Individual data points represent 1 mouse. Welch’s t tests were performed to compare differences in quantifications of epithelial thickness, percent basal zone, dilated intercellular spaces, and Ki67+ cells / mm epithelium between WT and Tg mice. * p < .05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Data represented as means ± SEMs of three independent experiments.
Figure 4:
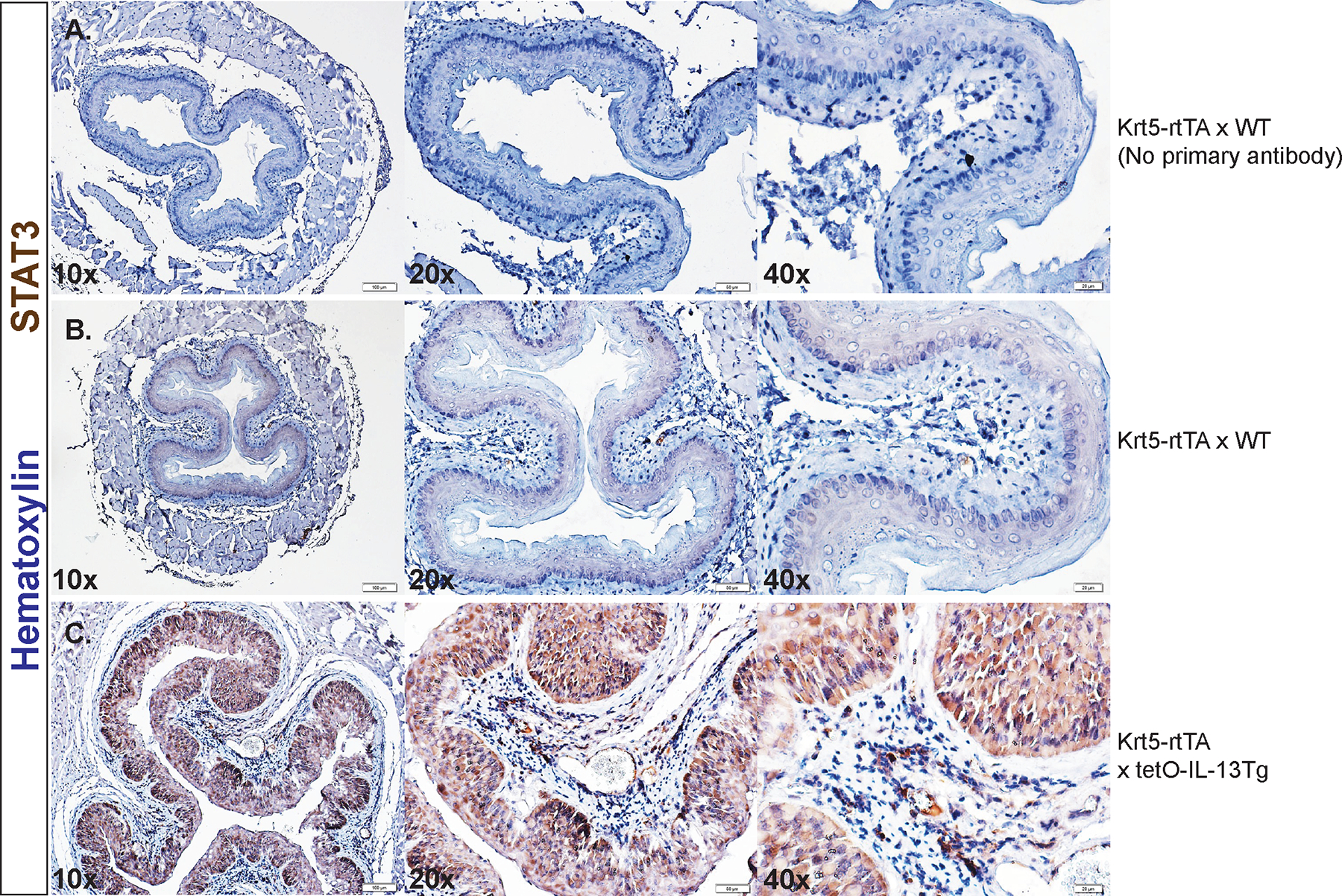
STAT3 protein expression in mice with inducible overexpression of IL-13 in esophageal epithelial cells. IHC staining of total STAT3 protein in A. Dox+ Krt5-rtTA x WT (WT) mouse esophagus with no primary antibody B. WT mouse esophagus and C. in Dox+ Krt5-rtTA x TetO-IL-13Tg (Tg) mouse esophagus. IHC staining of phosphorylated STAT3 (pSTAT3-Y705) in D. WT mouse esophagus and E. Tg mouse esophagus. All issues were counterstained with nuclear hematoxylin stain. Staining was performed on mouse tissues extracted from three independent experiments.
IL-13-induced esophageal epithelial proliferation is STAT3-dependent.
To determine the requirement of STAT3 in esophageal epithelial proliferation, we examined BrdU incorporation in IL-13-stimulated EPC2-ALI with shRNA-mediated knockdown of STAT3 (EPC2-ALIΔSTAT3) or empty-vector shRNA (EPC2-ALICTRL). Under steady state conditions we observed no difference in BrdU incorporation in EPC2-ALICTRL and EPC2-ALIΔSTAT3, however IL-13-stimulation induced a significant increase in BrdU incorporation in EPC2-ALICTRL cells (Fig 5A–B). In contrast, IL-13-induced proliferation was abrogated in EPC2-ALIΔSTAT3 cells (Fig 5A–B). Moreover, the level of proliferation following IL-13 stimulation was significantly diminished in EPC2-ALIΔSTAT3 cells compared with EPC2-ALICTRL cells (Fig 5A–B). To determine whether this was the consequence of developmental loss of STAT3 in EPC2 cell functionality, we next utilized the selective STAT3 protein degrader, SD-36 (10uM)51. SD-36 is a potent and selective PROTAC (proteolysis-targeting chimera) degrader which serves as an endogenous protein degradation tool. SD-36 serves as a “linker” molecule which tethers STAT3 to an E3 ligase for ubiquitin-mediated proteasomal degradation51. Treatment of EPC2-ALI cells with SD-36 led to degradation of STAT3 at steady state and following IL-13-stimulation (100ng/mL) (Fig 5C). SD36 degradation was associated with total loss of IL-13 induced phosphorylation of STAT3 Y705 and Y727 residues (Fig 5C). Importantly, SD36 demonstrated selectivity and did not degrade STAT6 protein at steady-state or following IL-13 stimulation. IL-13 exposure of EPC2-ALI leads to epithelial remodeling including basal cell proliferation (BZH), as evidenced by Ki67+ staining and DIS (Fig 5D–F; BZH indicated by yellow bracket, DIS indicated by green arrows). Consistent with our genetic approach, the IL-13-induced increase in EPC2-ALI Ki67+ cells were attenuated following STAT3 degradation (Fig 5F). To determine the effect of loss of STAT3 on IL-13-induction of key EoE genes we performed RT-qPCR for mRNA expression of surrogate EoE proliferation genes, ANO1 and TP63, and canonical EoE inflammatory genes, CCL26, SLC9A3, and CAPN14, in the presence and absence of SD36. We observed that IL-13 significantly induced mRNA expression of ANO1 (Fig 5G) and TP63 (Fig 5H), and this effect was abolished in the presence of SD-36. Surprisingly, IL-13 induction of canonical EoE inflammatory genes was not significantly inhibited by STAT3 degradation (Fig 5I–K). To determine the role of STAT6 in IL-13-induced esophageal epithelial proliferation we examined the BrdU incorporation in IL-13-stimulated EPC2-ALI with shRNA-mediated knockdown of STAT6 (EPC2-ALIΔSTAT6) or empty-vector shRNA (EPC2-ALICTRL). We observed an IL-13-induced increase in BrdU+ cells in both EPC2-ALICTRL and EPC2-ALIΔSTAT6 cells. Notably, the level of BrdU incorporation between IL-13 stimulated EPC2-ALICTRL and EPC2-ALIΔSTAT6 cells was not significantly different (Supplementary Fig 1B–C). Collectively, these data suggest that IL-13-induced esophageal epithelial proliferation is STAT3-dependent and STAT6-independent.
Figure 5:
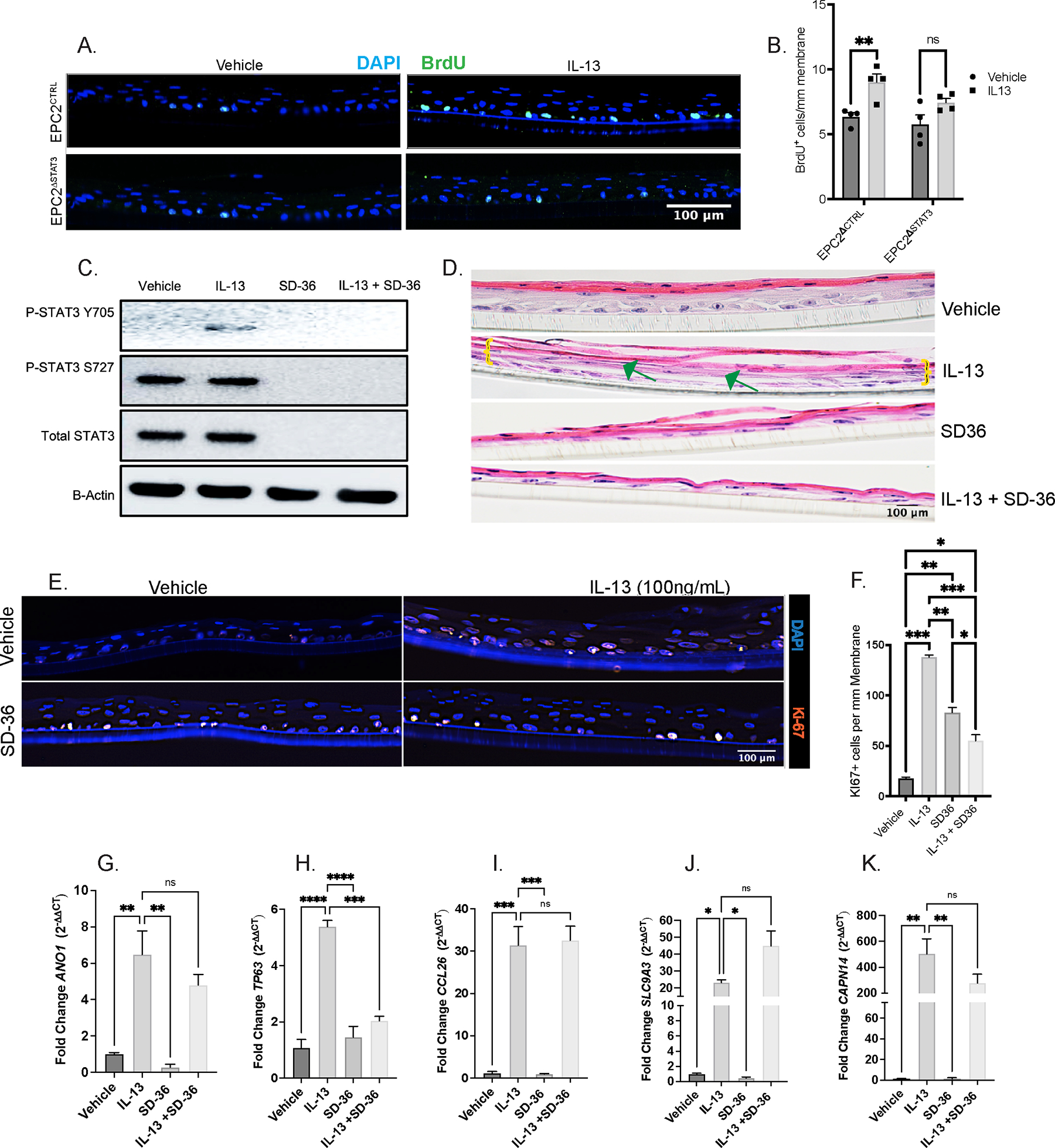
IL-13-induced esophageal epithelial proliferation requires STAT3. A. Immunofluorescence staining of Brdu (green) and DAPI of EPC2CTRL and EPC2ΔSTAT3 cells under Vehicle and IL-13 stimulation B. Quantification of Brdu+ cells/mm membrane C. Western blot of EPC2-ALI probing for total (STAT3), phosphorylated STAT3 (pSTAT3-Y705 and pSTAT3-S727), and total STAT6 (STAT6) in the presence and absence of IL-13 and selective STAT3 degrader, SD-36 D. Hematoxylin & Eosin staining showing epithelial remodeling (BZH, DIS) in EPC2-ALI in the presence and absence of IL-13 and SD-36; green arrow points to DIS and yellow brackets mark basal cell expansion E. IF staining for Ki67 (red) for basal cell proliferation and DAPI (blue) in EPC2-ALI in the presence and absence of IL-13 and SD-36 F. Quantification of Ki67+ cells/mm membrane G-K. qPCR for mRNA fold-change of ANO1, TP63, CCL26, SLC9A3, and CAPN14, respectively, in the presence and absence of IL-13 and SD-36. Ordinary one-way ANOVA with Tukey’s multiple comparisons test were performed on qPCR analyses and quantification of Brdu+ cells/mm membrane of the mean fold-change expression values across groups. Data represented as means ± SEMs of three independent experiments, with n = 3 samples per group. * p < .05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
SFRP1 is a novel candidate gene which is associated with cell proliferation and is a STAT3 target.
To gain a mechanistic understanding for how IL-13-induced STAT3-dependent signaling drives esophageal epithelial proliferation, we performed advanced in silico analysis on the 82 DEGs identified from our RNAseq analyses in Fig 1A (Fig 6A). We mapped the 32 putative STAT3 targets identified from n = 82 common DEGs onto the “Cell Proliferation” Gene Ontology (GO:0008283; n = 2148 genes) and identified n = 4 genes, secreted frizzled-related protein-1 (SFRP1), melanocyte-inducing transcription factor (MITF), LBH regulator of WNT signaling pathway (LBH), and RAS guanyl releasing protein-1 (RASGRP1) (Fig 6B). SFRP1 plays a critical role in the regulation of canonical WNT signaling and cell proliferation; therefore, we examined the role of SFRP1 in IL-13-induced STAT3-dependent esophageal epithelial proliferation. Notably, IL-13-stimulation of EPC2-ALI induced a significant increase in SFRP1 mRNA at 4- and 48-hours of stimulation (Fig 6C), and that IL-13 induction of SFRP1 mRNA was STAT3-dependent (Fig 6D). In addition, Dox+ IL-13 expression in esophageal epithelial cells in Krt5-rtTA x tetO-IL-13Tg mice led to an increase in SFRP1 protein expression as compared to the Dox+ Krt5-rtTA x WT mice (Fig 6E–F). These data suggest a functional role for SFRP1 in regulating IL-13-induced STAT3-dependent esophageal epithelial proliferation.
Figure 6:
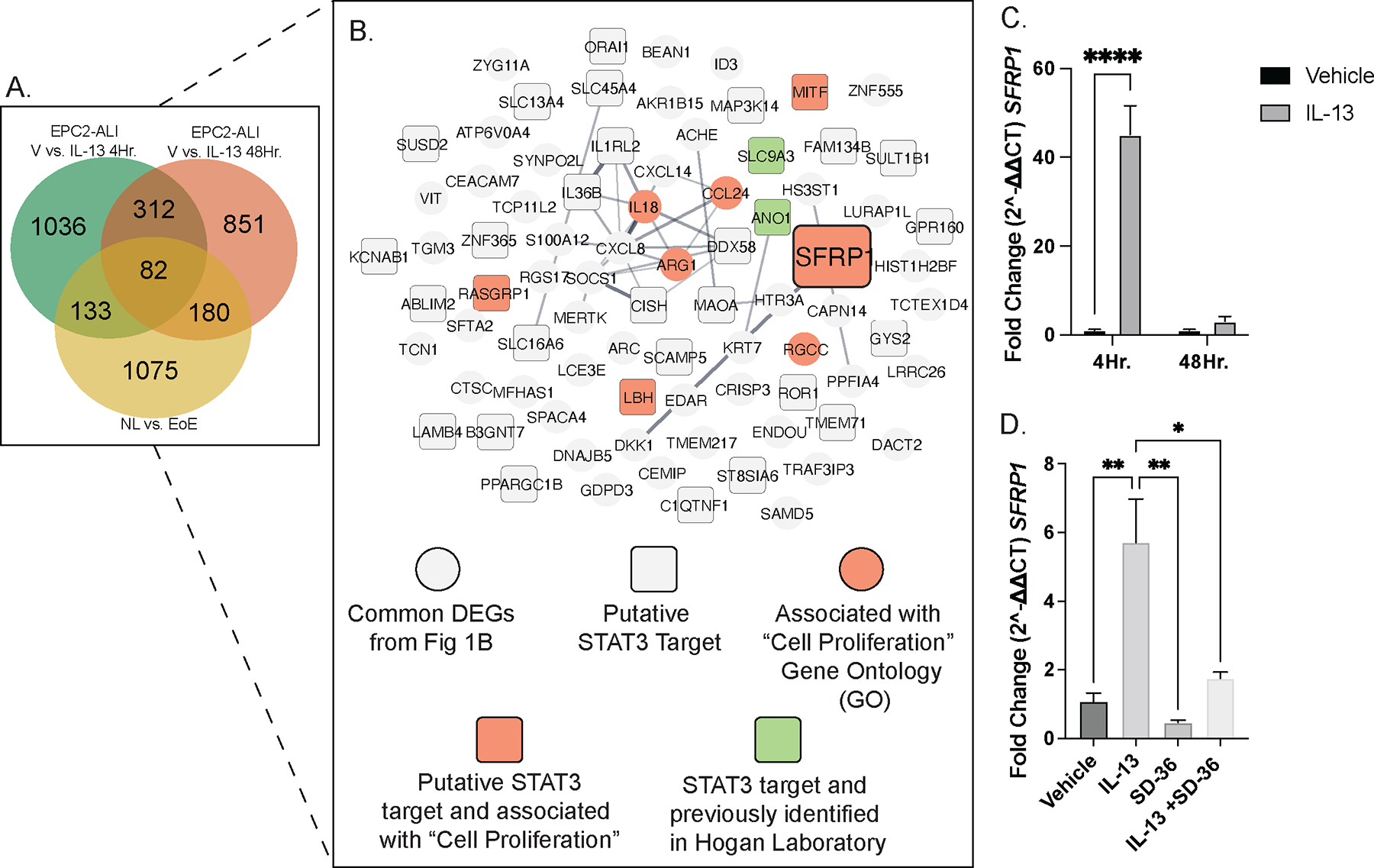
SFRP1 is a novel candidate gene which is associated with cell proliferation and is a STAT3 target. A. Venn diagram of common and unique DEGs from three independent RNAseq datasets, showing 82 common esophageal epithelial-specific EoE genes B. network map of the esophageal epithelial-specific EoE DEGs identifying candidate gene, SFRP1; gray circle: common DEGs from Fig 1B, gray square: putative STAT3 targets, purple circle: associated with “Cell Proliferation” gene ontology (GO), orange square: putative STAT3 target and associated with “Cell Proliferation” GO, and pink square: putative STAT3 target and previously studied in Hogan Laboratory C. qPCR mRNA fold-change of SFRP1 in EPC2-ALI cells stimulated with Vehicle and IL-13 at 4- and 48-hours. D. qPCR mRNA fold-change of SFRP1 in EPC2-ALI cells stimulated with Vehicle and IL-13 in the presence and absence of SD-36. E-F. IHC staining of SFRP1 protein in D. Krt5-rtTA x WT (WT) mouse esophagus and E. Krt5-rtTA x TetO-IL-13Tg (Tg) mouse esophagus. qPCR data represented as means ± SEMs of three independent experiments, with n = 3 samples per group. Staining was performed on mouse tissues extracted from three independent experiments. * p < .05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Esophageal epithelial cells are cellular sources of SFRP1 expression and IL-13-induced esophageal proliferation is regulated by SFRP1.
To determine the predominant cellular sources of SFRP1 we used publicly available single-cell RNAseq data of esophageal biopsies from healthy control, disease remission, and active disease patients (GSE201153; n = 2 healthy control, n = 3 remission, n = 5 active disease)52. Importantly, we identified that there was increased SFRP1 mRNA expression in biopsies of patients with active disease, specifically within the epithelial cluster, compared to biopsies of patients in remission or healthy controls (Fig 7A–F). To define the role of SFRP1 in esophageal epithelial proliferation we stimulated EPC2-ALI cells with IL-13 in the presence of a pharmacologic agonist (rSFRP1) and antagonist of SFRP1 (WAY316606, SFRP1inh). As we have previously shown, IL-13-stimulation of EPC2-ALI cells induced proliferation (Fig 7G–H). In contrast, IL-13-stimulation of EPC2-ALI cells in the presence of rSFRP1 resulted in a decrease in Ki67+ EPC2-ALI cells (Fig 7G–H). Conversely, IL-13-stimulation of EPC2-ALI in the presence of SFRP1inh resulted in an increase in Ki67+ cells suggesting that SFRP1 blockade enhances IL-13-induced esophageal epithelial proliferation. (Fig 7G–H). Intriguingly, treatment of EPC2-ALI cells with rSFRP1 alone was sufficient to inhibit esophageal epithelial proliferation; conversely, treatment of EPC2-ALI with SFRP1inh resulted in an increase in Ki67+ cells compared to steady-state (vehicle) (Fig 7G–H). Taken together, these data suggest that SFRP1 is predominantly produced in esophageal epithelial cells in active disease, and that SFRP1 regulates steady state and IL-13-induced esophageal epithelial proliferation in vitro.
Figure 7:
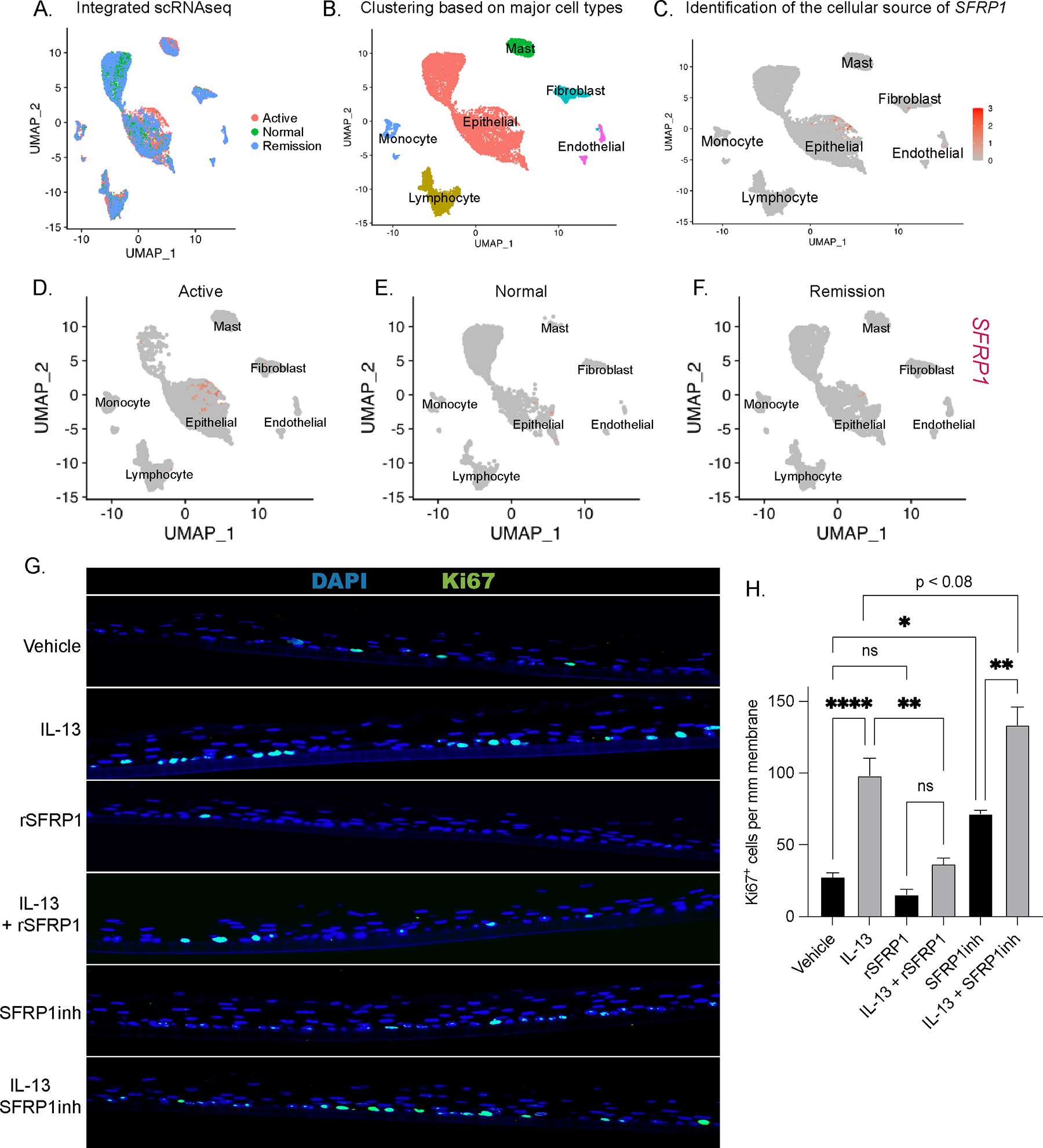
Esophageal epithelial cells are cellular sources of SFRP1 expression and IL-13-induced esophageal proliferation is regulated by SFRP1. A. Seurat-integrated single-cell RNA sequencing (scRNAseq) samples consisting of n = 5 Active, n = 3 Remission, and n = 2 Normal esophageal patient biopsies B. scRNAseq clustering of integrated samples based on major cell types which are annotated as Epithelial, Mast, Endothelial, Fibroblast, Monocyte, and Lymphocyte C. mRNA expression of SFRP1 amongst the six annotated clusters D-F. cell-type-specific mRNA expression of SFRP1 separated by disease state (Active, Normal, and Remission) G-H. IF staining of Ki67 and quantification in EPC2-ALI in the presence or absence of IL-13, recombinant SFRP1 protein (rSFRP1) and SFRP1 pharmacologic antagonist, WAY316606 (SFRP1inh). Ordinary one-way ANOVA with Tukey’s multiple comparisons test were performed on quantification of Ki67+ cells/mm membrane of the mean fold-change expression values across groups. Data represented as means ± SEMs of three independent experiments, with n = 3 samples per group. * p < .05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
SFRP1 expression is localized to an esophageal epithelial cell cluster restricted to active EoE.
Given that SFRP1 expression was predominantly restricted to the esophageal epithelial cluster of individuals with active disease (Fig 7D), we further sub-clustered the epithelial cells to provide insight into the SFRP1+ esophageal epithelial cell phenotype and proliferative signaling pathways that are co-expressed with SFRP1. Sub clustering of the epithelial populations within the ten integrated samples derived from the three disease states (healthy, remission, active disease) led to the generation of n = 13 communities that were categorized into six main epithelial clusters, defined by Rochman et al., 2023, including “Quiescent,” “Proliferating,” “Transdifferentiated 1,” “Transdifferentiated 2,” “Differentiated HI,” and “Differentiated LO” (Fig 8A–B)52. Stratification of the integrated data into the three disease states (healthy, remission, active disease) revealed a seventh epithelial sub-cluster that was restricted to the active disease (Fig 8A–B). Mapping SFRP1 expression on the sub-clustered epithelial populations, identified that SFRP1 expression was predominantly restricted to the seventh epithelial population that was unique to the active disease group (Fig 8C). Gene expression analyses of the active disease specific SFRP1+ cell population identified expression of 560 unique genes, of which 120 were differentially expressed. This seventh epithelial sub-cluster consisted of a suprabasal epithelial cell signature (n = 8 basal markers and n = 25 suprabasal markers; Fig 8D). Intriguingly, the SFRP1+ suprabasal epithelial cell population highly differentially expressed the canonical EoE disease-driving inflammatory genes such as TNFAIP6, POSTN, CCL26, and ALOX15 as compared with the n = 6 other epithelial clusters (Fig 8E–I, Supplementary Table 7A–C)53. Collectively, these studies identified SFRP1+ disease-associated esophageal epithelial cells as a suprabasal epithelial population that expressed the EoE-inflammatory signature.
Figure 8: A novel esophageal epithelial cell cluster present exclusively in Active EoE scRNAseq samples is a potent source of SFRP1.
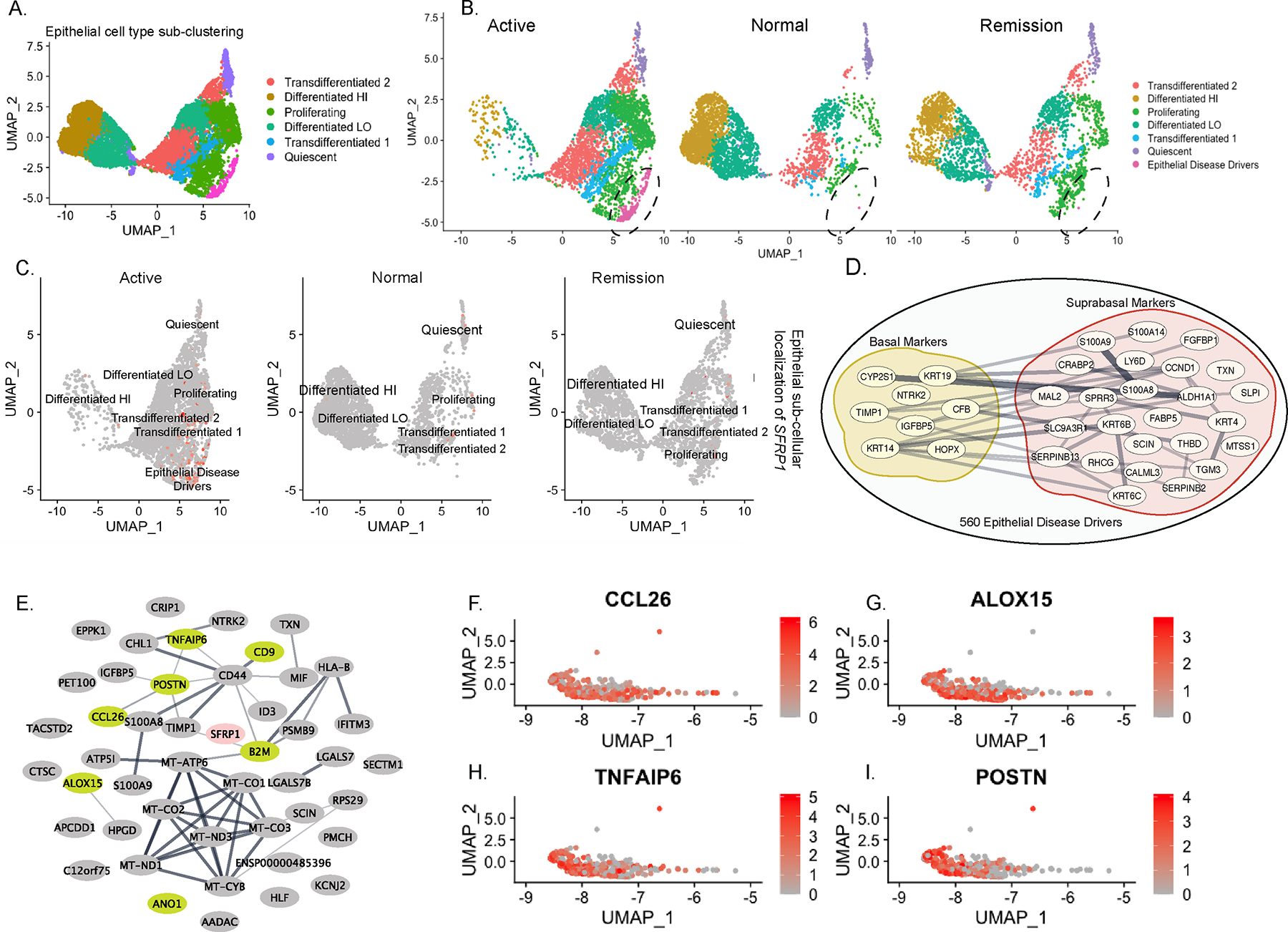
A. Sub-clustering of the integrated and combined “Epithelial Cell” cluster identified in Fig 7B into six predominant subpopulations B. Sub-clustered epithelial populations separated by disease state (Active, Normal, Remission) and identification of a seventh sub-population in the “Active” samples. C. SFRP1 mRNA expression in epithelial subpopulations separated by disease state (Active, Normal, Remission) D. Mapping of 560 genes expressed in the seventh epithelial subpopulation present in Active disease, upon the top 100 “Basal” and “Suprabasal” markers E. Network of top 50 differentially expressed genes present in the Disease-Associated epithelial population, depicting canonical genes highly differentially expressed in EoE (green). F-I. mRNA expression of EoE pro-inflammatory genes CCL26, ALOX15, TNFAIP6, and POSTN in the Disease-Associated epithelial population.
DISCUSSION
In this study, we utilized in vitro culture systems, a mouse model, and in silico analyses of human datasets to demonstrate a critical role for SFRP1 in IL-13-induced STAT3-dependent esophageal epithelial proliferation in EoE. We show that 1) IL-13 induces STAT3 activation and STAT3-driven transcriptional programs in esophageal epithelial cells; 2) IL-13-induced proliferation is regulated by a STAT3-dependent mechanism; 3) that inducible IL-13 overexpression in Krt5+ esophageal cells increased total STAT3 protein expression and resulted in esophageal epithelial remodeling (epithelial thickness, BZH and DIS); 4) IL-13 induces SFRP1 mRNA expression via STAT3-dependent mechanism; 5) SFRP1 counter-regulates IL-13-induced esophageal epithelial proliferative response; 6) SFRP1 is expressed by esophageal suprabasal epithelial cells; and 7) SFRP1+ suprabasal epithelial cells are a subset of epithelial cells that present in active disease and express the core EoE pro-inflammatory and pro-proliferative transcriptional programs.
Clinical and experimental studies provide corroborative evidence of a central role for IL-13 in the esophageal allergic inflammatory response and histopathological features of EoE21, 54. IL-13 is thought to dysregulate expression of key inflammatory and epithelial barrier regulatory genes that alter the esophageal epithelial barrier function and promote an eosinophilic inflammatory response, leading to esophageal remodeling and clinical symptoms of EoE33, 55 7, 20, 26. IL-13 is known to activate both STAT3- and STAT6-transcriptional programs in both hematopoietic and non-hematopoietic cell compartments27, 56. However, majority of studies have focused on delineating IL-13-induced STAT6 transcriptional programs and the role this pathway plays in driving CD4+ Th2 cells and downstream allergic effector cell populations (basophils, eosinophils, and mast cells)34, 57, 58. Consistent with this, investigators have demonstrated that esophageal eosinophilia in experimental EoE is dependent on IL-13 signaling through the Type II IL-4 receptor (IL-4Rα1 / IL-13Rα1) and STAT634, 59. We show that IL-13 induces phosphorylation of STAT3 and STAT3-transcriptional programs in esophageal epithelial cells and that IL-13-induced esophageal proliferation is STAT3-dependent and STAT6-independent. In support of a dominant role for STAT3 in esophageal epithelial proliferative response we showed that 32 of 82 common DEGs expressed by IL-13-stimulated esophageal cells and in EoE possessed putative STAT3 motifs and only 8 of 82 DEGs to possess putative STAT6 motifs. Notably, ~ 50% of n = 82 DEGs did not possess any putative STAT motifs and analyses revealed these genes were enriched for other transcription factors known to regulate cell proliferation including JUN and GATA2 (Supplementary Table 8). STAT3 has been shown to interact with JUN and GATA2 through protein-protein interaction and participate in cooperative gene transcription 60, 61. While there are limitations within the in-silico analyses specific to the limited knowledge of the quality control of the ChIPseq datasets used in the ChEA assessment, we validated these analyses by the demonstration that IL-13-induction of proliferative genes ANO1, TP63 and SFRP1 was STAT3-dependent. Collectively, these studies demonstrate a dominant role for IL-13-STAT3-axis in esophageal epithelial proliferative response.
Previous studies have demonstrated that the eosinophilic inflammatory response in EoE is mediated by STAT6-dependent expression of CCL26. JAK-STAT6 pathway inhibitors block IL-13-induced CCL26 expression both in esophageal fibroblasts and epithelial cells58. Consistent with this genetic variation in STAT6 have been associated with EoE62–64 and STAT6 genetic variant (rs324011) synergizes with the gain of function allele variant CYP2C19∗17 to predict a proton pump inhibitor-unresponsive eosinophilic esophagitis outcome62. We show that IL-13 stimulation of esophageal epithelial cells induces both STAT3 and STAT6 activation and CCl26 and CAPN14 mRNA expression. Blockade of STAT3 activity while inhibiting expression of pro-proliferative genes (ANO1 and TP63) did not impact IL-13 induction of core EoE pro-inflammatory gene expression including CCL26 and CAPN14. Conversely, we show that that STAT6 knockdown did not impact IL-13-induced esophageal epithelial proliferation. Collectively these studies support the emerging concept that IL-13-induced STAT3 signal pathways regulate esophageal epithelial proliferative response whereas IL-13-induced STAT6 signals regulate esophageal pro-inflammatory response. In support of this concept, treatment with Dupliumab, a fully human monoclonal antibody targeting interleukin 4 receptor alpha (IL4Rα), improved both inflammatory response (esophageal eosinophilia) and esophageal remodeling (BZH) in adults65, 66.
To gain insight into the IL-13-STAT3-dependent regulation of esophageal epithelial proliferation, we performed comparative in silico analyses and identified SFRP1, a putative STAT3 target and a soluble modulator of the Wnt signaling pathway, as a common DEG amongst our three RNAseq datasets. SFRP1 is a secreted glycoprotein that is member of the secreted frizzled-related receptor family (1–5) which act as soluble modulators of Wnt signaling. In the canonical Wnt/ β-catenin pathway, Wnt binds to Wnt receptor, Frizzled (Fzd), leading to inactivation of the β-catenin destruction complex and dissociation of β-catenin enabling b-catenin to traffic to the nucleus and induce transcription of target proliferation genes such as c-myc and Cyclin-D1 (CDK1) 51, 67–69. In the absence of Wnt, β-catenin is phosphorylated and bound to the destruction complex where it undergoes ubiquitin-mediated proteasomal degradation preventing transactivation of proliferative genes 51, 67–69. SFRP1 acts as a competitive antagonist binding to Wnt ligand through the netrin (NTR) domain and preventing Wnt binding to its receptor, Fzd. SFRP1 can also indirectly inhibit Wnt by binding to cytoplasmic β-catenin and preventing downstream gene transcription or by binding to the Fzd receptor through the cysteine-rich (CRD) domain and preventing binding of Wnt ligands to the receptor. These three antagonistic mechanisms of SFRP1 promote downstream proteasomal degradation of β-catenin and negative regulation of β-catenin-induced pro-survival and pro-proliferation transcriptional programs.
We show that IL-13 induced a rapid increase in SFRP1 mRNA in esophageal epithelial cells via a STAT3-dependent process. Furthermore, show that In vivo Dox-induced IL-13 overexpression resulted in an increase in SFRP1 protein expression within the esophageal epithelial compartment. To assess the role of SFRP1 in driving IL-13-induced esophageal epithelial proliferation, we employed a pharmacologic agonist and antagonist of SFRP1. Recombinant SFRP1 inhibited IL-13-induced esophageal epithelial proliferation and in contrast, SFRP1 inhibition enhanced IL-13-induced esophageal epithelial proliferation. Interestingly, Sinnberg et al. revealed an interaction between β-catenin and STAT3 which enhances resistance and tumor relapse in human melanomas and cancers with BRAF mutations. Importantly, they identified the β-catenin and STAT3 interactome as one of the important shared interactants70. Diminished SFRP1 has been associated with aberrant cell proliferation dynamics through hyperactivation of canonical Wnt signaling71. This supports our prediction for the involvement of the STAT3-SFRP1 signaling interactions, possibly through β-catenin and STAT3 in the regulation of esophageal epithelial proliferation. Given the canonical inhibitory roles of SFRP1 in Wnt-induced proliferative signaling, we speculate that SFRP1 expression is being induced in esophageal epithelial cells to serve a regulatory role in IL-13-induced esophageal epithelial proliferation. Interestingly, a study probing the epigenetics of SFRP1 shows that SFRP1 can undergo several genomic alterations depending on the tissue and disease context72. In a survey of genomic alterations across various cancers in the TCGA Pan-Cancer, the esophagus had the fifth highest degree of genomic alteration of SFRP1, where amplification was the most frequent genomic event, followed by mutation, and deep deletion72. Taken together, these data suggest that there is an important regulatory role of IL-13-STAT3-dependent SFRP1 in esophageal epithelial proliferation.
Utilizing the mouse Krt5-rtTA x tetO-IL-13Tg model we show that IL-13 induction led to increased esophageal epithelial thickening and remodeling, and esophageal epithelial proliferation. In Dox+ Krt5-rtTA x tetO-IL-13Tg mice, IL-13 expression resulted in an atopic dermatitis phenotype affecting the cutaneous tissues including ears, snout, and flank. Dox+ Krt5-rtTA x tetO-IL-13Tg mice demonstrated increased STAT3 expression and activation (pSTAT3-Y705) within the epithelial compartment of the esophagus, and this increase this was associated with esophageal epithelial proliferation and BZH. These studies suggest that IL-13-induction of STAT3 and the esophageal proliferative response contributes to esophageal epithelial remodeling such as BZH and DIS. In EPC2-ALI cells we show that degradation of STAT3 protein abrogated IL-13-induced esophageal epithelial proliferation and expression of EoE proliferation genes ANO1 and TP63. We have previously demonstrated that biopsies from patients with EoE had increased mRNA expression of ANO1 and the level of ANO1 expression correlates with BZH73. Furthermore, we showed that ANO1 played an important role in esophageal epithelial chloride transport and that loss of ANO1 dependent chloride transport reduced esophageal epithelial proliferation 73. ANO1 was required for phosphorylation of cyclin dependent kinase 2 (p-CDK2) and transition through G1/S phase cell cycle check point to permit esophageal epithelial proliferation. The functional relevance of BZH and ANO1 expression and relationship to human EoE is unclear. Shoda et al. identified an EoE endotype (EoE Endotype 3)74 that was associated with a fibrostenotic (rings, narrowing, strictures) phenotype and the highest degree of endoscopic and histological severity. Molecular analyses revealed that ANO1 was one of two discriminatory genes (ANO1 and UPK1A) to provide 98% PPV of EoE endotype 3 suggesting a link between ANO1 and the fibrostenotic and histologic phenotype74. We speculate that IL13-STAT3-proliferative response contributes to functional outcomes in EoE such as fibrostenosis not through directly driving esophageal dysfunction but rather indirectly through development of secondary associated histopathologic manifestations such as BZH and DIS formation.
We show that SFRP1 was expressed in a disease-associated esophageal epithelial population that was restricted to active disease. Notably, this population expressed canonical genes consistent with basal (KRT14, KRT19) / suprabasal (KRT4, KRT6B) epithelial cell phenotype suggesting that this population is undergoing cellular proliferation and basolateral-to-apical differentiation and maturation. Most notably, the SFRP1+ population differentially expressed key canonical EoE inflammatory genes including CCL2675, TNFAIP676 and ALOX15. This implicates this subset of suprabasal epithelial cells as the key cell population that promotes esophageal eosinophilia and the pro-inflammatory esophageal landscape. Given the canonical expression of key genes associated with epithelial remodeling (BZH and DIS) including ANO1 and SLC9A3 also raises an intriguing question of whether cellular proliferation within this basal / suprabasal epithelial cell population promotes esophageal epithelial remodeling (BZH and DIS) and that the inflammatory response is secondary to the esophageal epithelial remodeling and barrier dysfunction. Further transcriptional and functional analyses of the SFRP1+ esophageal epithelial population will be important in providing insight into whether EoE is a primary inflammation-driven disease or an esophageal epithelial proliferative disorder.
Identification of the IL-13-STAT3-SFRP1 signaling axis and its implications on BZH and EoE esophageal remodeling are indeed novel, however, there are limitations to our study, which we aim to address in follow-up studies. Here, we do not probe the correlation of STAT3 and SFRP1 protein expression and histopathological manifestation of EoE disease in patients (Eos/HPF, BZH and DIS score. Future studies are required to define the clinical relevance of SFRP1 as a surrogate marker for STAT3 aberration in EoE and to gain a deeper understanding of the IL-13-STAT3-SFRP1 signaling axis and its contribution to esophageal epithelial proliferation and remodeling in EoE. In summary, we identified a relationship between IL-13, STAT3 activation, and diminishment of SFRP1 towards the development of BZH in EoE. Mechanistically, we identified a previously undescribed role for IL-13-induced and STAT3-dependent SFRP1 in esophageal epithelial proliferation and provide data to indicate that STAT3 is required for IL-13 induced proliferation and has a role in the regulation of SFRP1. Collectively, these studies provide a rationale for the therapeutic use of STAT3 antagonists for the reduction in BZH and esophageal remodeling, and for the use of SFRP1 as a surrogate marker to indicate STAT3-mediated aberrations in epithelial proliferation signaling in EoE.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants AI138177, and AI140133; M-FARA; and the Mary H. Weiser Food Allergy Center supported (to S.P.H.).
Footnotes
Conflict of Interests: SPH receives research grant support from Regeneron Pharmaceuticals. MC: Consultant: Regeneron, Allakos, Adare/Ellodi, Shire/Takeda, AstraZeneca, Sanofi, Bristol Myers Squibb, Phathom. Research funding: Regeneron, Allakos, Shire/Takeda, AstraZeneca, Adare/Ellodi, Danone. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
REFERENCES
- 1.Aceves SS, Ackerman SJ. Relationships between eosinophilic inflammation, tissue remodeling, and fibrosis in eosinophilic esophagitis. Immunol Allergy Clin North Am 2009; 29:197–211, xiii-xiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collins MH. Histopathology of eosinophilic esophagitis. Dig Dis 2014; 32:68–73. [DOI] [PubMed] [Google Scholar]
- 3.Dellon ES. Cost-effective care in eosinophilic esophagitis. Ann Allergy Asthma Immunol 2019; 123:166–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furuta GT, Katzka DA. Eosinophilic Esophagitis. N Engl J Med 2015; 373:1640–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonsalves NP, Aceves SS. Diagnosis and treatment of eosinophilic esophagitis. Journal of Allergy and Clinical Immunology 2020; 145:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Warners MJ, van Rhijn BD, Verheij J, Smout A, Bredenoord AJ. Disease activity in eosinophilic esophagitis is associated with impaired esophageal barrier integrity. Am J Physiol Gastrointest Liver Physiol 2017; 313:G230–g8. [DOI] [PubMed] [Google Scholar]
- 7.Blanchard C, Mingler MK, Vicario M, Abonia JP, Wu YY, Lu TX, et al. IL-13 involvement in eosinophilic esophagitis: transcriptome analysis and reversibility with glucocorticoids. J Allergy Clin Immunol 2007; 120:1292–300. [DOI] [PubMed] [Google Scholar]
- 8.Cheng E, Souza RF, Spechler SJ. Tissue remodeling in eosinophilic esophagitis. Am J Physiol Gastrointest Liver Physiol 2012; 303:G1175–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanoni S, Zeng C, Marella S, Uddin J, Wu D, Arora K, et al. Identification of anoctamin 1 (ANO1) as a key driver of esophageal epithelial proliferation in eosinophilic esophagitis. J Allergy Clin Immunol 2020; 145:239–54 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aceves SS. Remodeling and fibrosis in chronic eosinophil inflammation. Dig Dis 2014; 32:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muir AB, Wang JX, Nakagawa H. Epithelial-stromal crosstalk and fibrosis in eosinophilic esophagitis. J Gastroenterol 2019; 54:10–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mulder DJ, Justinich CJ. Understanding eosinophilic esophagitis: the cellular and molecular mechanisms of an emerging disease. Mucosal Immunol 2011; 4:139–47. [DOI] [PubMed] [Google Scholar]
- 13.Warners MJ, Vlieg-Boerstra BJ, Verheij J, van Hamersveld PHP, van Rhijn BD, Van Ampting MTJ, et al. Esophageal and Small Intestinal Mucosal Integrity in Eosinophilic Esophagitis and Response to an Elemental Diet. Am J Gastroenterol 2017; 112:1061–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akei HS, Mishra A, Blanchard C, Rothenberg ME. Epicutaneous Antigen Exposure Primes for Experimental Eosinophilic Esophagitis in Mice. Gastroenterology 2005; 129:985–94. [DOI] [PubMed] [Google Scholar]
- 15.Collins MH. Histopathologic features of eosinophilic esophagitis. Gastrointest Endosc Clin N Am 2008; 18:59–71; viii-ix. [DOI] [PubMed] [Google Scholar]
- 16.Doherty TA, Baum R, Newbury RO, Yang T, Dohil R, Aquino M, et al. Group 2 innate lymphocytes (ILC2) are enriched in active eosinophilic esophagitis. J Allergy Clin Immunol 2015; 136:792–4.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bullock JZ, Villanueva JM, Blanchard C, Filipovich AH, Putnam PE, Collins MH, et al. Interplay of adaptive th2 immunity with eotaxin-3/c-C chemokine receptor 3 in eosinophilic esophagitis. J Pediatr Gastroenterol Nutr 2007; 45:22–31. [DOI] [PubMed] [Google Scholar]
- 18.Cianferoni A, Ruffner MA, Guzek R, Guan S, Brown-Whitehorn T, Muir A, et al. Elevated expression of activated T(H)2 cells and milk-specific T(H)2 cells in milk-induced eosinophilic esophagitis. Ann Allergy Asthma Immunol 2018; 120:177–83.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wen T, Aronow BJ, Rochman Y, Rochman M, Kc K, Dexheimer PJ, et al. Single-cell RNA sequencing identifies inflammatory tissue T cells in eosinophilic esophagitis. J Clin Invest 2019; 129:2014–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D’Mello RJ, Caldwell JM, Azouz NP, Wen T, Sherrill JD, Hogan SP, et al. LRRC31 is induced by IL-13 and regulates kallikrein expression and barrier function in the esophageal epithelium. Mucosal Immunol 2016; 9:744–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zuo L, Fulkerson PC, Finkelman FD, Mingler M, Fischetti CA, Blanchard C, et al. IL-13 induces esophageal remodeling and gene expression by an eosinophil-independent, IL-13R alpha 2-inhibited pathway. J Immunol 2010; 185:660–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirano I, Collins MH, Assouline-Dayan Y, Evans L, Gupta S, Schoepfer AM, et al. RPC4046, a Monoclonal Antibody Against IL13, Reduces Histologic and Endoscopic Activity in Patients With Eosinophilic Esophagitis. Gastroenterology 2019; 156:592–603.e10. [DOI] [PubMed] [Google Scholar]
- 23.Dellon ES, Collins MH, Rothenberg ME, Assouline-Dayan Y, Evans L, Gupta S, et al. Long-term Efficacy and Tolerability of RPC4046 in an Open-Label Extension Trial of Patients With Eosinophilic Esophagitis. Clin Gastroenterol Hepatol 2021; 19:473–83.e17. [DOI] [PubMed] [Google Scholar]
- 24.Hirano I, Dellon ES, Hamilton JD, Collins MH, Peterson K, Chehade M, et al. Efficacy of Dupilumab in a Phase 2 Randomized Trial of Adults With Active Eosinophilic Esophagitis. Gastroenterology 2020; 158:111–22.e10. [DOI] [PubMed] [Google Scholar]
- 25.Dellon ES, Rothenberg ME, Collins MH, Hirano I, Chehade M, Bredenoord AJ, et al. A Phase 3, Randomized, 3-Part Study to Investigate the Efficacy and Safety of Dupilumab in Adult and Adolescent Patients With Eosinophilic Esophagitis: Results From Part A. American College of Gastroenterology 2020 Annual Meeting. Virtual, 2020. [Google Scholar]
- 26.Sherrill JD, Kiran KC, Blanchard C, Stucke EM, Kemme KA, Collins MH, et al. Analysis and expansion of the eosinophilic esophagitis transcriptome by RNA sequencing. Genes Immun 2014; 15:361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhattacharjee A, Shukla M, Yakubenko VP, Mulya A, Kundu S, Cathcart MK. IL-4 and IL-13 employ discrete signaling pathways for target gene expression in alternatively activated monocytes/macrophages. Free Radic Biol Med 2013; 54:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gann PH, Deaton RJ, McMahon N, Collins MH, Dellon ES, Hirano I, et al. An anti-IL-13 antibody reverses epithelial-mesenchymal transition biomarkers in eosinophilic esophagitis: Phase 2 trial results. J Allergy Clin Immunol 2020; 146:367–76 e3. [DOI] [PubMed] [Google Scholar]
- 29.Gour N, Wills-Karp M. IL-4 and IL-13 signaling in allergic airway disease. Cytokine 2015; 75:68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rothenberg ME, Wen T, Greenberg A, Alpan O, Enav B, Hirano I, et al. Intravenous anti-IL-13 mAb QAX576 for the treatment of eosinophilic esophagitis. J Allergy Clin Immunol 2015; 135:500–7. [DOI] [PubMed] [Google Scholar]
- 31.Roy S, Liu HY, Jaeson MI, Deimel LP, Ranasinghe C. Unique IL-13Ralpha2/STAT3 mediated IL-13 regulation detected in lung conventional dendritic cells, 24 h post viral vector vaccination. Sci Rep 2020; 10:1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidt H, Braubach P, Schilpp C, Lochbaum R, Neuland K, Thompson K, et al. IL-13 Impairs Tight Junctions in Airway Epithelia. Int J Mol Sci 2019; 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller DE, Forney C, Rochman M, Cranert S, Habel J, Rymer J, et al. Genetic, Inflammatory, and Epithelial Cell Differentiation Factors Control Expression of Human Calpain-14. G3 (Bethesda) 2019; 9:729–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mishra A, Rothenberg ME. Intratracheal IL-13 induces eosinophilic esophagitis by an IL-5, eotaxin-1, and STAT6-dependent mechanism. Gastroenterology 2003; 125:1419–27. [DOI] [PubMed] [Google Scholar]
- 35.Miloux B, Laurent P, Bonnin O, Lupker J, Caput D, Vita N, et al. Cloning of the human IL-13R alpha1 chain and reconstitution with the IL4R alpha of a functional IL-4/IL-13 receptor complex. FEBS Lett 1997; 401:163–6. [DOI] [PubMed] [Google Scholar]
- 36.Schnyder B, Lugli S, Feng N, Etter H, Lutz RA, Ryffel B, et al. Interleukin-4 (IL-4) and IL-13 bind to a shared heterodimeric complex on endothelial cells mediating vascular cell adhesion molecule-1 induction in the absence of the common gamma chain. Blood 1996; 87:4286–95. [PubMed] [Google Scholar]
- 37.Johnston PA, Grandis JR. STAT3 signaling: anticancer strategies and challenges. Mol Interv 2011; 11:18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Umeshita-Suyama R, Sugimoto R, Akaiwa M, Arima K, Yu B, Wada M, et al. Characterization of IL-4 and IL-13 signals dependent on the human IL-13 receptor alpha chain 1: redundancy of requirement of tyrosine residue for STAT3 activation. Int Immunol 2000; 12:1499–509. [DOI] [PubMed] [Google Scholar]
- 39.Dellon ES, Liacouras CA, Molina-Infante J, Furuta GT, Spergel JM, Zevit N, et al. Updated International Consensus Diagnostic Criteria for Eosinophilic Esophagitis: Proceedings of the AGREE Conference. Gastroenterology 2018; 155:1022–33 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spergel JM, Dellon ES, Liacouras CA, Hirano I, Molina-Infante J, Bredenoord AJ, et al. Summary of the updated international consensus diagnostic criteria for eosinophilic esophagitis: AGREE conference. Ann Allergy Asthma Immunol 2018; 121:281–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeng C, Vanoni S, Wu D, Caldwell JM, Wheeler JC, Arora K, et al. Solute carrier family 9, subfamily A, member 3 (SLC9A3)/sodium-hydrogen exchanger member 3 (NHE3) dysregulation and dilated intercellular spaces in patients with eosinophilic esophagitis. J Allergy Clin Immunol 2018; 142:1843–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferrer-Torres D, Wu JH, Zhang CJ, Hammer MA, Dame MK, Wu A, et al. Mapping the adult human esophagus in vivo and in vitro. Development 2022; 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu TX, Sherrill JD, Wen T, Plassard AJ, Besse JA, Abonia JP, et al. MicroRNA signature in patients with eosinophilic esophagitis, reversibility with glucocorticoids, and assessment as disease biomarkers. J Allergy Clin Immunol 2012; 129:1064–75 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leisengang S, Heilen LB, Klymiuk MC, Nurnberger F, Ott D, Wolf-Hofmann K, et al. Neuroinflammation in Primary Cultures of the Rat Spinal Dorsal Horn Is Attenuated in the Presence of Adipose Tissue-Derived Medicinal Signalling Cells (AdMSCs) in a Co-cultivation Model. Mol Neurobiol 2022; 59:475–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nurnberger F, Ott D, Classen R, Rummel C, Roth J, Leisengang S. Systemic Lipopolysaccharide Challenge Induces Inflammatory Changes in Rat Dorsal Root Ganglia: An Ex Vivo Study. Int J Mol Sci 2022; 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qiu Y, Liu P, Ma X, Ma X, Zhu L, Lin Y, et al. TRIM50 acts as a novel Src suppressor and inhibits ovarian cancer progression. Biochim Biophys Acta Mol Cell Res 2019; 1866:1412–20. [DOI] [PubMed] [Google Scholar]
- 47.Srivastava M, Duan G, Kershaw NJ, Athanasopoulos V, Yeo JH, Ose T, et al. Roquin binds microRNA-146a and Argonaute2 to regulate microRNA homeostasis. Nat Commun 2015; 6:6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Diamond I, Owolabi T, Marco M, Lam C, Glick A. Conditional gene expression in the epidermis of transgenic mice using the tetracycline-regulated transactivators tTA and rTA linked to the keratin 5 promoter. J Invest Dermatol 2000; 115:788–94. [DOI] [PubMed] [Google Scholar]
- 49.Zheng T, Oh MH, Oh SY, Schroeder JT, Glick AB, Zhu Z. Transgenic expression of interleukin-13 in the skin induces a pruritic dermatitis and skin remodeling. J Invest Dermatol 2009; 129:742–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang M, Ku WY, Zhou Z, Dellon ES, Falk GW, Nakagawa H, et al. BMP-driven NRF2 activation in esophageal basal cell differentiation and eosinophilic esophagitis. J Clin Invest 2015; 125:1557–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bai L, Zhou H, Xu R, Zhao Y, Chinnaswamy K, McEachern D, et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019; 36:498–511 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rochman M, Wen T, Kotliar M, Dexheimer PJ, Ben-Baruch Morgenstern N, Caldwell JM, et al. Single-cell RNA-Seq of human esophageal epithelium in homeostasis and allergic inflammation. JCI Insight 2022; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sherrill JD, Kiran K, Blanchard C, Stucke EM, Kemme KA, Collins MH, et al. Analysis and expansion of the eosinophilic esophagitis transcriptome by RNA sequencing. Genes Immun 2014; 15:361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khokhar D, Marella S, Idelman G, Chang JW, Chehade M, Hogan SP. Eosinophilic esophagitis: Immune mechanisms and therapeutic targets. Clin Exp Allergy 2022; 52:1142–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hebenstreit D, Luft P, Schmiedlechner A, Duschl A, Horejs-Hoeck J. SOCS-1 and SOCS-3 inhibit IL-4 and IL-13 induced activation of Eotaxin-3/CCL26 gene expression in HEK293 cells. Mol Immunol 2005; 42:295–303. [DOI] [PubMed] [Google Scholar]
- 56.Furue M Regulation of Skin Barrier Function via Competition between AHR Axis versus IL-13/IL-4‒JAK‒STAT6/STAT3 Axis: Pathogenic and Therapeutic Implications in Atopic Dermatitis. J Clin Med 2020; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Niranjan R, Rayapudi M, Mishra A, Dutt P, Dynda S, Mishra A. Pathogenesis of allergen-induced eosinophilic esophagitis is independent of interleukin (IL)-13. Immunol Cell Biol 2013; 91:408–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cheng E, Zhang X, Wilson KS, Wang DH, Park JY, Huo X, et al. JAK-STAT6 Pathway Inhibitors Block Eotaxin-3 Secretion by Epithelial Cells and Fibroblasts from Esophageal Eosinophilia Patients: Promising Agents to Improve Inflammation and Prevent Fibrosis in EoE. PLoS One 2016; 11:e0157376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Avlas S, Shani G, Rhone N, Itan M, Dolitzky A, Hazut I, et al. Epithelial cell-expressed type II IL-4 receptor mediates eosinophilic esophagitis. Allergy 2023; 78:464–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang X, Wrzeszczynska MH, Horvath CM, Darnell JE Jr. Interacting regions in Stat3 and c-Jun that participate in cooperative transcriptional activation. Mol Cell Biol 1999; 19:7138–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ezoe S, Matsumura I, Gale K, Satoh Y, Ishikawa J, Mizuki M, et al. GATA transcription factors inhibit cytokine-dependent growth and survival of a hematopoietic cell line through the inhibition of STAT3 activity. J Biol Chem 2005; 280:13163–70. [DOI] [PubMed] [Google Scholar]
- 62.Mougey EB, Williams A, Coyne AJK, Gutierrez-Junquera C, Fernandez-Fernandez S, Cilleruelo ML, et al. CYP2C19 and STAT6 Variants Influence the Outcome of Proton Pump Inhibitor Therapy in Pediatric Eosinophilic Esophagitis. J Pediatr Gastroenterol Nutr 2019; 69:581–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rothenberg ME, Spergel JM, Sherrill JD, Annaiah K, Martin LJ, Cianferoni A, et al. Common variants at 5q22 associate with pediatric eosinophilic esophagitis. Nat Genet 2010; 42:289–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sleiman PM, Wang ML, Cianferoni A, Aceves S, Gonsalves N, Nadeau K, et al. GWAS identifies four novel eosinophilic esophagitis loci. Nat Commun 2014; 5:5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cao H, Zhang J, Liu H, Wan L, Zhang H, Huang Q, et al. IL-13/STAT6 signaling plays a critical role in the epithelial-mesenchymal transition of colorectal cancer cells. Oncotarget 2016; 7:61183–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hirano I, Dellon ES, Hamilton JD, Collins MH, Peterson K, Chehade M, et al. Efficacy of Dupilumab in a Phase 2 Randomized Trial of Adults With Active Eosinophilic Esophagitis. Gastroenterology 2020; 158:111–22 e10. [DOI] [PubMed] [Google Scholar]
- 67.Dong J, Cheng XD, Zhang WD, Qin JJ. Recent Update on Development of Small-Molecule STAT3 Inhibitors for Cancer Therapy: From Phosphorylation Inhibition to Protein Degradation. J Med Chem 2021; 64:8884–915. [DOI] [PubMed] [Google Scholar]
- 68.Lavergne E, Hendaoui I, Coulouarn C, Ribault C, Leseur J, Eliat PA, et al. Blocking Wnt signaling by SFRP-like molecules inhibits in vivo cell proliferation and tumor growth in cells carrying active beta-catenin. Oncogene 2011; 30:423–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang H, Sun D, Qiu J, Yao L. SFRP1 inhibited the epithelial ovarian cancer through inhibiting Wnt/beta-catenin signaling. Acta Biochim Pol 2019; 66:393–400. [DOI] [PubMed] [Google Scholar]
- 70.Sinnberg T, Makino E, Krueger MA, Velic A, Macek B, Rothbauer U, et al. A Nexus Consisting of Beta-Catenin and Stat3 Attenuates BRAF Inhibitor Efficacy and Mediates Acquired Resistance to Vemurafenib. EBioMedicine 2016; 8:132–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang Z, Li R, He Y, Huang S. Effects of secreted frizzled-related protein 1 on proliferation, migration, invasion, and apoptosis of colorectal cancer cells. Cancer Cell Int 2018; 18:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baharudin R, Tieng FYF, Lee LH, Ab Mutalib NS. Epigenetics of SFRP1: The Dual Roles in Human Cancers. Cancers (Basel) 2020; 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vanoni S, Zeng C, Marella S, Uddin J, Wu D, Arora K, et al. Identification of anoctamin 1 (ANO1) as a key driver of esophageal epithelial proliferation in eosinophilic esophagitis. J Allergy Clin Immunol 2020; 145:239–54.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shoda T, Wen T, Aceves SS, Abonia JP, Atkins D, Bonis PA, et al. Eosinophilic oesophagitis endotype classification by molecular, clinical, and histopathological analyses: a cross-sectional study. Lancet Gastroenterol Hepatol 2018; 3:477–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bhattacharya B, Carlsten J, Sabo E, Kethu S, Meitner P, Tavares R, et al. Increased expression of eotaxin-3 distinguishes between eosinophilic esophagitis and gastroesophageal reflux disease. Hum Pathol 2007; 38:1744–53. [DOI] [PubMed] [Google Scholar]
- 76.Matoso A, Mukkada VA, Lu S, Monahan R, Cleveland K, Noble L, et al. Expression microarray analysis identifies novel epithelial-derived protein markers in eosinophilic esophagitis. Mod Pathol 2013; 26:665–76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.