

Abstract
INTRODUCTION:
An estimated 160 million years have passed since the first placental mammals evolved. These eutherians are categorized into 19 orders consisting of nearly 4000 extant species, with ~70% being bats or rodents. Broad, in-depth, and comparative genomic studies across Eutheria have previously been unachievable because of the lack of genomic resources. The collaboration of the Zoonomia Consortium made available hundreds of high-quality genome assemblies for comparative analysis. Our focus within the consortium was to investigate the evolution of transposable elements (TEs) among placental mammals. Using these data, we identified previously known TEs, described previously unknown TEs, and analyzed the TE distribution among multiple taxonomic levels.
RATIONALE:
The emergence of accurate and affordable sequencing technology has propelled efforts to sequence increasingly more non-model mammalian genomes in the past decade. Most of these efforts have traditionally focused on genic regions searching for patterns of selection or variation in gene regulation. The common trend of ignoring or trivializing TE annotation with newly published genomes has resulted in severe lag of TE analyses, leading to extensive undiscovered TE variation. This oversight has neglected an important source of evolution because the accumulation of TEs is attributable to drastic alterations in genome architecture, including insertions, deletions, duplications, translocations, and inversions. Our approach to the Zoonomia dataset was to provide future inquirers accurate and meticulous TE curations and to describe taxonomic variation among eutherians.
RESULTS:
We annotated the TE content of 248 mammalian genome assemblies, which yielded a library of 25,676 consensus TE sequences, 8263 of which were previously unidentified TE sequences (available at https://dfam.org). We affirmed that the largest component of a typical mammalian genome is comprised of TEs average 45.6%). Of the 248 assemblies, the lowest genomic percentage of TEs was found in the star-nosed mole (27.6%), and the largest percentage was seen in the aardvark (74.5%), whose increase in TE accumulation drove a corresponding increase in genome size—a correlation we observed across Eutheria. The overall genomic proportions of recently accumulated TEs were roughly similar across most mammals in the dataset, with a few notable exceptions (see the figure). Diversity of recently accumulated TEs is highest among multiple families of bats, mostly driven by substantial DNA transposon activity. Our data also exhibit an increase of recently accumulated DNA transposons among carnivore lineages over their herbivorous counterparts, which suggests that diet may play a role in determining the genomic content of TEs.
CONCLUSION:
The copious TE data provided in this work emanated from the largest comprehensive TE curation effort to date. Considering the wide-ranging effects that TEs impose on genomic architecture, these data are an important resource for future inquiries into mammalian genomics and evolution and suggest avenues for continued study of these important yet understudied genomic denizens.
We examined transposable element (TE) content of 248 placental mammal genome assemblies, the largest de novo TE curation effort in eukaryotes to date. We found that although mammals resemble one another in total TE content and diversity, they show substantial differences with regard to recent TE accumulation. This includes multiple recent expansion and quiescence events across the mammalian tree. Young TEs, particularly long interspersed elements, drive increases in genome size, whereas DNA transposons are associated with smaller genomes. Mammals tend to accumulate only a few types of TEs at any given time, with one TE type dominating. We also found association between dietary habit and the presence of DNA transposon invasions. These detailed annotations will serve as a benchmark for future comparative TE analyses among placental mammals.
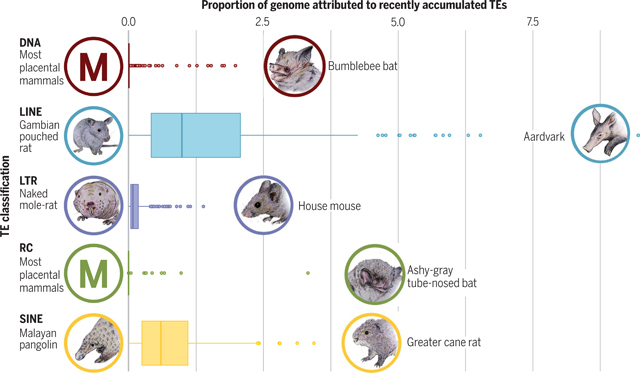
Boxplots depicting the range of recently accumulated TEs among mammals (by proportion of genome). Five categories of TE were examined: DNA transposons, long interspersed elements (LINEs), long terminal repeat (LTR) retrotransposons, rolling circle (RC) transposons, and short interspersed elements (SINEs). Species with the highest and lowest proportions for each TE type are indicated by a picture of the organism and its common name. With regard to RC and DNA transposons, we found that most mammalian genome assemblies exhibit essentially zero recent accumulation (RC: 240 of 248 mammals had <0.1%; DNA: 210 of 248 mammals had <0.1%).
Barbara McClintock became a scientific pioneer in the field of genomics with her Nobel Prize–winning discovery of transposable elements (TEs)—DNA sequences that can mobilize themselves in host genomes (1). A ubiquitous component of nearly all eukaryotes (2), TEs are typically classified into two major groups on the basis of their mobilization mechanism (3). Class I elements, also known as retrotransposons, use an RNA intermediate during transposition, allowing replication throughout the genome in a copy-and-paste style of mobility (4). Class I elements can be sorted further into three subcategories: short interspersed elements (SINEs), long interspersed elements (LINEs), and long terminal repeat (LTR) retrotransposons (5). SINEs are nonautonomous elements and depend on the presence of functional LINE elements, which contain anywhere from one to three open reading frames (ORFs) encoding the necessary proteins for mobilization. Class II elements, also known as DNA transposons, use a DNA intermediate and can also be subdivided. Terminal inverted repeat (TIR)–like DNA transposons, such as hATs, piggyBacs, and TcMariner transposons, use a cut-and-paste mechanism by using transposase enzymes to catalyze the TE’s relocation (6). Helitrons, a second subcategory of class II elements, use a rolling circle mechanism (7). The final subcategory of known DNA transposons are Maverick elements, which are thought to be derived from viruses because they have homologous genes coding for DNA polymerase and retroviral-like integrase (8).
An increase in activity from either class of elements can lead to marked alterations in genome architecture (9). A variety of changes, including insertions, duplications, translocations, deletions, and inversions, can result from TE mobilization and accumulation (9). For instance, the AMAC1 (acyl-malonyl condensing enzyme 1) gene, coding for a protein that is essential for breaking down phytanic acid from meat and dairy foods, has undergone multiple recent gene duplications mediated by SVA retrotransposons in the human genome (10, 11). In addition to these structural variants, the proliferative mechanisms of TE mobilization tend to cause eukaryotic genome sizes to linearly correlate with TE abundance (2).
Increasing evidence indicates that TE-derived sequences have substantially influenced the evolutionary histories of the organisms they occupy, even contributing to major evolutionary innovations benefiting host organisms. Examples include recent TE insertions into genes involved with insecticide resistance of the cotton bollworm (12), the rapid adaptation leading to melanistic phenotypes of peppered moths in the soot-ridden environment of British industrialization (13), and the myriad endogenous retroviruses that have contributed regulatory functions to the development and evolution of the mammalian placenta (9, 14). Most TE insertions, however, result in selectively neutral alterations in genome architecture, often showing no perceptible effect on host fitness (15). That being said, deleterious insertions do occur, and impairments in gene function are possible outcomes of TE mobilization, which can lead to a wide variety of genetic diseases (9).
As a result, numerous genomic TE defense mechanisms have evolved to combat TE activity by either regulating TE transcription or by targeting their intermediates to prevent integration into the genome (3). These defense mechanisms explain, in part and in some organisms, why few TE families retain the ability to mobilize over long periods of evolutionary time (16). For example, among the ~868,000 L1 insertions in the human genome, few are thought to be retrotransposition competent, and many of these exhibit cell type–specific mobilization profiles (3, 17). Alternatively to or in conjunction with the aforementioned scenario of low numbers of functionally mobile TEs among some categories of elements, genomic drift and the corresponding effects of fixation events among bottlenecked populations give rise to another explanation for varying levels of TE accumulation in different genome assemblies (18).
All these facets suggest that determining TE dynamics is key to understanding how genomes evolve and function. Thus, TE curation and annotation is one of the most important initial investigative steps in any description of a de novo genome assembly. Unfortunately, this step is often relegated to an afterthought rather than performing a time-intensive, de novo TE curation effort (19). As a result, many genome assemblies are misunderstood from a TE perspective (19). As the scientific community improves genome sequencing and assembly, the lack of thorough and accurate TE annotation promises to become a major problem, especially in the face of the number of large-scale genome sequencing initiatives now underway (20–24).
The Zoonomia project, described in (24), represents an opportunity to gain substantial knowledge about the diversity of TEs in an important vertebrate clade, Mammalia. We fill this knowledge gap by providing complete, de novo TE annotations of 248 Zoonomia mammalian genome assemblies using homology, de novo, and manual annotation approaches.
General TE trends among mammals
RepeatModeler (25), a de novo TE discovery tool, was used to examine 248 mammalian genome assemblies yielding 25,025 putative TE starting queries. After initial curation and elimination of duplicates, an iterative curation process consisting of between 1 and 19 rounds of detailed curation (19) depending on the species (see Materials and methods) yielded a library consisting of 8263 previously unidentified consensus sequences. That library was combined with known TEs to create a comprehensive mammalian TE library. This library, consisting of 25,676 consensus sequences, was used to mask all assemblies. The dynamics of TE biology and intricacies of TE detection lend themselves to a degree of false detection. For example, some TE families are chimeras of multiple elements, or they may contain similar core sequence components. To evaluate the potential for false positives, we took advantage of an idiosyncrasy of TE biology in bats. A family of bats, the Vespertilionidae, is, to our knowledge, the sole mammalian family to have incorporated a type of rolling circle transposon, Helitrons, into their TE repertoire (3). True Helitrons in mammals have not been detected outside of Vespertilionidae. Thus, any Helitrons detected outside of vesper bats would likely be a false positive. RepeatMasker (26) detected Helitrons in nonvesper mammals at a rate of 0.0013 ± 0.0019, suggesting a low false positive rate.
Previous work has suggested that the largest single classifiable component of a typical mammalian genome is TEs (27), and our data (Fig. 1) corroborate this. As noted previously by Elliott and Gregory in 2015 (2), genome size linearly correlates with the percentage of TE content within a genome, and this is again supported by our data (Fig. 1 and table S1). Overall, TE content in each of the examined species ranges from a low of 27.6% in the star-nosed mole (Condylura cristata) to 74.5% in the aardvark (Orycteropus afer) (table S2 and Fig. 1), with a distinct tendency to cluster in the middle of that range (average TE proportion: 45.6%, average genome size: 2.67 Gb). The hazel dormouse (Muscardinus avellanarius) and the Brazilian guinea pig (Cavia aperea) represent the extremes of this middle cluster, with 65.8 and 28.1% total TE contents, respectively. Assembly quality may affect the accuracy of TE annotation, but we could find no statistically significant trend among taxa. For example, lower-quality assemblies as measured by N50 or BUSCO completeness did not yield lower or higher rates of observed TE accumulation (figs. S1 and S2).
Fig. 1. Correlation of total genomic TE content and the size, in base pairs, of the genome.

Because of the log transformation and scaling of assembly size for the hierarchical Bayesian analysis and the resulting back-transformation, the x-axis values are approximately rendered. The blue line indicates the line of best-fit, and the shaded area is the 95% high probability density of the fit. The R2 for this relationship was estimated at 0.54 (95% high probability density, 0.42 to 0.64).
TE variation among mammals
When examining TE content from all categories across the mammalian tree, we find some general trends. For example, SINEs and LTR retrotransposons are more prevalent in Euarchontoglires, whereas LINEs dominate most other lineages, especially the bovids (Fig. 2). However, we find that placental mammals are generally similar with regard to overall TE proportions, reflecting the tendency to retain older insertions that occurred in the common ancestor of mammals. LINEs and SINEs always make up most TE abundance both in copy number and in total genomic percentage. LINEs occupy between 8.2 and 52.8% of the genomes examined, averaging 22.6%. SINEs occupy on average 10.5% of the mammalian genome (range, 0.4 to 32.1%) (table S3), whereas LTR retrotransposons, DNA transposons, and rolling circle transposons are substantially rarer—7.8% (range, 2.0 to 17.8%), 3.5% (range, 0.5 to 8.4%), and 0.5% (range, 0.01 to 19.7%), respectively.
Fig. 2. Total and young TE genomic proportions by species within a phylogenetic context.

Dots at branch tips indicate the TE class most prevalent among recent TE insertions (insertions with <4% divergence from the relevant consensus TE). The ring immediately following the branch tip dots indicates the mammalian order for each respective species. Orders represented by numbers are as follows: 1, Cingulata; 2, Pilosa; 3, Sirenia; 4, Proboscidea; 5, Hyracoidea; 6, Macroscelidea; 7, Tubulidentata; 8, Afrosoricida; 9, Scandentia; 10, Dermoptera; 11, Lagomorpha; 12, Eulipotyphla; 13, Perissodactyla; and 14, Pholidota. The inner ring of stacked-bar data depicts the total percentage of the genome attributed to the five main categories of TEs: DNA transposons, LINEs, SINEs, LTRs, and rolling circle transposons. The outer ring of stacked-bar data shows the percentage of the genome derived from recently inserted TEs. Cladogram adapted from (65).
Examination of younger insertions—those with divergences averaging <4% from their respective consensus—provides a picture of these genomes that is more dynamic, revealing substantial differences in accumulation from each category of TE (table S4). Some lineages, such as the pteropodid bats (Pteropus alecto, Pteropus vampyrus, Eidolon helvum, and Rousettus aegyptiacus in Fig. 2), exhibit essentially no recent accumulation by any TE category, whereas others have experienced massive expansions in one or more categories. The aardvark (Orycteropus afer) and musk deer (Moschus moschus), for instance, show substantial LINE accumulation over the past ~20 million years.
To examine these trends more closely, we conducted a redundancy analysis (RDA) for both orders and families to identify the major axes of variation in TE composition that were related to either order or family affiliation of taxa (Fig. 3). This analysis suggests a strong phylogenetic component to variation in TE composition among clades at the levels of order and family. Eleven orders of mammals were significantly correlated with at least one of the two axes, and these orders were quite variable in terms of association with different TE types. The first two major axes of variation in TE accumulation in analyses examining orders accounted for ~27.2% of the variation, and this was highly significant (P < 0.001). The first major axis was positively related to the number of young TEs generally and to young LINEs, LTRs, and SINEs, which are all obligately replicative. Unsurprisingly given this characteristic, genome size was also positively correlated with this axis. This axis was negatively related to young DNA transposons and young rolling circle transposons. The second major axis of TE composition related to ordinal affiliation was positively related to the number of young DNA transposons, rolling circle transposons, LINEs, and young TEs more generally, but it was negatively related to young LTRs, SINEs, and genome size.
Fig. 3. Redundancy analyses examining major axes of variation in TE accumulation and genome size related to orders and families of mammals.

Arrows represent significant correlations of TE types with the first two RDA axes. Each axis reflects changes in TE composition related to ordinal (top) or familial (bottom) affiliation of taxa used in analyses. Gray circles represent orders or families that were not significantly correlated to at least one of the RDA axes, whereas black circles represent orders or families with significant correlations.
Similar associations are seen at the family level. Families of mammals accounted for ~49.9% of variation in TE composition, and this was highly significant (Fig. 3; P < 0.001). As with orders, the first major axis of variation was positively related to the same categories of TE and to genome size. Correlations of young DNA transposons and young rolling circle TEs were weaker than for orders, likely because of the lineage specificity of those element types (see next section), whereas positive associations of all other TE types were stronger. The second major axis was positively related to the number of young DNA transposons, rolling circle transposons, LINEs, and young TEs generally and was negatively related to genome size. Fourteen families of mammals were significantly correlated with at least one of these two axes, and these families were variable in terms of association with different TE types.
TE diversity
An increasingly useful avenue of inquiry among whole-genome TE analyses draws from community ecology (28). The application of community diversity measures rendered on a genomic scale is of particular interest (29). We followed these lines of inquiry by investigating the diversity of recent TEs in each genome by calculating two diversity indices and applying them to our data—the Shannon diversity index (30) and Pielou’s J (31). Shannon diversity (H) is a measure of overall diversity in a population of objects, and Pielou’s J measures evenness by incorporating the relative numbers of each object—in this case, TE types (table S5). Species with the highest diversity values include bats and rodents. Bat TE diversity was driven primarily by recent expansions of DNA transposons among Craseonycteridae, Vespertilionidae, Hipposideridae, Rhinolophidae, and Mollossidae and recent accumulations of both DNA transposons and rolling circle transposons in Vespertilionidae (Fig. 4).
Fig. 4. Stacked bar charts depicting proportions of recently accumulated TEs (<4% kimura divergence from consensus TE) in bats.

Data are organized by TE classification and plotted onto the tips of the chiropteran portion of the mammalian tree, adapted from (65).
In rodents, higher diversity among recently inserted TEs was driven by accumulations in LTR retrotransposons, which made up 10 to 53% of recent TE accumulation. The highest rate of recent LTR accumulation among the rodents was seen in members of Cricetidae and Cricetomys gambianus.
To investigate general trends in diversity index values in relation to TE accumulation patterns, we plotted values from recently deposited TEs versus each diversity index (Fig. 5). Hierarchical Bayesian analyses indicate that both Shannon diversity and Pielou’s J exhibit significant negative relationships with increasing recent TE content [Shannon H (Fig. 5 and table S6) and Pielou’s J (Fig. 5, table S7, and fig. S3)]. Thus, the downward trend in Pielou’s J suggests that mammalian genomes tend to accumulate individual TE types at any given period rather than multiple TE types accumulating simultaneously. This is exemplified in the aardvark, where LINEs are currently dominating the recently active mobilome, whereas SINEs are the major recent contributor to the greater cane rat (Thryonomys swinderianus) genome (Fig. 2). However, clades of bats with recent DNA accumulation tend to refute this pattern.
Fig. 5. Recent mammalian TE diversity in relation to Shannon H and Pielou’s J.

The blue lines indicate the lines of best-fit, and the shaded areas are the 95% high probability density of the fit. The R2 for H (left) was estimated at 0.67 (95% high probability density, 0.52 to 0.78), and for J (right), the R2 was 0.69 (95% high probability density, 0.56 to 0.79).
DNA transposons and diet
The lineage specificity of the DNA transposon diversity described above suggests horizontal transfer (HT) as a potential source for TE invasions in certain mammalian genomes. To investigate patterns that may explain how such HT events might occur, we examined the potential for life history to play a role. We hypothesized that differences in diet may allow select species to come into contact with vectors for TEs (14, 32), which increase the likelihood of successful invasion of mammalian genomes. DNA transposon–rich food sources, such as many arthropods and nonmammalian vertebrates, may offer greater potential for HT to some species compared with those that eat plants. Hierarchical Bayesian analyses indicate that carnivorous mammals tend to accumulate more recent DNA transposons in their genomes compared with noncarnivores (Fig. 6A and table S8). This pattern is best exemplified in the cetartiodactyls (Fig. 6B). Recent DNA transposon accumulation is seen on average 20 times as much among the cetaceans compared with other artiodactyls. Carnivorous bats, however, did not have statistically higher accumulations of recent DNA transposons compared with herbivorous bats (Fig. 6C). Our datasets of primates and rodents did not reveal any statistical difference in recent DNA transposon accumulation between herbivores and omnivores (Fig. 6, D and E).
Fig. 6. Half eye plots depicting fold differences in recent DNA transposon accumulation among three dietary phenotypes: carnivore, herbivore, and omnivore.

Instead of showing the estimated values for each of the diets, these plots depict the fold ratio between each diet pair, so that the plot itself shows statistical significance. Comparisons for which the thin line does not overlap with 1 are significant (indicated by asterisks). Plots correspond to the following taxonomic groups: (A) placental mammals [R2 estimated at 0.92 (95% high probability density, 0.79 to 0.97)], (B) Artiodactyla [R2 estimated at 0.64 (95% high probability density, 0.32 to 0.78)], (C) Chiroptera [R2 estimated at 0.34 (95% high probability density, 0.02 to 0.86)], (D) Primates [R2 estimated at 0.18 (95% high probability density, 0.00 to 0.58)], and (E) Rodentia [R2 estimated at 0.07 (95% high probability density, 0.00 to 0.28)].
Discussion
As our ability to generate high-quality genome assemblies in rapid succession improves, the need to curate TEs in those assemblies will only increase. Toward that end, we performed a de novo assessment of the TE content of 248 mammal genome assemblies in what is, to our knowledge, the largest comprehensive TE curation effort to date. This represents an increase of ~58% compared with known mammalian TEs in RepBase as of 2019, when we began. Given the numerous effects that TEs are known to have at multiple levels of genome organization and function, this increased knowledge will serve as a particularly valuable resource for anyone interested in mammalian genomics and evolution. The full set of TE consensus sequences is available for download from the Dfam (33) database.
Previous work has noted that genome size among mammals is relatively constrained (34), and this work does not contradict that observation. Despite this constraint, our work reveals that there is substantial variation in rates of accumulation in the recent mammalian past. We found that there is substantial diversity in TE accumulation patterns among mammals, which suggests distinct TE-induced pressures on those genomes over evolutionary time and, likely, distinct differences in the ability of eutherians to defend their genomes against TEs. These differences represent an excellent opportunity for future researchers to investigate how TE defenses evolve and respond to differing TE loads.
Another avenue of such research is to further investigate TE accumulation through the lens of ecology and environment, an idea that has been discussed previously (14). Our data demonstrate that carnivorous lineages tend to harbor an excess of recently accumulated DNA transposons when compared with herbivorous taxa. The tendency of meat-eating mammals to have more recent DNA transposon accumulation compared with their non-carnivorous counterparts suggests that diet may play a significant role in a genome’s likelihood of experiencing HT from class II TEs. This scenario is supported in part by a recent analysis of HT in predator-prey pairs and their shared parasites (32). Nevertheless, this finding is not uniform across mammalian orders, and those varying patterns may reflect defenses against TE invasion (3), less availability of TEs in order-specific dietary items, or some combination of both.
Investigating mammalian TEs through the ecological lens also suggests that single TE types tend to dominate the mobilome during any given period (Fig. 5). This scenario is consistent with our current understanding of TE defense mechanisms. The current model of PIWI-mediated TE defense suggests that a heretofore unencountered TE may invade or arise in a genome and enjoy a period of relatively unfettered mobilization. Eventually, the PIWI-interacting RNA (piRNA) defenses generate an effective response and dampen the invading TE’s effects (16, 35, 36).
With regard to the prevalence of HT of DNA transposons in carnivores, our data support the hypothesis that the prevalence of HT of DNA transposons may be a consequence of the similar cellular environments of predator and prey and their necessarily shared environments and frequent interactions. Recent research has demonstrated the role that viruses and blood-feeding arthropods play in facilitating HT (14, 32). Frequent interactions would further facilitate HT by bringing such vectors into contact with both predator and prey. The similar cellular environments among animals (as opposed to mammals with plant-based diets) would further encourage the ready transfer of DNA transposons, which are already more amenable to HT because of their relatively weak dependence on a host’s cellular machinery to mobilize (37).
In conclusion, the annotation data provided in this work are essential for answering future questions related to emerging hypotheses around speciation, such as the TE-thrust hypothesis, the epi-transposon hypotheses, or the carrier subpopulation hypothesis (3, 38). As anthropogenic change exacerbates the decline in effective population size for many of the species in our dataset, TEs might be the reservoir of genomic mutagens that future populations or species rely on.
Materials and methods
Generating the mammalian TE library
A total of 248 genome assemblies of placental mammals were initially presented for analysis (table S2). For six species, higher-quality assemblies were available via Bat1k, a similar, large-scale genome sequencing and assembly effort (21). In those cases, we replaced the Zoonomia assembly with the higher-quality version. Some assemblies were not used in the development of our final mammalian TE library because of one or more of the following reasons: (i) the assembly exhibited a low N50 value (<20,000) resulting in short contigs, which are unsuitable for identifying longer TEs; (ii) multiple artifacts of assembly error were observed at TE sites, which yielded implausible consensus sequences; or (iii) a thorough, species-specific TE annotation had already been performed and is available from RepBase (Genetic Information Research Institute) (39), previous work from our own laboratory, or work conducted by a collaborator. This left us with 205 species as substrates for TE curation (table S2).
Mammalian genomes have only a minimal tendency to remove older TE insertions from the genome (40). Thus, most older TE families that mobilized in the common ancestor or early in the mammalian diversification were likely already characterized through efforts that focused on any of several model organisms, such as human, mouse, rat, pig, dog, cat, and horse (41–47). To avoid wasted effort on recuration of these shared and previously described TEs, we focused our manual curation efforts on identifying newer putative TEs that underwent relatively recent accumulation. We defined such young insertions as TEs with sequences with K2P genetic distances <4% when compared with their respective consensus. For temporal orientation, a kimura divergence of 4% approximates 20 million years or less since insertion, based on a general mammalian neutral mutation rate of 2.2 × 10−9 (48). The use of a general mutation rate allowed for consistency among K2P values in analyses; however, it limits the accuracy of species-specific temporal estimations due to varying neutral mutation rates among placental mammals. Thus, results with divergence values of <4% are considered young and do not provide exact dates. This approach yielded mostly lineage specific TEs, many of which were yet to be described, but some previously identified and shared elements were occasionally encountered (i.e., the Tigger family of Tc Mariner transpsosons and others), suggesting that we did not miss older but unidentified elements. Custom scripts associated with the identification of younger elements are available on Zenodo (49).
For details of the curation process, see previous work from Platt et al. (19). Briefly, for each iteration of manual TE curation, de novo consensus sequences were generated from the 50 BLAST hits that shared the highest sequence identity to the consensus used in our BLAST query for that iteration. Custom pipelines accomplished this by aligning BLAST hits with MUSCLE (50), trimming alignments with trimAl (-gt 0.6 -cons 60) (51), and estimating a consensus sequence with EBMOSS (cons -plurality 3 -identity 3) (52). Files that resulted in <10 BLAST hits were discarded. To consider a consensus sequence complete, the alignment needed to exhibit a pattern of random sequence at both the 5′ and 3′ ends or after extension to a length of 7 kb or greater, whichever came first.
Because the ubiquitous LINE-1 can introduce copies of any transcript into the genome, mammalian genomes have an unusually high number of processed pseudogenes (53–55). Including these in a repeat database would result in annotation of functional genes as TE copies. Comparisons with protein (domain) databases (https://www.ncbi.nlm.nih.gov/protein/, https://useast.ensembl.org/index.html) we found and removed 152 such entries, most characterized by a poly A tail. Small structural RNAs often occur in higher copy numbers partially because they are also substrates of LINE1 (56), and a further 49 entries were dismissed as models created from their genes and pseudogenes.
Two or three copies of interspersed repeats with very high copy numbers, usually but not exclusively SINEs, can often be found in tandem clusters. This occurs more than by chance due to target site preferences. For example, LINE-1–dependent SINEs insert in A-rich DNA, and such sites are introduced by their own poly A tails (57). These artifacts are often identified by de novo repeat finders but can be recognized when studying the seed alignments. Models will also have been built for the individual units, and many copies will end at the joining region between the units—the joining region is more variable than the rest of the model. More than 210 models were such artifacts and were eliminated.
Because in mammals most LTR elements are represented by solo LTRs (58), Dfam (33) and Repbase (39) harbor separate models for the LTRs and the internal sequences. De novo repeat finders like RepeatModeler often produce full elements or reconstruct a (partial) LTR and a fragment of the internal sequence. We split these models into their components, based on homology to well-defined LTRs and the presence of tRNA primer binding sites.
The combined original library contained several redundant models. Recognizing that models represent (fragments of) the same TE is complicated by incorrect base calls, indels, overextension, and incompleteness of the reconstruction as well as by the evolution of class I TEs in the genome: Copies created at different evolutionary times or from different descendants of the ancestral TE (sometimes subtly) differ. A solid test for redundancy is to match the genome to all related models simultaneously and find that some models are always outcompeted by others or that models converge to the same consensus sequence. This could only be accomplished once the database was finalized, so we applied arbitrary but informed cutoffs. Before comparison with each other, the low-complexity tails of SINEs and LINEs were set to a standard length and short overextensions were trimmed based on the expected signatures of terminal bases or target site duplications. Differences between models at possible (highly mutagenic) CpG sites were ignored. Dependent on class and age, elements were removed with alignment scores against another model with a more complete sequence or a better seed alignment that were between 90 and 95% of the score against itself. Partially overlapping fragments of potentially the same TE were not addressed at this point.
We eliminated duplicated entries only when they were built from the same assembly. The same TE can be reconstructed from the genomes of different species if it was active before their speciation time, but with our current approach we could not estimate if a repeat was shared or lineage-specific and merely similar. Thus in Dfam (33), each of the models of this study currently is associated with only one species and will not be matched when a same model is present in another species library.
To confirm the TE type, each sequence in the library was subjected to a custom pipeline (49), which used blastx to confirm the presence of known ORFs in autonomous elements, RepBase (39) to identify known elements, and TEclass (59) to predict the TE type. We also used structural criteria for categorizing TEs. DNA transposons were identified as elements with visible TIRs. Rolling circle transposons were required to have identifiable ACTAG at one end. Putative SINEs were inspected for a repetitive tail as well as A and B boxes. SINEs were also classified by comparison with a database of SINE modules (33): 800 small RNA class III promoter regions, 150 core regions, and 5500 3′ ends of LINE elements (which SINEs often share). LTR retrotransposons and solo LTRs were required to have recognizable hallmarks, such as TG, TGT, or TGTT at their 5′ and the inverse at the 3′ ends and the presence of a polyadenylation signal. LTR classes could often be assigned by (indirect) sequence homology to a coding internal sequence, when present. After this process, 8263 models and their seed alignments were submitted to Dfam (33).
Once the final mammalian TE library was created, we used RepeatMasker-4.1.0 to mask the genome assemblies. Postprocessing of output was performed using the rm2bed.py utility included with RepeatMasker, which merges overlapping hits and converts the output to bed format.
Plotting TE variation using ordination
To characterize the major axes of variation of young TE accumulation among taxa, we conducted a redundancy analysis for both orders and families. In these analyses, the number of base pairs attributed to each TE type as well as the genome size for each taxon (order or family) were the dependent matrix and dummy variables (60), and assigning a species to either family or order was the independent matrix. Redundancy is a multivariate regression that aims to examine the amount of variation and its statistical significance in the dependent matrix that can be accounted for by the independent matrix. Associations among variables where quantified based on a correlation matrix, and significance was determined based on 9999 permutations of the original datasets. Redundancy analyses were performed in Canoco version 5 (61).
Test for association between TE proportions and assembly size, two diversity indices, and diets
The three objectives of these analyses included (i) quantifying the association, if any, between the total TE proportion in genome and assembly size; (ii) estimating the difference in proportions of recently accumulated DNA transposons within a genome among species with different diets; and (iii) quantifying the association, if any, between recent TE proportion in a genome and two diversity indices.
Diversity indices
An increasingly useful avenue for characterizing TE accumulation draws on community ecology (28). Of particular interest is the application of community diversity measures rendered on a genomic scale (29). We followed these lines of inquiry by investigating recent TE diversity within each genome of our dataset by calculating the Shannon diversity index of TE classes. Focusing on recently inserted TEs, we summed the bases that were attributed to TEs with K2P values <4%. We then generated the proportions for each TE class attributed to the overall base pair total of recently inserted TEs. To calculate the Shannon diversity index, , we used the equation
To calculate the evenness of recent TE accumulation among the five main categories of TEs, we used the ecological metric, Pielou’s —a measure of species evenness. Here, was equal to the total number of recent TE hits found within an assembly
Dietary data
We gathered diet classification from the Animal Diversity Web (https://animaldiversity.org/) for 178 available mammals on the public database (table S8). The young DNA transposon dataset was then compared against three diet types: carnivore, herbivore, and omnivore.
Hierarchical Bayesian analyses
A hierarchical Bayesian approach was adopted to simultaneously estimate the species-specific structure of errors while estimating error for the beta-distributed proportion of TEs in the genome. A hierarchical approach is often called a mixed model in the literature, with cluster-specific effects called random and sample-wide effects called fixed. Because different fields apply random and fixed to different levels of the hierarchy, we adopt the language of cluster-specific and sample-wide effects (62). Analyses begin by modeling the proportion of genome as a function of the genome assembly size as a beta-distributed variable (63)
in which is the mean and relates to the variance such that
Given observations and covariate assembly size
Instead of a typical regression, in which observations are presumed to be independent, our analyses account for the phylogenetic structure of the errors by including normally distributed, species-specific effects with phylogenetic errors (64), such that
in which the phylogenetic relationship matrix (65) replaces the identity of observations for the residuals. The same distribution of the response and its phylogenetic errors was applied across all regressions.
Assembly sizes in base pairs were on the order of 109. To enable efficient modeling, this predictor was log10 transformed and then scaled (subtracting the mean and dividing by one standard deviation). No other predictor variables were transformed. Analyses of the association between diet and TE proportions used diet as a group-specific predictor.
To implement Bayesian sampling for these analyses, we used brms (66), a package that enables coding models in R for implementation in the stan statistical language (67). We ran separate univariate models for each set of predictors (assembly size, diet, Shannon diversity index, and Pielou’s evenness index), with the proportion of TE in the genome as the response. The covariance matrix A was obtained from the variance covariance matrix of the dated phylogeny (65) of sampled species. Models ran four separate Markov chain Monte Carlo chains using a Hamiltonian Monte Carlo (HMC) approach. Compared with other Bayesian implementations, the HMC approach saves time in sampling parameter spaces by generating efficient transitions spanning the posterior based on derivatives of the density function of the model. We used the approach of Gelman et al. (68) to estimate the coefficient of determination (R2) from hierarchical Bayesian models. This approach divides the variance of the predicted values by the variance of predicted values plus the expected variance of the errors.
Supplementary Material
ACKNOWLEDGMENTS
We thank the High-Performance Computing Center at Texas Tech University for providing computer resources and technical support throughout the project. This work was also made possible by the SeaWulf computing system from Stony Brook Research Computing and Cyberinfrastructure and by the Institute for Advanced Computational Science at Stony Brook University, funded by the National Science Foundation (NSF) (OAC 1531492). We also thank B. A. Hale for providing artistic renditions of mammalian taxa for our figures.
Funding:
This project was partially supported by NSF grant DEB 1838283 (D.D.M.-S. and D.A.R.), NSF grant IOS 2032006 (D.D.M.-S. and D.A.R.), National Institutes of Health (NIH) grant R01HG002939 (J.M.S., R.H., A.F.A.S., and J.Ros.), NIH grant U24HG010136 (J.M.S., R.H., A.F.A.S., and J.Ros.), NSF grant DEB 1838273 (L.M.D.), NSF grant DGE 1633299 (L.M.D.), NIH grant NHGRI R01HG008742 (Zoonomia Consortium), and a Swedish Research Council Distinguished Professor Award (Zoonomia Consortium).
Zoonomia Consortium
Gregory Andrews1, Joel C. Armstrong2, Matteo Bianchi3, Bruce W. Birren4, Kevin R. Bredemeyer5, Ana M. Breit6, Matthew J. Christmas3, Hiram Clawson2, Joana Damas7, Federica Di Palma8,9, Mark Diekhans2, Michael X. Dong3, Eduardo Eizirik10, Kaili Fan1, Cornelia Fanter11, Nicole M. Foley5, Karin Forsberg-Nilsson12,13, Carlos J. Garcia14, John Gatesy15, Steven Gazal16, Diane P. Genereux4, Linda Goodman17, Jenna Grimshaw14, Michaela K. Halsey14, Andrew J. Harris5, Glenn Hickey18, Michael Hiller19,20,21, Allyson G. Hindle11, Robert M. Hubley22, Graham M. Hughes23, Jeremy Johnson4, David Juan24, Irene M. Kaplow25,26, Elinor K. Karlsson1,4,27, Kathleen C. Keough17,28,29, Bogdan Kirilenko19,20,21, Klaus-Peter Koepfli30,31,32, Jennifer M. Korstian14, Amanda Kowalczyk25,26, Sergey V. Kozyrev3, Alyssa J. Lawler4,26,33, Colleen Lawless23, Thomas Lehmann34, Danielle L. Levesque6, Harris A. Lewin7,35,36, Xue Li1,4,37, Abigail Lind28,29, Kerstin Lindblad-Toh3,4, Ava Mackay-Smith38, Voichita D. Marinescu3, Tomas Marques-Bonet39,40,41,42, Victor C. Mason43, Jennifer R. S. Meadows3, Wynn K. Meyer44, Jill E. Moore1, Lucas R. Moreira1,4, Diana D. Moreno-Santillan14, Kathleen M. Morrill1,4,37, Gerard Muntané24, William J. Murphy5, Arcadi Navarro39,41,45,46, Martin Nweeia47,48,49,50, Sylvia Ortmann51, Austin Osmanski14, Benedict Paten2, Nicole S. Paulat14, Andreas R. Pfenning25,26, BaDoi N. Phan25,26,52, Katherine S. Pollard28,29,53, Henry E. Pratt1, David A. Ray14, Steven K. Reilly38, Jeb R. Rosen22, Irina Ruf54, Louise Ryan23, Oliver A. Ryder55,56, Pardis C. Sabeti4,57,58, Daniel E. Schäffer25, Aitor Serres24, Beth Shapiro59,60, Arian F. A. Smit22, Mark Springer61, Chaitanya Srinivasan25, Cynthia Steiner55, Jessica M. Storer22, Kevin A. M. Sullivan14, Patrick F. Sullivan62,63, Elisabeth Sundström3, Megan A. Supple59, Ross Swofford4, Joy-El Talbot64, Emma Teeling23, Jason Turner-Maier4, Alejandro Valenzuela24, Franziska Wagner65, Ola Wallerman3, Chao Wang3, Juehan Wang16, Zhiping Weng1, Aryn P. Wilder55, Morgan E. Wirthlin25,26,66, James R. Xue4,57, Xiaomeng Zhang4,25,26
1Program in Bioinformatics and Integrative Biology, UMass Chan Medical School, Worcester, MA 01605, USA. 2Genomics Institute, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 3Department of Medical Biochemistry and Microbiology, Science for Life Laboratory, Uppsala University, Uppsala 751 32, Sweden. 4Broad Institute of MIT and Harvard, Cambridge, MA 02139, USA. 5Veterinary Integrative Biosciences, Texas A&M University, College Station, TX 77843, USA. 6School of Biology and Ecology, University of Maine, Orono, ME 04469, USA. 7The Genome Center, University of California Davis, Davis, CA 95616, USA. 8Genome British Columbia, Vancouver, BC, Canada. 9School of Biological Sciences, University of East Anglia, Norwich, UK. 10School of Health and Life Sciences, Pontifical Catholic University of Rio Grande do Sul, Porto Alegre 90619–900, Brazil. 11School of Life Sciences, University of Nevada Las Vegas, Las Vegas, NV 89154, USA. 12Biodiscovery Institute, University of Nottingham, Nottingham, UK. 13Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala 751 85, Sweden. 14Department of Biological Sciences, Texas Tech University, Lubbock, TX 79409, USA. 15Division of Vertebrate Zoology, American Museum of Natural History, New York, NY 10024, USA. 16Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA. 17Fauna Bio Incorporated, Emeryville, CA 94608, USA. 18Baskin School of Engineering, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 19Faculty of Biosciences, Goethe-University, 60438 Frankfurt, Germany. 20LOEWE Centre for Translational Biodiversity Genomics, 60325 Frankfurt, Germany. 21Senckenberg Research Institute, 60325 Frankfurt, Germany. 22Institute for Systems Biology, Seattle, WA 98109, USA. 23School of Biology and Environmental Science, University College Dublin, Belfield, Dublin 4, Ireland. 24Department of Experimental and Health Sciences, Institute of Evolutionary Biology (UPF-CSIC), Universitat Pompeu Fabra, Barcelona 08003, Spain. 25Department of Computational Biology, School of Computer Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA. 26Neuroscience Institute, Carnegie Mellon University, Pittsburgh, PA 15213, USA. 27Program in Molecular Medicine, UMass Chan Medical School, Worcester, MA 01605, USA. 28Department of Epidemiology & Biostatistics, University of California San Francisco, San Francisco, CA 94158, USA. 29Glad-stone Institutes, San Francisco, CA 94158, USA. 30Center for Species Survival, Smithsonian’s National Zoo and Conservation Biology Institute, Washington, DC 20008, USA. 31Computer Technologies Laboratory, ITMO University, St. Petersburg 197101, Russia. 32Smithsonian-Mason School of Conservation, George Mason University, Front Royal, VA 22630, USA. 33Department of Biological Sciences, Mellon College of Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA. 34Senckenberg Research Institute and Natural History Museum Frankfurt, 60325 Frankfurt am Main, Germany. 35Department of Evolution and Ecology, University of California Davis, Davis, CA 95616, USA. 36John Muir Institute for the Environment, University of California Davis, Davis, CA 95616, USA. 37Morningside Graduate School of Biomedical Sciences, UMass Chan Medical School, Worcester, MA 01605, USA. 38Department of Genetics, Yale School of Medicine, New Haven, CT 06510, USA. 39Catalan Institution of Research and Advanced Studies (ICREA), Barcelona 08010, Spain. 40CNAG-CRG, Centre for Genomic Regulation, Barcelona Institute of Science and Technology (BIST), Barcelona 08036, Spain. 41Department of Medicine and Life Sciences, Institute of Evolutionary Biology (UPF-CSIC), Universitat Pompeu Fabra, Barcelona 08003, Spain. 42Institut Català de Paleontologia Miquel Crusafont, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Barcelona, Spain. 43Institute of Cell Biology, University of Bern, 3012 Bern, Switzerland. 44Department of Biological Sciences, Lehigh University, Bethlehem, PA 18015, USA. 45BarcelonaBeta Brain Research Center, Pasqual Maragall Foundation, Barcelona 08005, Spain. 46CRG, Centre for Genomic Regulation, Barcelona Institute of Science and Technology (BIST), Barcelona 08003, Spain. 47Department of Comprehensive Care, School of Dental Medicine, Case Western Reserve University, Cleveland, OH 44106, USA. 48Department of Vertebrate Zoology, Canadian Museum of Nature, Ottawa, ON K2P 2R1, Canada. 49Department of Vertebrate Zoology, Smithsonian Institution, Washington, DC 20002, USA. 50Narwhal Genome Initiative, Department of Restorative Dentistry and Biomaterials Sciences, Harvard School of Dental Medicine, Boston, MA 02115, USA. 51Department of Evolutionary Ecology, Leibniz Institute for Zoo and Wildlife Research, 10315 Berlin, Germany. 52Medical Scientist Training Program, University of Pittsburgh School of Medicine, Pittsburgh, PA 15261, USA. 53Chan Zuckerberg Biohub, San Francisco, CA 94158, USA. 54Division of Messel Research and Mammalogy, Senckenberg Research Institute and Natural History Museum Frankfurt, 60325 Frankfurt am Main, Germany. 55Conservation Genetics, San Diego Zoo Wildlife Alliance, Escondido, CA 92027, USA. 56Department of Evolution, Behavior and Ecology, School of Biological Sciences, University of California San Diego, La Jolla, CA 92039, USA. 57Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA 02138, USA. 58Howard Hughes Medical Institute, Harvard University, Cambridge, MA 02138, USA. 59Department of Ecology and Evolutionary Biology, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 60Howard Hughes Medical Institute, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 61Department of Evolution, Ecology and Organismal Biology, University of California Riverside, Riverside, CA 92521, USA. 62Department of Genetics, University of North Carolina Medical School, Chapel Hill, NC 27599, USA. 63Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden. 64Iris Data Solutions, LLC, Orono, ME 04473, USA. 65Museum of Zoology, Senckenberg Natural History Collections Dresden, 01109 Dresden, Germany. 66Allen Institute for Brain Science, Seattle, WA 98109, USA.
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability:
All assemblies are available in GenBank, and TE consensus sequences are available through the Dfam database. All other data are available in the supplementary materials. Code used in the analysis is available on Zenodo (49).
REFERENCES AND NOTES
- 1.McClintock B, The origin and behavior of mutable loci in maize. Proc. Natl. Acad. Sci. U.S.A. 36, 344–355 (1950). doi: 10.1073/pnas.36.6.344; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elliott TA, Gregory TR, Do larger genomes contain more diverse transposable elements? BMC Evol. Biol. 15, 69 (2015). doi: 10.1186/s12862-015-0339-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Platt RN 2nd, Vandewege MW, Ray DA, Mammalian transposable elements and their impacts on genome evolution. Chromosome Res. 26, 25–43 (2018). doi: 10.1007/s10577-017-9570-z; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eickbush TH, Jamburuthugoda VK, The diversity of retrotransposons and the properties of their reverse transcriptases. Virus Res. 134, 221–234 (2008). doi: 10.1016/j.virusres.2007.12.010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bourque G et al. , Ten things you should know about transposable elements. Genome Biol. 19, 199 (2018). doi: 10.1186/s13059-018-1577-z; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kapitonov VV, Jurka J, Self-synthesizing DNA transposons in eukaryotes. Proc. Natl. Acad. Sci. U.S.A. 103, 4540–4545 (2006). doi: 10.1073/pnas.0600833103; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas J, Pritham EJ, Helitrons, the Eukaryotic Rolling-circle Transposable Elements. Microbiol. Spectr. 3, 3.4.03 (2015). doi: 10.1128/microbiolspec.MDNA3-0049-2014; [DOI] [PubMed] [Google Scholar]
- 8.Pritham EJ, Putliwala T, Feschotte C, Mavericks, a novel class of giant transposable elements widespread in eukaryotes and related to DNA viruses. Gene 390, 3–17 (2007).doi: 10.1016/j.gene.2006.08.008; [DOI] [PubMed] [Google Scholar]
- 9.Senft AD, Macfarlan TS, Transposable elements shape the evolution of mammalian development. Nat. Rev. Genet. 22, 691–711 (2021). doi: 10.1038/s41576-021-00385-1; [DOI] [PubMed] [Google Scholar]
- 10.Xing J et al. , Emergence of primate genes by retrotransposon-mediated sequence transduction. Proc. Natl. Acad. Sci. U.S.A. 103, 17608–17613 (2006). doi: 10.1073/pnas.0603224103; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takata Y et al. , Phytanic acid in dairy products and risk of cancer: Current evidence and future directions. FASEB J. 31,790.37 (2017). doi: 10.1096/fasebj.31.1_supplement.790.37 [DOI] [Google Scholar]
- 12.Klai K et al. , Screening of Helicoverpa armigera Mobilome Revealed Transposable Element Insertions in Insecticide Resistance Genes. Insects 11, 879 (2020). doi: 10.3390/insects11120879; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van’t Hof AE et al. , The industrial melanism mutation in British peppered moths is a transposable element. Nature 534, 102–105 (2016). doi: 10.1038/nature17951; [DOI] [PubMed] [Google Scholar]
- 14.Gilbert C, Feschotte C, Horizontal acquisition of transposable elements and viral sequences: Patterns and consequences. Curr. Opin. Genet. Dev. 49, 15–24 (2018). doi: 10.1016/j.gde.2018.02.007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arkhipova IR, Neutral Theory, Transposable Elements, and Eukaryotic Genome Evolution. Mol. Biol. Evol. 35, 1332–1337 (2018). doi: 10.1093/molbev/msy083; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kofler R, Senti KA, Nolte V, Tobler R, Schlötterer C, Molecular dissection of a natural transposable element invasion. Genome Res. 28, 824–835 (2018). doi: 10.1101/gr.228627.117; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Philippe C et al. , Activation of individual L1 retrotransposon instances is restricted to cell-type dependent permissive loci. eLife 5, e13926 (2016). doi: 10.7554/eLife.13926; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Rouzic A, Capy P, The first steps of transposable elements invasion: Parasitic strategy vs. genetic drift. Genetics 169, 1033–1043 (2005). doi: 10.1534/genetics.104.031211; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Platt RN 2nd, Blanco-Berdugo L, Ray DA, Accurate Transposable Element Annotation Is Vital When Analyzing New Genome Assemblies. Genome Biol. Evol. 8, 403–410 (2016). doi: 10.1093/gbe/evw009; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Genome 10K Community of Scientists, Genome 10K: A proposal to obtain whole-genome sequence for 10,000 vertebrate species. J. Hered. 100, 659–674 (2009). doi: 10.1093/jhered/esp086; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teeling EC et al. , Bat Biology, Genomes, and the Bat1K Project: To Generate Chromosome-Level Genomes for All Living Bat Species. Annu. Rev. Anim. Biosci. 6, 23–46 (2018). doi: 10.1146/annurev-animal-022516-022811; [DOI] [PubMed] [Google Scholar]
- 22.Robinson GE et al. , Creating a buzz about insect genomes. Science 331, 1386–1386 (2011). doi: 10.1126/science.331.6023.1386; [DOI] [PubMed] [Google Scholar]
- 23.Threlfall J, Blaxter M, Launching the Tree of Life Gateway. Wellcome Open Res. 6, 125–125 (2021). doi: 10.12688/wellcomeopenres.16913.1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zoonomia Consortium A comparative genomics multitool for scientific discovery and conservation. Nature 587, 240–245 (2020). doi: 10.1038/s41586-020-2876-6; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smit AF, Hubley R, RepeatModeler Open-1.0 (2008–2015); http://www.repeatmasker.org/RepeatModeler/.
- 26.Smit AF, Hubley R, Green P, Repeat-Masker Open-3.0 (2004); http://www.repeatmasker.org/RepeatMasker/.
- 27.Smit AF, Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr. Opin. Genet. Dev. 9, 657–663 (1999). doi: 10.1016/S0959-437X(99)00031-3; [DOI] [PubMed] [Google Scholar]
- 28.Venner S, Feschotte C, Biémont C, Dynamics of transposable elements: Towards a community ecology of the genome. Trends Genet. 25, 317–323 (2009). doi: 10.1016/j.tig.2009.05.003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J et al. , Gigantic Genomes Provide Empirical Tests of Transposable Element Dynamics Models. Genomics Proteomics Bioinformatics 19, 123–139 (2021). doi: 10.1016/j.gpb.2020.11.005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spellerberg IF, Fedor PJ, A tribute to Claude Shannon (1916–2001) and a plea for more rigorous use of species richness, species diversity and the ‘Shannon–Wiener’ Index. Glob. Ecol. Biogeogr. 12, 177–179 (2003). doi: 10.1046/j.1466-822X.2003.00015.x [DOI] [Google Scholar]
- 31.Pielou EC, The measurement of diversity in different types of biological collections. J. Theor. Biol. 13, 131–144 (1966). doi: 10.1016/0022-5193(66)90013-0 [DOI] [Google Scholar]
- 32.Kambayashi C et al. , Geography-Dependent Horizontal Gene Transfer from Vertebrate Predators to Their Prey. Mol. Biol. Evol. 39, msac052 (2022). doi: 10.1093/molbev/msac052; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Storer J, Hubley R, Rosen J, Wheeler TJ, Smit AF, The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob. DNA 12, 2 (2021). doi: 10.1186/s13100-020-00230-y; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bachmann K, Genome size in mammals. Chromosoma 37, 85–93 (1972). doi: 10.1007/BF00329560; [DOI] [PubMed] [Google Scholar]
- 35.Kofler R, Dynamics of Transposable Element Invasions with piRNA Clusters. Mol. Biol. Evol. 36, 1457–1472 (2019). doi: 10.1093/molbev/msz079; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo S et al. , The evolutionary arms race between transposable elements and piRNAs in Drosophila melanogaster. BMC Evol. Biol. 20, 14 (2020). doi: 10.1186/s12862-020-1580-3; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lampe DJ, Churchill ME, Robertson HM, A purified mariner transposase is sufficient to mediate transposition in vitro. EMBO J. 15, 5470–5479 (1996). doi: 10.1002/j.1460-2075.1996.tb00930.x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jurka J, Bao W, Kojima KK, Families of transposable elements, population structure and the origin of species. Biol. Direct 6, 44 (2011). doi: 10.1186/1745-6150-6-44; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bao W, Kojima KK, Kohany O, Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015). doi: 10.1186/s13100-015-0041-9; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kapusta A, Suh A, Feschotte C, Dynamics of genome size evolution in birds and mammals. Proc. Natl. Acad. Sci. U.S.A. 114, E1460–E1469 (2017). doi: 10.1073/pnas.1616702114; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.International Human Genome Sequencing Consortium, Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004). doi: 10.1038/nature03001; [DOI] [PubMed] [Google Scholar]
- 42.Kirkness EF et al. , The dog genome: Survey sequencing and comparative analysis. Science 301, 1898–1903 (2003). doi: 10.1126/science.1086432; [DOI] [PubMed] [Google Scholar]
- 43.Mouse Genome Sequencing Consortium, Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 (2002). doi: 10.1038/nature01262; [DOI] [PubMed] [Google Scholar]
- 44.Pontius JU et al. , Initial sequence and comparative analysis of the cat genome. Genome Res. 17, 1675–1689 (2007). doi: 10.1101/gr.6380007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rat Genome Sequencing Project Consortium, Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428, 493–521 (2004). doi: 10.1038/nature02426; [DOI] [PubMed] [Google Scholar]
- 46.Groenen MAM et al. , Analyses of pig genomes provide insight into porcine demography and evolution. Nature 491, 393–398 (2012). doi: 10.1038/nature11622; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adelson DL, Raison JM, Garber M, Edgar RC, Interspersed repeats in the horse (Equus caballus); spatial correlations highlight conserved chromosomal domains. Anim. Genet. 41, 91–99 (2010). doi: 10.1111/j.1365-2052.2010.02115.x; [DOI] [PubMed] [Google Scholar]
- 48.Kumar S, Subramanian S, Mutation rates in mammalian genomes. Proc. Natl. Acad. Sci. U.S.A. 99, 803–808 (2002). doi: 10.1073/pnas.022629899; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Osmanski AB, aosmanski/Zoonomia_TEs: Zoonomia_TEs_Release_v1.0.0, version 1.0.0, Zenodo (2022); 10.5281/zenodo.6498977. [DOI]
- 50.Edgar RC, MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). doi: 10.1093/nar/gkh340; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T, trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009). doi: 10.1093/bioinformatics/btp348; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rice P, Longden I, Bleasby A, EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 (2000). doi: 10.1016/S0168-9525(00)02024-2; [DOI] [PubMed] [Google Scholar]
- 53.Pickeral OK, Makałowski W, Boguski MS, Boeke JD, Frequent human genomic DNA transduction driven by LINE-1 retrotransposition. Genome Res. 10, 411–415 (2000). doi: 10.1101/gr.10.4.411; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goodier JL, Ostertag EM, Kazazian HH Jr., Transduction of 3′-flanking sequences is common in L1 retrotransposition. Hum. Mol. Genet. 9, 653–657 (2000). doi: 10.1093/hmg/9.4.653; [DOI] [PubMed] [Google Scholar]
- 55.Esnault C, Maestre J, Heidmann T, Human LINE retrotransposons generate processed pseudogenes. Nat. Genet. 24, 363–367 (2000). doi: 10.1038/74184; [DOI] [PubMed] [Google Scholar]
- 56.Beck CR, Garcia-Perez JL, Badge RM, Moran JV, LINE-1 elements in structural variation and disease. Annu. Rev. Genomics Hum. Genet. 12, 187–215 (2011). doi: 10.1146/annurev-genom-082509-141802; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.El-Sawy M, Deininger P, Tandem insertions of Alu elements. Cytogenet. Genome Res. 108, 58–62 (2005). doi: 10.1159/000080802; [DOI] [PubMed] [Google Scholar]
- 58.Ma J, Devos KM, Bennetzen JL, Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 14, 860–869 (2004). doi: 10.1101/gr.1466204; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abrusán G, Grundmann N, DeMester L, Makalowski W, TEclass—A tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25, 1329–1330 (2009). doi: 10.1093/bioinformatics/btp084; [DOI] [PubMed] [Google Scholar]
- 60.Legendre P, Legendre L, Numerical Ecology, vol. 24 of Developments in Environmental Modelling (Elsevier, ed. 3, 2012). [Google Scholar]
- 61.ter Braak CJF, Šmilauer P, Canoco Reference Manual and User’s Guide: Software for Ordination, Version 5.0 (Microcomputer Power, 2012). [Google Scholar]
- 62.Gelman A, Analysis of variance—Why it is more important than ever. Ann. Statist. 33, 1–53 (2005). doi: 10.1214/009053604000001048 [DOI] [Google Scholar]
- 63.Douma JC, Weedon JT, Analysing continuous proportions in ecology and evolution: A practical introduction to beta and Dirichlet regression. Methods Ecol. Evol. 10, 1412–1430 (2019). doi: 10.1111/2041-210X.13234 [DOI] [Google Scholar]
- 64.Hadfield JD, Nakagawa S, General quantitative genetic methods for comparative biology: Phylogenies, taxonomies and multi-trait models for continuous and categorical characters. J. Evol. Biol. 23, 494–508 (2010). doi: 10.1111/j.1420-9101.2009.01915.x; [DOI] [PubMed] [Google Scholar]
- 65.Foley NM et al. , A genomic time scale for placental mammal evolution. Science 380, eabl8189 (2023). doi: 10.1126/science.abl8189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bürkner P-C, brms: An R Package for Bayesian Multilevel Models Using Stan. J. Stat. Softw. 80, 1–28 (2017). doi: 10.18637/jss.v080.i01 [DOI] [Google Scholar]
- 67.Carpenter B et al. , Stan: A Probabilistic Programming Language. J. Stat. Softw. 76, 1–32 (2017). doi: 10.18637/jss.v076.i01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gelman A, Goodrich B, Gabry J, Vehtari A, R-squared for Bayesian Regression Models. Am. Stat. 73, 307–309 (2019). doi: 10.1080/00031305.2018.1549100 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All assemblies are available in GenBank, and TE consensus sequences are available through the Dfam database. All other data are available in the supplementary materials. Code used in the analysis is available on Zenodo (49).