


Abstract
The TΨC stem and loop (TSL) of tRNA contains highly conserved nucleoside modifications, m5C49, T54, Ψ55 and m1A58. U54 is methylated to m5U (T) by m5U54 methyltransferase (RUMT); A58 is methylated to m1A by m1A58 tRNA methyltransferase (RAMT). RUMT recognizes and methylates a minimal TSL heptadecamer and RAMT has previously been reported to recognize and methylate the 3′-half of the tRNA molecule. We report that RAMT can recognize and methylate a TSL heptadecamer. To better understand the sensitivity of RAMT and RUMT to TSL conformation, we have designed and synthesized variously modified TSL constructs with altered local conformations and stabilities. TSLs were synthesized with natural modifications (T54 and Ψ55), naturally occurring modifications at unnatural positions (m5C60), altered sugar puckers (dU54 and/or dU55) or with disrupted U-turn interactions (m1Ψ55 or m1m3Ψ55). The unmodified heptadecamer TSL was a substrate of both RAMT and RUMT. The presence of T54 increased thermal stability of the TSL and dramatically reduced RAMT activity toward the substrate. Local conformation around U54 was found to be an important determinant for the activities of both RAMT and RUMT.
INTRODUCTION
Transfer RNA contains a large number of modified nucleosides. Four site-specific modifications, m5C49, T54, Ψ55 and m1A58, occur so prevalently in the TΨC stem and loop domain (TSL) of tRNAs that this part of the molecule is the most consistently modified (1). T is found at nucleoside position 54 in over 60% of all tRNA sequences and Ψ is found at position 55 in more than 90% of all tRNAs (Fig. 1) (2). The methylation of A58 to m1A58 in the T loop occurs in ~25% of all eukaryotic tRNAs. In addition, the tRNAs of many organisms are methylated at C49 to produce m5C49 in the stem of the TSL. The effects of specific TSL modifications on the enzymatic formation of other modified nucleosides are not known.
Figure 1.

Structures of TSL domains. (A) Native TSL sequence derived from yeast tRNAPhe. Shaded boxes indicate nucleosides modified by RAMT and RUMT. (B) Unmodified TSL.
Biosynthesis of a number of modifications has been shown to require the entire architecture of the tRNA (3). However, the modification enzymes responsible for the synthesis of T54, Ψ55 and m1A58 in the TSL loop do not require the entire tRNA for substrate recognition and synthesis of the modifications (4–7). The unmodified TSL heptadecamer, composed of a 5 bp stem and seven-membered loop, is modified by both m5U54 tRNA methyltransferase [S-adenosyl-l-methionine:tRNA (uracil-5)-methyltransferase, EC 2.1.1.35] (RUMT), which produces T54 (4,5), and Ψ55 tRNA synthase, which uniquely converts U55 to Ψ55 (6).
The recognition elements required for RUMT appear to reside in the TSL, yet recognition of the TSL by Escherichia coli RUMT is not highly sequence dependent (6,8). Eukaryotic m1A58 tRNA methyltransferase (RAMT) (EC 2.1.1.36) recognizes the 3′-half of the tRNA molecule for methylation of A58 to m1A58 (3,7), yet a heptadecamer substrate corresponding to that for T54 and Ψ55 synthesis has not been reported for m1A58 tRNA methyltransferase.
We report that RAMT, purified from Tetrahymena pyriformis, recognizes and methylates A58 in an unmodified heptadecamer RNA corresponding to the TSL. To better understand the recognition elements of RUMT and RAMT within the TSL, we have assayed the ability of E.coli RUMT to methylate U54 and T.pyriformis RAMT to methylate A58 in yeast tRNAPhe TSL loops constructed with local conformational perturbations. Local conformation around nucleoside 54, across the loop from A58, affected both RUMT and RAMT activity.
MATERIALS AND METHODS
Oligonucleotide synthesis and purification
Unmodified, modified and stable isotope-labeled heptadecamer RNAs were synthesized with the base sequence corresponding to the yeast tRNAPhe TSL (Fig. 1). Synthesis was accomplished with standard RNA phosphoramidite chemistry (9) on an Applied Biosystems Model 394 automated synthesizer. Modified and stable isotope-labeled nucleoside phosphoramidites [m5U (T), Ψ, m1Ψ, m1m3Ψ and m5C, 15N1,3-U and 15NH2-C] were prepared as previously described (10–12). Standard ribonucleoside phosphoramidites and dU phosphoramidite were purchased from Glen Research and ChemGenes. TSLs were purified by ion exchange HPLC (10). Successful incorporation of the modified nucleosides was verified by quantitative nucleoside composition analysis (13). Concentrations were determined spectrophotometrically at 260 nm with an extinction coefficient that had been calculated from the nearest neighbor approximation (14).
Preparation and assay of E.coli m5U54 tRNA methyltransferase (RUMT) activity
RUMT was extracted from strain GRB822 carrying the cloned trmA gene. This strain was a gift from Dr Glenn Björk (University of Umea, Sweden). The trmA gene was cloned under control of the inducible lac promoter, DH5α. Enzyme was prepared as described previously (15).
Enzymatic activity was confirmed using unfractionated methyl-deficient E.coli tRNA (16) and T-deficient E.coli tRNA (17) as substrates. Methylations of the TSL were conducted at 15°C with [methyl-3H]S-adenosyl-l-methionine (SAM) (~15 Ci/mmol; New England Nuclear). Assay mixtures (75 µl) contained either 3 µM tRNA or 24 µM TSL, 10 µM [methyl-3H]SAM, 50 µM unlabeled SAM (iodine salt) (Sigma), 6.25 µl of 10× methylation buffer (500 mM Tricine, pH 8.4, 50 mM DTT, 20 mM MgCl2, 10 mM EDTA, 400 mM NH4Cl, 200 mM spermidine) and 2 µl RNase inhibitor (Recombinant RNasin RNase Inhibitor; Promega). Reactions were initiated by the addition of 5 µl of enzyme preparation. Activity was determined with a filter binding assay as described previously (5). Radioactivity was counted for 5 min and non-specific binding was corrected by subtracting the results of parallel experiments in the absence of substrate. The data, analyzed by both non-linear regression and Michelis–Menten methods, produced values of Km and Vmax with errors of ~15 and 10%, respectively.
Preparation and assay of T.pyriformis m1A58 tRNA methyltransferase (RAMT) activity
m1A58 tRNA methyltransferase (RAMT) was purified from T.pyriformis (strain GL, amicronuclear) (18). Cells were grown to log phase at 26°C with aeration in a complex medium (0.7% proteoso peptone, 0.7% yeast extract, 1 mM MgSO4, 50 µM CaCl2, 100 µM Fe2+ citrate, pH 6.5). Purification was accomplished at 4°C. Cells (40 g) were harvested by centrifugation at 1000 g, washed and resuspended in an equal volume of buffer A (50 mM Tris–HCl, pH 8.0, 0.5 mM EDTA, 10 mM 2-mercaptoethanol, 0.5 mM phenylmethylsulfonyl fluoride, 10 µM antipain, 20% w/v glycerol) containing 80 mM NaCl. The cells were disrupted by ultrasonication (three times for 15 s each) and cell debris removed by centrifugation at 10 000 g for 50 min. The resulting supernatant (S-10) was centrifuged at 100 000 g for 90 min. S-100 supernatant was applied to a DEAE–cellulose (DE-52; Whatman) column (150 ml) that had been equilibrated with buffer A containing 80 mM NaCl. The column was washed with 300 ml of the equilibration buffer and 200 ml of buffer A containing 100 mM NaCl. Enzyme activity eluted with buffer A containing 250 mM NaCl. UV absorbing peaks (at 280 nm) were collected and dialyzed against 2 l of buffer B (80 mM potassium phosphate, pH 6.5, 0.5 mM EDTA, 10 mM 2-mercaptoethanol, 0.5 mM phenylmethylsulfonyl fluoride, 20% w/v glycerol). The eluted enzyme was subjected to chromatography on a P11 phosphocellulose (Whatman) column (100 ml) which had been equilibrated with buffer B. The enzyme was eluted with a 300 ml linear gradient of 0.08–0.5 M potassium phosphate. Fractions with m1A58 methyltransferase activity (0.3–0.35 M potassium phosphate) were collected and dialyzed against 1.5 l of buffer A containing 50 mM KCl. The dialyzed enzyme preparation was applied to a tRNA–Sepharose column (8 ml) that had been equilibrated with buffer A plus 50 mM KCl. The column was washed with 80 ml of equilibration buffer and the protein was eluted with 40 ml of a linear gradient of 0.1–0.5 M KCl in buffer A. The enzyme eluted at 0.35–0.40 M KCl. The specific activity of the m1A methylase was enriched in the purified fraction 390-fold over the whole cell cytosol fraction and contained no other specific tRNA methylase activities, as verified by modified nucleotide analysis. Fractions containing RAMT activity were concentrated in an Amicon Centricon 10 microconcentrator and used for methylation assays.
The RAMT assay reaction mixture contained 0.15 M NaCl, 30 mM Tris–HCl pH 7.5, 3 mM DTT, 0.5 mM EDTA, 10 mM putrescine, 40 µM [14C]SAM (54 mCi/mmol), 0.05–10 µM substrate and 20 µl of enzyme in a total volume of 150 µl. Before being used for the assay, the tRNA and TSL substrates were denatured by heating at 42°C for 3 min and allowed to renature at room temperature. Both E.coli tRNAPhe and tRNAVal (Sigma) were methylated at 30°C under these assay conditions. Methylation of the TSLs was conducted at 25°C and, in addition to the above, 5 mM MgCl2 was added to the assay mixtures. Methylation was assessed with a filter binding assay. Aliquots, taken at 15, 30, 60 and 90 min, were placed on DE-81 paper (Whatman) and the filters washed with sodium phosphate, pH 6.5. The disks were dried and counted in a scintillation counter. To verify the site of methylation on the base, methylated TSLs were hydrolyzed with nuclease P1 and bacterial alkaline phosphatase. The resulting nucleosides were separated by HPLC on an Ultrasphere ODS column and the elution of radioactive methylated nucleosides was compared to that of nucleoside standards. Kinetics of the reaction (Km and Vmax) were determined with E.coli tRNAPhe and TSLs. Km and Vmax were determined using both non-linear regression and Michelis–Menten analyses and had errors of 15 and 10%, respectively.
Determination of TSL thermal stability
TSL samples were diluted in a pH 7.2 buffer (10 mM sodium phosphate, pH 7.2, 100 mM NaCl, 0.1 mM EDTA) to concentrations ranging from 0.40 to 82 µM. Thermal denaturation was monitored by UV absorbance using a Cary 3 spectrophotometer with 2 and 10 mm cuvettes to maintain adequate optical response. The absorbance at 260 nm was recorded every 0.3°C from 5 to 90°C at a rate of 1.0°C/min. Each TSL was heated and cooled at least four times. No hysteresis was observed. The melting curves for denaturation and renaturation were treated similarly for data analysis. Thermodynamic parameters were calculated with a Van’t Hoff analysis of the data as described by Marky and Breslauer (19) using Origin software. The Tm of each TSL was concentration independent; thus, unimolecular denaturations were observed. Because monophasic melting curves were observed, a two-state transition was assumed.
Nuclear magnetic resonance (NMR) spectroscopy
NMR spectra were collected on a Bruker DRX 500 NMR Spectrometer and processed using Felix97 (Biosym/MSI, San Diego, CA). TSLs were prepared for spectroscopy by extensive dialysis with the NMR buffer (10 mM sodium phosphate, 0.1 mM EDTA, 94% H2O/6% D2O, pH 6.0) using Amicon Centricon 3 concentrators. All spectra were collected at 1°C, with a spectral width of 12 019 Hz and 8192 points. The number of scans varied from 1024 to 29 696 and was dependent on the sample concentration. Water suppression was accomplished using the Watergate pulse sequence (20). Further elimination of the residual water was achieved post-acquisition using a time domain convolution routine (21). The spectra were apodized using an exponential multiplication with a line broadening of 1 Hz and were zero filled to 8192 real points. Baselines were flattened using a fifth degree polynomial. Imino and amino proton resonances were assigned using standard methods (22) and site-selected incorporation of 15N1,3-U and 15NH2-C (10). Because of sample limitations, we were unable to determine the one-dimensional NMR spectra for TSLs T54Ψ55, dU55, dU54dU55 and m1Ψ55.
RESULTS
Design of TSLs
Escherichia coli RUMT modification of U54 in the unmodified TSL is not highly dependent on the nucleoside sequence of the TSL (6,8). Likewise, RAMT activity is probably not highly dependent on nucleoside sequence because A58 N1 methylation is frequently found in TSLs of different sequences. To better understand the substrate requirements for these modifying enzymes, we designed and synthesized TSLs with non-natural and naturally occurring modified nucleosides based on the sequence of the yeast tRNAPhe TSL (Fig. 1A). TSLs were designed to have altered loop conformations without perturbation of the TSL stem. Table 1 lists site-specific nucleoside modifications of TSL domain constructs with the expected structural contributions of the modified nucleosides.
Table 1. Contributions of nucleoside substitutions and thermal stabilitiesa of TSL constructs.
| TSL | Substitution contribution | Tm (°C) | ΔH (kcal/mol) | ΔS (cal/mol) | ΔG37 (kcal/mol) |
|---|---|---|---|---|---|
| Unmodified | Precursor | 54.5 ± 0.3 | –49.0 ± 1.0 | –149.0 ± 4.0 | –2.6 ± 0.1 |
| T54 | Natural modification | 56.7 ± 0.4 | –52.0 ± 2.0 | –157.0 ± 7.0 | –3.1 ± 0.1 |
| T54 m5C60b | Natural modification in unnatural position | 57.2 ± 0.2 | –49.0 ± 1.0 | –149.0 ± 3.0 | –3.0 ± 0.1 |
| T54Ψ55 | Natural modifications | NDc | ND | ND | ND |
| dU54 | Sugar pucker in C2′-endo conformation | 53.2 ± 0.5 | –48.0 ± 2.0 | –146.0 ± 8.0 | –2.4 ± 0.1 |
| dU55 | Sugar pucker prefers C2′-endo | 53.7 ± 0.6 | –49.0 ± 2.0 | –151.0 ± 6.0 | –2.5 ± 0.1 |
| dU54dU55 | Sugar pucker prefers C2′-endo | 52.9 ± 0.4 | –48.0 ± 2.0 | –146.0 ± 6.0 | –2.3 ± 0.2 |
| Ψ55 | Natural modification | 53.8 ± 0.8 | –49.0 ± 5.0 | –149.0 ± 20.0 | –2.5 ± 0.3 |
| m1Ψ55d | Negates hydrogen bonding at N1 position | 53.8 ± 0.3 | –47.0 ± 1.0 | –143.0 ± 3.0 | –2.4 ± 0.1 |
| m1m3Ψ55 | Negates hydrogen bonding capabilities | 53.2 ± 0.8 | –48.0 ± 3.0 | –148.0 ± 8.0 | –2.4 ± 0.2 |
aValues are averages based on four different RNA concentrations for most oligomers. Errors are standard deviations derived from multiple iterations of denaturation and renaturation. ΔG values were calculated at 37°C.
bValues based on two different RNA concentrations.
cND, not determined.
dValues based on one RNA concentration.
The unmodified TSL was used to confirm RUMT activity. To assay the sensitivity of RUMT to the local loop conformation of the substrate, deoxy forms of uridine were positioned at U54 and U55. While ribonucleosides tend to adopt a C3′-endo conformation, deoxynucleosides exist predominantly in the C2′-endo conformation (23). Introduction of dU54 and dU55 therefore may locally perturb both the U54 nucleoside, which RUMT methylates, and the U55 nucleoside involved in the TΨC domain U-turn.
T54 was incorporated into the unmodified TSL to mimic the natural modification and allow an investigation of the ability of RAMT to methylate A58 in the presence of a natural modification. Likewise, Ψ55 was incorporated into the unmodified TSL to mimic the natural modification and allow an investigation of the ability of both RAMT and RUMT to methylate their respective nucleoside substrates in the presence of this ubiquitous, natural modification. The methylated Ψ-substituted TSLs m1Ψ55 and m1m3Ψ55 were used to examine enzyme sensitivity to a conformational perturbation at the TSL U-turn. The amino protons of the loop nucleoside C60 within the T54-modified TSL interact with base paired stem nucleosides, particularly U50, in what could be a base triple (24). Therefore, C60 interactions are critical to the RNA loop conformation and substitution of m5C60 subtly alters these interactions (24). The modification m5C occurs naturally in tRNAs, but has never been found at position 60. Thus, the doubly modified TSL T54m5C60 allowed an examination of the sensitivity of RAMT to the natural modification T54 in the presence of a modification across the loop, m5C60, which altered loop conformation. The effects of TSL modifications (dU54, dU55, T54, m1Ψ55, m1m3Ψ55 and m5C60) on the stability, conformation and enzymatic activities of RAMT and RUMT were assessed.
Substrate activity for RAMT
Both E.coli tRNAPhe and tRNAVal were substrates for the T.pyriformis RAMT, as has been shown with other eukaryotic RAMTs (7,25–27). The enzyme was active with tRNAPhe as a substrate under the previously published conditions for RUMT (28), but in the absence of Mg2+ and with putresine substituting for spermidine. Methylation of A58 of E.coli tRNAPhe was characterized by a Km of 1.7 µM and a Vmax of 2.6 pmol/min. These kinetics compare favorably with those of a partially purified RAMT from Thermus flavus (Km 0.4–0.5 µM for E.coli tRNAGln2; 29), but are significantly poorer than that of a highly purified rat liver enzyme (Km 33 nM for E.coli tRNAGlu2; 25).
To determine if the heptadecamer TSL of yeast tRNAPhe could be a substrate for RAMT, the unmodified TSL was used as a substrate. Under assay conditions suitable for E.coli tRNAPhe, the unmodified TSL was not methylated by T.pyriformis RAMT, however, after the addition of 5 mM Mg2+ to the assay, the unmodified TSL was a substrate (Fig. 2B). Tetrahymena pyriformis RAMT methylation of an A in the TSL was verified by HPLC analysis of nucleoside composition. Compared to full-length tRNA, the unmodified TSL was ~10% as effective a substrate for RAMT. This is also true for the TSL when compared to full-length tRNA as substrates for RUMT (4).
Figure 2.
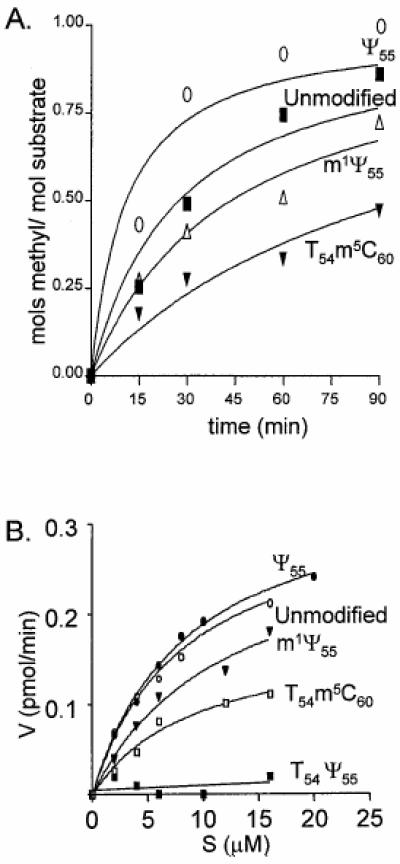
Activity of m1A58 methyltransferase. TSL was incubated with m1A methyltransferase and [3H]SAM under standard methylation conditions. (A) Time course showing methylation of TSLs Ψ55 (open circle), unmodified (closed square), m1Ψ55 (open triangle) and T54m5C60 (closed triangle). Aliquots were removed at 15, 30, 60 and 90 min. Moles of methyl group incorporated into TSLs are normalized to moles of methyl group incorporated into TSL Ψ55. (B) TSL substrate concentration versus reaction velocity. TSLs Ψ55 (closed circle), unmodified (open circle), m1Ψ55 (closed triangle), T54m5C60 (open square) and T54Ψ55 (closed square). Although not pictured, TSL dU54 produced similar results to T54Ψ55.
TSL Ψ55 (Km 8.6 µM, Vmax 0.35 pmol/min) was as good a substrate for RAMT as the unmodified TSL (Km 8.1 µM, Vmax 0.32 pmol/min) (Fig. 2). TSLs with m1Ψ55 (Km 17.3 µM, Vmax 0.36 pmol/min) (Fig. 2) and m1m3Ψ55 (data not shown) were also substrates for RAMT. Surprisingly, substrate activity was restricted to TSLs with uridine at position 54; constructs with T54, dU54 or dU54dU55 were not substrates for RAMT (data not shown). However, TSL T54m5C60 (Km 9.6 µM, Vmax 0.18 pmol/min) was the one exception to this restricted set of substrates (Fig. 2). We believe this is likely the result of an altered loop conformation induced upon addition of m5C60 to the T54-containing TSL.
Activity of RUMT
The unmodified and variously modified TSLs were assayed for their ability to be substrates for RUMT. The unmodified yeast tRNAPhe TSL was a substrate of RUMT, as has been reported for the E.coli tRNAVal heptadecamer TSL and the unmodified 76mer transcript of yeast tRNAPhe (4,8). A TSL with T54 served as a negative control and indirectly confirmed that RUMT in vitro only methylated the uridine at position 54, not U55 or U59. The unmodified TSL had a Km of 15.6 µM and a Vmax of 4.2 pmol/min. The Km compares favorably with that previously reported for the unmodified heptadecamer TSL of E.coli tRNAVal (5 µM) and an undecamer version of the same sequence (4 µM) (4). The Km for the heptadecamer TSL was ~20-fold higher than that reported for the complete 76mer unmodified T7 transcript of yeast tRNAPhe (0.8 µM) (4). TSL Ψ55 was a good substrate for the enzyme (Fig. 3), while other substitutions of either U54 (dU54) or U55 (dU55, m1Ψ55 and m1m3Ψ55) negated substrate activity (Fig. 3).
Figure 3.
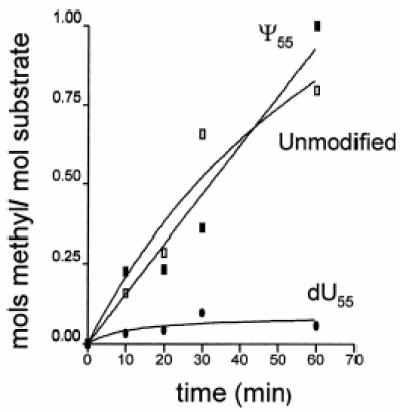
Methyl incorporation by m5U54 methyltransferase. TSLs Ψ55 (closed square), unmodified (open square) and dU55 (closed circle) were used as substrates for m5U54 methyltransferase as described in the text. Aliquots were taken at 10, 20, 30 and 60 min. Moles of methyl group incorporated into TSLs are normalized to moles of methyl group incorporated into TSL Ψ55. Although not pictured, TSLs m1Ψ55, m1m3Ψ55, dU54 and dU54dU55 produced similar results to dU55.
TSL thermal stability and global conformation
To determine the effects, if any, of modified nucleosides on stability of the TSL constructs used for enzymatic studies and to confirm that the TSLs were all adopting a hairpin conformation, thermal stability and thermodynamic parameters were assessed. Denaturation, monitored by UV absorbance, was concentration independent over a range of 0.4–82 µM, indicating unimolecular denaturation. Because monophasic melting curves were observed, a two-state approximation was assumed and applied to analysis of the data. Thermodynamic parameters derived from reiterated denaturations and renaturations are listed in Table 1. The general shape of the melting curves indicated that each TSL construct formed a hairpin (not shown). As would be expected of a RNA hairpin, incorporation of modified nucleosides into the TSL loop produced only modest effects on thermal stability. Introduction of the natural modification T at position 54 added stability to the TSL, as evidenced by a 2.2°C increase in Tm and a 0.4 kcal/mol decrease in ΔG, compared to that of the unmodified TSL (Table 1). Thermodynamic parameters of the doubly modified TSL T54m5C60 were similar to those of TSL T54. However, introduction of dU54 and methylated Ψ at position 55 slightly destabilized the RNA, whereas dU55 and Ψ55 had little or no effect on TSL stability (Table 1). Because of a limited quantity of the TSL containing the two natural modifications T54 and Ψ55 in combination, thermal denaturation could not be conducted for this construct.
NMR analysis of TSL constructs
To evaluate the effects of the various modified nucleosides on the conformation of the TSL, one-dimensional NMR spectra were collected for six of the variously modified TSLs in H2O. The imino and amino protons (9–15 p.p.m.), with the exception of one resonance at 10.7 p.p.m., were assigned for TSL T54, the most stable TSL with a natural modification (24). The base paired imino proton signals of the unmodified TSL stem region were quite similar to that of TSL T54 (24), suggesting that all of the TSL constructs formed a 5 bp stem and seven-membered loop. The signals emanating from the loop nucleosides C60, U59 and U55 are quite similar in the unmodified TSL construct and the TSL constructs containing T54, dU54 and Ψ55 (Fig. 4a–d). Therefore, any conformational fluctuations from the introduction of these modifications must be quite local or not detectable by one-dimensional NMR. The introduction of m1m3Ψ55 altered the spectrum of the TSL locally around U55 compared to that of TSL Ψ55. Because the doubly methylated Ψ (m1m3Ψ55) does not allow hydrogen bonding compared to Ψ, the disappearance of the chemical shifts around U55 is probably a result of methylation, while the general loop structure is similar to TSL Ψ55. The spectrum of TSL T54m5C60 (Fig. 4f) suggests that the introduction of m5C60 into the TSL containing T54 appeared to cause a more pronounced effect on loop conformation when compared to the spectrum of the TSL containing T54 alone (Fig. 4a). The chemical shifts from U59 and U55 are shifted and almost superimposed, suggesting that the introduction of m5C60 into the TSL containing T54 may alter loop conformation more globally across the loop instead of the more localized conformational alterations which probably occur with the introduction of the other modifications shown (Fig. 4a–e).
Figure 4.
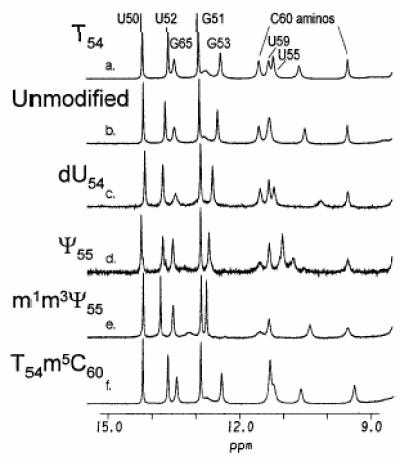
NMR spectra of the unmodified and modified TSLs. Proton NMR spectra (one-dimensional) were obtained for each TSL in 94% H2O/6% D2O at 1°C. Only the portion of the spectra exhibiting the base paired imino and loop imino and amino proton signals is shown. The imino and amino signals of the TSL T54 were assigned unambiguously (24) and are shown in the first spectrum (a). TSL: (a) T54; (b) unmodified; (c) dU54; (d) Ψ55; (e) m1m3Ψ55; (f) T54m5C60.
DISCUSSION
Using TSL constructs, we were able to determine that a heptadecamer corresponding to the TSL was a substrate for RAMT, as has been shown for two other modifying enzymes, RUMT and Ψ55 tRNA synthase, which act upon the same loop in tRNA. For all three enzymes, the activity is reduced when using only the heptadecamer compared to using the complete tRNA. However, because the enzymes recognize the TSL alone, the recognition determinants for these modifying enzymes are probably contained in the stem and loop.
Previous work using TSL constructs has shown that E.coli RUMT is not highly sequence specific (6,8), as may be expected from the highly conserved modifications occurring in tRNAs of differing sequence. Therefore, the substrate recognition determinants of RUMT are probably contained within the TSL tertiary conformation. To better understand the substrate conformational requirements of RUMT, we have site-specifically incorporated modified nucleosides that would be expected to produce local perturbations in the TΨC domain loop. In addition, TSL constructs with site-specific substitutions were also used to assess the effect that previous modifications and local loop perturbations had on the ability of RAMT to recognize the TSL as a substrate.
The unmodified TSL, TSL Ψ55 and TSL m1Ψ55 were substrates for m1A58 methyltransferase (RAMT). In contrast, TSLs containing T54 alone or T54Ψ55 were not substrates of RAMT. Incorporation of T54 increased the RNA Tm by ~2°C and stabilized the TSL by ~0.4 kcal/mol as compared to the unmodified TSL (Table 1). Although the one-dimensional spectrum of the TSL containing T54 does not suggest that there are any global conformational differences compared to the unmodified TSL, introduction of the natural modification T54 could cause local conformational restructuring of the loop around A58 not apparent from one-dimensional NMR. Rescue of the RAMT inactivity with the T54-containing TSL was achieved with incorporation of the second modification m5C60 (TSL T54m5C60). The melting curve for TSL T54m5C60 suggests that this construct forms a hairpin (not shown) and its thermodynamic parameters are not considerably different from those of TSL T54 (Table 1). The NMR spectrum of TSL T54m5C60 suggests that the stem conformation is quite similar to that of TSL T54. However, the chemical shifts from U55 and U59 are shifted, unlike any other TSL spectrum obtained (Fig. 4). This perhaps indicates that the m5C60 modification at position 60 more globally alters the conformation of the loop and allows TSL T54m5C60 to be a substrate for RAMT, while other T54-containing TSLs were not substrates. Because fully modified E.coli tRNAs were substrates for T.pyriformis RAMT (Fig. 2A) and other eukaryotic RAMTs (7,25–27), we cannot conclude from our results that modification events are ordered with, for instance, the synthesis of Ψ55 preceding that of m1A58 followed by synthesis of T54. In our determination of the structure of TSL T54, we noted that A58 was highly restricted and stacked on C61 (24). The unusual structure of TSL T54 may represent an intermediate in pre-tRNA folding (24) and preclude RAMT access to the N1 of A58. The absence of T54 could decrease the base stacking and partially disrupt the tertiary structure, explaining the lower Tm observed. The results presented here suggest that local conformation of the loop must be a critical determinant of RAMT activity.
Previously published results (8,30,31) suggest that properties other than the primary structure of the TSL are determinants for methylation by RUMT. Our results show that RUMT methylated only the unmodified TSL and the TSL containing the natural modification Ψ55. RUMT did not methylate TSLs with loop substitutions dU54, dU55 or the combination dU54dU55. In addition, TSL constructs containing methylated pseudouridine substitutions (m1Ψ55 and m1m3Ψ55) were not substrates for RUMT. There was little difference in thermodynamics between the TSLs with uracil at position 54 that were substrates for RUMT (the unmodified TSL and TSL Ψ55) and those that were not (TSLs dU54, dU55, dU54dU55, m1Ψ55 and m1m3Ψ55). The modest thermodynamic differences contributed by modification of the TSL loop indicate that the global conformations of substrates and non-substrates were similar (Table 1). The NMR spectrum obtained for TSL dU54 does not suggest conformational changes compared to the unmodified TSL. Therefore, structural differences must have been confined to the local conformation at or near U54 and U55, yet no obvious perturbation was apparent from the one-dimensional NMR spectra.
The recognition elements for RAMT are contained within tRNA nucleosides 49–65. As with RUMT, the 5 bp stem and seven-membered loop of the TSL are sufficient for recognition by RAMT. Our results strongly indicate that the primary and specific conformational determinants for RAMT and RUMT are contained within the sequence, modification and secondary structure of the TSL. RUMT activity is sensitive to conformational perturbations at or adjacent to the site of methylation, positions 54 and 55. However, RAMT activity is sensitive to conformational changes across the loop at position 54.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Glenn Björk for the E.coli strain GRB822 carrying the cloned trmA gene, Winnell Newman, as director of the NCSU Nucleic Acids Facility, for her synthesis of the TSLs and Guihua Liu for her expert technical assistance in purifying the TSLs. This work was supported by a USPHS grant GM-23037 (P.F.A.), Polish Committee for Scientific Research grant PBO506/P3/93/05 (A.M.) and the Lithuanian State Research Program, Molecular Background of Biotechnology (S.V.).
REFERENCES
- 1.Agris P.F. (1996) Prog. Nucleic Acid Res. Mol. Biol., 53, 79–129. [DOI] [PubMed] [Google Scholar]
- 2.Sprinzl M., Horn,C., Brown,M., Ioudovitch,A. and Steinberg,S. (1998) Nucleic Acids Res., 26, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grosjean H., Edqvist,J., Straby,K.B. and Giege,R. (1996) J. Mol. Biol., 255, 67–85. [DOI] [PubMed] [Google Scholar]
- 4.Gu X.R. and Santi,D.V. (1991) Biochemistry, 30, 2999–3002. [DOI] [PubMed] [Google Scholar]
- 5.Guenther R.H., Bakal,R.S., Forrest,B., Chen,Y., Sengupta,R., Nawrot,B., Sochacka,E., Jankowska,J., Kraszewski,A., Malkiewicz,A. et al. (1994) Biochimie, 76, 1143–1151. [DOI] [PubMed] [Google Scholar]
- 6.Gu X., Yu,M., Ivanetich,K.M. and Santi,D.V. (1998) Biochemistry, 37, 339–343. [DOI] [PubMed] [Google Scholar]
- 7.Yamazaki N., Hori,H., Ozawa,K., Nakanishi,S., Ueda,T., Kumagai,I., Watanabe,K. and Nishikawa,K. (1994) Biosci. Biotechnol. Biochem., 58, 1128–1133. [DOI] [PubMed] [Google Scholar]
- 8.Gu X., Ivanetich,K.M. and Santi,D.V. (1996) Biochemistry, 35, 11652–11659. [DOI] [PubMed] [Google Scholar]
- 9.Ogilvie K.K., Usman,N., Nicoghosian,K. and Cedergren,R.J. (1988) Proc. Natl Acad. Sci. USA, 85, 5764–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Agris P.F., Malkiewicz,A., Kraszewski,A., Everett,K., Nawrot,B., Sochacka,E., Jankowska,J. and Guenther,R. (1995) Biochimie, 77, 125–134. [DOI] [PubMed] [Google Scholar]
- 11.Guenther R., Forrest,B., Newman,W., Malkiewicz,A. and Agris,P.F. (1998) Acta Biochim. Pol., 45, 13–18. [PubMed] [Google Scholar]
- 12.Ariza X., Bou,V. and Vilarras,J. (1995) J. Am. Chem. Soc., 117, 3665–3669. [Google Scholar]
- 13.Gehrke C.W. and Kuo,K.C.T. (1990) In Kuo,K.C.T. and Gehrke,C.W. (eds), Chromatography and Modification of Nucleosides. Elsevier, Amsterdam, The Netherlands, Vol. 45A, pp. A3–A71.
- 14.Cantor C.R., Warshaw,M.M. and Shapiro,H. (1970) Biopolymers, 9, 1059–1077. [DOI] [PubMed] [Google Scholar]
- 15.Ny T., Lindstrom,H.R., Hagervall,T.G. and Bjork,G.R. (1988) Eur. J. Biochem., 177, 467–475. [DOI] [PubMed] [Google Scholar]
- 16.Revel M. and Littauer,U.Z. (1965) Biochem. Biophys. Res. Commun., 20, 187–194. [DOI] [PubMed] [Google Scholar]
- 17.Johnson L., Hayashi,H. and Söll,D. (1970) Biochemistry, 9, 2823–2831. [DOI] [PubMed] [Google Scholar]
- 18.Stribinskis V. (1993), PhD Thesis, Vilnius University, Lithuania.
- 19.Marky L.A. and Breslauer,K.J. (1987) Biopolymers, 26, 1601–1620. [DOI] [PubMed] [Google Scholar]
- 20.Piotto M., Saudek,V. and Sklenar,V. (1992) J. Biomol. NMR, 2, 661–665. [DOI] [PubMed] [Google Scholar]
- 21.Marion D., Ikura,M. and Bax,A. (1989) J. Magn. Reson., 84, 425–430. [Google Scholar]
- 22.Varani G. and Tinoco,I.,Jr (1991) Q. Rev. Biophys., 24, 479–532. [DOI] [PubMed] [Google Scholar]
- 23.Saenger W. (1984) Principles of Nucleic Acid Structure. Springer-Verlag, New York, NY.
- 24.Koshlap K.M., Guenther,R.H., Sochacka,E., Malkiewicz,A. and Agris,P.F. (1999) Biochemistry, 38, 8647–8656. [DOI] [PubMed] [Google Scholar]
- 25.Glick J.M. and Leboy,P.S. (1977) J. Biol. Chem., 252, 4790–4795. [PubMed] [Google Scholar]
- 26.Agris P.F., Spremulli,L.L. and Brown,G.M. (1974) Arch. Biochem. Biophys., 162, 38–47. [DOI] [PubMed] [Google Scholar]
- 27.Mutzel R., Malchow,D., Meyer,D. and Kersten,H. (1986) Eur. J. Biochem., 160, 101–108. [DOI] [PubMed] [Google Scholar]
- 28.Santi D.V. and Hardy,L.W. (1987) Biochemistry, 26, 8599–8606. [DOI] [PubMed] [Google Scholar]
- 29.Morozov I.A., Gambaryan,A.S., Lvova,T.N., Nedospasov,A.A. and Venkstern,T.V. (1982) Eur. J. Biochem., 129, 429–436. [DOI] [PubMed] [Google Scholar]
- 30.Becker H.F., Motorin,Y., Sissler,M., Florentz,C. and Grosjean,H. (1997) J. Mol. Biol., 274, 505–518. [DOI] [PubMed] [Google Scholar]
- 31.Gu X., Ofengand,J. and Santi,D.V. (1994) Biochemistry, 33, 2255–2261. [DOI] [PubMed] [Google Scholar]