


Abstract
An enzyme activity introducing an alkali-labile site at 2-hydroxyadenine (2-OH-A) in double-stranded oligonucleotides was detected in nuclear extracts of Jurkat cells. This activity co-eluted with activities toward adenine paired with guanine and 8-oxo-7,8-dihydroguanine (8-oxoG) as a single peak corresponding to a 55 kDa molecular mass on gel filtration chromatography. Further co-purification was then done. Western blotting revealed that these activities also co-purified with a 52 kDa polypeptide which reacted with antibodies against human MYH (anti-hMYH). Recombinant hMYH has essentially similar activities to the partially purified enzyme. Thus, hMYH is likely to possess both adenine and 2-OH-A DNA glycosylase activities. In nuclear extracts from Jurkat cells, a 52 kDa polypeptide was detected with a small amount of 53 kDa polypeptide, while in mitochondrial extracts a 57 kDa polypeptide was detected using anti-hMYH. With amplification of the 5′-regions of the hMYH cDNA, 10 forms of hMYH transcripts were identified and subgrouped into three types, each with a unique 5′ sequence. These hMYH transcripts are likely to encode multiple authentic hMYH polypeptides including the 52, 53 and 57 kDa polypeptides detected in Jurkat cells.
INTRODUCTION
Among many classes of DNA damage caused by oxygen radicals, an oxidized form of the guanine base, 8-oxo-7,8-dihydroguanine (8-oxoG), is considered to be pertinent to mutagenesis and carcinogenesis (1–3), because 8-oxoG can pair with adenine as well as cytosine with almost equal efficiency during DNA replication and thus has the potential to cause a high frequency of mutation (4,5). Accumulation of 8-oxoG in DNA is due to either incorporation of 8-oxo-dGTP, an oxidized form of dGTP, into DNA as a precursor or direct oxidation of the guanine base in DNA (6).
Studies on mutator mutants of Escherichia coli revealed that E.coli has several error avoiding mechanisms to minimize the deleterious effects of 8-oxoG, in which MutT, FPG (MutM) and MutY play important roles (7,8). MutT hydrolyzes 8-oxo-dGTP to 8-oxo-dGMP and pyrophosphate (5), thus avoiding the spontaneous occurrence of A:T→C:G transversion mutations. FPG (MutM), originally identified as a formamidopyrimidine DNA glycosylase, removes 8-oxoG paired with cytosine and introduces a single strand gap as a result of the accompanying apurinic/apyrimidinic (AP) lyase activity (9,10). MutY, with its DNA glycosylase activity, excises adenine paired with guanine or 8-oxoG (11,12). The rate of spontaneous occurrence of G:C→T:A transversion mutations in fpg(mutM)- or mutY-deficient strains is 10–50 times higher than that in the wild-type strain (13,14).
It has been shown that mammalian cells also come equipped with similar error avoiding mechanisms. MTH1 (MutT homolog-1) and MYH (MutY homolog) are likely to play roles as mammalian orthologs of E.coli MutT and MutY, respectively (8,15–20). An ortholog for yeast 8-oxoG DNA glycosylase, Ogg1, a functional homolog of E.coli FPG (MutM), has also been identified in humans and mice (21–27). We found that hMTH1 is mostly localized in the cytoplasm with some in the mitochondria of human cells (28) and that the hOGG1 gene produces more than seven transcripts by alternative splicing, one of which encodes a nuclear form of 8-oxoG DNA glycosylase while another encodes its mitochondrial form (29). Therefore, oxidation of nucleotides as well as DNA in mitochondria is indeed a crucial event.
hMTH1 hydrolyzes oxidized purine nucleoside triphosphates such as 8-oxo-dGTP, 8-oxo-GTP, 8-oxo-7,8-dihydro-dATP (8-oxo-dATP) and 2-hydroxy-dATP (2-OH-dATP), while MutT hydrolyzes only 8-oxo-dGTP and 8-oxo-GTP (30,31). It has been shown that 2-hydroxyadenine (2-OH-A) in plasmid DNA causes various base substitutions and deletion mutations in E.coli and in mammalian cells (32,33) and that introduction of 2-OH-dATP into E.coli cells induces mostly G:C→T:A transversion mutations (34). The mutagenic potential of 8-oxoadenine (8-oxoA) is limited (35,36). The existence of hMYH and hOGG1 suggests that cells may come equipped with an enzyme with 2-OH-A-specific DNA glycosylase activity to excise 2-OH-A incorporated into DNA.
In the present study, we detected and purified a repair enzyme for 2-OH-A in DNA from human cells. The enzyme was co-purified with authentic hMYH and recombinant hMYH has the same repair activity, as well as adenine DNA glycosylase activity. In human cells at least three isoforms (p52, p53 and p57) of hMYH were identified. p52 and p53 are localized in the nucleus and p57 locates in the mitochondria. Ten different forms of cDNA for hMYH were isolated, hence multiple forms of hMYH are present in human cells.
MATERIALS AND METHODS
Chemicals
[α-32P]dCTP and prepacked columns for FPLC were purchased from Amersham Pharmacia Biotech. Recombinant Taq DNA polymerase, restriction enzymes, T4 DNA polymerase and T4 DNA ligase were obtained from Takara (Japan) and Toyobo (Japan). DNA labeling kits were obtained from Nippon Gene (Japan). The 1 kb DNA and 0.24–9.5 kb RNA ladders as size standards, 10× RPMI medium, DMEM and formamide were purchased from Life Technologies. Other chemicals were obtained from Wako (Japan). Sources of other materials are shown at appropriate places in the text.
Oligonucleotides
A phosphoramidite derivative of 2-OH-A (isodG-CE phosphoramidite) was obtained from Glen Research and 2-OH-A (AO)-containing oligonucleotides were synthesized using a model 392 DNA synthesizer (PE Applied Biosystems) and purified by reverse phase HPLC. Other oligonucleotides, shown in Table 1, were obtained from Greiner Japan and Takara.
Table 1. Oligomers used in this study.

GO, 8-oxoG; AO, 2-OH-A; FAM, N-(3-fluorfluoranthyl)maleimide.
Isolation of hMYH cDNAs and reverse transcription (RT)–PCR analysis
hMYH cDNAs were amplified by RT–PCR as described (29). Two sets of primers (5-1 and 3-3 and 5-3 and 3-1) were used to amplify the hMYHα3 cDNA as two separate fragments, hMYH-N and hMYH-C, and each fragment was cloned into vector pT7Blue(R)T (Novagen), to construct pT7Blue:hMYH-N and pT7Blue:hMYH-C, respectively. A HindIII–AgeI fragment from pT7Blue:hMYH-N was inserted into the HindIII and AgeI sites of pT7Blue:hMYH-C, to obtain pT7Blue:hMYHα3.
Construction of plasmids
To construct plasmid pYN3103:TrpE-HhH-FeS, encoding the fusion protein TrpE-HhH-FeS, a cDNA fragment encoding amino acid residues 148–310 of hMYHα3, amplified from pT7Blue:hMYHα3 using primers 5′-HhH and 3′-FeS, was inserted into the SalI and BamHI sites of pYN3103:TrpE (37). Then, a BstXI–BamHI fragment prepared from the plasmid was transferred into the BstXI and BamHI sites of pET3d-TrpE (37) to construct pET3d:TrpE-HhH-FeS. The hMYH expression plasmid pXC35:hMYHα3-2 was constructed as follows. The 14-3-3 insert in plasmid pXC35:14-3-3 (38) was replaced with a fragment derived from hMYHα3 cDNA containing the open reading frame of hMYHα3 starting at the second ATG. pT7Blue:hMYHα3-2 carrying the same fragment was also constructed.
Northern blot analysis
Northern blot analysis was done as described (29,39). RNAs prepared from various human tissues were purchased from Clontech. A fragment of hMYH cDNA was prepared by PCR using a set of oligonucleotide primers (5-3 and 3-1) from plasmid pT7Blue:hMYH-C and was labeled with [α-32P]dCTP, using a DNA labeling kit. This was then used as a hybridization probe. To confirm the amounts of RNA loaded, the blot was reprobed with a 1.0 kb EcoRI–BamHI fragment of the 18S rRNA gene prepared from pRγcHE (40), obtained from the Japanese Cancer Research Resources Bank.
Partial purification of the nuclear form of hMYH
Nuclear extracts were prepared from Jurkat cells (2 × 1010 cells) according to a method described previously (29,41). Nuclear extracts (56 ml) were applied to an activated P11 cellulose phosphate column (bed volume 35 ml) (Whatman) equilibrated with buffer C (29) containing 50 mM KCl, then the column was eluted with a 480 ml linear gradient of KCl (50–750 mM). Fractions containing the 2-OH-A/adenine repair activities, which eluted at 400–510 mM KCl, were pooled (75 ml). The pooled fraction was concentrated with Ficoll to 5 ml, then applied to a HiLoad 26/60 Superdex 75 pg gel filtration column (bed volume 320 ml) equilibrated with buffer C containing 200 mM KCl. Active fractions were pooled (15 ml) and diluted by adding an equal volume of buffer C containing 50 mM KCl. The fraction was applied to a RESOURCE Q column (bed volume 1 ml) equilibrated with buffer C containing 125 mM KCl. The flow-through fraction was collected (25 ml) and applied to a RESOURCE S column (bed volume 1 ml) equilibrated with the same buffer. The column was eluted with a 6 ml linear gradient of KCl (125–500 mM) and 200 µl of each fraction was collected.
Nicking assay
Double-stranded oligonucleotide substrates for the nicking assay were prepared and the standard nicking assay was performed as described (29). One unit of nicking activity was defined as the activity which cleaved 1 fmol substrate DNA in 1 h at 37°C.
Preparation of antibodies against hMYH
Rabbit polyclonal antibodies were raised against the fusion protein TrpE-HhH-FeS, in which the helix–hairpin–helix (HhH) and iron–sulfur cluster (FeS) of hMYH (amino acids 148–310) were fused to E.coli TrpE, as described (37). Antibodies were purified with the aid of TrpE-HhH-FeS–Sepharose and TrpE–Sepharose columns (37,42). The affinity-purified antibodies were designated anti-hMYH. Preadsorption of anti-hMYH was done as described previously (29,37).
Preparation of extracts from E.coli cells expressing recombinant hMYH
Plasmids pXC35:hMYHα3-2 and pXC35:14-3-3 were introduced into E.coli strain TAP56 (38) and were grown in 10 ml of LB broth containing 100 µg/ml ampicillin and 0.2 mM ammonium iron(II) sulfate hexahydrate and 0.2 mM ammonium sulfide to an OD600 of 0.3–0.4 at 30°C, then the cultures were shifted to 52°C for 1 min and were further incubated at 42°C for 2 h. Cells were collected by centrifugation at 10 000 r.p.m. at 4°C for 10 min and cell pellets were resuspended in 250 µl of buffer A (50 mM HEPES–KOH, pH 7.4, 200 mM NaCl, 2 mM β-mercaptoethanol, 0.1 mM EDTA, 10% glycerol). After sonication, soluble fractions were collected by centrifugation at 15 000 r.p.m. at 4°C for 20 min and stored at –80°C.
Other methods
Nucleotide sequences were determined using Dye Terminator Cycle Sequencing FS Ready Reaction kits and a model 377 automated DNA sequencer (PE Applied Biosystems). Cell cultures, subcellular fractionation and western blotting were performed as described (29). Thin sections of isolated mitochondria were prepared and processed for electron microscopic immunocytochemistry as described (28). Protein concentration was determined using a Bio-Rad protein assay kit with bovine serum albumin as the standard. In vitro transcription and translation was done using the Single Tube Protein System 2 kit (Novagen). Gel shift analysis was done as described previously (43).
RESULTS
Identification of a repair activity for 2-OH-A in nuclear extracts prepared from human cells
An enzyme activity which introduces an alkali-labile site at 2-OH-A paired with guanine in double-stranded oligonucleotides (*AO:G) was detected in nuclear extracts prepared from Jurkat cells and was partially purified by successive chromatographies (Table 2). Gel filtration chromatography revealed that the estimated molecular mass of the enzyme was ~55 kDa, as shown in Figure 1. Fractions containing the repair activity for *AO:G also contained repair activities for *A:G and *A:GO, but not for *GO:C. An activity that introduces a nick into substrates with *GO:C was eluted as an enzyme with a molecular mass of ~40 kDa or less, which proved to be a nuclear form of hOGG1 (hOGG1-1a) (Fig. 1, *GO:C; 29).
Table 2. Purification of repair activities for 2-OH-A and adenine paired with guanine or 8-oxoG from Jurkat cells.

aOne unit is defined as the activity cleaving 1 fmol of DNA substrate containing *A:G in 1 h at 37°C. *Fluorescent dye-labeled strand.
bThe activity specific for DNA substrate containing *A:G was quantitatively detected first in fraction II.
cRelative ratio of the activity in each peak fraction able to cleave DNA substrate containing *A:G, *A:GO and *AO:G was calculated.
dND, not determined.
Figure 1.

Co-purification of repair activities for 2-OH-A and adenine paired with guanine or 8-oxoG. Nuclear extracts prepared from Jurkat cells were applied to successive chromatographies, as summarized in Table 1. Each fraction from gel filtration chromatography was incubated with various double-stranded oligonucleotides containing the abnormal base pair shown in the figure and nicking activities were determined as described in Materials and Methods. * indicates a strand labeled at its 5′-end with FAM. AO, 2-OH-A; GO, 8-oxoG.
As shown in Figure 2A, activities introducing an alkali-labile site into substrates with *AO:G and *A:G or *A:GO were co-purified even after two more chromatographies and the ratio of these activities to each substrate remained constant after gel filtration chromatography (Table 2).
Figure 2.
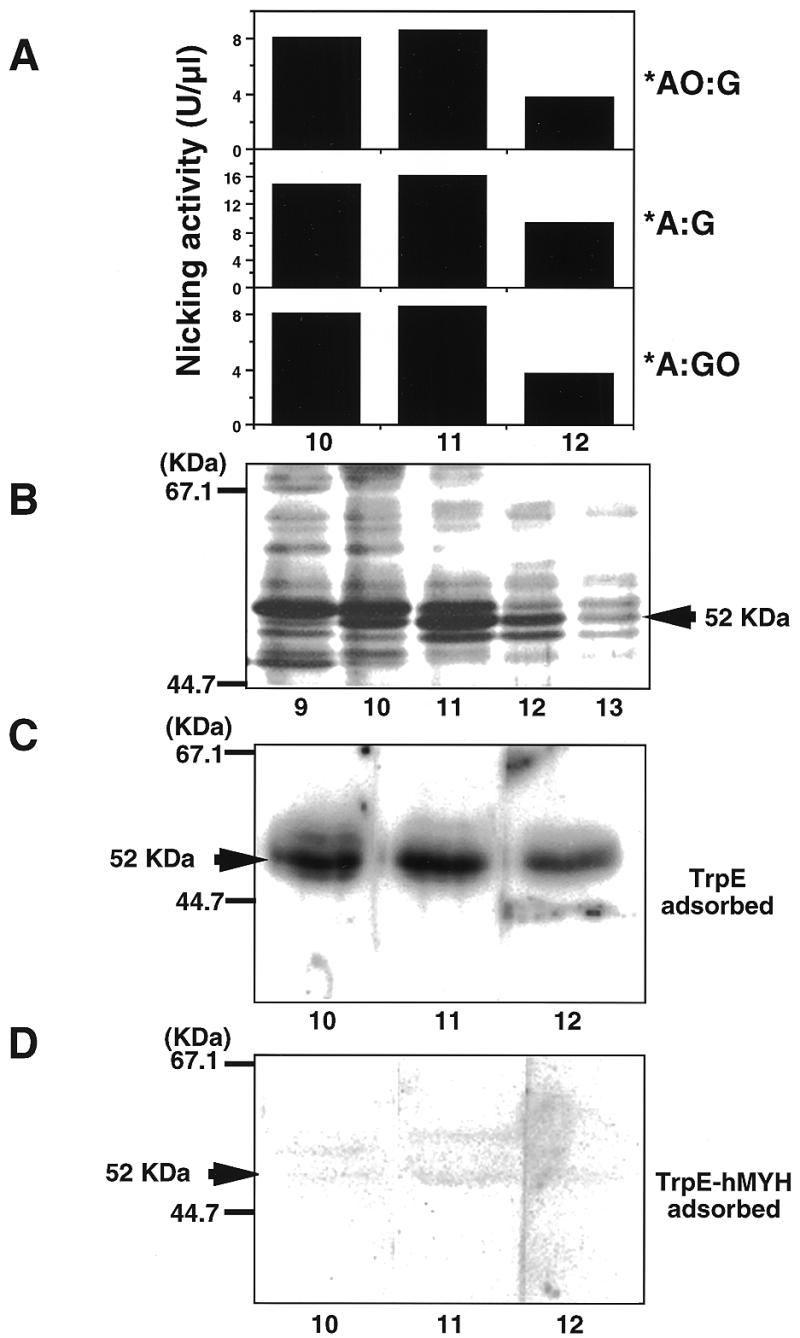
Authentic hMYH possesses repair activity for 2-OH-A and adenine paired with guanine or 8-oxoG. The RESOURCE S fraction was subjected to nicking assay for various double-stranded oligonucleotides (A). The active fractions were subjected to SDS–PAGE and then the gel was stained with silver (B) or was further analyzed by western blotting, using anti-hMYH which had been pre-adsorbed with TrpE–Sepharose (C) or TrpE-hMYH–Sepharose (D).
Authentic hMYH possesses the repair activity for 2-OH-A in DNA
Peak fractions from the RESOURCE S column containing activities introducing an alkali-labile site into substrates with *AO:G and *A:G or *A:GO pairs were subjected to SDS–PAGE and proteins in each fraction were detected by silver staining and western blotting using anti-hMYH. As shown in Figure 2B, two major bands (52 and 55 kDa) were detected. Only the 52 kDa polypeptide co-eluted with the enzyme activities (lanes 10–12). Anti-hMYH specifically reacted with the 52 kDa polypeptide in active fractions and the reaction was abolished by pretreatment of antibodies with the TrpE-hMYH antigen but not the carrier protein, TrpE (Fig. 2C and D).
Substrate specificity of authentic hMYH
The RESOURCE S fraction containing hMYH was incubated with various combinations of double-stranded oligonucleotides (Fig. 3A). Substrates containing *A:G and *A:GO, but not *A:A, *A:T and *A:C, were incised by the hMYH fraction with alkaline treatment. The enzyme acts most efficiently on substrates containing *A:G and then on those containing *AO:GO, *A:GO, *AO:G and *AO:A, with almost 40–50% efficiency for the former, with less activity on those containing *AO:T and *AO:C. With a mild alkaline treatment (0.17 N NaOH at 37°C for 30 min), two new bands, one migrating slightly faster than a 20mer and the other migrating at the position of a 19mer, were produced from these substrates, indicating that the former may correspond to a 19mer with α,β-unsaturated aldehyde generated by β-elimination of the abasic site and the other to a 19mer with 5′-phosphate generated by δ-elimination.
Figure 3.

Characterization of the enzymatic properties of partially purified hMYH. (A) Substrate specificity. A partially purified fraction of hMYH (the RESOURCE S fraction) was incubated with various double-stranded oligonucleotides. Reaction products were fractionated on a 6% LongRanger gel containing 7 M urea, after mild alikaline treatment. Relative amounts of cleaved product of each substrate to that of A:G substrate are shown. 19-P, FAM-labeled control oligonucleotide corresponding to the cleaved product at the 5′-side of adenine or 2-OH-A, with 3′-phosphate; 20-OH, FAM-labeled control oligonucleotide corresponding to the cleaved product at the 3′-side of adenine or 2-OH-A, without 3′-phosphate. (B) NaOH-dependent nicking of oligonucleotide containing adenine paired with 8-oxoG by partially purified hMYH fraction. Various amounts of the enzyme fraction were incubated with double-stranded oligonucleotides containing adenine paired with 8-oxoG and the mixture was fractionated with or without NaOH treatment. (C) Fluorescence intensities of bands corresponding to the cleavage products produced by hMYH were obtained from data in (B) and plotted. (D) AP lyase activity in partially purified hMYH fraction. Double-stranded oligonucleotides containing uracil paired with guanine were first treated with uracil DNA glycosylase, then incubated with or without the enzyme fraction. The products were fractionated with or without NaOH treatment.
Since we found no incision activity on substrates containing *A:GO without alkaline treatment (Fig. 3B and C) and substrates containing an abasic site were poorly incised by the RESOURCE S fraction (Fig. 3D), we may conclude that authentic hMYH has 2-OH-A and adenine DNA glycosylase activities with little AP lyase activity.
Optimum reaction of hMYH with a substrate containing *A:GO was obtained at pH 7.4 in the presence of 25 mM NaCl, with linear kinetics up to 60 min, and no metal ion (Mg, Zn or Co) was required. In the presence of 150 mM NaCl the activity was lowered to 30% of the optimum level. The enzyme activity in the partially purified fraction gradually decreased during storage at –80°C, indicating that hMYH is rather unstable. Adenine DNA glycosylase activity was inhibited neither by preincubation with anti-hMYH nor with adenine or 2-OH-A free base (data not shown). We also detected DNA-binding activity in the fraction, which preferentially binds to oligonucleotides containing *A:GO and very weakly to those containing *A:G (data not shown).
Recombinant hMYH possesses repair activitiy for 2-OH-A paired with guanine
hMYH encoded by an open reading frame of hMYH cDNA starting at the second ATG was expressed in E.coli cells. As shown in Figure 4A, a 53 kDa polypeptide was detected only in E.coli cells harboring hMYH cDNA, by western blotting with anti-hMYH; however, hMYH was likely to be highly susceptible to proteolysis in E.coli cells. Repair activities in extracts prepared from E.coli cells expressing recombinant hMYH and 14-3-3 protein as a negative control were examined (Fig. 4B). Activities introducing a single strand nick into substrates containing *A:GO or *A:G were detected in extracts from hMYH-expressing cells and the level was 10-fold higher than the level in extracts from 14-3-3-expressing cells. The hMYH extract also contained an activity introducing nicks into substrates containing *AO:G, which were not evident in the control extract from 14-3-3-expressing cells. However the reaction products from the *AO:G substrate were heterogeneous in size, even with heating at 95°C for 10 min in the presence of 0.17 N NaOH, a condition yielding a single nucleotide gap flanked by 3′- and 5′-phosphate termini by δ-elimination, as seen in the product from the *A:GO substrate. This result suggests that exonuclease activities in crude extracts degrade only the reaction products from the *AO:G substrate. Thus, we propose that hMYH has DNA glycosylase activity excising 2-OH-A as well as adenine.
Figure 4.
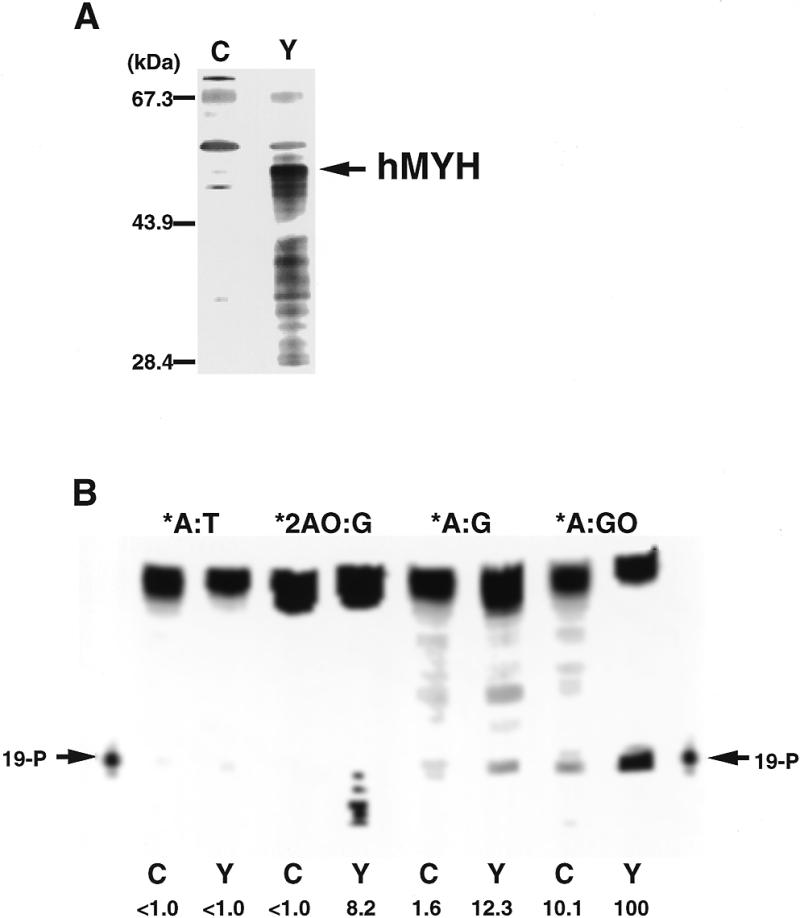
Recombinant hMYH possesses DNA repair activity for 2-OH-A and adenine paired with guanine or 8-oxoG. (A) Western blotting. Extracts (5 µl) prepared from E.coli cells carrying plasmid pXC35:14-3-3 (lane C) or pXC35:hMYHα3-2 (lane Y) were subjected to western blot analysis, using anti-hMYH. (B) DNA repair activities of recombinant hMYH. Various double-stranded oligonucleotides (150 fmol/15 µl reaction) were incubated with extracts (5 µl) prepared from E.coli cells carrying plasmid pXC35:14-3-3 (lane C) or pXC35:hMYHα3-2 (lane Y) for 30 min at 37°C, then 3 µl of 1 N NaOH was added, followed by heating at 95°C for 10 min. Then the mixture was fractionated on an 8% LongRanger gel containing 7 M urea. Relative amounts of cleaved products of each substrate produced by hMYH to that of A:GO substrate are shown.
Immunological detection of multiple forms of hMYH polypeptides in human cells
The molecular weight of the hMYH predicted from its cDNA sequence is 59 034 (18) and it has been reported that a 65 kDa polypeptide was identified as a mammalian homolog of MutY by western blotting using antibodies against E.coli MutY (amino acids 192–211) (44). Since only a 52 kDa polypeptide was detected by anti-hMYH in the partially purified fraction, as shown in Figure 2, we carried out western blotting analysis of isolated nuclei and mitochondria prepared from Jurkat cells to detect authentic hMYH in human cells. As shown in Figure 5A, two bands, a major one corresponding to p52 found in the partially purified fraction and a minor band corresponding to a 53 kDa (p53) polypeptide, were detected in the isolated nuclei. On the other hand, a 57 kDa polypeptide (p57) was specifically detected in the mitochondrial fraction.
Figure 5.
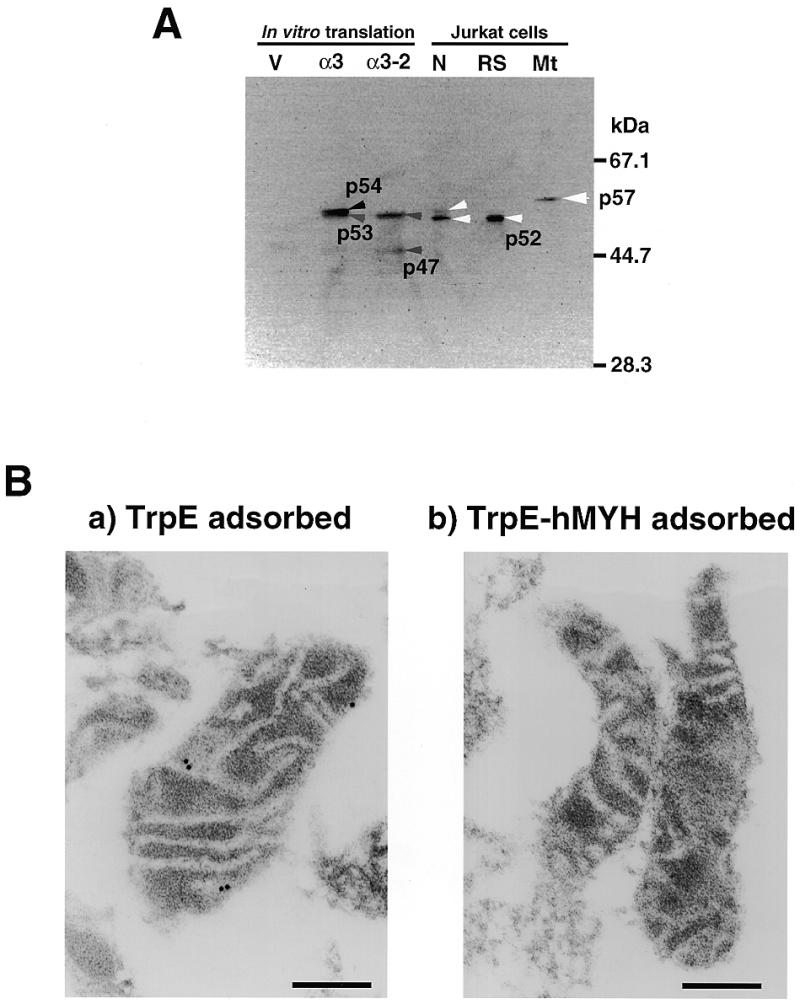
5. Immunological detection of authentic hMYH in human cells. (A) Western blotting. In vitro translation products of the pT7Blue vector (V), pT7Blue:hMYHα3 (α3) and pT7Blue:hMYHα3-2 (α3-2), and isolated nuclei (N) and mitochondria (Mt) (equivalent to 50 µg of protein) from Jurkat cells and partially purified hMYH in the RESOURCE S fraction (RS) were subjected to western blot analysis, using anti-hMYH. (B) Submitochondrial localization of hMYH, determined by electron microscopic immunocytochemistry. After mitochondria had been isolated from Jurkat cells, thin sections (~0.1 µm) were prepared for electron microscopic immunocytochemistry with anti-hMYH preadsorbed with TrpE–Sepharose or with TrpE-hMYH–Sepharose in combination with protein A–gold. Bars indicate 0.2 µm.
When the in vitro translation product of hMYHα3 cDNA was subjected to western blotting with anti-hMYH as control, two bands corresponding to 53 and 54 kDa polypeptides were detected (Fig. 5A, α3), indicating that translation may occur from two different initiation codons. A plasmid carrying an open reading frame from the second initiation codon of the hMYHα3 cDNA was then constructed and subjected to in vitro translation and western blotting. As shown in Figure 5A, two bands corresponding to 47 and 53 kDa polypeptides were detected, thus confirming that hMYH mRNA encodes three polypeptides translated from the first and second initiation codons and possibly also from the third initiation codon, at least in vitro.
We prepared thin sections from fixed mitochondrial pellets prepared from Jurkat cells and examined the localization of p57 by electron microscopic immunocytochemistry, using anti-hMYH in combination with protein A–gold (Fig. 5B). In all the mitochondrial sections, several specific signals were observed in low electron density areas (Fig. 5Ba), which correspond to the inner membrane of the mitochondria. When anti-hMYH preadsorbed by TrpE-hMYH–Sepharose was applied to sections, no signal was observed (Fig. 5Bb), indicating that the signals in mitochondria from Jurkat cells represent authentic p57 hMYH polypeptide.
Identification of multiple forms of hMYH mRNAs in human cells
Based on the DNA sequence of the hMYH gene reported (18), we amplified the cDNA sequence for hMYH mRNA by RT–PCR from HeLa cells. We identified four clones with different sequences: one corresponds to the cDNA sequence reported (18) and the other three are likely to be alternatively spliced variants (Fig. 6). To confirm these findings, we further performed 5′-RACE using a cDNA library from Jurkat cells as template and obtained two new forms of cDNA clones with novel 5′ sequences. Based on the cDNA sequences, three sets of type-specific primers were synthesized and at least 10 forms of hMYH cDNAs amplified from HeLa S3 cells were subcloned and their sequences determined (Fig. 6A and 7B). We classified hMYH mRNA into three types (α, β and γ) based on their 5′ sequences. Type α has the same 5′ sequence as does the cDNA reported (18) and there are four subtypes of type α, namely type α1, α2, α3 and α4. Type α3 corresponds to the cDNA sequence previously identified (18), α2 has an insertion sequence, CAG, while α1 has a much longer insertion sequence (33 bp) with the CAG, between exons 2 and 3, which is derived from the intron 2 sequence just in front of exon 3. Type α4 is missing 64 bases of the 5′-region of exon 3. Types β and γ have novel 5′ sequences, both of which differ from each other, indicating that the hMYH gene has at least three independent transcription initiation sites. There are also several splicing variants of type β and γ hMYH mRNAs; β1, β3 and β5 and γ2, γ3 and γ4. The common sequence of the 5′-ends of type β cDNAs was identical to that reported by Takao et al. (19) as an alternative 5′ sequence of hMYH cDNA.
Figure 6.

6. Multiple forms of hMYH transcripts and predicted polypeptides. (A) Sequences of 5′-regions (exons 1–3) of 10 different forms of hMYH cDNA. Sequences corresponding to the reported exons 2 and 3 are shown in box (18). Two putative initiation codons, ATG in exons 1 and 2, are shown in italic. (B) Predicted polypeptides encoded by the various hMYH transcripts. * indicates a methionine residue. The gray box indicates the mitochondrial targeting signal (19,56) and the hatched box indicates the 11 amino acid insertion.
Figure 7.
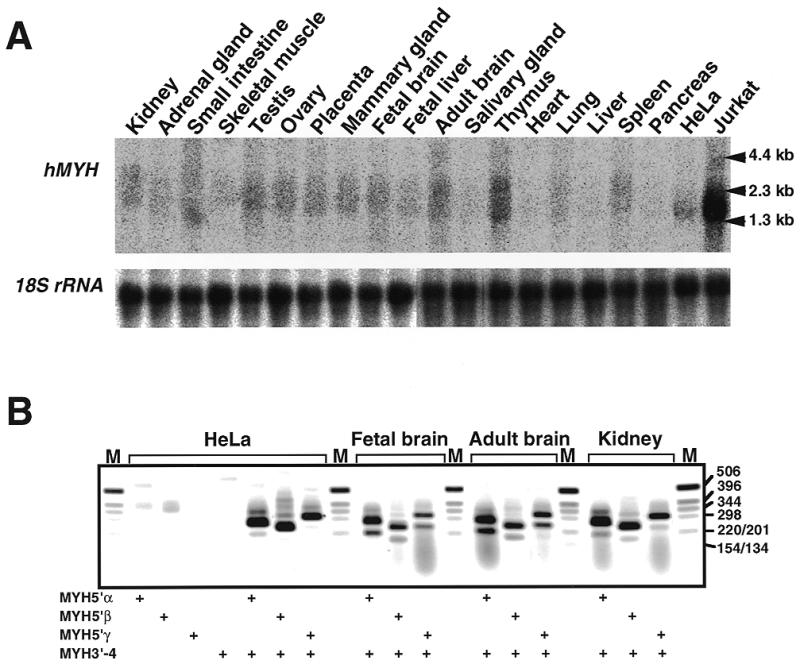
Expression of hMYH mRNAs in various human tissues and cell lines. (A) Northern blot analysis. Total RNAs (16 µg each) extracted from various human tissues and Jurkat and HeLa S3 cells were electrophoresed, transferred to nitrocellulose membrane and probed with 32P-labeled hMYH cDNA. (B) RT–PCR analysis. cDNAs were synthesized from total RNA prepared from HeLa S3 cells, fetal brain, adult brain and kidney, using oligo(dT)18 primer. The cDNAs were amplified using a common 3′ primer (MYH3′-4) and a 5′ primer specific for each type of hMYH mRNA (types 5′α, 5′β and 5′γ). PCR products were analyzed by 1.5% agarose gel electrophoresis. The presence of primers in the reaction mixture is shown by +.
A major form of hMYH mRNA, type α3, has an open reading frame for a polypeptide with a molecular weight (MW) of 59 034, type α1 may encode a polypeptide with MW 60 031, with an insertion of 11 amino acid residues, and type α2 may encode a polypeptide with MW 59 105, with insertion of an amino acid residue (Fig. 6B). In type α4 mRNA there is a termination codon in the open reading frame from the first initiation codon commonly used for the type α1–3 polypeptides. There is a polypeptide with MW 47 000 which may be translated from an open reading frame initiating at the third AUG and which may correspond to the p47 translation product from a plasmid carrying an open reading frame from the third initiation codon of the hMYHα3 cDNA shown in Figure 5 (lane α3-2). In type β and γ hMYH transcripts, there are open reading frames initiating at an AUG codon corresponding to the second AUG in the open reading frames found in type α1, α2, and α3 transcripts. Thus, types β3, β5 and γ3 may encode the same polypeptide with MW 57 410, type β1 one with MW 58 407, while type γ2 may encode a polypeptide with MW 57 481 and type γ4 one with MW 47 000.
Expression of hMYH mRNAs in various human tissues
We then examined levels of hMYH mRNA in various human tissues and cell lines using northern blot analysis (Fig. 7A). Radioactive bands corresponding to mRNAs with sizes from 1.3 to 2.3 kb were detected. The largest amounts of hMYH mRNA, the size of which was mostly 1.8 kb, were detected in Jurkat cells. HeLa cells express much smaller amounts of the 1.8 kb mRNA. Two major bands corresponding to 1.8 and 2.3 kb mRNAs were detected in almost all tissues examined; however, the radioactivity of the bands was lower than that in Jurkat cells and varied considerably. Among various human tissues, the largest amounts of hMYH mRNAs were found in the thymus, then in adult brain, testis and kidney. The amounts of hMYH mRNA in the heart, salivary gland, liver and pancreas were low compared to amounts in other tissues. It is interesting that adult brain, a post-mitotic tissue, has large amounts of hMYH mRNA, but liver and pancreas have small amounts.
RNAs from HeLa S3 cells, human fetal brain, adult brain and kidney were also analyzed by RT–PCR, using three sets of type-specific primers (Fig. 7B). Three bands (210, 275 and 310 bp) were amplified from all samples with the set of primers for type α mRNAs and sequence analysis of cloned fragments obtained from HeLa S3 cells revealed that type α3 mRNA (274 bp) was the major form among the four type α hMYH mRNAs, based on the number of clones identified by sequencing. Type α4 mRNA (210 bp) was minor in HeLa S3 cells and kidney; however, in brain tissues its relative amount was higher than that of type α1 mRNA (307 bp). With the set of primers for type β mRNAs, four or more bands were amplified from all samples, and a 245 bp fragment corresponding to the PCR product from type β3 (245 bp) was major in all cases. In samples from human tissues, substantial amounts of a 180 bp fragment, which may correspond to the PCR product from type β mRNAs missing 64 bases in the 5′-region of exon 3, as do types α4 and γ4, was amplified. For type γ mRNAs, a 315 bp fragment which may correspond to type γ2 (316 bp) or γ3 (313 bp) was the major form in all samples analyzed. Again, a 250 bp fragment corresponding to the PCR product from type γ4 mRNA (249 bp) was evident in brain tissues, but much less so in samples from HeLa S3 cells and kidney.
DISCUSSION
Among many oxidized bases, 8-oxoG has been intensively studied as it is implicated in various diseases, such as cancer, neuronal degeneration and teratogenicity. 8-oxoG has mutagenic potential and various enzymes specifically act on the lesion (3,7,8,45,46). In the present study, we propose that 2-OH-A, an oxidized form of adenine, must also be involved in biological responses, as 2-OH-A in DNA can be efficiently excised by a DNA glycosylase encoded by the hMYH gene.
2-OH-A, also known as isoguanine, has been detected in DNA from isolated human tissues and from experimental animals and its amount was increased by exposure to various sources of reactive oxygen species, both in vitro and in vivo (47–49). Furthermore, it has been shown that in certain human cancerous tissues, amounts of 2-OH-A are increased several-fold compared to findings in normal non-cancerous tissues (50). Concerning the origin of 2-OH-A, the extent of oxidation of the 2 position of the adenine base by hydroxyl radicals is higher in its free nucleotide form than in DNA (47), thereby indicating that misincorporation of 2-OH-dATP from the nucleotide pool is the major source of 2-OH-A in DNA. 2-OH-A in DNA thus incorporated forms with a relatively stable base pair with thymine and cytosine (51,52), and may also pair with the syn forms of guanine and adenine (47).
We reported that 2-OH-dATP is efficiently hydrolyzed to 2-OH-dAMP by hMTH1, originally identified as 8-oxo-dGTPase (31), indicating that organisms come equipped with an enzyme to eliminate 2-OH-dATP from their nucleotide pools. Since hMTH1 has the lowest Km value with 2-OH-dATP among its substrates and E.coli MutT, a prototype of 8-oxo-dGTPase, has little ability to hydrolyze 2-OH-dATP, one could argue that human and mammalian cells are exposed to a higher risk of incorporation of 2-OH-dATP into their genome than are E.coli cells, thus they eliminate the precursor from the nucleotide pool. Since 2-OH-A in DNA forms a stable Watson–Crick base pair with thymine, it has been speculated that 2-OH-A paired with thymine in DNA may escape repair (51). However, Jaruga and Dizdaroglu (53) reported that 2-OH-A detected in DNA of human cells after H2O2 exposure is repaired within 4 h to background level, indicating that human cells possess repair activity for 2-OH-A in DNA.
In the present work, we examined an enzyme activity that introduces an alkali-labile site into 2-OH-A-containing oligonucleotides and found evidence for such a DNA repair activity that is likely to act as a DNA glycosylase. Furthermore, we showed that the repair activity is co-purified with hMYH and that recombinant hMYH itself has such activity. hMYH lacks AP lyase activity, thus we suspect that one could not detect the repair activity for 2-OH-A without alkaline treatment (54).
Our data indicate that hMYH (2-OH-A/adenine DNA glycosylase) recognizes 2-OH-A paired with 8-oxoG or purines as well as adenine paired with guanine or 8-oxoG. hMYH in partially purified fractions preferentially acts on adenine paired with guanine in DNA rather than that paired with 8-oxoG; however, it binds more efficiently to substrates containing the latter. These characteristics of authentic hMYH are the same as those of calf MYH (44). However, recombinant hMYH in crude extracts exhibited strongest activity for oligonucleotides containing adenine paired with 8-oxoG, as reported by others (19,20), but substantially less nicking activity for oligonucleotides containing adenine or 2-OH-A paired with guanine. Thus, the partially purified fraction of hMYH from Jurkat cells may contain some other factors which modify its substrate specificity.
It has been shown that the C-terminal region of E.coli MutY (amino acids 227–350), the sequence of which is homologous to hMYH (amino acids 352–535 in hMYHα3) and partly to MutT (Fig. 8), specifically binds 8-oxoG opposite adenine or an abasic site (55). hMYH binds strongly to oligonucleotides containing adenine paired with 8-oxoG and to a lesser extent when paired with guanine, but it is likely that hMYH quickly dissociates from those with 2-OH-A paired with guanine after excising 2-OH-A. This may explain why the 2-OH-A-containing oligonucleotides but not the others were degraded by E.coli nucleases after incubation with the recombinant hMYH extract, as seen in Figure 4B.
Figure 8.
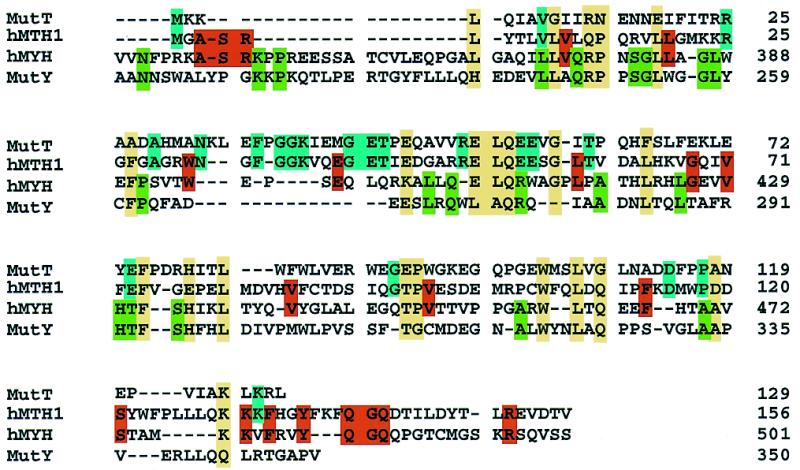
Sequence alignment of the E.coli MutT, hMTH1 and hMYH C-terminal regions (amino acids 341–501) and the E.coli MutY C-terminal region (amino acids 211–350). The alignments for MutT, hMYH and MutY are based on the data of Noll et al. (55) and those for hMTH1 and MutT are based on the data of Fujii et al. (45). Residues conserved among more than three proteins are shown in a yellow box and residues conserved only between MutT and hMTH1 or hMYH and MutY are shown in a blue or green box, respectively. Residues conserved only between hMTH1 and hMYH, which may be involved in 2-OH-A recognition, are shown in a red box.
In crude extracts from E.coli cells, we found no activity on substrates containing 2-OH-A (Fig. 4B). We then asked whether purified MutY acts on 2-OH-A, but no such activity was detected (T.Ohtsubo, H.Kamiya, H.Kasai and Y.Nakabeppu, unpublished results). Thus, hMYH but not MutY has a novel repair activity for 2-OH-A in DNA, as does hMTH1 for 2-OH-dATP (31). Interestingly, we found that there are significantly conserved residues between hMYH (amino acids 369–499 in hMYHα3) and the entire hMTH1 molecule, as shown in Figure 8. Thirty-nine residues in hMTH1 (156 amino acids) are identical to those in the hMYH C-terminal region and 18 of them are also conserved in E.coli MutT and MutY, suggesting that the 18 residues are involved in binding to 8-oxoG. The 21 residues conserved only between hMYH and hMTH1 may determine the specificities of the two proteins for 2-OH-A and 2-OH-dATP, in addition to 8-oxoG and 8-oxo-dGTP.
In human cells, authentic hMYH was detected in nuclei and mitochondria by western blotting and their molecular weights differed, thereby indicating that there are multiple forms of hMYH. Based on 5′-RACE and RT–PCR of hMYH transcripts, we conclude that there are three major hMYH transcripts, namely hMYH α, β and γ, with a different 5′ sequence or first exon, and further that each transcript is alternatively spliced, thus forming over 10 mature transcripts. A major transcript, hMYHα3, essentially corresponds to the hMYH cDNA originally reported (18) and encodes two polypeptides, p54 and p53, the former translated from the first and the latter from the second AUG.
It has been reported that the translation product of the hMYHα3 transcript is exclusively localized in mitochondria and that the N-terminal sequence of hMYH translated from the first AUG functions as a mitochondrial targeting signal (19,56); however, the authentic hMYH polypeptide detected in mitochondria is p57. The hMYHα1 transcript has a 33 nt insertion into hMYHα3 and thus encodes a polypeptide with an expected molecular mass of 60 031; this may be the p57 detected in mitochondria. Moreover, the nuclear form of hMYH, p52, partially purified from Jurkat cells, is likely to correspond to p53 translated from the second AUG and the hMYH β1, β3 and γ2 transcripts, which are missing the first AUG, may encode the nuclear form of p52 hMYH. Of course, it is also possible that the hMYHα3 transcript produces p53 and p54 by alternative translation initiation, and one of them may correspond to the p52 detected in nuclei of Jurkat cells. Determination of the primary sequences of purified hMYH will reveal which polypeptide is encoded by each hMYH transcript. It is likely that hMYH in mitochondria associates with the inner membrane structure, as does hOGG1-2a (29), suggesting that the machinery needed for base excision repair in mitochondria generally associates with the inner membrane to achieve efficient repair of mitochondrial DNA.
To date, human cells have been found to be equipped with a repair enzyme, hMYH, for 2-OH-A, as well as hMTH1 for 2-OH-dATP (31). We will now extend our search for 2-OH-A DNA glycosylase and 2-OH-dATPase activities to other mammalian cells in order to determine whether MYH and MTH1 have the same activities in other mammalian cells.
Acknowledgments
ACKNOWLEDGEMENTS
We extend special thanks to Drs M. Furuichi, K. Sakumi and M. Sekiguchi for helpful discussions, to Dr Neil Clarke for generous gifts of pXC35:14-3-3 and TAP56 and to M. Ohara for language assistance. This work was supported in part by Kyushu University Interdisciplinary Programs in Education and Projects in Research Development and a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan.
DDBJ/EMBL/GenBank accession nos AB032920–AB032929
REFERENCES
- 1.Kasai H. and Nishimura,S. (1984) Nucleic Acids Res., 12, 2137–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ames B.N. and Gold,L.S. (1991) Mutat. Res., 250, 3–16. [DOI] [PubMed] [Google Scholar]
- 3.Ames B.N., Shigenaga,M.K. and Hagen,T.M. (1993) Proc. Natl Acad. Sci. USA, 90, 7915–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shibutani S., Takeshita,M. and Grollman,A.P. (1991) Nature, 349, 431–434. [DOI] [PubMed] [Google Scholar]
- 5.Maki H. and Sekiguchi,M. (1992) Nature, 355, 273–275. [DOI] [PubMed] [Google Scholar]
- 6.Tajiri T., Maki,H. and Sekiguchi,M. (1995) Mutat. Res., 336, 257–267. [DOI] [PubMed] [Google Scholar]
- 7.Michaels M.L. and Miller,J.H. (1992) J. Bacteriol., 174, 6321–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sekiguchi M. (1996) Genes Cells, 1, 139–145. [DOI] [PubMed] [Google Scholar]
- 9.Bailly V., Verly,W.G., O’Connor,T. and Laval,J. (1989) Biochem. J., 262, 581–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tchou J., Kasai,H., Shibutani,S., Chung,M.H., Laval,J., Grollman,A.P. and Nishimura,S. (1991) Proc. Natl Acad. Sci. USA, 88, 4690–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Au K.G., Clark,S., Miller,J.H. and Modrich,P. (1989) Proc. Natl Acad. Sci. USA, 86, 8877–8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Michaels M.L., Tchou,J., Grollman,A.P. and Miller,J.H. (1992) Biochemistry, 31, 10964–10968. [DOI] [PubMed] [Google Scholar]
- 13.Cabrera M., Nghiem,Y. and Miller,J.H. (1988) J. Bacteriol., 170, 5405–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nghiem Y., Cabrera,M., Cupples,C.G. and Miller,J.H. (1988) Proc. Natl Acad. Sci. USA, 85, 2709–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakumi K., Furuichi,M., Tsuzuki,T., Kakuma,T., Kawabata,S., Maki,H. and Sekiguchi,M. (1993) J. Biol. Chem., 268, 23524–23530. [PubMed] [Google Scholar]
- 16.Kakuma T., Nishida,J., Tsuzuki,T. and Sekiguchi,M. (1995) J. Biol. Chem., 270, 25942–25948. [DOI] [PubMed] [Google Scholar]
- 17.Cai J.P., Kakuma,T., Tsuzuki,T. and Sekiguchi,M. (1995) Carcinogenesis, 16, 2343–2350. [DOI] [PubMed] [Google Scholar]
- 18.Slupska M.M., Baikalov,C., Luther,W.M., Chiang,J.-H., Wei,Y.-F. and Miller,J.H. (1996) J. Bacteriol., 178, 3885–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takao T., Zhang,Q.-M., Yonei,S. and Yasui,A. (1999) Nucleic Acids Res. 27, 3638–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slupska M.M., Luther,W.M., Chiang,J.-H., Yang,H. and Miller,J.H. (1999) J. Bacteriol., 181, 6210–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aburatani H., Hippo,Y., Ishida,T., Takashima,R., Matsuba,C., Kodama,T., Takao,M., Yasui,A., Yamamoto,K., Asano,M. et al. (1997) Cancer Res., 57, 2151–2156. [PubMed] [Google Scholar]
- 22.Arai K., Morishita,K., Shinmura,K., Kohno,T., Kim,S.-R., Nohmi,T., Taniwaki,M., Ohwada,S. and Yokota,J. (1997) Oncogene, 14, 2857–2861. [DOI] [PubMed] [Google Scholar]
- 23.Bjørås M., Luna,L., Johnsen,B., Hoff,E., Haug,T., Robnes,T. and Seeberg,E. (1997) EMBO J., 16, 6314–6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu R., Nash,H.M. and Verdine,G.L. (1997) Curr. Biol., 7, 397–407. [DOI] [PubMed] [Google Scholar]
- 25.Radicella J.P., Dherin,C., Desmaze,C., Fox,M.S. and Boiteux,S. (1997) Proc. Natl Acad. Sci. USA, 94, 8010–8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Róldan-Arjona T., Wei,Y.F., Carter,K.C., Klungland,A., Anselmino,C., Wang,R.P., Augustus,M. and Lindahl,T. (1997) Proc. Natl Acad. Sci. USA, 94, 8016–8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenquist T.A., Zharkov,D.O. and Grollman,A.P. (1997) Proc. Natl Acad. Sci. USA, 94, 7429–7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang D., Nishida,J., Iyama,A., Nakabeppu,Y., Furuichi,M., Fujiwara,T., Sekiguchi,M. and Takeshige,K. (1995) J. Biol. Chem., 270, 14659–14665. [DOI] [PubMed] [Google Scholar]
- 29.Nishioka K., Ohtsubo,T., Oda,H., Fujiwara,T., Kang,D., Sugimachi,K. and Nakabeppu,Y. (1999) Mol. Biol. Cell, 10, 1637–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hayakawa H., Hofer,A., Thelander,L., Kitajima,S., Cai,Y., Oshiro,S., Yakushiji,H., Nakabeppu,Y., Kuwano,M. and Sekiguchi,M. (1999) Biochemistry, 38, 3610–3614. [DOI] [PubMed] [Google Scholar]
- 31.Fujikawa K., Kamiya,H., Yakushiji,H., Fujii,Y., Nakabeppu,Y. and Kasai,H. (1999) J. Biol. Chem., 274, 18201–18205. [DOI] [PubMed] [Google Scholar]
- 32.Kamiya H. and Kasai,H. (1997) Biochemistry, 36, 11125–11130. [DOI] [PubMed] [Google Scholar]
- 33.Kamiya H. and Kasai,H. (1997) Nucleic Acids Res., 25, 304–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inoue M., Kamiya,H., Fujikawa,K. Ootsuyama,Y., Murata-Kamiya,N., Osaki,T., Yasumoto,K. and Kasai,H. (1998) J. Biol. Chem., 273, 11069–11074. [DOI] [PubMed] [Google Scholar]
- 35.Wood M.L., Esteve,A., Morningster,M.L., Kuziemko,G.M. and Essigman,J.M. (1992) Nucleic Acids Res., 22, 6023–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shibutani S., Bodepudi,V. and Grollman,A.P. (1993) Biochemistry, 32, 4615–4621. [DOI] [PubMed] [Google Scholar]
- 37.Nakabeppu Y. and Nathans,D. (1991) Cell, 64, 751–759. [DOI] [PubMed] [Google Scholar]
- 38.Cheng X. and Patterson,T.A. (1992) Nucleic Acids Res. 20, 4591–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oda H., Nakabeppu,Y., Furuichi,M. and Sekiguchi,M. (1997) J. Biol. Chem., 272, 17843–17850. [DOI] [PubMed] [Google Scholar]
- 40.Financsek I., Mizumoto,K. and Muramatsu,M. (1982) Gene, 18, 115–122. [DOI] [PubMed] [Google Scholar]
- 41.Dignam J.D., Lebovitz,R.M. and Roeder,R.G. (1983) Nucleic Acids Res., 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakabeppu Y., Oda,S. and Sekiguchi,M. (1993) Mol. Cell. Biol., 13, 4157–4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohtsubo T., Matsuda,O., Iba,K., Terashima,I., Sekiguchi,M. and Nakabeppu,Y. (1998) Mol. Gen. Genet., 259, 577–590. [DOI] [PubMed] [Google Scholar]
- 44.McGoldrick J.P., Yeh,Y.C., Solomon,M., Essigmann,J.M. and Lu,A.L. (1995) Mol. Cell. Biol., 15, 989–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fujii Y., Shimokawa,H., Sekiguchi,M. and Nakabeppu,Y. (1999) J. Biol. Chem., 274, 38251–38259. [DOI] [PubMed] [Google Scholar]
- 46.Boiteux S. and Radicella,J.P. (1999) Biochimie, 81, 56–67. [Google Scholar]
- 47.Kamiya H. and Kasai,H. (1995) J. Biol. Chem., 270, 19446–19450. [DOI] [PubMed] [Google Scholar]
- 48.Nackerdien Z., Karsprzak,K.S., Rao,G., Halliwell,B. and Dizdaroglu,M. (1991) Cancer Res., 51, 5837–5842. [PubMed] [Google Scholar]
- 49.Olinski R., Zastawny,T., Budzbon,J., Skokowski,J., Zegarski,W. and Dizdaroglu,M. (1992) FEBS Lett., 309, 193–198. [DOI] [PubMed] [Google Scholar]
- 50.Toyokuni S., Mori,T. and Dizdaroglu,M. (1994) Int. J. Cancer, 57, 123–128. [DOI] [PubMed] [Google Scholar]
- 51.Robinson H., Gao,Y.-G., Bauer,C., Roberts,C., Switzer,C. and Wang,A.H.-J. (1998) Biochemistry, 37, 10897–10905. [DOI] [PubMed] [Google Scholar]
- 52.Yang X.-L., Sugiyama,H., Ikeda,S., Saito,I. and Wang,A.H.-J. (1998) Biophys. J., 75, 1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jaruga P. and Dizdaroglu,M. (1996) Nucleic Acids Res., 24, 1389–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsurudome Y., Hirano,T., Kamiya,H., Yamaguchi,R., Asami,S., Itoh,H. and Kasai,H. (1998) Mutat. Res., 408, 121–127. [DOI] [PubMed] [Google Scholar]
- 55.Noll D., Gogos,A., Granek,J.A. and Clarke,N.D. (1999) Biochemistry, 38, 6374–6379. [DOI] [PubMed] [Google Scholar]
- 56.Takao M., Aburatani,H., Kobayashi,K. and Yasui,A. (1998) Nucleic Acids Res., 26, 2917–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]