

Abstract
Metabolic diseases, such as type 2 diabetes or obesity, are the consequence of the disruption of the organism’s metabolic pathways. The discovery of small non-coding RNAs—microRNAs (miRNAs)—as post-transcriptional gene regulators opened new doors for the development of novel strategies to combat said diseases. The two strands of miR-378a, miR-378a-3p, and miR-378a-5p are encoded in the Ppargc1b gene and have an active role in the regulation of several metabolic pathways such as mitochondrial metabolism and autophagy. Recent studies recognized miR-378a as an important regulator of energy and glucose homeostasis, highlighting it as a potential target for the improvement of metabolic dysregulation. In the present review, the current knowledge on miR-378a will be discussed with a particular emphasis on its biological functions and mechanisms of action in metabolism, mitochondria, and autophagy.
Keywords: miR-378a, Metabolic diseases, Mitochondria, Metabolism, Autophagy
Introduction
In recent years, microRNAs (miRNAs) have emerged as a new class of small non-coding RNAs and were revealed to be important players in gene regulation being involved in several biological processes. These small molecules are composed of about 22 nucleotides in length and mainly bind to the 3′ untranslated region (3′ UTR) of mRNAs to post-transcriptionally inhibit gene expression through two different mechanisms, either by degrading the targeted transcript or by inhibiting its translation. Our knowledge about gene expression regulation has greatly increased, since miRNAs were first discovered. Even so, new miRNAs are currently being found and their respective biological functions are being further explored. A growing body of evidence strongly supports the involvement of miRNAs in complex biological processes such as metabolism [1], cancer [2], programmed cell death [3], or cell differentiation [4].
miRNAs are synthesized in the cell nucleus and are subsequently translocated to the cytoplasm where they are matured and interfere with mRNAs. Although this has been well established, recent studies have found miRNAs in mitochondria, where they are likely to have a significant role [5–8]. However, the mechanisms supporting their translocation to mitochondria are far from being fully understood. Nevertheless, a recently found miRNA belonging to the family of miR-378 was found inside mitochondria and was able to interfere with mitochondrial DNA-encoded mRNAs [9]. miR-378a is an intronic miRNA located in the Ppargc1b gene and has been involved in a wide range of research fields. Recently, miR-378a has been extensively associated with metabolism. This miRNA favours tumour growth and angiogenesis by stimulating its glycolytic metabolism and by inhibiting tumour suppressors [10, 11], while it interferes with metabolic and signalling pathways in the liver and skeletal muscle proving to be a key regulator of lipid metabolism, and glucose and energy metabolism [12, 13]. In addition, miRNAs roles in the regulation of programmed cell death are mostly unexplored, although miR-378a was recently shown to be important in the maintenance of apoptosis and autophagy in skeletal muscle [14]. Here, we review the main roles of miR-378a in the regulation of metabolism and autophagy and discuss it as a promising therapeutic strategy to use against metabolic diseases.
Regulation of metabolism by miR-378a
miR-378a origin and structure
Peroxisome proliferator-activated receptor-γ (PPARγ) coactivator-1 beta (PGC-1β) is encoded by the gene Ppargc1b and embedded in its first intron are two members of the miR-378 family (Fig. 1) [10]. miR-378a-3p (usually identified as miR-378 and previously named miR-422b [15]) and miR-378a-5p (usually identified as miR-378*) are considered to be the guide and passenger strands of miR-378a, respectively, and are highly conserved between human (hsa-miR-378a) and mice (mmu-miR-378a) (reviewed in [16]). Both strands are thought to be simultaneously transcribed with PGC-1β [10, 17, 18], although it was recently found an exception to this rule. Liver receptor alpha (LXRα) reportedly activated miR-378a transcription, while at the same time inhibited Ppargc1b transcription [19]; therefore, hinting that the mechanisms ruling Ppargc1b and miR-378a transcription might be different. miR-378a-3p and miR-378a-5p were found to be expressed in mitochondria-enriched tissues, such as skeletal muscle, heart, liver, and brown adipose tissue (BAT) [14, 17] and it was suggested that it might be involved in metabolic pathways regulated by PGC-1β, such as in glucose and systemic energy homeostasis, fatty acid oxidation, and mitochondria metabolism. Furthermore, these miRNAs were also implicated in cancer metabolism and tissue differentiation, a topic that has been extensively reviewed in [16]. For example, miR-378a induced the differentiation of myoblasts by repressing the myogenic repressor MyoR [20], of bovine preadipocytes [21], and of 3T3-L1 cells by leading to the downregulation of Mapk1 [22]. Interestingly, miR-378a-3p levels were observed to increase throughout brown adipocytes differentiation [23] which is consistent with the previously observed PGC-1β increased mRNA levels during BAT differentiation [24] and during C2C12 myoblast cells’ differentiation [25].
Fig. 1.
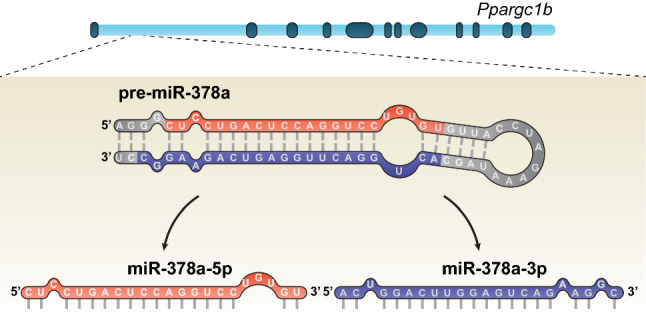
Genomic localization of miR-378a in Ppargc1b. miR-378a is located in the first intron of Ppargc1b and the processing of its pre-miRNA gives origin to two strands, miR-378a-5p and miR-378a-3p
miR-378a role in tumour progression
Unlike normal cells, under aerobic conditions, cancer cells privilege glycolysis as their main energy metabolic pathway over oxidative phosphorylation. This metabolic shift was named Warburg effect and its main purpose is to favour cancer cells’ proliferation and growth by conducting glycolytic intermediates into biosynthetic pathways. However, as in every biological process, cancer cell metabolism can be regulated by several factors, such as miRNAs. In fact, it was reported that miR-378a is involved in cancer metabolism, tumour growth and angiogenesis. For instance, the overexpression of miR-378a-5p in glioblastoma cells reduced the activity of caspase-3 (CASP3), therefore, impairing apoptosis and improving their survival. Moreover, the injection of miR-378a-5p-transfected cells into mice revealed that this miRNA promoted tumour growth and angiogenesis by suppressing the tumour suppressors FUS-1 and suppressor of fused homolog (SUFU) [11]. This study motivated the employment of more efforts to explore the molecular mechanism behind miR-378a’s action in cancer cells. Eichner et al. suggested that miR-378a might be co-expressed with its host gene, PPARGC1B, being both regulated by the oncogene ERBB2 in human breast cancer cells. Furthermore, the overexpression of miR-378a-5p led to the decrease of the mRNA levels of the estrogen-related receptor gamma (ERRγ) and of the transcription factor GA-binding protein α (GABPA), revealing the direct involvement of an intronic miRNA in the regulation of its host gene partners. miR-378a-5p was also found to be a major player in the Warburg effect in breast cancer cells. Its overexpression favoured the glycolytic pathways by downregulating metabolic genes encoding tricarboxylic acid (TCA) cycle’s metabolites targeted by ERRγ while reducing aerobic respiration [10].
miR-378a role in metabolic dysregulation
Metabolism entails several pathways involved in the production and consumption of energy to satisfy the organism’s needs, guaranteeing whole-body energetic homeostasis. However, metabolism is far more complex, and its roles surpass energy levels’ regulation. Metabolic pathways are intertwined with important signalling pathways, as the ones related to cell growth and proliferation. That is the case of phosphoinositide 3-kinase (PI3K)/AKT/mTOR and adenosine monophosphate-activated protein kinase (AMPK)-signalling pathways, two master players in metabolism [26, 27]. Indeed, PI3K/AKT/mTOR pathway stimulates anabolic metabolism by increasing glycolytic enzymes’ expression and by promoting glucose uptake [27]. In addition, metabolites are able to regulate elements of those signalling pathways through feedback mechanisms [27]. AMPK is a key enzyme in the regulation of metabolism, being involved in processes such as cell growth, autophagy, and mitochondrial homeostasis (reviewed in [28]). This protein is activated accordingly with the metabolic state of the organism to restore energy homeostasis. During stressful conditions, AMPK favours energy production and suppresses energy consumption by inducing catabolic pathways, such as glucose uptake and glycolysis, and suppressing anabolic pathways, such as gluconeogenesis or de novo lipid synthesis, respectively [26]. Herein, perturbations at the metabolism level and at the signal transduction level constitute the core of metabolic diseases, such as obesity, type 2 diabetes mellitus (T2DM), or non-alcoholic fatty liver disease (NAFLD). Currently, efforts are being employed to unravel new methods for the prevention or treatment of these diseases, and new possible therapeutic targets are being discovered. Recently, miR-378a-3p and miR-378a-5p were implicated in the regulation of metabolic disorders being found to interfere with a vast array of cellular processes (Table 1).
Table 1.
Metabolic and autophagy-related targets of miR-378a
| miR-378a strand | Target genes | Model | Function | Mechanism of action | References |
|---|---|---|---|---|---|
| Liver | |||||
| -3p | Crat | HFD, Mouse, KO | Mitochondrial fatty acid metabolism |
↓ Mitochondrial oxidative capacity ↓ Fatty acid oxidation |
[17] |
| -5p | Med13 | HFD, Mouse, KO | Mitochondrial fatty acid metabolism |
↓ Mitochondrial oxidative capacity ↓ Energy homeostasis |
[17] |
| -3p | Prkag2 | Chow Diet, HFD, Mouse; Human | Hepatic lipid metabolism | ↓ AMPK-signalling pathway | [40] |
| -3p | Gli3 | CCl4-treated mice, HSC cells, LSEC cells; Human | Hepatic lipid metabolism | ↓ Hedgehog-signalling pathway | [41] |
| -3p | p110α | Mouse (ob/ob), KO | Glucose and lipid metabolism, hepatic insulin signalling |
↑ Hepatic gluconeogenesis ↓ PI3K/AKT-signalling pathway |
[12] |
| -3p | Nrf1 | HFD, Mouse, Hepa1-6 cells | Mitochondrial metabolism, hepatic lipid metabolism | ↓ Fatty acid oxidation | [38, 39] |
| Skeletal muscle | |||||
| -3p | Akt1 | HFD, Mouse, C2C12 cells | Energy and glucose metabolism |
↑ AKT1/FOXO1/PEPCK pathway ↑ Pyruvate-PEP futile cycle |
[13] |
| -3p | Casp9 | Mouse, KO, C2C12 cells | Apoptosis | ↓ Intrinsic pathway of apoptosis | [14] |
| -3p | Pdk1 | Mouse, KO, C2C12 cells | Autophagy | ↓ AKT-signalling pathway | [14] |
| Adipose tissue | |||||
| -3p | Pde1b | HFD, Mouse, Mouse (ob/ob) | Adipogenesis | ↑ Brown preadipocytes differentiation | [22] |
| -3p | Scd1 | HFD, Mouse, 3T3-L1 cells | Lipogenesis | ↑ Lipolysis | [13] |
| Heart | |||||
| -3p | Ldha | Mouse, H9c2 cells | Cell proliferation, energy metabolism | ↓ Cell proliferation and survival | [32] |
| -3p | IGF1R | Mouse, H9c2 cells | Apoptosis | ↓ AKT-signalling pathway | [63] |
| mitomiR-378a-3p | ATP6 | Mouse, Mouse (db/db), HL-1 cells; Human | Mitochondrial metabolism | ↓ ATP synthase activity | [8, 9] |
ATP6 ATP synthase FO subunit 6, CASP9 caspase-9, CRAT carnitine-O-acetyltransferase, IGF1R insulin-like growth factor 1 receptor, LDHA lactate dehydrogenase A, MED13 mediator complex subunit 13, NRF-1 nuclear respiratory factor-1, P110α phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha, PDK1 phosphoinositide-dependent kinase-1, PDE1B phosphodiesterase 1B, PRKAG2 protein kinase AMP-activated non-catalytic subunit gamma 2, SCD1 stearoyl-CoA desaturase-1
It was discovered that miR-378a-3p and PGC-1β levels were increased in subcutaneous white adipose tissue (sc WAT) in response to pioglitazone [18], a thiazolidinedione that is an agonist of the adipogenesis inducer PPARγ. Moreover, sc WAT cells transfected with miR-378a-3p were revealed to have upregulated adipogenic genes (HSL and FASN). PPARγ is a nuclear receptor that is crucial for the regulation of lipid metabolism and adipose tissue differentiation, being mostly expressed in the adipose tissue [29]. Its activity can be regulated through many coactivators being PGC-1α and PGC-1β amongst them. Remarkably, PPARγ and PGC-1β appear to be involved in a positive feedback loop, where PPARγ can be coactivated by PGC-1β, and simultaneously, it was shown to induce the expression of PGC-1β [30]. Since miR-378a is included in Ppargc1b, it may also be implicated in the regulation of PPARγ. In fact, differentiated 3T3-L1 adipocytes treated with rosiglitazone had both Ppargc1b and miR-378a expression increased, being both identified as possible targets of PPARγ [31]. Hence, it is suggested that PPARγ, PGC-1β, and miR-378a are regulators of lipid metabolism and adipogenesis through a synergistic mechanism.
Furthermore, both strands of miR-378a were found to be upregulated in mice’s liver in response to a high-fat diet (HFD), alongside with its host gene Ppargc1b [17]. This finding hinted that miR-378a was possibly involved in oxidative metabolism and suggested that it might be a possible therapeutic target against metabolic diseases. To clarify this hypothesis, the authors knocked out both miR-378a strands in mice and fed them an HFD, finding that these mice were resilient to HFD-induced obesity. Furthermore, it was found that both strands interact with different factors from a PGC-1β-signalling pathway. miR-378a-3p and miR-378a-5p inhibit carnitine-O-acetyltransferase (CRAT), an enzyme that is involved in fatty acid metabolism, and the mediator complex subunit 13 (MED13), respectively, interfering with the systemic energy homeostasis. Finally, mice lacking both strands of miR-378a had an improvement on mitochondrial function, being characterized by having increased oxygen consumption and oxidative capacity, thus providing new evidences for a probable role of miR-378a in mitochondrial dysfunction.
More recently, transgenic mice overexpressing miR-378a were generated and fed with an HFD [13, 23]. Surprisingly, the phenotype observed in these transgenic mice was similar to HFD-fed mice with miR-378a knocked out [17]. Suggesting that mice globally overexpressing or lacking miR-378a had decreased WAT mass and body weight in comparison with their controls. In fact, a study has reported that both decrease and increase of a serine/threonine protein kinase (SRPK1) leads to the same tumorigenic phenotype through activation of AKT [32]. Thus, it might be possible that a similar mechanism is also present on the regulation of metabolism by miR-378a. Nevertheless, in HFD-fed mice, the KO of miR-378a-3p/miR-378a-5p was reported to improve hepatic mitochondrial oxidative capacity [17], whereas overexpression of both miR-378a strands was found to promote BAT expansion [23] and to improve systemic energy homeostasis [13]. In fact, the overexpression of miR-378a-3p in the adipose tissue of HFD-fed mice improved their obese phenotype by directly targeting and inhibiting Pde1b in BAT [23]. This gene encodes the protein phosphodiesterase 1B (PDE1B) that is responsible for the degradation of the signalling molecules cAMP and cGMP, both crucial in adipogenesis [33]. PDE1B inhibition resulted in increased levels of cAMP that further promoted the differentiation of brown preadipocytes. Thus, the observed BAT expansion was responsible for the prevention of HFD-induced obesity in mice.
Furthermore, Zhang and others reported that the overexpression of miR-378a-3p in mice resulted in the activation of a skeletal muscle futile cycle while at the same time improved lipolysis in adipose tissue [13]. It is well established that skeletal muscle has a key role in the maintenance of the organism’s glucose and energy homeostasis [34], not only because it is the most abundant tissue in the organism able to uptake most of the glucose in an insulin-dependent mechanism, but also because it is involved in a balanced crosstalk between liver and adipose tissues [34]. In cases of insulin resistance usually observed in metabolic diseases, insulin-stimulated glucose uptake in skeletal muscle is very reduced leading to hyperglycaemia and impaired glucose homeostasis [35]. Further experiments identified Akt1 as the main target of miR-378a-3p being found to activate pyruvate–phosphoenolpyruvate futile cycle through the AKT1/FOXO1/PEPCK pathway (Fig. 2a) [13]. The activation of this futile cycle in skeletal muscle prevents the entry of pyruvate in TCA cycle that is instead converted back to phosphoenolpyruvate in two reactions catalysed by pyruvate kinase and phosphoenolpyruvate carboxykinase (PEPCK) and involving the cost of ATP molecules. Pyruvate could be directly reduced to lactate instead of being used to fuel the futile cycle; however, the authors showed that miR-378a-3p overexpression did not affect lactate levels or lactate dehydrogenase A (LDHA) transcript levels [13]. Consistent with these results, it was reported in another study that miR-378a-3p targeted and was able to inhibit Ldha in H9c2 cardiac cells [36]. Clearly, miR-378a-3p causes a disruption in glucose metabolism leading to an energy-deficient state in skeletal muscle that was proved to be independent of insulin signalling [13]. However, it was shown that this is counterbalanced in adipose tissue by targeting Scd1 that encodes stearoyl-CoA desaturase-1 (SCD1). This enzyme is known to catalyse the conversion of saturated fatty acids, such as stearate and palmitate, into monounsaturated fatty acids, such as oleate and palmitoleate. In adipocytes transfected with miR-378a-3p, Scd1 expression levels were shown to be reduced alongside with increased levels of the lipolysis-related genes Hsl and Atgl. In addition, HFD-treated mice overexpressing miR-378a-3p were shown to have reduced expression of Scd1 in BAT. Thus, by targeting both Akt1 and Scd1 in skeletal muscle and adipose tissue, respectively, miR-378a-3p was able to positively regulate whole-body energy homeostasis and prevent obesity in mice. Furthermore, this study provided novel insights for the action of a miRNA in the regulation of metabolism by activating a futile cycle that proved to be essential in the maintenance of an energy balance mediated by inter-organ crosstalk.
Fig. 2.

Regulation of metabolic and mitochondrial pathways by miR-378a-3p. a miR-378a-3p has a crucial role in the regulation of oxidative and mitochondrial metabolism, being found to inhibit important elements of the insulin pathway, PI3K and AKT1. Furthermore, the inhibition of AKT1 stimulates the pyruvate-PEP futile cycle, in skeletal muscle. Moreover, miR-378a-3p supressed key regulators of oxidative metabolism such as NRF-1 and CRAT [12, 13, 17, 44, 45]. b Model of the miR-378a-3p transport into mitochondria where it interferes with the transcription of mitochondrial genes, such as ATP6 [8, 9]. AGO2 argonaute-2, ALCAR acetyl-carnitine, ATP6 ATP synthase FO subunit 6, CRAT carnitine-O-acetyltransferase, FAO fatty acid oxidation, FOXO1 forkhead box O1, IMS intermembrane space, NRF-1 nuclear respiratory factor-1, OAA oxaloacetate, PEP phosphoenolpyruvate, PEPCK phosphoenolpyruvate carboxykinase, PI3K phosphoinositide 3-kinase, PNPase polynucleotide phosphorylase
In addition, miR-378a-3p/miR-378a-5p overexpression in obese mice improved hepatic steatosis without aggravating hyperglycaemia. Moreover, these miRNAs proved to be regulators of glucose and lipid homeostasis by targeting the p110α gene that encodes the hepatic PI3K p110α subunit [12]. In this study, fasted mice exhibited higher levels of miR-378a-3p and miR-378-5p in the liver than fed and refed mice, following the same expression pattern of PGC-1β and gluconeogenic genes. In fact, the overexpression of the miR-378a duplex in mice upregulated several hepatic gluconeogenic genes, such as Pck1, G6pd, and Ppargc1a, acting as an inducer of liver gluconeogenesis. Furthermore, miR-378a duplex overexpression resulted in the reduction of phosphorylated AKT and phosphorylated forkhead box O1 (FOXO1) levels, both implicated in insulin signalling. When phosphorylated by AKT1, FOXO1 is excluded from the cell nucleus. Thus, a decrease of FOXO1 phosphorylated form is associated with an increase of gluconeogenic genes expression [37]. Following this study, miR-378a-3p was again identified as an inhibitor of PI3K in diabetic mice [38]. In this study, miR-378a-3p was downregulated in preosteoblast cells incubated with high glucose for 5 days. Nevertheless, miR-378a-3p transfection in diabetic mice restored osteogenic differentiation that was shown to be suppressed by hyperglycaemia. This miRNA repressed Casp3 and activated the PI3K/AKT-signalling pathway. Previously, Akt1 was also identified as a target of miR-378a-3p, thus reinforcing the action of miR-378a-3p in the PI3K/AKT-signalling pathway [39].
Finally, miR-378a-3p was previously found to induce adipose tissue lipolysis in human cancer cachexia [40]. In this study, cancer cachexia-derived adipose tissue was reported to have increased expression levels of miR-378a-3p as well as increased catecholamine-stimulated lipolysis. Furthermore, miR-378a-3p inhibition on adipocytes led to the downregulation of lipolysis regulator genes, such as LIPE, PLIN1, and PNPLA2 and to the reduction of their correspondent encoded protein levels, hormone-sensitive lipase (HSL), perilipin, and adipose triglyceride lipase (ATGL), respectively. However, opposed to the aforementioned studies, Gerin et al. [41] reported that in adipocytes, miR-378a-3p/miR-378a-5p promoted adipogenesis and lipogenesis. Their overexpression led to the upregulation of lipogenic genes encoding fatty acid synthase (FAS) and SCD1. However, the authors failed to demonstrate which miR-378a strand was responsible for this effect. Nevertheless, miR-378a-3p/miR-378a-5p overexpression led to the activation of the glucose transporter type 4 (GLUT4) promoter mediated by CCAAT/enhancer-binding α (C/EBPα) and C/EBPβ. Furthermore, a more recent study identified C/EBP to be part of an inflammatory pathway that promotes the expression of miR-378a. Actually, in adipose tissue, adipokines and cytokines were able to increase miR-378a levels through the sterol regulatory element binding factor (SREBP) and C/EBP that were found to have binding sites in the promoter region of this miRNA [42].
An emerging role of miR-378a in mitochondrial dysfunction
Some metabolic diseases are characterized by mitochondrial dysfunction. Mitochondria are important organelles involved in a series of processes, including regulation of cellular metabolism, apoptosis, or the maintenance of cellular redox state [43]. In fact, disturbances in mitochondrial quality can have serious effects in the whole organism. As mentioned above, miR-378a appears to be involved in the regulation of oxidative metabolism in mitochondria, having been shown that its inhibition improved oxidative capacity and increased oxygen consumption [17]. Furthermore, it was reported that miR-378a-3p targeted Nrf1 [44]. Nuclear respiratory factor-1 (NRF-1) is an important mitochondrial factor that interacts with mitochondrial transcription factor (TFAM) regulating the expression of mitochondrial genes encoding for respiratory chain’s proteins and is also a regulator of fatty acid oxidation. Jeon et al. found miR-378a-3p to be upregulated in the liver of HFD-fed mice where it inhibited NRF-1, leading to the exacerbation of hepatosteatosis. The same was observed in HEPA1-6 cells, where the overexpression of miR-378a-3p repressed Nrf1 promoting lipid accumulation and impairing fatty acid oxidation, resulting in the exacerbation of hepatosteatosis (Fig. 2a) [45]. In addition, NRF-1 and miR-378a-3p appear to be involved in a feedforward loop, where NRF-1 transcriptionally represses the miRNA.
miR-378a-3p was also reported to be an inhibitor of the subunit AMPKγ2 by targeting its encoding gene Prkag2 in HFD-treated mice liver [46]. In fact, miR-378a-3p transfection in HFD-treated mice resulted in sirtuin 1 (SIRT1) decreased activity due to AMPKγ2 inhibition, which ultimately led to the stimulation of the NF-kB/TNFα inflammatory pathway. The elevated miR-378a-3p levels observed in NAFLD were shown to contribute for the development of hepatic inflammation and fibrosis that led to the exacerbation of the disease. However, in another study, the targeted delivery of a miR-378a-3p mimic in liver fibrosis-induced mice ameliorated the damage caused on the liver [47]. It was identified that miR-378a-3p targeted and inhibited Gli3, that is responsible for the activation of the Hedgehog-signalling pathway, known to induce liver fibrosis. Furthermore, miR-378a-3p was found to be repressed by NF-kB p65 subunit that is activated by Smoothened, a component of the Hedgehog-signalling pathway [47]. Hence, it seems that miR-378a-3p has a dual-role in fibrosis, where it favours liver fibrosis in obese mice, whereas when administered in animals with hepatic fibrosis has a therapeutic effect [46, 47].
mitomiR-378a-3p, a regulator of mitochondrial genome
Although mature miRNAs are highly concentrated in cytoplasm their presence in cell organelles, such as nucleus or mitochondria, should not be ignored. In fact, it was reported that functional miRNAs are located inside mitochondria being designated mitochondria-located miRNAs (mitomiRs) and were shown to have an active role in the regulation of mitochondrial gene expression [5, 9, 48]. Indeed, mitochondria can be internally regulated by mitomiRs that are able to interfere with mRNA transcribed from mitochondrial DNA. However, little is known regarding the molecular mechanisms underlying the translocation of nuclear-encoded miRNAs into the mitochondrial matrix. Currently, there are many hypothesis attempting to explain those mechanisms which are in serious need to be clarified and confirmed. Those hypothesis have been extensively reviewed elsewhere [49, 50] and, therefore, will only be mentioned here briefly. Some authors report that argonaute-2 (AGO2), a component of the miRNA biogenesis’ RNA-induced silencing complex (RISC), is also a key player of mitomiR function and transport into mitochondria being imported alongside with miRNAs in the form of complexes [8, 9, 50–53]. In addition, polynucleotide phosphorylase (PNPase), that is located in the mitochondrial intermembrane space and is best known for the degradation of RNA molecules, has emerged as a potential player of mitochondrial RNA [54] and miRNA [8] import mechanisms. Furthermore, some studies also suggest that components of mitochondrial transport machinery may also be accountable for the translocation of miRNAs (reviewed in [6, 51]). For example, the translocases of the outer and inner membranes (TOM and TIM) have been suggested to mediate the transport of miRNA-AGO2 complexes [52]. Nonetheless, other potential miRNA–AGO2 transport mechanisms have also been taken into mind such as, translocation through the voltage-dependent anion channel (VDAC) [51] or its facilitation through interaction with P-bodies and endoplasmic reticulum [55].
miR-378a-3p was identified as a mitomiR (mitomiR-378a-3p) and it was found to interfere with mitochondrial ATP6 that encodes ATP synthase FO subunit 6 [9]. In this study, obese mice were reported to have higher expression levels of mitomiR-378a-3p in heart interfibrillar mitochondria, and interestingly, the analysis of whole heart’s miR-378a-3p expression did not show significant differences between non-diabetic and diabetic mice, meaning that most of miR-378a-3p induced by obesity was translocated into mitochondria. Furthermore, the overexpression of miR-378a-3p decreased ATP6 protein levels and activity in mitochondria that were further restored upon treatment with a miR-378a-3p inhibitor. Further studies began to establish miR-378a-3p translocation mechanism into mitochondria. It was reported that PNPase, in association with AGO2, is the importer of miR-378a [8] (Fig. 2b). In this study, the authors observed that in diabetic human and mouse models, miR-378a and PNPase levels are increased in mitochondria while compared with a non-diabetic condition. In addition, the ATP synthase activity is decreased due to miR-378a. Moreover, the overexpression of PNPase increased miR-378a levels and decreased ATP6 protein and mRNA levels, whereas PNPase knockdown restored them.
Complex I inhibitors regulate miR-378a expression
Mitochondrial complex I is one of the main complexes that composes the electron transport chain making it a suitable target to interfere with oxidative phosphorylation. Metformin is a common biguanide drug used for the treatment of T2DM being reported to inhibit hepatic gluconeogenesis followed by the decrease of glucose production in liver [56, 57]. Metformin exerts its action by specifically inhibiting respiratory chain’s complex I which causes a rise in the cellular AMP/ATP ratio, thus triggering the activation of AMPK and its related signalling pathways. The isoquinoline alkaloid berberine was also reported to improve T2DM condition either through the inhibition of complex I and activation of AMPK [58] or through the activation of SIRT1 and SIRT3 in skeletal muscle and liver, respectively [59, 60]. Nevertheless, these two anti-diabetic compounds were shown to be involved in the regulation of miRNAs. miRNA profiling using locked nucleic acid (LNA) technology revealed that miR-378a expression appears to be induced upon berberine treatment in MIHA and HepG2 cells [61]. Moreover, it was reported that metformin was able to increase the expression levels of miR-378a-3p [62]. In this study, miR-378a-3p was shown to be an important player in the suppression of cell proliferation in hepatocellular carcinoma by targeting cyclin-dependent kinase 1 (CDK1) and downregulating this important cell division factor, further leading to the inhibition of tumour growth. This is a remarkable finding, since miR-378a-3p stimulates hepatic gluconeogenic genes expression and metformin is known to inhibit hepatic gluconeogenesis. Therefore, this issue requires to be further elucidated to understand how miR-378a-3p can be upregulated by metformin and to what extent it participates in the mechanism of action of metformin.
miR-378a as an endocrine regulator
In an organism, miRNAs are not strictly confined within its cells, but instead, they can be secreted and released into the bloodstream. Indeed, some extracellular miRNAs were reported to be present in blood plasma and serum, and were even found to be differently expressed in healthy and diseased individuals [63–65]. These findings not only suggest the role of miRNAs as endocrine molecules able to regulate the gene expression of distant target cells, but also point towards their potential use as disease biomarkers.
Once outside the cells, it was expected that such as other RNA molecules, miRNAs would be degraded by ribonucleases (RNases); however, there are mechanisms that prevent their degradation. Some of these protector mechanisms consist in the enclosure of miRNAs in extracellular vesicles originated from the plasma membranes, such as exosomes and microvesicles [66]. In addition, there were also detected circulating miRNAs associated with AGO2 [64] and with high-density lipoprotein (HDL) particles [67]. Furthermore, they are internalized by their target cells and actively participate in the genome regulation of these cells [1, 68]. Their internalization can be mediated by interaction with membrane receptors [69]. However, the endocrine function of extracellular miRNAs is still a matter of debate [70], since for the development of a physiologically relevant response, a certain threshold is needed to be achieved and this is utterly dependent of the concentration of the miRNA in circulation. Nevertheless, several studies have provided evidences supporting the biological function of extracellular miRNAs in cancer [71], cardiovascular diseases [63], or metabolic diseases [1].
Recent studies reported that miR-378a is found amongst several other miRNAs in biological fluids. For instance, Assmann and others reported that miR-378a-5p is upregulated in the peripheral blood of severe diabetic kidney disease (DKD) patients, while compared with type 1 diabetes mellitus and moderate DKD patients [72]. Likewise, miR-378a-3p and miR-378a-5p were reported to be upregulated in the plasma of obese and insulin-resistance patients [73, 74]. In addition, it was shown that miR-378a expression was increased in the plasma of obese mice fed with a high-fat high-sugar diet [74]. In parallel, this miRNA was upregulated in their pericardial adipose tissue and downregulated in their visceral adipose tissue [74]. As it was mentioned here before, other studies have also reported the differential expression of miR-378a in different tissues [14, 17]. Thus, in the future, it is essential to determine if these modifications are tissue specific or if they are the result of miRNA uptake from circulation and release from tissues. At the present, the existing studies are very focused in the evaluation of a large set of extracellular miRNAs in a certain disease, but do not explore the physiological function of extracellular miR-378a in detail. Thus, some important questions for the understanding of the endocrine function of miR-378a still need to be further accessed. For example, how miR-378a manage to be in organisms’ circulation? Is it by association with AGO2, or due to being incorporated into extracellular vesicles? In fact, recently, miR-378a-3p was found to be inside exosomes secreted by H9c2 cells under hypoxia [75]. In addition, how is extracellular miR-378a released and incorporated into cells?
Regulation of programmed cell death by miR-378a
Cells can perish through different mechanisms: apoptosis, autophagy, or necrosis. Cells that suffer apoptosis are characterized by shrinkage, chromatin condensation, nuclear fragmentation, and plasma membrane blebbing. Apoptosis is dependent of caspase activation and can be regulated by an extrinsic pathway, activated due to extracellular signals, and an intrinsic pathway, that is initiated due to intracellular stress. On the other hand, autophagy involves three different sets of pathways (macroautophagy, microautophagy and chaperone-mediated autophagy) that direct organelles and other cytoplasmic components to be degraded in lysosomes and subsequently recycled. In macroautophagy, the cellular components are incorporated on autophagosomes delivering them to lysosomes. Cellular and tissue homeostasis is guaranteed with the coordination between the two cell death programs and their regulation. For instance, dysregulated autophagy favours the development of pathogenesis such as metabolic disorders [76, 77]. Therefore, targeting those two mechanisms is an appealing strategy for the improvement of pathogenesis complications. In the last years, miRNAs have been associated with autophagy being found to interfere with components from different phases of the macroautophagy pathway, such as in macroautophagy initiation where unc-51 like autophagy activating kinase (ULK1/2) was affected by miR-20a, miR-106b, and miR-26b [78, 79]; in macroautophagy nucleation by interfering with beclin 1 (miR-30a, miR-376a, and miR-376b) [80–82]; in macroautophagy elongation by regulating ATG7 (miR-375 and miR-17) [83, 84] and LC3-II (miR-204) [85]; and in lysosome fusion, where RAB proteins were inhibited by miR-502 and miR-373 [86, 87].
Apoptosis and autophagy can also be regulated by miR-378a. Some studies with the purpose of studying the effect of miR-378a in tumour growth and survival found that this miRNA inhibits CASP3 [11, 38]. CASP3 is the ultimate pro-apoptotic factor implicated in both extrinsic and intrinsic pathways of apoptosis. In addition, it was reported that the transfection of hypoxic H9c2 cells with miR-378a-3p inhibited apoptosis and improved cell survival by decreasing CASP3 protein levels, whereas miR-378a-3p inhibition exacerbated apoptosis [88]. Interestingly, unlike the above-mentioned studies, miR-378a-3p was found to induce apoptosis in postnatal mice cardiomyocytes [89]. Insulin-like growth factor 1 receptor (IGF1R) was identified to be a direct target of miR-378a-3p and through it the AKT signalling is inhibited, consequently leading to the upregulation of pro-apoptotic proteins and the promotion of cell death.
Moreover, a role for miR-378a in macroautophagy was recently identified for the first time. It was shown that miR-378a-3p was important for the maintenance of cell death programs such as apoptosis and autophagy in skeletal muscle [14]. The authors reported that miR-378a was upregulated upon fasting or starvation stresses and was inhibited by inflammation. Further experiments revealed that miR-378a-3p repressed apoptosis through CASP9 inhibition, and it was able to enhance macroautophagy by targeting and inhibiting phosphoinositide-dependent kinase-1 (PDK1) that normally activates AKT. At that point, macroautophagy could be regulated by three distinct factors that interact with AKT: FOXO1, FOXO3, and mammalian target of rapamycin complex 1 (mTORC1). On one hand, miR-378a-3p led to the activation of FOXO1 and FOXO3, positively regulating macroautophagy. On the other hand, mTORC1 was consequently inhibited by the miRNA leading to the activation of the autophagy inducer ULK1 complex. In addition, mice lacking miR-378a-3p/miR-378a-5p were found to have accumulation of abnormal swollen mitochondria in gastrocnemius muscle. It would have been interesting to evaluate mitochondria functionality to further confirm that oxidative mitochondria metabolism was negatively affected by the absence of miR-378a. Regardless, this work established a dual role for miR-378a in the regulation of two important mechanisms that guarantee full organism homeostasis.
Conclusions
Present studies show miR-378a as a mediator of a wide range of biological processes involved in cancer and angiogenesis or hepatosteatosis and T2DM. miR-378a is differentially expressed in metabolic diseases such as during the progression of hepatosteatosis, thus being a potential biomarker. In fact, metabolic diseases’ prevalence is still increasing, and new efficient strategies are urged to control and treat them. The current reports on miR-378a point towards a possible new therapeutic target in the shape of this microRNA, and studies are already underway to assess this hypothesis. A bioengineered synthetic RNA was able to arrest miR-378a function by leading to its misprocessing [90], a strategy that could be useful against tumour growth and angiogenesis. Still, the challenge remains to develop an effective targeting system for the delivery of miRNAs mimics or inhibitors that is able to protect them from degradation. This could be surpassed with the incorporation of miRNAs in nanoparticles. In the last years, this approach is growing with the incorporation of miRNAs in PEI-PLGA nanoparticles [91] or in PLGA nanoparticles [92] that were confirmed to have a functional role in vitro. In addition, in vivo studies positively verified that LTU2a nanoparticles encapsulated with miR-378a-3p mimics were able to efficiently deliver them to the liver leading to the amelioration of hepatic fibrosis [47].
Further studies are needed to clarify the mechanisms of action of miR-378a in mitochondrial dysfunction, since miR-378a-3p was reported to be involved in the regulation of mitochondrial function [17] and in the regulation of cell death programs in skeletal muscle, such as apoptosis and autophagy, contributing for the removal of abnormal mitochondria [14]. Thus, a more in-depth study of miR-378a action in mitochondria metabolism and in the selective removal of mitochondria through autophagy programmes will certainly provide more insights into this subject. Finally, besides the knowledge acquired, the clarification of miR-378a roles in metabolism can lead to the development of attractive therapeutic strategies against metabolic diseases.
Author contributions
IFM wrote the manuscript; JST, CMP, and APR supervised and revised the work. All authors have read and approved the final version of this review.
Funding
This work was financed by the European Regional Development Fund (ERDF), through the Centro 2020 Regional Operational Programme: project CENTRO-01-0145-FEDER-000012-HealthyAging2020, the Portugal 2020-Operational Programme for Competitiveness and Internationalisation, and the Portuguese national funds via FCT–Fundação para a Ciência e a Tecnologia, I.P.: project POCI-01-0145-FEDER-016770, as well as by UID/NEU/04539/2013 (CNC.IBILI Consortium strategic project). JST is a research fellow under the CEEC2017 programme (CEECIND/04400/2017).
Compliance with ethical standards
Conflict of interest
The authors declare that this review has no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Rottiers V, Näär AM. MicroRNAs in metabolism and metabolic disorders. Nat Rev Mol Cell Biol. 2012;13:239–251. doi: 10.1038/nrm3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peng Y, Croce CM. The role of microRNAs in human cancer. Signal Transduct Target Ther. 2016;1:15004. doi: 10.1038/sigtrans.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Su Z, Yang Z, Xu Y, et al. MicroRNAs in apoptosis, autophagy and necroptosis. Oncotarget. 2015;6:8474–8490. doi: 10.18632/oncotarget.3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ivey KN, Srivastava D. MicroRNAs as regulators of differentiation and cell fate decisions. Cell Stem Cell. 2010;7:36–41. doi: 10.1016/j.stem.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 5.Kren BT, Wong PY-P, Sarver A, et al. MicroRNAs identified in highly purified liver-derived mitochondria may play a role in apoptosis. RNA Biol. 2009;6:65–72. doi: 10.4161/rna.6.1.7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sripada L, Tomar D, Prajapati P, et al. Systematic analysis of small RNAs associated with human mitochondria by deep sequencing: detailed analysis of mitochondrial associated miRNA. PLoS One. 2012;7:e44873. doi: 10.1371/journal.pone.0044873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barrey E, Saint-Auret G, Bonnamy B, et al. Pre-microRNA and mature microRNA in human mitochondria. PLoS One. 2011;6:e20220. doi: 10.1371/journal.pone.0020220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shepherd DL, Hathaway QA, Pinti MV, et al. Exploring the mitochondrial microRNA import pathway through polynucleotide phosphorylase (PNPase) J Mol Cell Cardiol. 2017;110:15–25. doi: 10.1016/j.yjmcc.2017.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jagannathan R, Thapa D, Nichols CE, et al. Translational regulation of the mitochondrial genome following redistribution of mitochondrial microRNA in the diabetic heart. Circ Cardiovasc Genet. 2015;8:785–802. doi: 10.1161/CIRCGENETICS.115.001067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eichner LJ, Perry M-C, Dufour CR, et al. miR-378* mediates metabolic shift in breast cancer cells via the PGC-1β/ERRγ transcriptional pathway. Cell Metab. 2010;12:352–361. doi: 10.1016/j.cmet.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Lee DY, Deng Z, Wang C, Yang BB. MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. Proc Natl Acad Sci USA. 2007;104:20350–20355. doi: 10.1073/pnas.0706901104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu W, Cao H, Ye C, et al. Hepatic miR-378 targets p110α and controls glucose and lipid homeostasis by modulating hepatic insulin signalling. Nat Commun. 2014;5:5684. doi: 10.1038/ncomms6684. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Li C, Li H, et al. MiR-378 activates the pyruvate-PEP futile cycle and enhances lipolysis to ameliorate obesity in mice. EBioMedicine. 2016;5:93–104. doi: 10.1016/j.ebiom.2016.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Jiang J, Liu W, et al. microRNA-378 promotes autophagy and inhibits apoptosis in skeletal muscle. Proc Natl Acad Sci USA. 2018;115:E10849–E10858. doi: 10.1073/pnas.1803377115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kasashima K, Nakamura Y, Kozu T. Altered expression profiles of microRNAs during TPA-induced differentiation of HL-60 cells. Biochem Biophys Res Commun. 2004;322:403–410. doi: 10.1016/j.bbrc.2004.07.130. [DOI] [PubMed] [Google Scholar]
- 16.Krist B, Florczyk U, Pietraszek-Gremplewicz K, et al. The role of miR-378a in metabolism, angiogenesis, and muscle biology. Int J Endocrinol. 2015;2015:281756. doi: 10.1155/2015/281756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carrer M, Liu N, Grueter CE, et al. Control of mitochondrial metabolism and systemic energy homeostasis by microRNAs 378 and 378*. Proc Natl Acad Sci USA. 2012;109:15330–15335. doi: 10.1073/pnas.1207605109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu J, Kong X, Liu J, et al. Expression profiling of PPARγ-regulated microRNAs in human subcutaneous and visceral adipogenesis in both genders. Endocrinology. 2014;155:2155–2165. doi: 10.1210/en.2013-2105. [DOI] [PubMed] [Google Scholar]
- 19.Zhang T, Duan J, Zhang L, et al. LXRα promotes hepatosteatosis in part through activation of microRNA-378 transcription and inhibition of Ppargc1β expression. Hepatology. 2019 doi: 10.1002/hep.30301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gagan J, Dey BK, Layer R, et al. MicroRNA-378 targets the myogenic repressor MyoR during myoblast differentiation. J Biol Chem. 2011;286:19431–19438. doi: 10.1074/jbc.M111.219006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu S-Y, Zhang Y-Y, Gao Y, et al. MiR-378 plays an important role in the differentiation of bovine preadipocytes. Cell Physiol Biochem. 2015;36:1552–1562. doi: 10.1159/000430318. [DOI] [PubMed] [Google Scholar]
- 22.Huang N, Wang J, Xie W, et al. MiR-378a-3p enhances adipogenesis by targeting mitogen-activated protein kinase 1. Biochem Biophys Res Commun. 2015;457:37–42. doi: 10.1016/j.bbrc.2014.12.055. [DOI] [PubMed] [Google Scholar]
- 23.Pan D, Mao C, Quattrochi B, et al. MicroRNA-378 controls classical brown fat expansion to counteract obesity. Nat Commun. 2014;5:4725. doi: 10.1038/ncomms5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin J, Puigserver P, Donovan J, et al. Peroxisome proliferator-activated receptor γ coactivator 1β (PGC-1β), a novel PGC-1-related transcription coactivator associated with host cell factor. J Biol Chem. 2002;277:1645–1648. doi: 10.1074/jbc.C100631200. [DOI] [PubMed] [Google Scholar]
- 25.Shao D, Liu Y, Liu X, et al. PGC-1β-regulated mitochondrial biogenesis and function in myotubes is mediated by NRF-1 and ERRα. Mitochondrion. 2010;10:516–527. doi: 10.1016/j.mito.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 26.Garcia D, Shaw RJ. AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell. 2017;66:789–800. doi: 10.1016/j.molcel.2017.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ward PS, Thompson CB. Signaling in control of cell growth and metabolism. Cold Spring Harb Perspect Biol. 2012;4:a006783. doi: 10.1101/cshperspect.a006783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARγ. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 30.Wei W, Wang X, Yang M, et al. PGC1β mediates PPARγ activation of osteoclastogenesis and rosiglitazone-induced bone loss. Cell Metab. 2010;11:503–516. doi: 10.1016/j.cmet.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.John E, Wienecke-Baldacchino A, Liivrand M, et al. Dataset integration identifies transcriptional regulation of microRNA genes by PPARc in differentiating mouse 3T3-L1 adipocytes. Nucleic Acids Res. 2012;40:4446–4460. doi: 10.1093/nar/gks025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang P, Zhou Z, Hu A, et al. Both decreased and increased SRPK1 levels promote cancer by interfering with PHLPP-mediated dephosphorylation of Akt. Mol Cell. 2014;54:378–391. doi: 10.1016/j.molcel.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hemmrich K, Gummersbach C, Paul NE, et al. Nitric oxide and downstream second messenger cGMP and cAMP enhance adipogenesis in primary human preadipocytes. Cytotherapy. 2010;12:547–553. doi: 10.3109/14653241003695042. [DOI] [PubMed] [Google Scholar]
- 34.DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. 2009;32(Suppl 2):S157–S163. doi: 10.2337/dc09-S302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pendergrass M, Bertoldo A, Bonadonna R, et al. Muscle glucose transport and phosphorylation in type 2 diabetic, obese nondiabetic, and genetically predisposed individuals. Am J Physiol Endocrinol Metab. 2007;292:E92–E100. doi: 10.1152/ajpendo.00617.2005. [DOI] [PubMed] [Google Scholar]
- 36.Mallat Y, Tritsch E, Ladouce R, et al. Proteome Modulation in H9c2 Cardiac Cells by microRNAs miR-378 and miR-378*. Mol Cell Proteom. 2014;13:18–29. doi: 10.1074/mcp.M113.030569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Puigserver P, Rhee J, Donovan J, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1–PGC-1α interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 38.You L, Gu W, Chen L, et al. MiR-378 overexpression attenuates high glucose-suppressed osteogenic differentiation through targeting CASP3 and activating PI3K/Akt signaling pathway. Int J Clin Exp Pathol. 2014;7:7249–7261. [PMC free article] [PubMed] [Google Scholar]
- 39.Rückerl D, Jenkins SJ, Laqtom NN, et al. Induction of IL-4Rα-dependent microRNAs identifies PI3K/Akt signaling as essential for IL-4-driven murine macrophage proliferation in vivo. Blood. 2012;120:2307–2316. doi: 10.1182/blood-2012-02-408252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kulyté A, Lorente-Cebrián S, Gao H, et al. MicroRNA profiling links miR-378 to enhanced adipocyte lipolysis in human cancer cachexia. Am J Physiol Endocrinol Metab. 2014;306:E267–E274. doi: 10.1152/ajpendo.00249.2013. [DOI] [PubMed] [Google Scholar]
- 41.Gerin I, Bommer GT, McCoin CS, et al. Roles for miRNA-378/378* in adipocyte gene expression and lipogenesis. Am J Physiol Endocrinol Metab. 2010;299:E198–E206. doi: 10.1152/ajpendo.00179.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang X, Xue M, Fu Z, et al. Insight into the effects of adipose tissue inflammation factors on miR-378 expression and the underlying mechanism. Cell Physiol Biochem. 2014;33:1778–1788. doi: 10.1159/000362957. [DOI] [PubMed] [Google Scholar]
- 43.Schatz G. Mitochondria: beyond oxidative phosphorylation. Biochim Biophys Acta Mol Basis Dis. 1995;1271:123–126. doi: 10.1016/0925-4439(95)00018-Y. [DOI] [PubMed] [Google Scholar]
- 44.Il Jeon T, Park JW, Ahn J, et al. Fisetin protects against hepatosteatosis in mice by inhibiting miR-378. Mol Nutr Food Res. 2013;57:1931–1937. doi: 10.1002/mnfr.201300071. [DOI] [PubMed] [Google Scholar]
- 45.Zhang T, Zhao X, Steer CJ, et al. A negative feedback loop between microRNA-378 and Nrf1 promotes the development of hepatosteatosis in mice treated with a high fat diet. Metab Clin Exp. 2018;85:183–191. doi: 10.1016/j.metabol.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang T, Hu J, Wang X, et al. MicroRNA-378 promotes hepatic inflammation and fibrosis via modulation of the NF-kB-TNFα pathway. J Hepatol. 2019;70:87–96. doi: 10.1016/j.jhep.2018.08.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hyun J, Wang S, Kim J, et al. MicroRNA-378 limits activation of hepatic stellate cells and liver fibrosis by suppressing Gli3 expression. Nat Commun. 2016;7:10993. doi: 10.1038/ncomms10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bian Z, Li L-M, Tang R, et al. Identification of mouse liver mitochondria-associated miRNAs and their potential biological functions. Cell Res. 2010;20:1076–1078. doi: 10.1038/cr.2010.119. [DOI] [PubMed] [Google Scholar]
- 49.Macgregor-Das AM, Das S. A microRNA’s journey to the center of the mitochondria. Am J Physiol Hear Circ Physiol. 2018;315:H206–H215. doi: 10.1152/ajpheart.00714.2017. [DOI] [PubMed] [Google Scholar]
- 50.Weber-Lotfi F, Dietrich A. Intercompartment RNA trafficking in mitochondrial function and communication. In: Cruz-Reyes J, Gray M, editors. RNA metabolism in mitochondria. Nucleic acids and molecular biology. Cham: Springer; 2018. [Google Scholar]
- 51.Bandiera S, Matégot R, Girard M, et al. MitomiRs delineating the intracellular localization of microRNAs at mitochondria. Free Radic Biol Med. 2013;64:12–19. doi: 10.1016/j.freeradbiomed.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 52.Srinivasan H, Das S. Mitochondrial miRNA (MitomiR): a new player in cardiovascular health. Can J Physiol Pharmacol. 2015;93:855–861. doi: 10.1139/cjpp-2014-0500. [DOI] [PubMed] [Google Scholar]
- 53.Zhang X, Zuo X, Yang B, et al. MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell. 2014;158:607–619. doi: 10.1016/j.cell.2014.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang G, Chen H-W, Oktay Y, et al. PNPASE regulates RNA import into mitochondria. Cell. 2010;142:456–467. doi: 10.1016/j.cell.2010.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Makarova JA, Shkurnikov MU, Wicklein D, et al. Intracellular and extracellular microRNA: an update on localization and biological role. Prog Histochem Cytochem. 2016;51:33–49. doi: 10.1016/j.proghi.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 56.Foretz M, Guigas B, Bertrand L, et al. Metformin: from mechanisms of action to therapies. Cell Metab. 2014;20:953–966. doi: 10.1016/j.cmet.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 57.Natali A, Ferrannini E. Effects of metformin and thiazolidinediones on suppression of hepatic glucose production and stimulation of glucose uptake in type 2 diabetes: a systematic review. Diabetologia. 2006;49:434–441. doi: 10.1007/s00125-006-0141-7. [DOI] [PubMed] [Google Scholar]
- 58.Lee YS, Kim WS, Kim KH, et al. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes. 2006;55:2256–2264. doi: 10.2337/db06-0006. [DOI] [PubMed] [Google Scholar]
- 59.Gomes AP, Duarte FV, Nunes P, et al. Berberine protects against high fat diet-induced dysfunction in muscle mitochondria by inducing SIRT1-dependent mitochondrial biogenesis. Biochim Biophys Acta Mol Basis Dis. 2012;1822:185–195. doi: 10.1016/j.bbadis.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Teodoro JS, Duarte FV, Gomes AP, et al. Berberine reverts hepatic mitochondrial dysfunction in high-fat fed rats: a possible role for SirT3 activation. Mitochondrion. 2013;13:637–646. doi: 10.1016/j.mito.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 61.Li CH, Tang SC, Wong CH, et al. Berberine induces miR-373 expression in hepatocytes to inactivate hepatic steatosis associated AKT-S6 kinase pathway. Eur J Pharmacol. 2018;825:107–118. doi: 10.1016/j.ejphar.2018.02.035. [DOI] [PubMed] [Google Scholar]
- 62.Zhou J, Han S, Qian W, et al. Metformin induces miR-378 to downregulate the CDK1, leading to suppression of cell proliferation in hepatocellular carcinoma. Onco Targets Ther. 2018;11:4451–4459. doi: 10.2147/OTT.S167614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gupta SK, Bang C, Thum T. Circulating MicroRNAs as biomarkers and potential paracrine mediators of cardiovascular disease. Circ Cardiovasc Genet. 2010;3:484–488. doi: 10.1161/CIRCGENETICS.110.958363. [DOI] [PubMed] [Google Scholar]
- 64.Arroyo JD, Chevillet JR, Kroh EM, et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci USA. 2011;108:5003–5008. doi: 10.1073/pnas.1019055108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen X, Ba Y, Ma L, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18:997–1006. doi: 10.1038/cr.2008.282. [DOI] [PubMed] [Google Scholar]
- 66.Valadi H, Ekström K, Bossios A, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 67.Vickers KC, Palmisano BT, Shoucri BM, et al. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. 2011;13:423–435. doi: 10.1038/ncb2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y, Liu D, Chen X, et al. Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol Cell. 2010;39:133–144. doi: 10.1016/j.molcel.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 69.Prud’homme GJ, Glinka Y, Lichner Z, Yousef GM. Neuropilin-1 is a receptor for extracellular miRNA and AGO2/miRNA complexes and mediates the internalization of miRNAs that modulate cell function. Oncotarget. 2016;7:68057–68071. doi: 10.18632/ONCOTARGET.10929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Turchinovich A, Weiz L, Burwinkel B. Extracellular miRNAs: the mystery of their origin and function. Trends Biochem Sci. 2012;37:460–465. doi: 10.1016/j.tibs.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 71.Bayraktar R, Van Roosbroeck K, Calin GA. Cell-to-cell communication: microRNAs as hormones. Mol Oncol. 2017;11:1673–1686. doi: 10.1002/1878-0261.12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Assmann TS, Recamonde-Mendoza M, Costa AR, et al. Circulating miRNAs in diabetic kidney disease: case–control study and in silico analyses. Acta Diabetol. 2019;56:55–65. doi: 10.1007/s00592-018-1216-x. [DOI] [PubMed] [Google Scholar]
- 73.Choi H, Koh HWL, Zhou L, et al. Plasma protein and microRNA biomarkers of insulin resistance: a network-based integrative -omics analysis. Front Physiol. 2019;10:379. doi: 10.3389/fphys.2019.00379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jones A, Danielson KM, Benton MC, et al. miRNA signatures of insulin resistance in obesity. Obesity. 2017;25:1734–1744. doi: 10.1002/oby.21950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang J, Ma J, Long K, et al. Overexpression of exosomal cardioprotective miRNAs mitigates hypoxia-induced H9c2 cells apoptosis. Int J Mol Sci. 2017;18:711. doi: 10.3390/ijms18040711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ryter SW, Koo JK, Choi AMK. Molecular regulation of autophagy and its implications for metabolic diseases. Curr Opin Clin Nutr Metab Care. 2014;17:329–337. doi: 10.1097/MCO.0000000000000068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang Y, Sowers JR, Ren J. Targeting autophagy in obesity: from pathophysiology to management. Nat Rev Endocrinol. 2018;14:356–376. doi: 10.1038/s41574-018-0009-1. [DOI] [PubMed] [Google Scholar]
- 78.Wu H, Wang F, Hu S, et al. MiR-20a and miR-106b negatively regulate autophagy induced by leucine deprivation via suppression of ULK1 expression in C2C12 myoblasts. Cell Signal. 2012;24:2179–2186. doi: 10.1016/j.cellsig.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 79.John Clotaire DZ, Zhang B, Wei N, et al. MiR-26b inhibits autophagy by targeting ULK2 in prostate cancer cells. Biochem Biophys Res Commun. 2016;472:194–200. doi: 10.1016/j.bbrc.2016.02.093. [DOI] [PubMed] [Google Scholar]
- 80.Zhu H, Wu H, Liu X, et al. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy. 2009;5:816–823. doi: 10.4161/auto.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Korkmaz G, Ayse Tekirdag K, Gulfem Ozturk D, et al. MIR376A is a regulator of starvation-induced autophagy. PLoS One. 2013;8:e82556. doi: 10.1371/journal.pone.0082556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Korkmaz G, le Sage C, Tekirdag KA, et al. miR-376b controls starvation and mTOR inhibition-related autophagy by targeting ATG4C and BECN1. Autophagy. 2012;8:165–176. doi: 10.4161/auto.8.2.18351. [DOI] [PubMed] [Google Scholar]
- 83.Chang Y, Yan W, He X, et al. miR-375 inhibits autophagy and reduces viability of hepatocellular carcinoma cells under hypoxic conditions. Gastroenterology. 2012;143:177–187. doi: 10.1053/j.gastro.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 84.Comincini S, Allavena G, Palumbo S, et al. microRNA-17 regulates the expression of ATG7 and modulates the autophagy process, improving the sensitivity to temozolomide and low-dose ionizing radiation treatments in human glioblastoma cells. Cancer Biol Ther. 2013;14:574–586. doi: 10.4161/cbt.24597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xiao J, Zhu X, He B, et al. MiR-204 regulates cardiomyocyte autophagy induced by ischemia-reperfusion through LC3-II. J Biomed Sci. 2011;18:35. doi: 10.1186/1423-0127-18-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang R, Wang Z-X, Yang J-S, et al. MicroRNA-451 functions as a tumor suppressor in human non-small cell lung cancer by targeting ras-related protein 14 (RAB14) Oncogene. 2011;30:2644–2658. doi: 10.1038/onc.2010.642. [DOI] [PubMed] [Google Scholar]
- 87.Zhai H, Song B, Xu X, et al. Inhibition of autophagy and tumor growth in colon cancer by miR-502. Oncogene. 2013;32:1570–1579. doi: 10.1038/onc.2012.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fang J, Song X-W, Tian J, et al. Overexpression of microRNA-378 attenuates ischemia-induced apoptosis by inhibiting caspase-3 expression in cardiac myocytes. Apoptosis. 2012;17:410–423. doi: 10.1007/s10495-011-0683-0. [DOI] [PubMed] [Google Scholar]
- 89.Knezevic I, Patel A, Sundaresan NR, et al. A novel cardiomyocyte-enriched microRNA, miR-378, targets insulin-like growth factor 1 receptor: implications in postnatal cardiac remodeling and cell survival. J Biol Chem. 2012;287:12913–12926. doi: 10.1074/jbc.M111.331751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Deng Z, Yang X, Fang L, et al. Misprocessing and functional arrest of microRNAs by miR-Pirate: roles of miR-378 and miR-17. Biochem J. 2013;450:375–386. doi: 10.1042/BJ20120722. [DOI] [PubMed] [Google Scholar]
- 91.Wang S, Zhang J, Wang Y, Chen M. Hyaluronic acid-coated PEI-PLGA nanoparticles mediated co-delivery of doxorubicin and miR-542-3p for triple negative breast cancer therapy. Nanomedicine. 2016;12:411–420. doi: 10.1016/j.nano.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 92.Saraiva C, Talhada D, Rai A, et al. MicroRNA-124-loaded nanoparticles increase survival and neuronal differentiation of neural stem cells in vitro but do not contribute to stroke outcome in vivo. PLoS One. 2018;13:e0193609. doi: 10.1371/journal.pone.0193609. [DOI] [PMC free article] [PubMed] [Google Scholar]