

Abstract
We describe deep analysis of the human proteome in less than 1 h. We achieve this expedited proteome characterization by leveraging state-of-the-art sample preparation, chromatographic separations, and data analysis tools, and by using the new Orbitrap Astral mass spectrometer equipped with a quadrupole mass filter, a high-field Orbitrap mass analyzer, and an asymmetric track lossless (Astral) mass analyzer. The system offers high tandem mass spectrometry acquisition speed of 200 Hz and detects hundreds of peptide sequences per second within data-independent acquisition or data-dependent acquisition modes of operation. The fast-switching capabilities of the new quadrupole complement the sensitivity and fast ion scanning of the Astral analyzer to enable narrow-bin data-independent analysis methods. Over a 30-min active chromatographic method consuming a total analysis time of 56 min, the Q-Orbitrap-Astral hybrid MS collects an average of 4319 MS1 scans and 438,062 tandem mass spectrometry scans per run, producing 235,916 peptide sequences (1% false discovery rate). On average, each 30-min analysis achieved detection of 10,411 protein groups (1% false discovery rate). We conclude, with these results and alongside other recent reports, that the 1-h human proteome is within reach.
Keywords: proteomics, mass spectrometry, technology development, instrumentation, proteome, data independent acquisition, single shot, high throughput, CRISPR
Graphical Abstract
Highlights
-
•
Deep analysis of the human proteome in less than 1 h.
-
•
State-of-the-art sample preparation, chromatography, data acquisition, and analysis.
-
•
Comparison of quantification in Orbitrap Astral and Orbitrap Tribrid platforms.
-
•
Analysis of a CRISPR-Cas9 mitochondrial gene KO in human HAP1 cells.
-
•
A new age of proteomics at the confluence of throughput, depth, and sensitivity.
In Brief
We present deep analysis of the human proteome in less than 1 h. The Orbitrap Astral mass spectrometer, alongside advances in sample preparation, method development, and data processing, is ushering in a new era of proteomics where a harmony of throughput, depth, and sensitivity are enable studies of unprecedented impact. We contextualize these results within the landscape of human proteomics. Comparisons with previous instrumentation in interrogating gene knockouts demonstrate the potential of proteomics to elevate biological investigation in years to come.
The human proteome, the collection of proteins and their isoforms encoded by the ∼20,000 coding genes in the human genome, acts as cellular machinery driving diverse biological phenotypes (1). Despite the importance of knowing which, and when, genes are expressed, comprehensive characterization of the human proteome remains a difficult and elusive proposition (2, 3, 4, 5).
Mass spectrometry (MS)-based proteomics remains the central technology for proteome measurement and, over the past few decades, has proven invaluable for elucidating the dynamic and heterogeneous proteomic states of cells and tissues, even if not at full proteome depth. Achievable proteomic depth, of course, varies with complexity of the system. Ten years ago, we reported the near-complete analysis of the yeast proteome within approximately 1 h. To do this, we leveraged a newly developed Tribrid mass spectrometer having a quadrupole mass filter, Orbitrap analyzer, and dual-cell linear ion trap analyzer (i.e., Thermo Fisher Scientific Orbitrap Fusion mass spectrometer). This system offered tandem mass spectrometry (MS/MS) scan rates of 20 Hz, allowing detection of nearly 4000 proteins after a 70-min liquid chromatography tandem mass spectrometry (LC-MS/MS) experiment (6, 7). Over the past decade, advances in sample preparation, peptide separations, and data processing strategies, and the advent of faster, more sensitive MS instrumentation have led to new frontiers of depth and throughput in human proteomics (8).
Ten years ago, Helm et al. reported the identification of ∼7500 human proteins from a tissue within 1 day of analysis using offline fractionation and traveling wave ion mobility separations (9). Pozniak et al. expanded upon this by quantifying 10,043 protein groups from MCF7 cells, leveraging deep offline fractionation and a new Q-Orbitrap instrument capable of greater sampling rates of precursor ions in data-dependent acquisition (DDA) schemes (10).
Numerous groups over the past decade have built upon the promise of deep MS-based human proteomics, with several reporting over 10,000 unique protein groups (9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22). To achieve these depths, however, offline fractionation and lengthy acquisition times have been necessary. Several of us, for example, recently reported global detection of human variants and isoforms, including 17,717 protein groups with median sequence coverage of 80% (20). As with other drafts of the human proteome (13, 14), such an outcome required multiple cell lines, multiple protease treatments, and extensive fractionation, in all comprising thousands of hours of sample preparation and LC-MS/MS acquisition time, and countless hours of data analysis (20).
Modern proteomics applications often necessitate rapid analysis of proteomes, and new instrumentation is enabling unprecedented precursor sampling rates. Several groups have reported efforts approaching 10,000 human proteins quantified at a rate of over 500 proteins per hour (15, 17, 18). Bekker-Jensen et al. used extensive offline fractionation (46 fractions) coupled with short, 30-min liquid chromatography (LC) gradients online with a Q-Orbitrap instrument capable of high-resolution MS/MS scans at rates up to 18 Hz using a high-field Orbitrap analyzer (15). Advances in MS instrumentation have enabled faster rates of acquisition and improved depth of coverage. The redesigned Q-Orbitrap platform, termed the Orbitrap Exploris MS, is capable of scan rates up to 40 Hz. Additionally, time-of-flight (TOF) analyzers have long been capable of fast scanning speeds in excess of 100 Hz, although the poor ion transmission efficiency to and within TOF analyzers has hampered sensitivity (23, 24). Improvements within Q-TOF instruments have prompted a rise in popularity in recent years (25, 26). Leveraging the timsTOF platform, Meier et al. demonstrated parallel accumulation-serial fragmentation (PASEF) in 2015, making use of the trapped ion mobility spectrometry (TIMS) cell’s ability to trap and separate ions on the basis of collisional cross-section prior to rapid TOF analysis (27). Using wide, nonoverlapping quadrupole bins after the TIMS separations to coisolate multiple peptides in a method termed dia-PASEF, Meier et al. later reported over 7600 proteins in only 2 h of acquisition time (28). More recently, Skowronek et al. leveraged dia-PASEF methods with very deep spectral libraries to identify over 7700 unique proteins in a single 44 min LC-MS/MS run (29).
With many researchers aiming to achieve high depth of coverage across a wide breadth of samples, a demand for novel, high-throughput methods have come into the forefront. Indeed, numerous reports highlight exceptionally fast acquisition schemes with microflow LC configurations, in some instances identifying over 5000 human proteins within 5 min of active LC-MS/MS time (30, 31, 32, 33).
We highlight the leap in performance made possible with new instrumentation in the Thermo Fisher Scientific Orbitrap asymmetric track lossless (Astral) mass spectrometer. Integrated alongside a high-resolution accurate mass (HRAM) Orbitrap analyzer, the instrument introduces a fast-switching quadrupole mass filter congruent with downstream speeds of ion detection, a dual-pressure ion processor cell which fragments ions in the high-pressure cell before they are moved to the low-pressure cell for further accumulation and orthogonal extraction to the Astral analyzer (34), and a high dynamic range detector (21). Altogether, ions can be manipulated in five stages of the MS at once (Orbitrap, ion routing multipole, two ion processor stages, Astral analyzer), allowing for a high degree of parallelization which enables MS/MS scan speeds of 200 Hz while HRAM MS1 full scans are synchronously acquired in the Orbitrap analyzer. To provide such fast scan speeds, the Astral analyzer is capable of single-ion detection sensitivity like a linear ion trap, all while enabling resolving powers of 80,000 at m/z 524 (21, 35, 36).
Here, in triplicate 7-min microflow active LC gradients on the Orbitrap Astral MS, we report 7852 protein groups from 94,267 peptides on average. When using 15-, 30-, and 60-min active, nano-LC gradients, triplicate experiments yield an average of 9,831, 10,411, and 10,645 unique protein groups from 195,612, 234,406, and 245,754 unique peptides, respectively. These nanoflow methods delivered a median sequence coverage of 38%, 42.4%, and 44.4% across identified proteins for each respective gradient length. Our 30-min method delivered approximately 347 proteins per minute. Finally, we applied our 30-min active gradient method to analyze CRISPR-Cas9 KO of the mitochondrial gene MGME1 in human HAP1 cells. There, we quantified an average of 10,353 proteins across three technical replicates and find 449 significantly upregulated or downregulated proteins relative to WT HAP1 proteins. Proteins from the same sample quantified using a 61-min active gradient Orbitrap Ascend data-independent acquisition (DIA) method show strong fold-change correlations to and gene ontology (GO) term agreement with our results.
Building upon the immense progress of the MS and proteomics communities of the past 10 years, here, we report alongside other recent works (21, 22, 37) that the 1-h human proteome is now within reach.
Experimental Procedures
Materials
Human HAP1 WT and KO cells (Horizon Discovery) were cultured in Iscove's modified Dulbecco's medium (Thermo Fisher Scientific, 12440053) with 10% fetal bovine serum (Sigma-Aldrich, F2442) and 1% penicillin–streptomycin (Thermo Fisher Scientific, 15140122) at 37 °C and 5% CO2, as described previously (38). Cells were lysed in a buffer of 6 M guanidine followed by probe sonication. Protein precipitation was performed with 90% methanol (6, 7). The supernatant was decanted, and proteinaceous pellets were suspended in 8 M urea, 100 mM Tris, pH 8.0, and 40 mM chloroacetamide with lysyl endopeptidase (LysC) (1:50 enzyme/protein, Wako Chemicals). After a 4 h room temperature incubation, digested samples were diluted to 1.5 M urea with 100 mM Tris, pH 8.0, and a second digestion was performed with trypsin (1:50 enzyme/protein, Promega) for 12 h. Quenching of trypsin activity was performed by adding trifluoroacetic acid (TFA) to a final pH below 2.0. Peptides were desalted using Strata-X columns (Phenomenex Strata-X 33 μm Polymeric Reverse Phase, 10 mg/ml), which were equilibrated using one column volume of 100% acetonitrile (ACN) followed by one column volume of 0.1% TFA. Acidified peptides were added to the column, rinsed with three column volumes of 0.1% TFA, and eluted with 500 μl of 80% ACN with 0.1% TFA. Protein concentration was determined using a Pierce Quantitative Colorimetric Peptide Assay (Thermo Fisher Scientific). ACN (HPLC grade), water (HPLC grade), and ammonium acetate (LC-MS grade) were obtained from Sigma-Aldrich. LC-MS applications were performed with HAP1 tryptic digest ranging in loading amount from 200 ng to 1 μg.
Liquid Chromatography
LC was performed using a Vanquish Neo ultra high-performance liquid chromatography system, configured either in a direct injection or trap-and-elute format. Peptides were separated on either an Easy-Spray PepMap Neo C18 100 Å, 2 μm, 150 μm × 15 cm ultra high-performance liquid chromatography column (Thermo Fisher Scientific) or a reverse-phase column made in-house. The in-house column was prepared using a 75 μm i.d., 360 μm o.d. bare fused silica capillary and packed with 1.7 μm diameter, 130 Å pore size Bridged Ethylene Hybrid C18 particles (Waters) to a length of 40 cm. A high-pressure packing station, capable of reaching pressures of 30,000 psi was used (39). During LC separations, mobile phase A (MPA) was 0.1% formic acid (FA) in water and MPB was 80% ACN in water with 0.1% FA. For rapid profiling of the proteome, a 7-min active gradient ramped, at a flow rate of 2.5 μl/min, from 4 to 8% MPB from 0 to 0.2 min, 8 to 20% MPB from 0.3 to 4.0 min, 20 to 35% MPB from 4 to 5.8 min, 35 to 99% MPB from 5.8 to 6.2 min, and held at 99% MPB to 6.7 min before the column was washed and reequilibrated at 0% MPB for 1.3 min.
The 15-min nano-LC active gradient ramped, at a flow rate of 300 nl/min, 0 to 8% MPB from 0 to 2 min, 8 to 52% MPB from 2 to 17 min, and 52 to 100% MPB from 17 to 17.1 min, where the gradient was held until 30 min at which time the column was reequilibrated for 11 min. The 30-min nano-LC active gradient ramped, at a flow rate of 300 nl/min, 0 to 8% MPB from 0 to 2 min, 8 to 51% MPB from 2 to 32 min, and 51 to 100% MPB from 32 to 32.1 min, where the gradient was held until 44 min at which time the column was reequilibrated for 11 min. The 60-min active gradient ramped, at a flow rate of 300 nl/min, 0 to 8% MPB from 0 to 2 min, 8 to 52% MPB from 2 to 62 min, and 52 to 100% MPB from 62 to 62.1 min, where the gradient was held until 74 min during which the column was reequilibrated for 11 min.
For KO HAP1 samples, the 30-min nano-LC active gradient ramped at a flow rate of 300 nl/min, 0 to 6% MPB from 0 to 2 min, 6 to 48% MPB from 2 to 32 min, and 48 to 100% MPB from 32 to 32.1 min, where the gradient was held until 44 min during which the column was reequilibrated for 11 min. The 61-min nano-LC active gradient ramped at a flow rate of 300 nl/min, 0 to 5% MPB from 0 to 1 min, 5 to 46% MPB from 1 to 61 min, and 46 to 100% MPB from 61 to 62 min, where the gradient was held until 67 min during which the column was reequilibrated for 18 min.
Mass Spectrometry
DIA MS experiments were either performed on a prototype of Thermo Fisher Scientific Orbitrap-Astral mass spectrometer which is depicted in Figure 1A, or a Thermo Fisher Scientific Orbitrap Ascend Tribrid hybrid mass spectrometer system (Orbitrap Ascend, Thermo Fisher Scientific). For analysis in the OT-Astral, precursor ions were ionized using electrospray ionization at 2 kV with respect to ground. The inlet capillary was held at 280 °C, and the ion funnel radio frequency was held at 40%. During DIA experiments, all MS1 survey scans were acquired at a resolving power of 240,000 in the Orbitrap analyzer with a scan range of m/z 380 to 980, maximum injection time of 50 ms, and automatic gain control (AGC) target of 1,000,000 charges. MS1 spectra were collected every 0.6 s. For OT-Astral DIA MS/MS, a range of isolation widths and mass ranges was tested, and all results reported herein used 2 Th isolation windows without overlap with a precursor mass range of m/z 380 to 980. MS/MS scans were acquired in the Astral analyzer from scan range m/z 150–2000 with high-energy collisional dissociation normalized collision energy of 25%, maximum injection time of 3.5 ms, and AGC target of 50,000 charges.
Fig. 1.

Instrument schematic and explanation of narrow-bin DIA methods.A, Orbitrap Astral instrument schematic. B, MS1 extracted ion chromatograms for two peptides of similar m/z which coelute and are coisolated, even with narrow DIA bins during the 15-min method. The monoisotopic and 13C isotope extracted ion chromatograms are shown for both peptides. C, MS1 spectrum at 16.76 min with inset showing two peptides coisolated within 2 Th gray box. D, MS/MS extracted ion chromatograms of product ions of the two coisolated peptides. E, MS/MS spectrum at 16.75 min with annotation of product ions for two identified peptide sequences. Vertical bars indicate frequency of Orbitrap MS1 and MS/MS scans in B and D, respectively. F–H, the complexity of DIA MS/MS spectra is greatly reduced using narrow quadrupole isolation. Each dot represents a unique MS/MS scan and color corresponds to the number of PSMs in each scan, as identified by Spectronaut; 4 Th and 6 Th bins are theoretical, generated via in silico widening of bins of the 2 Th data processing. DIA, data-independent acquisition; MS, mass spectrometry; MS/MS, tandem mass spectrometry; PSM, peptide-spectrum match.
For analysis in the OT-Ascend, precursor ions were ionized using electrospray ionization at 2 kV with respect to ground. The inlet capillary was held at 275 °C, and the ion funnel radio frequencywas held at 30%. All MS1 survey scans were acquired at a resolving power of 30,000 in the Orbitrap analyzer with a scan range of m/z 380 to 980, maximum injection time of 59 ms, and AGC target of 1,000,000 charges; 10 Th isolation windows overlapping by 1 Th were collected across m/z 380 to 980. Normalized collision energy was set to 28%. MS/MS scans were acquired in the Orbitrap at a resolving power of 15,000 and required an injection time of 27 ms or 10,000 charges.
High-pH Peptide Fractionation and Spectral Library Generation
For spectral library generation, tryptic peptides were fractionated via high-pH reversed-phase liquid chromatography. A Waters XBridge, Peptide Bridged Ethylene Hybrid C18, 3.5 μm, 130 Å, 4.6 mm × 150 mm column was utilized with 10 mM ammonium formate (Sigma-Aldrich, >99/0%, LC-MS grade, 70221-100GF; pH 10) and 20% 10 mM ammonium formate (pH 10)/80% methanol (Optima LC/MS grade, Thermo Fisher Scientific) for MPA and MPB, respectively. Peptides were separated using an Agilent 1260 Infinity Bio-Inert LC at a flow rate of 0.8 ml/min, and fractions were collected using an automated fraction collector. The gradient ramped from 0 to 35% B over 2 min, 35 to 75% B over 6 min, and 75 to 100% B for 5 min, followed by washing at 100% B for 2 min and equilibrating for 5 min at 0% B. Fractions were collected from 5 to 18 min with a total of 16 fractions. Fractions were concatenated by combining 1 and 9, 2 and 10, and so on to yield a total of eight fractions for analysis. Samples were resuspended in 0.1% FA (Thermo Fisher Scientific, LC-MS grade) after drying in a SpeedVac (Thermo Fisher Scientific). Fractions were either analyzed with the OT-Astral over a 60-min active gradient method with 4 Th DIA windows overlapping by 1 Th or in the OT-Ascend over a 61-min active gradient with 4 Th DIA windows overlapping by 1 Th. A spectral library was generated in Spectronaut 18.5.231110.55695 for each method with the human proteome database (full database including Swiss-Prot and TrEMBL downloaded from UniProt on 4/19/2023) consisting of 103,859 protein entries. All settings were set to default library generation settings. Carbamidomethylated cysteine was set as a fixed modification and acetylated N terminus and oxidized methionine were set as variable modifications. Spectronaut first searches with a precursor and fragment mass tolerance of 40 ppm to determine a narrower, dynamic tolerance for a second search used for further analysis. Protein and peptide identifications were filtered to maintain a 1% false discovery rate (FDR). The OT-Astral-derived library consists of 14,036 protein groups from 318,764 peptides, and the OT-Ascend-derived library consists of 11,565 proteins from 210,185 peptides. Note that to create the library used in the analysis of these experiments, we required the prior analysis of a fractionated sample (taking ∼ 8 h). Of course, in the context of a large-scale experiment, this added library generation time is negligible. In cases where only a small number of experiments are required, one can use a library-free approach, for example, directDIA.
Data Analysis
DIA.raw files were searched against either the OT or Astral-derived spectral library using Spectronaut 18.5.231110.55695 default settings. Protein and peptide identifications were filtered to maintain 1% FDR. Mass tolerances for precursors and peptides were dynamically adjusted depending on an initial search set to a 40 ppm tolerance. For our OT-Astral data, the average mass tolerance for extraction from the 60-min gradient data was 5.5 ppm and 4.9 for MS1 and MS2, respectively. These values for the 30-min and 15-min raw data were 7.9 (MS1) and 5.7 (MS2) and 13.2 (MS1) and 6.9 (MS2), respectively. Proteins identified with only one peptide were filtered out of the final dataset. For 7-min experiments, .raw files were searched using directDIA.
Two-sided t tests were performed using the SciPy python package (v. 1.10.0) function “scipy.stats.ttest_ind” using the default parameters. p-Values were corrected for multiple hypotheses testing using the statsmodels package to apply Benjamini-Hochberg FDR filtering (https://www.statsmodels.org/stable/index.html). Linear regression was performed using the SciPy python package (v. 1.10.0) function “scipy.stats.linregress” using default parameters. GO enrichment analysis was performed using the GOATOOLS python package (40). GOs were derived from the 2023-04-01 GO Consortium release (http://purl.obolibrary.org/obo/go/go-basic.obo) (41, 42). The gene association to GO term was made using a database downloaded 2023-05-04 from NCBI.
Results and Discussion
To understand the expression of gene products and to uncover the underlying mechanisms of biological systems and human disease, achieving high coverage and depth for human samples remains an outstanding goal. The Orbitrap Astral offers scans speeds of 200 Hz, single-ion detection sensitivity, and high resolving power (80,000 at m/z 524) which should enable high coverage of the human proteome in short analysis times. To test this hypothesis, we optimized a LC-MS/MS method designed for rapid proteome profiling and a second set of longer methods aimed at probing deep into the human proteome. To examine very fast analysis times, we used a trap-and-elute, microflow LC configuration for fast peptide loading and equilibrations and a 7-min active gradient to separate 200 ng of peptide and consuming 8 min of total LC-MS/MS time.
Our methods for deeper coverage used a direct injection configuration and a 40-cm nanocapillary column to separate 1 μg peptide over a 7, 15, 30, or 60-min gradient, consuming 8, 41, 56, or 85 min of total LC-MS/MS (injection-to-injection) time, respectively. Coupling our separations to the new Orbitrap Astral system enabled the exploration of methods with unprecedented speed and depth. By integrating the Astral analyzer together with a quadrupole-Orbitrap (Q-OT) instrument, tandem MS spectra are acquired at nearly 200 Hz while HRAM MS1 scans are synchronously acquired in the Orbitrap analyzer.
Over each chromatographic method for 1 μg peptide, the Q-OT-Astral hybrid MS in the DIA mode collected 2847 MS1 scans, 288,422 MS/MS scans (15-min), 4319 MS1 scans and 438,062 DIA MS/MS (30-min), and 7264 MS1 scans, 737,451 MS/MS scans (60-min), on average. The optimal DDA method over the same gradient length generated an average of 67.5% of the MS/MS scans collected with the DIA method. Over the 7 min, through a microflow chromatographic method, an average of 787 MS1 scans and 82,117 DIA MS/MS scans were collected. Fast scan speeds and HRAM measurements are crucial for LC-MS experiments comprising of fast gradients. As multiple features elute in quick succession, the instrument must be capable of sampling each ion packet quick enough and with enough purity for quantitative accuracy without sacrificing depth of coverage. To that end, we explored how narrow-window DIA methods, which give spectral purity comparable to DDA schemes, could benefit analyses of complex proteomic mixtures separated by short gradients (Fig. 1, B–E and Supplemental Fig. S1). The increased sensitivity of the Astral analyzer enables injection times of 3 ms, compatible with narrow 2 Th window DIA methods which must raster through hundreds of windows per second. Although the typical 2 Th window in the 15 min method coisolated four or fewer peptides, extending this window in silico to 4 Th and 6 Th increased the number of coisolated precursors to over ten, greatly increasing spectral complexity (Fig. 1, F–H).
Figure 2 presents an example of Q-OT-Astral DIA across a 30 min active LC gradient with high-resolution Orbitrap MS1 scans collected in parallel with narrow-bin, high-resolution Astral MS/MS. At a retention time of 22.02 min, an Orbitrap MS1 was acquired (resolving power of 240,000 at m/z 200), whereas more than 100 DIA MS/MS were synchronously acquired in the Astral mass analyzer. For the precursor m/z range from 650 to 700, 25 DIA scans, each 2 Th in isolation width, were acquired over ∼150 ms resulting in 38 unique peptide identifications attributed to those scans via Spectronaut 18.1 (1% FDR). Within the 7-min method, a similar 25 scan slice from m/z 650 to 700 was performed in ∼142 ms and produced 48 unique peptide identifications (1% FDR) (Supplemental Fig. S2). Hundreds of spectra were manually annotated to provide confidence for both peptide-centric and spectrum-centric identifications. Despite the rapid Q-OT-Astral scan rate, we conclude that the platform regularly provides high-quality peptide spectra without the need for spectral averaging.
Fig. 2.

Example of Q-Orbitrap-Astral data-independent acquisition. Data-independent acquisition with high-resolution Orbitrap MS1 scans collected in parallel with narrow-bin, high-resolution Astral MS/MS scans. At a retention time of 22.02 min, scan #230,122, an Orbitrap full scan, was acquired while hundreds of DIA MS/MS were acquired in the Astral mass analyzer. For the m/z range from 650 to 700, 25 DIA scans were acquired over 150 ms resulting in 38 unique peptide identifications. The b- and y-type fragment ions are denoted in blue and red, respectively. Spectra are shown zoomed relative to the base beak due to limited fragmentation of coisolated, singly charged ions. DIA, data-independent acquisition; MS, mass spectrometry; MS/MS, tandem mass spectrometry.
As researchers continue to focus on benchmarking identification rates across diverse experimental paradigms, it is imperative to underscore the pivotal role of quantifiable peptides as the primary metric for evaluating the efficacy of proteomic methods (43). The ability to consistently sample a high number of reproducible, quantifiable peptides within an experiment serves as a cornerstone for the appraisal of these methodologies.
In triplicate 7-min active microflow LC gradients on Orbitrap Astral system, we report 7852 protein groups from 94,267 peptides on average with good quantitative reproducibility (Supplemental Fig. S3, A–E, Supplemental Data 1). Yielding relative standard deviation (RSD) values below 7%, on average (Supplemental Fig. S3C), three replicate injections of the 7-min gradient also show solid reproducibility of the MS1 total ion current (Supplemental Fig. S2). When using a longer 30-min active gradient, nanoflow method, triplicate experiments yield an average of 10,411 unique protein groups from 234,406 peptides for a median sequence coverage of 42.4% across the proteome (Fig. 3, A–E, Supplemental Data 2). This provided a notable boost in sequence coverage over the 7-min microflow method which covered a median of 27.4% of all amino acids among the 7852 identified protein groups (Supplemental Fig. S3H). The 15-min and 60-min nanoflow methods delivered an average of 195,613 and 254,754 unique peptides from 9831 and 10,645 protein groups for an average sequence coverage of 36.0% and 44.4%, respectively (Fig. 3, A–E, Supplemental Data 3, 6). Cumulative peptides and proteins identified over time reveal the rate of identifications over each gradient (Fig. 3, C and D).
Fig. 3.

Quantitative reproducibility and protein identification metrics for LC-MS gradients. Average (A) unique peptides and (B) protein groups for each experiment. Black bars denote total unique peptides or protein groups across all three replicates. Cumulative (C) peptides and (D) protein groups and (E) protein sequence coverage distributions for each LC-MS/MS method. Duplicate Q-Orbitrap-Astral data-independent acquisition analyses of HAP1 lysate with (F) 15-min, (G) 30-min, and (H) 60-min gradients and (I) comparison of protein label-free quantitation (LFQ) between 15- and 60-min gradients. J, relative standard deviation (RSD) of LC-MS/MS experiments performed in triplicate. K, distribution of LC peak width, (L) MS1 data points per peak, and (M) MS/MS data points per peak at baseline resolution. N, peak resolution relative to m/z of all b- and y-type product ions detected by the Astral analyzer, within 5-ppm of expected m/z, during the 15-min gradient method. LC-MS/MS, liquid chromatography with tandem mass spectrometry.
Replicate experiments of 15-, 30-, and 60-min nanoflow methods yield high quantitative reproducibility, illustrated by comparing log2 (protein label-free quantitation [LFQ]) values (Fig. 3, F–H). Between technical replicates, r2 values of 0.983, 0.990, and 0.992 for the 15-, 30-, and 60-min methods, respectively, are attained. When comparing protein LFQ between 15- and 60-min methods, an r2 value of 0.976 is attained for the 9820 shared protein groups (Fig. 3I). We monitored the RSD of protein LFQ between triplicate experiments and found that increasing active gradient length decreased the RSD appreciably, from a median of 13.6% for the 15-min method to 9.9% for the 30-min method and 8.9% for the 60-min method (Fig. 3J).
Fast LC gradients typically elute peptides with narrow peak widths, limiting the number of times that each precursor or mass range can be sampled. Chromatographic peak widths of 6.0, 8.2, and 9.7 s were observed for the 15-, 30, and 60-min nanoflow gradients, respectively (Fig. 3K). In our DIA scheme, MS1 spectra are acquired every 0.6 s, yielding a median of 10 MS1 data points per peak at baseline for our 15-min method, 14 MS1 data points per peak for our 30-min gradient, and 15 MS1 points per peak using a 60-in method (Fig. 3L). As our 2 Th DIA methods comprise 300 bins, each bin is sampled roughly every 1.67 s, yielding a median of 3, 3, and 5 MS/MS data points per peak for the 15-, 30-, and 60-min methods, respectively (Fig. 3M). One must be wary of relying on so few data points per peak when quantitation is calculated solely from the MS/MS-level. A method with fewer DIA bins (through wider bins or selection from a narrower mass range), shorter MS/MS maximum ion injection times, and/or lower AGC ion target would enable more MS/MS data points per peptide elution peak.
To verify the quality of MS/MS spectra beyond hand annotation of several hundred spectra, we globally profiled the resolution and ppm mass accuracy of all b- and y-type product ions for each experimental condition. The Astral MS/MS spectra produced fragment ion peaks with resolutions at or approaching 100,000 across the m/z distribution in a somewhat intensity-dependent manner (Fig. 3N and Supplemental Fig. S4) (44). For the 15-, 30-, and 60-min nanoflow methods, the majority (82.1%, 83.2%, and 84.0%, respectively) of all fragment ions’ mass error was within 5 ppm of the median mass error per experiment (Supplemental Fig. S5).
Next, we applied the 30-min nanoflow method to reanalyze a CRISPR-Cas9 KO of the mitochondrial gene MGME1 in human HAP1 cells (38). We quantified an average of 10,353 protein groups (Fig. 4A, Supplemental Data 5) from 9946 genes using our fractionated library-based Spectronaut workflow. For differential expression analysis, we removed any proteins that were not present in all three technical KO and WT replicates, rendering 9462 genes after filtering. To provide a benchmark to the previous generation Orbitrap instrumentation, we analyzed the same set of samples using a 61-min active gradient DIA method on an Orbitrap Ascend instrument. From these data, we quantified an average of 8010 (KO) and 7978 (WT) protein groups from 7889 (KO) and 7860 (WT) genes (Fig. 4A, Supplemental Data 6, all using the same Spectronaut workflow, see Experimental procedures for details). When these results are filtered to remove proteins not present in all technical KO and WT replicates, as done above, the dataset contained 7348 genes—of which 99.4% were also present in the Astral results. Figure 4B presents a comparison of relative fold changes between WT and KO of the 7304 overlapping protein measurements as measured by each MS platform. Overall, there is a strong correlation (r = 0.94), as one would expect, between these measurements.
Fig. 4.

Comparison of differential expression analyses upon CRISPR-Cas9 MGME1 KO between 30-min Orbitrap Astral and 61-min Orbitrap Ascend DIA methods.A, unique WT and ΔMGME1 KO HAP1 protein groups identified across methods. B, correlation of log2-transformed fold changes measured by OT-Ascend and that measured by OT-Astral have a Pearson correlation coefficient of 0.939 (n = 7304). C, two-sided t tests for each quantified protein present in three KO and three WT replicates (n = 9462) were performed assuming equal variance. Negative log10-transformed q-values were plotted against log2-transformed fold changes between KO and WT for the OT-Astral-quantified proteins (green and yellow). OT-Astral delivered 449 significantly upregulated or downregulated proteins. Subsequently, 234 significantly changed proteins from the OT-Astral dataset were also determined to be significantly upregulated or downregulated by the OT-Ascend method (yellow). D, subset of proteins that pass the significance threshold within OT-Ascend dataset but not the OT-Astral dataset (143 of 380) have a higher median number of precursors that were used for protein quantitation in the Astral dataset. E-F, GO-term enrichment was performed on proteins quantified in all three KO and WT replicates that were determined to have a fold-change of at least two and a q-value <0.05. There were 40 shared significantly enriched (p-value <0.01) GO-terms (red), and the top 11 enriched in each dataset were corroborated in the comparison dataset. DIA, data-independent acquisition; GO, gene ontology.
Impressively, the Orbitrap Astral instrument detected 449 significantly upregulated or downregulated proteins in comparing the WT to KO samples, following these single-shot, 30-min experiments. By comparison, the Orbitrap Ascend detected 380 significantly regulated proteins following the 61-min single-shot method. 61.6% of these, i.e., 234, overlap with the Orbitrap Astral results (Fig. 4C, yellow dots). To investigate cases where the Orbitrap Ascend and Astral instrument generated results that disagreed on whether a protein had significantly changed in KO versus WT, we plotted the number of precursors utilized in each protein measurement (Fig. 4D). From these data we observe that the Orbitrap Astral generates, on average, over two times the number of precursor measurements for each quantified protein. We suggest that this is one key factor that likely leads to the Orbitrap Astral system generating more statistically significant measurements. Finally, we examined the GO enrichment obtained from both of these datasets (Fig. 4, E and F), overlapping terms shown in red). From these results, we observe the top 11 terms for each dataset are shared in both, and beyond that, there is a high degree of overlap.
The Confluence of Proteomic Depth and Throughput
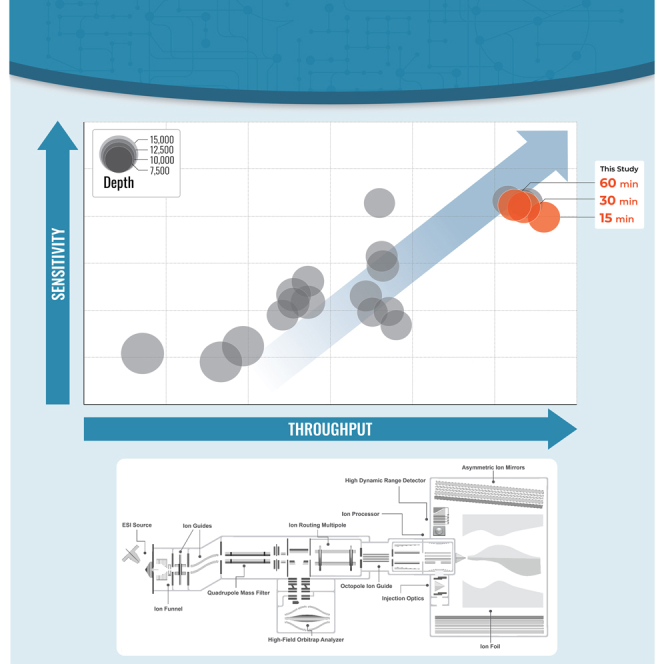
Our understanding of the human proteome is reaching new heights, and we report here, alongside other recent reports, that the analysis of human proteomes at of level of ∼10,000 proteins can now be achieved in less than 1 h. Figure 5 illustrates the leap in throughput and sensitivity that studies adopting the Orbitrap Astral system have reached relative to the previous studies that have aimed to deeply profile the human proteome (9, 10, 12, 13, 14, 15, 16, 17, 18, 19, 20, 28, 30, 31, 32, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55). We report here more than 10,000 protein groups quantified using a 30-min active gradient, in total requiring less than 1 h between injections. This milestone achieved by the Orbitrap Astral system heralds the advent of a new era of high-throughput human proteomics, one that aims to ultimately achieve detection of the entire proteome within a single experiment. Using this system, Stewart et al. recently reported over 10,000 protein groups using only 48 min of active MS analysis time (21), Guzman et al. reported an average of 9619 protein groups from 1 μg of HEK293 cell lysate with coefficient of variation < 3% in 28 min active gradients (22), and Heil et al. reported comparable protein metrics using short gradients with exceptional quantitative analysis (37), all performed on an Orbitrap Astral system and highlighted in green (Fig. 5 and Supplemental Fig. S6).
Fig. 5.

Throughput and depth are shown for several LC-MS/MS-based human proteomics investigations spanning the past decade. The number of unique human proteins identified are plotted against the total LC-MS/MS acquisition time. Squares denote fractionated experiments, and circles denote single-shot experiments. Note, these data report protein groups identified, not necessarily quantified. LC-MS/MS, liquid chromatography with tandem mass spectrometry.
A balance exists at the confluence of proteomic depth and throughput where researchers must make decisions regarding instrument demand at project inception. Nevertheless, in the past, researchers have needed to devote tens to hundreds of hours of LC-MS/MS analysis time of deep offline separated peptide fractions to eclipse 10,000 protein groups identified, represented by squares (Fig. 5). Only with the introduction of highly sensitive instrumentation, in addition to fast scan rates and high resolving power mass analyzers, has it been possible to measure this number of proteins in less than 1 h. As well, studies of single-cell proteomes (56, 57) and neat and depleted plasma proteomes (58, 59) are reaching impressive depth enabled by huge strides in sample preparation, especially nanoparticle bead approaches (59, 60, 61, 62, 63), paired with faster, more sensitive instrumentation. For many years, the dynamic range of human plasma stymied researchers to profile samples to only a few hundred proteins; studies now routinely report several thousand unique, quantified plasma proteins (58, 59, 60).
Conclusion
The human proteome represents a massive hurdle of biological complexity and researchers have long been stymied by its spatial and temporal fluctuations, the many protein isoforms and PTMs which can arise, and the extensive dynamic range of human protein abundances (5). These processes doubtless generate a tremendously complex proteome with each gene product likely existing in dozens or hundreds of variations—i.e., proteoforms. Still, the progress of the shotgun proteomics field has been rapid, spurred on by the development of faster, more sensitive instrumentation; cheaper, more efficient, and more facile sample preparation methods; new liquid chromatography separations technologies; and breakthroughs in data processing which dramatically reduce the time spent for analyzing MS data outputs.
Building on these efforts, we report that progress toward deeper and higher throughput proteomic efforts has led to comprehensive analysis of the majority of the expressed human proteome in less than 1 h of instrument time (Supplemental Fig. S7) (64, 65). We use narrow, 2 Th DIA bins collected in the Astral mass analyzer in parallel with high-resolution Orbitrap full scans. In 30-min active gradients, we report 10,411 human protein groups quantified from 234,406 peptides, on average. With a 60-min gradient, we detect 10,645 protein groups from 254,754 unique peptides, constituting a majority (57.9%) of the 18,397 human proteins for which protein expression has been credibly detected (66). Using a rapid trap-and-elute chromatographic platform, we report 7852 protein groups from 94,267 peptides with a 7-min gradient. Studies of single-cell proteomes, plasma proteomes, and posttranslational modifications also benefit from this high-resolution, fast scanning, and sensitive platform (37, 56, 59, 67). The Orbitrap Astral MS system, alongside other state-of-the-art instrumentation and paired with the many incredible advances in sample preparation, method development, and data processing, is ushering in a new era of proteomics where a harmony of throughput, depth, and sensitivity are enabling unprecedented studies of biological impact.
Data Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [1] partner repository with the dataset identifier PXD049028.
Supplemental data
This article contains supplemental data.
Conflict of interest
The authors declare the following competing financial interest(s): J. J. C. is a consultant for Thermo Fisher Scientific, Seer, and 908 Devices. T. N. A., E. D., A. P., M. Z., D. H., H. S., C. H., A. M., and V. Z. are employees of Thermo Fisher Scientific.
Acknowledgments
The authors thank other members of the Coon Lab and the many employees of Thermo Fisher Scientific without whom this work would not be possible.
Funding and additional information
This work was supported in part by a sponsored research agreement with Thermo Fisher Scientific (J. J. C.), the National Institute of General Medical Sciences of the National Institutes of Health (grants P41GM108538 and R35GM118110 to J. J. C.), and the National Human genome Research Institution through a training grant to the Genomic Science Training Program (grant T32HG002760 to L. R. S.). T. M. P.-C. acknowledges the ACS Division of Analytical Chemistry and Agilent for support through a graduate fellowship. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions
L. R. S., T. M. P.-C., T. N. A., E. D., E. S., A. P., P. S., D. R. B., S. T. Q., A. C. P., M. Z., D. H., H. S., C. H., A. M., V. Z., and J. J. C. validation; L. R. S., T. M. P.-C., T. N. A., E. D., E. S., A. P., P. S., A. C. P., M. Z., H. S., and J. J. C. methodology; L. R. S., T. M. P.-C, and P. S. formal analysis; L. R. S. and T. M. P.-C. data curation; L. R. S. and T. M. P.-C. software; L. R. S. and T. M. P.-C. visualization; L. R. S., T. M. P.-C., and J. J. C. writing–original draft; L. R. S., T. M. P.-C., and J. J. C. writing–review and editing; L. R. S., T. M. P.-C., M. L. R., N. M. L., and C. M. investigation; T. M. P.-C. and J. J. C. conceptualization; J. J. C. funding acquisition; J. J. C. supervision.
Supplemental Data
References
- 1.Liu Y., Beyer A., Aebersold R. On the dependency of cellular protein levels on mRNA abundance. Cell. 2016;165:535–550. doi: 10.1016/j.cell.2016.03.014. [DOI] [PubMed] [Google Scholar]
- 2.Aebersold R., Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 3.Han X., Aslanian A., Yates J.R. Mass spectrometry for proteomics. Curr. Opin. Chem. Biol. 2008;12:483–490. doi: 10.1016/j.cbpa.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yates J.R., Ruse C.I., Nakorchevsky A. Proteomics by mass spectrometry: approaches, advances, and applications. Annu. Rev. Biomed. Eng. 2009;11:49–79. doi: 10.1146/annurev-bioeng-061008-124934. [DOI] [PubMed] [Google Scholar]
- 5.Omenn G.S., Lane L., Overall C.M., Pineau C., Packer N.H., Cristea I.M., et al. The 2022 report on the human proteome from the HUPO human proteome project. J. Proteome Res. 2022;22:1024–1042. doi: 10.1021/acs.jproteome.2c00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hebert A.S., Richards A.L., Bailey D.J., Ulbrich A., Coughlin E.E., Westphall M.S., et al. The one hour yeast proteome. Mol. Cell. Proteomics. 2014;13:339–347. doi: 10.1074/mcp.M113.034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richards A.L., Hebert A.S., Ulbrich A., Bailey D.J., Coughlin E.E., Westphall M.S., et al. One-hour proteome analysis in yeast. Nat. Protoc. 2015;10:701–714. doi: 10.1038/nprot.2015.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peters-Clarke T.M., Coon J.J., Riley N.M. Instrumentation at the leading edge of proteomics. chemRxiv. 2023 doi: 10.26434/chemrxiv-2023-8l72m. [preprint] [DOI] [PubMed] [Google Scholar]
- 9.Helm D., Vissers J.P.C., Hughes C.J., Hahne H., Ruprecht B., Pachl F., et al. Ion mobility tandem mass spectrometry enhances performance of bottom-up proteomics. Mol. Cell. Proteomics. 2014;13:3709–3715. doi: 10.1074/mcp.M114.041038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pozniak Y., Balint-Lahat N., Rudolph J.D., Lindskog C., Katzir R., Avivi C., et al. System-wide clinical proteomics of breast cancer reveals global remodeling of tissue homeostasis. Cell Syst. 2016;2:172–184. doi: 10.1016/j.cels.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Beck M., Schmidt A., Malmstroem J., Claassen M., Ori A., Szymborska A., et al. The quantitative proteome of a human cell line. Mol. Syst. Biol. 2011;7:549. doi: 10.1038/msb.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagaraj N., Wisniewski J.R., Geiger T., Cox J., Kircher M., Kelso J., et al. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 2011;7:548. doi: 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim M.S., Pinto S.M., Getnet D., Nirujogi R.S., Manda S.S., Chaerkady R., et al. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilhelm M., Schlegl J., Hahne H., Gholami A.M., Lieberenz M., Savitski M.M., et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509:582–587. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- 15.Bekker-Jensen D.B., Kelstrup C.D., Batth T.S., Larsen S.C., Haldrup C., Bramsen J.B., et al. An optimized shotgun Strategy for the rapid generation of comprehensive human proteomes. Cell Syst. 2017;4:587–599.e4. doi: 10.1016/j.cels.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelstrup C.D., Bekker-Jensen D.B., Arrey T.N., Hogrebe A., Harder A., Olsen J.V. Performance evaluation of the Q Exactive HF-X for shotgun proteomics. J. Proteome Res. 2018;17:727–738. doi: 10.1021/acs.jproteome.7b00602. [DOI] [PubMed] [Google Scholar]
- 17.Bache N., Geyer P.E., Bekker-Jensen D.B., Hoerning O., Falkenby L., Treit P.V., et al. A novel LC system Embeds Analytes in Pre-formed gradients for rapid, Ultra-robust proteomics. Mol. Cell. Proteomics. 2018;17:2284–2296. doi: 10.1074/mcp.TIR118.000853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bian Y., Zheng R., Bayer F.P., Wong C., Chang Y.C., Meng C., et al. Robust, reproducible and quantitative analysis of thousands of proteomes by micro-flow LC–MS/MS. Nat. Commun. 2020;11:1–12. doi: 10.1038/s41467-019-13973-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Müller J.B., Geyer P.E., Colaço A.R., Treit P.V., Strauss M.T., Oroshi M., et al. The proteome landscape of the kingdoms of life. Nature. 2020;582:592–596. doi: 10.1038/s41586-020-2402-x. [DOI] [PubMed] [Google Scholar]
- 20.Sinitcyn P., Richards A.L., Weatheritt R.J., Brademan D.R., Marx H., Shishkova E., et al. Global detection of human variants and isoforms by deep proteome sequencing. Nat. Biotechnol. 2023;41:1776–1786. doi: 10.1038/s41587-023-01714-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stewart H.I., Grinfeld D., Giannakopulos A., Petzoldt J., Shanley T., Garland M., et al. Parallelized acquisition of orbitrap and astral analyzers enables high-throughput quantitative analysis. Anal. Chem. 2023;95:15656–15664. doi: 10.1021/acs.analchem.3c02856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guzman U.H., Martinez-Val A., Ye Z., Damoc E., Arrey T.N., Pashkova A., et al. Ultra-fast label-free quantification and comprehensive proteome coverage with narrow-window data-independent acquisition. Nat. Biotechnol. 2024 doi: 10.1038/s41587-023-02099-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chernushevich I.V. Duty cycle improvement for a quadrupole—time-of-flight mass spectrometer and its use for precursor ion scans. Eur. J. Mass Spectrom. 2000;6 doi: 10.1255/ejms.377. [DOI] [Google Scholar]
- 24.Hardman M., Makarov A.A. Interfacing the orbitrap mass analyzer to an electrospray ion source. Anal. Chem. 2003;75:1699–1705. doi: 10.1021/ac0258047. [DOI] [PubMed] [Google Scholar]
- 25.Baba T., Ryumin P., Duchoslav E., Chen K., Chelur A., Loyd B., et al. Dissociation of Biomolecules by an Intense low-energy Electron Beam in a high sensitivity time-of-flight mass spectrometer. J. Am. Soc. Mass Spectrom. 2021;32:1964–1975. doi: 10.1021/jasms.0c00425. [DOI] [PubMed] [Google Scholar]
- 26.Baba T., Campbell J.L., Le Blanc J.C.Y., Hager J.W., Thomson B.A. Electron capture dissociation in a branched radio-frequency ion trap. Anal. Chem. 2015;87:785–792. doi: 10.1021/ac503773y. [DOI] [PubMed] [Google Scholar]
- 27.Meier F., Beck S., Grassl N., Lubeck M., Park M.A., Raether O., et al. Parallel accumulation-serial fragmentation (PASEF): multiplying sequencing speed and sensitivity by synchronized scans in a trapped ion mobility device. J. Proteome Res. 2015;14:5378–5387. doi: 10.1021/acs.jproteome.5b00932. [DOI] [PubMed] [Google Scholar]
- 28.Meier F., Brunner A.D., Frank M., Ha A., Bludau I., Voytik E., et al. diaPASEF: parallel accumulation–serial fragmentation combined with data-independent acquisition. Nat. Methods. 2020;17:1229–1236. doi: 10.1038/s41592-020-00998-0. [DOI] [PubMed] [Google Scholar]
- 29.Skowronek P., Thielert M., Voytik E., Tanzer M.C., Hansen F.M., Willems S., et al. Rapid and in-depth coverage of the (phospho-)proteome with deep libraries and optimal window design for dia-PASEF. Mol. Cell. Proteomics. 2022;21:100279. doi: 10.1016/j.mcpro.2022.100279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Messner C.B., Demichev V., Bloomfield N., Yu J.S.L., White M., Kreidl M., et al. Ultra-fast proteomics with scanning SWATH. Nat. Biotechnol. 2021;39:846–854. doi: 10.1038/s41587-021-00860-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishikawa M., Konno R., Nakajima D., Gotoh M., Fukasawa K., Sato H., et al. Optimization of Ultrafast proteomics using an LC-quadrupole-orbitrap mass spectrometer with data-independent acquisition. J. Proteome Res. 2022;21:2085–2093. doi: 10.1021/acs.jproteome.2c00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Messner C.B., Demichev V., Wendisch D., Michalick L., White M., Freiwald A., et al. Ultra-high-throughput clinical proteomics reveals classifiers of COVID-19 Infection. Cell Syst. 2020;11:11–24.e4. doi: 10.1016/j.cels.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Demichev V., Messner C.B., Vernardis S.I., Lilley K.S., Ralser M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods. 2019;17:41–44. doi: 10.1038/s41592-019-0638-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stewart H., Grinfeld D., Wagner A., Kholomeev A., Biel M., Giannakopulos A., et al. A conjoined rectilinear collision cell and pulsed extraction ion trap with Auxiliary DC electrodes. J. Am. Soc. Mass Spectrom. 2023;35:74–81. doi: 10.1021/jasms.3c00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grinfeld D., Stewart H., Balschun W., Skoblin M., Hock C., Makarov A. Multi-reflection Astral mass spectrometer with isochronous drift in elongated ion mirrors. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrom. Detect. Assoc. Equip. 2024;1060:169017. [Google Scholar]
- 36.Stewart H., Grinfeld D., Hagedorn B., Ostermann R., Makarov A., Hock C. Proof of principle for enhanced resolution multi-pass methods for the astral analyzer. Int. J. Mass Spectrom. 2024;498 doi: 10.1002/jms.5006. [DOI] [PubMed] [Google Scholar]
- 37.Heil L.R., Damoc E., Arrey T.N., Pashkova A., Denisov E., Petzoldt J., et al. Evaluating the performance of the astral mass analyzer for quantitative proteomics using data-independent acquisition. J. Proteome Res. 2023;22:3290–3300. doi: 10.1021/acs.jproteome.3c00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rensvold J.W., Shishkova E., Sverchkov Y., Miller I.J., Cetinkaya A., Pyle A., et al. Defining mitochondrial protein functions through deep multiomic profiling. Nature. 2022;606:382–388. doi: 10.1038/s41586-022-04765-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shishkova E., Hebert A.S., Westphall M.S., Coon J.J. Ultra-high pressure (>30,000 psi) packing of capillary columns enhancing depth of shotgun proteomic analyses. Anal. Chem. 2018;90:11503–11508. doi: 10.1021/acs.analchem.8b02766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klopfenstein D.V., Zhang L., Pedersen B.S., Ramírez F., Warwick Vesztrocy A., Naldi A., et al. GOATOOLS: a python library for gene ontology analyses. Sci. Rep. 2018;8 doi: 10.1038/s41598-018-28948-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., et al. Gene ontology: tool for the unification of biology the gene ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Consortium T.G.O., Aleksander S.A., Balhoff J., Carbon S., Cherry J.M., Drabkin H.J., et al. The gene ontology knowledgebase in 2023. Genetics. 2023;224 doi: 10.1093/genetics/iyad031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pino L.K., Searle B.C., Bollinger J.G., Nunn B., MacLean B., MacCoss M.J. The skyline ecosystem: informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 2020;39:229–244. doi: 10.1002/mas.21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stewart H., Grinfeld D., Petzoldt J., Hagedorn B., Skoblin M., Makarov A., et al. Crowd control of ions in the astral analyzer. chemRxiv. 2023 doi: 10.26434/chemrxiv-2023-p6zln. [preprint] [DOI] [PubMed] [Google Scholar]
- 45.Cristobal A., Hennrich M.L., Giansanti P., Goerdayal S.S., Heck A.J.R., Mohammed S. In-house construction of a UHPLC system enabling the identification of over 4000 protein groups in a single analysis. Analyst. 2012;137:3541–3548. doi: 10.1039/c2an35445d. [DOI] [PubMed] [Google Scholar]
- 46.Geiger T., Wehner A., Schaab C., Cox J., Mann M. Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol. Cell. Proteomics. 2012;11:1–11. doi: 10.1074/mcp.M111.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pirmoradian M., Budamgunta H., Chingin K., Zhang B., Astorga-Wells J., Zubarev R.A. Rapid and deep human proteome analysis by single-dimension shotgun proteomics. Mol. Cell. Proteomics. 2013;12:3330–3338. doi: 10.1074/mcp.O113.028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scheltema R.A., Hauschild J.P., Lange O., Hornburg D., Denisov E., Damoc E., et al. The Q Exactive HF, a Benchtop mass spectrometer with a Pre-filter, high-performance quadrupole and an Ultra-high-field orbitrap analyzer. Mol. Cell. Proteomics. 2014;13:3698–3708. doi: 10.1074/mcp.M114.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thakur S.S., Geiger T., Chatterjee B., Bandilla P., Froḧlich F., Cox J., et al. Deep and highly sensitive proteome coverage by LC-MS/MS without prefractionation. Mol. Cell. Proteomics. 2011;10:1–9. doi: 10.1074/mcp.M110.003699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sacco F., Silvestri A., Posca D., Pirrò S., Gherardini P.F., Castagnoli L., et al. Deep proteomics of breast cancer cells reveals that metformin rewires signaling networks away from a pro-growth state. Cell Syst. 2016;2:159–171. doi: 10.1016/j.cels.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 51.Ivanov M.V., Bubis J.A., Gorshkov V., Abdrakhimov D.A., Kjeldsen F., Gorshkov M.V. Boosting MS1-only proteomics with machine Learning Allows 2000 protein identifications in single-shot human proteome analysis using 5 min HPLC gradient. J. Proteome Res. 2021;20:1864–1873. doi: 10.1021/acs.jproteome.0c00863. [DOI] [PubMed] [Google Scholar]
- 52.Ivanov M.V., Bubis J.A., Gorshkov V., Tarasova I.A., Levitsky L.I., Solovyeva E.M., et al. DirectMS1Quant: ultrafast quantitative proteomics with MS/MS-Free mass spectrometry. Anal. Chem. 2022;94:13068–13075. doi: 10.1021/acs.analchem.2c02255. [DOI] [PubMed] [Google Scholar]
- 53.Derks J., Leduc A., Wallmann G., Huffman R.G., Willetts M., Khan S., et al. Increasing the throughput of sensitive proteomics by plexDIA. Nat. Biotechnol. 2023;41:50–59. doi: 10.1038/s41587-022-01389-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li J., Van Vranken J.G., Pontano Vaites L., Schweppe D.K., Huttlin E.L., Etienne C., et al. TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat. Methods. 2020;17:399–404. doi: 10.1038/s41592-020-0781-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li J., Cai Z., Bomgarden R.D., Pike I., Kuhn K., Rogers J.C., et al. TMTpro-18plex: the expanded and complete set of TMTpro reagents for sample Multiplexing. J. Proteome Res. 2021;20:2964–2972. doi: 10.1021/acs.jproteome.1c00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petrosius V., Aragon-fernandez P., Arrey T.N., Üresin N., Furtwängler B., Stewart H., et al. Evaluating the capabilities of the Astral mass analyzer for single- cell proteomics. bioRxiv. 2023 doi: 10.1101/2023.06.06.543943. [preprint] [DOI] [Google Scholar]
- 57.Ye Z., Sabatier P., Martin-Gonzalez J., Eguchi A., Bekker-Jensen D.B., Bache N., et al. One-Tip enables comprehensive proteome coverage in minimal cells and single zygotes. bioRxiv. 2023 doi: 10.1101/2023.08.10.552756. [preprint] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu C.C., Tsantilas K.J., Park J., Plubell D.L., Naicker P., Govender I., et al. Mag-Net: rapid enrichment of membrane-bound particles enables high coverage quantitative analysis of the plasma proteome. bioRxiv. 2023 doi: 10.1101/2023.06.10.544439. [preprint] [DOI] [Google Scholar]
- 59.Huang T., Wang J., Stukalov A., Donovan M.K.R., Ferdosi S., Williamson L., et al. Protein Coronas on Functionalized nanoparticles enable quantitative and precise large-scale deep plasma proteomics. bioRxiv. 2023 doi: 10.1101/2023.08.28.555225. [preprint] [DOI] [Google Scholar]
- 60.Blume J.E., Manning W.C., Troiano G., Hornburg D., Figa M., Hesterberg L., et al. Rapid, deep and precise profiling of the plasma proteome with multi-nanoparticle protein corona. Nat. Commun. 2020;11:3662. doi: 10.1038/s41467-020-17033-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ferdosi S., Stukalov A., Hasan M., Tangeysh B., Brown T.R., Wang T., et al. Enhanced Competition at the nano–Bio Interface enables comprehensive characterization of protein corona dynamics and deep coverage of proteomes. Adv. Mater. 2022;34 doi: 10.1002/adma.202206008. [DOI] [PubMed] [Google Scholar]
- 62.Donovan M.K.R., Huang Y., Blume J.E., Wang J., Hornburg D., Ferdosi S., et al. Functionally distinct BMP1 isoforms show an opposite pattern of abundance in plasma from non-small cell lung cancer subjects and controls. PLoS One. 2023;18 doi: 10.1371/journal.pone.0282821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Suhre K., Venkataraman G.R., Guturu H., Halama A., Stephan N., Thareja G., et al. Nanoparticle enrichment mass-spectrometry proteomics Identifies protein Altering variants for precise pQTL mapping. bioRxiv. 2023 doi: 10.1101/2023.04.20.537640. [preprint] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ghandi M., Huang F.W., Jané-Valbuena J., Kryukov G.V., Lo C.C., McDonald E.R., et al. Next-generation characterization of the cancer cell line Encyclopedia. Nature. 2019;569:503–508. doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Omenn G.S., Lane L., Overall C.M., Lindskog C., Pineau C., Packer N.H., et al. The 2023 report on the proteome from the HUPO human proteome project. J. Proteome Res. 2024;23:532–549. doi: 10.1021/acs.jproteome.3c00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lancaster N.M., Sinitcyn P., Forny P., Peters-Clarke T.M., Fecher C., Smith A.J., et al. Fast and deep Phosphoproteome analysis with the orbitrap astral mass spectrometer. bioRxiv. 2023 doi: 10.1101/2023.11.21.568149. [preprint] [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [1] partner repository with the dataset identifier PXD049028.