


Abstract
We analyzed 10 genome expression data sets by large-scale cross-referencing against broad structural and functional categories. The data sets, generated by different techniques (e.g. SAGE and gene chips), provide various representations of the yeast transcriptome (the set of all yeast genes, weighted by transcript abundance). Our analysis enabled us to determine features more prevalent in the transcriptome than the genome: i.e. those that are common to highly expressed proteins. Starting with simplest categories, we find that, relative to the genome, the transcriptome is enriched in Ala and Gly and depleted in Asn and very long proteins. We find, furthermore, that protein length and maximum expression level have a roughly inverse relationship. To relate expression level and protein structure, we assigned transmembrane helices and known folds (using PSI-blast) to each protein in the genome; this allowed us to determine that the transcriptome is enriched in mixed α–β structures and depleted in membrane proteins relative to the genome. In particular, some enzymatic folds, such as the TIM barrel and the G3P dehydrogenase fold, are much more prevalent in the transcriptome than the genome, whereas others, such as the protein-kinase and leucine-zipper folds, are depleted. The TIM barrel, in fact, is overwhelmingly the ‘top fold’ in the transcriptome, while it only ranks fifth in the genome. The most highly enriched functional categories in the transcriptome (based on the MIPS system) are energy production and protein synthesis, while categories such as transcription, transport and signaling are depleted. Furthermore, for a given functional category, transcriptome enrichment varies quite substantially between the different expression data sets, with a variation an order of magnitude larger than for the other categories cross-referenced (e.g. amino acids). One can readily see how the enrichment and depletion of the various functional categories relates directly to that of particular folds. Further information can be found at http://bioinfo.mbb.yale.edu/genome/expression
INTRODUCTION
Whole-genome expression experiments have become important tools in functional genomics. The result of these experiments, the expression levels of all the genes in the genome, has been dubbed the transcriptome (1). Many of the initial expression experiments have focused on the eukaryote yeast for technical reasons as well as the fact that it is a widely studied model organism with a known genome sequence (2). Quantitative profiles of the yeast transcriptome have been determined for a variety of conditions using serial analysis of gene expression (SAGE) (1) as well as gene chip technology (8–11). Brown and colleagues have developed cDNA microarrays to conduct time-course experiments measuring the expression changes of yeast genes in response to a variety of conditions (3–7). Researchers have also started to investigate quantitative protein abundance profiles for yeast, using such approaches as fusion proteins (12) and two-dimensional gels (13).
Various approaches have been proposed to interpret the wealth of data generated by these experiments. Algorithms to cluster genes into functionally related groups have been proposed (14–17). Roth et al. (10), van Helden et al. (18) and Brazma et al. (19) have introduced new ways to identify regulatory regions located upstream of genes. Gerstein (20) proposed an initial ranking of protein folds in terms of their expression levels. A number of proposals have been made for the archiving and management of expression data (21).
Here we present another way to interpret gene expression data. We perform large-scale ‘cross-referencing’ of expression data against a number of structural and functional categories. These categories include (i) simple characteristics shared by all proteins, their amino acid composition and length, (ii) aspects of protein structure, fold family and number of transmembrane helices and (iii) broad functional classes. The correlation of expression level with these categories gives us insight into the characteristics of highly expressed proteins and also suggests some interesting conclusions about the overall biochemistry of the yeast cell. More specifically, we compare the composition of all our categories in the transcriptome with that in the genome. We find that the transcriptome is notably enriched with certain types of proteins (e.g. those rich in Ala and Gly, those with a mixed α–β structure and those associated with energy production and protein synthesis) and depleted in others (e.g. Asn-rich proteins, membrane proteins, very long proteins, and transcription factors and transport proteins).
Expression data
We based our analysis of the yeast transcriptome on a diverse set of publicly available expression experiments, which are summarized in Table 1a. Including data sets derived from different experimental techniques potentially reduces the bias introduced by focusing on one particular experiment. We focused more on data from DNA chips and SAGE technology rather than cDNA microarray experiments, since DNA chips and SAGE allow a better measurement of the absolute number of transcript copies for an open reading frame (ORF), facilitating direct comparisons between ORFs. In contrast, cDNA microarrays mainly measure expression level changes of a given ORF as ratios to a reference point, and ORF-to-ORF comparisons at a given time point are more problematic.
Table 1. Overview of data and methods.


(a) Overview of the expression data sets used in our analysis. The columns ‘reference’ and ‘URL’ provide the literature reference and the Internet address of the data sets. Column ‘# ORFs covered’ shows for how many different yeast ORFs expression levels were measured in the respective experiment. The column labeled ‘technology’ shows the technology with which the data sets were obtained. All the data from the expression experiments as well as the soluble and membrane fold assignments were homogenized and relationalized and stored in a simple database. We focused more on data from DNA chips (9–11) and the SAGE technology (1) than that from cDNA microarray experiments (5) since the former techniques allow a better measurement of the absolute number of transcript copies for a gene. In presenting our data, we decided, for convenience, to use the data set generated by Holstege et al. (9) as the main reference. For the SAGE data set we only considered SAGE tags that occur at most once per genome and fall into an ORF (rather than upstream regions) (1). (b) The general approach in our calculations. First, we calculate the genome composition of a specific feature F, G(F). Then, we compute the composition of feature F in the transcriptome, T(F); this is achieved by weighting the count of feature F with the expression level ei of the corresponding ORF i. Finally, D(F) yields the transcriptome enrichment of feature F, the relative difference between its transcriptome and genome compositions. The table shows the calculation of the transcriptome enrichment D(F) for the amino acid Ala and the TIM barrel fold as examples based on the data set by Holstege et al. (9). To be consistent, we include only those ORFs in our calculations (of both the transcriptome and the genome composition) for which an expression level ei exists. Because the set of ORFs for which expression levels were measured vary between the different experiments [see (a)], different genome compositions are obtained for each experiment. However, these differences are generally very small and do not influence the results significantly.
In presenting our data, we decided, for convenience, to use the data set generated by Holstege et al. (9) as the main reference. This data set represents the average of two transcript abundance level measurements for most yeast genes. Furthermore, the authors report that 99% of these transcripts exhibited a less than 2-fold change in the two measurements. We also extensively used the SAGE data sets (1), which give expression profiles of a large but less complete subset of the yeast genome in different conditions, the gene chip data generated by Roth et al. (10), which represent profiles of the yeast transcriptome for different conditions, and the gene chip data by Jelinsky et al. (11), who investigated expression profiles before and after the yeast cell is subjected to an alkylating agent (we only used the first, more typical, profile).
General approach
Most of our analyses have the same basic structure, which is schematized in Table 1b. First, we compute the genome composition of a specific category, then we compute its composition in the transcriptome and finally we determine its enrichment in the transcriptome, that is, the relative difference between transcriptome and genome composition. For computing transcriptome compositions we weight each gene with its respective expression level. With the term ‘genome’ we refer, strictly speaking, only to the set of ORFs which are covered by each particular expression experiment. In this sense, the ‘genomes’ covered by two different expression experiments might include different yeast ORFs, and therefore their composition (of a particular amino acid, for instance) might be different, though in practice these differences are generally very small.
Our complete results and additional information (such as genome and transcriptome compositions, and number of proteins per category) are available at http://bioinfo.mbb.yale.edu/genome/expression
RESULTS
Transcriptome composition of amino acids
One of the simplest attributes associated with a protein is its amino acid composition. The amino acid compositions of the genome and the transcriptome differ significantly for some amino acids (shown in Fig. 1a). The amino acids are ordered along the x-axis in the order of increasing transcriptome enrichment for the reference data set by Holstege et al. (9). Although the results vary between the different expression data sets, they all follow a general trend. Most notably, the composition of Ala increases by ~30–40% whereas the composition of Asn decreases by ~20%. The transcriptome is also significantly enriched in Gly and Val and the positively charged amino acids, Arg and Lys.
Figure 1.

Transcriptome enrichment of amino acids. (a) Amino acids are ordered along the x-axis according to the transcriptome enrichment found for the reference data set of Holstege et al. (9). Although the results vary between the different expression data sets, they all follow a general trend. Most notably, the composition of Ala increases by ~30–40% whereas the composition of Asn decreases by ~20%. The transcriptome is also significantly enriched in Gly and the positively charged amino acids, Arg and Lys. (b) Transcriptome enrichment calculated for the cDNA microarray expression data of the diauxic shift in yeast (5). The data from this experiment is primarily used for the measurement of expression level changes and we show the transcriptome enrichment only for purely illustrative purposes. Here we use the red fluorescence intensity minus the background intensity as measured by DeRisi et al. (5) as a crude approximation of the absolute expression level of a given ORF. We look at both time point 1 (fermentation) and time point 7 (respiration) of the experiment.
As mentioned previously, the data from cDNA microarrays, as given by ratios of red and green fluorescence intensities, is primarily used for the measurement of expression level changes. These data are less suitable for absolute expression level measurements. For purely illustrative purposes, we analyzed amino acid enrichment in the transcriptome using the red fluorescence intensity less the background intensity of the cDNA microarray data set as a crude approximation of the absolute expression level (Fig. 1b). Although the results for the enrichment of amino acid composition have a trend similar to that in Figure 1a, the magnitudes are much smaller (as expected). It can also be observed that there appears to be little difference in the amino acid composition of the transcriptome for different time points measured during the diauxic shift experiment, suggesting that even though the precise proteins that make up the transcriptome change in different conditions, the overall amino acid composition remains very similar. This is also suggested by the fact that there is little variance in transcriptome amino acid composition between DNA chip experiments in different conditions: i.e. between the different data sets of Roth et al. (10).
Relationship between gene length and expression level
Figure 2 shows the relationship between protein length (measured by the number of residues in the sequence) and expression level for the reference data set. It is obvious that there is no direct relationship between these two quantities. However, it seems that protein length is in some way an upper limit for the expression level of the corresponding gene. The straight line in Figure 2 represents the fit of a hyperbolic function through the maximum protein length at a given expression level. If the maximum protein length for a given expression level was inversely proportional to the expression level, the slope of this line would be equal to about –1. We find the slope to be about –0.7 for the data set of Holstege et al. (9). We find similar relationships for the other data sets (details in the legend to Fig. 2). The expression level of a short gene is dependent on the rate of transcription of RNA polymerase in relation to the rate of mRNA degradation. However, for a long gene, the overall rate of transcription might also be affected by the processivity of RNA polymerase: i.e. by the chance that the polymerase falls off.
Figure 2.


Dependence of expression level on gene length. We plotted protein length versus expression level for the reference data set of Holstege et al. (9) (for the other data sets, see http://bioinfo.mbb.yale.edu/genome/expression ). Each point on the graph represents one ORF and the axes of the graph are on a logarithmic scale. It is obvious that there is no strong positive or negative correlation between protein length and expression level (correlation coefficient is –0.16). However, it seems that protein length is related to the upper limit of the expression level possible for a given group of ORFs. A rough way to characterize this upper limit is to fit the hypberbolic function L = (K/E)A through the maximum protein lengths L (in units of amino acid residues) at given expression levels E (in units of transcripts per cell); K and A are constants. For the reference set of Holstege et al., parameter A was determined to be ~0.7 and K ~4.7 × 104. The following table lists the values for parameters A and K for all data sets. As can be seen in Figure 2 (especially on the left-hand side), the expression data is discrete, which makes the functional fit possible; this is due to the resolution limit of the experimental data [0.1 copies per cell for the data set of Holstege et al. (9)]. Different data discretizations affect the slope of the straight line somewhat (that is, parameter A), but the general trend can always be observed.
Transcriptome composition of membrane proteins
Another aspect of protein structure we analyzed was the occurrence of membrane proteins in the transcriptome. Membrane proteins are often classified in terms of the number of hydrophobic transmembrane (TM) helices they contain. We identified yeast ORFs coding for membrane proteins using a standard hydropathy scale and a sliding window, as described previously (20) (further details in the legend to Fig. 3a). Based on their most hydrophobic segment, we divided the predicted membrane proteins into ‘sure’ and ‘marginal’ candidates (using the MaxH approach also described in the legend) and then classified them further based on the number of TM helices they contain. Figure 3a shows how the composition of ORFs with ‘sure’ transmembrane regions changes from genome to transcriptome. For comparison we also show the relative enrichment of soluble proteins (for which no transmembrane region is predicted). The results show that, in general, helical membrane proteins are underrepresented in the transcriptome relative to the genome, whereas soluble proteins are enriched by ~22%. Furthermore, some classes of membrane proteins are more highly enriched than others: for instance, those with four TM helices are more enriched than those with one or two TM helices. However, for many of the membrane structure categories there is considerable variation between the different experiments.
Figure 3.
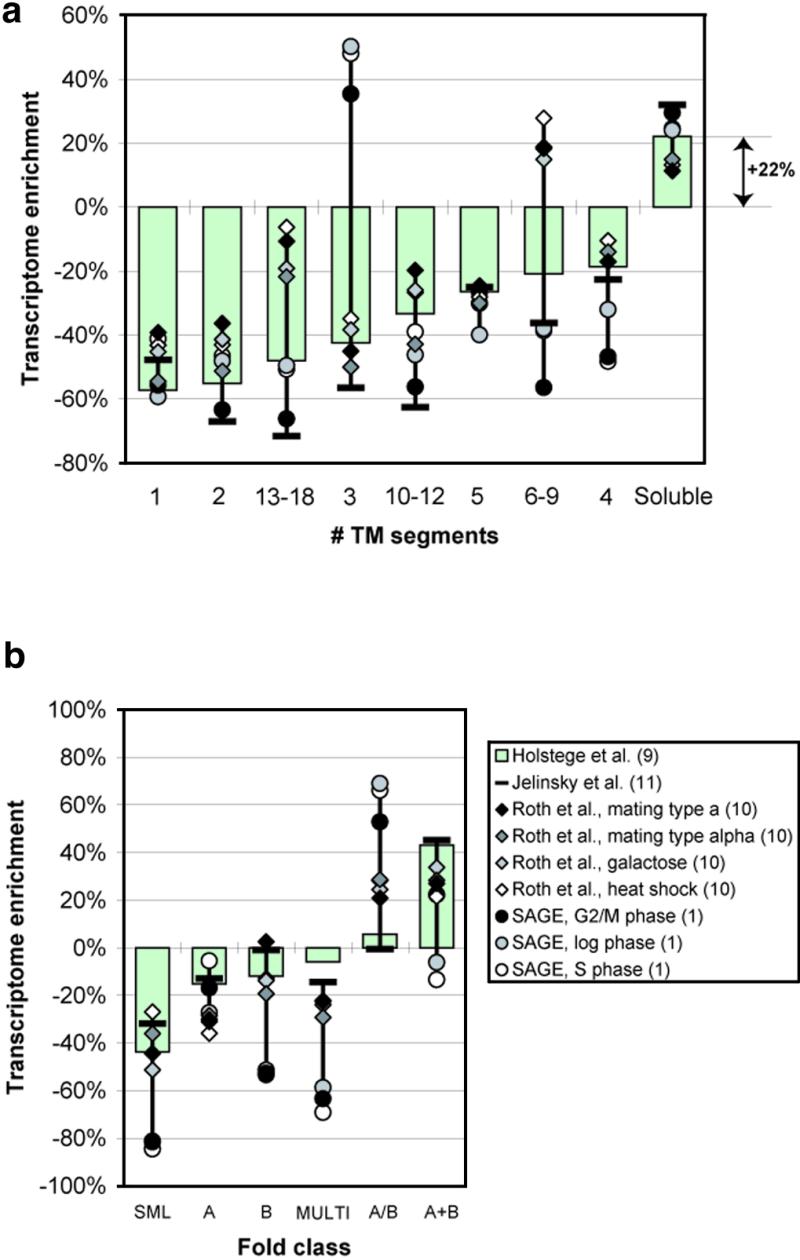
Transcriptome enrichment of structural classes. (a) Transcriptome enrichment of membrane proteins compared with soluble proteins. We identified yeast ORFs coding for membrane proteins using the GES hydrophobicity scale (33). The values from this scale in a window of size 20 (the typical size of a transmembrane helix) were averaged and then compared against a cut-off of –1 kcal/mol. A value under this cut-off was taken to indicate the existence of a transmembrane helix. Initial hydrophobic stretches corresponding to signal sequences for membrane insertion were excluded (these have the pattern of a charged residue within the first seven, followed by a stretch of 14 with an average hydrophobicity under the cut-off). These parameters have been used, tested and refined in surveys of membrane proteins in genomes (20,34–36). ‘Sure’ membrane proteins had at least one TM segment with an average hydrophobicity less than –2 kcal/mol. ‘Marginal’ membrane proteins had GES-identified TM helices but did not fulfil this ‘MinH’ criteria. This approach is similar to Boyd and Beckwith’s MaxH criteria (37) and to the approach of Klein et al. (38). (b) Transcriptome enrichment of soluble fold classes. The fold classes are sorted along the x-axis in the order of increasing transcriptome enrichment for the reference data set. To assign folds to the yeast genome, we followed a protocol similar to the one described previously, matching the PDB structure database against the yeast genome using both PSI-blast and FASTA (23,31,39–43). We used the following parameters in our PSI-blast searches: an inclusion threshold (h) of 10–5, the maximum number of iterations (j) of 10 and a final e-value cut-off of 10–4. These parameters are somewhat stricter than those used in previous PSI-blast analyses: e.g. our inclusion parameter is ~1/20 of that in Teichmann et al. (1998) (44) (who used 5 × 10–4 and j = 20); the inclusion parameter determines to which degree further homologs of a sequence are included at the next PSI-blast iteration. (A higher value leads to the inclusion of more sequences and greater coverage. However, an inclusion too high can lead to a corrupted profile and spurious matches.) We monitored our parameter settings by looking at how many domains were assigned to two different protein folds (obviously an erroneous assignment) and made sure this number was virtually nil. For the FASTA searches we used the usual e-value cut-off of 10–2 used in previous analyses (43).
Transcriptome composition of fold classes
In the previous section we compared the transcriptome enrichment of membrane and soluble proteins. Here we subdivide soluble proteins further according to their folds. To do this, we matched the PDB structure database (22) against the yeast genome using an iterative database search program (PSI-blast) (23) (see legend to Fig. 3b for more details on our fold assignment methods). Overall we found a total of 2305 domain level matches in 1710 distinct ORFs (~27% of the genome). We classified these structure matches into one of 344 folds using the structural classification of proteins (SCOP) (24). In addition, each fold is further grouped into one of six soluble protein classes: for instance, all α, all β, α/β, etc. (25).
For each domain match we looked at the expression level of the corresponding ORF. Figure 3b shows the relative differences of the composition of protein fold classes between genome and transcriptome. The fold classes are sorted along the x-axis in the order of increasing transcriptome enrichment for the reference data set. We observe an increase in the fraction of mixed α and β folds (α+β and α/β) while the fraction of the other fold classes decreases. It is also interesting to note that while the all α class is depleted in the transcriptome, the most helix-favoring amino acid, Ala (26), is greatly enriched (see Fig. 1a).
For fold class composition, the results for the SAGE experiments and the gene chip experiments differ significantly. This may be attributed to the substantially smaller number of ORFs covered by the SAGE experiments, which sample the structure matches in a somewhat biased fashion. Furthermore, the much greater enrichment of mixed helix and sheet structures in the SAGE experiments may, to some degree, result from the fact that these proteins tend to be longer (27) and the SAGE experiment is somewhat weighted towards longer proteins.
Top folds in the transcriptome
Figure 4 shows the 10 most highly expressed protein folds in yeast. Their exact fractions in the transcriptome are listed for the reference data set of Holstege et al. (9) and schematized with rankings for the other sets. The ranking of the most common folds in the transcriptome and the genome are very different. The most common transcriptome fold, by a large margin, is the TIM barrel (8 versus 5% for the second ranked fold), which is by contrast only ranked fifth in the genome. Many of the other common folds in the transcriptome also have a mixed α/β structure and are associated with enzymatic functions: for instance, the P-loop NTP hydrolase, ferrodoxin, Rossmann, thioredoxin and G3P dehydrogenase folds. In particular, the G3P dehydrogenase fold is greatly enriched in the transcriptome relative to the genome, increasing from 0.2 to 2.7%. Common folds in the genome that are depleted in the transcriptome include the protein kinase (catalytic core), long helix oligomers (the Leu-zipper fold) and the Zn2–C6 DNA binding domain. This makes sense since these folds act as ‘switches’ in signaling and transcription-factor functionality and thus do not need to be present in large quantities. In contrast, cytosolic enzymes are needed in bulk to ensure high throughput in synthetic and energy-producing pathways (see Fig. 5).
Figure 4.

The 10 most highly expressed protein folds in yeast. The folds are listed from top to bottom in the order of decreasing transcriptome composition for the reference data set of Holstege et al. (9). In the left half of the table we first list the protein fold, then its fold class and the identifier for a representative structure in the Protein Data Bank (PDB) (22). In the columns ‘genome’, ‘transcriptome’ and ‘transcriptome enrichment’ we list the genome and transcriptome compositions and the transcriptome enrichment of each fold, respectively. The right half of the table shows the rankings of each fold based on its transcriptome composition in the different expression data sets. For comparison we also show the ranking in the genome: i.e. based purely on the level of duplication within the genome. The genome compositions are calculated with respect to the ORFs for which expression levels in the reference data set exist. Their exact fractions in the transcriptome are listed for the reference data set and are schematized with rankings for the other sets. The ranking of the most common folds in the transcriptome and the genome are different. For instance, the most common transcriptome fold by a large margin (8 versus 5% for the 2nd ranked fold) is the TIM barrel, which is only ranked fifth in the genome. The second domain of this two-domain protein represents a G3P dehydrogenase-like fold.
Figure 5.

5. Transcriptome enrichment of MIPS categories. To analyze the transcriptome in terms of broad functional categories, we categorized the yeast ORFs using the functional categorization provided by MIPS (28–30). The functional categories are sorted along the x-axis in the order of increasing transcriptome enrichment for the reference data set.
The top-folds analysis is a relatively ‘fine-grained’ measurement, dividing the transcriptome into many categories, thus making the differences between the various experiments more apparent. Some of these differences may be explained by the different conditions probed by each experiment; others may reflect the natural variability of the experiments. However, in all cases the most common transcriptome fold that always remains is the TIM barrel. These fine-grained differences are also evident in the analysis of cDNA microarray data for the diauxic shift in yeast (5), which shows that the fold class composition does not change much over the time course of the experiment, but the ranking of the most common folds by expression level does (data not shown, related data in 19; absolute expression levels are approximated as explained in the legend to Fig. 1b).
It is well known that protein abundance can vary quite significantly for a given mRNA transcript abundance level. Recent large-scale studies suggest that there is only a weak linear relationship between mRNA and protein abundance for many genes, especially for weakly expressed genes (13). On the other hand, mRNA abundance is certainly still a better measure of protein abundance than genome content. From our results it seems clear that the distribution of folds in the cell’s proteins is very different from that in the genome complement.
Transcriptome composition of functions
To analyze the transcriptome in terms of broad functional categories, we used the functional categorization of the Munich Information Center for Protein Sequences (MIPS) (28–30), which divides proteins amongst a hierarchy of functional categories (for instance, ‘synthesis’, ‘metabolism’ etc. on the top level of the hierarchy).
Figure 5 shows the transcriptome enrichment of the various functional categories at the top level of the MIPS system. The functional categories are sorted along the x-axis in the order of increasing transcriptome enrichment for the reference data set. We observe an increase in the number of the proteins in the category ‘protein synthesis’ of ~200–500% depending on the data set. This is considerably larger than the change for the structural categories or simpler categories such as amino acid composition (5-fold versus 40%). The transcriptome is also notably enriched in proteins associated with energy production, cell structure and protein synthesis (most often ribosomal proteins). None of the other broad categories are as greatly depleted as these are enriched. However, it is worth noting that the depleted categories include transcription factors and signaling and transport proteins. Furthermore, the fraction of unclassified proteins in the transcriptome is lower than in the genome, perhaps because the more highly expressed genes are easier to study experimentally. There is also great variability between the different experiments; depending on the experiment, the most highly enriched MIPS category is different [for instance, the most highly enriched category is ‘protein synthesis’ for the reference data set by Holstege et al. (9), but ‘energy’ for some of the SAGE data sets].
DISCUSSION AND CONCLUSION
It is clear from our results that the structural and functional categories we investigated are differently distributed in the transcriptome and the genome. That is, the proteins of highly transcribed genes have on average different characteristics than the unweighted protein complement in the genome. There are variations between the different expression experiments, but we can observe some general trends in how structural and functional features occur in the transcriptome. In particular, we find that the transcriptome is enriched in Ala, Gly and, to a lesser extent, positively charged residues, soluble folds with combinations of helices and sheets, and proteins involved in protein synthesis (in particular ribosomal proteins), cell structure and energy production. Likewise, it is depleted in membrane proteins, transport, transcription and signaling proteins, very long proteins and those rich in Asn. Common sense, as well as a number of previous surveys, suggests that many of these structural and functional categories are interrelated (31,32). Thus, for instance, proteins involved with protein synthesis or energy production are often enzymes, which tend to be associated with α/β architectures. Likewise, membrane proteins tend to have less charged residues than soluble ones and also tend to have transport or signaling functions.
Looking at the variability of the transcriptome enrichment between experiments, it is particularly interesting to note that the greatest variability can be observed for the MIPS functional categories while the variability of amino acid composition is an order of magnitude lower. It seems that the usage of amino acids is very similar even when differential gene expression occurs to accommodate different functional tasks in the cell. This indicates that the cell might have to meet general requirements in its amino acid usage.
One requirement might be energy expenditure. In the metabolism of the yeast cell, Ala, which is the most enriched amino acid in the transcriptome, is synthesized directly in one step from pyruvate, a precursor of the TCA cycle. In contrast, Asn, the most depleted amino acid in the transcriptome, follows a more involved route. It is synthesized in two steps from oxaloacetate, the last component in the TCA cycle; in addition, the conversion of Asp to Asn involves conversion of ATP to AMP. This is the only step in amino acid biosynthesis in which two pyrophosphates are consumed at the same time. Thus, by strongly favoring Ala over Asn in highly expressed proteins, it seems that the cell has adapted to these energetic realities in the course of evolution. Further research could elaborate on this anecdotal evidence by looking comprehensively at the metabolic network in the cell.
In the context of Asn, it is also interesting to note that in some organisms (notably some archeons) Asn-tRNA is produced by an alternative pathway (transamidation) from Asp-tRNA (45). In the mitochondria of yeast, Gln-tRNA is synthesized by transamidation from Glu-tRNA; this might be related to the depletion of Gln in the yeast transcriptome.
It is worth emphasizing that this study uses mRNA abundance rather than protein abundance in the cell. It is to be hoped that techniques for large-scale protein abundance measurement will be developed that will provide us with a better view of the cellular machinery.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Hedi Hegyi for help with matching PDB structures to the yeast genome. M.G. would like to thank the Keck foundation and the NIH (grant 2P01GM54160-04) for support.
REFERENCES
- 1.Velculescu V.E., Zhang,L., Zhou,W., Vogelstein,J., Basrai,M.A., Bassett,D.E.,Jr, Hieter,P., Vogelstein,B. and Kinzler,K.W. (1997) Cell, 88, 243–251. [DOI] [PubMed] [Google Scholar]
- 2.Goffeau A., Barrell,B.G., Bussey,H., Davis,R.W., Dujon,B., Feldmann,H., Galibert,F., Hoheisel,J.D., Jacq,C., Johnston,M., Louis,E.J., Mewes,H.W., Murakami,Y., Philippsen,P., Tettelin,H. and Oliver,S.G. (1996) Science, 274, 546, 563–567. [DOI] [PubMed] [Google Scholar]
- 3.Schena M., Shalon,D., Davis,R.W. and Brown,P.O. (1995) Science, 270, 467–470. [DOI] [PubMed] [Google Scholar]
- 4.Shalon D., Smith,S.J. and Brown,P.O. (1996) Genome Res., 6, 639–645. [DOI] [PubMed] [Google Scholar]
- 5.DeRisi J.L., Iyer,V.R. and Brown,P.O. (1997) Science, 278, 680–686. [DOI] [PubMed] [Google Scholar]
- 6.Chu S., DeRisi,J., Eisen,M., Mulholland,J., Botstein,D., Brown,P.O. and Herskowitz,I. (1998) Science, 282, 699–705. [DOI] [PubMed] [Google Scholar]
- 7.Spellman P.T., Sherlock,G., Zhang,M.Q., Iyer,V.R., Anders,K., Eisen,M.B., Brown,P.O., Botstein,D. and Futcher,B. (1998) Mol. Biol. Cell, 9, 3273–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lockhart D.J., Dong,H., Byrne,M.C., Follettie,M.T., Gallo,M.V., Chee,M.S., Mittmann,M., Wang,C., Kobayashi,M., Horton,H. and Brown,E.L. (1996) Nat. Biotechnol., 14, 1675–1680. [DOI] [PubMed] [Google Scholar]
- 9.Holstege F.C., Jennings,E.G., Wyrick,J.J., Lee,T.I., Hengartner,C.J., Green,M.R., Golub,T.R., Lander,E.S. and Young,R.A. (1998) Cell, 95, 717–728. [DOI] [PubMed] [Google Scholar]
- 10.Roth F.P., Hughes,J.D., Estep,P.W. and Church,G.M. (1998) Nat. Biotechnol., 16, 939–945. [DOI] [PubMed] [Google Scholar]
- 11.Jelinsky S.A. and Samson,L.D. (1999) Proc. Natl Acad. Sci. USA, 96, 1486–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ross-Macdonald P., Sheehan,A., Roeder,G.S. and Snyder,M. (1997) Proc. Natl Acad. Sci. USA, 94, 190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gygi S.P., Rochon,Y., Franza,B.R. and Aebersold,R. (1999) Mol. Cell. Biol., 19, 1720–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michaels G.S., Carr,D.B., Askenazi,M., Fuhrman,S., Wen,X. and Somogyi,R. (1998) Proceedings of the Pacific Symposium of Biocomputing, 3, 42–53. [PubMed]
- 15.Eisen M.B., Spellman,P.T., Brown,P.O. and Botstein,D. (1998) Proc. Natl Acad. Sci. USA, 95, 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tamayo P., Slonim,D., Mesirov,J., Zhu,Q., Kitareewan,S., Dmitrovsky,E., Lander,E.S. and Golub,T.R. (1999) Proc. Natl Acad. Sci. USA, 96, 2907–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tavazoie S., Hughes,J.D., Campbell,M.J., Cho,R.J. and Church,G.M. (1999) Nat. Genet., 22, 281–285. [DOI] [PubMed] [Google Scholar]
- 18.van Helden J., Andre,B. and Collado-Vides,J. (1998) J. Mol. Biol., 281, 827–842. [DOI] [PubMed] [Google Scholar]
- 19.Brazma A., Jonassen,I., Vilo,J. and Ukkonen,E. (1998) Genome Res., 8, 1202–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerstein M. (1998) Proteins, 33, 518–534. [DOI] [PubMed] [Google Scholar]
- 21.Ermolaeva O., Rastogi,M., Pruitt,K.D., Schuler,G.D., Bittner,M.L., Chen,Y., Simon,R., Meltzer,P., Trent,J.M. and Boguski,M.S. (1998) Nat. Genet., 20, 19–23. [DOI] [PubMed] [Google Scholar]
- 22.Sussman J.L., Lin,D., Jiang,J., Manning,N.O., Prilusky,J., Ritter,O. and Abola,E.E. (1998) Acta Crystallogr. D, 54, 1078–1084. [DOI] [PubMed] [Google Scholar]
- 23.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murzin A.G., Brenner,S.E., Hubbard,T. and Chothia,C. (1995) J. Mol. Biol., 247, 536–540. [DOI] [PubMed] [Google Scholar]
- 25.Levitt M. and Chothia,C. (1976) Nature, 261, 552–558. [DOI] [PubMed] [Google Scholar]
- 26.Chakrabartty A., Kortemme,T. and Baldwin,R.L. (1994) Protein Sci., 3, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerstein M. (1998) Fold Des., 3, 497–512. [DOI] [PubMed] [Google Scholar]
- 28.Mewes H.W., Hani,J., Pfeiffer,F. and Frishman,D. (1998) Nucleic Acids Res., 26, 33–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frishman D., Heumann,K., Lesk,A. and Mewes,H.W. (1998) Bioinformatics, 14, 551–561. [DOI] [PubMed] [Google Scholar]
- 30.Mewes H.W., Heumann,K., Kaps,A., Mayer,K., Pfeiffer,F., Stocker,S. and Frishman,D. (1999) Nucleic Acids Res., 27, 44–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hegyi H. and Gerstein,M. (1999) J. Mol. Biol., 288, 147–164. [DOI] [PubMed] [Google Scholar]
- 32.Martin A.C., Orengo,C.A., Hutchinson,E.G., Jones,S., Karmirantzou,M., Laskowski,R.A., Mitchell,J.B., Taroni,C. and Thornton,J.M. (1998) Structure, 6, 875–884. [DOI] [PubMed] [Google Scholar]
- 33.Engelman D.M., Steitz,T.A. and Goldman,A. (1986) Annu. Rev. Biophys. Biophys. Chem., 15, 321–353. [DOI] [PubMed] [Google Scholar]
- 34.Arkin I., Brunger,A. and Engelman,D. (1997) Proteins, 28, 465–466. [DOI] [PubMed] [Google Scholar]
- 35.Wallin E. and von Heijne,G. (1998) Protein Sci., 7, 1029–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tomb J.-F., White,O., Kerlavage,A.R., Clayton,R.A., Sutton,G.G., Fleischmann,R.D., Ketchum,K.A., Klenk,H.P., Gill,S., Dougherty,B.A., Nelson,K., Quackenbush,J., Zhou,L., Kirkness,E.F., Peterson,S., Loftus,B., Richardson,D., Dodson,R., Khalak,H.G., Glodek,A., McKenney,K., Fitzegerald,L.M., Lee,N., Adams,M.D., Hickey,E.K., Berg,D.E., Gocayne,J.D., Utterback,T.R., Peterson,J.D., Kelley,J.M., Cotton,M.D., Weidman,J.M., Fujii,C., Bowman,C., Watthey,L., Wallin,E., Hayes,W.S., Borodovsky,M., Karpk,P.D., Smith,H.O., Fraser,C.M. and Venter,J.C. (1997) Nature, 388, 539–547. [DOI] [PubMed] [Google Scholar]
- 37.Boyd D., Schierle,C. and Beckwith,J. (1998) Protein Sci., 7, 201–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klein P., Kanehisa,M. and DeLisi,C. (1985) Biochim. Biophys. Acta, 815, 468–476. [DOI] [PubMed] [Google Scholar]
- 39.Lipman D.J. and Pearson,W.R. (1985) Science, 227, 1435–1441. [DOI] [PubMed] [Google Scholar]
- 40.Altschul S., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 41.Pearson W.R. (1997) Comput. Appl. Biosci., 13, 325–332. [DOI] [PubMed] [Google Scholar]
- 42.Pearson W.R. (1998) J. Mol. Biol., 276, 71–84. [DOI] [PubMed] [Google Scholar]
- 43.Gerstein M.G. (1997) J. Mol. Biol., 274, 562–576. [DOI] [PubMed] [Google Scholar]
- 44.Teichmann S., Park,J. and Chothia,C. (1998) Proc. Natl Acad. Sci. USA, 95, 14658–14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tumbula D., Vothknecht,U.C., Kim,H.S., Ibba,M., Min,B., Li,T., Pelaschier,J., Stathopoulos,C., Becker,H. and Soll,D. (1999) Genetics, 152, 1269–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]