

Abstract
Acute pancreatitis is a common inflammatory gastrointestinal disease without any successful treatment. Pancreatic exocrine acinar cells have high rates of protein synthesis to produce and secrete large amounts of digestive enzymes. When the regulation of organelle and protein homeostasis is disrupted, it can lead to endoplasmic reticulum (ER) stress, damage to the mitochondria and improper intracellular trypsinogen activation, ultimately resulting in acinar cell damage and the onset of pancreatitis. To balance the homeostasis of organelles and adapt to protect themselves from organelle stress, cells use protective mechanisms such as autophagy. In the mouse pancreas, defective basal autophagy disrupts ER homoeostasis, leading to ER stress and trypsinogen activation, resulting in spontaneous pancreatitis. In this review, we discuss the regulation of autophagy and its physiological role in maintaining acinar cell homeostasis and function. We also summarise the current understanding of the mechanisms and the role of defective autophagy at multiple stages in experimental pancreatitis induced by cerulein or alcohol.
INTRODUCTION
Acute pancreatitis (AP) is a common gastrointestinal disease with an annual incidence rate of 34 per 100 000 person-years in developed countries. This disease is characterised by an increased necrosis of acinar cells, followed by a local and systemic inflammatory response, and has a varied clinical course. Most patients present with mild acute pancreatitis, which self-resolves within a week. However, approximately one-fifth of patients develop moderate or severe acute pancreatitis, which has a substantial mortality rate of 20%–40%.1 2 Gallstone disease and alcohol consumption are the most common causes of acute pancreatitis. Other risk factors include hypertriglyceridaemia, hypercalcaemia, viral infection, genetics and autoimmune disease.1 2
Chronic alcohol consumption is responsible for 17%–25% of acute pancreatitis cases worldwide. Alcohol-associated pancreatitis usually affects individuals who have been consuming substantial amounts of alcohol on a daily basis for over 5 years, typically around four to five drinks per day. Isolated episodes of heavy drinking are rarely the cause of AP. However, when combined with other risk factors such as genetics, high-fat diet, cigarette smoking and exposure to infectious agents, alcohol consumption can further compound the damage to the pancreas.1 3 Notably, only less than 10% of heavy drinkers develop alcohol-associated pancreatitis, whereas most heavy drinkers have mild and autolimited pathological changes, suggesting that a refined adaptation underlies the autolimited induction of alcohol-associated pancreatitis.4 Pancreatic exocrine acinar cells possess high protein synthetic rates to produce, store and secrete large amounts of digestive enzymes. To meet the high demands of protein synthesis and trafficking, acinar cells are exceptionally enriched with endoplasmic reticulum (ER) and other organelle machinery, including the mitochondria, secretory vesicles, endosomes, autophagy and lysosomes. Dysregulation of ER homeostasis can lead to ER stress and acinar cell damage, resulting in the onset of pancreatitis.5 6 Cells use protective mechanisms, such as autophagy, to balance the homeostasis of organelles so that the ER and the mitochondria can adapt and protect themselves from ER stress and mitochondrial damage. Here, we summarise the current understanding of the roles and mechanisms of autophagy in the pathogenesis of pancreatitis.
CELLULAR AND MOLECULAR EVENTS OF AUTOPHAGY
Autophagy is a cellular degradation process that delivers excess or damaged cellular components to lysosomes, an evolutionarily conserved process.7 8 Autophagy degrades proteins, lipids and organelles, such as excess ER, ribosomes and damaged mitochondria. This provides nutrients and building blocks to maintain cellular homeostasis and enable cell survival.9–13 There are three main types of autophagy: macroautophagy, microautophagy and chaperone-mediated autophagy (CMA). These types differ in how cargos are delivered to lysosomes.14 Macroautophagy involves the formation of a double-membrane autophagosome that surrounds the autophagic cargo and transports it to a lysosome, where the membranes fuse to form an autolysosome that degrades autophagic cargos by lysosomal acidic hydrolytic enzymes.8 During microautophagy, lysosomes directly engulf autophagic cargos, bypassing autophagosome formation.15 16 The process of CMA involves the recognition of cellular proteins that contain the pentapeptide motif (KFERQ) by cytosolic chaperones, such as the HSC70 (heat shock cognate protein of 70 kDa). The chaperone proteins then bind to the lysosome-associated membrane protein type 2A (LAMP-2A), triggering LAMP-2A multimerisation. This process results in the formation of a translocation complex, which transports the CMA substrates across the lysosomal membrane for degradation.17–19
Microautophagy and CMA have been previously reviewed for their mechanisms and role in pathophysiology.20 21 However, their connection with pancreatitis has not been extensively studied. Therefore, in this review, we will focus on the current understanding of autophagy mechanisms and their potential role in pancreatitis. At present, more than 40 autophagy-related (ATG) proteins have been identified, which regulate the six key steps of autophagy and are detailed in the following.
Initiation
The initiation of autophagosome formation is regulated by the uncoordinated 51-like kinase (ULK) complex, consisting of ULK1 (or ULK2), FAK family-interacting protein of 200 kDa (FIP200), ATG13 and ATG101.22 Serine/threonine-protein kinase ULK1 is the sole kinase discovered among all ATG proteins. Its activity is inhibited by a nutrient sensor called the mechanistic target of rapamycin complex 1 (mTORC1) and stimulated by an energy sensor called AMP-activated protein kinase (AMPK).23 When there is a shortage of nutrients, mTORC1 is inhibited. This leads to the activation of ULK1, which is phosphorylated and activated. This results in the formation of an isolation membrane also called a phagophore. The exact source of the membrane for the isolation membrane is still being debated. However, it is suggested that the ER, omegasomes (a portion of the ER membrane extension), ER–mitochondria contact site, ER–Golgi intermediate compartment (ERGIC), and plasma membrane or endocytosis-derived vesicles could be contributing to the formation of autophagosomes.24
Autophagosome membrane biogenesis
The second autophagy-specific kinase complex, PIK3C3 complex I (PIK3C3-CI), consists of VPS34 (vacuolar protein sorting 34), BECLIN-1, VPS15 and ATG14L (ATG14-like). PIK3C3-CI is recruited to the initiation sites of the autophagosome by the activated ULK complex. The phosphorylation of several PIK3C3-CI subunits, including BECLIN-1 and VPS34, and the direct binding of ATG13 in the ULK complex with ATG14L in PIK3C3-CI achieve this process.25 26 VPS34 is an enzyme that belongs to class III phosphatidylinositol 3-phosphate kinase family. It produces a molecule called phosphatidylinositol 3-phosphate (PI3-P), which acts as a signalling molecule. This molecule then recruits other proteins, such as WD repeat protein interacting with phosphoinositide (WIPI) and double FYVE containing protein 1 (DFCP1), that bind to PI3-P. DFCP1 and WIPI then work together to form the omegasome, which is a structure that looks like the Greek letter omega (Ω). The omegasome is formed at a specific subdomain of the ER enriched with PI3-P.27 Concurrently, there are two ubiquitin-like conjugation systems that operate in the process of autophagy. These systems are the ATG7–ATG3–ATG8/microtubule-associated protein 1A/1B-light chain 3 (LC3) and ATG12–ATG5–ATG16L1 complexes. The function of these complexes is to conjugate the cytosolic form of LC3 (also known as LC3-I) to phosphatidylethanolamine (PE). This process creates the membrane-anchored LC3-II, a widely known autophagy marker protein. LC3-II is functionally essential for the recognition of cargo, expansion of the autophagosome membrane, closure of the autophagosome and possible fusion with the lysosome. In the ubiquitin-like conjugation system, ATG proteins behave similarly to ubiquitin enzymes. The process begins with the covalent binding of ATG12 to ATG5. This reaction is catalysed by ATG7, acting as an E1-like enzyme, and ATG10, acting as an E2-like enzyme. The resulting ATG12–ATG5 conjugate then interacts with ATG16L1, forming a non-covalent bond. This complex acts as an E3 ubiquitin ligase on the isolation membrane for LC3 lipidation. Before entering the system, newly synthesised pro-LC3 is cleaved by ATG4 proteases, exposing a free C-terminal glycine residue and transforming into the cytosolic form LC3-I. The E3-like ATG12–ATG5–ATG16L1 complex, together with the E1-like ATG7 and the E2-like ATG3, then mediates the covalent binding of LC3-I to the PE, forming the lipidated LC3-II that resides on the autophagosome membrane.28
Autophagosome membrane expansion and elongation
Autophagosome membrane expansion requires lipid delivery. ATG2A and VPS13 family proteins are two lipid transport proteins that possess high-capacity lipid-binding surfaces. They act as a non-selective lipid tunnel which is most likely located at a contact site between the edge of the expanding isolation membrane and the exit site of the endoplasmic reticulum.29 ATG9 is a unique ATG protein with multimembrane spanning domains and lipid scramblase activity. It plays a crucial role in transporting lipids with the help of proteins. Additionally, ATG9 is responsible for recycling membrane components from other organelles like endosomes and the ERGIC to the site of autophagosome biogenesis. This process is dependent on the ULK complex. Although there is no direct evidence, lipid extrusions from the ER and mitochondria have also been proposed as potential sources for phagophore expansion.25
Autophagosome closure and maturation
As the autophagosome membrane expands, it eventually envelops the cargo and detaches from the ER to form a self-contained autophagosome. The endosomal sorting complexes required for transport (ESCRT) machinery play a crucial role in regulating phagophore closure, as evidenced by the accumulation of autophagosomes containing holes in cells depleted of ESCRT.30 31
CHAMP2A, an ESCRT-III component, is required for autophagosome closure. This is shown by a HaloTag-LC3 assay that distinguishes unclosed from closed autophagosome membranes and an optogenetic assay for sealed versus open mitophagosomes.32 33 The mechanism by which the ESCRT machinery is directed towards unsealed phagophores is not yet fully understood. However, in the yeast system, it has been shown that the interaction of CHMP4 and ATG17 (the human counterpart of FIP200) may play a role in recruiting the ULK complex to ESCRT-III.34 The ER-localised metazoan-specific protein vacuole membrane protein 1 (VMP1; also known as transmembrane protein 49/TMEM49) and its interactor TMEM41B are phospholipid scramblases that may play roles in ER–membrane communication and autophagosome closure.35 VMP1 may also regulate organelle–organelle contact and the departure of closed autophagosomes from the ER. Deletion of VMP1 leads to tight isolation membrane–ER contacts, thus resulting in failed fusion of the autophagosome with the lysosome.36 More recent evidence indicates that VMP1 and TMEM41B also regulate ER–mitochondria contact and lipoprotein secretion independent of their autophagy functions.37–40
Fusion of autophagosomes with lysosomes
After the autophagosome has been closed, it joins together with the lysosome to create an autolysosome. The autolysosome is responsible for breaking down the enclosed contents using acidic lysosomal hydrolases. The fusion process is facilitated by SNARE (soluble NSF (N-ethylmaleimide-sensitive fusion protein) attachment protein receptor) family proteins, which include vesicle-associated membrane protein 7 (VAMP7), VAMP8, VAMP9, synaptosomal-associated protein 29 (SNAP29) and syntaxin 17 (STX17).41–43 Before fusion occurs, a specific group of small GTPase proteins, such as RAB7 and RAB2A, move to the late endosomes and autophagosomes. This movement connects them to motor proteins associated with the cytoskeleton, enabling successful encounters. In addition, tethering factors like the HOPS (homotypic fusion and protein sorting) complex can bind to RAB7 or RAB2A and help anchor autophagosomes to lysosomes, facilitating the fusion process.44
Termination of autophagy and lysosome biogenesis
After autophagy is complete, the process is terminated, and lysosomes are reformed through a process called autophagic lysosome reformation (ALR). During ALR, proto-lysosomes are generated from the tubulation, scission and budding of autolysosomes.45 Additionally, new lysosomes can be generated through a transcriptional programme mediated by the master lysosomal biogenesis transcription factor EB (TFEB).46 This process helps meet the needs of autophagic degradation in various tissues and cells, including the pancreas.47 48 The mammalian autophagy machinery is summarised in figure 1.
Figure 1.
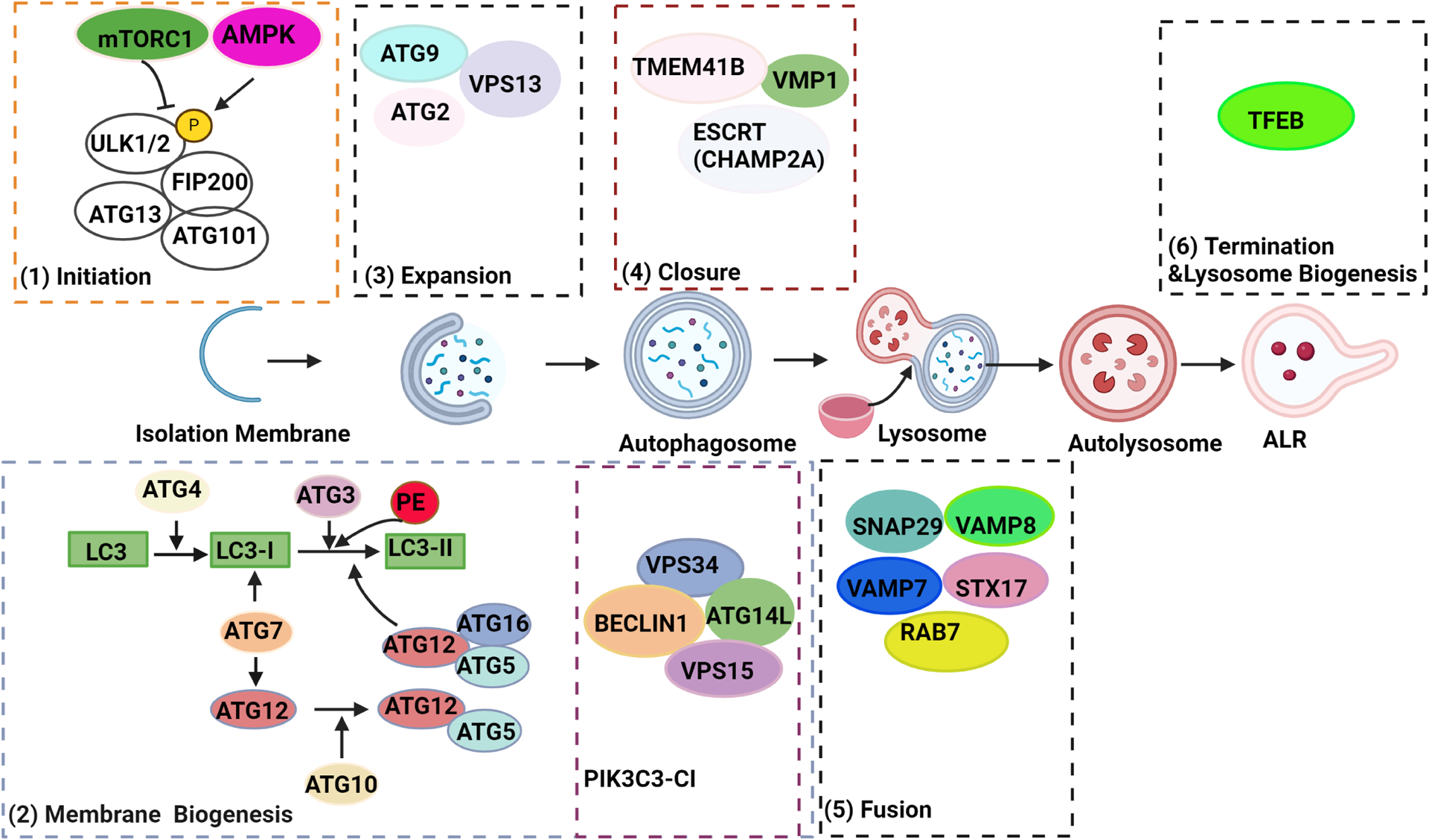
A scheme for the autophagy machinery in mammalian cells. Nutrient deprivation inhibits mTORC1 and activates AMPK, which then dephosphorylates or phosphorylates ULK1 at different sites, leading to the activation of the ULK complex and initiation of the isolation membrane (phagophore) formation. The activated ULK complex recruits the PIK3C3 complex I (PIK3C3-CI) to the initiation sites of the autophagosome, producing the signalling molecule phosphatidylinositol 3-phosphate to promote phagophore membrane biogenesis. Concurrently, two ubiquitin-like conjugation systems, the ATG7–ATG3–ATG8/microtubule-associated protein 1A/1B-light chain 3 (LC3) and ATG12–ATG5–ATG16L1 complexes, conjugate the cytosolic form LC3 (called LC3-I) to phosphatidylethanolamine (PE), forming an autophagosome membrane-anchored LC3-II. By supplying membranes from donor sources, ATG9-mediated cycling systems, which comprise the core proteins ATG9, ATG2, VPS13D and WIPI1/2, facilitate the elongation of phagophore. A mature autophagosome will be formed when the extending phagophore closes, mediated by the ESCRT protein CHAMP2A and the ER transmembrane protein VMP1 and TMEM41B. Autophagosomes eventually fuse with the lysosome mediated by STX17–VAMP7/8–SNAP29 complex and the GTPase RAB7 and complex HOPS, resulting in the degradation of cargos within autolysosomes. Autolysosomes then undergo ALR and lysosome biogenesis mediated by TFEB. ALR, autophagic lysosome reformation; ER, endoplasmic reticulum; ESCRT, endosomal sorting complexes required for transport; TFEB, transcription factor EB.
CELLULAR ORGANELLE STRESS AND AUTOPHAGY IN PANCREATITIS
Pancreas physiology
The pancreas is a complex metabolic organ with both endocrine and exocrine components. The endocrine component makes up only 2% of the pancreatic mass. It regulates blood glucose homeostasis by various types of endocrine cells clustered in islets (Langerhans) dispersed throughout the pancreatic parenchyma (figure 2A, arrow). These cell types include α cells, β cells, δ cells and pancreatic polypeptide or γ cells. They secrete different hormones such as glucagon, insulin, somatostatin and pancreatic polypeptide in response to the status of fasting or food intake. After a meal, insulin-producing β cells, the most common cell type in the islet, increase insulin secretion to lower blood glucose levels. Conversely, during the fasting stage, glucagon-producing α cells increase glucagon secretion to promote hepatic glycogen breakdown and glucose release.49
Figure 2.
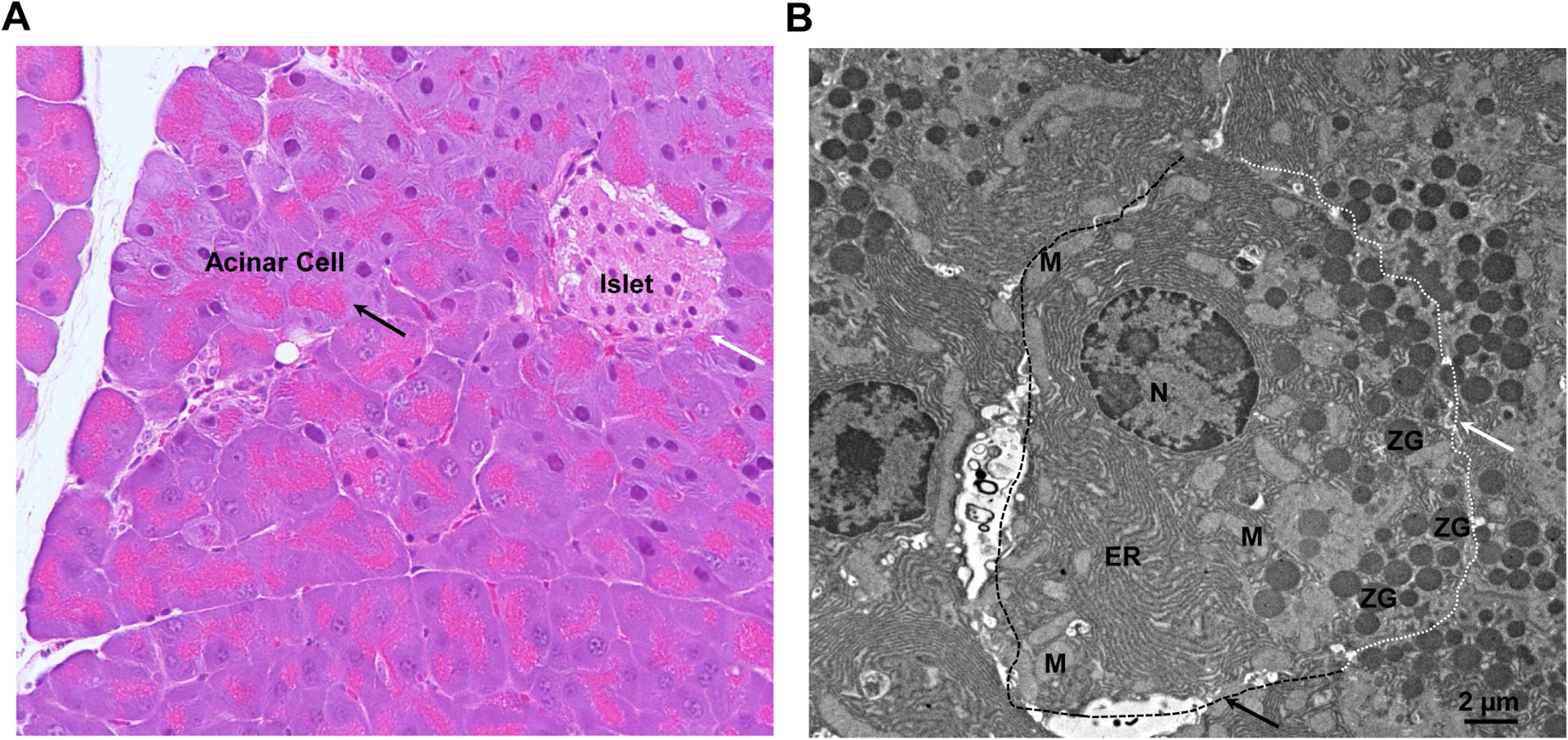
Histology of endocrine and exocrine pancreas and ultrastructure of exocrine acinar cells. (A) Representative H&E staining image of a mouse pancreas. The black arrow denotes the acinar cells and the white arrow denotes the islet. (B) Representative image of electron microscopy analysis of mouse pancreatic acinar cells. The white dotted line and the arrow denote the apical membrane. The black dotted line and the arrow denote the basal lateral membrane. ER, endoplasmic reticulum; M, mitochondria; N, nucleus; ZG, zymogen granule.
The exocrine pancreas, which accounts for more than 95%–98% of the pancreatic mass, is the main contributor to pancreatitis. Structurally, the exocrine pancreas comprised lobules, with acinar cells surrounding a duct system. The exocrine compartment contains two major cell types: the acinar cells, which produce enzymes that are transported to the gut via a ductal system that is lined up with the other prominent cell type, and the ductal cells (figure 2A). The primary function of the exocrine pancreas is to facilitate food digestion by secreting digestive enzymes from acinar cells into the pancreatic ducts and reaching the intestine. Owing to its functions for producing and secreting digestive enzymes in a timely manner to meet the demand after food consumption, acinar cells have developed complex intracellular protein synthesis processes as well as storage and trafficking machineries. Pancreatic acinar cells are highly polarised cells with distinct basal lateral and apical membranes. Acinar cells have remarkably abundant ER adjacent to the basal lateral membrane, whereas zymogen granules (ZGs) cluster around the apical membrane. These granules contain and store trypsinogen, the inactive precursor to trypsin, a key digestive protease (figure 2B). After the meal, the contents of ZGs are secreted to the pancreatic duct, and trypsinogen is activated to trypsin in the duodenum by enteropeptidase for protein digestion.50
Pancreatitis pathogenesis has been traditionally believed to be triggered by intra-acinar cell trypsin activation, which leads to acinar cell ‘autodigestion’, necrosis and subsequent pancreatic inflammation. This hypothesis is based on two key presumptions: (1) pancreatic trypsinogen is prematurely activated in the pancreas early in the course of pancreatitis, and (2) the pathologically activated enzymes are responsible for ‘auto digesting the pancreas’, resulting in acinar cell necrosis and progression of pancreatitis. Experimental pancreatitis models have well documented the increased acinar cell trypsinogen activation.1 51 52 In addition, genetic mutations of serine protease 1 (PRSS1) and the serine protease inhibitor gene (SPINK1) are linked to hereditary pancreatitis.53 54 The PRSS1 gene encodes cationic trypsinogen, and most of the PRSS1 variants convert trypsinogen to trypsin prematurely within the pancreas, whereas others prevent the degradation of trypsin. SPINK1 encodes a trypsin inhibitor that can inhibit 20% of trypsin activity, which is expressed in the pancreatic acinar cells and binds to activated trypsin. SPINK1 may cause chronic pancreatitis in the setting of autosomal recessive inheritance by promoting premature trypsinogen activation.53 54
Recent research has challenged the traditional belief that trypsinogen activation is the primary cause of pancreatitis.55 Studies using mice genetically modified to lack trypsinogen isoform 7 (T7) have shown that while these mice do not exhibit pathological trypsinogen activation and are partially protected against early acinar cell damage induced by cerulein, they still show an inflammatory response in acute pancreatitis. Additionally, when exposed to a chronic pancreatitis model induced by cerulein, T7 knockout mice still develop pancreatic atrophy, chronic inflammation and other histological features of chronic pancreatitis similar to those of wild-type mice.56 57 This suggests that the development of chronic pancreatitis may be independent of trypsinogen activation. Further research may be necessary to determine whether intra-acinar trypsin activation is a prerequisite for the pathogenesis of pancreatitis, both acute and chronic, in various animal or more clinically relevant pancreatitis models.
Acinar cell damage is believed to be the major trigger for acute pancreatitis. The main function of the pancreatic acinar cell is to synthesise, transport, store and secrete digestive enzymes. To do this, several intracellular organelles, such as the ER, mitochondria, Golgi apparatus, endolysosomal and autophagy system, and storage and secretory organelles, must work together. If these organelles do not function properly, it can lead to pancreatitis. This is a common problem in both humans and experimental animals and has been well documented in recent excellent review papers.58–60 In the following sections, we will focus on the latest research regarding ER homeostasis, mitochondrial damage, lysosomal dysfunction and impaired autophagy in the development of pancreatitis.
ER homeostasis and ER stress
Acinar cells, which produce zymogens, are prone to ER stress due to the high risk of protein misfolding. To counteract this, acinar cells use the unfolded protein response (UPR) to restore cellular homeostasis. This involves decreasing general protein translation and increasing the translation of chaperone proteins to facilitate protein folding. There are three branches of functional UPR: inositol-requiring enzyme 1α (IRE1α)-the spliced X-box binding protein 1 (XBP1s), protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK)-eukaryotic initiation factor 2α (eIF2α)-activating transcription factor 4 (ATF4), and the ATF6 axis that mediates the transcription of ER stress-inducible genes.61 If the UPR is insufficient to relieve ER stress, ER stress can further activate ER-associated proteasomal degradation (ERAD) or autophagic degradation as another layer of adaptive response.61 62 Failure to adapt to ER stress can lead to acinar cell death and thus pancreatitis.6 Thus, UPR and autophagy can act as the homeostatic adaptive processes to balance the effect of proteostasis in the exocrine pancreas. Activation of XBP1 via increased levels of XBP1s, a key transcription factor, increases the expression of genes involving oxidative protein folding, ERAD and lipid synthesis. Alcohol feeding increases ER stress and activation of the PERK-eIF2α UPR branch as well as induction of ATF4 and C/EBP homologous protein (CHOP), resulting in more severe pancreatitis in Xbp1+/− mice, but does not induce obvious pancreatitis in wild-type mice that only have mild ER stress.63 64 Nuclear protein 1 (NUPR1) is a stress response protein induced on cell injury in virtually all organs, including the exocrine pancreas. Loss of NUPR1 in mice remarkably prevents ER stress inducer tunicamycin-induced ER dilation in acinar cells from wild-type mice. Mechanistically, NUPR1 interacts and promotes dephosphorylation of p-eIF2α, and thus loss of NUPR1 promotes the recovery of normal protein synthesis by maintaining eIF2α phosphorylation.65
The ER is crucial for proper protein folding, stability and movement. These processes involve both co-translational and post-translational modifications of nascent proteins in the ER. One important post-translational modification is protein acetylation via ER-resident acetyltransferases. This process requires the import of cytosolic acetyl-CoA into the ER lumen through the ER acetyl-CoA transporter AT-1 (also known as SLC33A1). Loss of AT-1 in mouse pancreas can lead to chronic ER stress, persistent activation of UPR,66 inflammation and fibrosis, which are similar to mild/moderate chronic pancreatitis-like phenotypes. These findings suggest that disrupting ER-associated post-translational protein modifications can increase the risk of ER stress and pancreatitis.
In addition to UPR and proteasome-mediated ERAD, selective autophagy for ER (ER-phagy) is another primary quality control mechanism to maintain ER homeostasis. The ER-phagy, specifically, helps degrade excess ER through autophagosomes that envelop ER with the help of a group of ER-phagy receptors such as RTN3L (a long isoform of RTN3), CCPG1, SEC62, ATL3 and TEX264 in mammalian cells.67 Studies have shown that loss of CCPG1, one of the ER-phagy receptors, leads to defective proteostasis, increased UPR and infiltration of inflammatory cells in the mouse pancreas, although there is no obvious generalised defect in pancreatic exocrine function.68 On the other hand, another study has found that piperine, the active phenolic component of black pepper, protects against L-arginine-induced acute pancreatitis in mice by enhancing ER-phagy. The protection is lost in either FAM134B and CCPG1 knockout mice,69 which supports the crucial role of ER-phagy in the pathogenesis of pancreatitis.
Notably, ER is also the primary storage site for intracellular Ca2+. In experimental pancreatitis, supraphysiological doses of cerulein increase intracellular Ca2+, resulting in mitochondrial damage, premature trypsinogen activation and NFκB activation.59 70 Loss of pancreatic VMP1 leads to ER stress, which promotes alcohol-induced or spontaneous pancreatitis (more discussions in the next sections). Taken together, it is clear that maintaining ER homeostasis is critical to exocrine pancreas physiology. Disruption of ER homeostasis may contribute to pancreatitis.
Mitochondrial damage
Mitochondria are intracellular organelles that act as ‘powerhouses’ in cells, generating ATP for cellular functions and survival. They also play a crucial role in intracellular signalling and metabolism, including calcium buffering, lipid and glucose metabolism, redox homeostasis, and cell death.71 In various acute experimental pancreatitis cases, it has been demonstrated that mitochondrial depolarisation is an early and common event. This is concurrent with the activation of the mitochondrial permeability transition pore (MPTP), which is associated with mitochondrial calcium overload. In rodent models of L-arginine-induced acute pancreatitis, swollen mitochondria with loss of mitochondrial cristae and increased mitochondrial fragmentation were observed in acinar cells. Cyclophilin D, an essential component of MPTP, was found to play a critical role in regulating mitochondrial ultrastructure and function during pancreatitis in mice. Genetic loss of cyclophilin D or pharmacological inhibition of MPTP markedly protects from various experimental pancreatitis by improving ER stress and autophagic flux.72 73 This suggests that mitochondria may crosstalk with other cellular organelles and autophagy to regulate acinar cell functions and prevent acinar cell damage, although how MPTP would regulate autophagy in acinar cells is not clear. Damaged mitochondria can be removed via selective mitophagy, either through PARKIN-PINK1 dependent or independent mechanisms, to maintain mitochondrial homeostasis.74 However, the role of mitophagy in the pathogenesis of pancreatitis remains largely unknown.
Lysosomal dysfunction and impaired autophagy in pancreatitis
Pancreatitis is an inflammatory disease resulting from damage to the exocrine acinar cells. Acinar cells have tremendous capacities for protein synthesis, trafficking and storage. Thus, properly regulating these processes is critical to maintaining acinar cell functions. As autophagy plays a vital role in the quality control of proteins and organelles for cellular homeostasis, it is unsurprising that defective autophagy would be detrimental to acinar cells. Indeed, the accumulation of large vacuoles has long been noted as a typical phenotype in experimental and human pancreatitis (figure 3, arrow).48 75 Research conducted by Gukovskaya’s team58 75 76 suggests that the large vacuoles found in acinar cells may be dysfunctional autolysosomes caused by defective lysosome and autophagy functions in various experimental pancreatitis induced by cerulein, L-arginine or choline-deficient or ethionine-supplemented diet in rodents, as well as in isolated acinar cells hyperstimulated with cholecystokinin-8 (CCK-8).77 Subsequent experimental evidence shows that these large vacuoles are often positive for LC3, an important marker of autophagosomes, and LAMP-1, a marker for lysosomes/autolysosomes,48 60 further supporting these large vacuoles are most likely autolysosomes. An earlier study also reveals abnormal subcellular distribution of the lysosomal enzymes cathepsin B and D among ZGs, lysosomes and mitochondria in cerulein-induced pancreatitis in rats.78
Figure 3.
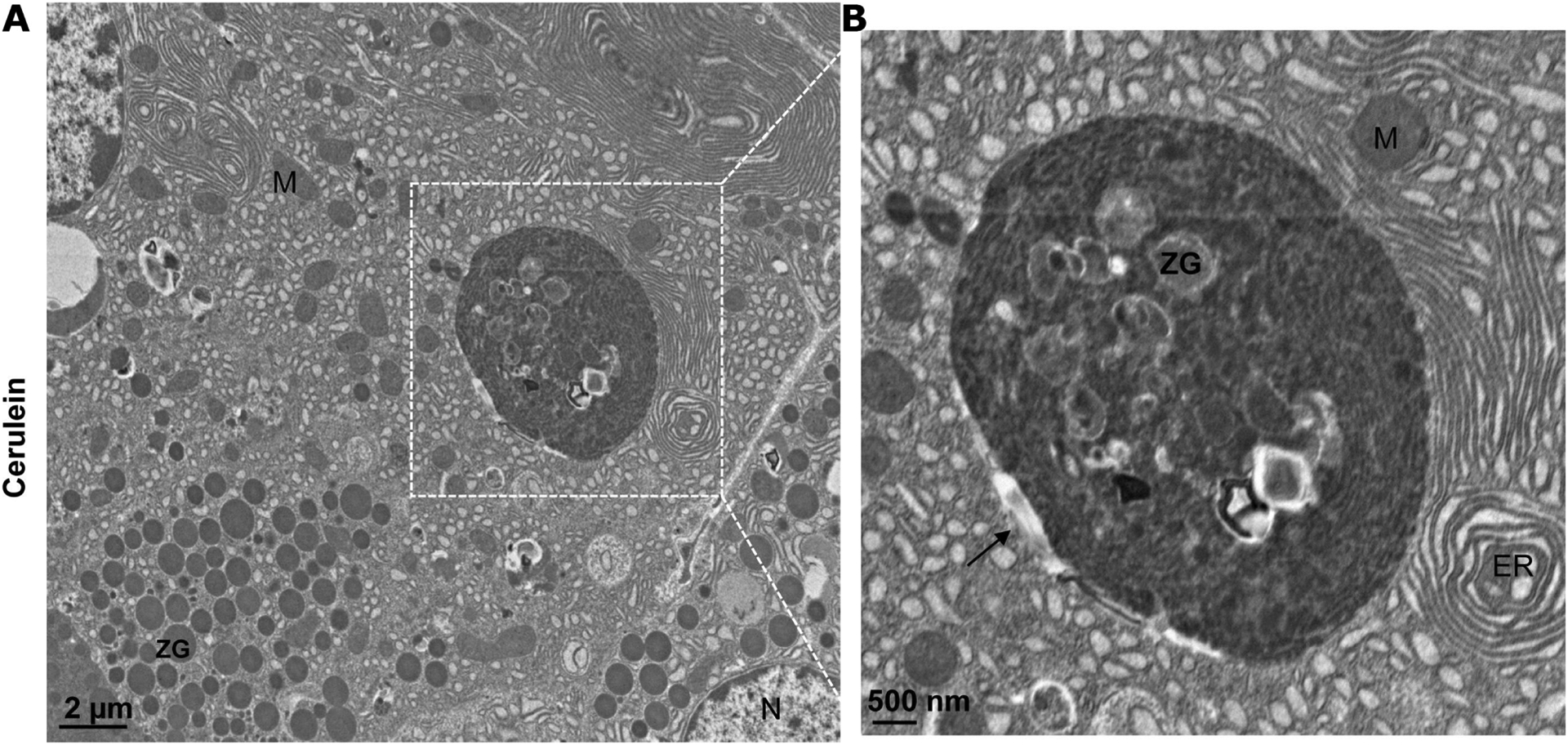
Electron microscopy photographs of a large vacuole in acute pancreatitis tissues. (A) Representative image of electron microscopy analysis of mouse acute pancreatitis tissues induced by cerulein treatment (for details, see Wang S et al48). (B) An enlarged photograph from the boxed area in (A) showing a large autophagic vacuole (black arrow). ER, endoplasmic reticulum; M, mitochondria; N, nucleus; ZG, zymogen granule.
The cause of lysosome dysfunction is likely due to impaired processing of the lysosomal proteases cathepsin L and cathepsin B into their fully active, mature forms in experimental pancreatitis.77 The mannose 6-phosphate (M6P) pathway plays a crucial role in delivering newly synthesised hydrolases to lysosomes. GNPTAB encodes the catalytic α/β subunits of N-acetylglucosamine-1-phosphotransferase, a key enzyme in the M6P-tag formation. Loss of Gnptab in mice leads to the accumulation of non-esterified cholesterol in late endosomes/lysosomes and autophagic vacuoles, resulting in spontaneous pancreatitis.79 Increased cellular free cholesterol impairs lysosome function and causes autolysosome accumulation in hepatocytes.80 Although it has yet to be studied, it is likely that increased cholesterol may also contribute to the dysfunction of endosomes/lysosomes, leading to impaired autophagic flux in acinar cells. Notably, decreased pancreatic LAMP-1/2 expression also contributes to lysosome dysfunction in experimental pancreatitis models.47 48 81 The enlarged lysosome/autolysosome compartments containing undegraded ZG and other partially degraded cellular contents are likely due to impaired lysosomal functions or decreased lysosome numbers resulting in insufficient autophagy.82 Indeed, TFEB directly regulates lysosomal biogenesis and indirectly autophagy, which is impaired by cerulein or alcohol, causing pancreatitis.47 48
As discussed above, VMP1 is an ER-resident multispanning transmembrane protein, which regulates autophagy by promoting the closure of autophagosomes.83 84 VMP1 also regulates soluble protein secretion in Drosophila cells, and lipoprotein secretion in zebrafish intestine and liver, independent of its autophagy activity.37–39 Recent studies suggest that VMP1 has phospholipid scramblase activity, which regulates the cellular distribution of cholesterol and phosphatidylserine. This activity is also involved in the formation of lipid droplets and is associated with SARS-CoV-2 and other coronavirus infections.85–87 Cerulein and alcohol feeding decrease VMP1 expression at both mRNA and protein levels.88 Damaged and excess ER can be removed via selective autophagy, termed as ER-phagy, which helps maintain ER homeostasis and relieve ER stress. Consequently, impaired autophagy/ER-phagy can lead to the accumulation of abnormal ER structures, ER dilation and ER stress (figure 4). Nonetheless, it remains unclear whether VMP1 may act as an ER-phagy receptor that regulates ER homeostasis in pancreatitis. It has been reported that fragile ZGs are removed by VMP1-p62-mediated selective autophagy to avoid intracellular activation of trypsinogen in cerulein-induced pancreatitis.89 Moreover, ZGs are colocalised with green fluorescence protein (GFP)-LC3 positive autophagosomes, and purified ZG fractions are enriched with LC3-II in alcohol-induced or cerulein-induced pancreatitis.47 48 Therefore, it is possible that one mechanism by which autophagy protects against the pathogenesis of pancreatitis is by the removal of damaged and fragile ZGs.
Figure 4.
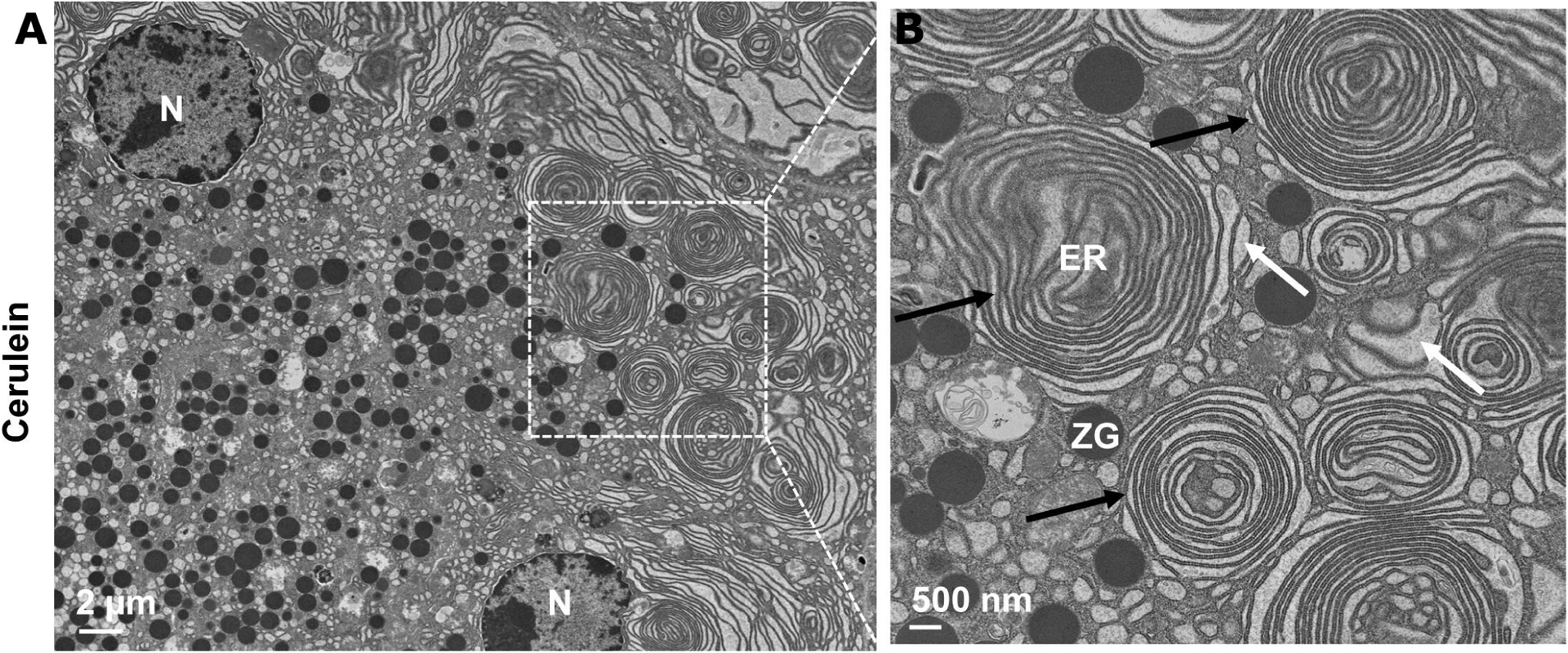
Electron microscopy photographs of abnormal ER structures in acute pancreatitis tissues. (A) Representative image of electron microscopy analysis of mouse acute pancreatitis tissues induced by cerulein treatment (for details, see Wang S et al48). (B) An enlarged photograph from the boxed area in (A) showing abnormal ER structures (black arrows) and ER dilation (white arrows). ER, endoplasmic reticulum; N, nucleus; ZG, zymogen granule.
The highly dynamic autophagy process involves the early biogenesis of autophagosomes and later fusion of autophagosomes with lysosomes for degradation. This is facilitated by membrane fusion, which is mainly mediated by the SNARE proteins.90 91 Among them, the SNARE protein VAMP7 and its partner SNAREs (STX7, STX8 and Vti1b) are necessary for the formation of autophagosomes.91 In contrast, STX17, VAMP8 and SNAP29 are crucial for the fusion of autophagosomes with lysosomes.92 93 STX2 is a target membrane-SNARE syntaxin and is usually located on the apical membrane of acinar cells. In experimental pancreatitis induced by the supraphysiological concentration of CCK-8 or a low concentration of CCK-8 together with ethanol, STX2 is decreased by cysteine protease-mediated cleavage, resulting in increased autophagosome formation by enhancing Atg16L/CHC (clathrin heavy chain) complex assembly. Deletion of STX2 in mouse acini increases susceptibility to pancreatitis associated with increased autophagy.94 SNAP23 is reported as part of ZG exocytotic SNARE complexes regulating apical and basolateral exocytosis. In cerulein and cerulein with ethanol-induced experimental pancreatitis, SNAP23 is phosphorylated by inhibitor of nuclear factor kappa B kinase β (IKKβ) and translocates to autophagosomes to bind STX17 in mediating excessive autolysosome formation. Pancreas-specific SNAP23 knockdown decreases autolysosome formation and protects against pancreatitis.95 An interesting concept has been proposed based on these findings that mild attenuation of autophagy and autolysosome formation could be beneficial, while enhancing autolysosome formation, but hindering its maturation could worsen pancreatitis. However, it should be noted that the assessment of autophagy activity is mainly based on the levels of LC3-II, which could increase due to either increased autophagosome biogenesis or impaired autolysosome/lysosome functions. Moreover, a recent study shows that cerulein also decreases STX17, which reduces the fusion of autophagosomes with lysosomes, and knockdown of Stx17 exacerbates cerulein-induced pancreatitis.96 Furthermore, as discussed in detail below, genetic deletion of Atg5, Atg7 or Tfeb in mouse pancreas leads to spontaneous pancreatitis. Therefore, it is still unclear whether autophagy is helpful or harmful in the development of pancreatitis, although all studies support the importance of autophagy in pancreatitis.97
Perhaps another direct connection between the autophagy–lysosomal pathway and pancreatitis is from the genetic mouse models, which was further demonstrated by the deletion of the essential Atg gene Atg5 or Atg7, a lysosomal gene lysosomal-associated membrane protein-2 (Lamp-2), as well as more recently the deletion of Vmp1 in mice, all of which led to spontaneous pancreatitis.81 88 98 Cre/LoxP has been widely used to generate genetic mouse models to study the role of autophagy in pancreatitis in mice. The Ptf1a-Cre or Pdx-Cre-driven recombination occurs during pancreas development, which impacts both exocrine acinar cells and endocrine islet cells.99 Ptf1a-Cre, Atg5, and Pdx-Cre, Atg7, conditional knockout mice deleted Atg5 and Atg7 in both endocrine and exocrine cells, and these mice developed spontaneous pancreatitis.98 100 However, concerns are raised on whether the pancreatitis phenotype in these knockout mice is also affected by impairing endocrine pancreatic functions. Indeed, controversial observations were also reported that deletion of Atg5 using an acinar cell-specific Ela Cre improves cerulein-induced acute pancreatitis in mice.101 We have recently created mice with a specific gene modification (acinar cell-specific Atg5 knockout or Tfeb knockout mice) by using a tamoxifen-inducible Cre, known as BAC-Ela-CreErT. This modification results in the deletion of Atg5 or Tfeb only in acinar cells of adult mice. These modified mice only develop mild pancreatitis when fed a regular chow diet. However, they display severe pancreatitis when fed a liquid ethanol diet or liquid control diet.47 In contrast, using the same BAC-Ela-CreErT Cre to delete Vmp1 in mouse acinar cells leads to severe spontaneous pancreatitis in mice fed a chow diet.88 This suggests that the loss of acinar cell Atg5 or Tfeb may not be enough to trigger spontaneous pancreatitis under normal conditions, but they become susceptible to stresses that can induce pancreatitis. Alternatively, targeting different phases of autophagy, such as early autophagosome formation (ATG5) versus late autophagosome closure (VMP1) or lysosome biogenesis (TFEB) and possible non-autophagic functions of these proteins, may have a different impact on acinar cell functions. Although the Cre/LoxP system is useful, caution must be taken as Cre transgenic mice can cause toxicity.102 Therefore, interpretation of the mouse data should include Cre transgenic mice for comparison purposes.
The above-mentioned ATG-mediated autophagy generally requires the conjugation of PE to the cytosolic LC3-I to form LC3-II on the double-membrane autophagosomes (so-called canonical autophagy). In contrast, in the absence of ATG5 or ATG7, double-membrane autophagosomes can still be formed for bulk degradation of intracellular proteins that is dependent on ULK1, BECLIN1 and RAB9 via alternative/non-canonical autophagy.103 Moreover, non-canonical autophagy can also occur without the formation of double-membrane autophagosomes in which LC3-II targets single-membrane vesicles or compartments of the endolysosomal system, which is a crucial distinction between canonical and non-canonical autophagy. Non-canonical autophagy shares some subsets of common ATGs, such as core ubiquitin-like conjugation systems that support LC3 lipidation to membranes, including ATG3, ATG4, ATG5, ATG7, ATG10, ATG12 and ATG16L1. However, the upstream autophagy machinery, including ULK1/2, FIP200, ATG13, ATG9, WIPI2 and ATG14L1, is not required for non-canonical autophagy.104 Recent findings show that pancreatic levels of RAB9 decrease in rodent and human pancreatitis. Moreover, overexpression of RAB9 switches canonical autophagy to non-canonical autophagy, exacerbating experimental pancreatitis.105 During CCK-induced experimental pancreatitis, increased LC3-positive single-membrane endocytic vacuoles reminiscent in its properties to LC3-associated phagocytosis (LAP) were found to be associated with pancreatitis.106 However, it remains to be determined whether LAP plays a causal role in the pathogenesis of pancreatitis.
Taken together, it appears that multiple steps of the autophagy process can be altered either at the early autophagosome formation/closure or late stage of fusion of the autophagosome with lysosome or lysosomal functions/numbers to promote the pathogenesis of pancreatitis. Moreover, canonical and non-canonical autophagy may be involved in exocrine pancreas homeostasis and pancreatitis.
Alcohol-associated pancreatitis
Alcohol-associated pancreatitis is a major, untreatable complication of alcohol abuse. Pancreatitis arises through alcohol-induced damage to pancreatic acinar cells. These cells metabolise alcohol through oxidative and non-oxidative pathways.76 107 108 Alcohol dehydrogenase catalyses the oxidative pathway of alcohol metabolism, leading to the production of acetaldehyde and increased reactive oxygen species (ROS), which are highly toxic. The isoform 2 of aldehyde dehydrogenase, found in mitochondria, further metabolises acetaldehyde to acetate. On the other hand, the non-oxidative pathway of alcohol metabolism in acinar cells is mediated by fatty acid ethyl ester (FAEE) synthases, which esterify alcohol to form FAEE. Both FAEE and products of oxidative metabolism, acetaldehyde and ROS can destabilise and damage lysosome and ZG membranes. Although no study has been conducted to examine the effects of alcohol metabolism on impaired autophagy in acinar cells, we have previously demonstrated that alcohol metabolism plays a crucial role in inducing autophagy in hepatocytes.109 Therefore, it may be worthwhile to further investigate this topic in future research. Alcohol consumption can cause lysosome dysfunction, leading to the premature intracellular activation of digestive enzymes and subsequent acinar cell death and pancreatitis.76 110–112 Timely removal of fragile and deleterious ZGs and damaged lysosomes is crucial for protecting against alcohol-induced pancreatic injury in both mice and humans. High alcohol consumption can increase the number of damaged ZGs and directly injure lysosomes, leading to the accumulation of large vacuoles in pancreatitis.60 113 Studies from cultured non-pancreatic cells indicate that damaged lysosomes/endosomes can also be selectively sequestered by autophagy (a process termed as lysophagy).114 115 Since lysosomes sit at the last step of autophagy by fusing with autophagosomes, the accumulation of dysfunctional lysosomes will lead to impaired autophagic degradation. Therefore, maintaining the quantity and quality of lysosomes through lysosomal biogenesis is critical to maintaining sufficient autophagic degradation to remove damaged lysosomes and fragile ZGs to protect against the pathogenesis of pancreatitis. Typically, pancreatitis is prevented by controlling the homeostasis and quality of ZGs and lysosomes through autophagy and lysosomal biogenesis. However, these normal protective autophagy processes are impaired by alcohol at multiple steps.
Lysosomes are the final stage of autophagy, where they merge with autophagosomes. If dysfunctional lysosomes accumulate, it can hinder proper autophagic degradation. Hence, it is crucial to maintain the quality and quantity of lysosomes through lysosomal biogenesis to ensure sufficient autophagic degradation. This helps in removing damaged lysosomes and fragile ZGs and protects against alcohol-associated pancreatitis. TFEB is a transcription factor that belongs to the CLEAR (coordinated lysosomal expression and regulation) gene network,116 which is a crucial regulator of genes involved in lysosome creation and autophagy.117 118 It coordinates a transcriptional programme to activate genes that are responsible for both the early (autophagosome formation) and late (lysosome biogenesis) phases of autophagy in response to increased degradation needs. TFEB is mainly regulated at the post-translational level through specific amino acid phosphorylation. TFEB phosphorylation at Ser142 and Ser211 by mTOR and the mitogen-activated protein kinase (MAPK) increases its binding with the cytosolic chaperone 14-3-3, resulting in TFEB sequestration in the cytosol and reduced TFEB transcription activity.116 Conversely, lysosomal Ca2+ release activates the phosphatase calcineurin, which dephosphorylates TFEB at Ser142 and Ser211 and promotes TFEB nuclear translocation.119
Our lab’s findings suggest that alcohol inhibits mTOR but increases the levels of phosphorylated MAPK in the mouse pancreas, indicating that impaired TFEB is mediated by MAPK activation but independent of mTOR.47 Alcohol feeding decreases both the mRNA and protein levels of TFEB in the mouse pancreas, suggesting that alcohol may regulate pancreatic TFEB at both transcriptional and post-translational levels. Acinar cell-specific TFEB knockout mice fed with a chow diet develop mild pancreatic oedema, but severe pancreatic changes resembling chronic pancreatitis in alcohol-fed mice. Interestingly, the Lieber-DeCarli control diet-fed acinar cell-specific TFEB knockout mice also developed severe pancreatitis with no apparent difference compared with alcohol-diet fed TFEB knockout mice.47 These results suggest that the administration of a liquid diet can trigger pancreatic damage in the absence of acinar cell TFEB. How the liquid diet potentiates pancreatic damage in the absence of TFEB is currently unknown. Increased mucosal permeability and translocation of intestinal bacteria to the pancreas have been implicated in the pathogenesis of pancreatitis,120 121 and it is likely that the Lieber-DeCarli diet may alter the microbiota and prime the TFEB knockout mice to be more sensitive to develop pancreatitis. Future studies are needed to test this hypothesis by profiling the gut microbiome and administering antibiotics to these Lieber-DeCarli diet and alcohol-fed TFEB knockout and their matched wild-type mice. In addition to impaired TFEB, alcohol feeding increases pancreatic ATG4B, a critical cysteine protease, by inhibiting its proteolytic degradation, resulting in increased LC3-II deconjugation and impaired autophagy.122 More recent findings from our lab show that alcohol also decreases VMP1 to impair the closure of autophagosomes.88 Notably, decreased pancreatic VMP1, TFEB and LAMP-1/2 is also found in human pancreatitis,48 81 88 123 indicating the relevance of impaired autophagy in pancreatitis in the clinical setting. More importantly, overexpression of TFEB or knockdown of ATG4B increases autophagic activity and alleviates alcohol-induced pancreatitis.47 122 Taken together, targeting TFEB and ATG4B-mediated autophagy may be beneficial for attenuating alcohol-associated pancreatitis.
SUMMARY AND FUTURE PERSPECTIVES
In summary, autophagy is a multistep process that plays a crucial role in regulating the health of pancreatic acinar cells. It serves as a self-degradative and quality control mechanism that helps remove damaged or fragile organelles such as ZGs, mitochondria, ER and lysosomes. Disruption of autophagy at any stage, including autophagosome formation, closure or fusion with lysosomes, as well as lysosomal functions and biogenesis, can lead to pancreatitis. Selective autophagy is essential for maintaining healthy acinar cells and protecting against pancreatitis. However, it is unclear whether a specific or general autophagy receptor is required for selective zymophagy, ER-phagy, mitophagy and lysophagy in acinar cells during pancreatitis (figure 5). Although cerulein has been widely used as an experimental pancreatitis model, it may not completely mimic the pathogenesis of human pancreatitis. Moreover, rodents are resistant to alcohol-induced pancreatitis, which has limited the development of proper animal models and therapeutic strategies for preventing and treating alcohol-associated pancreatitis. While recent progress has greatly enriched our understanding of the role and mechanisms of autophagy in the pathogenesis of pancreatitis, many important questions remain unanswered. Pancreatitis is an inflammatory disease, and it is known that immune cells play critical roles in its development and progression. An increased number of macrophages and neutrophils is well documented in pancreatitis. Studies in mice have shown that myeloid-specific deletion of Atg5 leads to more proinflammatory polarisation, resulting in increased secretion of inflammatory cytokines.124 However, no study has investigated the direct impact of autophagy deficiency in immune cells in the development of pancreatitis. Changes in ER structure, ER stress and damaged mitochondria are involved in the development of pancreatitis. However, it is still unclear how these changes in ER structure and damaged mitochondria contribute to the production of membrane sources for autophagosome biogenesis. Further research is needed to understand this process thoroughly. Furthermore, future studies are needed to determine the translational value of targeting autophagy for treating pancreatitis. It remains to be tested whether a pharmacological boost of autophagy or lysosomal activities would be beneficial for treating pancreatitis.
Figure 5.
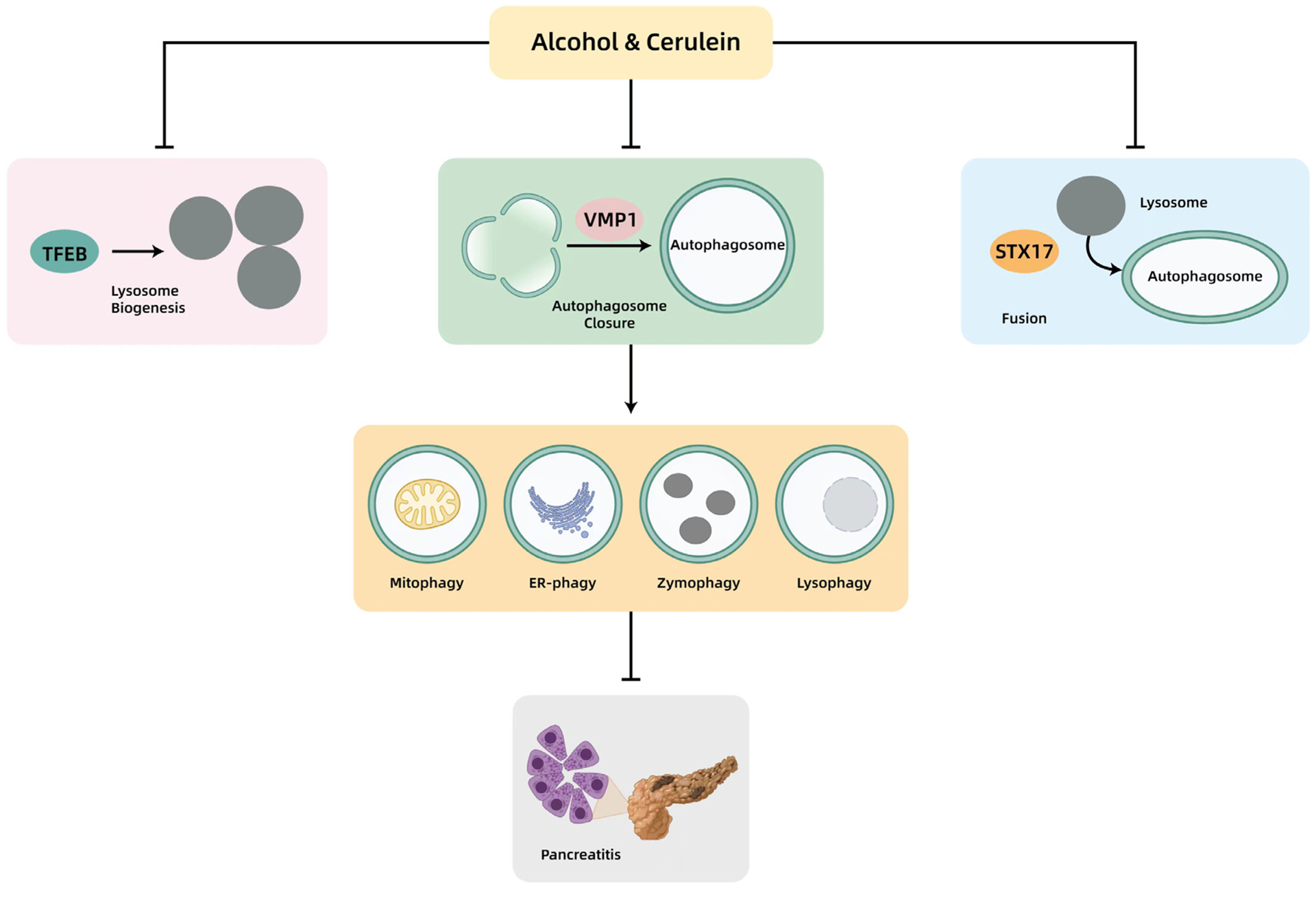
A proposed model of impaired autophagy in experimental acute pancreatitis induced by cerulein or alcohol. Cerulein or alcohol impairs autophagy at multiple steps to promote pancreatitis. Cerulein or alcohol decreases pancreatic VMP1 to impair the closure of autophagosomes. Cerulein also decreases STX17 to impair the fusion of the autophagosome with a lysosome. Cerulein or alcohol decreases pancreatic TFEB, resulting in impaired lysosomal biogenesis and insufficient autophagy. Autophagy may selectively remove damaged mitochondria (mitophagy), abnormal ER (ER-phagy), leaky ZGs (zymophagy) and damaged lysosomes (lysophagy) to protect against acinar cell death and pancreatitis. ER, endoplasmic reticulum; STX17, syntaxin 17; TFEB, transcription factor EB; VMP1, vacuole membrane protein 1.
Funding
This study was supported in part by the National Institutes of Health (NIH) funds R37 AA020518, R21 AA030617 (W-XD) and R01DK134737 (H-MN).
Footnotes
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
REFERENCES
- 1.Mederos MA, Reber HA, Girgis MD. Acute pancreatitis: a review. JAMA 2021;325:382–90. [DOI] [PubMed] [Google Scholar]
- 2.Boxhoorn L, Voermans RP, Bouwense SA, et al. Acute pancreatitis. Lancet 2020;396:726–34. [DOI] [PubMed] [Google Scholar]
- 3.Żorniak M, Sirtl S, Mayerle J, et al. What do we currently know about the pathophysiology of alcoholic pancreatitis: a brief review. Visc Med 2020;36:182–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Apte MV, Pirola RC, Wilson JS. Individual susceptibility to alcoholic pancreatitis. J Gastroenterol Hepatol 2008;23 Suppl 1:S63–8. [DOI] [PubMed] [Google Scholar]
- 5.Li H, Wen W, Luo J. Targeting endoplasmic reticulum stress as an effective treatment for alcoholic pancreatitis. Biomedicines 2022;10:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pandol SJ, Gorelick FS, Gerloff A, et al. Alcohol abuse, endoplasmic reticulum stress and pancreatitis. Dig Dis 2010;28:776–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature 2008;451:1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000;290:1717–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mizushima N A brief history of autophagy from cell biology to physiology and disease. Nat Cell Biol 2018;20:521–7. [DOI] [PubMed] [Google Scholar]
- 10.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011;147:728–41. [DOI] [PubMed] [Google Scholar]
- 11.Liu K, Czaja MJ. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ 2013;20:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011;12:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chino H, Mizushima N. ER-Phagy: quality control and turnover of endoplasmic reticulum. Trends Cell Biol 2020;30:384–98. [DOI] [PubMed] [Google Scholar]
- 14.Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 2014;20:460–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schuck S Microautophagy - distinct molecular mechanisms handle cargoes of many sizes. J Cell Sci 2020;133:jcs246322. [DOI] [PubMed] [Google Scholar]
- 16.Marzella L, Ahlberg J, Glaumann H. Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Archiv B Cell Pathol 1981;36:219–34. [DOI] [PubMed] [Google Scholar]
- 17.Arias E, Cuervo AM. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol 2011;23:184–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qian H, Chao X, Williams J, et al. Autophagy in liver diseases: a review. Mol Aspects Med 2021;82:100973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem 2000;275:31505–13. [DOI] [PubMed] [Google Scholar]
- 20.Wang L, Klionsky DJ, Shen HM. The emerging mechanisms and functions of microautophagy. Nat Rev Mol Cell Biol 2023;24:186–203. [DOI] [PubMed] [Google Scholar]
- 21.Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 2018;19:365–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zachari M, Ganley IG. The mammalian ULK1 complex and autophagy initiation. Essays Biochem 2017;61:585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011;13:132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei Y, Liu M, Li X, et al. Origin of the autophagosome membrane in mammals. Biomed Res Int 2018;2018:1012789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Melia TJ, Lystad AH, Simonsen A. Autophagosome biogenesis: from membrane growth to closure. J Cell Biol 2020;219:e202002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park J-M, Jung CH, Seo M, et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy 2016;12:547–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoon MS. Vps34 and PLD1 take center stage in nutrient signaling: their dual roles in regulating autophagy. Cell Commun Signal 2015;13:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizushima N The ATG conjugation systems in autophagy. Curr Opin Cell Biol 2020;63:1–10. [DOI] [PubMed] [Google Scholar]
- 29.Osawa T, Noda NN. Atg2: A novel phospholipid transfer protein that mediates de novo autophagosome biogenesis. Protein Sci 2019;28:1005–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Filimonenko M, Stuffers S, Raiborg C, et al. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol 2007;179:485–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee J-A, Beigneux A, Ahmad ST, et al. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr Biol 2007;17:1561–7. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi Y, He H, Tang Z, et al. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat Commun 2018;9:2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhen Y, Spangenberg H, Munson MJ, et al. ESCRT-mediated phagophore sealing during mitophagy. Autophagy 2020;16:826–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou F, Wu Z, Zhao M, et al. Rab5-dependent autophagosome closure by ESCRT. J Cell Biol 2019;218:1908–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang PP, Kou DQ, Le WD. Roles of VMP1 in autophagy and ER–membrane contact: potential implications in neurodegenerative disorders. Front Mol Neurosci 2020;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao YG, Chen Y, Miao G, et al. The ER-localized transmembrane protein EPG-3/VMP1 regulates SERCA activity to control ER-isolation membrane contacts for autophagosome formation. Mol Cell 2017;67:974–89. [DOI] [PubMed] [Google Scholar]
- 37.Morishita H, Zhao YG, Tamura N, et al. A critical role of VMP1 in lipoprotein secretion. Elife 2019;8:e48834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang X, Fulte S, Deng F, et al. Lack of VMP1 impairs hepatic lipoprotein secretion and promotes non-alcoholic steatohepatitis. J Hepatol 2022;77:619–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang X, Chen A, Ding WX, et al. VMP1 regulates hepatic lipoprotein secretion and NASH independent of autophagy. Autophagy 2023;19:367–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang D, Xu B, Liu L, et al. TMEM41B acts as an ER scramblase required for lipoprotein biogenesis and lipid homeostasis. Cell Metab 2021;33:1655–70. [DOI] [PubMed] [Google Scholar]
- 41.Fader CM, Sánchez DG, Mestre MB, et al. TI-VAMP/VAMP7 and VAMP3/cellubrevin: two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim Biophys Acta 2009;1793:1901–16. [DOI] [PubMed] [Google Scholar]
- 42.Furuta N, Fujita N, Noda T, et al. Combinational soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins VAMP8 and Vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes. Mol Biol Cell 2010;21:1001–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012;151:1256–69. [DOI] [PubMed] [Google Scholar]
- 44.Lőrincz P, Juhász G. Autophagosome-lysosome fusion. J Mol Biol 2020;432:2462–82. [DOI] [PubMed] [Google Scholar]
- 45.Yu L, McPhee CK, Zheng L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010;465:942–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011;332:1429–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang S, Ni H-M, Chao X, et al. Critical role of TFEB-mediated lysosomal biogenesis in alcohol-induced pancreatitis in mice and humans. Cell Mol Gastroenterol Hepatol 2020;10:59–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang S, Ni H-M, Chao X, et al. Impaired TFEB-mediated lysosomal biogenesis promotes the development of pancreatitis in mice and is associated with human pancreatitis. Autophagy 2019;15:1954–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mastracci TL, Apte M, Amundadottir LT, et al. integrated physiology of the exocrine and endocrine compartments in pancreatic diseases: workshop proceedings. Diabetes 2023;72:433–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pandiri AR. Overview of exocrine pancreatic pathobiology. Toxicol Pathol 2014;42:207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lerch MM, Gorelick FS. Early trypsinogen activation in acute pancreatitis. Med Clin North Am 2000;84:549–63. [DOI] [PubMed] [Google Scholar]
- 52.Buchler M, Uhl W. Trypsinogen activation peptides assay in the early prediction of severity of acute-pancreatitis. Z Gastroenterol 1991;29:79–81. [PubMed] [Google Scholar]
- 53.Fu Y, Lucas AL. Genetic evaluation of pancreatitis. Gastrointest Endosc Clin N Am 2022;32:27–43. [DOI] [PubMed] [Google Scholar]
- 54.Jalaly NY, Moran RA, Fargahi F, et al. An evaluation of factors associated with pathogenic PRSS1, SPINK1, CTFR, and/or CTRC genetic variants in patients with idiopathic pancreatitis. Am J Gastroenterol 2017;112:1320–9. [DOI] [PubMed] [Google Scholar]
- 55.Sah RP, Dawra RK, Saluja AK. New insights into the pathogenesis of pancreatitis. Curr Opin Gastroenterol 2013;29:523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sah RP, Dudeja V, Dawra RK, et al. Cerulein-induced chronic pancreatitis does not require intra-acinar activation of trypsinogen in mice. Gastroenterology 2013;144:1076–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dawra R, Sah RP, Dudeja V, et al. Intra-acinar trypsinogen activation mediates early stages of pancreatic injury but not inflammation in mice with acute pancreatitis. Gastroenterology 2011;141:2210–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gukovskaya AS, Gorelick FS, Groblewski GE, et al. Recent insights into the pathogenic mechanism of pancreatitis: role of acinar cell organelle disorders. Pancreas 2019;48:459–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Habtezion A, Gukovskaya AS, Pandol SJ. Acute pancreatitis: a multifaceted set of organelle and cellular interactions. Gastroenterology 2019;156:1941–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gukovsky I, Pandol SJ, Mareninova OA, et al. Impaired autophagy and organellar dysfunction in pancreatitis. J Gastroenterol Hepatol 2012;27 Suppl 2(Suppl 2):27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hwang JW, Qi L. Quality control in the endoplasmic reticulum: crosstalk between ERAD and UPR pathways. Trends Biochem Sci 2018;43:593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy 2008;4:141–50. [DOI] [PubMed] [Google Scholar]
- 63.Lugea A, Waldron RT, French SW, et al. Drinking and driving pancreatitis: links between endoplasmic reticulum stress and autophagy. Autophagy 2011;7:783–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lugea A, Tischler D, Nguyen J, et al. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology 2011;140:987–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Borrello MT, Santofimia-Castaño P, Bocchio M, et al. NUPR1 interacts with eIF2α and is required for resolution of the ER stress response in pancreatic tissue. FEBS J 2021;288:4081–97. [DOI] [PubMed] [Google Scholar]
- 66.Cooley MM, Thomas DDH, Deans K, et al. Deficient endoplasmic reticulum acetyl-coA import in pancreatic acinar cells leads to chronic pancreatitis. Cell Mol Gastroenterol Hepatol 2021;11:725–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chino H, Mizushima N. ER-phagy: quality control and turnover of endoplasmic reticulum. Trends Cell Biol 2020;30:384–98. [DOI] [PubMed] [Google Scholar]
- 68.Smith MD, Harley ME, Kemp AJ, et al. CCPG1 is a non-canonical autophagy cargo receptor essential for ER-phagy and pancreatic er proteostasis. Dev Cell 2018;44:217–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang W, Zhang J, Jin W, et al. Piperine alleviates acute pancreatitis: a possible role for FAM134B and CCPG1 dependent ER-phagy. Phytomedicine 2022;105:154361. [DOI] [PubMed] [Google Scholar]
- 70.Saluja A, Dudeja V, Dawra R, et al. Early intra-acinar events in pathogenesis of pancreatitis. Gastroenterology 2019;156:1979–93. [DOI] [PubMed] [Google Scholar]
- 71.Ma X, McKeen T, Zhang J, et al. Role and mechanisms of mitophagy in liver diseases. Cells 2020;9:837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Biczo G, Vegh ET, Shalbueva N, et al. Mitochondrial dysfunction, through impaired autophagy, leads to endoplasmic reticulum stress, deregulated lipid metabolism, and pancreatitis in animal models. Gastroenterology 2018;154:689–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mukherjee R, Mareninova OA, Odinokova IV, et al. Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: inhibition prevents acute pancreatitis by protecting production of ATP. Gut 2016;65:1333–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem 2012;393:547–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gukovskaya AS, Gukovsky I. Autophagy and pancreatitis. Am J Physiol Gastrointest Liver Physiol 2012;303:G993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gukovskaya AS, Mouria M, Gukovsky I, et al. Ethanol metabolism and transcription factor activation in pancreatic acinar cells in rats. Gastroenterology 2002;122:106–18. [DOI] [PubMed] [Google Scholar]
- 77.Mareninova OA, Hermann K, French SW, et al. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest 2009;119:3340–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saluja A, Hashimoto S, Saluja M, et al. Subcellular redistribution of lysosomal enzymes during caerulein-induced pancreatitis. Am J Physiol 1987;253(4 Pt 1):G508–16. [DOI] [PubMed] [Google Scholar]
- 79.Mareninova OA, Vegh ET, Shalbueva N, et al. Dysregulation of mannose-6-phosphate-dependent cholesterol homeostasis in acinar cells mediates pancreatitis. J Clin Invest 2021;131:e146870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang Y, Ding Y, Li J, et al. Targeting the enterohepatic bile acid signaling induces hepatic autophagy via a CYP7A1-AKT-mTOR axis in mice. Cell Mol Gastroenterol Hepatol 2017;3:245–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mareninova OA, Sendler M, Malla SR, et al. Lysosome associated membrane proteins maintain pancreatic acinar cell homeostasis: LAMP-2 deficient mice develop pancreatitis. Cell Mol Gastroenterol Hepatol 2015;1:678–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chao X, Ni HM, Ding WX. Insufficient autophagy: a novel autophagic flux scenario uncovered by impaired liver TFEB-mediated lysosomal biogenesis from chronic alcohol-drinking mice. Autophagy 2018;14:1646–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010;6:764–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tian Y, Li Z, Hu W, et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 2010;141:1042–55. [DOI] [PubMed] [Google Scholar]
- 85.Li YE, Wang Y, Du X, et al. TMEM41B and VMP1 are scramblases and regulate the distribution of cholesterol and phosphatidylserine. J Cell Biol 2021;220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schneider WM, Luna JM, Hoffmann H-H, et al. Genome-scale identification of SARS-CoV-2 and pan-Coronavirus host factor networks. Cell 2021;184:120–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ghanbarpour A, Valverde DP, Melia TJ, et al. A model for a partnership of lipid transfer proteins and scramblases in membrane expansion and organelle biogenesis. Proc Natl Acad Sci U S A 2021;118:e2101562118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang S, Chao X, Jiang X, et al. Loss of acinar cell VMP1 triggers spontaneous pancreatitis in mice. Autophagy 2022;18:1572–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Grasso D, Ropolo A, Lo Ré A, et al. Zymophagy, a novel selective autophagy pathway mediated by VMP1-USP9x-p62, prevents pancreatic cell death. J Biol Chem 2011;286:8308–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nair U, Jotwani A, Geng J, et al. SNARE proteins are required for macroautophagy. Cell 2011;146:290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Moreau K, Ravikumar B, Renna M, et al. Autophagosome precursor maturation requires homotypic fusion. Cell 2011;146:303–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang Y, Li L, Hou C, et al. SNARE-mediated membrane fusion in autophagy. Semin Cell Dev Biol 2016;60:97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tian XY, Teng JL, Chen JG. New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy 2021;17:2680–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dolai S, Liang T, Orabi AI, et al. Pancreatitis-induced depletion of syntaxin 2 promotes autophagy and increases basolateral exocytosis. Gastroenterology 2018;154:1805–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dolai S, Takahashi T, Qin T, et al. Pancreas-specific SNAP23 depletion prevents pancreatitis by attenuating pathological basolateral exocytosis and formation of trypsin-activating autolysosomes. Autophagy 2021;17:3068–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang T-T, Zhang L-C, Qin Z, et al. Decreased syntaxin17 expression contributes to the pathogenesis of acute pancreatitis in murine models by impairing autophagic degradation. Acta Pharmacol Sin 2023;44:2445–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang SG, Ding WX. Does autophagy promote or protect against the pathogenesis of pancreatitis? Gastroenterology 2018;155:1273–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Diakopoulos KN, Lesina M, Wörmann S, et al. Impaired autophagy induces chronic atrophic pancreatitis in mice via sex- and nutrition-dependent processes. Gastroenterology 2015;148:626–38. [DOI] [PubMed] [Google Scholar]
- 99.Magnuson MA, Osipovich AB. Pancreas-specific Cre driver lines and considerations for their prudent use. Cell Metab 2013;18:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Antonucci L, Fagman JB, Kim JY, et al. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc Natl Acad Sci U S A 2015;112:E6166–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hashimoto D, Ohmuraya M, Hirota M, et al. Involvement of autophagy in trypsinogen activation within the pancreatic acinar cells. J Cell Biol 2008;181:1065–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rashbrook VS, Brash JT, Ruhrberg C. Cre toxicity in mouse models of cardiovascular physiology and disease. Nat Cardiovasc Res 2022;1:806–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nishida Y, Arakawa S, Fujitani K, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009;461:654–8. [DOI] [PubMed] [Google Scholar]
- 104.Durgan J, Florey O. Many roads lead to CASM: diverse stimuli of noncanonical autophagy share a unifying molecular mechanism. Sci Adv 2022;8:eabo1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mareninova OA, Dillon DL, Wightman CJM, et al. Rab9 mediates pancreatic autophagy switch from canonical to noncanonical, aggravating experimental pancreatitis. Cell Mol Gastroenterol Hepatol 2022;13:599–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.De Faveri F, Chvanov M, Voronina S, et al. LAP-like non-canonical autophagy and evolution of endocytic vacuoles in pancreatic acinar cells. Autophagy 2020;16:1314–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jelski W, Kutylowska E, Laniewska-Dunaj M, et al. Alcohol dehydrogenase (ADH) isoenzymes and aldehyde dehydrogenase (ALDH) activity in the sera of patients with acute and chronic pancreatitis. Exp Mol Pathol 2011;91:631–5. [DOI] [PubMed] [Google Scholar]
- 108.Rasineni K, Srinivasan MP, Balamurugan AN, et al. Recent advances in understanding the complexity of alcohol-induced pancreatic dysfunction and pancreatitis development. Biomolecules 2020;10:669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ding W-X, Li M, Chen X, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010;139:1740–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Apte MV, Pirola RC, Wilson JS. Mechanisms of alcoholic pancreatitis. J Gastroenterol Hepatol 2010;25:1816–26. [DOI] [PubMed] [Google Scholar]
- 111.Shalbueva N, Mareninova OA, Gerloff A, et al. Effects of oxidative alcohol metabolism on the mitochondrial permeability transition pore and necrosis in a mouse model of alcoholic pancreatitis. Gastroenterology 2013;144:437–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kaphalia BS, Bhopale KK, Kondraganti S, et al. Pancreatic injury in hepatic alcohol dehydrogenase-deficient deer mice after subchronic exposure to ethanol. Toxicol Appl Pharmacol 2010;246:154–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pandol SJ, Lugea A, Mareninova OA, et al. Investigating the pathobiology of alcoholic pancreatitis. Alcohol Clin Exp Res 2011;35:830–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Maejima I, Takahashi A, Omori H, et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 2013;32:2336–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen X, Khambu B, Zhang H, et al. Autophagy induced by calcium phosphate precipitates targets damaged endosomes. J Biol Chem 2014;289:11162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Settembre C, Fraldi A, Medina DL, et al. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 2013;14:283–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Settembre C, Zoncu R, Medina DL, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 2012;31:1095–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Settembre C, Ballabio A. TFEB regulates autophagy: an integrated coordination of cellular degradation and recycling processes. Autophagy 2011;7:1379–81. [DOI] [PubMed] [Google Scholar]
- 119.Medina DL, Di Paola S, Peluso I, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 2015;17:288–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhu Y, He C, Li X, et al. Gut microbiota dysbiosis worsens the severity of acute pancreatitis in patients and mice. J Gastroenterol 2019;54:347–58. [DOI] [PubMed] [Google Scholar]
- 121.Patel BK, Patel KH, Bhatia M, et al. Gut microbiome in acute pancreatitis: a review based on current literature. World J Gastroenterol 2021;27:5019–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mareninova OA, Gretler SR, Lee GE, et al. Ethanol inhibits pancreatic acinar cell autophagy through upregulation of ATG4B, mediating pathological responses of alcoholic pancreatitis. Am J Physiol Gastrointest Liver Physiol 2023;325:G265–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fortunato F, Bürgers H, Bergmann F, et al. Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology 2009;137:350–60, [DOI] [PubMed] [Google Scholar]
- 124.Liu K, Zhao E, Ilyas G, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015;11:271–84. [DOI] [PMC free article] [PubMed] [Google Scholar]