

Abstract
Purpose of review
The mutational landscape of acute myeloid leukemia (AML) has revised diagnostic, prognostic, and therapeutic schemata over the past decade. Recurrently mutated AML genes have functional consequences beyond typical oncogene-driven growth and loss of tumor suppresser function.
Recent findings
Large-scale genomic sequencing efforts have mapped the complexity of AML and trials of mutation-based targeted therapy has led to several FDA-approved drugs for mutant-specific AML. However, many recurrent mutations have been identified across a spectrum from clonal hematopoiesis to myelodysplasia to overt AML, such as effectors of DNA methylation, chromatin modifiers, and spliceosomal machinery. The functional effects of these mutations are the basis for substantial discovery.
Summary
Understanding the molecular and pathophysiologic functions of key genes that exert leukemogenic potential is essential towards translating these findings into better treatment for AML.
Keywords: acute myeloid leukemia driver mutations, epigenetics, methylation, splicing, transcription factors
INTRODUCTION
Acute myeloid leukemia (AML) is a heterogenous and complex disease characterized by differentiation blockade and clonal proliferation of hematopoietic stem and progenitor cells (HSPCs) at the expense of normal hematopoiesis. The application of next generation of sequencing technologies has led to the identification of 40–50 genes harbor recurrent somatic mutations in various AML subtypes [1–4]. These discovery studies have led to a greater understanding of AML biology, especially the striking frequency of epigenetic dysregulation in AML pathogenesis. In this review, we will provide an overview of genomic and epigenomic alterations in AML by addressing the role of underlying molecular events, which contribute to AML (Fig. 1). Consequently, our evolving understanding of the molecular basis of AML is refining prognostic schema and leading to new therapeutic approaches.
FIGURE 1.
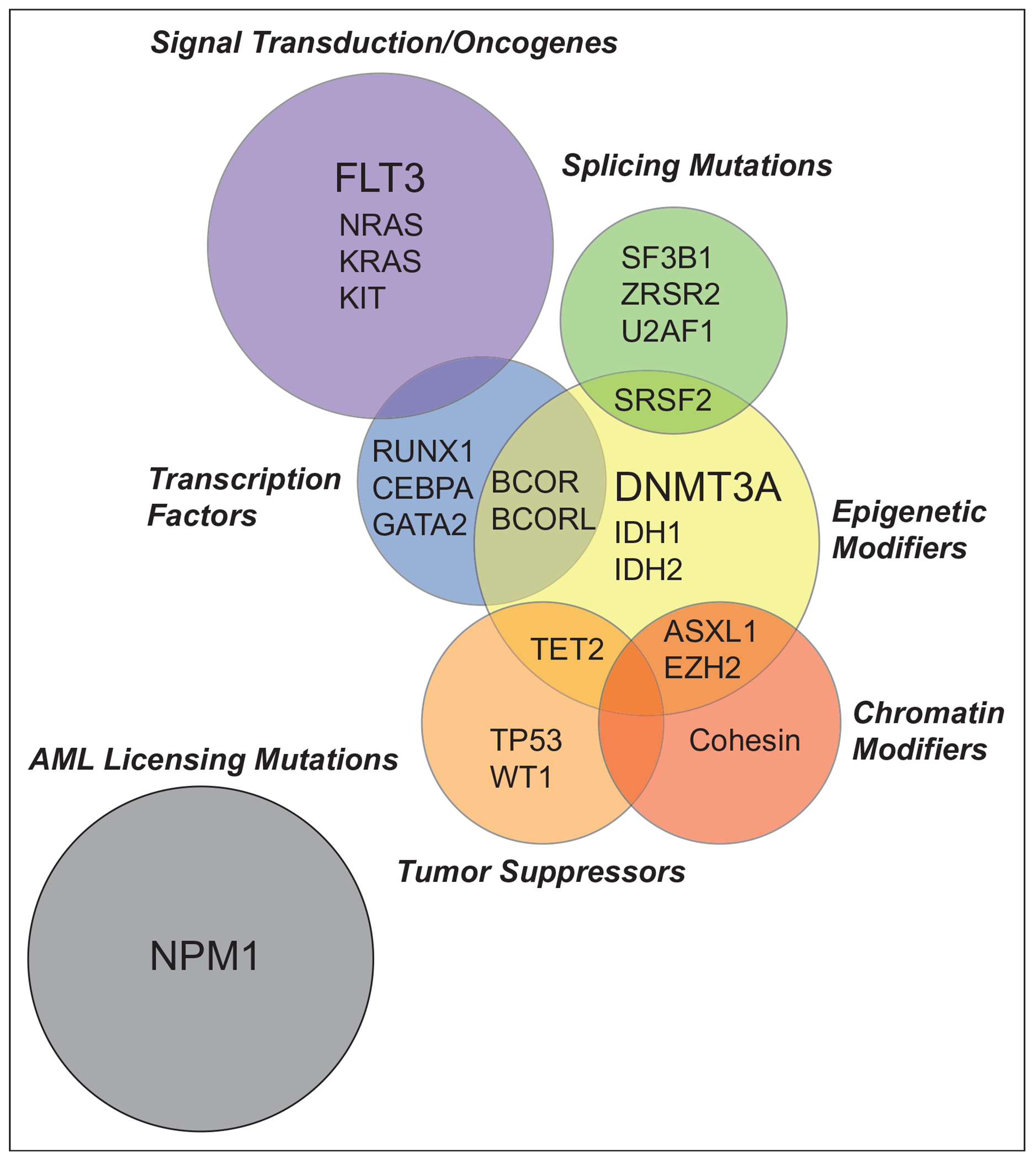
Functional overlap of acute myeloid leukemia driver mutations. Driver mutations in AML stratified by mechanistic consequence. Size of circles reflect mutational frequency with three most common mutations in bold. AML, acute myeloid leukemia.
DRIVER MUTATIONS IN ACUTE MYELOID LEUKEMIA
Acute myeloid leukemia-licensing mutations
NPM1
Mutations in nucleophosmin 1 (NPM1) represents the largest of these genetically defined AML groups, constituting ~30% of AMLs. NPM1 is a multifunctional protein involved in histone chaperoning, ribosome biogenesis, centrosome duplication, and the DNA damage response [5]. NPM1-mutant AML requires nuclear export of NPM1 to the cytoplasm, and the resultant overexpression of stem cell gene signature including HOXA, HOXB genes, and MEIS1 [6▪,7]. A potential explanation of this unique gene regulatory network in NPM1 mutation AML is that NPM1c chaperones PU.1 (SPI1) into the cytoplasm, which releases PU.1-mediated repression of HOX/MEIS1 [7]. AML with NPM1 mutations is a distinct entity in the classification and prognostication of myeloid neoplasms and are associated with high remission rates with intensive chemotherapy [8,9▪▪]. AML patients with NPM1 mutations generally carry a favorable prognosis in the absence of FLT3-ITD, or when the FLT3-ITD allelic ratio is low [9▪▪] (Table 1). The molecular underpinnings of the favorable prognostic effect of NPM1c is unclear, though the observations that NPM1c does not occur in clonal hematopoiesis and the infrequent occurrence of NPM1c in myelodysplastic syndrome, both suggest that the unique licensing effect of Npm1c for transformation carries a shorter lead time to acquire additional mutational hits and may result in less complex clonal architecture.
Table 1.
The European LeukemiaNET 2017 risk stratification of acute myeloid leukemia
| Risk category | Genetic abnormality |
|---|---|
| Favorable | t(8;21)(q22;q22.1); RUNX1-RUNX1T1 inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11 Mutated NPM1 without FLT3-ITD or with FLT3-ITDlow = allelic ratio < 0.5 Biallelic mutated CEBPA |
| Intermediate | Mutated NPM1 and FLT3-ITDhigh = allelic ratio > 0.5 Wild-type NPM1 without FLT3-ITD or with FLT3-ITDlow (without adverse-risk genetic lesions) t(9;11)(p21.3;q23.3); MLLT3-KMT2A Cytogenetic abnormalities not classified as favorable or adverse |
| Adverse | t(6;9)(p23;q34.1); DEK-NUP214 t(v;11q23.3); KMT2A rearranged t(9;22)(q34.1;q11.2); BCR-ABL1 inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2); GATA2,MECOM(EVI1) −5 or del(5q); −7; −17/abn(17p) Complex karyotype monosomal karyotype Wild-type NPM1 and FLT3-ITDhigh Mutated RUNX1 Mutated ASXL1 Mutated TP53 |
Data from [9▪▪].
Mutations altering signal transduction
FLT3-ITD and FLT3-TKD
Mutations in the transmembrane growth factor receptor fms-related tyrosine kinase 3 (FLT3) can occur either as an in-frame internal tandem duplication within the juxtamembrane domain of the receptor (FLT3-ITD), seen in ~20 to 25% of AML, or as point mutations most commonly in the activation loop of the tyrosine kinase domain (FLT3-TKD), seen in ~7 to 8% of AML patients. These mutations lead to autoactivation of kinase activity and constitutive activation of downstream signaling pathways, including PI3K/AKT/mTOR, RAS/MAPK, and STAT5 [10]. FLT3-ITD mutations are frequently accompanied with leukocytosis, high percentage of blasts in the bone marrow and presages a poor prognosis, with most of these patients relapse after chemotherapy and allogeneic-HSCT [11,12,13▪▪]. The poor prognosis is particularly significant in the presence of DNMT3A, the absence of NPM1, or when a high ITD allele burden is present, and shows inferior overall survival (OS) compared with FLT3-TKD or wild-type FLT3 [2,4] (Table 1). The development of the FLT3 inhibitors represents a paradigm of targeted therapy in AML and have changed clinical practice. There are currently two Food and Drug Administration (FDA)-approved small-molecule-targeted inhibitors (midostaurin and gilteritinib) of FLT3 kinase activity [14▪▪,15▪▪]. Although improved outcomes have been observed with the use of these agents, the magnitude of the effects on survival have only been modest, suggesting either the immense strength of subclonal resistance or the need for agents with better pharmacokinetic properties. There is recent evidence suggesting the measurement of fms-related tyrosine kinase 3 ligand (Flt3L) during induction chemotherapy and follow-up provides prognostic information and can serve as a biomarker with the potential to inform management of these patients [16].
KIT
KIT, also known as CD117, is a transmembrane glycoprotein type III receptor tyrosine kinase. Upon binding of stem cell factor (KIT ligand), the monomeric KIT receptor dimerizes and becomes autophosphorylated at key tyrosine sites and activates downstream signaling pathways (Ras/ERK, PI3K, and JAK/STAT pathways) important for cell proliferation, differentiation, and survival [17]. Gain-of-function mutations in the KIT proto-oncogene results in ligand-independent constitutive activation. KIT mutations occur in less than 10% of patients, but they are enriched in patients with AML with core-binding factor [t(8;21)/RUNX1/RUNX1T1, inv(16)/CBFB-MYH11] rearrangements and portend a poorer outcome in this otherwise favorable disease group [18].
NRAS and KRAS
The Ras family of small GTPases, NRAS and KRAS, activate downstream signaling effectors, such as Raf and PI3K, thereby transducing signals from activated growth factor receptors. NRAS and KRAS encode proteins that accumulate in the active GTP-bound conformation, leading to constitutive activation [19]. These mutations have been reported in ~12% (NRAS) and ~5% (KRAS) of AML patients [20]. Mutations in epigenetic modifiers (TET2/IDH/WT1) often co-occur and cooperate with NRAS mutations in AML [4] and show preferential sensitivity to MAPK kinase (MEK) inhibition in mouse models and patient samples [21▪]. Although RAS mutations tend to be mutually exclusive with FLT3 mutations, N/KRAS mutations are a common and clinically important mechanism of resistance to FLT3 inhibitors including gilteritinib and crenolinib [22▪].
Mutations disrupting transcription factor function in acute myeloid leukemia
RUNX1
The master hematopoietic transcription factor Runt-related transcription factor 1 (RUNX1) is an essential regulator of cell lineage specification, proliferation, and differentiation [23]. RUNX1 contains a runt-homology domain (RHD), a protein motif responsible for both DNA-binding and heterodimerization with CBFβ. Somatic mutations and chromosomal rearrangements involving RUNX1 are relatively common in AML. RUNX1 mutations occur in approximately 5–15% of all patients with AML and are enriched in intermediate risk (including normal karyotype AML) disease [2,4]. RUNX1 mutations are mutually exclusive with NPM1 and CEBPA mutations, and are associated with lower CR rates and inferior OS in both younger and older adults with AML [24]. Germline mutations in RUNX1 are associated with familial platelet disorder with propensity to myeloid malignancy (FPDMM), with a 20–60% estimated rate of transformation to myeloid neoplasm [25].
CEBPA
The lineage master hematopoietic transcription factor CCAAT/enhancer-binding protein α (CEBPA) is involved in cell fate decisions including a key role in governing myeloid differentiation [26]. Loss-of-function mutations in CEBPA are reported in ~10% of AML patients and are enriched in younger patients and normal karyotype. CEBPA mutations can occur in either the N-terminus, leading to expression of a truncated dominant negative protein, which retains the DNA-binding domain, or in the C-terminus, leading to impaired DNA binding and disrupted protein–protein interactions [27]. Biallelic mutations in CEBPA constitute a defined patient subgroup and are associated with a specific gene expression signature with a markedly favorable prognosis in normal karyotype AML owing to disease chemosensitivity [28] (Table 1). Germline mutations in CEBPA are associated with autosomal dominant familial AML with near complete penetrance [29].
GATA2
The GATA transcription factor family members, GATA1 and GATA2, play critical roles in hematopoiesis. The GATA2 gene encodes a zinc-finger transcription factor involved in transcriptional regulation of hematopoietic stem/progenitor cell differentiation [30]. Somatic mutations in GATA2 are relatively rare in AML, occurring in less than 5% of cases overall, and are described in normal karyotype AML arising in the context of bi-allelic mutations in CEBPA [31]. These mutations are most often missense mutations that target the zinc finger domains impairing DNA binding and affecting transcriptional activity. Heterozygous germline mutations in GATA2 cause a spectrum of disorders with overlapping features and predisposition to MDS and AML [32,33].
Mutations in epigenetic modifiers
DNMT3A
DNA methylation is a key epigenetic modification involved in normal hematopoiesis, which is altered during leukemogenesis. DNA methyltransferase 3A (DNMT3A) is a highly conserved member of the DNA methyltransferase family. DNMT3A catalyzes de novo methylation of cytosine residues in DNA. Mutations in DNMT3A are present in ~30% of AML cases, mostly in AML patients who present with a normal karyotype [3,34]. Mutations include nonsense, frameshift, and missense alterations throughout the open-reading frame, with a significant enrichment (~40 to 60% of DNMT3A) for mutations at codon R882 [3]. This mutation has been shown to exert a dominant-negative effect on the wild-type DNMT3A and DNMT3B that reduces DNA methylation activity by ~80% in vitro [35,36]. DNMT3A-mutant AMLs frequently have co-occurring mutations NPM1 and FLT3-ITD and confer adverse-risk [37], and have been shown to promote anthracy-cline-resistance through impaired DNA damaging sensing [38]. Clonal hierarchy studies show that mutations in DNMT3A are one of the earliest events in leukemogenesis [39], making this an attractive therapeutic target for novel therapeutic approaches. This is underscored by studies showing that the most common mutations seen in preleukemic clonal hematopoiesis are in DNMT3A and that these mutations are often initiating events in myeloid malignancies [40–43]. Though important insights into the role of DNMT3A mutations in AML have emerged from human/preclinical studies, the fundamental mechanism by which these mutations lead to enhanced AML and increased self-renewal has not been delineated.
TET2
Ten-eleven-translocation (TET) proteins are α-ketoglutarate dependent DNA dioxygenases (TET1–3), which in the presence of oxygen, Fe2+ and ascorbic acid, catalyze the successive oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) and other oxidation products down to 5-carboxylcytosine (5caC) and promote passive and active DNA demethylation [44–47]. TET2 inactivation (but not TET1 or TET3) through loss-of-function mutations is a common clonal event in myeloid neoplasms [48,49], indicating that TET2 functions as a tumor suppressor. Mutations in TET2 have been identified in ~20% of patients with AML, and are enriched in patients with prior MDS or MPN, and has been found to be a variable prognostic indicator [50]. TET2 mutations co-occur with NPM1, FLT3-ITD, and DNMT3A, and largely mutually exclusive with IDH1/2 mutations [4]. The majority of the evidence indicates that TET2 is an early event (‘first hit’) in the multihit model of leukemogenesis, although additional hits are necessary for further progression. Consistent with this observation, TET2 mutations are also common in clonal hematopoiesis [51].
IDH1/2 mutations
Neomorphic mutations in the genes encoding isocitrate dehydrogenase 1 and 2 (IDH1 at R132 or in IDH2 at R140 or R172) in AML have been shown to lead to the production of the oncometabolite R enantiomer of 2-hydroxyglutratrate (R-2-HG), which inhibits dioxygenases-including TET family of enzymes-by competing with α-ketoglutarate [1,52,53]. The oncometabolite R-2-HG leads to DNA and histone hypermethylation, leading to a repressive chromatin landscape that disrupts cellular differentiation and contributes to leukemogenesis [54,55]. IDH1/2 mutations are respectively found in ~5 to 10% and ~15 to 20% of patients with AML, and are enriched in normal karyotype [56]. The prognostic significance of these mutations is controversial but appears to be influenced by co-mutational status (NPM1) and the specific location of the mutation. Ivosidenib and enasidenib are first-in-class, oral, selective inhibitors of the IDH1 and IDH2, respectively, and are FDA-approved for the management of adults with refractory or relapsed AML [57▪,58▪].
ASXL1
Additional sex combs-like 1 (ASXL1) is 1 of 3 mammalian homologs of the Drosophila additional sex combs (Asx), a protein that was originally identified as an enhancer of trithorax and polycomb genes and plays a critical role in regulating Hox gene expression [59]. ASXL1 forms the polycomb repressive deubiquitinase complex with BRCA1-associated protein 1 (BAP1), which deubiquitinates H2AK119Ub [60], a repressive mark. ASXL1 mutations promote myeloid transformation, at least in part, through attenuated PRC2-mediated histone H3K27 trimethylation [61], a repressive mark. It is not clear whether mutant-ASXL1 in myeloid malignancies are loss-of-function, dominant-negative, or gain-of-function mutations, which promote BAP1 deubiquitinase activity. ASXL1 mutations occur in ~10 to 20% of patients with AML and are enriched in those with underlying myelodysplasia and confer a poor prognosis [62]. These mutations are more common in older patients and coexist with RUNX1 mutations [63].
EZH2
The enhancer of zeste homolog 2 (EZH2) is a histone methyltransferase and functional core subunit of the PRC2, a key epigenetic regulator, which catalyzes the methylation of histone H3 at lysine 27 (H3K27me2/3) [64]. EZH2 play a critical role in epigenetic regulation during hematopoiesis. EZH2 mutation exert context-specific and sometimes opposing effects to the development of hematologic malignancies. Oncogenic gain-of-function EZH2 mutations are reported in lymphoid malignancies. In contrast, loss-of-function EZH2 mutations resulting in abrogation of histone methyltransferase activity occur in MDS/AML, suggesting that EZH2 functions as a tumor suppressor for myeloid malignancies [65,66]. These mutations are associated with shorter OS and event-free-survival and are enriched in AML patients with a previous history of MDS/MPN. The effects of loss-of-function EZH2 mutations on epigenetic and metabolic reprogramming (branch chain amino acid metabolism) may be exploited as potential therapeutic targets [67▪].
BCOR and BCORL1
BCL6 corepressor (BCOR) and BCL6 corepressor like 1 (BCORL1), are key transcription factors and function as components of PRC1.1, a noncanonical PRC1 complex, which monoubiquitinates histone 2A at lysine 119 (H2AK119ub) and mediates transcriptional repression [68▪]. Loss of Bcor function results in expansion of myeloid progenitor cells and cooperates with KrasG12D to drive leukemogenesis [69]. Mutations in BCOR and BCORL1 are reported in ~4% of AML patients with normal karyotype [70] and carriers an unfavorable prognostic significance.
Cohesin complex
The cohesin core complex constitutes a tripartite ring composed of SMC1A, SMC3, and RAD21 that are bound to a HEAT-repeat protein (STAG1 or STAG2). The cohesin complex functions to stabilize sister chromatids during metaphase, stabilize the replication fork, and structure the chromatin of the interphase genome [71]. Recurrent loss-of-function cohesin mutations are heterozygous and mutually exclusive from each other with a reported frequency of 6–12% of AML and are more prevalent in Downs syndrome-associated acute megakaryo-blastic leukemia [72] and secondary AML [73]. These mutations are not associated with aneuploidy and are not independently prognostic [73]. Functional studies in hematopoietic cells have demonstrated that cohesin mutations occur in preleukemic clones [74] and affect transcriptional regulation and lineage priming. This leads to enhanced self-renewal and defective differentiation of HSPCs [75–77]. STAG2 is commonly mutated across many hematopoietic and solid tumors. A recent study describing the consequences of Stag2 ablation in the HSPCs demonstrated a specific role for STAG2 in balancing self-renewal and differentiation in HSPCs, a critical feature of leukemogenesis [78▪].
Mutations in splicing factors
Alternative premRNA splicing is a primary source of diversity in messenger RNA species orchestrated by the macromolecular spliceosome complex [79]. Somatic mutations in the genes encoding splicing factors have been discovered at high frequency in patients with hematologic malignancies, including MDS and AML [80]. The most common mutations occur in SF3B1, SRSF2, U2AF1, and ZRSR2 and they tend to be mutually exclusive to one another, suggests synthetic lethal interactions when coexpressed and/or convergent biological effects of these mutations to hyperactivate innate immune signaling [81,82]. These mutations are most common in secondary AML [83]. A more recent study including 1540 patients with AML performed targeted sequencing of 111 genes and cytogenetic analysis and classified 11 subgroups including ~18% of AML patients with mutations in chromatin modifiers and spliceosome genes. The chromatin–spliceosome group was the second largest subgroup and was composed of older patients with lower white blood cell counts, a lower percentage of blasts, decreased responsiveness to chemotherapy, and overall poor survival [4]. A recent study analyzed the transcriptomes of 982 AML patients and found an overlap of mutations between SRSF2 and IDH2. This study demonstrated the synergy between RNA splicing and epigenetic regulation, which is partially because of co-regulation of the gene INTS3, a member of the integrator complex [84▪▪]. The deeper understanding of underlying mechanisms through which these mutations promote leukemogenesis are still being investigated, whereas several groups have demonstrated therapeutic targeting using genetic or pharmacologic perturbation of splicing [85–87].
Mutations in tumor-suppressor genes
TP53
The tumor suppressor gene TP53 encodes for the transcription factor p53 and is the most frequently mutated gene in human cancer. It plays a central role in multiple pathways in response to cellular stress, including cell cycle arrest, senescence and apoptosis [88]. Mutations in TP53 occur in less than 10% of patients with AML but are enriched in AML cases with genomic instability, including therapy-related, complex karyotype, and relapsed disease. TP53 mutations are mutually exclusive with mutations in NPM1, RUNX1, FLT3-ITD, and CEBPA. The majority of TP53 mutations are missense mutations and occur in the DNA-binding domain. A recent study has shown that missense TP53 mutation in myeloid malignancies do not lead to neomorphic gain-of-function activities but instead drive leukemogenesis through a dominant negative effect [89▪]. The resulting loss of activity of p53 favors genomic instability and resistance to chemotherapy. AML patients with TP53 mutations have been associated with a poor response to chemotherapy and dismal overall survival rates (median of 5–9 months) [90].
WT1
Wilm’s tumor 1 (WT1) is a zinger finger transcription factor that is mutated in less than 10% of patients with AML and the wild-type WT1 protein is often overexpressed in AML [91]. Loss-of-function mutations in WT1 led to marked reduction in 5hmC levels and a defect in hematopoietic differentiation, similar to that observed in TET2 mutation [92]. WT1 physically interacts and recruits TET2 to WT1-target genes to activate their expression [93]. Mutations in WT1 are mutually exclusive with TET2 and IDH1/2 mutations suggesting that these genes function in the same epigenetic pathway. These mutations are associated with younger age and adverse prognostic significance likely secondary to chemoresistance [94].
CONCLUSION
The heterogeneity of the genetic landscape of AML hallmarks the 40-year old notion of clonal evolution in cancer proposed by Peter Nowell. These genetic events have variance in their strength of leukemic drive and disease constitution is a reflection of either few strong drivers (i.e. NPM1) or serial acquisition of mutations of additive lower tumorigenic potential. Co-mutational interaction also has a major influence that is challenging to decode. For example, while DNMT3A and NPM1 mutations often cooccur, the presence of an NRAS mutation as compared with a FLT3-ITD have dramatically different effects on the prognosis and chemosensitivity of the resultant disease.
The toolbox of clinical leukemia physicians has recently been augmented by targeted agents with inhibitors against FLT3 and IDH1/2. Although both have improved outcomes, the response has not been akin to inhibitors of onco-fusion proteins, such as BCR-ABL with Imatinib. One challenge ahead is how to handle the complexity and multigenic nature of AML. Better still, as the detection of many of the disease alleles are found in clonal hematopoiesis, can we identify indications for earlier intervention, most notably in clonal cytopenia of undetermined significance. These targeted agents may find use in, as Peter Nowell once urged, ‘controlling the evolutionary process in tumors before it reaches the late stage’.
KEY POINTS.
Recurrent mutations in acute myeloid leukemia have functional consequences beyond gain of oncogene/loss of tumor suppressor.
Mutations, such as NPM1c often are sufficient for transformation.
Other alleles require cooperating mutations and are frequently found in preleukemic entities, such as myelodysplastic syndrome and clonal hematopoiesis.
Acknowledgements
This work was supported by a National Cancer Institute career development grant K08 CA215317 (A.D.V), the William Raveis Charitable Fund Fellowship of the Damon Runyon Cancer Research Foundation (DRG 117–15) (A.D.V), and an Evans MDS Young Investigator grant from the Edward P. Evans 35 Foundation (A.D.V). A.K. is supported by the VeloSano Catalyst Award. R.L.L. was supported by National Cancer Institute awards R35 CA197594-01A1 (R.L.L.), R01 CA216421 (R.L.L.), and PS-OC U54 CA143869-05 (R.L.L.) and a grant from Leukemia & Lymphoma Society Translational Research Foundation 6499-17 (R.L.L.).
Conflicts of interest
A.D.V. received travel support from Mission Bio and is on the Editorial Advisory Board of Hematology News. R.L.L. is on the supervisory board of QIAGEN and is a scientific advisor to Loxo, Imago, C4 Therapeutics, and Isoplexis. He receives research support from and consulted for Celgene and Roche and has consulted for Janssen, Astellas, Morphosys, and Novartis. He has received honoraria from Roche, Lilly, and Amgen for invited lectures and from Gilead for grant reviews. A.K. has no competing interests to disclose.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
- 1.Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. New Engl J Med 2009; 361:1058–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012; 366:1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research Network. Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013; 368:2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016; 374:2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falini B, Mecucci C, Tiacci E, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. New Engl J Med 2005; 352:254–266. [DOI] [PubMed] [Google Scholar]
- 6.▪.Brunetti L, Gundry MC, Sorcini D, et al. Mutant NPM1 maintains the leukemic state through HOX expression. Cancer Cell 2018; 34:499–512.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first article to elucidate the role of mutant NPM1 and its cytoplasmic mislocalization in AML.
- 7.Gu X, Ebrahem Q, Mahfouz RZ, et al. Leukemogenic nucleophosmin mutation disrupts the transcription factor hub that regulates granulomonocytic fates. J Clin Invest 2018; 128:4260–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127:2391–2405. [DOI] [PubMed] [Google Scholar]
- 9.▪▪.Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017; 129:424–447. [DOI] [PMC free article] [PubMed] [Google Scholar]; Recommended guidelines for prognostication, diagnosis and suggested treatments in AML.
- 10.Meshinchi S, Appelbaum FR. Structural and functional alterations of FLT3 in acute myeloid leukemia. Clinical Cancer Res 2009; 15:4263–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002; 99:4326–4335. [DOI] [PubMed] [Google Scholar]
- 12.Sengsayadeth SM, Jagasia M, Engelhardt BG, et al. Allo-SCT for high-risk AML-CR1 in the molecular era: impact of FLT3/ITD outweighs the conventional markers. Bone Marrow Transplant 2012; 47:1535–1537. [DOI] [PubMed] [Google Scholar]
- 13.▪▪.Hourigan C, Dillon LW, Gui G, et al. Impact of conditioning intensity of allogeneic transplantation for acute myeloid leukemia with genomic evidence of residual disease. J Clin Oncol 2019; In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]; Prognostic impact of minimal residual disease depends on the mutated gene with myeloablative conditioning failing to improve transplant outcomes with clonal persistence.
- 14.▪▪.Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. New Engl J Med 2017; 377:454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]; A large study, which for the first time incorporates targeted therapy with conventional chemotherapy in AML.
- 15.▪.Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. New EnglJ Med 2019; 381: 1728–1740. [DOI] [PubMed] [Google Scholar]; The phase 3 study that led to the FDA-approval of gilteritinib in relapsed or refractory FLT3-mutated AML.
- 16.Milne P, Wilhelm-Benartzi C, Grunwald MR, et al. Serum Flt3 ligand is a biomarker of progenitor cell mass and prognosis in acute myeloid leukemia. Blood Adv 2019; 3:3052–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malaise M, Steinbach D, Corbacioglu S. Clinical implications of c-Kit mutations in acute myelogenous leukemia. Curr Hematol Malig Rep 2009; 4:77–82. [DOI] [PubMed] [Google Scholar]
- 18.Paschka P, Marcucci G, Ruppert AS, et al. , Cancer and Leukemia Group B. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): a Cancer and Leukemia Group B Study. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2006; 24:3904–3911. [DOI] [PubMed] [Google Scholar]
- 19.Li S, Balmain A, Counter CM. A model for RAS mutation patterns in cancers: finding the sweet spot. Nature reviews Cancer 2018; 18:767–777. [DOI] [PubMed] [Google Scholar]
- 20.Bacher U, Haferlach T, Schoch C, et al. Implications of NRAS mutations in AML: a study of 2502 patients. Blood 2006; 107:3847–3853. [DOI] [PubMed] [Google Scholar]
- 21.▪.Kunimoto H, Meydan C, Nazir A, et al. Cooperative epigenetic remodeling by TET2 loss and nras mutation drives myeloid transformation and MEK inhibitor sensitivity. Cancer cell 2018; 33:44.e8–59.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]; Mutations in epigenetic modifiers and signaling factors often co-occur in AML. The study showed that this cooperativity between these mutations can be exploited for therapeutic vulnerabilities.
- 22.▪.McMahon CM, Ferng T, Canaani J, et al. Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov 2019; 9:1050–1063. [DOI] [PubMed] [Google Scholar]; This study illustrates dynamic and complex changes in clonal architecture unerlying response and resistance to FLT3 kinase inhibition.
- 23.Ito Y, Bae SC, Chuang LS. The RUNX family: developmental regulators in cancer. Nat Rev Cancer 2015; 15:81–95. [DOI] [PubMed] [Google Scholar]
- 24.Mendler JH, Maharry K, Radmacher MD, et al. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and MicroRNA expression signatures. J Clin Oncol 2012; 30:3109–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song WJ, Sullivan MG, Legare RD, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet 1999; 23:166–175. [DOI] [PubMed] [Google Scholar]
- 26.Avellino R, Delwel R. Expression and regulation of C/EBPalpha in normal myelopoiesis and in malignant transformation. Blood 2017; 129: 2083–2091. [DOI] [PubMed] [Google Scholar]
- 27.Pabst T, Mueller BU, Zhang P, et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nature Genet 2001; 27:263–270. [DOI] [PubMed] [Google Scholar]
- 28.Fasan A, Haferlach C, Alpermann T, et al. The role of different genetic subtypes of CEBPA mutated AML. Leukemia 2014; 28:794–803. [DOI] [PubMed] [Google Scholar]
- 29.Smith ML, Cavenagh JD, Lister TA, Fitzgibbon J.Mutation of CEBPA in familial acute myeloid leukemia. New Engl J Med 2004; 351:2403–2407. [DOI] [PubMed] [Google Scholar]
- 30.Crispino JD, Horwitz MS. GATA factor mutations in hematologic disease. Blood 2017; 129:2103–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greif PA, Dufour A, Konstandin NP, et al. GATA2 zinc finger 1 mutations associated with biallelic CEBPA mutations define a unique genetic entity of acute myeloid leukemia. Blood 2012; 120:395–403. [DOI] [PubMed] [Google Scholar]
- 32.Hahn CN, Chong CE, Carmichael CL, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet 2011; 43:1012–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ostergaard P, Simpson MA, Connell FC, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat Genet 2011; 43:929–931. [DOI] [PubMed] [Google Scholar]
- 34.Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. New Engl J Med 2010; 363:2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Russler-Germain DA, Spencer DH, Young MA, et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer cell 2014; 25:442–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holz-Schietinger C, Matje DM, Reich NO. Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J Biol Chem 2012; 287:30941–30951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marcucci G, Metzeler KH, Schwind S, et al. Age-related prognostic impact of different types of DNMT3A mutations in adults with primary cytogenetically normal acute myeloid leukemia. J Clin Oncol 2012; 30:742–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guryanova OA, Shank K, Spitzer B, et al. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat Med 2016; 22:1488–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012; 150:264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. New Engl J Med 2014; 371:2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. New Engl J Med 2014; 371:2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014; 20: 1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shlush LI, Zandi S, Mitchell A, et al. Identification of preleukaemic haematopoietic stem cells in acute leukaemia. Nature 2014; 506:328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ko M, Huang Y, Jankowska AM, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010; 468:839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009; 324:930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ito S, Shen L, Dai Q, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011; 333:1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He YF, Li BZ, Li Z, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011; 333: 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delhommeau F, Dupont S, Della Valle V, et al. Mutation in TET2 in myeloid cancers. The New England journal of medicine 2009; 360:2289–2301. [DOI] [PubMed] [Google Scholar]
- 49.Jankowska AM, Szpurka H, Tiu RV, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 2009; 113:6403–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abdel-Wahab O, Mullally A, Hedvat C, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009; 114:144–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Busque L, Patel JP, Figueroa ME, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet 2012; 44:1179–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010; 18:553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011; 19:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012; 483:474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Losman JA, Looper RE, Koivunen P, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013; 339:1621–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Medeiros BC, Fathi AT, DiNardo CD, et al. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 2017; 31:272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.▪.Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017; 130:722–731. [DOI] [PMC free article] [PubMed] [Google Scholar]; Monotherapy targeting the IDH2 mutation demonstrates efficacy with a favorable safety profile. This study led to the FDA-approval of enasidenib in relapsed or refractory IDH2-mutated AML.
- 58.▪.DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. New Engl J Med 2018; 378:2386–2398. [DOI] [PubMed] [Google Scholar]; Monotherapy targeting the IDH1 mutation demonstrates efficacy with a favorable safety profile. This study led to the FDA approval of ivosidenib in relapsed or referactory IDH1-mutated AML.
- 59.Micol JB, Abdel-Wahab O. The role of additional sex combs-like proteins in cancer. Cold Spring Harb Perspect Med 2016; 6:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Scheuermann JC, de Ayala Alonso AG, Oktaba K, et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 2010; 465:243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abdel-Wahab O, Adli M, LaFave LM, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer cell 2012; 22:180–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Metzeler KH, Becker H, Maharry K, et al. ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood 2011; 118:6920–6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schnittger S, Eder C, Jeromin S, et al. ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia 2013; 27:82–91. [DOI] [PubMed] [Google Scholar]
- 64.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life Nature 2011; 469:343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ernst T, Chase AJ, Score J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nature Genet 2010; 42:722–726. [DOI] [PubMed] [Google Scholar]
- 66.Nikoloski G, Langemeijer SM, Kuiper RP, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nature Genet 2010; 42:665–667. [DOI] [PubMed] [Google Scholar]
- 67.▪.Gu Z, Liu Y, Cai F, et al. Loss of EZH2 reprograms BCAA metabolism to drive leukemic transformation. Cancer Discov 2019; 9:1228–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uncovered a mechanism by which epigenetic alterations rewire metabolism during leukemogenesis, causing epigenetic and metabolic liabilities that may be exploited as potential therapeutics.
- 68.▪.Di Carlo V, Mocavini I, Di Croce L. Polycomb complexes in normal and malignant hematopoiesis. J Cell Biol 2019; 218:55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]; An excellent review on Polycomb group protein complexes, which are frequently disregulated in hematologic malignancies.
- 69.Kelly MJ, So J, Rogers AJ, et al. Bcor loss perturbs myeloid differentiation and promotes leukaemogenesis. Nat Commun 2019; 10:1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grossmann V, Tiacci E, Holmes AB, et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood 2011; 118:6153–6163. [DOI] [PubMed] [Google Scholar]
- 71.Nasmyth K, Haering CH. Cohesin: its roles and mechanisms. Annu Rev Genet 2009; 43:525–558. [DOI] [PubMed] [Google Scholar]
- 72.Kon A, Shih LY, Minamino M, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet 2013; 45:1232–1237. [DOI] [PubMed] [Google Scholar]
- 73.Thota S, Viny AD, Makishima H, et al. Genetic alterations of the cohesion complex genes in myeloid malignancies. Blood 2014; 124:1790–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jan M, Snyder TM, Corces-Zimmerman MR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med 2012; 4:149ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mazumdar C, Shen Y, Xavy S, et al. Leukemia-associated cohesin mutants dominantly enforce stem cell programs and impair human hematopoietic progenitor differentiation. Cell Stem Cell 2015; 17:675–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Viny AD, Ott CJ, Spitzer B, et al. Dose-dependent role of the cohesin complex in normal and malignant hematopoiesis. J Exp Med 2015; 212:1819–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mullenders J, Aranda-Orgilles B, Lhoumaud P, et al. Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J Exp Med 2015; 212:1833–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.▪.Viny AD, Bowman RL, Liu Y, et al. Cohesin members Stag1 and Stag2 display distinct roles in chromatin accessibility and topological control of HSC self-renewal and differentiation. Cell Stem Cell 2019; 25: 682.e8–696.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study elucidated the role of STAG2 in hematopoiesis. The study described the consequences of Stag2 ablation in the HSPCs demonstrated a specific role for STAG2 in balancing self-renewal and differentiation in HSPCs, a critical feature of leukemogenesis.
- 79.Lee Y, Rio DC. Mechanisms and regulation of alternative PremRNA splicing. Annu Rev Biochem 2015; 84:291–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Anczukow O, Krainer AR. Splicing-factor alterations in cancers. RNA 2016; 22:1285–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011; 478:64–69. [DOI] [PubMed] [Google Scholar]
- 82.Lee SC, North K, Kim E, et al. Synthetic lethal and convergent biological effects of cancer-associated spliceosomal gene mutations. Cancer cell 2018; 34:225.e8–241.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015; 125:1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.▪▪.Yoshimi A, Lin KT, Wiseman DH, et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature 2019; 273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]; An elegant study identified a pathogenic crosstalk between altered epigenetic state and splicing in AML, providing functional evidence that mutations in splicing factors drive myeloid malignancy development.
- 85.Obeng EA, Chappell RJ, Seiler M, et al. Physiologic expression of Sf3b1(K700E) causes impaired erythropoiesis, aberrant splicing, andsensitivity to therapeutic spliceosome modulation. Cancer Cell 2016; 30:404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee SC, Dvinge H, Kim E, et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med 2016; 22:672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Seiler M, Yoshimi A, Darman R, et al. H3B-8800, an orally available smallmolecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med 2018; 24:497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell 2017; 170:1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.▪.Boettcher S, Miller PG, Sharma R, et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 2019; 365:599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed in myeloid cancers, missense mutations in TP53 are selected because of their dominant-negative effects.
- 90.Stengel A, Kern W, Haferlach T, et al. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: an analysis of 3307 cases. Leukemia 2017; 31:705–711. [DOI] [PubMed] [Google Scholar]
- 91.Rampal R, Figueroa ME. Wilms tumor 1 mutations in the pathogenesis of acute myeloid leukemia. Haematologica 2016; 101:672–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rampal R, Alkalin A, Madzo J, et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep 2014; 9:1841–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang Y, Xiao M, Chen X, et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol Cell 2015; 57:662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hou HA, Huang TC, Lin LI, et al. WT1 mutation in 470 adult patients with acute myeloid leukemia: stability during disease evolution and implication of its incorporation into a survival scoring system. Blood 2010; 115: 5222–5231. [DOI] [PubMed] [Google Scholar]