


Abstract
Variable gene expression amongst transgenic lines occurs due to copy number and to random associations of incoming DNA with chromosomal elements at the site of integration. Here we describe a method of identifying sites permissive for transgene expression and their use for efficient introduction of single copy transgenes by homologous recombination. ES clones were selected in HAT medium for expression of a randomly integrated HPRT marker lying 5′ to an Oct4/lacZ transgene. 794 clones were assessed in vitro for appropriate down-regulation of lacZ following differentiation. Two clones were chosen for further analysis which displayed appropriate and inappropriate gene regulation (clones 710 and 91, respectively). Three developmental promoters (thyroglobulin, Hox2.6 and Myf5) were then sequentially introduced into the original insertion sites in each clone (710 and 91) by homologous recombination, to drive expression of lacZ. Transgenic embryos were assessed for their ability to direct lacZ expression to tissues in which the respective promoter sequences are normally active. The site which appropriately down-regulated lacZ in vitro (710) also showed appropriate in vivo regulation of lacZ from the three developmental promoters. Site 91, however, directed an additional pattern of ectopic expression, which was common to all four promoters. Pre-selection of genomic sites for the introduction of transgenes by gene targeting improves the repeatability of transgene expression and provides an efficient means of single copy transgene introduction by homologous recombination.
INTRODUCTION
Tissue-specific and position-independent expression of transgenes in animals is frequently confounded by unpredictable effects of elements at the site of chromosomal integration (1) and by variable transgene copy number (2). Solutions to these problems will be critical for successful correction of genetic defects in somatic cells by gene therapy and for large animal transgenic programmes. In transgenic research generally it is highly desirable that a series of gene alterations be compared without the experimental variation arising from differences due to incorporation site. Microinjection of DNA into the pronuclei of mouse zygotes (3) usually results in multiple copies of the transgene being integrated in large arrays. In most cases the level of expression of the transgene is not correlated with the number of copies. Exceptions to this are transgenes which contain all of the elements necessary for position-independent gene expression. For example, study of the human β-globin genes (4,5) has identified a locus control region (LCR) which confers position-independent and copy number-dependent expression. Only a few transgenic constructs have displayed such an efficient expression profile, for example, the chicken lysozyme locus (6) and ovine β-lactoglobulin locus (7).
Variable gene expression among transgenic lines can be manifest as silencing, low expression or ectopic expression (8). In practice, up to 10 transgenic lines may have to be analysed to obtain a single line in which the transgene is expressed in the desired temporal and spatial manner. One way to address this issue is to include an LCR in the transgenic construct (4,6). However, LCRs have not been defined for most genes and this approach does not always confer position-independence upon a contiguous, but unrelated, transgene (9). The complementary approach is to introduce the transgene into chosen sites in the genome by homologous recombination. This latter strategy has been used successfully to introduce a lacZ cassette into the hypoxanthine phosphoribosyl transferase (HPRT) locus (10), to replace the β-globin (11) and α-lactalbumin genes (12) and to introduce a bcl-2 minigene into the HPRT locus in embryonic stem (ES) cells (13). Other approaches are the co-introduction of the transgene with fragments which ‘rescue’ genes from positional silencing (14) and the flanking of transgenes with inverted terminal repeat sequences from adeno-associated virus (15), although the latter method is not yet exemplified in mammalian cells.
ES cells can also be modified by the introduction of a loxP site which is recognised by the site-specific recombinase Cre (16,17). In a subsequent step, and in the presence of Cre recombinase, this allows site-specific insertion of trangenes which carry flanking loxP sites. This method combines the advantages of homologous and site-specific recombination and has been used to introduce a marker into the whey acidic protein locus (18) and a promotorless luciferase gene into the β-casein locus (19).
All of the above methods allow transgenes to be targeted to a site known to be permissive for gene expression and eliminate the silencing effects of multi-copy arrays. In general, these strategies can be characterised as based upon selection of a ‘candidate locus’. Instead of selecting specific target loci with properties tailored to each transgene, we have sought to identify sites which are broadly permissive for a range of transgenic applications, which we define as ‘neutral’ sites. The method employs an in vitro screen for sites in the mouse genome which allow expression of an HPRT marker and which appropriately down-regulate a second, contiguous lacZ reporter gene following in vitro differentiation. The lacZ gene is placed under the transcriptional control of upstream sequences taken from the murine Oct4 gene (also termed Oct3 or Oct3/4), which is tightly down-regulated in differentiating ES cells (20). We demonstrate that a site which provided correct developmental regulation of the Oct4 promoter in vitro also allowed appropriate expression from three other tissue-specific promoters in vivo. In contrast, a site showing inappropriate expression of an Oct4-regulated lacZ gene in vitro also showed inappropriate expression when lacZ was regulated by tissue-specific promoters in vivo. These results provide a method for the rapid introduction of a series of constructs to the same site in the mouse genome, thus eliminating a major source of experimental variation. An important advantage of this approach is that a single site may provide an appropriate environment for the position-independent and high level expression of a variety of transgenes expressed from different promoters.
MATERIALS AND METHODS
Constructs
The plasmid pOct1.9 was constructed by insertion of a 2.7 kb SalI fragment from PGK/pDWM1 (21) comprising an HPRT cassette into the SalI site of pOctlacZ (22). A nuclear localisation signal was introduced into the lacZ 5′-UTR by exchanging a 2 kb KpnI–SacI fragment of the lacZ gene with that of pnLacF (23). pOct1.9 contains Oct4 sequences extending from –1876 to +29.
The plasmid pOct8.5 was prepared by NotI digestion and Klenow treatment of pGOF18 (24) followed by partial HindIII digestion to remove a 12 kb OctlacZ fragment. This was then cloned into the EcoRV and HindIII sites of the vector pPolyIII (25). The 12 kb Oct–lacZ fragment was then re-isolated as a NotI(blunt)–SalI fragment and was cloned into SalI and EcoRV digested PGK/pDWM1. The resulting plasmid pOct8.5 carries Oct4 upstream sequences extending from nucleotide –8457 to +43.
The plasmid pMR was generated by HindIII digestion of pOct1.9, treatment with Klenow and religation to generate a unique NheI site. An NheI–KpnI fragment from the upstream sequence of c-mos was isolated from pIG500 (26) and was inserted between the NheI and KpnI sites of pMR to prevent transcriptional read-through from the truncated HPRT gene. A XhoI–XbaI fragment of plasmid 6P-T3-IresNeo-BS-MO (P. Mountford), which contained an IRESneo cassette, was then blunt-end inserted into the NheI site of pMR.
Addition of tissue-specific promoters to pMR was as follows.
pMR/Myf. A SalI fragment from pMyf5/57.3 (J. McWhir) containing the 5.5 kb promoter sequences of the Myf5 gene was ligated into the SalI site in the pMR polylinker.
pMR/TG. The plasmid pTG-tk2 (27) was linearised with BglII and partially filled in using Klenow with dGTP and dATP. Subsequent KpnI digestion released a 3 kb fragment of bovine thyroglobin upstream sequence. This was then subcloned into pMR. The pMR vector was prepared for ligation by digestion with SalI, partial filling in with Klenow in the presence of dTTP and dCTP only and KpnI digestion.
pMR/Hox. The Hox enhancer element A was isolated from plasmid p2.6A (28) as a 3 kb KpnI–SalI fragment and ligated into the KpnI and SalI sites pf pMR. The 1.3 kb XhoI–NcoI fragment containing the Hox2.6A promoter was isolated from plasmid p2.6A and subcloned into SalI and NcoI digested pnLacF. This fragment was then re-isolated as a SmaI–EcoRV fragment and ligated into the SalI (blunted) and EcoRV sites of plasmid pMR (now containing the Hox enhancer element A).
ES cell culture and transfection
ES cells were maintained without feeders in Glasgow’s modification of Eagle’s medium (Life Technologies) supplemented with 400 U/ml recombinant murine leukaemia inhibitory factor (LIF; Life Technologies), 0.1 mM MEM non-essential amino acids (Life Technologies), 5% newborn bovine serum, 5% foetal bovine serum and 0.1 mM 2-mercaptoethanol (Sigma).
HM-1 ES cells (29) were electroporated by mixing 2 × 107 cells with 100 µg of DNA fragment (random integrations) or 300 µg of linearised DNA (targeting experiments) in 0.8 ml of HEPES-buffered saline (pH 7.5), giving a single pulse at 800 V, 3 µF (Bio-Rad gene pulser) and plating at 2 × 106 cells/10 cm dish. Prior to electroporation the insert from pOct1.9 was excised by NotI digestion and that from pOct8.5 by NotI and SalI digestion. Selection was applied 20–24 h after electroporation. Electroporation with pOct1.9 and pOct8.5 was followed by selection in HAT medium (1 mM hypoxanthine, 0.8 µM aminopterin and 20 µM thymidine) for 10 days. Selection of targeted clones following electroporation with pMR/Myf, pMR/TG and pMR/Hox were selected in G418 (300 µg/ml) for 8 days followed by selection in 6-thioguanine (6TG) (5 µg/ml) for a further 8 days.
Differentiation of ES cells
ES cells were lightly trypsinised to give clumps of cells and were induced to differentiate by culturing as aggregates in suspension in the absence of LIF. After 6 days the aggregates were outgrown on gelatinised tissue culture plates in the absence of LIF for a further 6 days.
Tetraploid rescue aggregation embryos
The derivation of tetraploid embryos was according to Nagy et al. (30). Embryo donors were C57/BL×CBA. Fusion occurred by passing 33 V for 200 µs across the two-cell embryo. The zona pellucidae were removed from fused tetraploid embryos that had developed to the four cell stage by treating with Tyrode’s acid. Clumps of ~15 ES cells were sandwiched between two tetraploid embryos, cultured overnight and transferred to the uterus of 2.5 day pseudopregnant recipient C57/BL×CBA mice. Diploid aggregations were carried out using single eight cell embryos and a clump of 10–15 ES cells.
lacZ staining
β-Galactosidase activity was determined in situ in cells or embryos fixed in 4% formaldehyde in phosphate-buffered saline followed by staining in phosphate buffer containing 35 mM potassium ferricyanide, 35 mM potassium ferrocyanide, 1.5 mM magnesium sulphate and 1 mg/ml X-gal overnight at 37°C. For embryos the wash and staining solutions also contained 0.1% Na desoxycholate, 0.02% HP4O and 0.05% bovine serum albumin.
DNA analysis
Cells were lysed and DNA extracted by standard methods (31). Following SacI digestion, DNA was subjected to electrophoresis through 0.8% agarose and transferred onto ZETAPROBE according to the manufacturer’s recommendations (Bio-Rad). Hybridisation was performed at 65°C using as probes: (i) a 0.2 kb SalI–XhoI fragment of pDWM1/5′Δ+63 (32) comprising HPRT coding sequence; (ii) a 4.7 kb HindIII–BglII fragment of pOct1.9 comprising lacZ coding sequence (used in Fig. 3Ba); (iii) a 1.2 kb HindIII–BamHI fragment of Oct4/lacZ (33) comprising Oct upstream sequence (used in Fig. 3Bb). Probes were labelled to a specific activity of >108 c.p.m./µg by random hexamer labelling (High Prime; Boehringer) in the presence of [α-32P]dCTP (3000 Ci/mmol). The final wash was with 40 mM phosphate, 1% SDS at 65°C.
Figure 3.
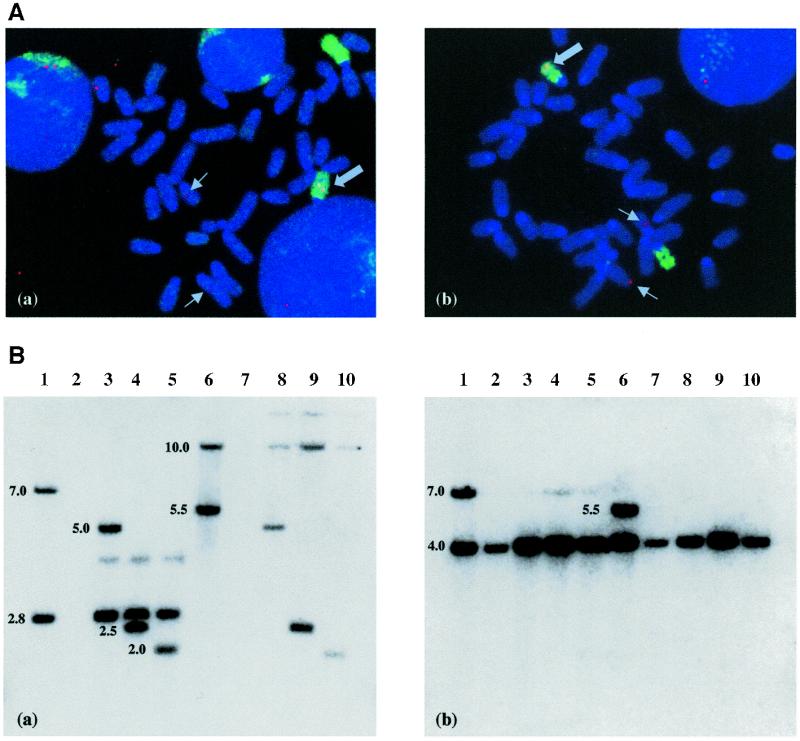
Structural analysis of primary integrants and secondary targeted cells. (A) FISH analysis of clones 91 (a) and 710 (b). Chromosome spreads were hybridised with a probe generated by nick translation of the plasmid pOct1.9 and painted with chromosome 7-specific (a) and chromosome 13-specific (b) fluorescent probes. FISH signals on chromosomes 7 (a) and 13 (b) are indicated with arrows. The signals detectable on non-painted chromosomes are the result of hybridisation with the endogenous Oct4 gene (chromosome 17). (B) Southern blot analysis of clones 91 and 710 and their targeted derivatives. The position of the probes is indicated in Figure 1. DNA was digested with SacI, blotted and probed with the lacZ coding sequence (a) and a fragment of the Oct promoter (b). Lane 1, clone 91; lane 2, HMI ES cells; lanes 3–5, clone 91 derivatives targeted with pOct/TG (3), pOct/Hox (4) and pOct/Myf (5), respectively; lane 6, clone 710; lane 7, HMI ES cells; lanes 8–10, clone 710 derivatives targeted with pOct/TG (8), pOct/Hox (9) and pOct/Myf (10), respectively. Sizes of bands are given to the left in kb. The faint bands in (a) (lanes 3–5 and 8–10) are consistent with those expected from partial digestion.
FISH analysis
ES cells were arrested at metaphase by adding 0.02 µg/ml colcemid (Life Technologies) 1 h before trypsinisation of cells. Metaphase spreads were prepared and hybridised with biotin-labelled pOct1.9. Chromosomes were revealed with specific paints as described (34).
RESULTS
Pre-screening random sites for lacZ expression in vitro
In order to identify permissive sites for transgene expression in vitro, an Oct4/lacZ reporter was constructed such that its transcriptional control could be regulated in ES cells in culture. The octamer binding transcription factor Oct4 is one of the earliest expressed genes which is developmentally regulated (20). It is expressed in ES cells, but is down-regulated when ES cells are induced to differentiate in the absence of LIF. A 1.9 kb upstream sequence from the Oct4 locus drives Oct4 expression in EC cells and at a low level in early embryos (35). A larger 8.5 kb upstream sequence contains a distal element, which imparts high level expression in ES cells and the germline (24). The constructs pOct1.9 and pOct8.5 (Fig. 1) contain the lacZ coding sequence under transcriptional control of the 1.9 and 8.5 kb Oct4 upstream sequences, respectively. Both constructs also contain an HPRT minigene transcribed from the murine phosphoglycerate kinase (PGK) promoter. Following transfection, selection was carried out in HAT for expression of HPRT in the ES line HM1, which carries an inactivating deletion in the endogenous HPRT locus (29).
Figure 1.

Structure of the primary constructs pOct1.9 and pOct8.5 and multi-targeting construct pMR. pOct1.9 and pOct8.5 contain a PGKhprt cassette 5′ to a lacZ reporter gene. lacZ expression is directed by 1.9 (pOct1.9) and 8.5 kb (pOct8.5) of 5′-flanking sequence from the Oct4 gene, respectively. In the targeting vector pMR, part of the HPRT gene has been replaced with IRESneo to allow selection for neo expression and against HPRT expression and a c-mos termination sequence to prevent read-through. A multiple cloning site (MCS) was included to allow the insertion of tissue-specific promoters or transgenes. The 5′ and 3′ homology arms are given by the PGK and part of the HPRT gene and by the lacZ gene, respectively. The position of the probes used for Southern blot analysis and the position of the SacI sites are indicated.
The permissive nature of integration sites for PGK/HPRT expression can be inferred by colony survival in HAT medium. A second indication of neutrality could be obtained by staining transfected ES clones for lacZ expression following in vitro differentiation. The native Oct4 gene is known to be expressed in undifferentiated ES cells and is tightly down-regulated following their differentiation in vitro (35). Hence the constructs pOct1.9 and pOct8.5 were designed to indicate whether particular sites allowed Oct controlling elements to respond appropriately to in vitro differentiation. Following transfection, cells were selected in HAT medium and colonies were expanded. Replicate cultures were maintained in conditions which allowed limited differentiation, such that both undifferentiated and differentiated cells were present. These were then stained for β-galactosidase activity and scored on the basis of expression in: only undifferentiated cells; only differentiated cells; both cell types; no staining (Fig. 2A).
Figure 2.
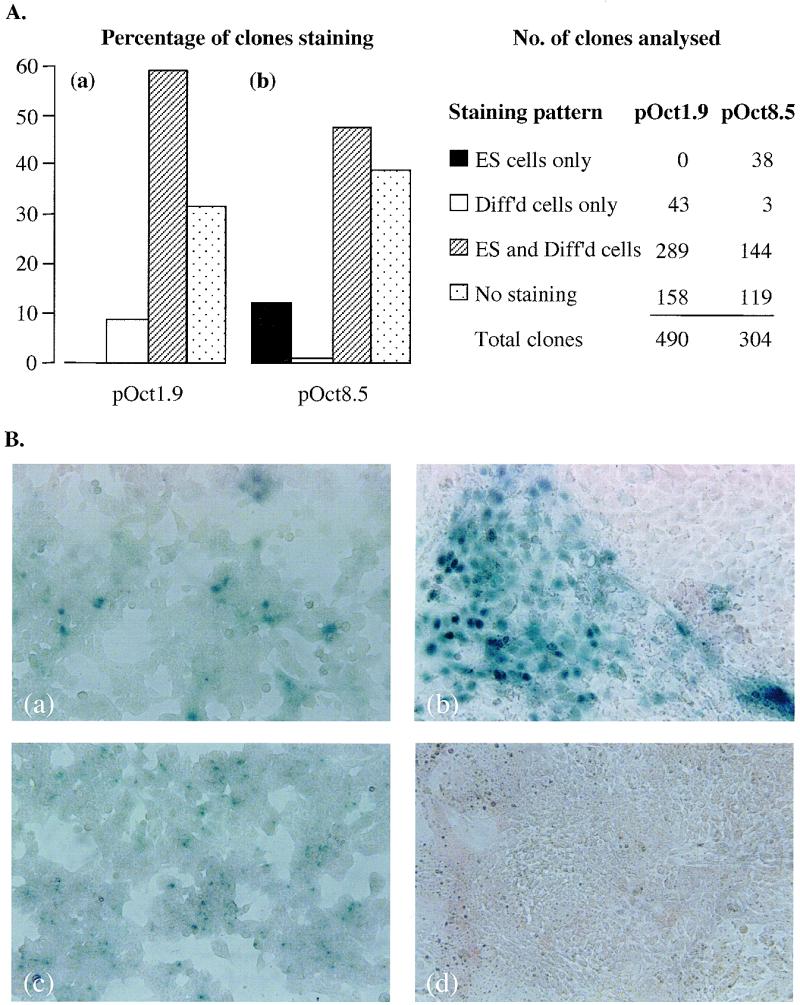
lacZ expression in ES cells in vitro. (A) lacZ expression with the randomly integrated constructs pOct1.9 (a) and pOct8.5 (b) in ES cells and their differentiated derivatives in vitro. The percentage of clones staining in ES cells only, in differentiated cells only, staining in both ES and differentiated cells and with no staining is given in histogram format (a) for clones transfected with pOct1.9 and (b) for clones transfected with pOct8.5. (B) Monolayer cultures of clone 91 (a and b) and clone 710 (c and d) grown as undifferentiated ES cells (a and c) and following differentiation (b and d). Clones 91 and 710 contain the constructs pOct1.9 and pOct8.5, respectively. Down-regulation of lacZ expression following differentiation of ES cells is apparent in clone 710 (c and d), while in clone 91 lacZ is expressed in both undifferentiated and differentiated cells (a and b).
The majority of clones obtained with pOct1.9 expressed lacZ in both differentiated and undifferentiated ES cells (Fig. 2Aa). In ~30% of clones lacZ expression was not observed in any cell type. As we have not done exhaustive Southern analyses on this subset, it is likely that they comprise both clones in which the transgene has been repressed and those in which the transgene is not intact. In ~10% of clones lacZ was expressed only in differentiated cells, a complete reversal of the native pattern of Oct expression. No clones were identified which stained exclusively in undifferentiated cells (ES cells). Hence, the 1.9 kb Oct4 promoter was not appropriately regulated in any site. As we had previously used the same 1.9 kb sequence to develop a selection procedure for ES isolation from genotypes that were non-permissive using standard ES isolation techniques (33), this was an unexpected result. Others have shown that the 1.9 kb fragment was sufficient to give ICM-specific lacZ staining in embryos (20). We suggest that while the 1.9 kb promoter may provide sufficient levels of transcription in ES cells to sustain G418 selection (33), these same levels may be insufficient for detection of lacZ activity in vitro. The patterns of expression we observed from pOct1.9 in vitro (Fig. 2Aa) therefore may be due to the activity of ‘trapped’ enhancers on a minimal promoter.
ES clones generated with the construct pOct8.5 were also subject to position effects on lacZ expression (Fig. 2Ab). In almost 50% of pOct8.5 clones staining was detected in both differentiated and undifferentiated cell types. As with the shorter promoter, ~30% showed no expression in any cell type. However, in contrast to the 1.9 kb Oct4 promoter, appropriate down-regulation of lacZ was observed in a subset (12%) of clones. These data are consistent with those of Yeom et al. (24) and suggest that the distal enhancer absent in pOct1.9 may be necessary to obtain transgene expression in ES cells at the levels required for lacZ detection. This result underlines the double requirements for a site which is permissive for gene expression and for a construct which includes all of the elements necessary for appropriate gene regulation. It should be noted that while appropriate lacZ regulation was observed in a subset of clones, within a clone staining was confined to a proportion of cells (see for example Fig. 2Bc).
Promoter replacement by gene targeting
We next sought to determine if lacZ expression in vitro would be predictive of in vivo expression when the Oct promoter sequences were removed and replaced with different promoters by homologous recombination. Two clones were selected for further analysis. One clone was chosen because it displayed β-galactosidase activity in the pre-screen exclusively in undifferentiated ES cells (clone 710). For comparison, a second was chosen which was characterised by weak expression in ES cells and by strong expression in differentiated cells (clone 91). Pre-screen staining patterns determined for clones 91 and 710 were confirmed by comparisons of lacZ expression in undifferentiated cultures with expression following embryoid body formation (Fig. 2B). Clones 91 and 710 contained the constructs pOct1.9 and pOct8.5, respectively. Note that in their targeted derivatives, all Oct sequences were removed and recombinants of clones 91 and 710 were therefore identical in all respects bar position. Southern blot analysis revealed that both clones 91 and 710 contained a single integration (Fig. 3B), and this was confirmed by FISH analysis, which placed the single integration site of clone 91 on chromosome 7 (Fig. 3Aa) and of site 710 on chromosome 13 (Fig. 3Ab).
The multiple replacement construct pMR (Fig. 1c) was constructed to allow rapid cloning of any promoter or transgene into a vector modified to facilitate identification of targeted clones. The homology for gene targeting is provided by sequences held in common between pOct1.9 and pOct8.5. Hence recombinant loci are identical internally whether they are derived from clone 91 or clone 710. pMR contains a truncated (non-functional) HPRT minigene which, following recombination, renders the cell HPRT-deficient. An additional feature of pMR is the inclusion of an IRES neomycin cassette downstream of the PGK promoter. A variant of pMR was also tested in which the PGK promoter was deleted (data not shown). In this variant, translation of neomycin phosphotransferase could occur only if the gene was integrated into the 5′-untranslated region (5′-UTR) of an expressed gene to acquire an active promoter. In practice, deletion of the PGK promoter did not lead to an improvement in the proportion of doubly resistant clones which were targeted, suggesting that the frequency with which random integration of neo is accompanied by spontaneous loss of HPRT approaches zero.
In selection for loss of HPRT function, cells are normally grown in the absence of selective medium for 5–8 days to allow previously synthesised HPRT enzyme to decay. Cells must often be replated in the presence of 6TG at low cell density to prevent the ‘kiss of death’ through metabolic cooperation. The neo cassette allows cells to be reduced to single cell clones in the course of G418 selection and prior to 6TG selection. This in turn reduces the number of manipulations and the amount of medium required and eliminates loss of targeted clones due to metabolic cooperation. Others have employed a similar strategy as a rapid screen for targeting efficiency (36).
The thyroglobulin (TG), Hox2.6 and Myf5 promoters were cloned into the multi-cloning site in pMR to generate three targeting constructs (pMR/TG, pMR/Hox and pMR/Myf). In their endogenous contexts, these promoters direct expression to the following tissues: pMR/TG, the thyroid gland from embryonic day 14 (E14) onwards (37); pMR/Hox, the developing neural tube from E9.5, with a distinct boundary between rhombomeres 6 and 7 (38); pMR/Myf5, the hyoid arch from E10.5 and, to a lesser extent, the somites and mandibular arches (39). The TG, Myf5 and Hox2.6 promoter fragments used in these constructs had previously been shown to be subject to position effects (39–42).
Efficient gene replacement
Six targeting experiments were undertaken in which ES clones 91 and 710 were each transfected with the three constructs pMR/TG, pMR/Hox and pMR/Myf5. In each experiment DNA from G418/6TG doubly resistant colonies was restricted with SacI and hybridised with a 3′ probe comprising the lacZ coding sequence and a 5′ probe comprising a portion of the PGKhprt cassette (Fig. 1). Prior to targeting, Southern analysis of clones carrying pOct1.9 with the 3′ lacZ probe reveals two flanking bands characteristic of each site (see for example Fig. 3Ba, lane 1). Analysis of clones carrying pOct8.5 reveals an internal band of 5.5 kb and a single 3′-flanking band characteristic of each site (see for example Fig. 3Ba, lane 6). Following targeting, clones which previously carried pOct1.9 are characterised by loss of the 5′-flanking band and its replacement with a 5.0 (pMR/TG), 2.5 (pMR/Hox) or 2.0 kb (pMR/Myf5) band (Fig. 3Ba, lanes 3–5). Clones which previously carried pOct8.5 are characterised by loss of the 5.5 kb internal band and its replacement with the same 5.0, 2.5 or 2.0 kb bands (Fig. 3Ba, lanes 8–10). Replacement of the Oct3/4 promoter was confirmed by rehybridisation of the stripped filter with an Oct3/4 probe to reveal loss of the Oct3/4 promoter in all targeted clones (Fig. 3Bb). The remaining 4.0 kb band (Fig. 3Bb) results from hybridisation of the probe with a fragment of the endogenous Oct locus. Reprobing with a 5′ probe (Fig. 1) also revealed predicted changes in the size of the 5′-flanking bands following targeting (data not shown). Since it was not possible to use flanking probes in uncharacterised sites, correct targeting is inferred by the following considerations: (i) the absence of extra flanking bands diagnostic for a second random insertion; (ii) coincident and predicted changes in the sizes of the original 5′-flanking bands with the 5′ probe (data not shown); (iii) coincident loss of the Oct promoter fragments.
Targeting efficiencies at sites 91 and 710 for the three tissue-specific promoters are given in Table 1. The targeting frequency was similar at both sites and for all three constructs, ranging from 10–3 to 10–4 when measured as a proportion of total transfectants. Of doubly resistant clones analysed, 50–86% (average 70%) were correctly targeted at both the 5′- and 3′-ends (Table 1). Although some clones had undergone re-arrangements at one end, >90% of those analysed were integrated within the target site, confirming that random integrants seldom gave rise to doubly resistant colonies.
Table 1. Targeting efficiencies at two random sites.
| Targeting | Site 91 (pOct1.9) | Site 710 (Oct8.5) | ||||||
|---|---|---|---|---|---|---|---|---|
| constructs | G4l8R coloniesa | G4l8R/6TGR colonies | G4l8R/6TGR G4l8R | Correctly targetedb G4l8R/6TGR | G4l8R coloniesa | G4l8R/6TGR colonies | G4l8R/6TGR G4l8R | Correctly targetedb G4l8R/6TGR |
| TG | nd | 208 | nd | 83% | 3312 | 3 | 9 × 10–4 | 50% |
| Myf | 3664 | 27 | 7.4 × 10–3 | 73% | 3808 | 13 | 3.4 × 10–3 | 73% |
| Hox 2.6A | 5184 | 25 | 4.8 × l0–3 | 54% | 3392 | 8 | 2.4 × 10–3 | 86% |
Numbers of G418-resistant and doubly (G418 and 6TG) resistant colonies in each of three targeting experiments. Adjusted targeting frequency reflects the proportion of doubly resistant clones which, on average, were correctly targeted and gave the predicted bands upon Southern analysis of both the 5′- and 3′-flanks. nd, not determined.
aDrug-resistant colonies per 2 × 107 cells.
bAs determined by Southern blot analysis.
Transgene expression in vivo
In order to characterise site effects on transgene expression in vivo, entirely ES-derived foetuses were generated by tetraploid rescue (43). Clone 710, however, could not be used successfully in tetraploid rescue experiments and chimeras were instead generated by aggregation of ES cells with diploid morulae. This technique has been shown to generate predominantly ES-derived embryos (43). Failure of clone 710 in tetraploid experiments may have been due to the presence of a subset of trisomic (41XY) cells, revealed by standard karyoptypic analysis (data not shown).
Embryos were obtained at E10.5 from ES clone 91 and its targeted derivatives and stained for lacZ activity (Fig. 4a–e). No variation was observed between embryos of the same stage obtained with the same ES clone (representative embryos are shown in Fig. 4). The 1.9 kb Oct promoter in site 91 is expressed in the ventral pharynx, gut and brain (Fig. 4a). This pattern is almost certainly a consequence of elements at the site of integration and is reiterated when the Oct sequences are replaced with three different promoters (Fig. 4a–d). The same pattern of ectopic expression is observed even when the promoter would normally be transcriptionally inactive, as in the case of the thyroglobulin promoter at E10.5 (Fig. 4d). Note that the lower gut expression in Figure 4a regresses as development proceeds from E9.5 to E10.5 (data not shown). Hence, the presence or absence of this sub-pattern is sensitive to developmental differences between embryos. In spite of additional ectopic expression, however, the Hox2.6 and Myf5 promoters (Fig. 4b and c, respectively) are still capable of directing expression of the lacZ gene in appropriate tissues and at the correct developmental stage. Similarly, the thyroglobulin promoter at E16.5 is active in the thyroid gland (Fig. 4e).
Figure 4.

lacZ expression in transgenic embryos. Representative embryos generated from ES clone 91 and its targeted derivatives and stained for β-galactosidase activity are shown in (a)–(d) (day 10.5 of development) and (e) (day 16). Arrows indicate sites of ectopic expression. Embryos generated from ES clone 710 and its targeted derivatives are shown in (f)–(i) (day 10.5) and (j) (day 16). No ectopic expression was observed. lacZ expression was directed by the Oct1.9 promoter (a) the Oct8.5 promoter (f) the Hox2.6 promoter (b and g), the Myf5 promoter (c and h) and the thyroglobulin promoter (d, e, i and j). Embryos in (a)–(e) were generated by tetraploid rescue and in (f)–(j) by diploid aggregation. Panels (e) and (j) are thyroid glands following removal of overlying tissue in day 16 embryos.
In contrast to clone 91, diploid aggregation embryos obtained with clone 710 and stained for lacZ activity showed no ectopic expression and reproduced the expected patterns of developmental expression (Fig. 4f–j). Embryos generated from clone 710 continued to show appropriate down-regulation of the 8.5 kb Oct promoter in vivo at embryonic day 10.5 (Fig. 4f). When the thyroglobulin promoter was targeted into site 710 no expression was observed at E10.5 (Fig. 4i) and expression was restricted to the thyroid gland at E16.5 (Fig. 4j). This is consistent with the onset of endogenous thyroglobulin expression at E14.5. When the Myf5 promoter was targeted into site 710, lacZ activity was observed in the hyoid arch (Fig. 4h), but not in somites nor in the mandibular arches. The 5.5 kb promoter used in pMR/Myf lacks an element now known to be required for somitic expression (40) and may provide insufficient levels of transcript to detect weak mandibular expression by lacZ staining. The Hox2.6 promoter (Fig. 4g) provides appropriate high levels of β-galactosidase activity in the developing spine with an R6/7 boundary of expression. In contrast to site 91, site 710 provides appropriate regulation of four different promoters in vivo, which parallels expression of the endogenous gene products.
Clone 710 contributes to the germline of chimeras
Six male chimeras were generated by blastocyst injection of clone 710 and tested for germline transmission. One of these chimeras was a partial transmitter, generating 2/12 pups with coat colour markers diagnostic for germline transmission.
DISCUSSION
Recent developments in nuclear transfer and cell technology make it possible to generate transgenic animals of several species from somatic cells and cell lines (44,45) and from their genetically modified derivatives (46). In large species where cost and time constraints weigh heavily, predictable high level transgene expression will be particularly critical. Existing elements at a chosen site of incorporation can be co-opted for the production of recombinant proteins by targeting transgenes to endogenous loci. However, in some instances this ‘candidate locus’ approach may be associated with instability of transgene expression (47), with unpredictable effects on neighbouring genes (48) or with hypomorphic phenotypes arising from the acquisition within a locus of cryptic splice acceptors (49,50). Hence, an attractive alternative would be a ‘neutral site’ in which a series of transgenes with relatively simple controlling elements could be inserted by homologous recombination and in which the properties of putative regulatory elements could be readily analysed. We have taken the first steps towards identifying such a neutral site in the mouse genome.
Detection of neutral sites in vitro
ES cells provide an in vitro model of differentiation in which changes in gene expression parallel those in normal development. We have used this model to show that a clone which appropriately down-regulates a randomly integrated Oct4/lacZ cassette in vitro identifies a useful integration site in which to express other transgenes in vivo. When the 1.9 kb Oct4 promoter was assessed in vitro, no clones were generated in which lacZ staining was exclusive to undifferentiated cells (Table 1). Only when the larger 8.5 kb promoter was used was it possible to identify a subset of clones with the appropriate staining pattern. This was consistent with the presence of an ES-specific enhancer element, necessary for high level expression in ES cells, upstream of the 1.9 kb Oct4 fragment (24).
Although pOct8.5 provided ES-specific expression in some clones, none were identified in which all undifferentiated cells were stained (Fig. 1). Shaw-White and colleagues have compared the expression of a single copy lacZ transgene in random sites with its expression when targeted to the HPRT locus (10) and report similar variegation within cell types, both in vitro and in teratocarcinomas. Since the variegation observed both in these experiments and in our own in vitro experiments occurs in all sites examined (including when targeted to the HPRT locus), it is unlikely that it is caused by genomic position. Variegation may arise due to an influence of cell cycle on transcription levels (10). However, for rapidly dividing ES cells, this explanation is inconsistent with the relatively long half-life of the lacZ gene product. A further explanation is offered by reports that neighbouring promoters occasionally inhibit one another (promoter occlusion), possibly as a consequence of transcriptional read-through (10,47–53). Consistent with this explanation, variegated expression in the present study was not apparent in embryos generated from targeted clones. Significantly, a transcription terminator sequence had been introduced into the targeting plasmid pMR in order to prevent read-through (Fig. 1). It remains to be seen if the apparent absence of variegation is borne out by detailed examination of lacZ staining in sectioned embryos.
The neutral site provides an efficient means of transgene insertion
There are now several options for targeting transgenes into predetermined sites in the genome (reviewed in 50), either by homologous recombination (10–13), by site-specific recombination (17–19) or by homologous recombination enhanced by prior insertion of the rare cutting endonuclease cleavage site I-SceI (23). In common with the neutral site approach described here, the site-specific and I-SceI-enhanced strategies require prior modification of a site by gene targeting and can incorporate features which render subsequent targeting easily detectable. Kolb et al. (19) employed a promoterless hygromycin marker, which acquires a promoter following site-specific recombination, and achieved a targeting efficiency of 1/6 hygromycin-resistant clones. By using mutated loxP half-sites which favour the integration event over the excision event, site-specific insertion efficiencies of up to 16% of cells transfected have been reported without any requirement for selection (54). Although targeting efficiency remains orders of magnitude lower for homology-based recombination, the pMR vector compares favourably when efficiency is measured as a proportion of clones analysed. Hence, both the Cre-based approach and the neutral site approach offer simplified targeting procedures appropriate for rapid and efficient introduction of transgenes to single sites.
We have also demonstrated that an ES clone (710) which has been genetically modified to facilitate ‘knock-in’ at a specific site remains capable of colonising the germline in chimeric mice. The frequency of germline transmission was relatively low (1/6 male chimeras), possibly reflecting a mixed karyotype. In order to improve the rate of germline colonisation, it is our intention to reclone this line either by selection and characterisation of single colonies or by re-isolation from transgenic embryos.
In vitro screening for neutral sites can prevent ectopic expression
Mice generated by pronuclear injection of a Myf5/lacZ transgene similar to that in pMR/Myf respond to early Myf5 activating signals in the visceral arches, but not in the somites (39). In addition, many of these lines also reveal additional ectopic expression. When introduced as single copies in predominantly ES-derived embryos, a similar transgene directs only limited skeletal muscle expression and was associated with ectopic expression in one of three lines (39). In the present work, pre-selecting a site for down-regulation of Oct/lacZ in vitro avoided ectopic expression from the Myf5 promoter. The same was true for two further promoters (thyroglobulin and Hox2.6) when recombined into the same ‘neutral site’ (site 710). Hence, the neutral site can provide a single substrate for a battery of transgenic applications.
Candidate locus approaches have the important advantage that the transgene acquires a complete complement of native regulatory elements. As a consequence this is the preferred technique for gene expression studies. For example, randomly situated Myf5/lacZ transgenes rarely reproduce endogenous Myf5 expression patterns. Only when a promoterless lacZ gene was targeted to the Myf5 locus was the complete repertoire of native Myf5 expression observed (39). Nevertheless, it is now emerging that in certain instances the targeted introduction of novel sequences into a candidate locus can have unexpected consequences on neighbouring genes and can lead to hypomorphic phenotypes (48,49). Where the objective is to maximise production of recombinant protein, then utilising natural controlling elements at a target locus places an upper limit on transgene expression which is determined by properties of the candidate locus and by a maximum diploid copy number of two. A further disadvantage of the candidate locus approach is that different target loci will be favoured for different applications. In contrast, the neutral site may provide a suitable environment for high level tissue-specific expression of a variety of transgenes.
To date we have characterised position effects for two sites chosen on the basis of appropriate (site 710) and inappropriate (site 91) transgene expression in an in vitro pre-screen. We have yet to establish unequivocally the status of site 710 as a ‘neutral site’; it remains possible that 710 actually lies within a locus. We anticipate that further analysis of a number of independent sites chosen for appropriate in vitro transgene regulation will identify new candidates for more comprehensive analysis. The present work, however, establishes the principle that in vitro selection for appropriate developmental regulation of a marker is informative for in vivo expression and demonstrates the practicality of using a marked ‘neutral site’ in repeat gene targeting to generate a variety of genetically modified mice. Finally, the neutral locus strategy can be considered a first step towards optimal transgene expression. Subsequent steps could involve modification of the site to introduce elements conducive to transgene expression. Although proven ES cells are presently unavailable in livestock species, we suggest that the present strategy is adaptable to ovine foetal fibroblast cells (46) and can be used in conjunction with nuclear transfer to generate genetically modified livestock. In mouse transgenics the neutral site approach may be particularly useful in reducing animal usage in gene expression studies and for the generation of reliable Cre-expressing ‘deletor’ strains.
Acknowledgments
ACKNOWLEDGEMENTS
We thank the staff of the small animal unit at Roslin Institute for animal care, Elliot Armstrong, Norrie Russell and Roddie Field for photography, Rob Krumlauf for the Hox2.6 promoter, Hans Scholer for the 8.5 kb Oct promoter, Maria Jasin for the nuclear localisation signal, Muriel Lee for teaching H.W. FISH analysis and Bruce Whitelaw and Ian Chambers for helpful discussions. This work was supported by a BBSRC ROPA award.
REFERENCES
- 1.Clark A.J., Bissinger,P., Bullock,D.W., Damak,S., Wallace,R., Whitelaw,C.B. and Yull,F. (1994) Reprod. Fertil. Dev., 6, 589–598. [DOI] [PubMed] [Google Scholar]
- 2.Dorer D.R. (1997) Transgenic Res., 6, 3–10. [DOI] [PubMed] [Google Scholar]
- 3.Gordon J.W., Scangos,G.A., Plotkin,D.J., Barbosa,J.A. and Ruddle,F.H. (1980) Proc. Natl Acad. Sci. USA, 77, 7380–7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grosveld F., Van Assendelft,G.B., Greaves,D.R. and Kollias,G. (1987) Cell, 51, 975–985. [DOI] [PubMed] [Google Scholar]
- 5.Ryan T.M., Behringer,R.D., Martin,N.C., Townes,T.M., Palmiter,R.D. and Brinster,R.L. (1989) Genes Dev., 3, 314–323. [DOI] [PubMed] [Google Scholar]
- 6.Bonifer C., Yannoutsos,N., Kruger,G., Grosveld,F. and Sippel,A.E. (1994) Nucleic Acids Res., 22, 4202–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitelaw C.A.B., Harris,S., McClenaghan,M., Simons,J.P. and Clark,A.J. (1992) Biochem. J., 286, 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonifer C., Huber,M.C., Jagle,U., Faust,N. and Sippel,A.E. (1996) J. Mol. Med., 74, 663–671. [DOI] [PubMed] [Google Scholar]
- 9.Guy L.-G., Kothary,R., DeRepentigny,Y., Delvoye,N., Ellis,J. and Wall,L. (1996) EMBO J., 15, 3713–3721. [PMC free article] [PubMed] [Google Scholar]
- 10.Shaw-White J.R., Denko,N., Albers,L., Doetschman,T.C. and Stringer,J.R. (1993) Transgenic Res., 2, 1–13. [DOI] [PubMed] [Google Scholar]
- 11.Detloff P.J., Lewis,J., John,S.W.M., Shehee,W.R., Lengenbach,R., Maeda,N. and Smithies,O. (1994) Mol. Cell. Biol., 14, 6936–6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stacey A., Schnieke,A., McWhir,J., Cooper,J., Colman,A. and Melton,D.W. (1994) Mol. Cell. Biol., 14, 1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bronson S.K., Plaehn,E.G., Kluckman,K.D., Hagaman,J.R., Maeda,N. and Smithies,O. (1996) Proc. Natl Acad. Sci. USA, 93, 9067–9072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark A.J., Couper,A., Wallace,R., Wright,G. and Simons,J.P. (1992) Biotechnology, 10, 1450–1454. [DOI] [PubMed] [Google Scholar]
- 15.Fu Y., Wang,Y. and Evans,S.M. (1998) Nature Biotechnol., 16, 253–257. [DOI] [PubMed] [Google Scholar]
- 16.Fukushige S. and Sauer,B. (1992) Proc. Natl Acad. Sci. USA, 89, 7905–7909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kolb A.F. and Siddel,S.G. (1997) Gene, 209, 209–216. [Google Scholar]
- 18.Rucker E.B. and Piedrahita,J.A. (1997) Mol. Reprod. Dev., 48, 324–331. [DOI] [PubMed] [Google Scholar]
- 19.Kolb A.F., Ansell,R., McWhir,J. and Siddel,S.G. (1998) Gene, 227, 21–31. [Google Scholar]
- 20.Okamoto K., Okazawa,H., Okuda,A., Sakai,M. and Muramatsu,H. (1990) Cell, 60, 461–472. [DOI] [PubMed] [Google Scholar]
- 21.Melton D.W., Ketchen,A. and Selfridge,J. (1997) Nucleic Acids Res., 25, 3937–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mercer E.H., Hoyle,G.W., Kapur,R.P., Brinster,R.L. and Palmiter,R.D. (1991) Neuron, 7, 703–716. [DOI] [PubMed] [Google Scholar]
- 23.Donoho G., Jasin,M. and Berg,P. (1998) Mol. Cell. Biol., 18, 4070–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeom Y., Fuhrmann,G., Ovitt,C.E., Brehm,A., Ohbo,K., Gross,M., Hubner,K. and Scholer,H. (1996) Development, 122, 881–894. [DOI] [PubMed] [Google Scholar]
- 25.Lathe R., Vilotte,J.L. and Clark,A.J. (1987) Gene, 57, 193–201. [DOI] [PubMed] [Google Scholar]
- 26.McGeady M.L., Wood,T.G., Maizel,J.V. and Vande Woude,G.F. (1986) DNA, 5, 289–298. [DOI] [PubMed] [Google Scholar]
- 27.Ellison A.R., Wallace,H., Al-Shawi,R. and Bishop,J.O. (1995) Mol. Reprod. Dev., 41, 425–434. [DOI] [PubMed] [Google Scholar]
- 28.Whiting J., Marshal,H., Cook,M., Krumlauf,R., Rigby,P.W.J., Stott,D. and Alleman,R.K. (1991) Genes Dev., 5, 2048–2059. [DOI] [PubMed] [Google Scholar]
- 29.Magin T.M., McWhir,J. and Melton,D.W. (1992) Nucleic Acids Res., 20, 3795–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagy A., Rossant,J., Nagy,R., Abramow-Newerly,W. and Roder,J.C. (1993) Proc. Natl Acad. Sci. USA, 90, 8424–8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 32.Asangla A., Monk,M., Lovell-Badge,R. and Melton,D.W. (1988) Development, 104, 465–471. [DOI] [PubMed] [Google Scholar]
- 33.McWhir J., Schnieke,A.E., Ansell,R., Wallace,H., Colman,A., Scott,A.R. and Kind,A.J. (1996) Nature Genet., 14, 223–226. [DOI] [PubMed] [Google Scholar]
- 34.Fantes J., Redeker,B., Breen,M., Boyle,S., Brown,J., Fletcher,J., Jones,S., Bickmore,W., Fukushima,Y., Mannens,M., Danes,S., van Heyningen,V. and Hanson,I. (1995) Hum. Mol. Genet., 4, 415–422. [DOI] [PubMed] [Google Scholar]
- 35.Okazawa H., Okamoto,K., Ishino,F., Ishino-Kaneko,T., Takeda,S., Toyoda,Y., Muramatsu,M. and Hamada,H. (1991) EMBO J., 10, 2997–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Te Reile H., Maandag,E.L. and Burns,A. (1992) Proc. Natl Acad. Sci. USA, 89, 5128–5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawaori A. and Tsuneda,M. (1985) Endocrinology, 108, 518–524. [Google Scholar]
- 38.Whiting J., Marshal,H., Cook,M., Krumlauf,R., Rigby,P.W.J., Stott,D. and Alleman,R.K. (1991) Genes Dev., 5, 2048–2059. [DOI] [PubMed] [Google Scholar]
- 39.Tajbakhsh S., Bober,E., Babinet,C., Pournin,S., Arnold,H. and Buckingham,M. (1996) Dev. Dyn., 206, 291–300. [DOI] [PubMed] [Google Scholar]
- 40.Ott M.-O., Bober,E., Arnold,H. and Buckingham,M. (1991) Development, 111, 1097–1107. [DOI] [PubMed] [Google Scholar]
- 41.Patapoutian A., Miner,J.H., Lyons,G.E. and Wold,B. (1993) Development, 118, 61–69. [DOI] [PubMed] [Google Scholar]
- 42.Wallace H., Ledent,C., Vassart,G., Bishop,J.O. and Al-Shawi,R. (1991) Endocrinology, 129, 3217–3226. [DOI] [PubMed] [Google Scholar]
- 43.Bradley A. (1987) In Robertson,E. (ed.), Teratocarcinomas and Embryonic Stem Cells. IRL Press, Oxford, UK, pp. 113–151.
- 44.Wakayama T., Perry,A.C.F., Zucotti,M., Johnson,K.R. and Yanagimachi,R. (1998) Nature, 394, 369–374. [DOI] [PubMed] [Google Scholar]
- 45.Wilmut I., Schnieke,A.E., McWhir,J., Kind,A.J. and Campbell,K.H.S. (1997) Nature, 385, 810–813. [DOI] [PubMed] [Google Scholar]
- 46.Schnieke A., Kind,A.J., Ritchie,W.A., Mycock,K., Scott,A.R., Ritchie,M., Wilmut,I., Colman,A. and Campbell,K.H.S. (1997) Science, 278, 2130–2133. [DOI] [PubMed] [Google Scholar]
- 47.Olsen E.N., Arnold,H.-H., Rigby,P.W.J. and Wold,B.J. (1996) Cell, 85, 1–4. [DOI] [PubMed] [Google Scholar]
- 48.Nagy A., Moens,C., Ivanyi,E., Pawling,J., Gertsenstein,M., Hadjantonakis,A.-K., Pirity,M. and Rossant,J. (1998) Curr. Biol., 8, 661–664. [DOI] [PubMed] [Google Scholar]
- 49.Meyers E.N., Lewandoski,M. and Martin,G.R. (1998) Nature Genet., 18, 136–141. [DOI] [PubMed] [Google Scholar]
- 50.Bartolomei M.S., Webber,A.L., Brunkow,M.E. and Tilghman,S.M. (1993) Genes Dev., 7, 1663–1673. [DOI] [PubMed] [Google Scholar]
- 51.Jasin M., Moynahan,M.E. and Richardson,C. (1996) Proc. Natl Acad. Sci. USA, 93, 8804–8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kadesch T. and Berg,P. (1986) Mol. Cell. Biol., 6, 2593–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim C.G., Epner,E.M., Forrester,W.C. and Groudine,M. (1992) Genes Dev., 6, 928–938. [DOI] [PubMed] [Google Scholar]
- 54.Araki K., Araki,M. and Yamamura,K. (1997) Nucleic Acids Res., 25, 868–872. [DOI] [PMC free article] [PubMed] [Google Scholar]