


Abstract
We have identified a 74 kDa double-stranded (ds)RNA-binding protein that shares extensive homology with the mouse spermatid perinuclear RNA-binding (Spnr) protein. p74 contains two dsRNA-binding motifs (dsRBMs) that are essential for preferential binding to dsRNA. Previously, dsRNA-binding proteins were shown to undergo homo- and heterodimerization, raising the possibility that regulation of activity could be controlled by interactions between different family members. Homodimerization is required to activate the dsRNA-dependent protein kinase PKR, whereas heterodimerization between PKR and other dsRNA-binding proteins can inhibit kinase activity. We have found that p74 also interacts with PKR, both the wild-type enzyme and a catalytically defective mutant (K296R). While co-expression of p74 and wild-type PKR in the yeast Saccharomyces cerevisiae did not alter PKR activity, co-expression of p74 and the catalytically defective K296R mutant surprisingly resulted in abnormal morphology and cell death in transformants that maintained a high level of p74 expression. These transformants could be rescued by overexpression of the α-subunit of wild-type eukaryotic translation initiation factor 2 (eIF2α), one of the known substrates for PKR. We hypothesize that competing heterodimers between p74–K296R PKR and eIF2α–K296R PKR may control cell growth such that stabilization of the p74–K296R PKR heterodimer induces abnormal morphology and cell death.
INTRODUCTION
Double-stranded (ds)RNA-binding proteins play important roles in translation, RNA processing, RNA editing and embryonic pattern formation, yet the characterization and regulation of these proteins has lagged considerably compared to single-stranded (ss)RNA-binding proteins. The family of dsRNA-binding proteins are unique in that they interact primarily with the A-form double helix with little or no observable binding to ssRNA, ssDNA or dsDNA (1–3). The A-form helix differs from the typical dsDNA B-form helix in that the minor groove is shallow and broad and the major groove is narrow and deep, precluding extensive base interactions unless the helix is distorted (4,5). Thus, it is thought that these proteins bind dsRNA non-specifically, however, within a given RNA the combination of single-stranded loops, bulged nucleotides and double-stranded stems seems likely to combine to promote substrate specificity.
The dsRNA-binding protein family is defined by the presence of one or more copies of a dsRNA-binding motif (dsRBM), consisting of ~70 amino acids with dispersed conservation of basic and hydrophobic residues throughout the region (1,2,6). Ryter and Schultz (7) crystallized the second dsRBM of the Xenopus protein Xlrbpa in complex with dsRNA and found that the dsRBM uniquely contacts the dsRNA helix in two successive minor grooves and once in the intervening major groove. Despite these minimal contacts, mutation of nearly all of the conserved residues within the consensus dsRBM decreases dsRNA binding (1,6,8–11).
Perhaps the most intensely studied dsRNA-binding protein is the mammalian interferon-induced, dsRNA-dependent protein kinase PKR, which is involved in the regulation of translation initiation in response to stress (for reviews see 12–14). PKR is a serine/threonine kinase that plays a key role in interferon-mediated antiviral and antiproliferative responses by interfering with virus propagation through phosphorylation of the α subunit of translation initiation factor 2 (eIF2α). Phosphorylation of eIF2α causes the sequestration of the guanine nucleotide exchange factor eIF2B by eIF2, leading to inhibition of protein synthesis at the level of translation initiation (15). PKR is activated by dsRNA, consistent with the presence of two copies of the dsRBM in the N-terminus of the protein (12,16–19). It has been proposed that upon binding dsRNA, PKR undergoes a conformational change and becomes autophosphorylated (20). This activation event is mediated through homodimerization and makes PKR competent to phosphorylate exogenous substrates such as eIF2α (21,22). Overexpression of mammalian PKR in the yeast Saccharomyces cerevisiae leads to a slow growth phenotype due to the hyperphosphorylation of eIF2α. Mutation of the dsRBMs, which impairs dsRNA binding, reduces the ability of PKR to phosphorylate eIF2α in yeast, consistent with the idea that the dsRBMs mediate the stimulatory effect of dsRNA on PKR kinase activity (23,24). In addition, activation of PKR in transfected mammalian cell lines leads to the inhibition of translation, whereas transfection of a catalytically defective PKR mutant (K296R) can induce a transformed phenotype in NIH 3T3 cells and generate tumors upon injection into nude mice (25–28). Therefore, it has been proposed that PKR has tumor suppressor activity, in addition to antiviral activity.
Consistent with a key role for PKR in regulating cell growth, several viral and cellular inhibitors have been identified. Many of these proteins interfere with the binding of dsRNA activators while others are pseudosubstrates or PKR-specific proteases (14). In addition, interference with homodimer formation through the creation of inactive heterodimers has been found to potently inhibit kinase activity. Interestingly, two such interacting proteins are themselves members of the dsRNA-binding protein family: Tar RNA-binding protein (TRBP) and the vaccinia virus E3 protein (29,30). We recently identified a new dsRNA-binding protein (p74) that contains two dsRBMs and shares extensive homology with mouse spermatid perinuclear RNA-binding (Spnr) protein (31). In this report, we characterize p74, describe its RNA-binding properties and show that p74 interacts with PKR. Interestingly, co-expression in yeast of a catalytically defective form of PKR with p74 resulted in abnormal morphology and cell death in transformants that maintained a high level of p74 expression.
MATERIALS AND METHODS
Cloning and sequencing of p74 cDNAs
Radiolabeled RNA probes were used to screen a rat smooth muscle cDNA expression library (Stratagene) as described (32). Among several RNA-binding proteins isolated in this screen, one clone contained an insert of 2.8 kb encoding the entire open reading frame (ORF) of p74 (accession no. AF226864). The EcoRI–XhoI fragment containing the p74 ORF was subcloned into a pBlueScript SK– vector to facilitate DNA sequencing. Both strands of each cDNA were sequenced using Sequenase 2.0 and appropriate primers. Deduced amino acid sequence and alignments were compiled and compared using Mac-DNASIS. Alignment of the dsRBMs between p74 and related family members was performed using Clustal V.
Deletion and truncation mutants of p74 were prepared following cloning of the BglII fragment of the p74 cDNA into the BamHI site of pcDNA Amp (Invitrogen). Reverse PCR (33) was then used to repair the first 6 nt and all subsequent deletion constructs were prepared from this vector. Deletions were made using standard reverse PCR conditions. Each deletion construct was verified by sequencing.
For antibody production, a BglII fragment of p74 was cloned into the BamHI site of the pGEX2T vector creating a glutathione S-transferase (GST) fusion protein. Reverse PCR was then used to delete all but the N-terminus of p74, including the two conserved dsRBMs. The resulting plasmid was transformed into the Escherichia coli strain BL21pLysS and cells were grown in medium containing 0.1% glucose to an OD600 of 0.8. GST fusion proteins were then induced with 0.4 mM IPTG for 1 h at 37°C. Following sonication in PP150 buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 0.5 mM EDTA, 0.2% NP-40, 0.5 mM DTT, 2 µg/ml leupeptin and aprotonin, 1 mM PMSF), lysates were passed over a glutathione–agarose column, washed four times with PP150 buffer and eluted in 2 mM glutathione in PP150. The purified protein was then used as antigen for antibody production in rabbits (East-Acres Biologicals).
RNA expression analysis
A multiple tissue poly(A)+ RNA northern blot (Clontech) was prehybridized for 2 h at 42°C in prehybridization/hybridization solution (5× SSPE, 2× Denhardt’s solution, 100 µg/ml freshly denatured salmon sperm DNA, 0.5% SDS). Oligonucleotide probes were radiolabeled by incorporating [γ-32P]CTP using the 3′-exonuclease-deficient mutant of the Klenow fragment of DNA polymerase I, random nonamer primers and the p74 cDNA (Prime-It Random Primer Labeling Kit; Stratagene). Following labeling, reactions were passed over a Sephadex G-50 spin column to remove unincorporated nucleotides. Hybridization was carried out overnight at 42°C in the buffer outlined above with the radiolabeled probe. After hybridization, the membrane was washed (2× SSC, 0.05% SDS) at room temperature and exposed to a phosphorimager screen overnight. To control for equivalent RNA loading between lanes, the blot was stripped of the p74 probe and reprobed with a β-actin probe.
Western blots
Equivalent protein concentrations of yeast extracts overexpressing either p74 and/or PKR (wild-type or K296R) were separated on a 9% SDS–PAGE gel and then transferred to an Immobilon-P transfer membrane (Millipore) using a Transblot-SD semidry transfer cell for 45 min at 300 mA. Membranes were blocked at room temperature for 1 h in TBST (20 mM Tris, pH 8.0, 150 mM NaCl, 0.1% Tween-20) containing 5% non-fat dry milk, washed with TBST and incubated with the primary antibody (anti-PKR was a generous gift from Bryan Williams’ laboratory) for 1 h in TBST containing 10% fetal bovine serum. After washing in TBST, membranes were incubated with ECL detection reagents (Amersham) and exposed to X-ray film. The levels of K296R PKR and p74 protein expression between viable and lethal cells were analyzed from the same samples for comparison to one another as ratios.
In vitro immunoprecipitations
In vitro transcription and translation of p74, p74ΔD1 and PKR were performed in rabbit reticulocyte lysates using the TnT system (Promega). Direct immunoprecipitation of in vitro translated p74 or p74ΔD1 was performed using a p74 antibody immobilized on protein A–Sepharose (Sigma). For co-immunoprecipitation, parallel TnT reactions were carried out for p74, one with [35S]methionine and one with unlabeled methionine. The 35S-labeled proteins were separated on a 9% SDS–PAGE gel and the amount of radioactive protein was quantitated using a phosphorimager. Assuming equivalent translation efficiencies, unlabeled p74 protein was incubated with equal amounts of either wild-type or K296R [35S]PKR. The mixed proteins were incubated at room temperature for 30 min, after which 20 µl of anti-p74 prebound to protein A–Sepharose in binding buffer (20 mM Tris, pH 8.0, 100 mM KCl, 0.01% Tween-20) was added along with 300 µl of binding buffer and rocked at room temperature for 1 h. The beads were spun down briefly and washed three times with binding buffer. Bound proteins were eluted with an equal volume of 2× Laemmli loading buffer, boiled for 5 min and then loaded on 9% SDS–PAGE gels.
Polynucleotide binding studies
In vitro transcription/translation reactions of p74 were carried out using the TnT coupled reticulocyte lysate extract system (Promega) in the presence of [35S]methionine. Radiolabeled protein was incubated with various resins (Pharmacia) which were first washed and then resuspended in wash buffer 1 [20 mM Tris, pH 7.5, 5 mM β-mercaptoethanol, 4 mM Mg(C2H3O2)2·4H2O, 200 mM KCl, 0.5% NP-40]. Equal aliquots of 35S-labeled protein were added to 40 µl of a 1:1 volume of each resin and allowed to incubate at room temperature for 30 min. The resins were then spun down and washed three times with 500 µl of wash buffer 1 and then washed twice with 500 µl of wash buffer 2 [20 mM Tris, pH 7.5, 5 mM β-mercaptoethanol, 4 mM Mg(C2H3O2)2·4H2O, 50 mM KCl]. Bound proteins were eluted by adding an equal volume of 2× Laemmli loading buffer and equal aliquots were subjected to scintillation counting to determine the amount of protein bound to each resin. For analysis, the percentage of protein bound to each resin was calculated taking into consideration the specific activity of each deletion construct and comparisons were performed relative to the levels of wild-type protein bound to each resin. For competition binding studies, analyses were performed as above in the presence of competitor polynucleotides. Various concentrations of competitor [either poly(I:C) of poly(dI:dC)] ranging from 0.05 to 100 µg were added to the in vitro translated 35S-labeled p74 protein followed by the addition of resin. Analysis of bound protein was carried out as above.
Yeast strains and plasmid constructs
A BglII fragment of the p74 cDNA was cloned into the BamHI site downstream of the GAL1-10 promoter of the 2µ URA3 pSEY18 plasmid. Reverse PCR (33) was used to alter the start site of translation to match the yeast consensus. The repaired p74 cDNA, along with the upstream GAL1-10 promoter, was excised from plasmid pSEY18 as one fragment with EcoRI and SalI, treated with Klenow (Promega) and then cloned into the PstI site of the pEMBL yEX4 vector downstream of either the wild-type PKR or the K296R PKR cDNAs (pEMBL yEX plasmids were a generous gift from Dr Bryan Williams; 23).
A GAL1-10 promoter was cloned as an EcoRI–BamHI fragment into the 2µ HIS3 pRS423 vector. BamHI fragments encoding either the wild-type (sui2) or a mutant of the eIF2α gene (sui2M, Ser51Ala) (generous gifts from Dr Thomas Donahue; 34) were cloned into the BamHI site of plasmid pRS423/GAL1-10.
The yeast strain PSY315 (MATa, leu2, ura3, his3) was transformed with various combinations of the p74, PKR and sui2/2M constructs as described (35). To assay for the induction of lethality, individual or combinations of constructs were transformed a total of 98 times and 380 individual colonies were tested. Induced lethality was only observed upon co-transformation of both p74 and K296R PKR, whereas no lethality was observed in a similar number of control transformations and testing of individual colonies.
For normal growth, cells were grown in the appropriate synthetic liquid or agar medium containing 2% glucose. For induction, cells were grown in the presence of 2% raffinose synthetic medium to an approximate OD600 of 0.8–1.0, after which they were diluted to an approximate OD600 of 0.1 in synthetic medium containing 2% raffinose and 10% galactose at 30°C. In order to cure the strain of the plasmid co-expressing K296R PKR and p74, cells were grown on minimal plates containing 2 mg/ml uracil and 0.1% 5-fluoroorotic acid. Cells were tested for loss of the plasmid based on the ability to survive on minimal plates lacking uracil. All phase contrast micrographs were taken with a Zeiss Axioplan fluorescence microscope at 100× magnification.
RESULTS
Identification of p74
Based on protocols designed to identify novel DNA-binding proteins by screening cDNA expression libraries with DNA probes (36,37), we devised a protocol to identify RNA-binding proteins using short RNA probes (32). During one of these screens, we fortuitously identified an uncharacterized 2.8 kb cDNA with an open reading frame of 671 amino acids encoding a protein with a predicted molecular weight of 74 kDa most closely related to members of the dsRNA-binding protein family (p74; Fig. 1). Searching GenBank with the p74 protein sequence, a number of proteins highly homologous to p74 were identified, including dsRNA-binding proteins that had been previously identified using similar strategies (Fig. 2; 2,31). All of these related proteins are members of the dsRNA-binding protein family with two distinct dsRNA-binding motifs (dsRBMs; 1–3). An alignment of the two dsRBMs of p74 to other known dsRNA-binding proteins and the derived consensus is shown in Figure 3. These proteins include: Spnr, a spermatid perinuclear dsRNA-binding protein of unknown function (31); 4F.1, a Xenopus dsRNA-binding protein also of unknown function (2); human nuclear factor 90 (NF90), a human protein that was originally identified based on its binding to promoter elements upstream of the interleukin-2 gene (38); and MPP4, a human mitosis-specific phosphoprotein (39) that is apparently identical to NF90.
Figure 1.

p74 cDNA sequence and encoded protein. (A) Sequence of the 2.8 kb p74 cDNA. The arrow indicates the start of translation and the polyadenylation signal is underlined. (B) Domain structure of p74. p74 is a 671 amino acid protein containing two dsRBMs (residues 389–453 and 510–575; hatched boxes), a putative leucine zipper that overlaps with the first dsRBM (black box) and an N-terminal region (USCR) (residues 185–280; gray box; see Fig. 2) that is highly conserved among p74 and a subset of other dsRNA-binding proteins.
Figure 2.

Amino acid comparison between p74 and related dsRNA-binding proteins. The amino acid sequences for p74 and related proteins in human (NF90/MPP4, accession nos U10324 and X98254), mouse (Spnr, accession no. X84692) and frog (4F.1, accession no. U463902) are aligned. Identical amino acids are indicated with an asterisk while conserved changes are noted with a dot. TheUSCR and the two dsRNA-binding motifs (dsRBM 1 and 2) are overlined. The leucines in the putative leucine zipper are underlined in the p74 sequence. The GenBank submission for the MPP4 amino acid sequence is used above. Despite reported amino acid differences, the submitted DNA sequences for NF90 and MPP4 are nearly identical.
Figure 3.

Alignment of dsRBMs. The two dsRBMs of p74 (p74-1 and p74-2) are aligned with the dsRBMs from other dsRNA-binding proteins with the consensus sequence shown at the bottom (1). Amino acids matching the consensus are underlined. # designates hydrophobic residues. The alignment was performed using Clustal V. Sequence designations and accession numbers are as follows: rat p74 motifs 1 and 2, residues 389–453 and 510–575; human PKR, P19525, motifs 1 and 2, residues 8–77 and 99–167; human ADAR 2, U10493, motifs 1–3, residues 502–571, 613–682 and 725–794; Xenopus Xlrbpa, M96370, motifs 1–2, 19–87, 111–180 and 224–293; Drosophila staufen, P25159, motifs 1–5, residues 310–378, 489–557, 577–645, 710–780 and 951–1018; human TRBP, M60801, motifs 1–3, residues 8–76, 137–206 and 273–340; E.coli RNase III, P05797, residues 154–225.
Apart from the two dsRBMs, a second region within the N-termini of these proteins was also found to be remarkably conserved (Figs 1B and 2). This region, which we have named the upstream conserved region (USCR), does not show any homology to other functional domains but its conservation is suggestive of functional importance. Between p74 and Spnr there is also conservation of a putative leucine zipper that overlaps with the first dsRBM consisting of four heptad repeats preceded by a highly charged region (Fig. 2). Leucine zippers have been shown to be involved in protein–protein interactions during the formation of both homo- and heterodimers (40,41).
p74 is ubiquitously expressed
Despite extensive similarity between p74 and Spnr, northern blot analysis of p74 showed that it is widely expressed (Fig. 4), in contrast to Spnr, which is expressed primarily in testis (31). A 32P-labeled probe complementary to the p74 cDNA hybridized to three transcripts of ~3.1, 2.8 and 1.0 kb. The 2.8 kb mRNA species corresponds to the cDNA shown in Figure 1A. All isoforms appear to be ubiquitously expressed with slight differences between tissues. More stringent hybridization and wash conditions resulted in the loss of all three bands, suggesting that the additional bands are all p74-related transcripts.
Figure 4.
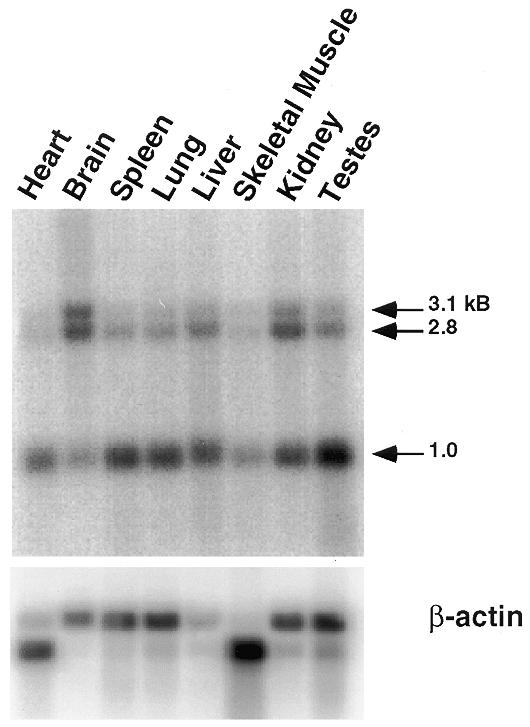
Northern blot analysis of p74 mRNA expression. A rat multiple tissue poly(A)+ RNA blot was probed with 32P-labeled random primed p74 cDNA (top blot). The same blot was stripped and reprobed with 32P-labeled random primed rat β-actin cDNA.
Nucleotide binding analysis of p74
To determine whether p74 actually binds dsRNA, 35S-labeled in vitro translated p74 was assayed for its ability to bind various polynucleotide resins. As shown in Figure 5B, wild-type p74 bound most efficiently to poly(I:C) RNA. However, to a lesser extent, wild-type p74 also bound poly(dI:dC). In contrast, wild-type p74 showed little or no ability to bind poly(A), poly(U) or poly(C) RNA, whereas efficient binding to poly(G) was detected. Given the propensity for poly(G) to adopt significant secondary structure, including extensive double-stranded character (42), this is not too surprising.
Figure 5.

Nucleotide binding analysis of the p74 protein. (A) Deletion constructs. The domain structure of wild-type p74 and various deletion mutants are shown with the USCR (gray) and dsRBMs (hatched) as noted. (B) Polynucleotide binding analysis. p74 was in vitro translated in rabbit reticulocyte lysates and the 35S-labeled translation products were incubated with various excess polynucleotide resins (as noted). Bound proteins were eluted and the amount of bound protein was determined relative to wild-type binding (see Materials and Methods). (C) Competition binding studies. In vitro translated wild-type p74 was incubated with various concentrations of competitor nucleic acid [either poly(I:C) or poly(dI:dC), as indicated] followed by incubation with dsRNA–agarose. Bound proteins were eluted and analyzed as in (B).
Competition binding studies were next performed to compare the binding of wild-type p74 to dsRNA versus dsDNA. For these experiments, in vitro translated p74 was incubated with poly(I:C)–agarose in the presence of either free poly(I:C) or poly(dI:dC) competitor and assayed for the ability to bind to the poly(I:C) resin (Fig. 5C). In the presence of increasing amounts of poly(I:C) RNA, the binding of wild-type p74 to poly(I:C)–agarose resulted in a 75% decrease in binding in the presence of the highest concentration of competitor. In contrast, poly(dI:dC) failed to compete for wild-type p74 binding to the poly(I:C) resin with the highest concentration of competitor resulting in only a 30% reduction in binding. From additional experiments using equimolar amounts of competitor (data not shown), it is clear that wild-type p74 preferentially binds dsRNA.
The two dsRBMs of p74 do not contribute equally to dsRNA-binding activity
St Johnston and co-workers (1) have defined regions of the Drosophila protein staufen and the Xenopus protein Xlrpba that are capable of independently binding dsRNA through an ~65 amino acid motif known as the dsRBM. Many dsRNA-binding proteins contain more than one dsRBM but the role of individual dsRBMs within these proteins is not entirely clear considering that not all dsRBMs within a particular protein display identical dsRNA-binding capability. For PKR, optimal binding of dsRNA apparently requires both of its dsRBMs but mutations in the first motif are more debilitating than those in the second (6). In contrast, only the second dsRBM of TRBP is needed for dsRNA-binding in vitro (8,10). Therefore, to determine if both dsRBMs of p74 were required for binding to dsRNA and/or whether additional regions of p74 might modulate dsRNA-binding, a series of deletion constructs of p74 were assayed for their ability to bind dsRNA (Fig. 5A and B). Deletion of the first dsRBM (ΔD1) resulted in a 50% decrease in dsRNA binding. However, when the second dsRBM was deleted (ΔD2) there was an even more substantial decrease in binding to ~20% of wild-type. When the USCR was deleted, no significant decrease in dsRNA binding was observed. However, when both the USCR and the first dsRBM were deleted (ΔU&D1), dsRNA-binding decreased by ~5-fold over the ΔD1 construct alone, suggesting that when only one dsRBM is present, the USCR might promote or stabilize dsRNA binding. Deletion of both dsRBMs or deletion of all three domains (ΔU/D1&2) completely abolished dsRNA binding. These results suggest that the dsRBMs are mostly responsible for the ability of p74 to bind dsRNA but the USCR may facilitate and/or stabilize such binding.
To examine binding specificity, the same set of deletion constructs were tested for their ability to bind dsDNA and ssRNA as above. The most interesting finding from these experiments was that when the second dsRBM or both dsRBMs were deleted (ΔD2 and ΔD1&2, respectively), the binding of p74 to dsDNA and poly(G) was greater than the binding observed for these constructs to dsRNA. This suggests that there may be information encoded within the USCR that increases the potential binding specificity of p74.
p74 interacts with the dsRNA-dependent protein kinase PKR
Activation of PKR has been found to require homodimer formation and disruption of such dimers inhibits kinase activity (29,43–45). Interestingly, two other dsRNA-binding proteins, TRBP and vaccinia virus E3 protein, have been found to inactivate PKR through heterodimerization (29,30). Recently, the human protein NF90/MPP4 has also been found to interact with PKR, although the effect of such an interaction remains to be defined (48,50). Therefore, we were interested to know if p74 might also interact with PKR, potentially regulating kinase activity. Initially, we tested whether these proteins might interact in rabbit reticulocyte lysates following in vitro translation and co-immunoprecipitation. Co-immunoprecipitations were performed with in vitro translated p74 and PKR (wild-type and K296R mutant) and an antibody directed against the N-terminus of p74. As shown in Figure 6B, anti-p74 antibodies did not immunoprecipitate either form of PKR in the absence of p74. In contrast, when lysates programmed with both p74 and PKR were mixed, PKR could be immunoprecipitated with antibodies against p74. Since both the wild-type and mutant forms of PKR could be immunoprecipitated with the anti-p74 antibodies, the interaction between these proteins is apparently not dependent upon an active kinase. When the converse experiment was performed using antibodies against PKR the same results were obtained (data not shown). Thus, p74, like TRBP, NF90 and E3, can interact with PKR.
Figure 6.

In vitro association between p74 and PKR. (A) In vitro translation products. In vitro translation of p74, p74ΔD1 (see Fig. 5), PKR and K296R PKR was performed in reticulocyte lysates. Radioactive proteins were separated by SDS–PAGE and visualized by autoradiography. (B) Co-immunoprecipitation of p74 and PKR. Immunoprecipitation reactions were performed on individual lysates (IPs) or mixed lysates (co-IPs) with antibodies against p74. For ease of identification, the input p74 translation product (wt or ΔD1) was unlabeled in the co-immunoprecipitation reactions and only PKR was labeled.
Since p74 contains a region resembling a leucine zipper and because leucine zippers have been broadly implicated in protein–protein interactions (40,41), we sought to determine whether this region might mediate the interaction with PKR. Thus, the first dsRBM, which overlaps with the putative leucine zipper domain, was deleted (ΔD1) and then assayed for interaction with PKR via co-immunoprecipitation. Both wild-type PKR and the K296R PKR mutant efficiently co-immunoprecipitated with the p74ΔD1 deletion construct (Fig. 6B). Therefore, neither the first dsRBM nor the overlapping leucine zipper motif encompass the p74–PKR interaction domain. Rather than simply continue to define which domains are essential for interaction, we next chose to determine whether such interaction might be functionally relevant.
Co-expression of p74 and K296R PKR in yeast results in abnormal morphology and cell death
Overexpression of wild-type PKR in S.cerevisiae has been found to result in hyperphosphorylation of the translation initiation factor eIF2α resulting in a slow growth phenotype due to the inhibition of translation. In contrast, overexpression of a catalytically defective form of PKR (K296R) does not lead to altered cell growth in yeast (23). Due to the fact that p74 and PKR were found to interact, we tested whether co-expression of p74 with PKR could ameliorate the suppressive effects of wild-type PKR on yeast growth. Accordingly, yeast expression vectors were created expressing p74 and either wild-type or mutant (K296R) PKR under the control of independent GAL promoters. Yeast transformed with these vectors showed no growth defects when grown in the presence of glucose, but upon shifting to galactose-containing medium the presence of wild-type PKR dramatically slowed growth, confirming that expression of PKR was responsible for the observed defect (Fig. 7). As expected, growth of yeast strains expressing the K296R PKR mutant grew equally well in the presence of glucose or galactose. Similarly, expression of p74 did not result in altered growth rates under either condition. Interestingly, when yeast were transformed with both p74 and wild-type PKR, co-expression of p74 did not rescue the slow growth phenotype induced by PKR whereas co-expression of p74 with the K296R PKR mutant surprisingly resulted in a lethal phenotype.
Figure 7.

Co-expression of p74 and PKR in S.cerevisiae. Wild-type PKR (wt PKR), mutant PKR (K296R PKR) and p74 were placed under the control of independent GAL promoters and overexpressed in S.cerevisiae either alone or in combination. (A) Representative glucose- and galactose-containing plates of the various constructs. (B) Liquid growth curves of the same constructs as in (A).
Closer examination of yeast strains exhibiting the lethal phenotype uncovered a dramatic morphological change as well. Upon induction of gene expression with galactose, these cells adopted a predominantly unbudded, uniformly spherical shape, approximately four times larger than their uninduced or wild-type counterparts (Fig. 8A, lane 4). Most of the increased size was taken up by large vacuoles that nearly obliterated other structures. When grown in the presence of glucose, these cells appeared identical to wild-type strains consisting of numerous budded and unbudded, elliptically shaped cells of normal size (Fig. 8A, lanes 1–3 and 5). To confirm that the abnormal morphology and lethal phenotype were due to the expression of both p74 and the K296R PKR mutant, strains were cured of the plasmid encoding the two proteins and re-examined (Fig. 8B). Importantly, removal of these genes resulted in a return to normal growth and morphology. Thus, the combination of p74 and K296R PKR is responsible for the observed abnormal morphology and lethal phenotype.
Figure 8.

Protein expression of p74 and K296R PKR in yeast. (A) After overnight growth of yeast in glucose-containing medium, K296R PKR and/or p74 were expressed in medium containing 2% raffinose and 10% galactose. Following induction, extracts were prepared and protein concentrations were determined. Equivalent amounts of protein were then loaded onto 9% SDS–PAGE gels and western blots were performed sequentially with anti-p74 antibodies (top blot) and anti-PKR antibodies (bottom blot). Lane 1, K296R PKR strain; lane 2, p74 strain; lane 3, viable K296R PKR and p74 strain; lane 4, lethal K296R PKR and p74 strain; lane 5, vector alone strain. The morphology of each of the strains at the same time point was examined using phase contrast micrographs of representative yeast cells (top). (B) Restoration of normal morphology by loss of the URA3 plasmid expressing both K296R PKR and p74. Yeast were plated onto medium containing 5-fluoroorotic acid (5FOA) which selects for loss of URA3 plasmids. Representative phase contrast micrographs, at 100× magnification, were taken as above.
Initial yeast transformation experiments suggested that the lethal phenotype observed upon overexpression of p74 and K296R PKR occurred in a majority of transformants. However, subsequent work has shown that induction of lethality is a relatively rare event, occurring in only 5.9% of transformed yeast colonies (see Materials and Methods). Nevertheless, the observed phenotype is consistent and is only observed upon co-expression of both p74 and K296R PKR; lethality has never been observed upon expression of either construct alone or with other combinations. We hypothesized that lethality might be triggered by a specific stoichiometry between p74 and K296R PKR. If this is true, one would expect differences in the levels of the two proteins in strains exhibiting the lethal phenotype versus those capable of continued growth. To test this hypothesis, the levels of the two proteins were analyzed by western blot analysis in both types of strains. Strains exhibiting the lethal phenotype contained ~2- to 5-fold higher levels of p74 than unaffected strains (Fig. 8A, lanes 3 and 4). Thus, there appears to be strong selective pressure on yeast strains to alter the expression level of p74 to enable continued growth. Since we have never observed changes in the levels of K296R PKR between the two different growth states (Fig. 8A and data not shown), it appears that down-regulation of p74 predominates to ensure survival.
Induction of lethality involves eIF2α
While it is formally possible that p74 and K296R PKR are disrupting unrelated pathways whose combination proves lethal, the simplest interpretation of the observed growth defects and the co-immunoprecipitation data are that these proteins interact in vivo. To further investigate the mechanistic basis of lethality upon overexpression of p74 and K296R PKR, we focused on the known role of PKR in the inhibition of translation initiation via phosphorylation of eIF2α. PKR phosphorylates eIF2α at Ser51 to prevent initiation of translation by hindering eIF2B activity. eIF2B mediates the guanine nucleotide exchange step required to regenerate active eIF2–GTP. Thus, if eIF2α is phosphorylated, translation initiation is inhibited due to a lack of eIF2–GTP (15). One hypothesis that focused on the inhibitory effect of PKR on translation initiation to explain the observed growth defect in the p74–K296R PKR strain was that eIF2α might be phosphorylated by the K296R PKR mutant leading to inhibition of translation. To directly test whether phosphorylation of eIF2α played a role in inhibiting growth in these strains, we co-transformed p74 and the K296R PKR mutant into yeast strains expressing a non-phosphorylatable form of eIF2α (Ser51Ala) under the control of a GAL promoter (23). This construct did not rescue the lethal phenotype (Fig. 9) nor did it alter the abnormal morphology seen in these cells (data not shown). Interestingly, however, when wild-type eIF2α, also under control of the GAL promoter, was overexpressed in the p74–K296R PKR strains, the lethal phenotype was never observed (Fig. 9) and the morphology of all such transformants resembled wild-type yeast (data not shown). Due to the fact that only wild-type eIF2α is competent to rescue the lethal phenotype, the phosphorylation state of Ser51 must be intricately involved in the induction of lethality. Furthermore, the ability to rescue lethality, both with wild-type eIF2α and by curing the strains of genes encoding p74 and K296R PKR (Fig. 8B), suggests that even though the induction of lethality is a rare event, it requires the presence of both proteins.
Figure 9.

Rescue of lethality by expression of eIF2α. Yeast strains expressing the indicated proteins were plated onto glucose- or galactose-containing plates as in Figure 7. Co-expression of wild-type eIF2α rescued the lethality induced by expression of p74 and K296R PKR whereas co-expression of non-phosphorylatable eIF2α(Ser51Ala) did not.
DISCUSSION
In this paper, we have identified a 74 kDa protein (p74) encoding a new member of the dsRNA-binding protein family. The protein contains two dsRBMs, an N-terminal auxiliary region of unknown function (USCR) and a region resembling a leucine zipper. p74 shares the highest level of amino acid identity (92%) with Spnr, a mouse spermatid perinuclear RNA-binding protein (31). While the degree of conservation between these two proteins suggests they may be homologous, p74 is widely expressed whereas Spnr is expressed primarily in the testis and localizes to the manchette, a spermatid-specific microtubule array. Both p74 and Spnr are similar to the Xenopus 4F.1 and NF90 proteins (54 and 50% identity, respectively, compared to p74), with the highest degree of similarity found in the USCR and the two dsRBMs. With the possible exception of NF90, the function of each of these proteins remains unknown. NF90 has been found to exist as a heterodimer with a protein of 45 kDa, nuclear factor 45 (NF45; 38). Together, these factors are proposed to be members of the NFAT family of transcription factors involved in the regulation of interleukin-2 gene transcription. More recently, NF90 has been found to preferentially bind the adenovirus-associated VA RNAII, suggesting a potential involvement in the regulation of viral infection (47). NF90 is apparently identical to MPP4 (M-phase phosphoprotein 4) which was identified based on its specific phosphorylation during mitosis, suggestive of a cell cycle-dependent function (39).
Substrate binding analyses with p74 have shown that it requires both dsRBMs to confer wild-type binding to dsRNA. However, a single dsRBM in the presence of the USCR is capable of binding dsRNA, suggesting that the USCR may be able to modulate or stabilize dsRNA binding. In the absence of dsRNA, we have found that p74 is capable of binding to dsDNA provided the UCSR is intact (Fig. 5B, see ΔD1&2 versus ΔU). The ability of p74 to bind dsDNA is consistent with the finding that other dsRNA-binding proteins, including NF90, which contains a USCR, are capable of binding dsDNA as well (38,46). However, competition binding studies have shown that p74 clearly prefers dsRNA to dsDNA (Fig. 5), as does NF90 (48). Depending on the relative levels of dsRNA versus dsDNA in cells, nuclei or other sub-compartments, differential binding to either dsDNA or dsRNA may allow these proteins to function in multiple pathways.
Growth control by dsRNA-binding proteins
The hypothesis that PKR may act as a tumor suppressor suggests that the normal cellular function of PKR must be tightly regulated to control translation and/or transcription of normally repressed growth-promoting or differentiation-specific genes. Previously, PKR has been shown to be inactivated by heterodimerization with the cellular protein TRBP and vaccinia virus E3 protein, both of which are dsRNA-binding proteins (29,45,49). We have now shown by co-immunoprecipitation that another dsRNA-binding protein, p74, can also interact with PKR. Thus, overall regulation of dsRNA-binding proteins could be due to the formation of both homo- and heterodimers between different family members.
To test whether heterodimer formation between PKR and p74 might regulate the activity of PKR, the two genes were overexpressed in the yeast S.cerevisiae. Expression of p74 in the presence of wild-type PKR was not able to rescue the growth-suppressive activity of PKR. However, when p74 was co-expressed with a catalytically defective form of PKR (K296R), a subset of the transformants displayed abnormal morphology and eventually died. Lethality always required the presence of both genes and excision of the plasmid containing these genes restored normal morphology and growth, indicating that the effects on yeast growth are solely due to the action of these two gene products. While only a minority of transformants exhibited the lethal phenotype, all arrested strains consistently expressed higher levels of p74, whereas the expression of K296R PKR was relatively constant (Fig. 8A). The inability of p74 to alter the slow growth phenotype of yeast expressing wild-type PKR could conceivably have been due to autorepression of PKR, thereby ablating stable p74–PKR interaction. However, western blots have shown that the ratios of p74 to either K296R or wild-type PKR were comparable, except in strains exhibiting the lethal phenotype, which maintained higher levels of p74 (Fig. 8A and data not shown). Since the genes for both proteins are contained within a single plasmid, the difference in p74 levels cannot be explained simply by variation in plasmid copy number. Rather, it appears that there is great selective pressure to down-regulate p74 expression or activity in order to ensure survival.
One possible mechanism by which yeast may be able to down-regulate p74 is through post-translational modification and targeted degradation. Using extracts prepared from yeast expressing p74, we have recently found that p74 is a phosphoprotein (data not shown), suggesting that the regulation of p74 function may be integrally linked to its phosphorylation state. Since the closely related protein NF90 has recently been shown to be a substrate for PKR (48), it is possible that phosphorylation of p74 by PKR might disrupt heterodimer formation whereas the inability of the K296R PKR mutant to phosphorylate p74 might stabilize the complex, potentially altering downstream signaling pathways eventually leading to lethality. Alternatively, stable complex formation between p74 and K296R PKR might block phosphorylation of p74 by an unknown yeast kinase, thereby affecting its half-life and/or disrupting downstream signaling cascades.
Rescue of lethality by eIF2α
Due to the well-characterized role of PKR in the phosphorylation of eIF2α and the inhibition of translation initiation, we initially investigated whether phosphorylation of eIF2α and/or potential sequestration of eIF2α by the p74–K296R PKR heterodimer might be responsible for cell death in strains co-expressing p74 and K296R PKR. We hypothesized that if lethality was a result of phosphorylation of eIF2α by the p74–K296R PKR heterodimer, then one would expect to be able to rescue lethality by overexpressing a non-phosphorylatable mutant of eIF2α (Ser51Ala). However, we found that lethality could only be rescued by overexpression of wild-type eIF2α and not by the Ser51Ala mutant. This observation argues against lethality being a result of the direct phosphorylation of eIF2α by the K296R PKR mutant. In addition, these results seem to argue against a simple sequestration model of eIF2α since both forms of eIF2α should have been able to titrate the levels of the p74–K296R PKR complex. Perhaps the most direct explanation for our results assumes simple competition between competing heterodimers containing either p74–K296R PKR or eIF2α–K296R PKR. In this model, eIF2α–K296R PKR heterodimers are the most stable, followed by heterodimeric complexes p74–K296R PKR and K296R PKR–eIF2α(Ser51Ala), such that excess wild-type eIF2α promotes dissociation of the p74–K296R PKR heterodimer, restoring normal morphology and growth. The relative stability of these different heterodimers may well be regulated by phosphorylation (see below).
Alternatively, p74, PKR and eIF2α may normally associate as part of a larger complex whose disruption is responsible for the growth defects we have observed and which may be unrelated to the regulation of translation initiation. In mammalian cells, there is support for a large complex consisting of NF45, NF90, PKR, the three subunits of eIF2 and DNA-dependent protein kinase (DNA-PK) (48,50). DNA-PK is a serine/threonine kinase that has been found to be involved in recombination and dsDNA break repair. The enzyme is composed of a large catalytic subunit (DNA-PKcs) and two DNA-binding subunits (Ku70 and Ku80). DNA-PK requires dsDNA for activity. Interestingly, NF90 and NF45 are substrates for both PKR and DNA-PK (50; M.B.Mathews, personal communication), raising the possibility that misregulation of phosphorylation of one or more of these proteins may lead to aberrant complex formation (or stability) which could in turn lead to the inability to repair double-strand breaks and/or accurately replicate DNA during S phase. Consistent with regulated phosphorylation of these subunits, MPP4, which is identical to NF90, was identified because its phosphorylation state changes during mitosis. Also, yeast strains lacking Ku70, one of the subunits of DNA-PK, are deficient in double-strand break repair and accumulate as large, unbudded yeast (51), reminiscent of the morphological defect we observe in yeast strains co-expressing p74 and K296R PKR. Thus, the mechanism of lethality in yeast strains overexpressing p74 and K296R PKR may be due not only to potential misregulation of translation, but also to potential misregulation of a complex that functions in DNA repair and/or monitors accurate DNA replication prior to mitosis. Future work will be needed to test these and other models to explain the morphological and growth defects we observe in yeast cells expressing p74 and the K296R PKR mutant, but the exciting possibility exists that these proteins participate in complexes that may link the regulation of translation with progression through the cell cycle.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Drs Bryan Williams and Thomas Donahue for yeast expression plasmids and antibodies and Dr Ronald Wek for yeast and bacterial strains and recombinant PKR. We would also like to thank Dr Michael Mathews and members of his laboratory for helpful discussions and communicating results prior to publication and Dr Douglas Cavener for critical reading of the manuscript.
DDBJ/EMBL/GenBank accession no. AF226864
REFERENCES
- 1.St Johnston D., Brown,N.H., Gall,J.G. and Jantsch,M. (1992) Proc. Natl Acad. Sci. USA, 89, 10979–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bass B.L., Hurst,S.R. and Singer,J.D. (1994) Curr. Biol., 4, 301–314. [DOI] [PubMed] [Google Scholar]
- 3.Bevilacqua P.C. and Cech,T.R. (1996) Biochemistry, 35, 9983–9994. [DOI] [PubMed] [Google Scholar]
- 4.Delarue M. and Moras,D. (1989) Nucleic Acids Mol. Biol., 3, 182–196. [Google Scholar]
- 5.Steitz T.A. (1993) In Gesteland,R.F. and Atkins,J.F. (eds), The RNA World. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 219–237.
- 6.Green S.R. and Mathews,M.B. (1992) Genes Dev., 6, 2478–2490. [DOI] [PubMed] [Google Scholar]
- 7.Ryter J.M. and Schultz,S.C. (1998) EMBO J., 17, 7505–7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gatignol A., Buckler-White,A., Berkhout,B. and Jeang,K.-T. (1991) Science, 251, 1597–1600. [DOI] [PubMed] [Google Scholar]
- 9.McCormack S.J., Thomis,D.C. and Samuel,C.E. (1992) Virology, 188, 47–56. [DOI] [PubMed] [Google Scholar]
- 10.Gatignol A., Buckler-White,A. and Jeang,K.-T. (1993) Mol. Cell. Biol., 13, 2193–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krovat B.C. and Jantsch,M.F. (1996) J. Biol. Chem., 271, 28112–28119. [DOI] [PubMed] [Google Scholar]
- 12.Clemens M.J. (1997) Int. J. Biochem. Cell Biol., 29, 945–949. [DOI] [PubMed] [Google Scholar]
- 13.Williams B.R.G. (1995) Semin. Virol., 6, 191–202. [Google Scholar]
- 14.Proud C.G. (1995) Trends Biochem. Sci., 20, 241–246. [DOI] [PubMed] [Google Scholar]
- 15.Merrick W.C. (1992) Microbiol. Rev., 56, 291–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maas S., Melcher,T., Herb,A., Seeburg,P.H., Keller,W., Krause,S., Higuchi,M. and O’Connell,M.A. (1996) J. Biol. Chem., 271, 12221–12226. [DOI] [PubMed] [Google Scholar]
- 17.Mathews M.B. (1993) Semin. Virol., 4, 247–257. [Google Scholar]
- 18.Safer B. (1983) Cell, 33, 6–8. [DOI] [PubMed] [Google Scholar]
- 19.Proud C.G. (1986) Trends Biochem. Sci., 11, 73–77. [Google Scholar]
- 20.Galabru J. and Hovanessian,A.G. (1987) J. Biol. Chem., 262, 15538–15544. [PubMed] [Google Scholar]
- 21.Langland J.O. and Jacobs,B.L. (1992) J. Biol. Chem., 267, 10729–10736. [PubMed] [Google Scholar]
- 22.Thomis D.C. and Samuel,C. (1993) J. Virol., 67, 7695–7700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chong K.L., Feng,L., Schappert,K., Meurs,E., Donahue,T.F., Friesen,J.D., Hovanessian,A.G. and Williams,B.R. (1992) EMBO J., 11, 1553–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Romano P.R., Green,S.R., Barber,G.N., Mathews,M.B. and Hinnebusch,A.G. (1995) Mol. Cell. Biol., 15, 365–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koromilas A.E., Roy,S., Barber,G.N., Katze,M.G. and Sonenberg,N. (1992) Science, 257, 1685–1689. [DOI] [PubMed] [Google Scholar]
- 26.Meurs E.F., Galabru,J., Barber,G.N., Katze,M.G. and Hovanessian,A.G. (1993) Proc. Natl Acad. Sci. USA, 90, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barber G.N., Wambach,M., Thompson,S., Jagus,R. and Katze,M.G. (1995) Mol. Cell. Biol., 15, 3138–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donze O., Jagus,R., Koromilas,A.E., Hershey,J.W. and Sonenberg,N. (1995) EMBO J., 14, 3828–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cosentino G.P., Venkatesan,S., Serluca,F.C., Green,S.R., Mathews,M.B. and Sonenberg,N. (1995) Proc. Natl Acad. Sci. USA, 92, 9445–9449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Romano P.R., Zhang,F., Tan,S.-L., Garcia-Barrio,M.T., Katze,M.G., Dever,T.E. and Hinnebusch,A.G. (1998) Mol. Cell. Biol., 18, 7304–7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schumacher J.M., Lee,K., Edelhoff,S. and Braun,R.E. (1995) J. Cell Biol., 129, 1023–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patton J.G., Dye,B.T., Barnard,D.C. and McAfee,J.G. (1997) In Richter,J.D. (ed.), Analysis of mRNA Formation and Function. Academic Press, San Diego, CA, pp. 55–78.
- 33.Imai Y., Matshushima,Y., Sugimura,T. and Terada,M. (1991) Nucleic Acids Res., 19, 2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cigan A.M., Pabich,E.K., Feng,L. and Donahue,T.F. (1989) Proc. Natl Acad. Sci. USA, 86, 2784–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ito H., Fukuda,Y., Murata,K. and Kimura,A. (1983) J. Bacteriol., 153, 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vinson C.R., LaMarco,K.L., Johnson,P.F., Landschulz,W.H. and McKnight,S.L. (1988) Genes Dev., 2, 801–806. [DOI] [PubMed] [Google Scholar]
- 37.Singh H., LeBowitz,J.H., Baldwin,A.S.J. and Sharp,P.A. (1988) Cell, 52, 415–423. [DOI] [PubMed] [Google Scholar]
- 38.Kao P.N., Chen,L., Brock,G., Ng,J., Kenny,J., Smith,A.J. and Corthesy,B. (1994) J. Biol. Chem., 269, 20691–20699. [PubMed] [Google Scholar]
- 39.Matsumoto-Taniura N., Pirollet,F., Monroe,R., Gerace,L. and Westendorf,J.M. (1996) Mol. Biol. Cell, 7, 1455–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Landshultz W.H., Johnson,P.F. and McKnight,S.L. (1988) Science, 240, 1759–1764. [DOI] [PubMed] [Google Scholar]
- 41.Busch S.J. and Sassone-Corsi,P. (1990) Oncogene, 5, 1549–1556. [PubMed] [Google Scholar]
- 42.Gellert M., Lipsett,M.N. and Davies,D.R. (1962) Proc. Natl Acad. Sci. USA, 48, 2013–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Polyak S.J., Tang,N., Wambach,M., Barber,G.N. and Katze,M.G. (1996) J. Biol. Chem., 271, 1702–1707. [DOI] [PubMed] [Google Scholar]
- 44.Gale M. Jr, Tan,S.L., Wambach,M. and Katze,M.G. (1996) Mol. Cell. Biol., 16, 4172–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Romano P.R., Garcia-Barrio,M.T., Zhang,X., Wang,Q., Taylor,D.R., Zhang,F., Herring,C., Mathews,M.B., Qin,J. and Hinnebusch,A.G. (1998) Mol. Cell. Biol., 18, 2282–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herbert A., Lowenhaupt,K., Spitzner,J. and Rich,A. (1995) Proc. Natl Acad. Sci. USA, 92, 7550–7554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liao H.-J., Kobayashi,R. and Mathews,M.B. (1998) Proc. Natl Acad. Sci. USA, 95, 8514–8519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langland J.O., Kao,P.N. and Jacobs,B.L. (1999) Biochemistry, 38, 6361–6368. [DOI] [PubMed] [Google Scholar]
- 49.Benkirane M., Neuveut,C., Chun,R.F., Smith,S.M., Samuel,C.E., Gatignol,A. and Jeang,K.-T. (1997) EMBO J., 16, 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ting N.S.Y., Kao,P.N., Chan,D.W., Lintott,L.G. and Lees-Miller,S.P. (1998) J. Biol. Chem., 273, 2136–2145. [DOI] [PubMed] [Google Scholar]
- 51.Barnes G. and Rio,D. (1997) Proc. Natl Acad. Sci. USA, 94, 867–872. [DOI] [PMC free article] [PubMed] [Google Scholar]