

Abstract
Kynureninases (KYNases) are enzymes that play a key role in tryptophan catabolism through the degradation of intermediate kynurenine and 3′-hydroxy-kynurenine metabolites (KYN and OH-KYN, respectively). Bacterial KYNases exhibit high catalytic efficiency toward KYN and moderate activity toward OH-KYN, whereas animal KYNases are highly selective for OH-KYN, exhibiting only minimal activity toward the smaller KYN substrate. These differences reflect divergent pathways for KYN and OH-KYN utilization in the respective kingdoms. We examined the Homo sapiens and Pseudomonas fluorescens KYNases (HsKYNase and PfKYNase respectively) using pre-steady-state and hydrogen–deuterium exchange mass spectrometry (HDX-MS) methodologies. We discovered that the activity of HsKYNase critically depends on formation of hydrogen bonds with the hydroxyl group of OH-KYN to stabilize the entire active site and allow productive substrate turnover. With the preferred OH-KYN substrate, stabilization is observed at the substrate-binding site and the region surrounding the PLP cofactor. With the nonpreferred KYN substrate, less stabilization occurs, revealing a direct correlation with activity. This correlation holds true for PfKYNases; however there is only a modest stabilization at the substrate-binding site, suggesting that substrate discrimination is simply achieved by steric hindrance. We speculate that eukaryotic KYNases use dynamic mobility as a mechanism of substrate specificity to commit OH-KYN to nicotinamide synthesis and avoid futile hydrolysis of KYN. These findings have important ramifications for the engineering of HsKynase with high KYN activity as required for clinical applications in cancer immunotherapy. Our study shows how homologous enzymes with conserved active sites can use dynamics to discriminate between two highly similar substrates.
Graphical Abstract

INTRODUCTION
In both prokaryotes and eukaryotes, de novo production of quinolinate (and subsequently NAD) from l-tryptophan occurs through the kynurenine pathway.1,2 KYNases are key enzymes in this pathway. They belong to the aminotransferase superfamily and utilize pyridoxal-5′-phosphate (PLP) to catalyze the retro-Claisen hydrolysis of the Cβ–Cγ bond of l-kynurenine (KYN) or 3′-OH-l-kynurenine (OH-KYN) yielding either anthranilic acid (AA) or 3′-OH-anthranilic acid (OH-AA), respectively, followed by release of l-alanine (ALA).3 Animal KYNases, and more specifically the mammalian enzymes including HsKYNase, accept OH-KYN with >400-fold higher catalytic selectivity over the slightly smaller KYN substrate (Figure S1A,B, Table 1). In contrast, prokaryotic KYNases and more specifically PfKYNase have the inverse selectivity, hydrolyzing KYN with a ~500-fold higher kcat/KM compared to their mammalian counterparts (Figure S1C, Table 1).4 Similarly, the kcat/KM for PfKYNase hydrolysis of the larger OH-KYN substrate is decreased, but only by 50-fold compared to HsKYNase (Figure S1D, Table 1).
Table 1.
Steady-State Kinetic Parametersa of HsKYNase and PfKYNase for Both KYN and OH-KYN in PBS, pH 7.4 at 37 °C
| kynurenine | OH-kynurenine | |||||
|---|---|---|---|---|---|---|
| enzyme | kcat (s−1) | KM (μM) | kcat/KM (M−1S1−) | kcat (s−1) | KM (μM) | kcat/KM (M−1S1−) |
| HsKYNase | 0.18 ± 0.02 | 1200 ± 200 | (1.5 ± 0.4) × 102 | 1.7 ± 0.1 | 29 ± 5 | (6 ± 1.3) × 104 |
| PfKYNase | 7 ± 0.35 | 90 ± 15 | (7.7 ± 1.4) × 104 | (1.5 ± 0.25) × 103 | ||
The distinct catalytic selectivity of bacterial and animal enzymes is presumably dictated by the physiological roles of both KYN and OH-KYN and their respective reaction products, AA and OH-AA. In prokaryotes, AA is an important precursor for 2-heptyl-3-hydroxy-4-quinolone and for other virulence factors,5 whereas in mammals it has no known physiological role and is readily secreted in urine.6 By contrast, the expression of OH-KYN-selective KYNases in mammals has several major consequences. First and foremost, under certain conditions, the nonpreferred substrate KYN accumulates at high micromolarity concentrations within the cell and is secreted to the extracellular milieu where it exerts potent, immune-suppressive effects;7,8 this process is subverted in cancer and is particularly relevant to immunotherapy.9,10 We recently showed that the systemic depletion of KYN in mice following administration of bacterial enzymes with high KYN hydrolysis activity is capable of mediating potent activation of the immune system resulting in tumor eradication.11 Second, mammalian KYNases produce OH-AA, which has an important role in cellular redox control and which, together with downstream KYN pathway metabolites, exert numerous neurophysiological functions12 and are of major relevance to neurological diseases.13
On the basis of the aforementioned observations, it is challenging to explain how the HsKYNase enzyme is able to discriminate between substrates that only differ by a single hydroxyl group. Steric hindrance from the larger OH-KYN substrate could potentially explain the substrate preference of PfKYNase. However, the profound selectivity of HsKYNase toward the hydrolysis of OH-KYN relative to KYN is hard to rationalize a priori. To determine the underlying molecular mechanisms that confer substrate discrimination in HsKYNase and PfKYNase, we performed extensive pre-steady-state kinetic analyses for both enzymes and identified rate-limiting steps for their reactions with KYN and OH-KYN. In addition, we carried out hydrogen–deuterium exchange coupled to mass spectrometry (HDX-MS) experiments in the presence of either KYN or OH-KYN in order to assess the conformational dynamics of the enzymes during catalysis. Our data provide strong evidence that the conformational dynamics of the HsKYNase active site facilitates OH-KYN binding and hydrolysis while discriminating against productive KYN turnover. Our study addresses a mechanism by which enzymes that belong to the same family and characterized by a highly conserved active site allows for strict discrimination between two structurally, very similar substrates.
RESULTS AND DISCUSSION
Presteady-State Kinetic Characterization and Determination of Rate-Limiting Steps for the Reactions of HsKYNase and PfKYNase with KYN and OH-KYN.
Interestingly, while bacterial and mammalian enzymes, including HsKYNase and PfKYNase, are close structural homologues (1.2 Å2 RMSD) their amino acid sequences are highly divergent (26% identity and 45% homology; Figure S2A), suggesting that significant changes are required to discriminate between KYN and OH-KYN. Philips and co-workers demonstrated that three-residue motifs within the first shell of the active site help coordinate the side chain of either OH-KYN or KYN, namely, a His–Ser–Asn triplet (H102/S332/N333, HsKYNase numbering) in mammalian enzymes or a Trp–Gly–Thr at the respective positions (W64/G281/T282 in PfKYNase) in prokaryotic enzymes, respectively.14,15 Both the mammalian and prokaryotic enzymes are active as homodimers, with one chain contributing the triplet while the other chain completes the first shell of the active site including the essential PLP cofactor. Aside from the triplet, this first shell is conserved within the KYNase family. Introduction of the KYN-preferring motif into the human enzyme (i.e., H102W/S332G/N333T) resulted in a 1000-fold loss in catalytic activity toward OH-KYN, and while it improved the KM for KYN by 3.5 times, the kcat was reduced by 13 times, resulting in 4-fold lower catalytic efficiency.14
Previous mechanistic studies of KYN hydrolysis by PfKYNase revealed that the rate-limiting step is the release of the second product ALA.16 To elucidate the molecular basis for the distinct catalytic selectivity of the prokaryotic and mammalian KYNases, we conducted in-depth pre-steady-state kinetic analyses by means of stopped-flow fluorescence spectroscopy. We determined the rate of formation of the first product (OH-AA) by monitoring the fluorescence at 407 nm (Figure S3) separately as functions of enzyme and substrate concentration and analyzed the data by the KinTek Explorer program17,18 (Figure 1A and B). Upon mixing of HsKYNase with OH-KYN, a pronounced burst of OH-AA was observed, indicating that a step after the formation of OH-AA was rate-limiting at the steady state (steps 6 and/or 7 in Scheme S1 and Table S1; rate constant of ~20 s−1 for OH-AA formation and a rate of ~7.85 s−1 for OH-AA release are shown in Scheme 1A, Table S2, and Figure S4).19 The OH-AA release rate (~7.85 s−1) is almost 5-fold greater than the steady-state turnover value for OH-KYN by HsKYNase (kcat ~1.7 s−1, Table 1) indicating that ALA release is rate-limiting as is the case for PfKYNase with its preferred substrate KYN.
Figure 1.

Pre-steady-state kinetic analysis of substrate binding and processing under multiple turnover conditions. (A) Stopped-flow transients monitoring OH-AA formation after mixing HsKYNase with 500 μM OH-KYN. (B) OH-AA monitored by stopped-flow after mixing HsKYNase with different concentrations of OH-KYN. Simulated traces are shown as smooth curves. (C) Fluorescent traces of anthranilate formation monitored after mixing 25 μM HsKYNase with different concentrations of KYN as shown; the inset shows the rates as a function of KYN concentration (see SI). (D) Stopped-flow fluorescent trace of the AA burst after mixing 5 μM PfKYNase with 800 μM KYN. (E) Fluorescent traces of AA formation after mixing PfKYNase with different concentrations of KYN. (F) Stopped-flow fluorescent trace of the OH-AA formation after mixing 25 μM PfKYNase with 500 μM OH-KYN. In all plots, each trace represents the average of five different measurements.
Scheme 1.

Four-Step Minimal Kinetic Model for the Reaction of (A) HsKYNase with OH-KYN and (B) PfKYNase with KYNa
a“S” indicates the substrate in either case (OH-KYN and KYN), and “P” is the second product alanine (see SI Methods for more details for the data fitting process and the model development).
In contrast, when HsKYNase was mixed with its nonpreferred substrate, KYN, no burst was observed, but instead, AA formation and release were linear over time, following a lag phase during the first 20–30 ms (we note that KYN substrate inhibition was observed at high concentrations in the steady-state and was also prominent in the pre-steady-state phase at similar KYN concentrations). Importantly, the normalized rate of AA release (~0.06 s−1; Figure 1C) matches the observed steady-state rates (~0.08 s−1; Figure S1A). Thus, in contrast to the reaction with the preferred OH-KYN, the rate-determining step in the hydrolysis of KYN is not ALA release but rather an earlier step in the catalytic reaction leading to the formation of the first product, AA. The incorporation of the bacterial KYNpreferred motif on HsKYNase (H102W–I331C–S332G–N333T) did not alter the profile of AA formation but rather made the reaction ~50-fold slower as compared to the wildtype HsKYNase. This finding suggests that while first shell triplet motif residues contribute to substrate preference, there must be other processes that dictate the formation rate of AA.
In close analogy to the human enzyme, we found that, whereas the rate of hydrolysis of the preferred substrate (KYN) by PfKYNase is limited by ALA release as reported earlier16 (presteady-state burst trace and parameters are shown in Figure 1D and Table S1, respectively), with the nonpreferred OH-KYN substrate, AA formation was linear with a rate ~0.5 ± 0.03 s−1 (Figure 1F). Resolving the intrinsic rate constants20 up to AA release (Figure 1E, Scheme 1B, Table S3, Figure S5) revealed that the AA release rate of 12.5 s−1 is almost 2-fold faster than the steady-state rate (~7 s−1, Table 1), further supporting the hypothesis that ALA release is the rate-determining step. In short, both HsKYNase and PfKYNase are rate limited by ALA release with preferred substrates (free energy profile analysis of the reaction steps of the enzymes with their preferred substrates showed that substrate binding and chemical steps have similar free energies, suggesting that optimal substrate binding commits the reaction to proceeding forward; Figure S6), and conversely with nonpreferred substrates the reaction is rate-limited by the formation and release of the first product (either OH-AA or AA).
HDX-MS of HsKYNase and PfKYNase in the Presence of Either Their Preferred or Nonpreferred Substrate.
It is plausible that in the catalysis of OH-KYN by PfKYNase the hydroxyl group partially sterically hinders the reaction, thus resulting in OH-AA formation being rate limiting. However, it is less clear why HsKYNase hydrolysis of the smaller KYN substrate is 10-fold more impaired and why introduction of the H102W/S332G/N333T KYN preferring motif even more drastically reduces activity. To address this question, we performed HDX-MS analyses21–26 of Hs- and PfKYNase incubated with OH-KYN, KYN or buffer. About 200 overlapping peptides were monitored for each enzyme after 1, 10, and 100 min exposure to D2O (D; Tables S4 and S5). Kinetic rates measured in D (Figure S10, Table 2) indicate reactions with preferred substrates were under steady-state conditions for the first two time-points. The difference in average D uptake (ΔHDX) between substrate and no substrate conditions was calculated for each peptide, and significance was defined as ΔHDX > 0.3 Da and the p value as <0.01 (Figures 2 and 4). Boundaries of regions with significant change were determined based on overlapping peptides (Figure S7) and assuming complete back-exchange of the N-terminal residue of each peptide.
Table 2.
Steady-State Kinetic Parametersa of HsKYNase and PfKYNase for Both KYN and OH-KYN in D2O-PBS, pD 7.4 at 37 °C
| kynurenine | OH-kynurenine | |||||
|---|---|---|---|---|---|---|
| enzyme | kcat (s−1) | KM (μM) | kcat/KM (M−1 S−1) | kcat (s−1) | KM (μM) | kcat/KM (M−1 S−1) |
| HsKYNase | 0.06 ± 0.01 | 2500 ± 300 | 25 ± 6.7 | 0.52 ± 0.03 | 10 ± 2 | (5.2 ± 1.3) × 104 |
| PfKYNase | 2 ± 0.2 | 240 ± 30 | (8.5 ± 1.8) × 103 | (0.5 ± 0.1) × 103 | ||
Parameters are shown as the best fit value ± standard error upon fitting the experimental data to either the Michaelis–Menten equation (eq 1 in Supporting Information) or to a linear function (y = ax + b) to calculate kcat/KM.
Figure 2.

Conformational changes measured by HDX-MS of HsKYNase are larger with preferred OH-KYN (A,B) than with nonpreferred substrate KYN (C,D). (A, C) Volcano plots showing the average ΔHDX calculated by subtracting either OH-KYN (A) or KYN (C) from no substrate after 1 and 10 min of D exposure (1′, 10′). p values were calculated using a Welch’s t test. Significance cutoffs are p value <0.01 and an average ΔHDX > 0.3 Da. Significant peptides are shown in blue (1′) or black (10′). Boundaries of significant peptides are labeled on the plot or listed on the right underneath regions narrowed down based on overlapping peptides (Figure S7) and assuming complete back exchange of the N-terminal residue of each peptide. Two significant peptides are not listed in A (315–327 and 313–329) as they lie outside the active site. * indicates that the region is contributed by the “second” KYNase chain. (B, D) HsKYNase homodimer (PDB code: 3E9K) colored by the difference in fractional D uptake (%) between no substrate and OH-KYN (B) or KYN (C). Figure was prepared using DynamX per residue output without statistics and Pymol. Residues without coverage are shown in gray. PLP is shown in yellow sticks, and a 3-hydroxy hippuric competitive inhibitor is shown in green sticks.
Figure 4.
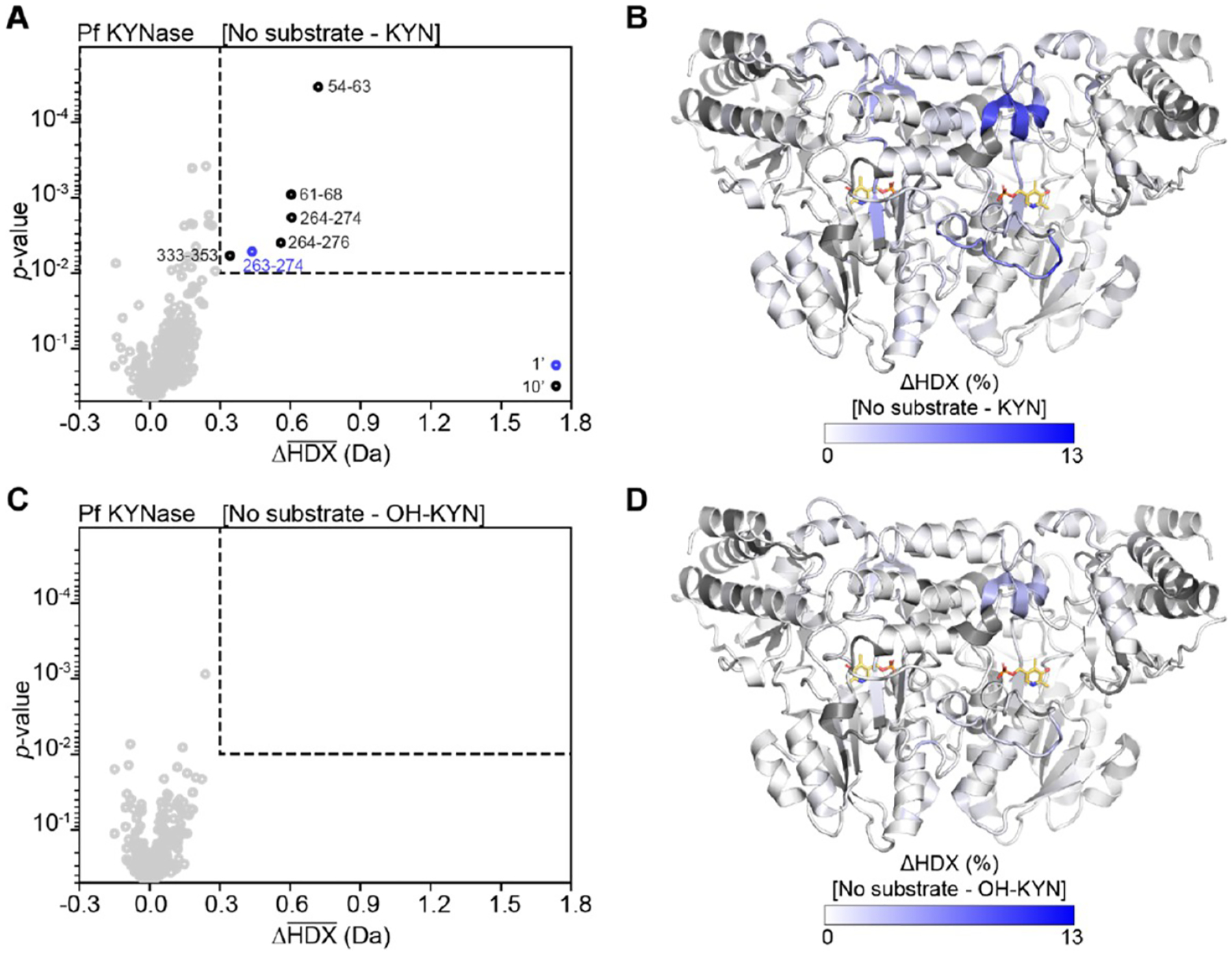
Conformational changes measured by HDX-MS of PfKYNase are larger with preferred KYN (A,B) than with nonpreferred substrate OH-KYN (C,D). (A,C) Volcano plots showing the average ΔHDX calculated by subtracting either KYN (A) or OH-KYN (C) from no substrate after 1 and 10 min D exposure (1′, 10′). p values were calculated using a Welch’s t test. Significance cutoffs are p value <0.01 and an average ΔHDX > 0.3 Da. Significant peptides are shown in blue (1′) or black (10′). Boundaries of significant peptides are labeled on the plot. In A, peptides 263–274, 264–274, and 264–276 lie outside the active site. (B,D) PfKYNase homodimer (PDB code: 1QZ9) colored by the difference in fractional D uptake (%) between no substrate and KYN (B) or OH-KYN (C). Figure was prepared using DynamX per residue output without statistics and Pymol. Residues without coverage are shown in gray; PLP is shown in yellow sticks.
For HsKYNase, the reaction with preferred OH-KYN caused a significant decrease in D uptake for residues Y100–I116 (Figure 2A,B). This was observed in five overlapping peptides and is consistent with substrate and product binding, as this region harbors the critical H102 of the first shell triplet thought to coordinate substrate via π-stacking interactions with its aromatic ring (Figure 3, e.g., peptide 100–112). A smaller and shorter-lived decrease in D uptake was induced by the nonpreferred KYN, and this reached significance in only one of the five overlapping peptides (Figure 2C,D). The dominant influence of substrate binding on D uptake has been observed for other enzymes.27,28 A similar trend is also observed for G304-T312, with OH-KYN causing a more pronounced decrease in D uptake than the nonpreferred KYN. Nine overlapping peptides were significant for OH-KYN, with only one peptide being significant for KYN. This region contains residues W305 and F306 that interact with the PLP cofactor (Figure 3, e.g., peptides 303–312). Reaction with either substrate thus causes a stabilization in the HsKYNase substrate-binding pocket and a region surrounding the PLP cofactor.
Figure 3.
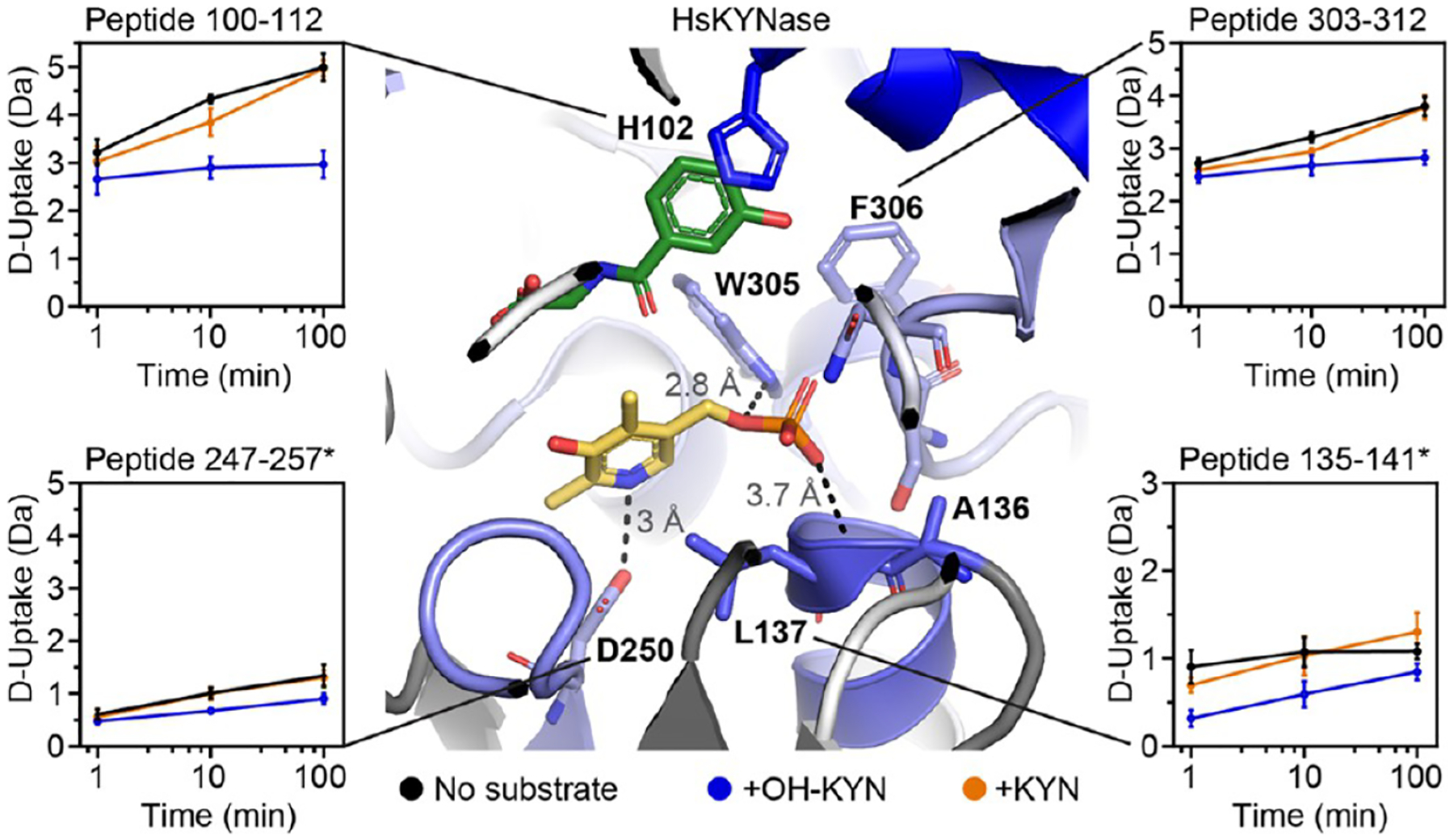
The substrate binding site and PLP-pocket of HsKYNase show altered D uptake in the presence of substrates. D-uptake plot traces are no substrate (black), OH-KYN (blue), or KYN (orange). The y axis range is 50% of the max D uptake assuming the N-terminal residue undergoes complete back-exchange. Data have not been corrected for back-exchange. * indicates the region is contributed by the “second” KYNase chain. Error bars are ±2σ from three technical replicates. The structure is colored as in Figure 2A with H102, A136, L137, D250, W305, F306, S332, and N333 shown as sticks. Image shows H102 side chain interacting with competitive inhibitor, and distance between PLP and the L137 backbone amide (3.7 Å), D250 side chain (3 Å), and W305 side chain (2.8 Å).
Two regions of HsKYNase showed a decrease in D uptake only in the reaction with OH-KYN (Figure 2A,B). These are A136-L137* and G248-L251*, which are starred as they come from the “second” KYNase chain that coordinates the internal aldimine. A136-L137* has altered D-uptake kinetics in the presence of either substrate but only a significant difference with the preferred OH-KYN. A136* and L137* have uncapped backbone amides at the end of an α-helix located near the PLP phosphate and the side-chain of S332, while G248–L251* contains D250* that directly coordinates the pyridine nitrogen of PLP (Figure 3, e.g., peptides 135–141 and 247–257, respectively). We only observed decreased D uptake for S332 and N333, the other two residues of the first shell triplet, at a late time point, where we cannot guarantee steady-state turnover events (Figure S8). Altogether, these data suggest that productive substrate turnover by HsKYNase upon formation of the external aldimine is strongly associated with stabilizing key interactions between PLP and the enzyme.
For PfKYNase, the reaction with preferred KYN similarly caused a decrease in D uptake around the aromatic residue of the inner shell triplet (W64) indicative of substrate binding. A significant decrease is observed in the single peptide containing W64 (peptide 61–68), as well as for the adjacent peptide 54–63 (Figure 4A,B). A smaller and shorter-lived decrease in D uptake was seen for the nonpreferred OH-KYN (Figure 4 peptide 61–68), although this did not pass our significance criteria (Figure 4C,D). We also did not see a difference in D-uptake around the other triplet residues G281 and T282 for either substrate, even when considering a late time point (Figure S9). Unlike HsKYNase, however, there were no significant decreases in D uptake around the cofactor upon reaction with preferred KYN. No difference was observed for the PLP-interacting residues (W256 and F257) or for regions homologous to HsKYNase A136–L137* and G248–L251* (Figure 5, e.g., PfKYNase peptides 250–262, 95–101*, and 198–209*, respectively). Overall, this suggests that the PfKYNase active site is a priori arranged for successful turnover once the substrate has bound. Hs- and PfKYNase clearly have distinct changes in dynamics upon substrate binding and turnover, potentially accounting for their distinct substrate preferences and rate-limiting steps.
Figure 5.
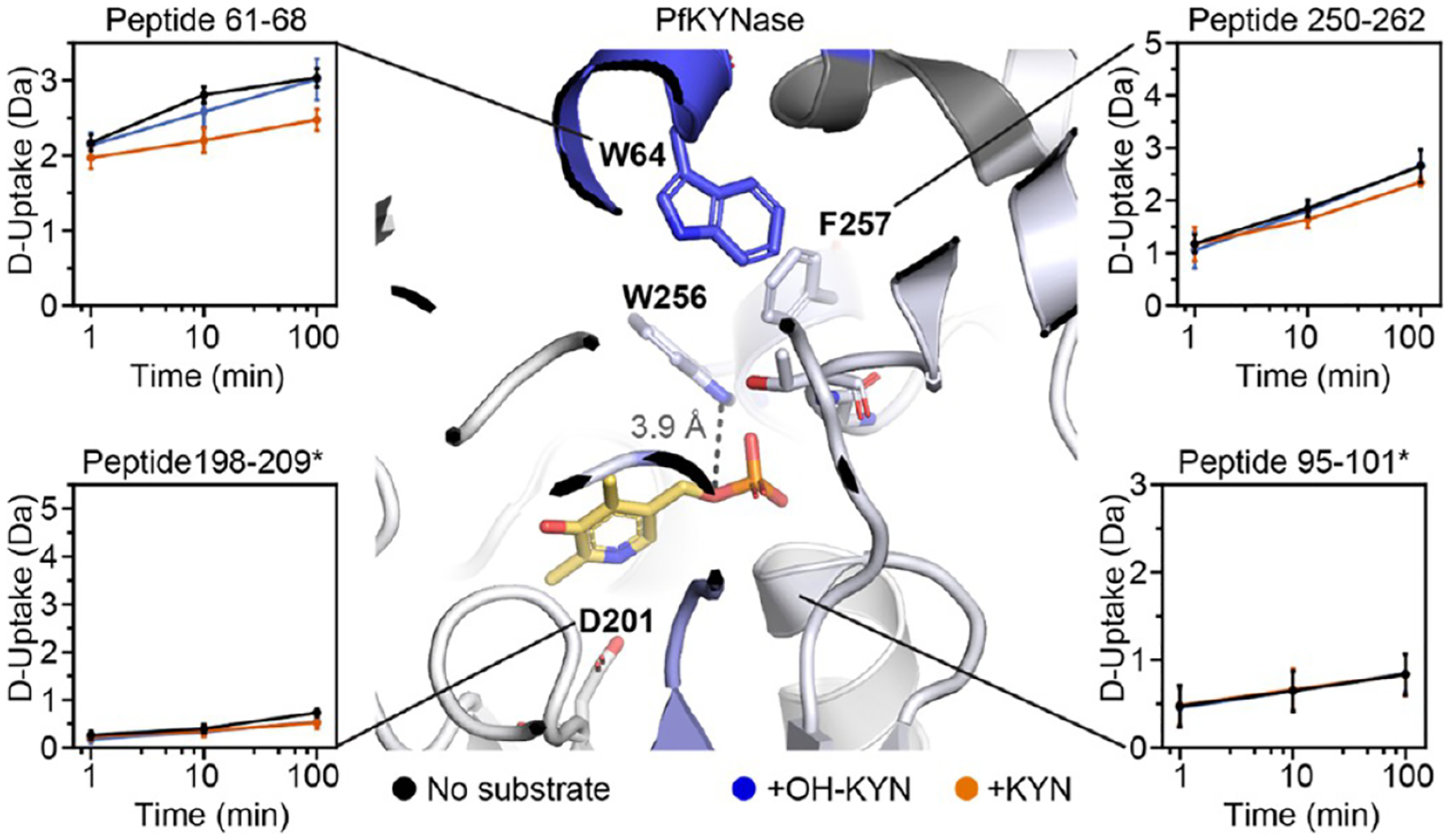
The substrate binding site (but not the PLP pocket) of PfKYNase shows altered D uptake in the presence of substrates. D-uptake plot traces are no substrate (black), OH-KYN (blue), or KYN (orange). The y axis range is 50% of max D uptake assuming the N-terminal residue undergoes complete back-exchange. Data have not been corrected for back-exchange. * indicates that the region is contributed by the “second” KYNase chain. Error bars are ±2σ from three technical replicates. The structure is colored as in Figure 4A with W64, D201, W256, F257, G281, and T282 shown as sticks. Image shows distance between PLP and the W256 side chain (3.9 Å).
CONCLUSIONS
We employed extensive pre-steady-state kinetics and HDX-MS experiments to elucidate why mammalian and prokaryotic KYNases have opposite catalytic selectivities for two physiologically relevant substrates that differ by only a hydroxyl group. Understanding the processes underpinning KYN catalytic specificity is important for the engineering of HsKYNase variants with high catalytic activity, as required for clinical applications in cancer immunotherapy.11 The active site of HsKYNase specifically accepts OH-KYN through formation of a hydrogen bond between the substrate hydroxyl and N333 in the active site. The HDX-MS analysis suggests that, upon formation of this critical hydrogen bond, the entire active site coordinating the external aldimine has stabilized hydrogen bonding that allows the subsequent chemical step and formation of reaction intermediates (manifest in the pre-steady-state OH-AA burst phase). In contrast, in the presence of KYN, there are no changes surrounding the PLP, showing a conformation similar to the quiescent enzyme, suggesting very inefficient formation of the external aldimine and, in turn, catalysis. Our HDX-MS analysis further reveals that PfKYNase has a more rigid and preorganized active site compared to HsKYNase with well-stabilized PLP-interacting residues prior to substrate binding, with OH-KYN discrimination occurring by steric hindrance. In fact, a number of studies have begun to reveal that for several other enzymes both catalysis and substrate specificity cannot be described by a classic “lock and key” mechanism but instead are controlled by conformational dynamics.29 Particularly, studies focusing on enzyme directed evolution and complemented by elegant molecular dynamics (MD) simulations further support the pivotal role of conformational dynamics in catalysis.30–34 In sum, we believe these findings demonstrate how mammalian KYNases commit OH-KYN hydrolysis products toward nicotinamide synthesis while avoiding futile hydrolysis of KYN and uncover a unique mechanism of substrate discrimination governed by conformational dynamics gated upon a single hydrogen bond.
EXPERIMENTAL PROCEDURES
Expression and Purification of Enzymes.
Detailed analysis of the expression and purification of both HsKYNase and PfKYNase is provided in the Supporting Information. Briefly, a codon-optimized gene of HsKYNase was expressed in the E. coli C43 strain using the pMAL c2X plasmid backbone (chloramphenicol resistant) with an N-terminal six-histidine tag (His6) to facilitate purification. The gene was cloned such that the produced protein was free of the maltose-binding protein. Due to very low expression yields of soluble HsKYNase in conventional shaking flasks (~2 mg/L), we developed a fed-batch fermentation protocol that yielded >1.5 g of soluble protein per liter of culture medium.
PfKYNase was expressed as an N-terminal His6 tagged protein using pET28 plasmid in BL21(DE3) E. coli strain and was purified by IMAC (Qiagen). The very high expression levels of PfKYNase in conventional shake flasks (>200 mg/L TB medium) allowed for one step IMAC purification, which was sufficient to yield a protein purity >95% as evidenced by SDS-PAGE analysis. A fraction of the IMAC-eluted proteins was subjected to size-exclusion chromatography using a Superdex 200 gel-filtration column attached to an Akta FPLC instrument to ensure the formation of dimers that represent the active oligomeric state of the enzyme. Ultimately, the purified enzymes were mixed with a 15% (v/v) final concentration of glycerol, flash frozen in liquid N2, and stored at −80 °C for future use.
Steady-State Kinetic Analysis of Kynureninase Enzymes.
The steady-state kinetic characterization of HsKYNase and PfKYNase for both substrates was performed either in H2O-PBS, pH 7.4, or in D2O-PBS, pD 7.4, at 37 °C using a BioTek Synergy H1 96-well microplate reader as described in detail in the Supporting Information.
Pre-Steady-State Stopped-Flow Fluorescence Spectroscopy of HsKYNase and PfKYNase.
The pre-steady-state formation of the first product of the reactions (anthranilate and OH-anthranilate) was monitored by means of stopped-flow fluorescence spectroscopy by capitalizing on the intrinsic fluorescent properties of these molecules. The exact excitation and emission wavelengths of AA and OH-AA were determined by recording the excitation and emission spectra for both compounds in PBS, pH 7.4, using a fluorospectrometer (Fluorescence System instrument from Photon Technology International). Subsequently, those wavelengths were used to follow the traces of either AA or OH-AA upon mixing the enzyme with the substrate in the stopped-flow instrument. Standard curves of fluorescence-vs-known concentrations of either AA or OH-AA served as a reference to quantify the amount of produced product during the actual enzymatic reactions and, thus, directly measure the rate of chemistry in the active site of the enzymes. Substrate concentration dependence experiments were performed for both HsKYNase and PfKYNase, and in the case of their preferred substrates, the data were fit to a four-step minimal kinetic model by numerical integration of the rate equations using the KinTek Explorer program. Thorough analysis of the pre-steady-state kinetic data, the global fitting process, and the model development, as well as additional information for all the different experimental steps, are provided in the Supporting Information.
Hydrogen–Deuterium Exchange Mass Spectrometry and Data Analysis.
Purified HsKYNase and PfKYNase were mixed with either their preferred, nonpreferred (under saturating conditions at 37 °C), or no substrate, and the labeling reactions were quenched with chilled 0.8% formic acid (v/v) at 1, 10, and 100 min for subsequent pepsin digestion using an Enzymate Pepsin Column (Waters, 300 Å, 5 μm, 2.1 × 30 mm for online digestion at 15 °C). The substrates were prepared in labeling buffer 1 × PBS in D2O, at a pD of 7.4. Following pepsin digestion and trapping, the peptides were desalted via reverse-phase trap (Waters Protein BEH C4 VanGuard Precolumn, 300 Å, 1.7 μm, 2.1 × 5 mm) and separated on a C18 column (Waters BEH C18 Column, 130 Å, 1.7 μm, 1 × 100 mm) for subsequent mass spectrometry analysis. The hydrogen–deuterium exchange data analysis was done by initially exporting the raw data to Protein Lynx Global Server 3.0.2 (Waters) for peptide identification, and finally the deuterium incorporation was analyzed by DynamX 3.0 (Waters). Peptides were manually curated in DynamX 3.0 after automated peptide assignment. More details for the liquid chromatography, mass spectrometry, hydrogen–deuterium exchange, and data analysis are provided in the Supporting Information.
Supplementary Material
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.0c00676.
Experimental details of protein expression and purification, steady-state kinetics, stopped-flow pre-steady-state fluorescence spectroscopy, model development and global fitting of pre-steady-state data, liquid chromatography, mass spectrometry, and hydrogen–deuterium exchange methods; Tables S1–S7, Scheme S1, and Figures S1–S10 (PDF)
PfKynase and HsKynase details (XLSX)
HDX Summary Table (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.0c00676
The authors declare the following competing financial interest(s): Kenneth A. Johnson is president of KinTek Corporation, which provided the stopped-flow instrument and KinTek Explorer software used in this study.
Contributor Information
Christos S. Karamitros, Department of Chemical Engineering, The University of Texas at Austin, Austin, Texas 78712, United States.
Kyle Murray, Department of Chemistry and Biochemistry, The University of Texas at Dallas, Richardson, Texas 75080, United States.
Yusuke Sugiyama, Department of Molecular Chemistry and Engineering, Graduate School of Science and Technology, Kyoto Institute of Technology, Kyoto, Japan.
Yoichi Kumada, Department of Molecular Chemistry and Engineering, Graduate School of Science and Technology, Kyoto Institute of Technology, Kyoto, Japan.
Kenneth A. Johnson, Department of Molecular Biosciences, The University of Texas at Austin, Austin, Texas 78712, United States
George Georgiou, Department of Chemical Engineering, Department of Molecular Biosciences, and Institute for Cellular and Molecular Biology, The University of Texas at Austin, Austin, Texas 78712, United States; Department of Oncology, University of Texas Dell Medical School, LiveSTRONG Cancer Institutes, Austin, Texas 78712, United States.
Sheena D’Arcy, Department of Chemistry and Biochemistry, The University of Texas at Dallas, Richardson, Texas 75080, United States.
Everett M. Stone, Department of Molecular Biosciences, The University of Texas at Austin, Austin, Texas 78712, United States Department of Oncology, University of Texas Dell Medical School, LiveSTRONG Cancer Institutes, Austin, Texas 78712, United States.
REFERENCES
- (1).Gazzaniga F, Stebbins R, Chang SZ, McPeek MA, and Brenner C (2009) Microbial NAD Metabolism: Lessons from Comparative Genomics. Microbiol. Mol. Biol. Rev 73 (3), 529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lima WC, Varani AM, and Menck CFM (2009) NAD Biosynthesis Evolution in Bacteria: Lateral Gene Transfer of Kynurenine Pathway in Xanthomonadales and Flavobacteriales. Mol. Biol. Evol 26 (2), 399–406. [DOI] [PubMed] [Google Scholar]
- (3).Phillips RS (2014) Structure and Mechanism of Kynureninase. Arch. Biochem. Biophys 544, 69–74. [DOI] [PubMed] [Google Scholar]
- (4).Koushik SV, Moore JA, Sundararaju B, and Phillips RS (1998) The Catalytic Mechanism of Kynureninase from Pseudomonas Fluorescens: Insights from the Effects of PH and Isotopic Substitution on Steady-State and Pre-Steady-State Kinetics. Biochemistry 37 (5), 1376–1382. [DOI] [PubMed] [Google Scholar]
- (5).Farrow JM, and Pesci EC (2007) Two Distinct Pathways Supply Anthranilate as a Precursor of the Pseudomonas Quinolone Signal. J. Bacteriol 189 (9), 3425–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Theofylaktopoulou D, Midttun Ø, Ulvik A, Ueland PM, Tell GS, Vollset SE, Nygård O, and Eussen SJP (2013) M. A Community-Based Study on Determinants of Circulating Markers of Cellular Immune Activation and Kynurenines: The Hordaland Health Study. Clin. Exp. Immunol 173 (1), 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Cervenka I, Agudelo LZ, and Ruas JL (2017) Kynurenines: Tryptophan’s Metabolites in Exercise, Inflammation, and Mental Health. Science 357 (6349), eaaf9794. [DOI] [PubMed] [Google Scholar]
- (8).Campesato LF, Budhu S, Tchaicha J, Pourpe S, Liu C, Manfredi MG, McGovern K, Wolchok JD, and Merghoub T (2018) Interaction between Kynurenine and the AhR Is an Effector Mechanism of Tumor Immunosuppression and Represents a Potential Immunotherapy Target. J. Immunol 200 (1), 177.5–177.5.29150567 [Google Scholar]
- (9).Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, and Prendergast GC (2005) Inhibition of Indoleamine 2,3-Dioxygenase, an Immunoregulatory Target of the Cancer Suppression Gene Bin1, Potentiates Cancer Chemotherapy. Nat. Med 11 (3), 312–319. [DOI] [PubMed] [Google Scholar]
- (10).Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, and Lou Y (2018) Next Generation of Immune Checkpoint Therapy in Cancer: New Developments and Challenges. J. Hematol. Oncol 11, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Triplett TA, Garrison KC, Marshall N, Donkor M, Blazeck J, Lamb C, Qerqez A, Dekker JD, Tanno Y, Lu W-C, Karamitros CS, Ford K, Tan B, Zhang XM, McGovern K, Coma S, Kumada Y, Yamany MS, Sentandreu E, Fromm G, Tiziani S, Schreiber TH, Manfredi M, Ehrlich LIR, Stone E, and Georgiou G (2018) Reversal of Indoleamine 2,3-Dioxygenase–Mediated Cancer Immune Suppression by Systemic Kynurenine Depletion with a Therapeutic Enzyme. Nat. Biotechnol 36 (8), 758–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Vécsei L, Szalárdy L, Fülöp F, and Toldi J (2013) Kynurenines in the CNS: Recent Advances and New Questions. Nat. Rev. Drug Discovery 12 (1), 64–82. [DOI] [PubMed] [Google Scholar]
- (13).Lovelace MD, Varney B, Sundaram G, Lennon MJ, Lim CK, Jacobs K, Guillemin GJ, and Brew BJ (2017) Recent Evidence for an Expanded Role of the Kynurenine Pathway of Tryptophan Metabolism in Neurological Diseases. Neuropharmacology 112 (B), 373–388. [DOI] [PubMed] [Google Scholar]
- (14).Lima S, Khristoforov R, Momany C, and Phillips RS (2007) Crystal Structure of Homo Sapiens Kynureninase. Biochemistry 46 (10), 2735–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Momany C, Levdikov V, Blagova L, Lima S, and Phillips RS (2004) Three-Dimensional Structure of Kynureninase from Pseudomonas Fluorescens. Biochemistry 43 (5), 1193–1203. [DOI] [PubMed] [Google Scholar]
- (16).Phillips RS, Sundararaju B, and Koushik SV (1998) The Catalytic Mechanism of Kynureninase from Pseudomonas Fluorescens: Evidence for Transient Quinonoid and Ketimine Intermediates from Rapid-Scanning Stopped-Flow Spectrophotometry. Biochemistry 37 (24), 8783–8789. [DOI] [PubMed] [Google Scholar]
- (17).Johnson KA (2009) Fitting Enzyme Kinetic Data with KinTek Global Kinetic Explorer. Methods Enzymol 467, 601–626. [DOI] [PubMed] [Google Scholar]
- (18).Johnson KA, Simpson ZB, and Blom T (2009) Global Kinetic Explorer: A New Computer Program for Dynamic Simulation and Fitting of Kinetic Data. Anal. Biochem 387 (1), 20–29. [DOI] [PubMed] [Google Scholar]
- (19).Johnson KA, Simpson ZB, and Blom T (2009) FitSpace Explorer: An Algorithm to Evaluate Multidimensional Parameter Space in Fitting Kinetic Data. Anal. Biochem 387 (1), 30–41. [DOI] [PubMed] [Google Scholar]
- (20).Johnson KA (1992) 1 Transient-State Kinetic Analysis of Enzyme Reaction Pathways, In The Enzymes (Sigman DS, Ed.), Vol. 20, pp 1–61, Academic Press, DOI: 10.1016/S1874-6047(08)60019-0. [DOI] [Google Scholar]
- (21).Englander SW (2006) Hydrogen Exchange and Mass Spectrometry: A Historical Perspective. J. Am. Soc. Mass Spectrom 17 (11), 1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Fast CS, Vahidi S, and Konermann L (2017) Changes in Enzyme Structural Dynamics Studied by Hydrogen Exchange-Mass Spectrometry: Ligand Binding Effects or Catalytically Relevant Motions? Anal. Chem 89 (24), 13326–13333. [DOI] [PubMed] [Google Scholar]
- (23).Hodge EA, Benhaim MA, and Lee KK (2020) Bridging Protein Structure, Dynamics, and Function Using Hydrogen/Deuterium-Exchange Mass Spectrometry. Protein Sci 29 (4), 843–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zheng J, Strutzenberg T, Pascal BD, and Griffin PR (2019) Protein Dynamics and Conformational Changes Explored by Hydrogen/Deuterium Exchange Mass Spectrometry. Curr. Opin. Struct. Biol 58, 305–313. [DOI] [PubMed] [Google Scholar]
- (25).Oganesyan I, Lento C, and Wilson DJ (2018) Contemporary Hydrogen Deuterium Exchange Mass Spectrometry. Methods 144, 27–42. [DOI] [PubMed] [Google Scholar]
- (26).Masson GR, Jenkins ML, and Burke JE (2017) An Overview of Hydrogen Deuterium Exchange Mass Spectrometry (HDX-MS) in Drug Discovery. Expert Opin. Drug Discovery 12 (10), 981–994. [DOI] [PubMed] [Google Scholar]
- (27).Sowole MA, Simpson S, Skovpen YV, Palmer DRJ, and Konermann L (2016) Evidence of Allosteric Enzyme Regulation via Changes in Conformational Dynamics: A Hydrogen/Deuterium Exchange Investigation of Dihydrodipicolinate Synthase. Biochemistry 55 (38), 5413–5422. [DOI] [PubMed] [Google Scholar]
- (28).Konermann L, Pan J, and Liu Y-H (2011) Hydrogen Exchange Mass Spectrometry for Studying Protein Structure and Dynamics. Chem. Soc. Rev 40 (3), 1224–1234. [DOI] [PubMed] [Google Scholar]
- (29).Rabe von Pappenheim F, Aldeghi M, Shome B, Begley T, de Groot BL, and Tittmann K (2020) Structural Basis for Antibiotic Action of the B 1 Antivitamin 2′-Methoxy-Thiamine. Nat. Chem. Biol 16 (11), 1237–1245. [DOI] [PubMed] [Google Scholar]
- (30).Pabis A, Risso VA, Sanchez-Ruiz JM, and Kamerlin SC (2018) Cooperativity and Flexibility in Enzyme Evolution. Curr. Opin. Struct. Biol 48, 83–92. [DOI] [PubMed] [Google Scholar]
- (31).Dusan P, and Shina Caroline Lynn K (2018) Molecular Modeling of Conformational Dynamics and Its Role in Enzyme Evolution. Curr. Opin. Struct. Biol 52, 50–57. [DOI] [PubMed] [Google Scholar]
- (32).Gobeil SMC, Ebert MCCJC, Park J, Gagné D, Doucet N, Berghuis AM, Pleiss J, and Pelletier JN (2019) The Structural Dynamics of Engineered β-Lactamases Vary Broadly on Three Timescales yet Sustain Native Function. Sci. Rep 9 (1), 6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Maria-Solano MA, Serrano-Hervás E, Romero-Rivera A, Iglesias-Fernández J, and Osuna S (2018) Role of Conformational Dynamics in the Evolution of Novel Enzyme Function. Chem. Commun 54 (50), 6622–6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Otten R, Liu L, Kenner LR, Clarkson MW, Mavor D, Tawfik DS, Kern D, and Fraser JS (2018) Rescue of Conformational Dynamics in Enzyme Catalysis by Directed Evolution. Nat. Commun 9 (1), 1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.