


Abstract
Thymidylate synthase (TS) functions as an RNA-binding protein by interacting with two different sequences on its own mRNA. One site is located in the 5′-upstream region of human TS mRNA while the second site is located within the protein coding region corresponding to nt 434–634. In this paper, a 70 nt RNA sequence, corresponding to nt 480–550, was identified that binds TS protein with an affinity similar to that of full-length TS mRNA and TS434–634 RNA. In vitro translation studies confirmed that this sequence is critical for the translational autoregulatory effects of TS. To document in vivo biological significance, TS sequences contained within this region were cloned onto the 5′-end of a luciferase reporter plasmid and transient transfection experiments were performed using H630 human colon cancer cells. In cells transfected with p644/TS434–634 or p644/TS480–550, luciferase activity was decreased 2.5-fold when compared to cells transfected with p644 plasmid alone. Luciferase mRNA levels were identical for each of these conditions as determined by RNase protection and RT–PCR analysis. Immunoprecipitation of TS ribonucleoprotein complexes revealed a direct interaction between TS protein and TS480–550 RNA in transfected H630 cells. Treatment with 5-fluorouridine resulted in a nearly 2-fold increase in luciferase activity only in cells transfected with p644/TS434–634 and p644/TS480–550. This study identifies a 70 nt TS response element in the protein coding region of TS mRNA with in vitro and in vivo translational regulatory activity.
INTRODUCTION
Thymidylate synthase (TS) catalyzes the conversion of 2′-deoxyuridine-5′-monophosphate (dUMP) and 5,10-methylenetetrahydrofolate (CH2THF) to 2′-deoxythymidine-5′-monophosphate (dTMP) and dihydrofolate (1). Because this reaction is the sole intracellular de novo source of dTMP, TS is essential for regulating the balanced supply of the four DNA precursors required for DNA replication. For this reason, significant efforts have focused on TS as an important target enzyme in cancer chemotherapy in the design and development of novel inhibitor compounds (2,3).
In addition to its role in enzyme catalysis, recent studies have shown that TS functions as an RNA-binding protein (4–8). Specifically, translation of human TS mRNA is negatively regulated by direct binding of TS to two different cis-acting elements on its cognate mRNA (4,5). The first element is a 36 nt sequence contained within the 5′-untranslated region (UTR) and includes the translational start site in a putative stem–loop structure. The second site is encompassed within a 200 nt sequence in the protein coding region corresponding to nt 434–634 (5). Subsequent work has revealed that binding of TS protein to either of these two sites is determined by several cellular factors. Treatment of TS with either of the nucleotides dUMP or FdUMP, the reduced folate CH2THF and/or antifolate analog inhibitors of TS, such as ZD1694, impairs RNA binding in a dose-dependent manner (9). In addition to the effects of ligand binding, the RNA-binding activity of TS is exquisitely sensitive to the redox state of the protein (9). In its maximally reduced state, TS interacts with high affinity with its target mRNA. In contrast, in the presence of an oxidizing agent, such as diamide, the RNA-binding activity of TS protein is significantly diminished. This marked dependence on cellular redox state suggests that certain cysteine sulfhydryl groups on TS may play a key role in mediating the process of RNA binding.
Previous studies have focused on characterizing the determinants of binding by TS for the 5′-upstream site on TS mRNA (5). In the present study, we performed an in vitro and in vivo analysis of the cis-acting element contained within the protein coding region. Using a series of in vitro RNA gel shift assays, in vitro translation assays and in vivo transient transfection experiments, we have identified a 70 nt sequence corresponding to TS480–550 that functions as a translational regulatory element. In addition, we present evidence that this sequence is able to form a ribonucleoprotein (RNP) complex with TS in intact human colon cancer H630 cells.
MATERIALS AND METHODS
Cell culture
The characteristics of the human colon cancer H630 cell line have been previously described (10). Cell lines were grown in 75 cm2 plastic tissue culture flasks in growth medium consisting of RPMI 1640 with 10% dialyzed fetal bovine serum (Gibco BRL, Grand Island, NY).
The human colon cancer cell line HCT-C18 is a subline of the HCT-C parent cell line that expresses an inactive human TS protein with a Ser216→Leu mutation at amino acid position 216 of human TS. This cell line was a kind gift from Dr Sondra Berger (University of South Carolina, Columbia, SC) (11). HCT-C18 cells were maintained in RPMI 1640 medium containing 10% dialyzed fetal bovine serum and supplemented with 10 µM thymidine. The HCT-C:His-TS(+) cell line was established by stable transfection of HCT-C18 cells with human His-tagged TS cDNA as previously described (12).
Synthesis of plasmid constructs
The luciferase reporter plasmid p644 was constructed by inserting an EGR-1 (early growth response gene) promoter at the BglII restriction site of plasmid pGL2-basic (Promega, Madison, WI). Plasmid pcEHTS containing the full-length TS cDNA was used as template for PCR amplification of various TS fragments. Sequences corresponding to TS434–634, TS434–534, TS534–634, TS480–580, TS480–550 and TS1024–1240 were PCR amplified, digested with HindIII and cloned into the HindIII restriction site of p644 to generate heterologous luciferase reporter constructs (Fig. 1). The TS480–550 sequence was PCR amplified, digested with StyI and then cloned into the StyI restriction site of p644 (Fig. 1). The sequence and orientation of the cloned TS inserts were confirmed by direct sequencing analysis. All of the inserts were documented to be in-frame with the luciferase gene.
Figure 1.


(A) TS cDNA sequence. (B) Structure of heterologous luciferase plasmids. Details of construction of the luciferase heterologous plasmids are presented in Materials and Methods. The solid lines represent sequences of the pGL-2 basic plasmid. The solid box indicates the EGR-1 promoter and the hatched box represents the protein coding region of the luciferase gene. The open box represents the TS RNA sequence cloned into the HindIII restriction site of the p644 recombinant plasmid.
All primers were synthesized on an Applied Biosystems model 391 DNA synthesizer. The oligonucleotide sequences were as follows (underlined bases represent the HindIII restriction enzyme site; italic bases represent the StyI restriction enzyme site): TS434–454 (sense), 5′-AAG CTT CCC GAG ACT TTT TGG ACA GCC T-3′; TS534–515 (antisense), 5′-AAG CTT CTG TAT TCT GCC CCA AAA TGC-3′; TS534–553 (sense), 5′-AAG CTT AGA TAT GGA ATC AGA TTA TTC-3′; TS480–500 (sense), 5′-AAG CTT GGA CTT GGG CCC AGT TTA TGG-3′; TS580–560 (antisense), 5′-AAG CTT AAA CGT CAA CCA GTT GAG GGA-3′; TS550–530 (antisense), 5′-AAG CTT AAT CTG ATT CCA TAT CTC TGT-3′; TS634–614 (antisense), 5′-AAG CTT CGC ACA TGA TGA TTC TTCTGTCGT-3′; TS480–504 (sense), 5′-CCA AGG GGA CTT GGG CCC AGT TTA TGG CTT C-3′; TS550–532 (antisense) 5′-CCA AGG AAT CTG ATT CCA TAT CTC TGT AT-3′.
A mutant human TS cDNA plasmid pcDNA3.1(+):TS(m) was designed by a PCR-mediated method and 21 conserved mutations were placed within the protein coding region TS480–550. These mutations were termed conserved as they did not alter the encoded protein sequence.
The plasmid pcDNA3.1(+):TS(m) was digested with BglII and AvaI restriction enzymes and the resulting 214 nt DNA fragment, which includes the 21 conserved mutation sequence within the protein coding region, was cloned into pcEHTS containing the full-length human TS cDNA, to give pcEHTS(m).
The oligonucleotide sequences were as follows (underlined bases represent the NarI, HindIII and XbaI restriction enzyme sites; bold italic bases represent mutant nucleotides):
pcDNA3.1(+):TS910–930 (sense), 5′-AAG CTT GGT ACC GAG CTC GG-3′; pcDNA3.1(+):TS1940–1920 (antisense), 5′-TCT AGA CTC GAG CGG CCG-3′; TS520–571(m) (sense), 5′-GGC GCC GAG TAT AGG GAC ATG GAG TCT GAC TAT TCA GGA CAG GGA GTT GAC C-3′; TS520–458 (m) (antisense), 5′-GGC GCC GAA GTG TCT CCA TTG AAA TCC GTA GAC AGG TCC TAA ATC CCC TTC TTC TCT GGT GGA GAA TC-3′.
In vitro mRNA transcription
A luciferase cDNA sequence corresponding to nt 446–675 (luc446–675) was PCR amplified using the pGEM-luc plasmid (Promega) as DNA template. The sequences of the primers were as follows (underlined bases represent the T7 promoter/primer sequence): luc (antisense), 5′-TAA TAC GAC TCA CTA TAG GGTTG CCA GGC TTA GGT CTA AAC-3′; luc (sense), 5′-GGA CCT CAG TTT GAC GCC AAC-3′.
A 32P-radiolabeled antisense luciferase RNA probe was synthesized by in vitro transcription using luc446–675 as template and T7 RNA polymerase. The 32P-radiolabeled β-actin antisense RNA probe was synthesized in vitro using the human pTR1-β-actin transcription template (Ambion, Austin, TX) and T7 RNA polymerase. Each radiolabeled antisense RNA probe was further purified on a denaturing 5% polyacrylamide–8 M urea gel.
Full-length TS mRNA (TS1–1524) and mutant full-length TS mRNA (TS1–1524m) were synthesized in vitro with SP6 RNA polymerase after linearization of the respective pcEHTS and pcEHTS(m) plasmids with HindIII. A truncated TS mRNA without the complete 5′-UTR and nearly all of the 3′-UTR (TS94–1053) and a corresponding truncated mutant TS RNA (TS94–1053m) were synthesized in vitro with T7 RNA polymerase using linearized plasmids pcDNA3.1(+):TS and pcDNA3.1(+):TS(m) as their respective templates.
TS cDNA sequences corresponding to TS434–634, TS434–534, TS534–634, TS480–580 and TS480–550 were PCR amplified using pcEHTS as DNA template. The mutant TS480–550(m) was PCR amplified using pcDNA3.1(+):TS(m) as template. The primers used for this PCR amplification reaction were as follows (underlined bases indicate the T7 promoter/primer sequence): TS-1(sense), 5′-TAA TAC GAC TCA CTA TAG GG AAG CTT CCC GAG ACT TTT TGG ACA GCC T-3′; TS-2 (antisense), 5′-CTG TAT TCT GCC CCA AAA TGC-3′; TS-3 (sense), 5′-TAA TAC GAC TCA CTA TAG GG AGA TAT GGA ATC AGA TTA TTC-3′; TS-4 (antisense), 5′-CGC ACA TGA TGA TTC TTC TGT CGT-3′; TS-5 (sense), 5′-TAA TAC GAC TCA CTA TAG GG GGA CTT GGG CCC AGT TTA TGG-3′; TS-6 (antisense), 5′-AAA CGT CAA CCA GTT GAG GGA-3′; TS-7 (antisense), 5′-AAT CTG ATT CCA TAT CTC TGT-3′; TS(m)-1 (sense), 5′-TAA TAC GAC TCA CTA TAG GGA TTT AGG ACC TGT CTA CGG-3′; TS(m)-2, (antisense), 5′-ACA GTC TGA GGT ACA GGG ATA-3′.
All RNA transcripts were synthesized in vitro with T7 RNA polymerase and the reagents included in the Ambion in vitro transcription kit (no. 13349; Ambion, Austin, TX). Once synthesized, the RNAs were resolved on a 5% polyacrylamide–8 M urea gel, excised after visualization by UV shadowing and gel purified. The concentration of RNA was quantitated by UV spectrometry.
RNase protection assay
Equal amounts of total RNA (10 µg) from each sample were used in the RNase protection assay according to the Ambion protocol (no. 1412). In brief, total RNA was incubated with 40 µg of yeast tRNA and with a molar excess of either radiolabeled luciferase antisense or β-actin antisense RNA probe (specific activity 2 × 108 c.p.m./µg, 2 × 105 c.p.m./reaction). The reaction sample was ethanol precipitated and resuspended in 10 µl HybSpeed hybridization buffer. All samples were heat denatured at 95°C for 3 min and then incubated at 68°C for 10 min. After digestion with an RNase A/T1 mix for 30 min at 37°C, samples were ethanol precipitated and resolved on a denaturing 5% polyacrylamide–8 M urea gel.
In vitro translation
Translation reactions were performed using a rabbit reticulocyte lysate system (Promega) as previously outlined (4). Reaction mixtures (final volume 20 µl) containing rabbit reticulocyte lysate (14 µl), amino acids without methionine (0.4 µl), RNase inhibitor (0.4 µl) and 12 µCi [35S]methionine were incubated with 0.4 pmol of the respective RNA transcripts at 30°C for 60 min. The translation products were analyzed by SDS–PAGE (15% acrylamide) and visualized by autoradiography.
Purification of human TS protein
Human recombinant TS protein was partially purified by ammonium sulfate fractionation (30–60%) followed by affinity chromotography with an Affi-Gel Blue Sepharose (Bio-Rad) column, according to a modification of previously described methods (13,14). The specific activity of this partially pure TS was 0.05 U/mg. The protein was further purified using anion exchange chromatography with a DE-52 column (Whatman International, UK) and the specific activity of the DE-52-purified protein was 0.08 U/mg. Human recombinant His-tagged TS protein (His-Tag-hTS) was purified as previously described (15).
RNA gel mobility shift assay
The RNA gel mobility shift assay was performed as previously described (4,5,16). Competition experiments were performed with 200 ng of recombinant human TS protein and 1 ng of 32P-radiolabeled TS RNA (1 × 105 c.p.m.). These conditions were selected based on experiments using a fixed amount of radiolabeled TS mRNA with increasing concentrations of TS protein to determine linearity of binding. Unlabeled TS RNAs including TS434–634, TS480–580, TS480–550, TS1040–1240 and full-length TS mRNA (TS1–1524), at concentrations of 0- to 100-fold molar excess, were then added to the reaction mixture prior to addition of TS protein. The relative binding affinities (IC50) of TS RNA sequences for TS protein were determined in terms of the concentration of unlabeled RNA at which specific binding of 32P-radiolabeled full-length TS mRNA was inhibited by 50%. Quantitation was performed using a Hewlett Packard ScanJet 4P Plus scanner and NIH Image 1.51 software.
Transient transfection and luciferase assay
In order to determine the effect of TS RNA sequences on expression of the heterologous luciferase construct, transient transfection experiments were performed as described by Felgner et al. (17). Briefly, cells were plated at 2 × 105 cells/ 60 mm dish and grown to ~50% confluence. In each transfection experiment, cells were incubated with 5 µg of plasmid DNA and 10 µg lipofectin (Gibco BRL). Cells were also incubated with 0.05 µg of pRL-SV40 plasmid DNA (Promega), which encodes renilla (sea pansy) luciferase. The expression of renilla luciferase served as an internal control for transfection efficiency. After incubation at 37°C for 24 h, cells were washed twice with serum-free RPMI 1640 medium, then incubated in 2 ml RPMI 1640 containing 10% dialyzed fetal bovine serum for an additional 48 h at 37°C. Cells were harvested using reagents from the Promega dual luciferase assay kit.
To examine the effect of 5-fluorouridine (5-FU) on luciferase activity, human colon cancer H630 cells were transiently transfected, as described above, with p644, p644/TS434–634, p644/TS480–550 and p644/TS1040–1240 and incubated for 24 h at 37°C. Transfected cells were then treated with 1 µM 5-FU for an additional 24 h and harvested as described.
Isolation of total cellular RNA
Human colon cancer cells were washed three times with ice-cold phosphate-buffered saline (PBS) and harvested from 75 cm2 tissue culture flasks with a rubber policeman. Total cellular RNA was extracted according to the method of Sacchi and Chomczynski (18). All RNAs were resolved on a 1% agarose–formaldehyde gel to verify their integrity and the RNA concentration was quantitated by UV spectrometry.
Whole cell extraction and immunoprecipitation
Whole cell extracts were prepared and immunoprecipitation of TS RNP complexes was performed as described previously (6,19). In brief, whole cell extracts from ~5 × 107 cells were first cleared of non-specific binding material by incubation with 300 µl protein A–agarose (Gibco BRL) for 30 min on ice and then centrifuged. The cleared cell extract was then incubated with an anti-TS monoclonal antibody for 30 min, to which 140 µl protein A–agarose, 50 µg yeast tRNA and 40 U Prime RNase Inhibitor (5Prime-3Prime, Boulder, CO) were added for an additional 30 min at 4°C. The protein A–agarose immune complex precipitates were centrifuged at 14 000 g for 15 min at 4°C and then washed five times with 500 µl of NET-2 buffer. After resuspension in 300 µl of NET buffer, the immunoprecipitated pellets were subjected to phenol/chloroform extraction.
RT–PCR analysis
Total RNA was isolated from human colon cancer H630 cells as described above. Synthesis of the first strand cDNA was performed as previously described (6). The resulting single strand cDNA was then used as template for PCR amplification. The reagents used were those outlined with the Perkin Elmer protocol (no. N808-0009; Perkin-Elmer, Foster City, CA) and included 1.5 mM MgCl2.
Amplification was performed using an aliquot of the single-strand cDNA reaction mixture and 100 ng of the respective DNA oligonucleotide primers synthesized on an Applied Biosystems model 391 DNA synthesizer. The sequences of the primers were as follows: luc (sense), 5′-TAC AGA TGC ACA TAT CGA GG-3′; luc (antisense), 5′-ATC CAC AAC CTT CGC TTC AA-3′; β-actin (sense), 5′-GCG GGA AAT CGT GCG TGC GTG ACA TT-3′; β-actin (antisense), 5′-GAT GGA GTT GAA GGT AGT TTC GTG-3′.
The RNA isolated from cellular TS RNP complexes was subjected to reverse transcription using the methods described above. The single-strand cDNA was then used as template for PCR amplification. The sequences of the primers were as follows: EGR-1 (sense), 5′-GAG TCG CGA GAG ATC CAG-3′; luc (antisense), 5′-CAA CAG TAC CGG AAT GCC-3′.
PCR reactions were carried out under standard conditions (95°C for 1 min, 60°C for 1 min, 72°C for 1.5 min, for 30 cycles). At the end of the reaction, samples were incubated at 72°C for an additional 10 min and then cooled to 4°C. PCR products were resolved on a 1–2% agarose gel stained with ethidium bromide. A series of control experiments were performed to determine the range of linear amplification for each gene. To relate the expression of luciferase to that of the internal standard control gene β-actin, a ratio was determined by comparing the amount of the PCR product within the linear amplification range of the luciferase gene and the β-actin gene.
RESULTS AND DISCUSSION
Previous work from this laboratory had identified a 200 nt sequence in the protein coding region of TS mRNA that bound with high affinity (1–3 nM) to human TS protein (5). For these studies, an RNA gel mobility shift assay system had been used to show that this sequence bound to TS with an affinity nearly identical to that observed for the 5′-upstream binding site corresponding to nt 80–110. In order to more precisely localize the binding site for TS within this 200 nt sequence, we initially performed a series of RNA competition, gel mobility shift experiments. Several TS RNA sequences, contained within nt 434–634, were synthesized in vitro and used as unlabeled RNA competitors in gel mobility shift assays to determine their relative binding affinity (IC50) for human TS protein (Table 1). Full-length, TS1–1524 RNA (IC50 1.5 nM) and TS434–634 RNA (IC50 3.2 nM) bound TS with relatively similar affinity. These results are consistent with those previously reported from this laboratory (6). In contrast, the affinities of TS for RNAs corresponding to TS434–534, TS534–634 and TS1040–1240, a sequence located in the 3′-UTR region, were significantly lower than for either of these two elements (IC50 >100 nM). The TS480–580 RNA sequence effectively competed with 32P-radiolabeled full-length TS mRNA for binding to TS protein (IC50 3.5 nM). When this RNA was shortened to 70 nt to give rise to the TS480–550 sequence, no loss in binding affinity was observed (IC50 3.8 nM) relative to the TS480–580 and TS434–634 RNA sequences. However, sequences shorter than this 70 nt element, including TS480–540, TS480–530, TS490–550 and TS500–550, were unable to compete for binding, even at concentrations of up to 100-fold molar excess of radiolabeled TS mRNA probe (data not shown).
Table 1. Relative binding affinity of TS RNA sequences.
| Sequence | IC50 (nM) |
|---|---|
| TS1–1524 | 1.5 ± 0.3 |
| TS434–634 | 3.2 ± 0.7 |
| TS434–534 | >100 |
| TS534–634 | >100 |
| TS480–580 | 3.5 ± 0.9 |
| TS480–550 | 3.8 ± 1.0 |
| TS1040–1240 | >100 |
Binding of human TS protein to human TS RNA sequences relative to full-length TS mRNA was determined by comparing the concentration of unlabeled RNA which inhibited specific binding of 32P-radiolabeled full-length TS mRNA by 50%. The details for in vitro synthesis of TS RNAs are presented in Materials and Methods. Each value represents the mean ± SE of three to five experiments.
To further demonstrate the specificity of interaction between TS and the TS480–550 RNA sequence, the effects of point mutations within this sequence on binding to TS protein were subsequently investigated (Table 2). The truncated TS94–1053 RNA showed similar binding affinity to TS protein (IC50 3.0 nM) when compared to full-length TS1–1524 RNA (IC50 1.5 nM). When 21 conserved nucleotide mutations were introduced into the TS480–550 RNA sequence, the binding affinity of TS480–550(m) RNA was significantly decreased (IC50 >100 nM). This same sequence of conserved mutations was placed in the TS94–1053 RNA and the relative binding affinity of this TS95–1053(m) RNA sequence was also dramatically decreased, yielding an IC50 of >100 nM.
Table 2. Relative binding affinity of mutant TS mRNA sequences.
| Sequence | IC50 (nM) |
|---|---|
| TS1–1524 | 1.5 ± 0.3 |
| TS480–550 | 3.8 ± 1.0 |
| TS94–1053 | 2.0 ± 0.8 |
| TS94–1053(m) | >100 |
| TS480–550(m) | >100 |
Binding of human TS protein to wild-type and mutant TS RNA sequences relative to full-length TS mRNA was determined by comparing the concentration of unlabeled RNA which inhibited specific binding of 32P-radiolabeled full-length TS mRNA by 50%. The details for in vitro synthesis of TS RNAs are presented in Materials and Methods. Each value represents the mean ± SE of three to five experiments.
These results provide further evidence that the TS480–550 sequence is an important cis-acting element for TS. However, we next investigated whether this sequence played a role in the translational autoregulation of TS. To specifically address this issue, a series of in vitro translation experiments were performed. For these studies, we investigated the effect of recombinant human His-tagged TS protein on the translation of various TS mRNA sequences, including full-length TS mRNA (TS1–1524), a truncated TS mRNA that started immediately at the AUG sequence and lacked both the 5′- and 3′-UTRs (TS94–1053) and a truncated mutant TS mRNA containing conserved point mutations within the TS480–550 sequence (TS94–1053m). In vitro translated TS protein products resolving at 35 kDa (Fig. 2A, lanes 1 and 2), His-tagged TS protein at 37 kDa (Fig. 2A, lanes 3–6) and luciferase at 61 kDa (Fig. 2A, lanes 1–6) were observed. Translation of both full-length TS1–1524 (Fig. 2A, lane 2) and truncated TS94–1053 (Fig. 2A, lane 4) mRNAs was nearly completely repressed upon addition of human His-tagged TS protein. In contrast, the translational efficiency of mutant truncated TS94–1053(m) mRNA was not affected by the presence of exogenous His-tagged TS protein (Fig. 2A, lane 6). As an important internal control, translation of luciferase mRNA remained unchanged in the absence and/or presence of exogenous TS protein (Fig. 2A, lanes 1–6). This initial series of experiments suggest that the TS480–550 sequence can function independently of the 5′-upstream binding site and, thus, appears to be sufficient to mediate the process of TS translational autoregulation.
Figure 2.
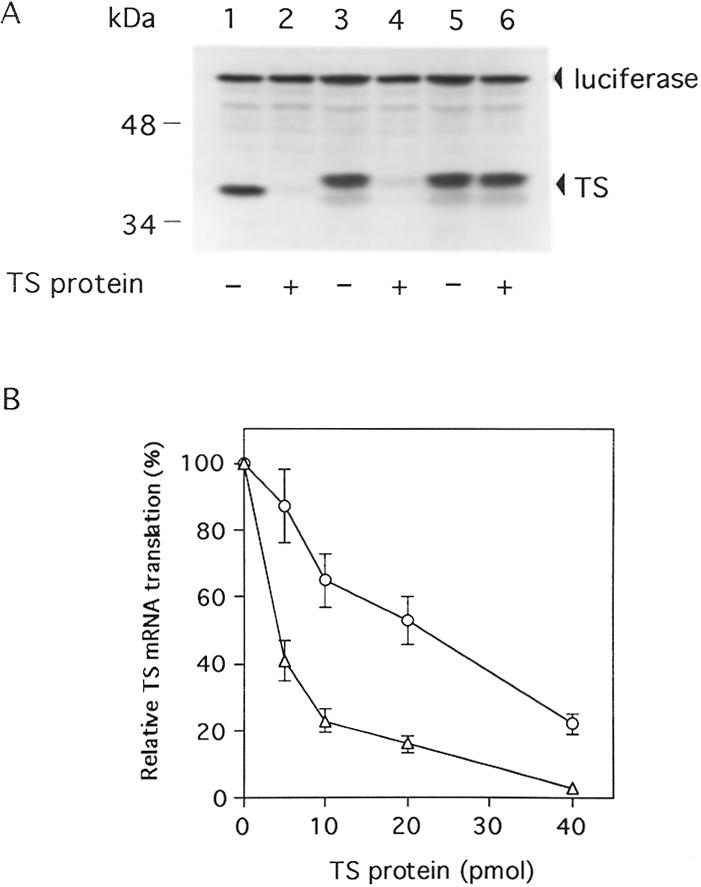
Effects of mutation in the TS480–550 RNA sequence on translational inhibition of human TS mRNA by TS. (A) Translation reactions containing rabbit lysate were incubated with TS1–1524 mRNA (0.4 pmol) (lanes 1 and 2), TS94–1053 (0.4 pmol) (lanes 3 and 4) or TS94–1053m (0.4 pmol) (lanes 5 and 6). Human His-tagged TS protein (11 pmol) was included in the reaction mixtures where indicated (lanes 2, 4 and 6). As an internal control for translation, luciferase mRNA (0.4 pmol) was included in each reaction (lanes 1–6). Translation reactions were incubated at 30°C for 60 min and protein products were analyzed by SDS–PAGE and autoradiography as described in Materials and Methods. TS and luciferase protein products are indicated by the arrows. (B) Translational inhibition of TS mRNA sequences by TS. TS1–1524 (0.4 pmol) (triangle) and TS1–1524(m) RNA sequences (0.4 pmol) (cirlce) were incubated with human His-tagged TS protein (0–40 pmol) in the rabbit reticulocyte translation system as described in Materials and Methods. Proteins were resolved by SDS–PAGE and then subjected to autoradiography. The level of TS mRNA translation in the absence of TS protein represents 100%. Points represent the mean ± SE from three experiments.
However, to further address this issue, we compared the effect of human recombinant His-tagged TS protein on the translational efficiency of full-length TS mRNA (TS1–1524) with full-length TS mRNA that contained the same 21 conserved mutations within nt 480–550 of the protein coding region. This mutant TS RNA sequence was designated TS1–1524(m). As seen in Figure 2B, translation of mutant full-length TS mRNA was inhibited in the presence of human TS protein. However, the concentration of TS protein required to inhibit by 50% translation of TS1–1524(m) RNA (IC50 21 pmol) was significantly higher when compared to that required to inhibit translation of wild-type TS1–1524 mRNA (IC50 2.1 pmol). Taken together, the results from these experiments suggest that the 5′-upstream binding sequence is insufficient to allow for complete translational repression by TS. It would appear then that the 5′-upstream sequence and the element in the protein coding region are both required for the process of TS translational autoregulation in vitro. Our preliminary findings suggest that these sequences function independently of one another. However, it is conceivable that these two sequences may interact with one another to form a complex secondary structure that then interacts with TS protein. Further studies are now in process to characterize the precise role of each of these elements in the in vivo expression of TS.
To begin to characterize the potential in vivo biological relevance of this second cis-acting element, we investigated whether the presence of this 70 nt sequence could confer the property of TS-dependent translational regulation onto an unrelated mRNA. For these studies, we made use of a heterologous luciferase p644 indicator plasmid that had been placed under control of an EGR-1 promoter element. The various heterologous constructs are outlined in schematic form in Figure 1. Various TS sequences contained within nt 434–634 were inserted immediately upstream of the sequence encoding the luciferase gene to yield several heterologous constructs. Both the parent p644 and the heterologous p644/TS434–634 plasmids were transiently expressed in human colon cancer H630 cells. There was a significantly decreased level of luciferase activity (2.5-fold) in cells transfected with p644/TS434–634 when compared to cells transfected with the parent plasmid p644 (Fig. 3). When TS sequences corresponding to nt 434–534 and 534–634 were placed immediately upstream of the luciferase gene, no changes in luciferase activity were observed. In contrast, transient expression of a heterologous luciferase plasmid containing a 100 nt sequence corresponding to nt 480–580 resulted in a significant reduction in luciferase activity. The repressive effect of this sequence on luciferase expression was nearly identical to that observed with the TS434–634 element (Fig. 3). Further truncation to a 70 nt sequence corresponding to nt 480–550 also resulted in markedly suppressed levels of luciferase activity (2.5-fold). However, when sequences shorter than this 70 nt element were placed upstream of the luciferase gene, including TS480–540, TS480–530, TS490–550 and TS 500–550, no changes in luciferase expression were observed (data not shown). These initial results suggest that the TS RNA sequence corresponding to nt 480–550 could, in fact, down-regulate expression of luciferase in an intact biological system.
Figure 3.

Effect of TS RNA sequences on luciferase expression in vivo. Luciferase heterologous constructs were transiently expressed in H630 cells. Luciferase activity was measured 48 h post-transfection using the dual luciferase reporter assay system as described in Materials and Methods. The activity in cells transfected with the parent p644 plasmid was defined as 100%. Luciferase expression in cells transfected with p644/TS434–634, p644/TS480–580, p644/TS480–550 and p644/TS480–550(3′) was significantly less than in cells transfected with p644 (paired Student’s t-test, *P < 0.01). Each value represents the mean ± SE of three to five experiments.
In addition to placing the TS480–550 element immediately upstream of the luciferase coding region, we designed a heterologous construct that contained the TS480–550 element downstream of the coding region, p644/TS480–550(3′). Luciferase activity was significantly decreased by 2.4-fold when the p644/TS480–550(3′) plasmid was transfected into H630 cells (Fig. 3). No such decrease was observed, however, when this sequence was placed in the antisense orientation (data not shown). This effect on luciferase activity is nearly identical to that observed when this same element was placed immediately upstream of the luciferase coding region. These results demonstrate that the TS480–550 element can affect the in vivo translation of luciferase when placed either upstream or downstream of the coding region. This finding would suggest that the translational repression mediated by this element may be mediated by a complex RNA secondary structure that renders the luciferase inaccessible to the machinery required for translational initiation. Studies are on-going to further characterize the specific mechanism(s) by which this translational repression is mediated.
To provide further evidence for the specificity of effect of the TS434–634 and TS480–550 sequences, we next investigated the effect of an unrelated TS RNA sequence on luciferase expression. Previous RNA gel shift experiments in this laboratory had shown that human TS protein did not interact with the 3′-UTR of TS mRNA (5). Given these earlier in vitro findings, the sequence corresponding to nt 1040–1240 was cloned onto the 5′-end of the luciferase gene. This p644/TS1040–1240 heterologous construct was then transiently expressed in H630 cells. As seen in Figure 3, no alterations in luciferase activity were observed when compared to cells transfected with the parent plasmid p644. This finding is consistent with that obtained in cell-free RNA-binding studies and provides further support for the specificity of the effect of the TS434–634 and TS480–550 sequences.
Previous studies demonstrated that the process of TS translational autoregulation is mediated directly by alterations in translational efficiency of TS mRNA in the absence of actual changes in TS mRNA levels (4,20). In order to identify the specific molecular event regulating the changes in luciferase activity in the H630 system, two different strategies were employed. The first was an RNase protection assay to determine the expression of luciferase mRNA in transfected H630 cells. Using this method, luciferase mRNA levels were shown to be identical in cells transfected with p644 (Fig. 4A, lane 2), p644/TS434–634 (Fig. 4A, lane 3), p644/TS480–550 (Fig. 4A, lane 4) and p644/TS1040–1240 (Fig. 4A, lane 5). As a complementary approach, an RT–PCR analysis was used to determine luciferase-specific mRNA levels in cells transfected with the luciferase reporter plasmids p644, p644/TS434–634, p644/TS480–550 and p644/TS1040–1240. As seen in Figure 4B, luciferase mRNA levels were identical in H630 cells transfected with each of these reporter constructs. A control RT–PCR experiment revealed no differences in the levels of β-actin mRNA (Fig. 4B). The ratio of luciferase to β-actin remained the same in cells transfected with p644, p644/TS434–634, p644/480–550 and p644/TS1040–1240. Together, these results suggest that the TS434–634 and TS480–550 elements exert their regulatory effects at the translational level and most likely interfere with the efficiency of translation of the luciferase mRNA. The specific mechanism(s) by which this event occurs remains to be characterized but may involve either interference with binding of critical translation initiation factors near or at the cap site or interference with the process of ribosomal scanning.
Figure 4.
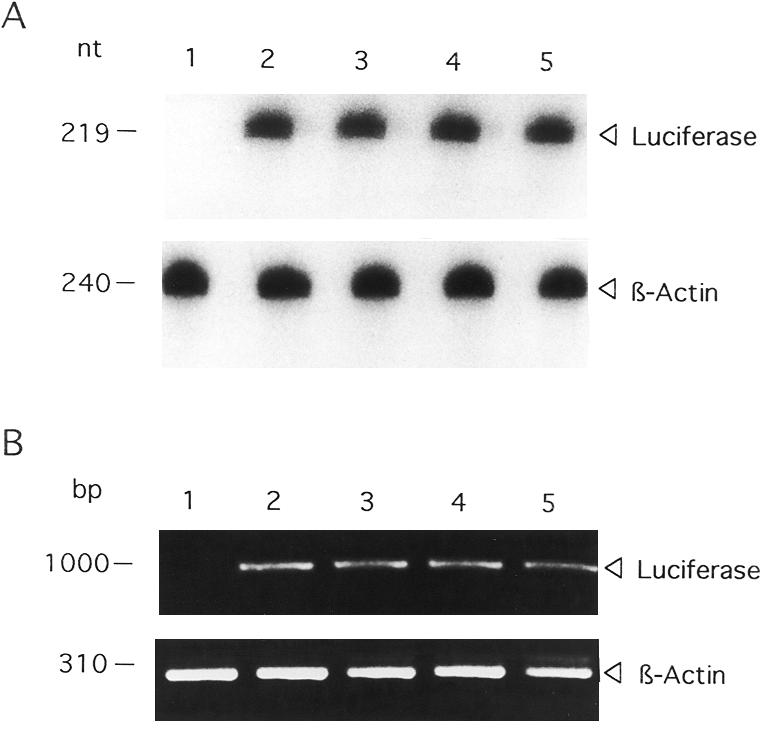
Effect of TS RNA sequences on luciferase mRNA expression. (A) RNase protection assay; (B) RT–PCR analysis. Human colon cancer H630 cells were transiently transfected in the absence (lane 1) or presence of p644 (lane 2), p644/TS434–634 (lane 3), p644/TS480–550 (lane 4) or p644/TS1040–1240 (lane 5). Total cellular RNA was isolated and then subjected to either RNase protection or RT–PCR analysis as described in Materials and Methods. The arrowheads indicate the positions of the luciferase- and β-actin-associated bands.
Our laboratory previously developed an immunoprecipitation/RT–PCR method to isolate RNP complexes composed of TS protein and TS mRNA from human colon cancer H630 cells (6). In the present report, we employed a similar strategy to isolate RNP complexes from H630 cells transfected with the heterologous reporter plasmid p644/TS480–550. TS RNP complexes were immunoprecipitated with an anti-TS monoclonal antibody, and the specific RNA fraction bound to TS was isolated and subjected to RT–PCR amplification. Using primers corresponding to sequences of the EGR-1 promoter and the luciferase gene as outlined in Figure 5B, a 123 bp DNA product was amplified from cells transfected with p644/TS480–550 (Fig. 5A, lane 1) but not from cells transfected with p644 (Fig. 5A, lane 2) or p644/TS1040–1240 (Fig. 5A, lane 3). This band resolved at the same position as the DNA product obtained from a control PCR reaction using the p644/TS480–550 plasmid as DNA template (Fig. 5A, lane 6). When the immunoprecipitation reaction was performed in the absence of antibody, no amplified DNA product was observed (Fig. 5A, lane 4). This finding offers support for the specificity of antibody recognition. To confirm that the nucleic acid origin of this band was indeed RNA, experiments were performed in which RNAs isolated from TS RNP complexes were directly PCR amplified in the absence of the RT step (Fig. 5A, lane 5). Under this condition, the 123 bp DNA product was not amplified. The results of the immunoprecipitation/RT–PCR analysis confirmed a direct interaction between TS protein and the TS480–550 RNA sequence in intact human colon cancer H630 cells.
Figure 5.
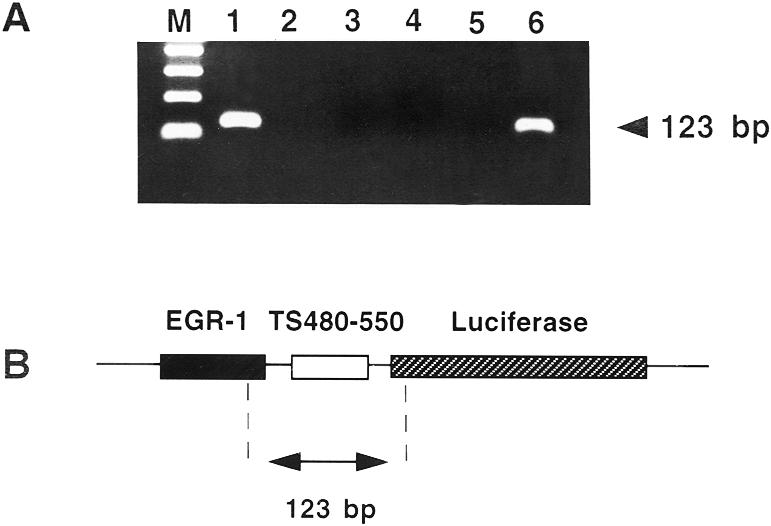
Isolation of TS RNP complexes in human colon cancer cells transfected with luciferase heterologous constructs. (A) Ethidium bromide stain. An immunoprecipitation/RT–PCR method was used to isolate TS RNP complexes, as described in Materials and Methods. H630 cells were transiently transfected with p644/TS480–550 (lanes 1, 4–6), p644 (lane 2) or p644/TS1040–1240 (lane 3). Whole cell extracts were immunoprecipitated with an anti-TS monoclonal antibody (lanes 1–3 and 5) or with no antibody (lane 4) as outlined in Materials and Methods. The isolated nucleic acid fraction was reverse transcribed and PCR amplified using EGR-1- and luciferase-specific primers. In lane 5, the immunoprecipitated nucleic acid fraction was not subjected to reverse transcription. Lane 6 represents a control PCR reaction in which the p644/TS480–550 DNA template was subjected to PCR amplification using the same primer set as used in lanes 1–5. Lane M, size marker. (B) p644/TS480–550 RNA sequence. The 123 bp fragment predicted to be RT–PCR amplified is shown in relation to its position on the full-length p644 plasmid. The positions of the EGR-1 promoter and the luciferase gene are indicated.
Earlier work from this laboratory and others showed that exposure of human colon and breast cancer cells to the fluoropyrimidine 5-FU and/or other TS inhibitor compounds resulted in a marked induction of both TS enzyme activity and TS protein expression (20–24). Cell-free RNA binding studies revealed that the presence of such TS inhibitor compounds prevented the binding of TS protein to its target TS mRNA, thereby resulting in enhanced translational efficiency and synthesis of new protein (4,9). In addition, recent work suggests that treatment of human colon cancer cells with the fluoropyrimidine analog 5-fluoro-2′-deoxyuridine results in increased levels of TS protein on the basis of enhanced stability of the protein (25). In either case, one would predict that following exposure to a TS inhibitor compound, luciferase activity would be increased in those cells transfected with luciferase reporter constructs expressing the TS RNA cis-acting elements. As seen in Figure 6, 5-FU treatment of cells transfected with either p644/TS434–634 or p644/TS480–550 resulted in a significant increase in the level of luciferase activity (P < 0.05). In contrast, 5-FU treatment did not alter luciferase activity in cells transfected with either the parent p644 or p644/TS1040–1240. The assay system used in the present study allows only determination of total enzyme activity. While antibodies to luciferase protein do exist, the level of luciferase expression in the transfected H630 cells was apparently below the level of detection as determined by western immunoblot analysis (data not shown). Thus, it is possible that the effects of TS on luciferase activity may not correspond directly to changes in luciferase protein expression and that these experiments underestimate the effects of TS on the absolute biosynthetic rate of luciferase. A similar discordance between enzyme activity and protein expression was observed with regard to the effects of 5-FU on the expression of TS in these same human colon cancer H630 cells. Treatment with 5-FU for 24 h resulted in a 1.5-fold increase in TS enzyme activity, as determined by the TS catalytic assay, but a significantly greater 5-fold increase in levels of TS protein expression, as determined by western immunoblot analysis (20,23). A second potential explanation is that the 5′-upstream element may be required in order for the full range of expression of TS to be exhibited in vivo following treatment with a TS inhibitor compound. Studies are now underway to address this second possibility.
Figure 6.

Effect of 5-FU treatment on luciferase expression in vivo. Plasmids p644, p644/TS434–634, p644/TS480–550 and p644/TS1040–1240 were each transiently expressed in H630 cells. Twenty-four hours after transfection, cells were incubated in the absence (open bars) or presence (closed bars) of 1 µM 5-FU for an additional 24 h. Luciferase activity in cells transfected with p644 in the absence of 5-FU was defined as 100%. 5-FU treatment of cells transfected with either p644/TS434–634 or p644/TS480–550 resulted in a significant increase (P < 0.05) in luciferase activity, as determined by paired Student’s t-test. Each value represents the mean ± SE of three to five experiments.
As further evidence for the role of the TS480–550 sequence as a specific TS response element, we investigated luciferase expression from the TS-responsive p644/TS480–550 heterologous construct in human colon cancer HCT-C18 cells. This cell line was selected since HCT-C18 cells express a functionally inactive TS protein (11). We recently established an HCT-C:His-TS(+) cell line by stable transfection of mutant HCT-C18 cells with a human His-tagged TS cDNA. Biochemical analysis using the TS catalytic assay confirmed that HCT-C18 cells express 24-fold less TS enzyme activity (0.33 pmol/min/mg protein) than TS-overexpressing HCT-C:His-TS(+) cells (8.03 pmol/min/mg protein). The p644 and p644/TS480–550 plasmids were each transiently expressed in HCT-C18 and HCT-C:His-TS(+) cells. As seen in Figure 7, luciferase activity in TS-deficient HCT-C18 cells was similar following transfection with either reporter construct. However, luciferase activity was nearly 3-fold less in HCT-C:His-TS(+) cells transfected with p644/TS480–550 compared to these same cells transfected with the parent p644 plasmid. This set of experiments provides further supportive evidence that the TS480–550 sequence is a TS response element and that a functionally active TS is required for its complete translational regulatory effects. Further studies are now underway to characterize the specific domain(s) on TS protein that recognizes this particular cis-acting sequence.
Figure 7.

Effect of TS480–550 on luciferase expression in HCT-C18 and HCT-C:His-TS(+) cells. Human colon cancer HCT-C18 and HCT-C:His-TS(+) cells were transiently transfected with either p644 (closed bars) or p644/TS480–550 (open bars). Luciferase activity was measured 48 h post-transfection using the dual luciferase assay system as described in Materials and Methods. The activity in cells transfected with the p644 plasmid was taken as 100%. Transfection with p644/TS480–550 resulted in a significant decrease in luciferase activity (P < 0.01), indicated by the asterisk, when compared to transfection with p644, as determined by paired Student’s t-test. Each value represents the mean ± SE of three to five experiments.
In contrast to previous studies from this laboratory which concentrated primarily on the 5′-upstream binding site on TS mRNA, the present study focused on characterizing the RNA sequence in the protein coding region. We provide evidence that the 200 nt cis-acting element in the protein coding region of TS mRNA, corresponding to nt 434–634, is sufficient to confer the property of translational regulation onto a heterologous luciferase reporter gene. Further studies using a series of in vitro RNA gel mobility shift and in vivo transfection experiments have localized this element to a 70 nt sequence corresponding to nt 480–550 of TS mRNA. In vitro translation experiments confirm that this specific RNA sequence is required along with the 5′-upstream element in order for human TS protein to exhibit its full range of translational autoregulatory activity. We have also demonstrated that in cells transfected with the TS480–550 element, treatment with the TS inhibitor compound 5-FU results in induced expression of luciferase. Furthermore, this element exerts biological activity only in human colon cancer cells expressing a functionally active TS protein. Taken together, these findings provide strong support for the TS480–550 cis-acting element serving as a specific binding site for TS in vitro and in vivo. Moreover, this work suggests that this particular sequence has biological activity and that it can function independently of the 5′-upstream cis-acting element.
One issue raised by the present study relates to how binding of TS to the cis-acting element in the protein coding region of TS mRNA results in translational repression. In order for such events to occur, some type of interaction between the 5′-end of the mRNA and the protein coding region must take place. Several groups have identified sequences within the protein coding region and the 3′-UTR as being critical for the translational control of various genes (26–31). While the specific molecular mechanism(s) by which the process of translational repression occurs remains to be determined, there is growing evidence to suggest that sequences in both the protein coding region and in the 3′-UTR play a key role in controlling the process of translational initiation (26–31). Sophisticated molecular modeling studies will be required, however, to more precisely characterize the molecular events mediating this complex process. As part of this effort, studies are now in progress to identify the domain(s) on TS protein required for RNA recognition. Our preliminary studies suggest that certain cysteine moieties, as well as the folate-binding region of TS, are key elements in mediating this RNA–protein interaction (7).
Careful sequence analysis has yet to reveal a consensus nucleotide sequence within the 5′-upstream and the protein coding region binding sites that is recognized by TS protein. However, one potential drawback to such a ‘consensus’ approach is that many of the RNA-binding proteins characterized to date seem to recognize a combination of sequence and structure (32,33). Thus, identification of a consensus RNA-binding site by simple sequence analysis may be inadequate. A preliminary analysis using the Zuker RNA fold algorithm predicts the existence of stable secondary structures in the TS434–634 and TS480–550 sequences. However, these structures appear to be somewhat different from the one predicted for the cis-acting element located within the 5′-upstream binding site. Studies are now in progress to perform a more detailed characterization of the sequence and/or structural determinants required for binding of TS protein to this specific element in the protein coding region. Once this analysis is performed, comparisons with the molecular elements involved in binding of TS to the 5′-upstream binding site can be more readily accomplished. This work should provide critical insight into the essential elements required for RNA recognition by TS protein and enhance our understanding of the specific molecular events mediating the translational regulation of TS. Finally, these studies contribute to a growing body of evidence regarding the critical role of translational control mechanisms in the regulation of cellular gene expression.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs Jing Ju and John Schmitz for helpful discussions and critical reading of the manuscript and Ms Edna McCarthy for editorial preparation of the manuscript. This work was supported, in part, by grants from the National Science Foundation (MCB97-24071 to G.F.M.) and the National Cancer Institute (CA 44355 to F.M.; CA16359 and CA75712 to E.C.).
REFERENCES
- 1.Friedkin M. and Kornberg A. (1957) Chemical Basis of Heredity. John Hopkins Press, Baltimore, MD, pp. 609–614.
- 2.Danenberg P.V. (1977) Biochim. Biophys. Acta, 473, 73–79. [DOI] [PubMed] [Google Scholar]
- 3.Carreras C. and Santi,D.V. (1995) Annu. Rev. Biochem., 64, 721–762. [DOI] [PubMed] [Google Scholar]
- 4.Chu E., Koeller,D.M., Casey,J.L., Drake,J.C., Chabner,B.A., Elwood,P.C., Zinn,S. and Allegra,C.J. (1991) Proc. Natl Acad. Sci. USA, 88, 8977–8981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chu E., Voeller,D., Koeller,D.M., Drake,J.C., Takimoto,C.H., Maley,G.F., Maley,F. and Allegra,C.J. (1993) Proc. Natl Acad. Sci. USA, 90, 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chu E., Voeller,D.M., Jones,K.L., Takechi,T., Maley,G.F., Maley,F., Segal,S. and Allegra,C.J. (1994) Mol. Cell. Biol., 14, 207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Voeller D.M., Changchien,L.M., Maley,G.F., Maley,F., Takechi,T., Turner,R.E., Montfort,W.R., Allegra,C.J. and Chu,E. (1995) Nucleic Acids Res., 23, 869–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu E. and Allegra,C.J. (1996) Bioessays, 18, 191–198. [DOI] [PubMed] [Google Scholar]
- 9.Chu E., Voeller,D.M., Morrison,P.F., Jones,K.L., Takechi,T., Maley,G.F., Maley,F. and Allegra,C.J. (1994) J. Biol. Chem., 269, 20289–20293. [PubMed] [Google Scholar]
- 10.Park J.G., Oie,H.K., Sugarbaker,P.H., Henslee,J.G., Chen,T.R., Johnson,B.E. and Gazdar,A. (1987) Cancer Res., 47, 6710–6718. [PubMed] [Google Scholar]
- 11.Hoganson D.K., Williams,A. and Berger,S. (1999) Biochem. Pharmacol., 58, 1529–1537. [DOI] [PubMed] [Google Scholar]
- 12.Ju J.F., Pedersen-Lane,J., Maley,F. and Chu,E. (1999) Proc. Natl Acad. Sci. USA, 96, 3769–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu Y.Z., Aiello,P.D. and Matthews,R.G. (1984) Biochemistry, 23, 6870–6876. [DOI] [PubMed] [Google Scholar]
- 14.Davisson V.J., Sirawaporn,W. and Santi,D.V. (1989) J. Biol. Chem., 264, 9145–9148. [PubMed] [Google Scholar]
- 15.Pedersen-Lane J., Maley,G.F., Chu,E. and Maley,F. (1997) Protein Expr. Purif., 10, 256–262. [DOI] [PubMed] [Google Scholar]
- 16.Liebold E. and Munro,H.N. (1988) Proc. Natl Acad. Sci. USA, 85, 2171–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Felgner P.L., Gadek,T.R., Holm,M., Roman,R., Chan,H.W., Wenz,M., Northrop,J.P., Ringold,G.M. and Danielsen,M. (1987) Proc. Natl Acad. Sci. USA, 84, 7413–7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chomczynski P. and Sacchi,N. (1987) Anal. Biochem., 162, 156–159. [DOI] [PubMed] [Google Scholar]
- 19.Steitz J.A. (1989) Methods Enzymol., 180, 468–481. [DOI] [PubMed] [Google Scholar]
- 20.Chu E., Koeller,D.M., Johnston,P.G., Zinn,S. and Allegra,C.J. (1993) Mol. Pharmacol., 43, 527–533. [PubMed] [Google Scholar]
- 21.Washtien W.L. (1984) Mol. Pharmacol., 25, 171–177. [PubMed] [Google Scholar]
- 22.Swain S.M., Lippman,M.E., Egan,E.F., Drake,J.C., Steinberg,S.M. and Allegra,C.J. (1989) J. Clin. Oncol., 7, 890–899. [DOI] [PubMed] [Google Scholar]
- 23.Chu E., Zinn,S., Boarman,D. and Allegra,C.J. (1990) Cancer Res., 50, 5834–5840. [PubMed] [Google Scholar]
- 24.Keyomarsi K., Samet,J., Molnar,G. and Pardee,A.B. (1993) J. Biol. Chem., 268, 15142–15149. [PubMed] [Google Scholar]
- 25.Kitchens M.E., Forsthoefel,A.M., Rafique,Z., Spencer,H.T. and Berger,F.G. (1999) J. Biol. Chem., 274, 12544–12547. [DOI] [PubMed] [Google Scholar]
- 26.Gallie D.R. (1991) Genes Dev., 5, 2108–2116. [DOI] [PubMed] [Google Scholar]
- 27.Jackson R.J. (1993) Cell, 74, 9–14. [DOI] [PubMed] [Google Scholar]
- 28.Leathers V., Tanguay,R., Koyayashi,M. and Gallie,D.R. (1993) Mol. Cell. Biol., 13, 5331–5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ostareck-Lederer A.D., Ostareck,D.H., Standard,N. and Thiele,B.J. (1994) EMBO J., 13, 1476–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chu E., Takechi,T., Jones,K.L., Voeller,D.M., Copur,S.M., Maley,G.F., Maley,F., Segal,S. and Allegra,C.J. (1995) Mol. Cell. Biol., 15, 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sonenberg N. (1994) Curr. Opin. Genet. Dev., 4, 310–315. [DOI] [PubMed] [Google Scholar]
- 32.Uhlenbeck O.C., Wu,H.N. and Sampson,J.R. (1987) Molecular Biology of RNA. Academic Press, New York, NY, pp. 285–294.