

Abstract
T cells orchestrate adaptive immunity against pathogens and other immune challenges, but their dysfunction can also mediate the pathogenesis of cancer and autoimmunity. Metabolic adaptation in response to immunological and microenvironmental cues contributes to T cell function and fate decision. Lipid metabolism has emerged as a key regulator of T cell responses, with selective lipid metabolites serving as metabolic rheostats to integrate environmental cues and interplay with intracellular signaling processes. Here, we discuss how extracellular, de novo synthesized, and membrane lipids orchestrate T cell biology. We also describe the roles of lipids as regulators of intracellular signaling at the levels of transcriptional, epigenetic, and post-translational regulation in T cells. Finally, we summarize therapeutic targeting of lipid metabolism and signaling, and conclude with a discussion of important future directions. Understanding the molecular and functional interplay between lipid metabolism and T cell biology will ultimately inform therapeutic intervention for human disease.
Introduction
T cells are a major component of the adaptive immune system by providing defense against pathogens and tumors. Upon activation by antigens, costimulatory signals and cytokines, naïve T cells proliferate and differentiate into effector cells. Specifically, naïve CD4+ T cells differentiate into conventional T helper (TH) cells, including TH1, TH2, TH17, and TFH cells, to mediate diverse immune functions. Naïve CD8+ T cells differentiate into effector CD8+ T cells to fight against infections and tumors via interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), and cytotoxic molecules. In addition, immunosuppressive CD4+ regulatory T (Treg) cells form during T cell development or are generated from naïve T cells to maintain self-tolerance, which is essential to prevent autoimmune responses but acts as a brake for tumor eradication. Therefore, there has been a great emphasis on understanding how T cell differentiation and function are programmed in different contexts, and to develop methods to harness the potent therapeutic properties of T cells.
Recent evidence suggests that nutrients and metabolic programs are key regulators of T cell fate decisions1, with lipids emerging as crucial mediators. Lipids are essential components of cellular membranes, and intracellular homeostasis of lipids is dynamically regulated by the uptake, de novo synthesis, and hydrolysis. Recent studies have identified the enzymatic nodes that alter lipid metabolism in T cells to promote or inhibit their functions in different disease contexts. Moreover, lipid molecules act as signaling messengers to impart their physiological and functional effects. Thus, lipid metabolism represents a key interface between physiology, chemical biology, and biochemical signaling.
In this review, we summarize how lipids contribute to T cell fate and function in different physiological and pathological settings. We first discuss the roles of extracellular lipids and intracellular lipid metabolism in orchestrating T cell fate choices in various immunological contexts and disease microenvironments. Then, we describe the functional effects of membrane lipids found in discrete subcellular compartments (e.g., plasma versus mitochondrial membrane). We also emphasize the interplay between intracellular signaling and lipid metabolism at multiple levels, including transcriptional and epigenetic regulation and post-translational modifications, in mediating T cell responses. Finally, we summarize preclinical and clinical data that support potential strategies to target lipid pathways for modulation of T cell function in disease therapy.
Lipids shape the fate and function of T cells
It has long been appreciated that lipid-derived molecules, such as prostaglandins, play important roles in regulating T cell responses2. Extracellular lipids, including fatty acids (FAs) and cholesterol, are emerging as important nutrient sources for the generation of cellular energy and biomass, and as signaling mediators for innate and adaptive immune responses (Table 1). The metabolic and signaling roles of extracellular lipids in conventional T cell responses are discussed below (Figure 1). The emerging roles of lipid metabolism in γδ T cells, natural killer (NK) cells, invariant natural killer T (iNKT) cells, and B cells are described in Box 1.
Table 1.
Overview of lipid metabolism in T cell subsets
| Lipid source or function | Lipids | Cell type | Ref. |
|---|---|---|---|
| Lipids in microenvironment | SCFAs | Effector, memory-like, or CXCR6+ CD8+ T cell (+), Treg (+), TH1 (+), TH17 (+) | 3–9 |
| LCFAs | TH1 (+), TH17 (+), Treg (+), and intratumoral CD8+ T cell (−) | 10–14 | |
| Cholesterol | Exhausted CD8+ T cell (+) and Tc9 (−) | 17,18 | |
| Oxysterols | TFH (+) | 19 | |
| Bile acids | TH17 (−), Treg (+), pTreg (+), and proinflammatory CD4+ T cell (+) | 21–25 | |
| Intracellular programs | De novo lipid synthesis | TH17 (+), memory CD4+ T cell (−), antigen-specific CD8+ T cell (+), Treg (+), and iTreg (−) | 31–36 |
| Ceramide | Antigen-specific CD8+ T cell (+) | 39 | |
| Mevalonate pathway | Conventional T cell (+), Treg (+), and TH17 (+) | 40–44 | |
| Mitochondrial fitness (lipid catabolism?) | TCM (+) and TRM (+) | 45–49 | |
| FABPs | TRM (+) and Treg (−) | 53–56 | |
| Lipid droplets | Treg (+), CD4+ T cell (−), and CD8+ T cell (−) | 58,59 | |
| Membrane lipids | Phosphatidic acid | Antigen-specific CD8+ T cell (−) and memory CD8+ T cell (+) | 60 |
| Phosphatidylethanolamine | TFH (+) | 63 | |
| Cardiolipin | CD8+ T cell (+) | 64 | |
| Signaling | PPARs and LXRs | VAT Treg (+) and TH17 (−) | 66,68 |
| SREBPs | CD8+ T cell (+) and intratumoral Treg (+) | 26,35 | |
| FAO-derived acetyl-CoA | Effector or memory CD8+ T cell (+), Treg (+), and TH1 (+) | 70–73 | |
| FPP and GGPP | eTreg (+) | 27 | |
| N-myristoyltransferase | Proinflammatory TH1 (−) and TH17 (−) | 76 |
Positive (+) or inhibitory (–) role in differentiation, persistence, or function of the indicated T cell subset (see main text for more details). CerS6, ceramide synthase 6; eTreg, effector Treg; FABPs, fatty acid binding proteins; FPP, farnesyl pyrophosphate; GGPP, geranylgeranyl pyrophosphate; iTreg, in vitro-derived Treg; LCFAs, long-chain fatty acids; LXRs, liver X receptors; PPARs, peroxisome proliferation activating receptors, SCFAs, short-chain fatty acids; SREBPs, sterol regulatory binding proteins; VAT, visceral adipose tissue.
Figure 1. Extracellular lipids in T cell differentiation and functional adaptation.
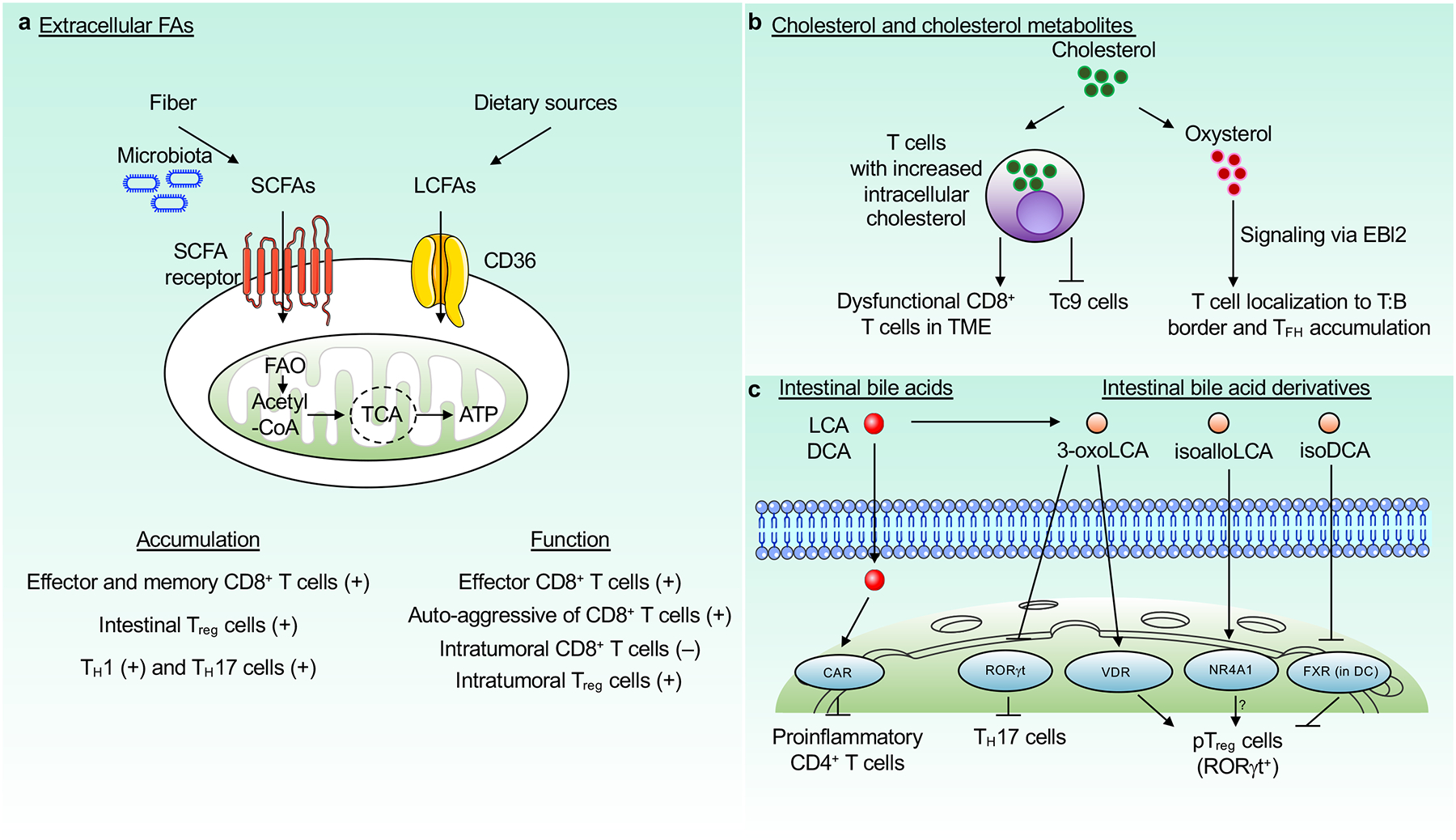
Extracellular lipids, such as FAs, cholesterol or cholesterol-derived metabolites, and bile acids regulate multiple aspects of T cell biology. a, SCFAs, which are derived from dietary fiber via microbiota-dependent fermentation, regulate T cell accumulation and function. Exogenous LCFAs such as oleic acid are taken up via CD36 from the TME and impair the function of CD8+ T cells but enhance the function of Treg cells. b, T cells take up cholesterol from the TME, which inhibits the function of CD8+ T cells. Cholesterol accumulation also impedes Tc9 cell generation. Oxysterols signal via EBI2 to promote migration of T cells to the T cell–B cell border and contribute to TFH cell generation. c, Intestinal bile acids (lithocholic acid (LCA) and deoxycholic acid (DCA)) and their derivatives (3-oxoLCA, isoalloLCA, and isoDCA) modulate T cell differentiation and maintenance. 3-oxoLCA inhibits TH17 and promotes pTreg (marked by RORγt expression) cell differentiation, by binding to RORγt and vitamin D receptor (VDR), respectively. IsoalloLCA promotes pTreg cell differentiation, likely in an NR4A1-dependent manner. IsoDCA promotes pTreg cell differentiation by suppressing FXR activity in dendritic cells (DCs). Intestinal bile acids are detoxified by constitutive androstane receptor (CAR) to prevent activation of proinflammatory CD4+ T cell response. FAs, fatty acids; SCFAs, short-chain FAs; LCFAs, long-chain FAs; TME, tumor microenvironment. Receptors, mitochondrial, and nuclear structures used in this and other figures were obtained from Servier Medical Art website (http://smart.servier.com).
Box 1. Lipid metabolism of additional lymphocytes in disease contexts.
Lipid metabolism orchestrates the function of multiple lymphocyte populations to modulate immune responses. γδ T cells and NK cells have crucial roles in protective immunity against tumors and viral infections. IL-17+ γδ T cells are expanded in obesity and display high capacity for lipid uptake and storage, associated with enhanced tumor growth94. Conversely, NK cell functions are impaired in fatty-acid-rich environments, such as obesity and certain TMEs, which occurs due to activation of PPARs95,96. iNKT cells are CD1d-restricted T cells that share features of both innate and adaptive immune cells. iNKT cells express PPAR-γ, which induces lipid metabolism and especially cholesterol synthesis to promote the activation, proliferation and antitumor function of iNKT cells97. In contrast, high levels of extracellular lipids suppress inflammatory cytokine production by iNKT cells in a murine model of rheumatoid arthritis98. B cells are adaptive immune cells specialized in antibody production. Glutathione peroxidase-4 is required for the maintenance of antibody responses by preventing lipid peroxidation and ferroptosis in B cells, which is associated with increased lipid metabolism99. IL-10-producing regulatory B cells restrict immune and inflammatory responses, and such regulatory function is dependent upon mevalonate metabolism. B cells from patients with mevalonate kinase deficiency, an inherited autoimmune disorder, have poor IL-10 induction, which is restored by exogenous GGPP treatment100. Thus, lipid metabolism regulates multiple facets of adaptive immune responses that may be targetable for disease treatment.
Extracellular FAs
FAs are divided into short-chain, medium-chain, long-chain and very-long-chain FAs. Short-chain FAs (SCFAs), such as propionate, acetate and butyrate, are generated by gut microbiota-derived bacterial fermentation from dietary fibers3. Diet high in fiber or butyrate boosts the effector function of CD8+ T cells to aid in the resolution of influenza infection, which is associated with enhanced glycolytic and mitochondrial respiratory capacities3. Similarly, SCFAs are capable of rewiring metabolism such that activated T cells take up and oxidize more FAs, which enables their transition to memory-like CD8+ T cells with long-term survival. Mechanistically, butyrate-derived acetyl-coenzyme A (acetyl-CoA) enters the tricarboxylic acid (TCA) cycle (also called Kreb’s cycle) and fuels oxidative phosphorylation4. Thus, certain SCFAs affect both effector and memory CD8+ T cell responses. In addition, CXCR6-expressing CD8+ T cells from a mouse model of nonalcoholic steatohepatitis (NASH) are auto-aggressive when exposed to acetate5. Further, SCFAs are also important regulators of Treg cell differentiation and function in the intestines6–8, which likely contributes to intestinal tissue homeostasis. In addition, SCFAs potentiate anti-CD3-induced differentiation of TH1 and TH17 cells in the spleen and mesenteric lymph nodes, and IL-10-producing T cells in the intestines9. Thus, SCFAs play multifactorial roles in T cell responses (Figure 1a).
Most long-chain FAs (LCFAs) are obtained from dietary sources, although certain LCFAs are synthesized de novo. LCFA catabolism via FA oxidation (FAO) generates acetyl-CoA to fuel mitochondrial function. LCFAs such as palmitate and oleate enhance TH1 and TH17 cell differentiation and proliferation through the p38–MAPK pathway10. Accordingly, LCFAs accelerate disease progression of experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis10. The tumor microenvironment (TME) is often high in cholesterol and FA content, which can alter the functionality of intratumoral T cells11–13. Uptake of lipids such as LCFAs and oxidized low density lipoprotein (LDL) by the scavenger receptor CD36 promotes the function of Treg cells but dampens the effector function of CD8+ T cells in the TME11–13. Mechanistically, CD36-mediated uptake of lipids in CD8+ T cells leads to increased lipid peroxidation, which promotes activation of p38–MAPK or induction of ferroptosis, both of which induce CD8+ T cell dysfunction12,13 (Figure 1a). In pancreatic ductal adenocarcinoma (PDAC), accumulation of LCFAs impairs the mitochondrial function of CD8+ T cells and reduces lipid catabolism. Of note, very-long-chain acyl CoA dehydrogenase (VLCAD), the enzyme that initiates catabolism of palmitate and oleate, is downregulated in CD8+ T cells from mouse models of PDAC, and overexpression of very-long-chain acyl-CoA dehydrogenase is associated with improved survival and persistence of the CD8+ T cells in the TME14. The differential effects of LCFAs on T cell function in EAE and tumors may be related to interplay with other environment-specific signals that impart selective metabolic, functional, and cellular alterations in T cells, in line with site-specific requirements for glucose metabolism in TH17 cells in the intestines and central nervous system during EAE1,15. Together, these studies illustrate the diverse effects of extracellular lipids on T cell biology in different contexts.
Exogenous cholesterol and cholesterol derivatives
The levels of cholesterol and its biosynthetic intermediates profoundly alter T cell function in disease settings. For example, high abundance of plasma cholesterol disrupts T cell homeostasis, which mediates inflammation in hypercholesterolemia16. Moreover, cholesterol in the TME induces CD8+ T cell exhaustion (a dysfunctional state defined by expression of certain coinhibitory molecules and impaired effector function) by triggering ER stress, leading to uncontrolled tumor growth17. Compared to IFN-γ-producing CD8+ T cells, IL-9-producing CD8+ T (Tc9) cells, which are potent inducers of antitumor immunity, have distinct gene profiles related to the synthesis and efflux of cholesterol, and have a lower cholesterol level. Further, cholesterol depletion enhances, and supplementation reduces, Tc9 cell generation by regulating the activity of the transcription factor liver X receptor (LXR)18. Similar effects are observed upon supplementation with oxysterols, which are oxidized cholesterol derivatives that can activate LXR signaling upon their intracellular accumulation18. Additionally, the oxysterol 7α,25-dihydroxycholesterol also signals via EBI2 (also called GPR183) to promote localization of activated CD4+ T cells to the interface between the B cell follicle and T cell zone of the spleen; this ultimately promotes TFH cell accumulation and induction of humoral immunity19 (Figure 1b). Of note, LDL, which acts as a carrier for cholesterol, can be taken up by LDL receptor (LDLR). LDL depletion or LDLR deficiency impairs CD8+ T cell function in vitro and their antitumor activity, although the antitumor effects may be partly independent of uptake for cholesterol or LDL20. Thus, sterols have multiple effects on T cell function that may be exploited for immunotherapies.
Bile acids
Bile acids are cholesterol-derived metabolites that are abundant in the mammalian gut and play important roles in lipid absorption. Liver-derived bile acids are metabolically modified by intestinal microbiota to generate intestinal bile acids (especially lithocholic acid (LCA) and deoxycholic acid (DCA)). These intestinal bile acids and their derivatives serve critical functions in T cell biology. Specifically, the LCA derivative 3-oxoLCA inhibits TH17 cell differentiation via binding to TH17 cell-specific transcription factor RORγt (retinoic acid receptor-related orphan receptor γt)21. In contrast, combined supplementation of LCA and its derivative 3-oxoLCA increases RORγt+ peripherally derived Treg (pTreg) cell accumulation via Treg cell-intrinsic engagement of vitamin D receptor, which limits susceptibility to intestinal inflammation and colitis22. The LCA derivative isoalloLCA also induces expression of the transcription factor Foxp3 and subsequent differentiation of pTreg cells by promoting generation of mitochondrial reactive oxygen species21. NR4A1, which is a nuclear hormone receptor, likely also mediates the effect of isoalloLCA on pTreg cells23. In addition, the deoxycholic acid derivative 3β-hydroxydeoxycholic acid (isoDCA) induces Foxp3 expression by inhibiting farnesoid X receptor (FXR, also called NR1H4) transcriptional activity in dendritic cells (DCs); consequently, these DCs have enhanced ability to prime pTreg cell differentiation from naïve CD4+ T cells24. However, upon loss of the nuclear xenobiotic receptor CAR (encoded by Nr1i3), intestinal bile acids drive the proinflammatory CD4+ T cell response and trigger inflammation in the lamina propria of the small intestine, owing to diminished detoxification of bile acids25 (Figure 1c). Thus, modulation of intestinal bile acid generation or signaling may provide beneficial effects, by tuning T cells responses to improve therapy for gastrointestinal disease and possibly other inflammatory disorders.
Intracellular reprogramming of lipid metabolism
Intracellular lipid homeostasis is balanced by lipid synthesis, catabolism and storage. Immunological signals are key regulators of lipid metabolism in T cells. T cell receptor (TCR) stimulation activates PI3K–Akt and mechanistic target of rapamycin (mTOR) signaling to induce FA and mevalonate synthesis26,27. Further, CD28 costimulatory signals transiently induce carnitine palmitoyltransferase-1 (CPT1a) expression before the first cell division28. However, CD28 costimulation upregulates TCR-dependent activation of mTORC1, an important positive regulator of effector T cell responses that promotes anabolic metabolism29,30, suggesting temporal effects of CD28 costimulation on lipid metabolic programs (Figure 2). In this section, we discuss how intracellular lipid metabolism directs T cell differentiation and function (Table 1).
Figure 2. Regulation of lipid metabolism by immunological signals.
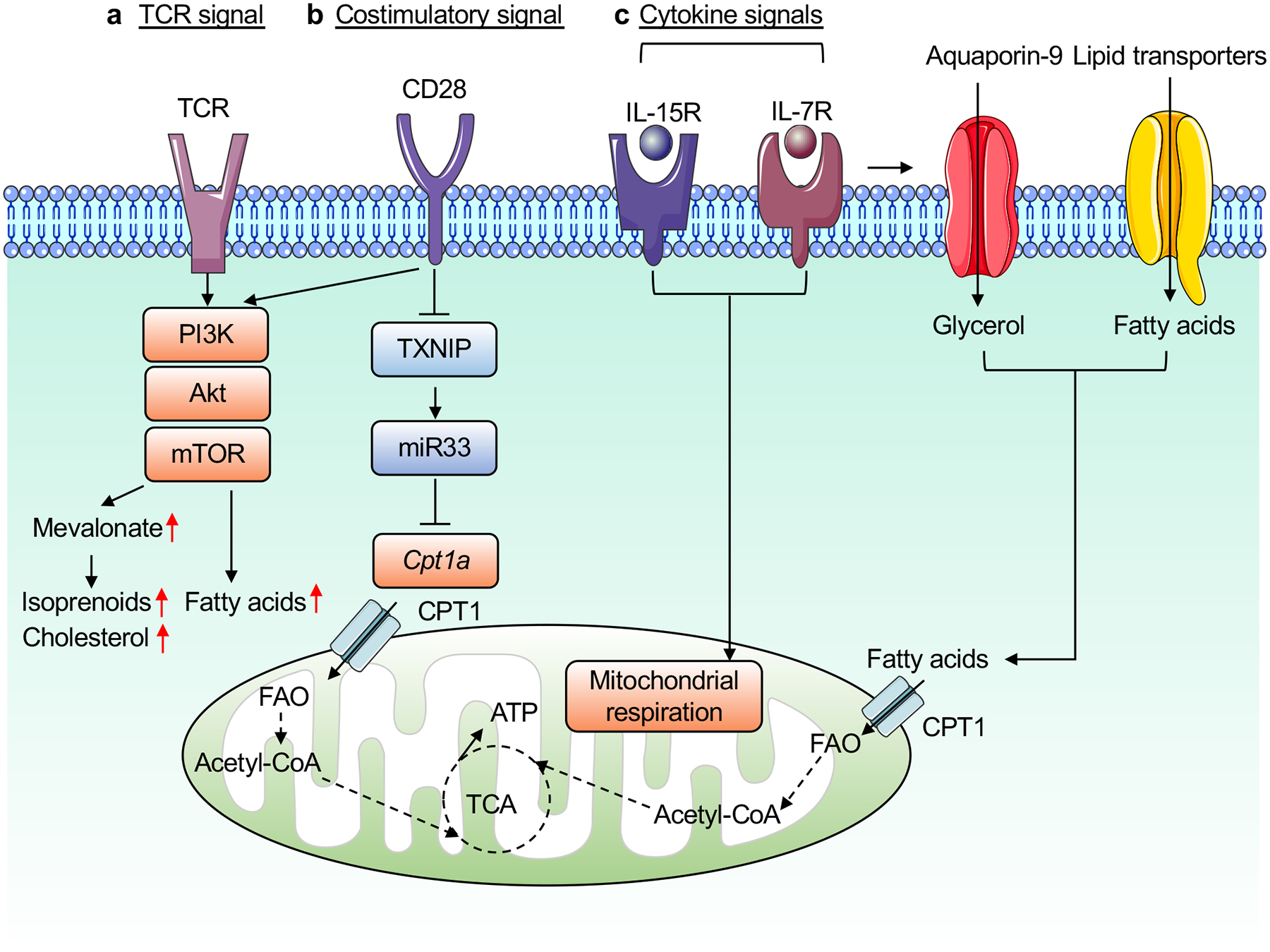
A summary of immunological signals for the regulation of lipid metabolism in T cells. a, Upon TCR engagement, T cells activates PI3K–Akt and mTOR signaling to induce FA synthesis and mevalonate metabolism. b, CD28 costimulation induces transient expression of CPT1a through thioredoxin-interacting protein (TXNIP) and miR33 (before first cell division), and may enhance mevalonate and lipid synthesis by boosting TCR-dependent mTORC1 activation. c, The cytokines IL-15 and IL-7 induce FAO, in part by enhancing expression of CPT1a and mediating glycerol uptake via aquaporin-9, respectively, to maintain mitochondrial respiration.
Lipid synthesis
Activated T cells upregulate de novo synthesis programs that lead to the generation of FA-and mevalonate-derived lipids. FA synthesis (FAS) is orchestrated by several enzymes, including acetyl-CoA citrate lyase (ACLY), acetyl-CoA carboxylase 1 (ACC1), and fatty acid synthase (FASN) (Figure 3a). Inhibition of ACC1 reduces TH17 cell differentiation31,32 but enhances memory CD4+ T cell formation33. ACC1 is also required for antigen-specific CD8+ T cell accumulation during bacterial infection34 and the suppressive function of Treg cells in the TME35. In addition, ACLY, which converts cytosolic citrate to acetyl-CoA, is a crucial enzyme in the regulation of FAS versus FAO. ACLY is actively degraded during the differentiation of in vitro Treg (iTreg) cells, leading to a downregulation of FAS and corresponding upregulation of FAO to support iTreg generation36. Overall, these studies establish important roles for FAS in the control of T cell differentiation and function.
Figure 3. Intracellular lipid metabolism in T cells.
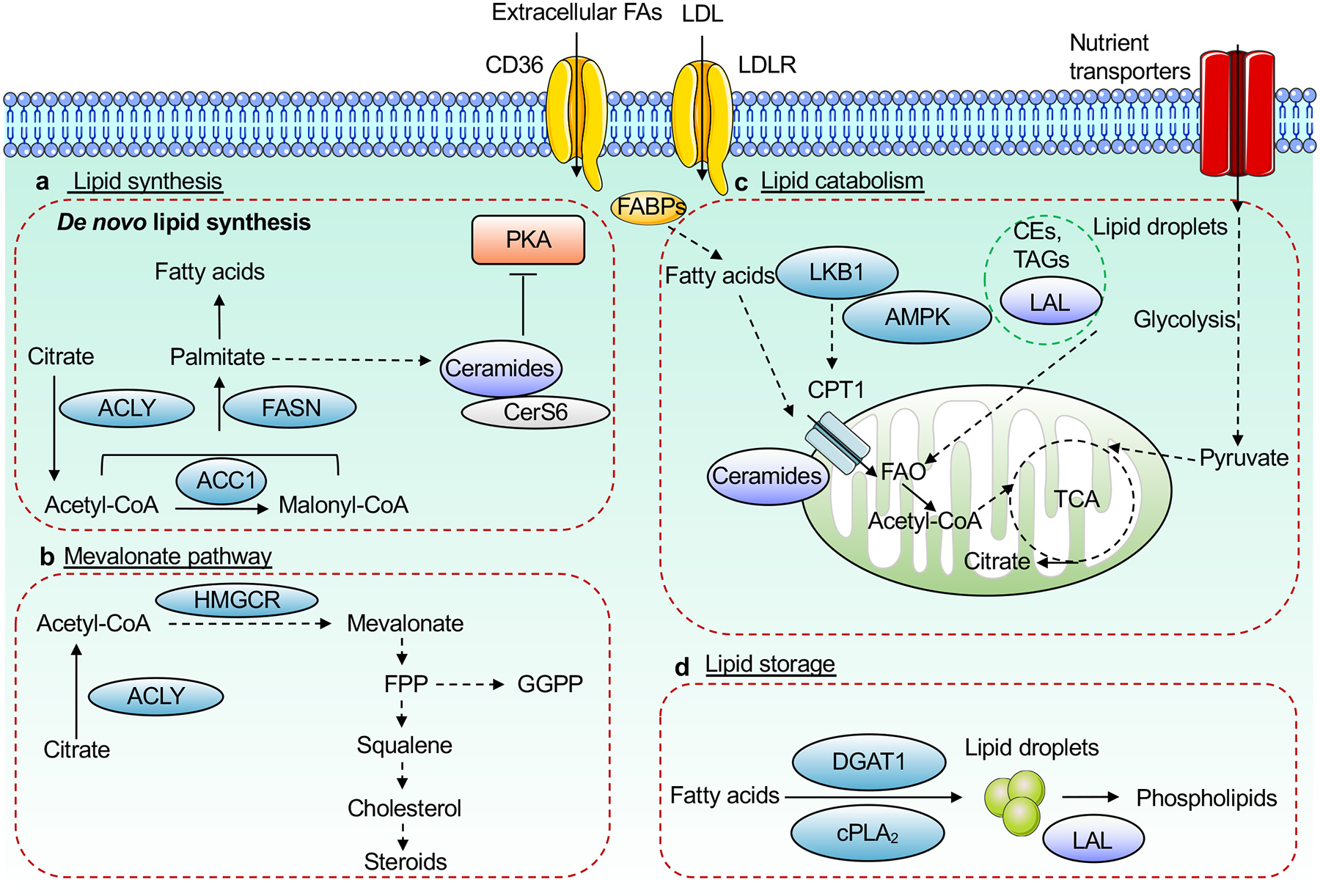
Intracellular lipid homeostasis is regulated by de novo lipid synthesis via the fatty acid (FA) and mevalonate pathways, catabolism, storage, and uptake (via specific transporters such as CD36 and LDLR). a, b, For de novo synthesis to occur, nutrients are catabolized into acetyl-CoA, which is used for synthesis of various FAs (via the enzymatic actions of ACLY, ACC1 and FASN, among others), ceramide (via CerS6), which suppresses PKA signaling (a), or mevalonate and downstream metabolites such as cholesterol and isoprenoids (via the mevalonate pathway including the rate-limiting enzyme HMGCR) (b). c, Lipid catabolism is initiated by FAO in the mitochondria, which is used to generate energy in certain T cell subsets. FAO can be mediated by exogenous lipids, whose intracellular levels are regulated by surface transporters (such as CD36 and LDLR) or intracellular FABPs, or lipid droplet-derived TAGs, which is mediated by LAL-dependent lipolysis. d, Lipid droplets form via cPLA2 or DGAT1-dependent mechanisms to store excess levels of triacylglycerides (TAGs) and cholesterol esters (CEs). TAGs and CEs stored in these lipid droplets undergo hydrolysis via LAL to generate phospholipids. CerS6, ceramide synthase 6; ACC1, acetyl-CoA carboxylase 1; FASN, FA synthase; PKA, protein kinase A; FABPs, FA binding proteins; LAL, lysosomal acid lipase; FAO, FA oxidation; HMGCR, HMG-CoA reductase; DGAT1, diacylglycerol acyltransferase 1; cPLA2, group IVA phospholipase A2.
Ceramides are composed of sphingosine and FA, and serve as lipid messengers for intracellular signaling cascades. Ceramide synthase 6 (CerS6), which is a rate-limiting enzyme for ceramide biosynthesis (Figure 3a), facilitates expansion and function of alloantigen-specific T cells and contributes to graft-versus-host disease (GVHD) in a mouse model of allogeneic hematopoietic cell transplantation (allo-HCT) therapy. Further, CerS6-deficient T cells have impaired proliferation and IFN-γ production, which is associated with a partial reduction of GVHD but retention of graft-versus-leukemia responses in an allo-HCT model, suggesting that targeting ceramide synthesis may improve allo-HCT therapy37. In T cells from aged mice, ceramides accumulate in the mitochondria and promote mitophagy through inhibition of protein kinase A. Consequently, the antitumor function of aged T cells is attenuated38. However, treatment of liver tumor-bearing mice with nanoliposomes containing C6-ceramide results in enhanced antigen-specific CD8+ T cell functionality and delayed tumor growth39, revealing possibly T cell-extrinsic effects of exogenous ceramide at impacting antitumor function of T cells. Collectively, these studies suggest that modulation of ceramide metabolism may be useful for tuning therapies against hematological and solid tumors.
The mevalonate pathway is essential for the generation of isoprenoids, cholesterol, and cholesterol derivatives. Lipid synthesis via the mevalonate pathway requires several key enzymes, including HMG-CoA reductase (HMGCR) (Figure 3b). T cell-specific deletion of HMGCR leads to a severe reduction of peripheral T cells, while Treg cell-specific deletion of HMGCR is associated with the development of systemic autoimmunity40. HMGCR-deficient T cells have a cell-survival defect, which is rectified by the addition of mevalonate or the isoprenoid geranylgeranyl pyrophosphate (GGPP)40,41. Moreover, deletion of Pggt1b (enzyme for protein geranylgeranylation via GGPP) or Fntb (enzyme for protein farnesylation via farnesyl pyrophosphate or FPP) in Treg cells results in a profound loss of Treg cell-suppressive function and development of fatal autoimmunity27, highlighting the importance of mevalonate metabolism and downstream protein prenylation (see below for more details). Similar defects in mevalonate- and GGPP-dependent Treg cell survival are also observed upon deletion of LKB1, a kinase that is essential to promote mevalonate biosynthesis41 and FAO in Treg cells42. However, mTORC1 orchestrates mevalonate metabolism in Treg cells to establish their suppressive activity and immunological tolerance43, suggesting complex regulation of mevalonate metabolism in Treg cells that may be associated with their activation or inflammatory state. The specific deletion of HMGCR in TH17 cells also protects mice from EAE44. Therefore, targeting of mevalonate metabolism is an attractive means to tune autoimmune and inflammatory responses mediated by CD4+ T cells.
Lipid catabolism
The induction of FAO is associated with elevated AMP-activated protein kinase (AMPK) activity (Figure 3c), which promotes the generation of central memory CD8+ T cells (TCM; a subset of memory CD8+ T cells that recirculate via secondary lymphoid organs)45,46. Compared with effector CD8+ T cells, TCM cells have increased FAO, which is driven by triacylglycerides that are synthesized de novo from glucose rather than extracellular FAs47. Mechanistically, intrinsic lipolysis of intracellular triacylglycerides is mediated by lysosomal acid lipase (LAL)47 (Figure 3c). IL-15 signaling promotes upregulation of CPT1a expression to facilitate FAO48, whereas IL-7 signaling induces glycerol uptake for triacylglyceride synthesis and FAO49, thereby supporting memory CD8+ T cell longevity (Figure 2). However, in contrast to the defective memory responses upon etomoxir treatment45,48, genetic deletion of CPT1a in T cells does not impair memory CD8+ T cell generation. Nonetheless, etomoxir treatment of CPT1a-deficient T cells reduces mitochondrial oxidative function and incorporation of extracellular palmitate into TCA cycle intermediates50, suggesting that memory T cell generation requires mitochondrial function. Such effect may be partly mediated by short-chain or medium-chain FAs whose oxidation does not require CPT1a, glucose, or amino acids, and future studies are required to test these or additional hypotheses. In an obesity-associated breast tumor model, STAT3 activation increases FAO and impairs the effector function of CD8+ T cells51. In contrast, although they also show CD8+ T cell dysfunction, FAO-associated genes are not upregulated in CD8+ T cells from an obesity-associated MC-38 tumor model52, suggesting TME-dependent regulation of lipid metabolism in tumor-infiltrating T cells in response to obesity. Thus, FAO is associated with memory T cell generation and functionality in inflammatory contexts.
Tissue-resident memory (TRM) cells reside in non-lymphoid tissues. Similar to the role of P2RX7 in TCM cells, activation of P2RX7 promotes AMPK signaling, mitochondrial function and accumulation of TRM cells46. Moreover, extracellular lipid uptake by TRM cells is increased compared with other memory T cell populations in vitro, and inhibition of CPT1a or FAO reduces the accumulation of skin TRM cells in vivo53. Also, deficiency in FA-binding proteins (FABPs) 4 and 5, which act as complex regulators of lipid transport and trafficking in cells, impairs mitochondrial oxygen consumption and virus-specific TRM cell accumulation in the skin53, whereas FABP1 is important for TRM accumulation in the liver54. Thus, FABPs orchestrate extracellular FA uptake to promote FAO for TRM generation or survival. Metabolic reprogramming toward FAO also prolongs TRM cell longevity, which is associated with enhanced antitumor immunity in gastric adenocarcinoma55. Finally, Treg cells express FABP5, which is important to restrain mitochondrial DNA-induced type I IFN signaling and downstream IL-10 production in Treg cells56. Collectively, FAO and FABPs tune T cell responses in tissue microenvironments, including tumors.
Lipid storage
Lipid droplets are an intracellular lipid storage depot, which are utilized by effector memory CD4+ T cells in nutrient-depleted environments57 such as the TME. Lipid droplets are also dynamically regulated in certain T cell subsets in nutrient-replete environments. The effector memory population of CD4+ T cells contain fewer lipid droplets than other CD4+ memory T cell subsets57. Also, Treg cells have enhanced lipid droplet content compared to conventional T cells58. Lipid droplet formation is mediated by diacylglycerol acyl transferase 1 (DGAT1) (Figure 3d), and DGAT1 inhibition impairs Foxp3 induction, suggesting a role for lipid droplets in Treg cell formation or maintenance58. Moreover, senescent T cells, which are in a dysfunctional state, in the TME display alterations in the composition of lipid species and droplet accumulation that is related to elevated group IVA phospholipase A2 activity. Inhibition of such activity reverses T cell senescence, thereby reducing tumor size and extending the survival of tumor-bearing mice59. Therefore, understanding the role of lipid droplets in T cell subsets and how to modulate lipid droplet-related metabolism may give rise to effective therapies for cancer and other diseases.
Regulation of T cell function by membrane lipids
The activation of T cells is associated with profound changes in the composition, distribution, and dynamics of membrane lipids. These processes affect T cell differentiation and function in different physiological contexts as discussed below (Figure 4 and Table 1).
Figure 4. Membrane lipids coordinate signaling in T cells.
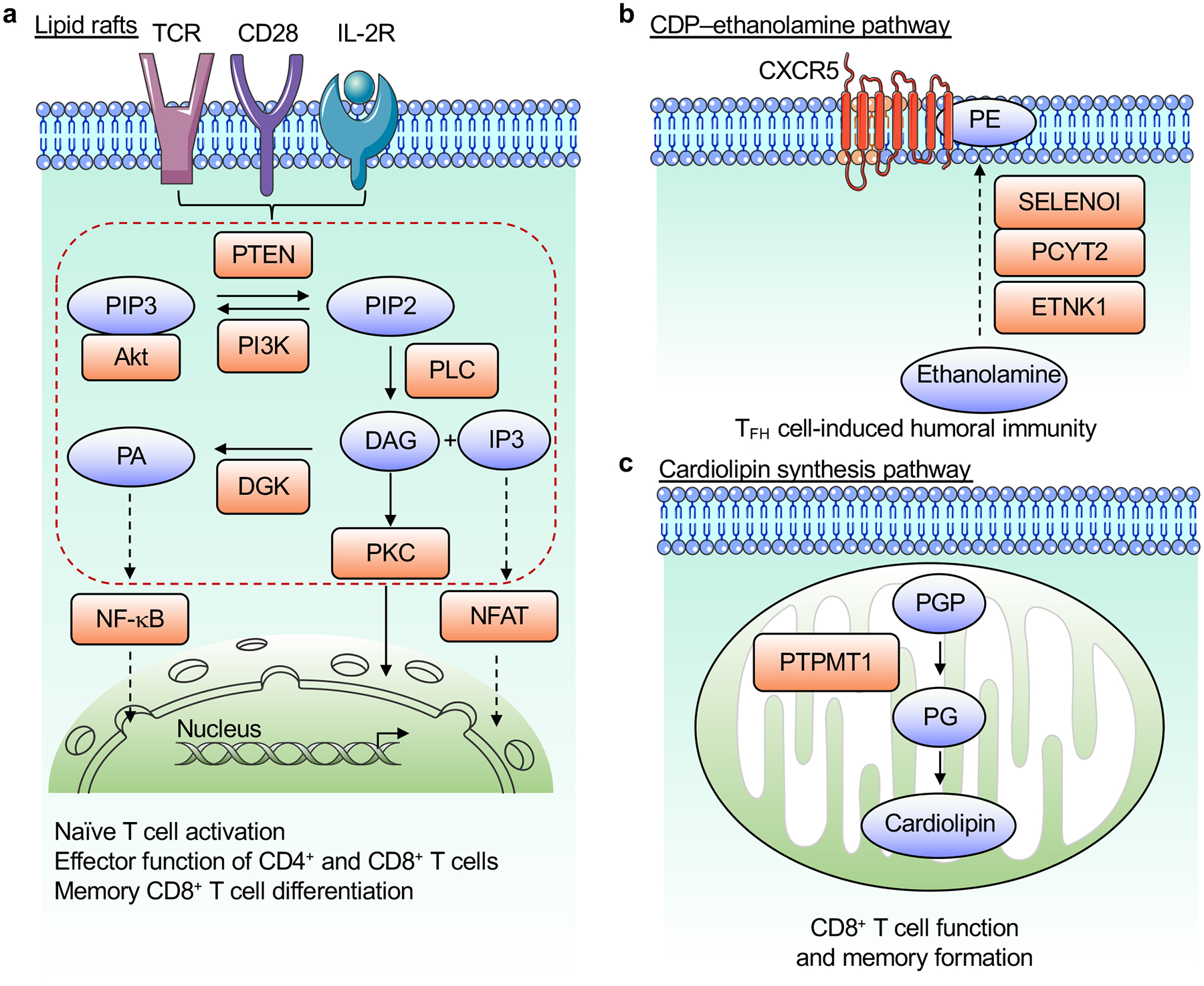
a, Antigens, costimulatory signals and IL-2 stimulation induce PI3K activation that generates PIP3 from PIP2, which is opposed by PTEN. Immunological signals induce PLC activation to produce DAG and IP3 from PIP2. These lipid molecules activate several signaling cascades, including those downstream of PKC, to promote activation of NFAT and NF-κB in the nucleus. DGK opposes DAG-dependent signaling by converting DAG to PA. These lipid-coordinated signaling events play multiple roles in T cell biology. b, De novo PE synthesis via the CDP–ethanolamine pathway, mediated by the enzymes ETNK1, PCYT2, and SELENOI, selectively regulates PE localization to the outer layer of the TFH cell membrane and promotes humoral immunity. c, De novo cardiolipin synthesis in the mitochondria, which depends upon PTPMT1, promotes the function and memory differentiation of CD8+ T cells. DAG, diacylglycerol; IP3, inositol trisphosphate; DGK, diacylglycerol kinase; PA, phosphatidic acid; PIP2, phospholipid phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-triphosphate; PKC, protein kinase C; PLC, phospholipase C; PE, phosphatidylethanolamine; PGP, phosphatidylglycerophosphate; PG, phosphatidylglycerol; PTPMT1, protein tyrosine phosphatase mitochondrial 1.
Membrane lipids transmit intracellular signaling
The plasma membrane is composed of various lipid molecules, which form heterogenous domains that act as signaling platforms. For example, specific lipids recruit signaling proteins that contain lipid-binding domains to the plasma membrane, such as the pleckstrin homology domain of Akt that binds to phosphatidylinositol 3,4,5-triphosphate (PIP3) to induce downstream signaling. Notably, TCR signaling, in combination with costimulation and IL-2 signaling, activate phosphatidylinositol-3-kinase (PI3K) to increase PIP3 generation from phosphatidylinositol 4,5-bisphosphate (PIP2), and this reaction is opposed by the lipid phosphatase and tumor suppressor PTEN29.
Lipids also function as second messengers with important immune-regulatory functions. Following TCR engagement, phospholipase C (PLC) hydrolyzes PIP2 to generate inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 and DAG act as signaling mediators to promote calcium release from intracellular stores and drive protein kinase C (PKC) activation, respectively. These events lead to activation of the transcription factors NFAT and NF-κB that are important for T cell functionality. The DAG-dependent signaling pathway is terminated by diacylglycerol kinase (DGK), which converts DAG to phosphatidic acid (PA). DGK-deficient antigen-specific CD8+ T cells show enhanced expansion and increased cytokine production, which enhances viral clearance. However, DGK-deficient memory CD8+ T cells have impaired expansion after viral re-challenge, suggesting opposing roles for DGK in effector and memory CD8+ T cells60 (Figure 4a).
Lipids spatially coordinate immune receptor signaling
The immunological synapse is a specialized structure that forms in the plasma membrane when TCRs and costimulatory molecules expressed on T cells bind their ligands on antigen-presenting cells. The immunological synapse serves important spatiotemporal roles in signal integration to tune T cell activation and function. Increasing the level of cholesterol in the plasma membrane through inhibition of ACAT1, a cholesterol esterification enzyme, enhances TCR clustering, maturation of the immunological synapse, and promotes cytolytic granule secretion in CD8+ T cells61. These alterations cause ACAT1-deficient CD8+ T cells to exhibit better antitumor effect. Moreover, cholesterol sulfate, which is a natural analog of cholesterol, impinges upon TCR signaling and triggers apoptosis in double-positive thymocytes undergoing positive selection62. Together, these results show that lipids are important mediators of activation-associated signaling events in T cells.
Context-specific regulation of membrane lipids
Recent studies have uncovered context-dependent roles for membrane lipid species in the regulation of T cell responses. Phosphatidylethanolamine (PE) is an essential phospholipid in the plasma membrane that is de novo synthesized via the CDP–ethanolamine pathway. CDP–ethanolamine pathway-dependent PE synthesis selectively promotes TFH cell generation to drive humoral immunity, with such regulation associated with PE distribution to the outer layer of TFH cell membrane and stabilization of surface CXCR5 expression63 (Figure 4b). Another phospholipid, cardiolipin, is synthesized and localized in inner mitochondrial membrane, and its de novo synthesis maintains CD8+ T cell function. Accordingly, T cells lacking PTPMT1 (protein tyrosine phosphatase mitochondrial 1), an enzyme essential for cardiolipin synthesis, respond poorly to antigenic stimulation and have impaired mitochondrial fitness that limits memory T cell differentiation64 (Figure 4c). Therefore, lipid synthetic pathways regulate context-dependent T cell responses, suggesting that membrane lipids may serve as actionable targets to tune T cell functionality in different diseases.
Lipids are metabolic rheostats of intracellular signaling
Lipids serve as central regulators of intracellular signaling processes at the transcriptional, epigenetic and post-translational levels. We summarize below recent advances of understanding the roles of lipids as signaling molecules, with a particular emphasis on lipid-dependent post-translational modifications (Figure 5 and Table 1).
Figure 5. Lipid-dependent post-translational modifications orchestrate T cell responses.
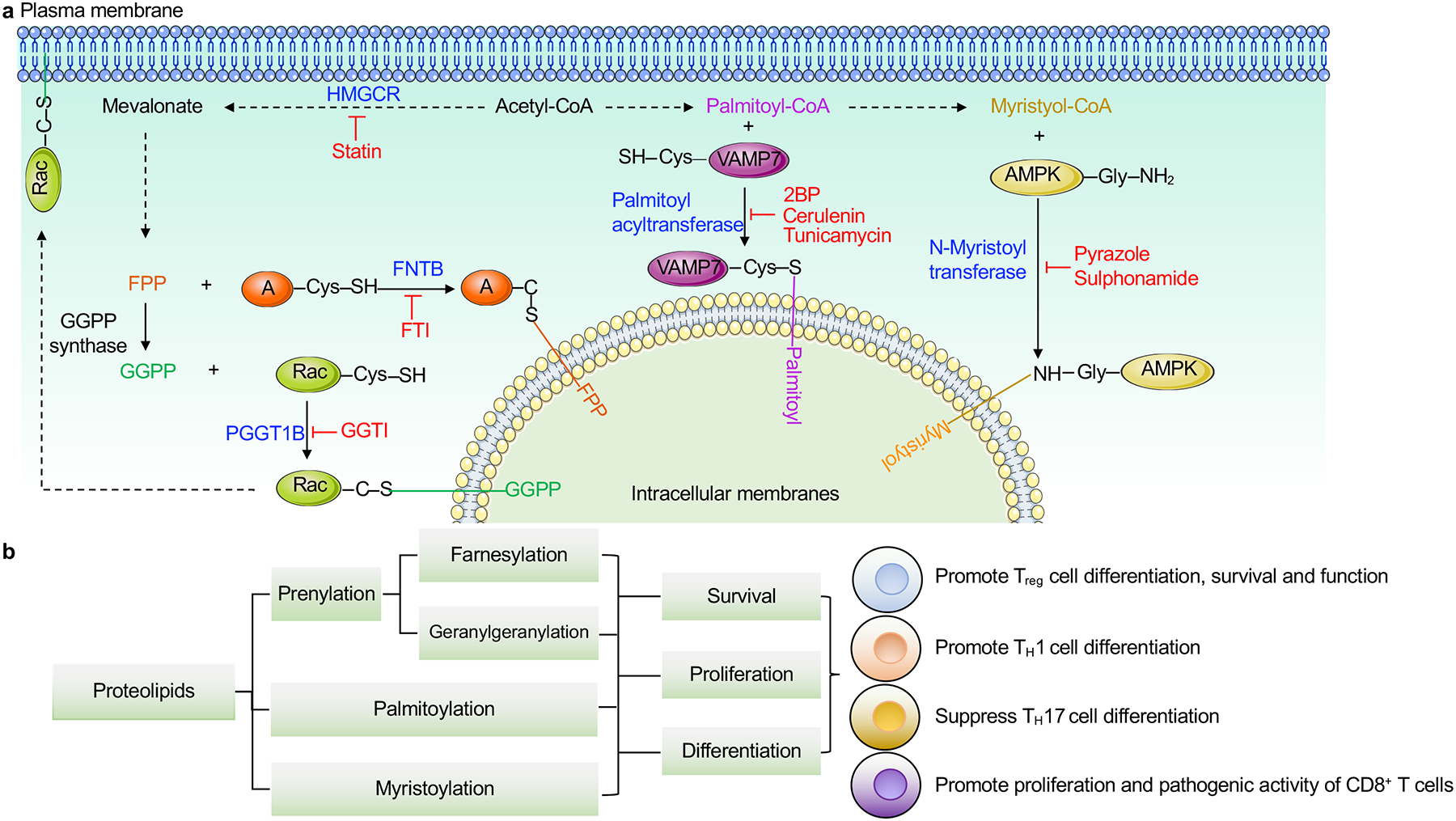
a, A summary of lipid-dependent post-translational modifications in T cells. Acetyl-CoA is used for synthesis of either mevalonate or FAs. Mevalonate-derived farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP) are covalently linked to specific cysteine residues in small G proteins, such as via Fntb-dependent protein farnesylation that attaches FPP to target proteins (denotated as A in the figure), and Pggt1b-dependent protein geranylgeranylation that conjugates GGPP to Rac. FA-derived palmitoyl-CoA and myristoyl-CoA are conjugated to glycine resides of certain proteins important for T cell biology. For example, palmitoyl acyltransferase DHHC18 promotes protein palmitoylation of VAMP7, while N-myristoyltransferase NMT leads to protein myristoylation of AMPK. These modifications serve important roles in establishing the localization of target proteins for the propagation of intracellular signaling. Inhibitors for the different lipid transferases are shown in red. b, Lipid-mediated post-translational modifications play critical roles in the survival, proliferation and differentiation of T cell subsets, including Treg, TH1, TH17, memory and effector T cells, as summarized in more detail in the figure. AMPK; AMP-activated protein kinase, GGTI; geranylgeranyl transferase type-1 inhibitor, FTI; farnesyltransferase inhibitor, HMGCR; HMG-CoA reductase, 2BP; 2-brompalmitate.
Lipids as metabolic modulators of gene transcription
Lipids bind to and influence the activation of transcription factors that affect T cell responses, including nuclear receptors peroxisome proliferation-activated receptors (PPARs) and LXRs. Specifically, FAs activate PPARs, and cholesterol and other sterols activate LXRs. Gain- and loss-of-function studies suggests that PPARs play complex roles in conventional T cell differentiation65. Also, deficiency of PPARγ impairs the accumulation and function of Treg cells in visceral adipose tissue (VAT)66, and as such contributes to systemic metabolic homeostasis. LXR signaling impairs T cell activation by inhibiting lipid and cholesterol synthesis via antagonism of the sterol regulatory binding proteins (SREBPs)67, which are discussed more below. However, LXRs can play both pro- and anti-inflammatory roles in T cell-driven disease severity. For instance, treatment with pharmacological agonists of LXRs dampens MOG-induced pathogenic TH17 cell differentiation in vitro and alleviates disease severity of EAE68 associated with reduced TH17 cell effector cytokines in vivo, while oxysterols, which are natural ligands that activate LXRs, inhibit IL-10 production by anti-inflammatory type 1 regulatory cells69. Overall, these complex roles of PPARs and LXRs appear to be dependent on the disease or tissue context and the subset of T cells analyzed.
SREBPs promote lipid synthesis and require SREBP cleavage-activating protein (SCAP) for activation. SCAP–SREBP signaling drives reprogramming of lipid metabolism toward synthesis and uptake, which causes T cells to exit from their quiescent state and undergo cellular proliferation26. Further, the SCAP–SREBP axis coordinates FA and mevalonate metabolism to promote the functional maturation and immunosuppressive function of Treg cells in the TME. Loss of SCAP or FASN in Treg cells results in the loss of suppressive function selectively in the TME, leading to enhanced tumor clearance35. Understanding the relationship between lipids and transcriptional programming in different contexts may provide insight for therapeutic targeting of T cells in diverse diseases.
FAO-derived acetyl-CoA can also be used as a substrate for histone acetyltransferases to promote histone acetylation. Indeed, acetate induces histone acetylation to promote IFN-γ production from CD8+ T cells in glucose-limiting conditions70 and promotes GAPDH acetylation and activation to augment glycolysis and recall responses of memory CD8+ T cells during acute viral infection71. Further, butyrate suppresses the activity of histone deacetylases, thereby boosting the effector function of CD8+ T cells72. Butyrate also enhances histone acetylation of Foxp3 and differentiation of Treg cells6,7. Consistent with the importance of acetyl-CoA in T cell responses, ACLY, which catalyzes synthesis of acetyl-CoA, is a positive regulator of histone acetylation and T cell immunity. Inhibition of ACLY impairs the function and proliferation of TH1 cells associated with reduced histone H3K9 acetylation73. Thus, acetyl-CoA serves context-dependent roles in epigenetic regulation. Future work is required to ascertain the signals that contribute to ACLY-dependent lipid synthesis versus histone acetylation, as well as the roles of these ACLY-dependent processes in fate decisions.
Post-translational modifications for modulation of intracellular signaling
Bidirectional metabolic signaling, which is the two-way communication between intracellular signaling networks and cell metabolic programs, is emerging as a crucial regulator of T cell responses29, with lipid modifications serving a key node underlying these processes. Certain lipid metabolites are post-translationally attached to proteins at specific amino acid residues to alter protein stability or membrane localization for modulation of T cell function (Figure 5a). For example, mevalonate pathway-derived isoprenoids farnesyl pyrophosphate (FPP) and GGPP are important for protein prenylation linked to specific cysteine residues of certain small G proteins such as the RAC family74. Inhibition of the mevalonate pathway or protein prenylation limits the activation, proliferation, survival and effector function of T cells74. Protein prenylation also controls the accumulation of activated Treg cells (also called effector Treg cells) by interplaying with immune receptor signaling that promotes the differentiation, proliferation and survival of Treg cells to prevent autoimmune reactions27. Further, downstream of SCAP–SREBP signaling, GGPP-mediated protein geranylgeranylation drives expression of the immune checkpoint molecule PD-1 to support the suppressive function of intratumoral Treg cells35 (Figure 5b). Therefore, post-translational modifications represent a regulatory axis that links lipid metabolism to intracellular signaling, which may be used as therapeutic targets for T cell-mediated diseases.
Additional lipid post-translational modifications, including palmitoylation and myristoylation, are implicated in T cell immunity. Palmitoylation is the process by which palmitate is conjugated to cysteine on a thioester linkage. Recent studies show that the palmitoyl acyltransferase DHHC18 promotes the palmitoylation of VAMP7 (a protein that is essential for intracellular vesicle fusion), thereby facilitating VAMP7 recruitment to the immunological synapse upon TCR activation to propagate TCR signaling75. Moreover, myristoylation occurs when the FA myristate is attached to glycine residues in proteins. One of the myristoylation targets is AMPK76 (Figure 5a), which has roles in memory T cell formation as discussed above45,46. Functional defect in N-myristoyltransferase, which mediates myristoylation, in T cells from rheumatoid arthritis impedes AMPK activity and instead, drives hyperactive mTOR and uncontrolled differentiation of TH1 and TH17 cells76 (Figure 5b). Thus, lipids are crucial modulators of intracellular signaling and play diverse roles in distinct T cell subsets (summarized in Table 1).
Targeting lipid metabolism in T cells for disease therapy
Given the important roles of T cells in human disease, there is growing interest in targeting T cells for disease therapy. Here, we highlight approaches to target lipid metabolism or signaling, and also summarize these studies in Table 2.
Table 2.
Targeting lipid metabolism in T cells for the treatment of diseases.
| Inhibitor or small molecule | Lipid category | Target | Function of target | T cells affected | Disease context | Ref. |
|---|---|---|---|---|---|---|
| Preclinical models | ||||||
| Etomoxir | Catabolism | CPT1a | FAO | iTreg and memory T cells | EAE | 50 |
| TOFA | Anabolism | ACC1 | Synthesis of FAs | Memory T cells | Chronic infection | 79 |
| C75 | Anabolism | FASN | Synthesis of FAs | Effector T cells | N/A | 80 |
| Statin | Anabolism | HMGCR | Synthesis of mevalonate | Treg and CD8+ T cells | EAE | 27,78 |
| FTI | Post-translational modification | Fntb | Protein farnesylation | Treg cells | N/A | 27 |
| GGTI | Post-translational modification | Pggt1b | Protein geranylgeranylation | Treg and CD8+ T cells | Autoimmune colitis | 27,78 |
| Ciglitazone | Agonist | PPARγ | Transcription factor | iTreg cells | N/A | 81 |
| Pioglitazone | Agonist | PPARγ | Transcription factor | VAT Treg cells | High fat diet-fed obese mice | 66 |
| Gemfibrozil | Agonist | PPARα | Transcription factor | TH2 cells | N/A | 82 |
| GW-0742 | Agonist | PPARβ/δ | Transcription factor | N/A | EAE | 83 |
| Bexarotene | Agonist | RXR | Transcription factor | iTreg cells | N/A | 65 |
| Clinical trials | ||||||
| Statin | Anabolism | HMGCR | Synthesis of mevalonate | N/A | Dyslipidemia92 | |
| PF-05221304 | Anabolism | ACC1 | Synthesis of FAs | N/A | NASH, NAFLD | 91 |
iTreg, in vitro-derived Treg; N/A: not applicable; NAFLD, nonalcoholic fatty liver disease.
Chemicals targeting of lipid metabolism or signaling alter T cell biology
Several inhibitors that target enzymes in lipid-related metabolic pathways have been developed (Table 2 and Figure 5a), with complementary genetic and chemical targeting approaches demonstrating their specificity and applicability to modulate T cell function. As noted above, etomoxir irreversibly inhibits CPT1a to suppress FAO, with etomoxir treatment inhibiting the survival or differentiation of Treg and memory T cells45,50. However, genetic deletion of CPT1a does not alter the homeostasis of Treg or memory T cells in vivo50,77. Instead, etomoxir treatment in vivo reduces mitochondrial respiration independent of CPT1a-mediated FAO50. Whether these effects are attributed to inhibition of CPT1a-independent oxidation of SCFAs or MCFAs, or other CPT isoforms that may promote FAO, remains uncertain. Similar to genetic deletion, pharmacological inhibition of HMGCR using statins impairs conventional T cell and Treg expansion and function27,40,41. Moreover, inhibition of protein farnesylation with a farnesyltransferase inhibitor (FTI) partially phenocopies the cellular features of statin-treated Treg cells, while suppression of protein geranylgeranylation using a geranylgeranyltransferase inhibitor (GGTI) fully recapitulates the statin-induced effects27. Additionally, both statin and GGTI treatments increase KLF2 expression that modulates inflammatory and pathogenic CD8+ T cells78. It would be insightful to examine the relative effects of inhibitors targeting the mevalonate pathways in individual cell types, given the central importance of this pathway in cellular physiology.
Studies have also begun to explore the effects of FAS-related inhibitors on T cell function. Inhibition of ACC1 by TOFA during early T cell priming enhances the pool of memory T cells in chronic parasitic infection79, which is consistent with genetic evidence in ACC1-deficient T cells in some systems33, with the caveat that ACC1 may be important for survival that maintains memory T cells over time34. Also, TOFA treatment limits TH17 and increases Treg cell generation31, with ACC1 deletion also reducing TH17 cell differentiation, especially in an obese environment, with no notable effects on Treg cell differentiation32. Treatment of human CD4+ T cells with the FASN inhibitor C75 prevents activation-induced cell death of effector cells80. However, aside from a role in intratumoral Treg cells35, genetic deletion of FASN in conventional T cells has not been explored. Comparative analyses of the effects of pharmacological and genetic perturbations of FASN will be insightful for identifying possible off-target or toxic effects of FASN inhibitors in T cells, and for potential clinical applications to modulate T cell responses in disease.
Lipid agonists that promote PPARs also modulate T cell fate. For instance, the PPARγ ligand ciglitazone enhances the conversion of naïve CD4+ T cells into Treg cells81. Further, pioglitazone, another PPARγ ligand, enhances Treg cell accumulation in the VAT66, while the PPARα agonist gemfibrozil skews naïve CD4+ T cell differentiation in favor of TH2 cells82. Collectively, lipid agonists may be instrumental for altering T cell subset differentiation.
Targeting lipid metabolism or signaling for disease therapeutics
Targeting lipid pathways or molecules in T cells holds enormous therapeutic potential (Table 2). In particular, modulation of lipid uptake or metabolism alters the antitumor function of T cells in mouse models of cancer11–14,17,35,59. In addition, treatment with GW0742, an agonist of PPARβ/δ, mitigates EAE83. The generation of specific lipids or induction of lipid metabolic pathways promote Treg cell generation and function, which may also be applied to the treatment of autoimmune diseases, including multiple sclerosis and type 1 diabetes84,85. Moreover, microbiota-derived lipids mitigate disease in a murine model of GVHD by modulating T cell responses86. Similarly, an interplay between the microbiome, dietary fiber, and SCFAs influences the response to cancer immunotherapies. Enhanced anti-PD-1 therapeutic responses are observed in melanoma patients with high microbiome diversity and an abundance of certain microbiota species, including Ruminococcaceae, Faecalibacterium, Bifidobacterium longum, Collinsella aerofaciens, and Enterococcus faecium87,88. In addition, reconstitution of germ-free mice with fecal material from anti-PD-1 responding patients improves tumor control and anti-PD-L1 therapy that is associated with enhanced T cell responses88. Moreover, higher intake of dietary fiber is correlated with improved responsiveness to immune checkpoint blockade treatment in melanoma patients89. The precise role of microbiota-derived SCFAs in reprogramming T cell metabolism and the effects on the antitumor response warrant further investigation.
Several lipid metabolism-related drugs are also being explored for clinical use (Table 2). Treatment with statins impedes the growth and survival of certain tumors. Moreover, statins are being explored for use in combination therapies for anticancer treatments, as reviewed elsewhere90. The ACC1 inhibitor PF-05221304 is being explored as a treatment for NASH and nonalcoholic fatty liver disease91. Due to certain adverse effects associated with statin treatment, novel drugs to decrease lipid content in patients with dyslipidemia or hypercholesterolemia have been developed, including inclisiran, bempedoic acid and icosapent ethyl92. These drugs inhibit PCSK9, whose expression in tumor cells hinders LDLR surface level and TCR signaling in intratumoral T cells20, further highlighting the clinical potential to target lipid metabolism.
Conclusions and future outlook
As summarized in this review, lipids are remarkably diverse and have crucial roles in the regulation of energy or biomass production, membrane structure, and signal transduction in T cells. By integrating immunological and environmental cues, T cells undergo rewiring of metabolic programs including intracellular lipid synthesis and extracellular lipid uptake and utilization, which contribute to their functional specialization and adaption by modulating metabolic and signaling events. Consequently, perturbation of lipid metabolism alters T cell responses in different physiological states and diseases. Therefore, lipid metabolism-based therapy offers an attractive means to modulate T cell function for the treatment of human disease.
Several key questions and directions remain to be explored regarding the roles of lipid metabolism and signaling in T cells. First, how do immune receptor-mediated processes orchestrate uptake, anabolism, catabolism and storage of lipids in discrete T cell subsets? Second, does nutrient competition or coordination between cell types in a microenvironment influence the acquisition of extracellular lipids by T cells in different contexts? Third, what role do environmental cues, including macronutrients (e.g. glucose or amino acids), cytokines, growth factors, and oxygen levels, play in modulating lipid metabolism to instruct T cell fate decisions? Finally, it will be essential to reconstruct T cell-specific lipid signaling networks, including signals that shape lipid metabolic rewiring and the downstream programs tuned by lipids at the transcriptional, epigenetic, and post-translational levels. These investigations will allow for the development of lipid-based therapies to target specific subsets of T cells in discrete settings and contexts.
Although context-dependent regulation of lipid metabolism is emerging for T cell responses63,64, lipid metabolism is a central biological process wherein conventional targeting methods, such as small molecule inhibitors, may lead to non-specific or adverse effects. Therefore, strategies to target lipid metabolic processes selectively in T cells will be of critical importance to exploit. Antibodies have high specificity and affinity for their ligands, and these can be conjugated to small molecules or nanoparticles to achieve site- or cell type-specific targeting of T cell subsets. Also, technologies that predict the molecular targets of lipid signaling, including the integration of advanced computational tools, machine learning, and robotic-aided high-throughput screening93, will likely rapidly advance drug development to target lipid metabolism. The combined use of innovative technologies and targeting strategies to modulate lipid metabolism or signaling is anticipated to advance next-generation therapies.
Acknowledgements
This work was supported by NIH grants AI105887, AI131703, AI140761, AI150241, AI150514 and CA253188, Alliance for Lupus Research grant, and ALSAC (to H.C.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing interests
H.C. is a consultant for Kumquat Biosciences.
References
- 1.Chapman NM & Chi H Metabolic adaptation of lymphocytes in immunity and disease. Immunity 55, 14–30, doi: 10.1016/j.immuni.2021.12.012 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maseda D, Ricciotti E & Crofford LJ Prostaglandin regulation of T cell biology. Pharmacol Res 149, 104456, doi: 10.1016/j.phrs.2019.104456 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Trompette A et al. Dietary Fiber Confers Protection against Flu by Shaping Ly6c(−) Patrolling Monocyte Hematopoiesis and CD8(+) T Cell Metabolism. Immunity 48, 992–1005 e1008, doi: 10.1016/j.immuni.2018.04.022 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Bachem A et al. Microbiota-Derived Short-Chain Fatty Acids Promote the Memory Potential of Antigen-Activated CD8(+) T Cells. Immunity 51, 285–297 e285, doi: 10.1016/j.immuni.2019.06.002 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Dudek M et al. Auto-aggressive CXCR6(+) CD8 T cells cause liver immune pathology in NASH. Nature 592, 444–449, doi: 10.1038/s41586-021-03233-8 (2021). [DOI] [PubMed] [Google Scholar]; This paper shows a pathogenic role for the SCFA acetate in CD8+ T cell-mediated autoinflammation and tissue damage in the liver, which is in contrast to the protective roles for SCFAs in tissue homeostasis or anti-pathogen immunity.
- 6.Furusawa Y et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450, doi: 10.1038/nature12721 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Arpaia N et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455, doi: 10.1038/nature12726 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith PM et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573, doi: 10.1126/science.1241165 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park J et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol 8, 80–93, doi: 10.1038/mi.2014.44 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haghikia A et al. Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity 43, 817–829, doi: 10.1016/j.immuni.2015.09.007 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Wang H et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat Immunol 21, 298–308, doi: 10.1038/s41590-019-0589-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study establishes a role for CD36-dependent lipid uptake in supporting Treg cell function in the TME.
- 12.Xu S et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity 54, 1561–1577 e1567, doi: 10.1016/j.immuni.2021.05.003 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study, along with Ma et al. 2021, shows that CD36-mediated lipid uptake triggers CD8+ T cell dysfunction in the TME.
- 13.Ma X et al. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab 33, 1001–1012 e1005, doi: 10.1016/j.cmet.2021.02.015 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study, along with Xu et al. 2021, reveals that CD36-mediated lipid uptake triggers CD8+ T cell dysfunction in the TME.
- 14.Manzo T et al. Accumulation of long-chain fatty acids in the tumor microenvironment drives dysfunction in intrapancreatic CD8+ T cells. J Exp Med 217, doi: 10.1084/jem.20191920 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu L et al. Niche-Selective Inhibition of Pathogenic Th17 Cells by Targeting Metabolic Redundancy. Cell 182, 641–654 e620, doi: 10.1016/j.cell.2020.06.014 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Proto JD et al. Hypercholesterolemia induces T cell expansion in humanized immune mice. J Clin Invest 128, 2370–2375, doi: 10.1172/JCI97785 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma X et al. Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab 30, 143–156 e145, doi: 10.1016/j.cmet.2019.04.002 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma X et al. Cholesterol negatively regulates IL-9-producing CD8(+) T cell differentiation and antitumor activity. J Exp Med 215, 1555–1569, doi: 10.1084/jem.20171576 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J, Lu E, Yi T & Cyster JG EBI2 augments Tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nature 533, 110–114, doi: 10.1038/nature17947 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yuan J et al. Potentiating CD8(+) T cell antitumor activity by inhibiting PCSK9 to promote LDLR-mediated TCR recycling and signaling. Protein Cell 12, 240–260, doi: 10.1007/s13238-021-00821-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hang S et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 576, 143–148, doi: 10.1038/s41586-019-1785-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study establishes that selective bile acids balance intestinal TH17 and Treg cell differentiation, which contributes to tissue homeostasis.
- 22.Song X et al. Microbial bile acid metabolites modulate gut RORgamma(+) regulatory T cell homeostasis. Nature 577, 410–415, doi: 10.1038/s41586-019-1865-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li W et al. A bacterial bile acid metabolite modulates Treg activity through the nuclear hormone receptor NR4A1. Cell Host Microbe 29, 1366–1377 e1369, doi: 10.1016/j.chom.2021.07.013 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campbell C et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 581, 475–479, doi: 10.1038/s41586-020-2193-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen ML et al. CAR directs T cell adaptation to bile acids in the small intestine. Nature 593, 147–151, doi: 10.1038/s41586-021-03421-6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kidani Y et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol 14, 489–499, doi: 10.1038/ni.2570 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su W et al. Protein Prenylation Drives Discrete Signaling Programs for the Differentiation and Maintenance of Effector Treg Cells. Cell Metab 32, 996–1011 e1017, doi: 10.1016/j.cmet.2020.10.022 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates that mevalonate metabolism-dependent post-translational modifications are essential for Treg cell activation and establishment of immunological tolerance.
- 28.Klein Geltink RI et al. Mitochondrial Priming by CD28. Cell 171, 385–397 e311, doi: 10.1016/j.cell.2017.08.018 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chapman NM, Boothby MR & Chi H Metabolic coordination of T cell quiescence and activation. Nat Rev Immunol 20, 55–70, doi: 10.1038/s41577-019-0203-y (2020). [DOI] [PubMed] [Google Scholar]
- 30.Yang K et al. T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming. Immunity 39, 1043–1056, doi: 10.1016/j.immuni.2013.09.015 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berod L et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med 20, 1327–1333, doi: 10.1038/nm.3704 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Endo Y et al. Obesity Drives Th17 Cell Differentiation by Inducing the Lipid Metabolic Kinase, ACC1. Cell Rep 12, 1042–1055, doi: 10.1016/j.celrep.2015.07.014 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Endo Y et al. ACC1 determines memory potential of individual CD4(+) T cells by regulating de novo fatty acid biosynthesis. Nat Metab 1, 261–275, doi: 10.1038/s42255-018-0025-4 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Lee J et al. Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity. J Immunol 192, 3190–3199, doi: 10.4049/jimmunol.1302985 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lim SA et al. Lipid signalling enforces functional specialization of Treg cells in tumours. Nature 591, 306–311, doi: 10.1038/s41586-021-03235-6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study establishes a relationship between lipid synthesis and PD-1 immune checkpoint signaling in Treg cell-mediated suppression of antitumor immunity.
- 36.Tian M et al. ACLY ubiquitination by CUL3-KLHL25 induces the reprogramming of fatty acid metabolism to facilitate iTreg differentiation. Elife 10, doi: 10.7554/eLife.62394 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sofi MH et al. Ceramide synthesis regulates T cell activity and GVHD development. JCI Insight 2, doi: 10.1172/jci.insight.91701 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vaena S et al. Aging-dependent mitochondrial dysfunction mediated by ceramide signaling inhibits antitumor T cell response. Cell Rep 35, 109076, doi: 10.1016/j.celrep.2021.109076 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li G et al. Nanoliposome C6-Ceramide Increases the Anti-tumor Immune Response and Slows Growth of Liver Tumors in Mice. Gastroenterology 154, 1024–1036 e1029, doi: 10.1053/j.gastro.2017.10.050 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lacher SM et al. HMG-CoA reductase promotes protein prenylation and therefore is indispensible for T-cell survival. Cell Death Dis 8, e2824, doi: 10.1038/cddis.2017.221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Timilshina M et al. Activation of Mevalonate Pathway via LKB1 Is Essential for Stability of Treg Cells. Cell Rep 27, 2948–2961 e2947, doi: 10.1016/j.celrep.2019.05.020 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Yang K et al. Homeostatic control of metabolic and functional fitness of Treg cells by LKB1 signalling. Nature 548, 602–606, doi: 10.1038/nature23665 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeng H et al. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 499, 485–490, doi: 10.1038/nature12297 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karmaus PWF et al. Metabolic heterogeneity underlies reciprocal fates of TH17 cell stemness and plasticity. Nature 565, 101–105, doi: 10.1038/s41586-018-0806-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pearce EL et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460, 103–107, doi: 10.1038/nature08097 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borges da Silva H et al. The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8(+) T cells. Nature 559, 264–268, doi: 10.1038/s41586-018-0282-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Sullivan D et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 41, 75–88, doi: 10.1016/j.immuni.2014.06.005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van der Windt GJ et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36, 68–78, doi: 10.1016/j.immuni.2011.12.007 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cui G et al. IL-7-Induced Glycerol Transport and TAG Synthesis Promotes Memory CD8+ T Cell Longevity. Cell 161, 750–761, doi: 10.1016/j.cell.2015.03.021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raud B et al. Etomoxir Actions on Regulatory and Memory T Cells Are Independent of Cpt1a-Mediated Fatty Acid Oxidation. Cell Metab 28, 504–515 e507, doi: 10.1016/j.cmet.2018.06.002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang C et al. STAT3 Activation-Induced Fatty Acid Oxidation in CD8(+) T Effector Cells Is Critical for Obesity-Promoted Breast Tumor Growth. Cell Metab 31, 148–161 e145, doi: 10.1016/j.cmet.2019.10.013 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ringel AE et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 183, 1848–1866 e1826, doi: 10.1016/j.cell.2020.11.009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pan Y et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543, 252–256, doi: 10.1038/nature21379 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper establishes that fatty acid binding proteins are essential for regulating TRM cell accumulaion in the skin, pointing to a role for extracellular fatty acid uptake and oxidiation in orchestrating TRM persistence.
- 54.Frizzell H et al. Organ-specific isoform selection of fatty acid-binding proteins in tissue-resident lymphocytes. Sci Immunol 5, doi: 10.1126/sciimmunol.aay9283 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Lin R et al. Fatty Acid Oxidation Controls CD8(+) Tissue-Resident Memory T-cell Survival in Gastric Adenocarcinoma. Cancer Immunol Res 8, 479–492, doi: 10.1158/2326-6066.CIR-19-0702 (2020). [DOI] [PubMed] [Google Scholar]
- 56.Field CS et al. Mitochondrial Integrity Regulated by Lipid Metabolism Is a Cell-Intrinsic Checkpoint for Treg Suppressive Function. Cell Metab 31, 422–437 e425, doi: 10.1016/j.cmet.2019.11.021 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ecker C et al. Differential Reliance on Lipid Metabolism as a Salvage Pathway Underlies Functional Differences of T Cell Subsets in Poor Nutrient Environments. Cell Rep 23, 741–755, doi: 10.1016/j.celrep.2018.03.084 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Howie D et al. A Novel Role for Triglyceride Metabolism in Foxp3 Expression. Front Immunol 10, 1860, doi: 10.3389/fimmu.2019.01860 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu X et al. Reprogramming lipid metabolism prevents effector T cell senescence and enhances tumor immunotherapy. Sci Transl Med 13, doi: 10.1126/scitranslmed.aaz6314 (2021). [DOI] [PubMed] [Google Scholar]
- 60.Shin J, O’Brien TF, Grayson JM & Zhong XP Differential regulation of primary and memory CD8 T cell immune responses by diacylglycerol kinases. J Immunol 188, 2111–2117, doi: 10.4049/jimmunol.1102265 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang W et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 531, 651–655, doi: 10.1038/nature17412 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang F, Beck-Garcia K, Zorzin C, Schamel WW & Davis MM Inhibition of T cell receptor signaling by cholesterol sulfate, a naturally occurring derivative of membrane cholesterol. Nat Immunol 17, 844–850, doi: 10.1038/ni.3462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fu G et al. Metabolic control of TFH cells and humoral immunity by phosphatidylethanolamine. Nature 595, 724–729, doi: 10.1038/s41586-021-03692-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study establishes a context-specific role for de novo phosphatidylethanolamine synthesis in driving TFH cell generation and humoral immunity.
- 64.Corrado M et al. Dynamic Cardiolipin Synthesis Is Required for CD8(+) T Cell Immunity. Cell Metab 32, 981–995 e987, doi: 10.1016/j.cmet.2020.11.003 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Howie D, Ten Bokum A, Necula AS, Cobbold SP & Waldmann H The Role of Lipid Metabolism in T Lymphocyte Differentiation and Survival. Front Immunol 8, 1949, doi: 10.3389/fimmu.2017.01949 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cipolletta D et al. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 486, 549–553, doi: 10.1038/nature11132 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bensinger SJ et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell 134, 97–111, doi: 10.1016/j.cell.2008.04.052 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu J, Wagoner G, Douglas JC & Drew PD Liver X receptor agonist regulation of Th17 lymphocyte function in autoimmunity. J Leukoc Biol 86, 401–409, doi: 10.1189/jlb.1008600 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vigne S et al. IL-27-Induced Type 1 Regulatory T-Cells Produce Oxysterols that Constrain IL-10 Production. Front Immunol 8, 1184, doi: 10.3389/fimmu.2017.01184 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Qiu J et al. Acetate Promotes T Cell Effector Function during Glucose Restriction. Cell Rep 27, 2063–2074 e2065, doi: 10.1016/j.celrep.2019.04.022 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Balmer ML et al. Memory CD8(+) T Cells Require Increased Concentrations of Acetate Induced by Stress for Optimal Function. Immunity 44, 1312–1324, doi: 10.1016/j.immuni.2016.03.016 (2016). [DOI] [PubMed] [Google Scholar]
- 72.Luu M et al. Regulation of the effector function of CD8(+) T cells by gut microbiota-derived metabolite butyrate. Sci Rep 8, 14430, doi: 10.1038/s41598-018-32860-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bailis W et al. Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 571, 403–407, doi: 10.1038/s41586-019-1311-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thurnher M & Gruenbacher G T lymphocyte regulation by mevalonate metabolism. Sci Signal 8, re4, doi: 10.1126/scisignal.2005970 (2015). [DOI] [PubMed] [Google Scholar]
- 75.Morrison E et al. Dynamic palmitoylation events following T-cell receptor signaling. Commun Biol 3, 368, doi: 10.1038/s42003-020-1063-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wen Z et al. N-myristoyltransferase deficiency impairs activation of kinase AMPK and promotes synovial tissue inflammation. Nat Immunol 20, 313–325, doi: 10.1038/s41590-018-0296-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Saravia J et al. Homeostasis and transitional activation of regulatory T cells require c-Myc. Sci Adv 6, eaaw6443, doi: 10.1126/sciadv.aaw6443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bu DX et al. Statin-induced Kruppel-like factor 2 expression in human and mouse T cells reduces inflammatory and pathogenic responses. J Clin Invest 120, 1961–1970, doi: 10.1172/JCI41384 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ibitokou SA et al. Early Inhibition of Fatty Acid Synthesis Reduces Generation of Memory Precursor Effector T Cells in Chronic Infection. J Immunol 200, 643–656, doi: 10.4049/jimmunol.1602110 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Voss K, Luthers CR, Pohida K & Snow AL Fatty Acid Synthase Contributes to Restimulation-Induced Cell Death of Human CD4 T Cells. Front Mol Biosci 6, 106, doi: 10.3389/fmolb.2019.00106 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wohlfert EA, Nichols FC, Nevius E & Clark RB Peroxisome proliferator-activated receptor gamma (PPARgamma) and immunoregulation: enhancement of regulatory T cells through PPARgamma-dependent and -independent mechanisms. J Immunol 178, 4129–4135, doi: 10.4049/jimmunol.178.7.4129 (2007). [DOI] [PubMed] [Google Scholar]
- 82.Gocke AR et al. Transcriptional modulation of the immune response by peroxisome proliferator-activated receptor-{alpha} agonists in autoimmune disease. J Immunol 182, 4479–4487, doi: 10.4049/jimmunol.0713927 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Polak PE et al. Protective effects of a peroxisome proliferator-activated receptor-beta/delta agonist in experimental autoimmune encephalomyelitis. J Neuroimmunol 168, 65–75, doi: 10.1016/j.jneuroim.2005.07.006 (2005). [DOI] [PubMed] [Google Scholar]
- 84.Marino E et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat Immunol 18, 552–562, doi: 10.1038/ni.3713 (2017). [DOI] [PubMed] [Google Scholar]
- 85.Pompura SL et al. Oleic acid restores suppressive defects in tissue-resident FOXP3 Tregs from patients with multiple sclerosis. J Clin Invest 131, doi: 10.1172/JCI138519 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mathewson ND et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat Immunol 17, 505–513, doi: 10.1038/ni.3400 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gopalakrishnan V et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103, doi: 10.1126/science.aan4236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Matson V et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359, 104–108, doi: 10.1126/science.aao3290 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Spencer CN et al. Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science 374, 1632–1640, doi: 10.1126/science.aaz7015 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study suggests that high dietary fiber intake improves responsiveness to immunotherapy via interplay with microbiota-derived production of the SCFA propionate.
- 90.Longo J, van Leeuwen JE, Elbaz M, Branchard E & Penn LZ Statins as Anticancer Agents in the Era of Precision Medicine. Clin Cancer Res 26, 5791–5800, doi: 10.1158/1078-0432.CCR-20-1967 (2020). [DOI] [PubMed] [Google Scholar]
- 91.Calle RA et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: two parallel, placebo-controlled, randomized phase 2a trials. Nat Med 27, 1836–1848, doi: 10.1038/s41591-021-01489-1 (2021). [DOI] [PubMed] [Google Scholar]
- 92.Pecin I & Reiner Z Novel Experimental Agents for the Treatment of Hypercholesterolemia. J Exp Pharmacol 13, 91–100, doi: 10.2147/JEP.S267376 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Payandeh J & Volgraf M Ligand binding at the protein-lipid interface: strategic considerations for drug design. Nat Rev Drug Discov 20, 710–722, doi: 10.1038/s41573-021-00240-2 (2021). [DOI] [PubMed] [Google Scholar]
- 94.Lopes N et al. Distinct metabolic programs established in the thymus control effector functions of gammadelta T cell subsets in tumor microenvironments. Nat Immunol 22, 179–192, doi: 10.1038/s41590-020-00848-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kobayashi T et al. Increased lipid metabolism impairs NK cell function and mediates adaptation to the lymphoma environment. Blood 136, 3004–3017, doi: 10.1182/blood.2020005602 (2020). [DOI] [PubMed] [Google Scholar]
- 96.Michelet X et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol 19, 1330–1340, doi: 10.1038/s41590-018-0251-7 (2018). [DOI] [PubMed] [Google Scholar]
- 97.Fu S et al. Impaired lipid biosynthesis hinders anti-tumor efficacy of intratumoral iNKT cells. Nat Commun 11, 438, doi: 10.1038/s41467-020-14332-x (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ko JS et al. Palmitate inhibits arthritis by inducing t-bet and gata-3 mRNA degradation in iNKT cells via IRE1alpha-dependent decay. Sci Rep 7, 14940, doi: 10.1038/s41598-017-14780-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Muri J, Thut H, Bornkamm GW & Kopf M B1 and Marginal Zone B Cells but Not Follicular B2 Cells Require Gpx4 to Prevent Lipid Peroxidation and Ferroptosis. Cell Rep 29, 2731–2744 e2734, doi: 10.1016/j.celrep.2019.10.070 (2019). [DOI] [PubMed] [Google Scholar]
- 100.Bibby JA et al. Cholesterol metabolism drives regulatory B cell IL-10 through provision of geranylgeranyl pyrophosphate. Nat Commun 11, 3412, doi: 10.1038/s41467-020-17179-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]