

Abstract
It has been suggested that the persistence of coxsackieviruses-B (CV-B) in pancreatic beta cells plays a role in the pathogenesis of type 1 diabetes (T1D). Yet, immunological effectors, especially natural killer (NK) cells, are supposed to clear virus-infected cells. Therefore, an evaluation of the response of NK cells to pancreatic beta cells persistently infected with CV-B4 was conducted. A persistent CV-B4 infection was established in 1.1B4 pancreatic beta cells. Infectious particles were found in supernatants throughout the culture period. The proportion of cells containing viral protein VP1 was low (< 5%), although a large proportion of cells harbored viral RNA (around 50%), whilst cell viability was preserved. HLA class I cell surface expression was downregulated in persistently infected cultures, but HLA class I mRNA levels were unchanged in comparison with mock-infected cells. The cytolytic activities of IL-2-activated non-adherent peripheral blood mononuclear cells (PBMCs) and of NK cells were higher towards persistently infected cells than towards mock-infected cells, as assessed by an LDH release assay. Impaired cytolytic activity of IL-2-activated non-adherent PBMCs from patients with T1D towards infected beta cells was observed. In conclusion, pancreatic beta cells persistently infected with CV-B4 can be lysed by NK cells, implying that impaired cytolytic activity of these effector cells may play a role in the persistence of CV-B in the host and thus in the viral pathogenesis of T1D.
Keywords: Enterovirus, Persistence, HLA class I, Type 1 diabetes, LDH assay
Introduction
Enteroviruses are small, non-enveloped, positive single-stranded RNA viruses belonging to the Picornaviridae family [1]. The genus Enterovirus encompasses seven human pathogenic species (Human enterovirus A-D and Human rhinoviruses). Enteroviruses, especially coxsackievirus B (CV-B) (Human enterovirus B), are suspected to play a role in the development of chronic diseases such as type 1 diabetes (T1D) [2, 3]. Indeed, the frequent detection of enteroviral components (protein and RNA) in the serum, intestinal mucosa, pancreas and peripheral blood cells of diabetic patients supports the role of persistent infection in the pathogenesis of the disease [4–10]. CV-Bs are able to establish a persistent infection in duct-like cells and pancreatic cells in vitro with structural and functional changes in these cells [11–13]. Persistent non-cytopathic CV-B infection of human pancreatic islets can induce the production of interferon (IFN)-α and hyper expression of HLA class I molecules by endocrine cells, resulting in insulitis and destruction of beta cells by cytotoxic T cells [7, 13, 14]. Virus-infected cells can escape recognition and destruction by cytotoxic T cells by a reduction of cell surface expression of HLA class I antigen, but they can nevertheless be killed by NK cells. The cytolytic activity of NK cells is modulated by activating and inhibiting signals provoked by interactions between target cells and NK cell receptors [15, 16]. NK cells represent a first defense line against viruses based on their cytolytic activity towards infected cells and their interactions with the innate and adaptive immune system through the release of cytokines such as IFN-γ [17, 18]. Pancreatic β cells of recent-onset type 1 diabetic patients were found to be infected with CV-B4, and nondestructive insulitis with a predominant NK cell infiltration was reported [19]. Other studies have reported the cytolytic activity of NK cells towards cells infected with various families of viruses including Picornaviridae [20, 21]. NK cells exhibited cytolytic activity against cells acutely infected with CV-B3 in mice [22, 23], but not in humans [24]. By contrast, the immune response to persistent CV-B infection remains an open issue. An improved understanding of the clearance of pancreatic β cells persistently infected with CV-B4 in patients with T1D could provide new insights into the enteroviral pathogenesis of the disease. The aim of this study was to evaluate the response of NK cells towards pancreatic beta cells persistently infected with CV-B4.
Materials and methods
Cell lines and virus
HEp-2 cells (BioWhittaker, Vervier, Belgium), MRC-5 cells (Human embryonic lung fibroblasts ATCC® CCL-171™, Biomerieux, Marcy l’Etoile, France) and 1.1B4 cells (Human pancreatic beta cell lines ECACC 10012801) were routinely cultured at 37 °C in 5% CO2 in, respectively, Eagle’s minimum essential medium (MEM) and in RPMI-1640 medium (Gibco®, Invitrogen, UK), supplemented with 10% inactivated fetal calf serum (FCS), 1% l-glutamine, 1% nonessential amino-acids (Gibco-BRL), penicillin (100 U/mL) and streptomycin (100 mg/mL) (Invitrogen, Saint Aubin, France). In our experiments 1.1B4 cells did not respond to glucose. Neither insulin nor pro-insulin could be detected in supernatants of cell cultures (mock-infected) and insulin mRNA was not found in cells. The human ductal cell line Panc-1 (ATCC® CRL-1469™) was cultured in Dulbecco’s modified Eagle’s medium DMEM 4.5 g/L glucose (Invitrogen, France) supplemented with 10% FCS, 1% l-glutamine and 1% penicillin and streptomycin [11]. CV-B4 E2, the diabetogenic strain of coxsackievirus B4, (kindly provided by Ji-Won Yoon, Julia McFarlane Diabetes Research Center, Calgary, Alberta, Canada) was propagated in HEp-2 cells as previously described [11]. Aliquots of virus preparations were stored at − 80 °C.
Chronic cell infections and viral progeny in supernatants
A persistent infection in 1.1B4 cells with CV-B4 was obtained as previously described by our team [11, 12]. Briefly, 1.1B4 cells were harvested and seeded at 1.25 × 105 cells per well in 24-well plates and infected with CV-B4 at a multiplicity of infection (MOI) of 0.01. After incubation for 24 h post-infection at 37 °C, in a humidified atmosphere with 5% CO2, cells were washed three times with cold RPMI-1640 medium to find equilibrium between viral replication and cell proliferation during the acute lytic infection. CV-B4 infected and mock-infected cell cultures were then seeded in vented tissue culture flasks (Becton–Dickinson, France). The cells were scraped and sub-cultured once a week and followed-up for more than 70 weeks. Harvested culture supernatants were stored at − 80 °C for viral progeny. The viral titers in supernatants of chronically infected cells were assessed on HEp-2 cell monolayers using the end-point dilution assay and the Reed–Muench statistical method was used to determine the tissue culture 50% infectious dose (TCID50). The results were expressed as TCID50/mL. The cells’ viability was assessed using Uptiblue™ viable cell counting reagent (Uptima, Interchim, Montluçon, France) and the trypan blue exclusion assay (Sigma), as routinely described.
Immunofluorescence
Mock-infected and CV-B4-infected 1.1B4 cells were harvested by scraping and seeded on sterile glass slides (105 cells/slide) and allowed to adhere for overnight at 37 °C and 5% CO2. The presence of intracellular viral capsid protein VP1 in CV-B4-infected 1.1B4 cells was investigated regularly by immunofluorescence as previously described [9, 11] using a primary mouse anti-enterovirus antibody, VP1 clone 5D8/1 (DAKO, Les Ulis, France), and a secondary rabbit anti-mouse antibody conjugated with Alexa Fluor™ 488 (Invitrogen, France).
Mock-infected and persistently infected 1.1B4 cells were double-stained for HLA class I antigen and for VP1 analysis with rabbit anti-human HLA class I ABC (1:50 dilution) (15240-1-AP, Proteintech, USA) and with mouse anti-enterovirus VP1 clone 5D8/1 antibodies (1:100 dilution), respectively. The respective secondary antibodies (1:400 dilutions): Alexa Fluor™ 488-conjugated goat anti-rabbit IgG, and rabbit anti-mouse conjugated with Alexa Fluor™ 647, were then applied sequentially. Cell nuclei were revealed by Hoescht dye solution (Sigma-Aldrich). The slides were then mounted with Permafluor (Coulter Immunotech, Marseille, France), and positive cells were visualized with specific detector filters using a Leica DMI8 videomicroscope.
Sn-RT-PCR applied directly on a few cells
Mock and CV-B4-infected 1.1B4 cells were washed eight times with cold PBS and harvested with trypsin–EDTA 5 min at 37 °C. Viable cells were counted with trypan blue and diluted to obtain an average of 105 cells/mL. Cell suspensions were then serially twofold diluted in PBS on 96-well microtiter plates, to obtain around 6, 5, 3, 2 and 1 cell per tube. Intracellular enteroviral RNA from a few cells was obtained without extraction and was detected by a RT-PCR step followed by a semi-nested (Sn)-PCR carried out using Perkin Elmer Applied Gene Amp PCR System 2400 thermocycler as previously described by our team [11].
Blood collection and effector cells isolation
Buffy coats were obtained from healthy donors at the Regional Blood Bank (Lille, France) after their informed and written consent. Seven patients (mean age 37.0 ± 16.7 years old) previously diagnosed with T1D and ten control subjects comparable for age (mean age 34.9 ± 13.2 years old) and sex, were enrolled after their informed and written consent. The disease duration was 13.2 ± 8.4 years. At the time of sampling, all patients were on insulin therapy and exhibited mean glycosylated hemoglobin (HbA1c) of 9.7 ± 2.4%. T1D patients with neurological or micro- and macro-vascular complications were excluded from this study. Participants with infectious, acute or chronic diseases, pregnancy or on medications were also excluded. Whole blood of subjects was collected by venipuncture into sterile BD Vacutainer® sodium heparin tubes (Becton–Dickinson, CA, USA). Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats and whole blood by density gradient centrifugation using Ficoll® Paque Plus (GE Healthcare, Vélizy-Villacoublay, France), as described previously [25]. Cells were then suspended in RPMI-1640 culture medium (Gibco BRL, Thermo Fischer Scientific, Villebon sur Yvette, France) supplemented with 10% complement-inactivated fetal calf serum (FCS), 1% non-essential amino acids, 50 µg/mL penicillin, 100 U/mL streptomycin and 2 mM l-glutamine. The concentration of viable cells was assessed by trypan blue staining and an average of 107 cells per well was distributed in a Falcon® polystyrene six-well plate (Fischer Scientific, Illkirch-Graffenstaden, France).
The plastic adherence method was used to deplete monocytes from the PBMCs as described previously [26]. Purified CD56+ (NK cells) and CD3+ (T lymphocytes) fractions were isolated from non-adherent PBMCs using EasySep™ human immunomagnetic cell isolation kits (StemCell Technologies, Grenoble, France), according to the manufacturer’s protocol. The purity of each separated cell subset and the proportion of NK cells in PBMCs of T1D patients and control subjects was assessed by flow cytometry using staining with anti-CD56-PE, anti-CD16-APC and anti-CD3-FITC-conjugated antibodies (Becton–Dickinson, Mountain View, CA, USA) (data not shown). In all experiments, the mean proportions of CD3−CD56+ and CD3+CD56− cells in the isolated fractions were 92.6 ± 4.9% and 98.2 ± 1.5%, respectively. To obtain activated cells, non-adherent PBMCs, CD56+ and CD3+ cells were cultured in RPMI-1640 medium for 72 h at 37 °C, 5% CO2 with 1000 U/mL of recombinant human IL-2 (PreproTech, Neuilly-Sur-Seine, France) prior to their use in cytotoxicity assays as effector cells.
Cytolytic assay and IFN-γ production
Cytolytic activity of effector cells against HEp-2, Panc-1, MRC-5 and 1.1B4 cell lines was evaluated with a lactic acid dehydrogenase (LDH) release assay. Mock and CV-B4-infected cells (target cells) were harvested 12 h and 23–58 weeks post-infection and 70 weeks post-infection and seeded at 2 × 104 cells per well into 96-well flat-bottom plates and allowed to adhere to plastic for 12 h at 37 °C and 5% CO2. Prior to the assay, effector and target cells were washed in RPMI-1640 medium without phenol red, supplemented with 1% FCS and 100 µL of this medium was added to target cells. 100 µL of effector cells was then added in triplicate to target cells with various effector: target cell ratios (E:T) (5:1, 10:1, 20:1 and 40:1) and plates were centrifuged at 500 rpm for 2 min and incubated at 37 °C in a humidified 5% CO2 atmosphere. After 5 h of incubation, 100 µL of supernatant was collected and the amount of LDH released from the cytosol of damaged cells was immediately measured using an LDH cytotoxicity detection kit Plus (Roche Applied sciences, Mannheim, Germany) according to the manufacturer’s instructions. Spontaneous LDH release was determined by incubating target cells in medium without effectors, and maximal LDH release was determined by lysis of target cells in 1% Triton X-100 (Sigma). The percentage of cell-mediated cytolysis was determined with average absorbance of the triplicate samples by applying the formula: % cytolysis = [(sample LDH release − spontaneous LDH release)/(maximal LDH release − spontaneous LDH release)] × 100.
IL-2-activated non-adherent PBMCs cells were cultured either in the absence or in the presence of mock-infected or persistently infected 1.1B4 cells 52 weeks post-infection at 37 °C, 5% CO2 (effector: target cell ratio = 40:1). 5 h and 24 h later, supernatants were harvested for quantification of IFN-γ with a Human IFN-γ ELISA kit (Peprotech®) according to the manufacturer’s instructions.
Flow cytometry and viability measurements
The cell surface expression of Human Leukocyte Antigens (HLA)-class I molecules on mock-infected and CV-B4-infected 1.1B4 cells was determined. Cells were trypsinized, washed twice in FACS buffer (PBS supplemented with 2% FCS and 1 mM EDTA), and incubated with monoclonal mouse anti-human HLA-ABC-FITC (Clone B9.12.1, Beckman Coulter, Marseille, France) for 30 min at 4 °C in the dark. The cells were then washed twice with PBS, fixed in 0.5% paraformaldehyde, and analyzed by flow cytometry on a Navios Flow cytometer (Beckman Coulter, Inc.). The influence of IFN-γ on HLA class I expression of 1.1B4 cells was evaluated. Mock and CV-B4-persistently infected 1.1B4 cells were cultured in presence or in absence of 100 U/mL of human recombinant IFN-γ (Peprotech®) for 48 h. The cell surface expression of HLA class I molecules on 1.1B4 cells was determined by flow cytometry as described above.
For intracellular detection of viral double-stranded RNA (dsRNA) and viral capsid protein VP1, trypsinized 1.1B4 cells were first fixed and permeabilized with BD Cytoperm/Cytofix for 20 min at 4 °C. Then, after two washes with BD Cytoperm/Wash, cells were incubated with primary antibodies diluted in BD Cytoperm/Wash: 1:200 of mouse monoclonal J2 anti-dsRNA IgG2a antibody (Scicons English & Scientific Consulting Bt, Hungary) or 1:4000 of mouse anti-enterovirus VP1 clone 5D8/1 (DAKO, Les Ulis, France). After two washes with Cytoperm/Wash, the cells were stained with a 1:400 dilution of Alexa Fluor™ 488-conjugated rabbit anti-mouse IgG antibody (Invitrogen), and analyzed by flow cytometry.
Adherent mock-infected or CV-B4-infected 1.1B4 cells 58 weeks post-infection were cultured for 1 h in the absence or in the presence of IL-2-activated non-adherent PBMCs at effector/target ratios of 10:1. Effector cells were subsequently removed by aspiration and 1.1B4 cells were washed twice with PBS. 1.1B4 cells were then harvested using Accutase™ Cell Detachment Solution (BD Biosciences). On the one hand, apoptotic or necrotic cells were evaluated with Annexin V APC (BD Pharmingen™) and propidium iodide (PI) (BD Pharmingen™) staining, according to the manufacturer’s protocol, and analyzed by flow cytometry. On the other hand, 3.106 1.1B4 cells were resuspended in cell lysis buffer and Caspase-3 activity was measured using ApoAlertTM Caspase-3 colorimetric Assay Kit (Clontech, Mountain View, CA). Briefly, 50 µL cell lysates were incubated with 50 µL 2X reaction buffer/DTT mix containing 5 µL caspase-3 substrate (DEVD-pNA) for 1 h at 37 °C. 1 µL Caspase-3 inhibitor (DEVD-fmk) was used as control to confirm the specificity of the assay. Absorbance was read at 405 nm with a microplate reader and caspase-3 activity (units) quantification was determined with pNA calibration curve according to the manufacturer’s instructions.
Mock and CV-B4-persistently infected 1.1B4 cells 58 weeks post-infection were grown on sterile glass slides. After 2 h of culture in the absence or the presence of IL-2-activated non-adherent PBMCs on the slides at effector/target ratios of 10:1, effector cells were then removed and 1.1B4 cells were washed, fixed with 4% paraformaldehyde, and permeabilized with cold methanol/acetone. Cells were incubated for 5 min at room temperature with 1 µg/mL Hoechst 33342 solution (Thermo Scientific™) to study morphological features of apoptosis (chromatin condensation and fragmentation of the nucleus). Stained cells were visualized with a fluorescence microscope (Leitz Diaplan, Wetzlar, Germany).
Quantification of HLA class I mRNA expression
Total RNA was extracted from mock and CV-B4-infected 1.1B4 cells using TriReagent® RNA isolation reagent/chloroform procedure (Sigma). Extracted RNA was dissolved in 50 μL of nuclease-free water and quantified with a Nanodrop® spectrophotometer (Thermofisher Scientific). After a DNA enzymatic digestion performed on RNA extracts as previously described [25, 26], the RNA retro-transcription was performed using Affinityscript® QPCR cDNA Synthesis kit (Agilent technologies, Les Ulis, France) on a Perkin Elmer 2400 thermocycler (Villebon-sur-Yvette, France).
HLA class I mRNA was quantified on the Mx3000p® (Stratagene) by real-time quantitative polymerase chain reaction (qPCR) using a Brilliant II SYBR® Green QPCR Master Mix (Agilent technologies, Les Ulis, France) according to the manufacturer’s instructions. The primer sequences used for HLA class I mRNA were as follows: forward, 5′-CCTACGACGGCAAGGATTAC-3′; reverse, 5′-TGCCAGGTCAGTGTGATCTC-3′. Those for beta-actin gene (used as endogenous control for normalization) were as follows: forward, 5′-TTGCCGACAGGATGCAGAA-3′; reverse, 5′-GCCGATCCACACGGAGTACT-3′. HLA class I mRNA relative expression in mock as compared to CV-B4 infected 1.1B4 cells was determined with the 2−ΔΔCt formula [27]. All reactions were performed in triplicate.
Data and statistical analyses
Data analyses were performed using Graph Pad Prism 6.0 (Graph Pad Inc, CA, USA). Results are presented as mean values ± standard deviation. The Mann–Whitney U test and the Kruskal–Wallis test were used when appropriate. p values < 0.05 were considered to indicate statistically significant differences.
Results
Persistent CV-B4 infection of 1.1B4 cells
Human pancreatic 1.1B4 cells were inoculated with CV-B4 at MOI 0.01. The viral titer increased gradually during the acute infection step, peaking at 8.9 log TCID50/mL on day 11 post-infection (p.i.) (Fig. 1a). Titers of infectious particles ranging from 4 to 7.8 logs TCID50/mL were found in supernatants throughout the culture period until day 300 p.i. (Fig. 1a). The viral capsid protein VP1 of CV-B4 was detected in infected 1.1B4 cells cultures by immunofluorescence as shown in Fig. 1c. The proportion of stained cells was higher (42.4 ± 3.7%; n = 3) on day 8 p.i. followed by a decrease (5%) on day 210 p.i. (Fig. 1a). There was no significant difference between CV-B4 and mock-infected cell viability monitored through metabolic activity evaluated by Uptiblue™ assay during the first 21 days p.i. (Fig. 1b). Beyond 21 days p.i., the viability of these cells was evaluated using the Trypan blue dye exclusion test which displayed a proportion ranging from 90 to 96% of viable cells throughout the follow-up.
Fig. 1.

Persistent infection of pancreatic beta cells with CV-B4. A persistent infection was established in pancreatic beta cells (1.1B4 cell line) with CV-B4 at multiplicity of infection (MOI) of 0.01. a Viral titers in supernatants of CV-B4 infected 1.1B4 cell culture at various times post-infection (p.i.). The results were expressed as TCID50/mL. The percentage of CV-B4 infected cells was determined by detection of viral capsid protein VP1 by indirect immunofluorescent staining. b Mock and CV-B4-infected 1.1B4 cells’ viability was assessed using Uptiblue™ viable cell reagent. The results are expressed as a viability index (ratio between the absorbance at a wavelength of 570 and 600 nm). c Indirect immunofluorescent staining. CV-B4 infected 1.1B4 cells (21 weeks p.i.) were stained for intracellular VP1 with mouse anti-enterovirus VP1 and Alexa Fluor™ 488-conjugated rabbit anti-mouse IgG antibody (original magnification × 20). Pictures from one representative experiment out of three are shown. d Intracellular detection of VP1 and viral dsRNA in CV-B4 infected 1.1B4 cells by flow cytometry, using mouse anti-enterovirus VP1 and mouse monoclonal J2 anti-dsRNA IgG2a antibodies, respectively. The percentage of VP1-positive and dsRNA-positive cells was determined at 24 h p.i. at MOI 1, 58 and 70 weeks p.i. The results (a, b, d) are mean ± SD of three independent persistent infections. e Semi-nested (Sn)-PCR for intracellular enteroviral RNA detection in a few cells harvested at 30 and 58 weeks p.i. and distributed to obtain around 6, 5, 3, 2 and 1 cell per tube. Intracellular enteroviral RNA from these cells was obtained without extraction and was detected by a specific RT-PCR step for the positive strand of EV genome and for beta actin followed by a Sn-PCR. The electrophoresis in agarose gel of specific amplicons is presented. Results from one representative experiment out of three are shown
The proportion of cells containing double-stranded viral RNA (dsRNA) and VP1 in CV-B4 infected cultures was analyzed by flow cytometry. The percentages of dsRNA-positive and VP1-positive cells at 24 h p.i. (MOI 1) were around 12.7 ± 2.6% and 10.8 ± 2.5%, respectively, but were lower at 58 weeks of persistent infection (3.6 ± 0.9% and 3.0 ± 0.8%) and at 70 weeks of persistent infection (4.0 ± 2.1% and 2.2 ± 0.9%). Considering the level of infectious particles in supernatants and the proportion of VP1-positive cells it can be estimated that less than 2 × 103 infectious particles were produced by each productively infected cell throughout the follow-up of cultures. The extent of CV-B4 infection in 1.1B4 cells was further investigated at 30 and 58 weeks p.i. in a few cells distributed to obtain around one to six cells per tube. The presence of intracellular enteroviral RNA in these cells was detected by RT-PCR without RNA extraction. Thus, viral RNA was detected in all tubes containing six, five, three and two cells (Fig. 1e). These data suggest that CV-B4 can establish a persistent infection in human pancreatic 1.1B4 cells with at least 50% of cells that can harbor CV-B4 RNA.
Lysis of CV-B4 persistently infected 1.1B4 cells by IL-2-activated non-adherent PBMCs
The specific lysis of HEp-2 and Panc-1 cells and of mock and CV-B4-acutely infected MRC-5 and 1.1B4 cells co-cultured with non-adherent PBMCs was investigated through the release of LDH in supernatants.
The extent of LDH release by mock-infected HEp-2 and Panc-1 cells co-cultured with IL-2-activated non-adherent PBMCs from healthy donors was higher compared with the other cells (Fig. 2a). There was no significant difference between mock and CV-B4-acutely infected MRC-5 and 1.1B4 cells (Fig. 2a).
Fig. 2.

Pancreatic beta cells persistently infected with CV-B4 can be lysed by IL-2-activated NK cells. a HEp-2, Panc-1 cells or CV-B4 and mock-infected MRC-5 and 1.1B4 cells 12 h post-infection at M0I 1 were co-cultured with IL-2-activated non-adherent PBMCs from 5 healthy donors at indicated Effector: target cell ratios. Five hours later, cell-mediated lysis was measured by LDH release assay. The results are expressed as percentage (%) of specific lysis and are presented as mean ± SD. b Mock or CV-B4-persistently infected 1.1B4 cells harvested 23–58 weeks post-infection were co-cultured with resting or IL-2-activated non-adherent PBMCs from 8 healthy donors and cell-mediated lysis was measured as described above. c Cytolysis of mock and persistently infected pancreatic beta cells (23–58 weeks post-infection) mediated by IL-2-activated CD56+ (NK cells) and CD3+ (T lymphocytes) cell fractions isolated from non-adherent PBMCs of 5 healthy subjects. d IL-2-activated non-adherent PBMCs were cultured in absence or presence of mock or persistently infected pancreatic beta cells 52 weeks post-infection. Supernatants were harvested after 5 h and 24 h for quantification of IFN-γ by ELISA. The limit of detection of the test is represented by the dashed line. *p < 0.05 versus mock-infected cells or controls
Mock and CV-B4-persistently infected 1.1B4 cells were co-cultured with resting or IL-2-activated non-adherent PBMCs. The specific lysis of mock or infected cells by resting non-adherent PBMCs was low (< 10% at effector: target cell ratio = 40:1), in contrast the specific lysis of infected cells by IL-2-activated non-adherent PBMCs was higher and there was a significant difference as compared to lysis of mock-infected cells in this condition (42.3 ± 11.2% vs 18.4 ± 7.8% p = 0.015 at effector: target cell ratio of 40:1) (Fig. 2b).
To identify effector cells in non-adherent PBMCs (encompassing 15.7 ± 6.5% CD3−CD56+ NK cells and 76.8 ± 5.5% CD3+CD56− T cells, n = 5) that mediated cytolysis towards 1.1B4 cells persistently infected with CV-B4, CD56+ (NK cells) and CD3+ (T cells) cell fractions isolated from healthy donors were cultured in the presence of IL-2, and used in cell-mediated lysis assays as effector cells. The specific lysis of infected cells was much higher when they were co-cultured with IL-2-stimulated CD56+ cells compared with IL-2-stimulated CD3+ cells (53.5 ± 11.1% vs 9.1 ± 2.3%; p = 0.016) (Fig. 2c). The specific lysis obtained with IL-2-activated CD56+ cells was significantly higher with infected cells than mock-infected cells (p = 0.028), but not with IL-2-stimulated CD3+ cells (p = 0.079) (Fig. 2c). In Fig. 2c, the effector cells were in a medium used to sort CD3−CD56+ and CD3+CD56− cells. It was observed that in this medium the cytolytic activity of effector cells was reduced compared with regular culture medium. In addition to cytolytic activity towards infected cells, NK cells can contribute to antiviral immune responses through production of cytokines such as IFN-γ. The levels of IFN-γ in supernatants of IL-2-activated non-adherent PBMCs cultured in the presence or absence of mock and infected 1.1B4 cells were determined. The levels of IFN-γ were higher in supernatants of co-cultures than in those of cultures of IL-2-activated non-adherent PBMCs; but the values were not significantly different in co-cultures of effectors with mock-infected 1.1B4 cells or infected 1.1B4 cells (Fig. 2d).
NK cells mediate target cell killing by caspase-dependent apoptosis through perforin/granzyme granule exocytosis or TNF death receptor family members [17]. 1.1B4 cells persistently infected with CV-B4 were cultured in the absence or presence of resting or IL-2-activated non-adherent PBMCs. Then, to determine the pathway of cell death, the proportion of apoptotic and necrotic cells was evaluated by flow cytometry, using Annexin V and propidium iodide (PI) stainings. In this experiment, early and late apoptosis were defined as annexin V positive/PI negative and annexin V positive/PI positive cells, respectively, while annexin V negative/PI positive cells defined necrotic cells. The proportions of apoptotic and necrotic CV-B4 infected 1.1B4 cells were low (1.4 ± 0.6% and 0.3 ± 0.1%, respectively; n = 5), which could be a result of the infection or cell detachment procedure (Fig. 3aa). The proportion of apoptotic CV-B4 infected 1.1B4 cells co-cultured with resting non-adherent PBMCs was low (1.9 ± 0.4%; n = 3); in contrast, the proportion was significantly higher when the cells were co-cultured with IL-2-activated non-adherent PBMCs (6.5 ± 0.8%; n = 3) (p = 0.03) (Fig. 3ab, ac). The number of gated cells is the same in Fig. 3aa, ab, ac. The results in Fig. 3c are representative of three independent experiments; at least five fields in each experiment were analyzed for quantification.
Fig. 3.

Markers of apoptosis investigated in pancreatic beta cells persistently infected with CV-B4 co-cultured with IL-2-activated non-adherent PBMCs. Pancreatic beta cells persistently infected with CV-B4 58 weeks post-infection were cultured in absence or presence of resting or IL-2-activated non adherent PBMCs at Effector: Target cell ratio = 10:1. One hour later, PBMCs were removed and pancreatic beta cells were then washed and harvested. a The proportion of apoptotic or necrotic pancreatic beta cells was evaluated by flow cytometry, using Annexin V APC and propidium iodide (PI) staining. Results from one representative experiment out of three are shown. b Caspase-3 activity was quantified in pancreatic beta cells (1.1B4) lysates using the colorimetric substrate DEVD-pNA. Caspase-3 inhibitor DEVD-fmk was used to confirm the specificity of the assay. Absorbance was read at 405 nm and caspase-3 activity (units) was determined with pNA calibration curve. c Pancreatic beta cells were cultured on slides in absence or in presence of IL-2-activated PBMCs for 2 h; then the slides were washed, fixed, permeabilized and incubated with Hoechst 33342 solution for nuclei staining. Stained cells were visualized with a Leitz Diaplan fluorescence microscope. Pictures from one representative experiment out of three are shown
Furthermore, caspase-3 activity of CV-B4 infected 1.1B4 cells was increased when these cells were co-cultured with IL-2-activated non-adherent PBMCs. However, caspase-3 activity of mock-infected 1.1B4 cells was increased to the same extent (Fig. 3b). In contrast, after co-culture with IL-2-activated non-adherent PBMCs, chromatin condensation was observed in a larger proportion of CV-B4 infected cells compared to mock-infected 1.1B4 cells (78% ± 16.5% vs 43 ± 11.9%) (Fig. 3c). It cannot be excluded that the features of nuclei of some cells were due to an incomplete cell attachment to the cover slide and further treatment and washing steps. Nevertheless, the proportion of cells with chromatin condensation was higher when the targets of PBMC treated with IL-2 were CV-B4-infected cells than when the targets were mock-infected cells.
Taken altogether these experiments show that IL-2-activated non-adherent PBMC, through NK cells, can lyse pancreatic beta cells persistently infected with CV-B4 and can induce apoptosis of these cells.
CVB4 infection downregulates cell surface expression of HLA class I molecules on 1.1B4 cells
NK cells express inhibitory receptors specific for classical HLA class I molecules on target cells [28]. Thus, altered HLA class I antigen expression on target cells can induce spontaneous NK cell-mediated killing [16]. Therefore, we reasoned that the cytolytic activity of NK cells towards infected 1.1B4 cells might be due to reduced cell surface expression of HLA class I molecules. The proportion of HLA class I positive CV-B4 infected 1.1B4 cells gradually declined during persistent infection from 90 ± 2.5% (n = 5) at 38 weeks post-infection (p.i.), to 28 ± 7.1% (n = 5) at 58 weeks p.i., to 17 ± 3.7% (n = 5) at 70 weeks p.i., contrasting with mock-infected cells (99 ± 0.4%; n = 5) and with CV-B4 infected cells 48 h p.i. at MOI 1 (98 ± 0.3%; n = 5) (Fig. 4a, b). A model of persistent infection of Panc-1 cells (human ductal cell line) with CV-B4 was previously reported by our team [11]. The proportion of HLA class I-positive cells in mock-infected and CV-B4-persistently infected Panc-1 cells at week 56 p.i. was not different (Fig. 4b). At 38 and 46 weeks p.i., 90.2 ± 2.0% and 76.1 ± 2.7% persistently infected 1.1B4 cells still expressed HLA class I molecules on their surface but the fluorescence intensity of HLA class I staining was reduced, compared to mock-infected cells (p < 0.05) (Fig. 4c). Thus these data showed that the fluorescence intensity of HLA class I staining was already reduced in CV-B4 persistently infected 1.1B4 cells while the proportion of HLA class I positive cells was still unchanged compared to controls.
Fig. 4.

Downregulation of HLA class I molecules at the surface of pancreatic beta cells persistently infected with CV-B4. a Pancreatic beta cells persistently infected with CV-B4 and mock-infected cells were analysed for cell surface HLA class I expression by flow cytometry at various weeks post-infection (p.i.), using anti-human HLA-ABC-FITC. Results from one representative experiment out of three are shown. b Percentage of cell surface HLA class I positive pancreatic beta cells (1.1B4) infected with CV-B4 harvested 48 h p.i., 52, 58 and 70 weeks p.i. and Panc-1 cells persistently infected with CV-B4 harvested 56 weeks p.i. The cells were analysed by flow cytometry using anti-human HLA-ABC-FITC. c Mean fluorescence intensity of cell surface HLA class I expression on mock and CV-B4-persistently infected 1.1B4 cells harvested 38 and 46 weeks p.i. was analysed by flow cytometry using anti-human HLA-ABC-FITC. Results presented in b and c are mean ± SD of three independent acute and persistent infections.*p < 0.05. d Mock and persistently infected 1.1B4 cells were stained with anti-human HLA-ABC-FITC and were evaluated for apoptosis by flow cytometry, using Annexin V APC and propidium iodide (PI) stainings. Results from one representative experiment out of three are shown. e Mock-infected and persistently infected 1.1B4 cells harvested 58 weeks p.i. were double-stained for HLA class I and for VP1 analysis by indirect immunofluorescence. Pictures from one representative experiment out of three are shown
It was decided to study the viability of persistently CV-B4 infected 1.1B4 cells, on which the expression of HLA class I molecules was reduced, by flow cytometry using Annexin V and propidium iodide (PI) staining. At 58 weeks p.i., around 78% of CV-B4 persistently infected 1.1B4 cells did not express HLA class I molecules at their surface; nevertheless, their viability was maintained, as shown by the low-level of Annexin V and/or PI-positive cells (less than 1.5%) (Fig. 4d).
The impact of CV-B4 on the expression of HLA class I at the surface of CV-B4 persistently infected cells was evaluated by indirect immunofluorescence staining for HLA class I and intracellular VP1. Figure 4e shows representative fields of view of three independent experiments of mock and persistently infected 1.1B4 cells with CV-B4 and at least five fields in each experiment were analyzed for quantification. At 58 weeks p.i., the proportion of HLA class I positive cells and the fluorescence intensity was low in CV-B4 infected cells compared to mock-infected cells, confirming the results of flow cytometry. Interestingly, the detection of HLA class I antigen in 2 out of 3 VP1 positive cells was negative whereas the detection was positive in the third one in CV-B4 persistently infected 1.1B4 cells (Fig. 4e). When many fields were observed under the microscope, an average of 1.6% cells were positive for VP1 and negative for HLA class I and 2% cells were positive for VP1 and positive for HLA class I. HLA class I can be internalized in CVB-infected cells. Indeed it was reported that CV-B3 2B and 2BC proteins upregulated the internalization of HLA class I [29]. However, in our experiments with persistently-infected cells the detection of intracellular HLA class I by immunofluorescence assay after cell permeabilization was negative in a large proportion of cells as shown by Fig. 4e.
It has been reported that viruses downregulate HLA class I gene transcription through binding of regulatory factors to promoters of HLA class I heavy chain [16]. Therefore, in our experiments the expression of HLA class I mRNA was assessed in CV-B4 persistently infected 1.1B4 cells using real-time RT-qPCR and the results were expressed as fold-change (2−∆∆Ct) as compared to mock-infected cells. There was no significant difference in expression of HLA class I mRNA at 38 and 58 weeks p.i. in CV-B4 persistently infected 1.1B4 cells as compared to mock-infected cells (Fig. 5a), suggesting that CV-B4-mediated reduction of HLA class I molecules on 1.1B4 cells was not due to a transcriptional downregulation process.
Fig. 5.

Expression of HLA class I mRNA in pancreatic beta cells persistently infected with CV-B4. a Levels of HLA class I mRNA. HLA class I mRNA expression was assessed in 1.1B4 cells persistently infected with CV-B4 38 and 58 weeks post-infection using real-time RT-qPCR. The results are expressed as fold-change (2−∆∆Ct) as compared to mock-infected cells. Results are mean ± SD of three independent experiments. b Cell surface expression of HLA class I on mock and persistently infected 1.1B4 cells harvested 70 weeks post-infection cultured in presence or absence of IFN-γ (100 U/mL) for 48 h.**p < 0.01
Further investigations assessed whether the downregulation of HLA class I expression in CV-B4 persistently infected 1.1B4 cells could be corrected by IFN-γ that is able to stimulate HLA class I expression in cell lines and human pancreatic beta-cells [30–32]. Mock and CV-B4-persistently infected 1.1B4 cells were cultured in the absence or presence of IFN-γ for 48 h, and the proportions and mean fluorescence intensities of HLA Class I-positive cells were evaluated by flow cytometry. In CV-B4-persistently infected 1.1B4 cell cultures at 70 weeks post infection, the viral titer in supernatants was 7.3 ± 0.8 log TCID50/mL and the specific lysis of infected cells and mock-infected cells by IL-2-activated non-adherent PBMCs at effector: target cell ratio 40:1 were 36.8 ± 5.4% and 15.2 ± 4.8%, respectively (p = 0.021, n = 3). The proportions of cells expressing HLA class I antigen in mock and CV-B4-persistently infected 1.1B4 cells harvested 70 weeks p.i. remained unchanged after exposure to IFN-γ (data not shown). Nevertheless, IFN-γ treatment resulted in an increased fluorescence intensity of HLA class I molecule staining at the surface of mock and persistently infected 1.1B4 cells with CV-B4 but at a lower extent (Fig. 5b).
IFN-γ is known to upregulate HLA class I expression; however, the impact of IFN-γ on the expression of HLA class I in infected cells was weak (at 48 h). HLA class I was downregulated in persistently infected cells, and the level of IFN-γ in co-cultures was low when the cytolysis was measured (at 5 h). Taken together our data suggest that the expression of HLA class I was not enhanced by IFN-γ or at a low extent in co-cultures and therefore it can be assumed that CV-B4 persistently infected 1.1B4 cells were readily lysed by NK cells.
Lysis of CV-B4 persistently infected 1.1B4 cells by IL-2-activated non-adherent PBMCs of patients with T1D
Mock or CV-B4-persistently infected 1.1B4 cells were co-cultured with IL-2-activated non-adherent PBMCs isolated from seven patients with T1D and ten control subjects. The specific cytolysis of CV-B4 infected 1.1B4 cells was low when they were co-cultured with IL-2-activated non-adherent PBMCs obtained from diabetic patients compared with IL-2-activated non-adherent PBMCs obtained from controls (p < 0.01) (Fig. 6a). In so far as the cytolysis of CV-B4-infected 1.1B4 cells by IL-2-activated non-adherent PBMCs depends on NK cells (see above), the proportion of NK cells (CD3−CD56+CD16+) in PBMCs from patients and controls was evaluated by flow cytometry. The percentage of NK cells was significantly lower in PBMCs from patients with T1D than in PBMCs from controls (p < 0.01) (Fig. 6b). These results highlight impaired cytolytic activity of IL-2-activated non-adherent PBMCs of patients with T1D towards CV-B4 persistently infected 1.1B4 cells.
Fig. 6.
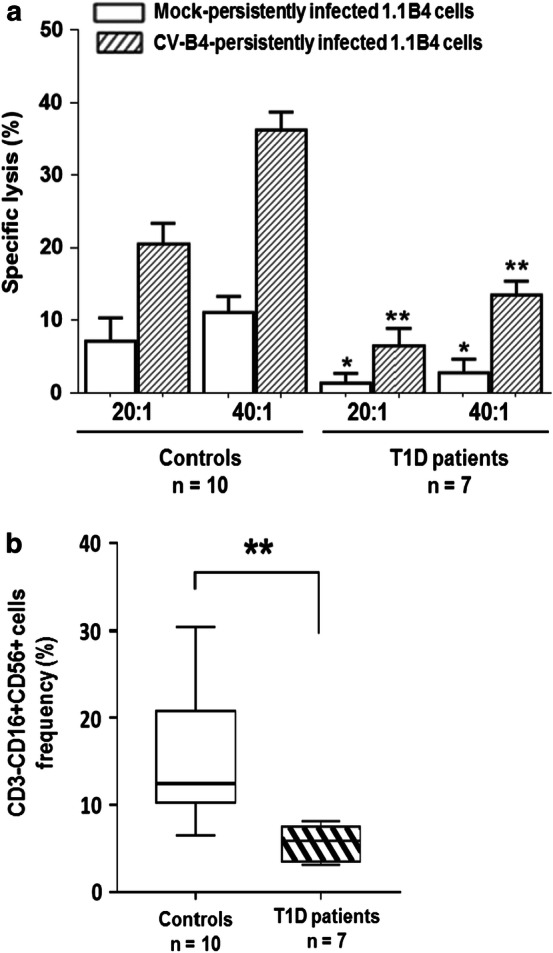
Lysis of CV-B4 persistently infected pancreatic beta cells by IL-2-activated non adherent PBMCs of patients with T1D. a Mock and persistently infected 1.1B4 cells with CV-B4 70 weeks post-infection were co-cultured for 5 h with IL-2-activated non-adherent PBMCs from 10 healthy donors and 7 T1D patients at Effector: target cell ratio 20:1 and 40:1. Cell-mediated lysis was measured by LDH release assay. The results are expressed as percentage (%) of specific lysis and are presented as mean ± SD. b Frequencies of peripheral blood NK cell subsets in T1D patients and control subjects. Peripheral blood mononuclear cells were stained with monoclonal antibodies, acquired using a flow cytometer, and analyzed using appropriate software. Box plots represent medians with 25th and 75th percentiles and whiskers illustrating 10th and 90th percentiles of CD3−CD16+CD56+ cell subset frequencies from 10 control subjects and 7 T1D patients.*p < 0.05 and **p < 0.01 versus controls
Discussion
The current study is different in many respects from those of other investigators; this study reports for the first-time human NK cell cytolytic activity towards human pancreatic beta cells persistently infected with CV-B4. Several points are worth noting. In vitro studies reported the infection of human pancreatic islets by CV-B [33–35], and confirmed that CV-B4 can establish a persistent infection in human pancreatic β and ductal cells, resulting in disturbance of the viability and function of these cells [10–13]; however, persistent infection of primary human islet cultures with CV-B4 is restricted by the difficulties of supplying and preparing viable islets and long-term maintenance of dissociated islet cultures [13]. Therefore, we took advantage of a 1.1B4 pancreatic beta cell line produced by electrofusion of fresh human pancreatic beta cells with a human pancreatic duct cell line (Panc-1) [36], to address the issue of the clearance of pancreatic beta cells persistently infected with CV-B4 by human PBMCs. In this system, cytolysis was measured by quantifying the level of LDH released in supernatants of co-cultures using a sensitive assay, allowing comparisons between the lysis of mock and infected pancreatic beta cells by non-adherent PBMCs. Compared to 1.1B4 cells, the level of specific cytolysis of HEp-2 and Panc-1 cells by IL-2-activated non-adherent PBMCs was high, therefore HEp-2 and Panc-1 cells were not used in the rest of this study. HEp-2 and Panc-1 are tumor cell lines that can be readily lysed by effector cells compared with either MRC-5, a human diploid cell line, or with 1.1B4, that is the result of a fusion between primary beta cell and Panc-1 cell line. Acutely infected MRC-5 and 1.1B4 cells were not suitable for investigation of the killing of pancreatic beta cells by PBMCs in our system, since the specific lysis of mock and infected cells was not different under such conditions. Pancreatic cell lines, although interesting, present limitations in terms of translation of results, and beta cells from healthy individuals, diabetic or at-risk patients could behave differently and could respond differently to the infection.
It has been shown that CV-B4 can infect and establish a persistent infection in human pancreatic 1.1B4 cells in vitro for up to 70 weeks post-infection. The persistent infection is characterized by an acute phase during the first week post-infection followed by a moderate and stable release of infectious particles by the cells subsequently. The proportion of cells positive for VP1 was small (less than 5%). Furthermore, the virus was cleared when cultures were treated with fluoxetine (data not shown). This is in agreement with the anti-CV-B4 effect of fluoxetine in persistently-infected cells reported by our team previously [37, 38]. Downregulation of HLA class I molecules was observed at the surface of a large proportion of 1.1B4 cells in cultures persistently infected with CV-B4. The proportion of cells harboring viral RNA was higher than predicted by the low levels of intracellular VP1 and viral dsRNA (< 5%) detected in CV-B4 infected 1.1B4 cells. These observations argue in favor of a low-grade viral replication compatible with a carrier-state persistent infection [39–41], and are in agreement with those of a previous study which reported that at least 50% of cells harbored enteroviral RNA in a model of pancreatic ductal-like cells persistently infected with CV-B4 [11]. In cells harboring viral RNA it cannot be excluded that viral RNA is latent in some of them and activated in some others, with a low-level replication under the limit of detection of dsRNA by immunostaining. Such characteristics could explain the large proportion of cells displaying reduced expression of HLA class I amongst 1.1B4 cells persistently infected with CV-B4 in this study.
It has been shown that HLA class I hyperexpression and VP1 immunostaining are usually present in islet beta cells [14, 42]. However, HLA class I expression is not directly related to VP1 expression, as many other insulin-containing islets can overexpress HLA class I without the presence of VP1 [42]. It has been observed that CV-B4-infected beta cells produce IFN-α [13] and that IFN-α can increase the expression of HLA class I in beta cells [43]. So far, the present study shows that the infection of beta cells with CV-B4 results in inhibition of HLA class I, it is likely that HLA class I hyperexpression in islet beta cells is due to an indirect mechanism mediated by IFN-α as suggested by other teams [14, 42]. It cannot be excluded that a few infected beta cells in islets of prediabetic and recent onset T1D individuals results in production of IFN-α that enhances the expression of HLA class I in non-infected beta cells.
The mechanism underlying CV-B4-mediated downregulation of HLA class I molecules on 1.1B4 cells was assessed using real-time RT-qPCR and immunofluorescence. The expression of HLA class I mRNA in persistently infected cells remained unchanged compared to mock-infected cells. There was a progressive inhibition of HLA class I in a first stage (HLA class I negative cells 10% at week 38) then the inhibition was more pronounced as the culture progressed (HLA class I negative cells 80% week 58). Internalization of HLA class I molecules to intracellular location, degradation of HLA class I molecules or disturbance of their intracellular trafficking cannot be excluded. Indeed, it has been shown that Picornaviruses are able to inhibit transit from endoplasmic reticulum to Golgi through non-structural viral proteins, resulting in an impairment of HLA class I expression [44–46]. In addition, protein 3A of CV-B3 disrupted the Golgi complex while 2B and 2BC proteins inhibited protein traffic through the Golgi complex, and upregulated HLA class I endocytosis [29, 47, 48]. Further studies are needed to elucidate the mechanism of inhibition of HLA class I in persistently-infected cells.
The proportion of HLA class I positive cells was high 48 h p.i. (as shown by Fig. 4b) and cytolytic killing of cells did not occur during this phase. Cytolytic killing of cells occurred after the acute phase. During week 38, when there was a higher proportion of HLA class I-positive cells compared with week 58, the cytolytic killing of cells was observed. Whether the reduced expression of HLA class I, reflected by a reduction of the fluorescence intensity of cell surface HLA class I analyzed by FACS, was involved in rendering the persistently-infected cells more sensitive to cytolytic killing by NK cells remains to be investigated.
The higher level of LDH in supernatants of infected cells co-cultured with IL-2-activated non-adherent PBMCs on the one hand, and with CD3−CD56+ cells on the other, supports the idea that NK cells are the major effector cells involved in the lysis of pancreatic beta cells persistently infected with CV-B4 in our experiments. Interestingly, as compared to mock-infected 1.1B4 cells, IL-2-activated non-adherent PBMCs did not lyse acutely CV-B4-infected 1.1B4 cells that comprised a similar proportion of HLA class I-positive cells. In contrast, previous studies reported that CV-B3 acute infection can downregulate HLA class I surface expression in HepG2, HUVECs and HeLa tumour cell lines acutely infected with CV-B3 [24, 29]. We have no clear explanation regarding the discrepancy between the results obtained by other researchers and by our group. It can be due to cell types used in these studies and/or to the virus. Taken together, our results show that the lysis of persistently infected 1.1B4 cells required activation of NK cells in our system and suggest that a downregulation of HLA class I molecules at the surface of infected cells was involved in the process.
In this study, the presence of annexin V positive cells, of Caspase-3 activity and of chromatin condensation in CV-B4 persistently infected 1.1B4 cells co-cultured with IL-2-activated non-adherent PBMCs, showed that apoptosis was induced in infected cells by activated PBMCs [49, 50]. There is increasing evidence indicating that apoptotic beta cells can orient immune response toward autoimmunity in T1D [51]. Indeed, apoptotic beta cells are the most important source of autoantigens and enhanced beta cells apoptosis or defective apoptotic beta cells clearance by phagocytes can contribute to the autoimmune process in T1D through permanent autoreactive lymphocyte activation [51–54].
We also observed that the cytolytic activity of IL-2-activated non-adherent PBMCs from patients with T1D towards pancreatic beta cells persistently infected with CV-B4 was reduced compared to PBMCs from controls. Such a reduced cytolytic activity of patients’ cells could be due to a lower proportion of NK cells compared to controls, as suggested by our results regarding the decreased number of NK cells (CD3−CD56+CD16+) in patients. Moreover it cannot be excluded that the cytolytic activity of NK cells from the patients with T1D was reduced since it was already observed that NK cells from such patients are not able to lyse NK-sensitive K562 tumor cells [28]. Several studies have reported a low NK cell frequency and impaired cytolytic activity of these cells in patients with T1D, irrespective of the duration of the disease, compared to control subjects [28, 55–59]. Furthermore, weak NK cell responses to IL-2 stimulation and aberrant signaling of the activating NK cell receptor, NKG2D, have been reported in patients with T1D [59, 60]. Taken together these reports and our observations support the hypothesis of a defect of circulating NK cells in patients with T1D. It has been suggested that disturbance of viral clearance and manipulation of the host antiviral immune response may be involved in the enteroviral pathogenesis of T1D [19, 61]. The results of the present study argue in favor of impaired viral clearance that could play a role in the persistence of CV-B4 reported in patients with T1D [7, 9, 19, 62–64]. The impaired cytolytic activity of PBMC from patients in our study may also be due to the exhaustion of NK cells, which can be observed in the course of chronic infection as previously reported [65].
In conclusion, the persistence of CV-B4 in pancreatic beta cells resulted in downregulation of HLA class I cell surface expression and increased their susceptibility to NK cell-mediated killing. The cytolytic activity of IL-2-activated non-adherent PBMCs of patients with T1D against pancreatic beta cells persistently infected with CV-B4 was reduced due to the low frequency of NK cells on the one hand and, possibly, to the dysfunctional state of the cells that are present on the other. The impact of NK cells on pancreatic beta cells persistently infected with CV-B4 can result in apoptosis of these cells, a process that is thought to play a role in triggering autoimmunity [51–54]. Furthermore the hypothesis of a role of NK cells at an early stage of the development of the disease is in agreement with a previous report showing that NK cells can induce T1D in mice [66]. Defective clearance of pancreatic beta cells persistently infected with CV-B4 in patients with T1D can nevertheless not be excluded [19, 61]. The in vitro culture system based on pancreatic beta cells persistently infected with CV-B4 developed in the present report could prove useful in assessing the cytolytic activity of IL-2-activated non-adherent PBMCs of individuals at risk for T1D, as well as at various stages of the disease to investigate further the role of NK cells in the viral pathogenesis of T1D. Further studies will be directed along this line in our laboratory.
Acknowledgements
This work was supported by Ministere de l’Education Nationale de la Recherche et de la Technologie, Universite Lille 2 (Equipe d’accueil 3610), Centre Hospitalier Regional et Universitaire de Lille, and by EU FP7 (GA-261441-PEVNET: Persistent virus infection as a cause of pathogenic inlammation in type 1 diabetes—an innovative research program of biobanks and expertise). M. P. N was supported by a “CABRI 2016” scholarship of Universite Lille 2 and a “Programme Eifel 2017” scholarship of Ministere des Afaires etrangeres et du Developpement International de la Republique Francaise. Funding was supported by Campus France (EIFFELDOCTORAT 2017/n°P714914K). The authors thank Dr Sarah Richardson (Exeter, UK) for helpful discussion. The authors thank Dr Adrian J. F. Luty for reading the manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Zell R, Delwart E, Gorbalenya AE, Hovi T, King AMQ, Knowles NJ, Lindberg AM, Pallansch MA, Palmenberg AC, Reuter G, Simmonds P, Skern T, Stanway G, Yamashita TIRC. ICTV virus taxonomy profile: Picornaviridae. J Gen Virol. 2017;98:2421–2422. doi: 10.1099/jgv.0.000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hober D, Alidjinou EK. Enteroviral pathogenesis of type 1 diabetes: queries and answers. Curr Opin Infect Dis. 2013;26:263–269. doi: 10.1097/QCO.0b013e3283608300. [DOI] [PubMed] [Google Scholar]
- 3.Hober D, Sauter P. Pathogenesis of type 1 diabetes mellitus: interplay between enterovirus and host. Nat Rev Endocrinol. 2010;6:279–289. doi: 10.1038/nrendo.2010.27. [DOI] [PubMed] [Google Scholar]
- 4.Oikarinen S, Martiskainen M, Tauriainen S, et al. Enterovirus RNA in blood is linked to the development of type 1 diabetes. Diabetes. 2011;60:276–279. doi: 10.2337/db10-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oikarinen M, Tauriainen S, Oikarinen S, et al. Type 1 diabetes is associated with enterovirus infection in gut mucosa. Diabetes. 2012;61:687–691. doi: 10.2337/db11-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeung W-CG, Rawlinson WD, Craig ME. Enterovirus infection and type 1 diabetes mellitus: systematic review and meta-analysis of observational molecular studies. BMJ. 2011;342:d35–d35. doi: 10.1136/bmj.d35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alidjinou EK, Sané F, Engelmann I, et al. Enterovirus persistence as a mechanism in the pathogenesis of type 1 diabetes. Discov Med. 2014;18:273–282. [PubMed] [Google Scholar]
- 8.Krogvold L, Edwin B, Buanes T, et al. Detection of a low-grade enteroviral infection in the islets of langerhans of living patients newly diagnosed with type 1 diabetes. Diabetes. 2015;64:1682–1687. doi: 10.2337/db14-1370. [DOI] [PubMed] [Google Scholar]
- 9.Alidjinou EK, Chehadeh W, Weill J, et al. Monocytes of patients with type 1 diabetes harbour enterovirus RNA. Eur J Clin Investig. 2015;45:918–924. doi: 10.1111/eci.12485. [DOI] [PubMed] [Google Scholar]
- 10.Yin H, Berg A-K, Tuvemo T, Frisk G. Enterovirus RNA is found in peripheral blood mononuclear cells in a majority of type 1 diabetic children at onset. Diabetes. 2002;51:1964–1971. doi: 10.2337/diabetes.51.6.1964. [DOI] [PubMed] [Google Scholar]
- 11.Sane F, Caloone D, Gmyr V, et al. Coxsackievirus B4 can infect human pancreas ductal cells and persist in ductal-like cell cultures which results in inhibition of Pdx1 expression and disturbed formation of islet-like cell aggregates. Cell Mol Life Sci. 2013;70:4169–4180. doi: 10.1007/s00018-013-1383-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alidjinou EK, Engelmann I, Bossu J, et al. Persistence of Coxsackievirus B4 in pancreatic ductal-like cells results in cellular and viral changes. Virulence. 2017;8:1229–1244. doi: 10.1080/21505594.2017.1284735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chehadeh W, Kerr-Conte J, Pattou F, et al. Persistent infection of human pancreatic islets by coxsackievirus B is associated with alpha interferon synthesis in beta cells. J Virol. 2000;74:10153–10164. doi: 10.1128/jvi.74.21.10153-10164.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richardson SJ, Morgan NG, Foulis AK. Pancreatic pathology in type 1 diabetes mellitus. Endocr Pathol. 2014;25:80–92. doi: 10.1007/s12022-014-9297-8. [DOI] [PubMed] [Google Scholar]
- 15.Warren H, Smyth M. NK cells and apoptosis. Immunol Cell Biol. 1999;77:64–75. doi: 10.1046/j.1440-1711.1999.00790.x. [DOI] [PubMed] [Google Scholar]
- 16.Seliger B, Ritz U, Ferrone S. Molecular mechanisms of HLA class I antigen abnormalities following viral infection and transformation. Int J Cancer. 2006;118:129–138. doi: 10.1002/ijc.21312. [DOI] [PubMed] [Google Scholar]
- 17.Smyth MJ, Cretney E, Kelly JM, et al. Activation of NK cell cytotoxicity. Mol Immunol. 2005;42:501–510. doi: 10.1016/j.molimm.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 18.Vitale C, Chiossone L, Morreale G, et al. Human natural killer cells undergoing in vivo differentiation after allogeneic bone marrow transplantation: analysis of the surface expression and function of activating NK receptors. Mol Immunol. 2005;42:405–411. doi: 10.1016/j.molimm.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 19.Dotta F, Censini S, van Halteren AGS, et al. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci USA. 2007;104:5115–5120. doi: 10.1073/pnas.0700442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baba M, Hasegawa H, Nakayabu M, et al. Cytolytic activity of natural killer cells and lymphokine activated killer cells against hepatitis A virus infected fibroblasts. J Clin Lab Immunol. 1993;40:47–60. [PubMed] [Google Scholar]
- 21.Biron CA, Nguyen KB, Pien GC, et al. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol. 1999;17:189–220. doi: 10.1146/annurev.immunol.17.1.189. [DOI] [PubMed] [Google Scholar]
- 22.Godeny EK, Gauntt CJ. Involvement of natural killer cells in coxsackievirus B3-induced murine myocarditis. J Immunol. 1986;137:1695–1702. [PubMed] [Google Scholar]
- 23.Godeny EK, Gauntt CJ. Murine natural killer cells limit coxsackievirus B3 replication. J Immunol. 1987;139:913–918. [PubMed] [Google Scholar]
- 24.Hühn MH, Hultcrantz M, Lind K, Ljunggren HG, Malmberg KJF-TM. IFN-γ production dominates the early human natural killer cell response to Coxsackievirus infection. Cell Microbiol. 2008;10:426–436. doi: 10.1111/j.1462-5822.2007.01056.x. [DOI] [PubMed] [Google Scholar]
- 25.Alidjinou EK, Sané F, Engelmann I, Hober D. Serum-dependent enhancement of Coxsackievirus B4-induced production of IFNα, IL-6 and TNFα by peripheral blood mononuclear cells. J Mol Biol. 2013;425:5020–5031. doi: 10.1016/j.jmb.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 26.Alidjinou EK, Sané F, Trauet J, et al. Coxsackievirus B4 can infect human peripheral blood-derived macrophages. Viruses. 2015;7:6067–6079. doi: 10.3390/v7112924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 28.Marca V, Gianchecchi E, Fierabracci A. Type 1 diabetes and its multi-factorial pathogenesis: the putative role of NK cells. Int J Mol Sci. 2018 doi: 10.3390/ijms19030794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cornell CT, Kiosses WB, Harkins S, Whitton JL. Coxsackievirus B3 proteins directionally complement each other to downregulate surface major histocompatibility complex class I. J Virol. 2007;81:6785–6797. doi: 10.1128/JVI.00198-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campbell IL, Bizilj K, Colman PG, et al. Interferon-gamma induces the expression of HLA-A, B, C but not HLA-DR on human pancreatic beta-cells. J Clin Endocr Metab. 1986;62:1101–1109. doi: 10.1210/jcem-62-6-1101. [DOI] [PubMed] [Google Scholar]
- 31.Rodríguez T, Méndez R, Del Campo A, et al. Distinct mechanisms of loss of IFN-gamma mediated HLA class I inducibility in two melanoma cell lines. BMC Cancer. 2007;7:34. doi: 10.1186/1471-2407-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao J, Brouwer NJ, Jordanova ES, et al. HLA class I antigen expression in conjunctival melanoma is not associated with PD-L1/PD-1 status. Investig Ophthalmol Vis Sci. 2018;59:1005–1015. doi: 10.1167/iovs.17-23209. [DOI] [PubMed] [Google Scholar]
- 33.Roivainen M, Rasilainen S, Ylipaasto P, et al. Mechanisms of coxsackievirus-induced damage to human pancreatic beta-cells. J Clin Endocr Metab. 2000;85:432–440. doi: 10.1210/jcem.85.1.6306. [DOI] [PubMed] [Google Scholar]
- 34.Vuorinen T, Nikolakaros G, Simell O, et al. Mumps and Coxsackie B3 virus infection of human fetal pancreatic islet-like cell clusters. Pancreas. 1992;7:460–464. doi: 10.1097/00006676-199207000-00007. [DOI] [PubMed] [Google Scholar]
- 35.Yoon JW, Onodera T, Jenson AB, Notkins AL. Virus-induced diabetes mellitus. XI. Replication of coxsackie B3 virus in human pancreatic beta cell cultures. Diabetes. 1978;27:778–781. doi: 10.2337/diab.27.7.778. [DOI] [PubMed] [Google Scholar]
- 36.McCluskey JT, Hamid M, Guo-Parke H, et al. Development and functional characterization of insulin-releasing human pancreatic beta cell lines produced by electrofusion. J Biol Chem. 2011;286:21982–21992. doi: 10.1074/jbc.M111.226795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benkahla MA, Alidjinou EK, Sane F, et al. Fluoxetine can inhibit coxsackievirus-B4 E2 in vitro and in vivo. Antiviral Res. 2018;159:130–133. doi: 10.1016/j.antiviral.2018.10.002. [DOI] [PubMed] [Google Scholar]
- 38.Alidjinou EK, Sané F, Bertin A, et al. Persistent infection of human pancreatic cells with Coxsackievirus B4 is cured by fluoxetine. Antiviral Res. 2015;116:51–54. doi: 10.1016/j.antiviral.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 39.Heim A, Canu A, Kirschner P, et al. Synergistic interaction of interferon-beta and interferon-gamma in coxsackievirus B3-infected carrier cultures of human myocardial fibroblasts. J Infect Dis. 1992;166:958–965. doi: 10.1093/infdis/166.5.985. [DOI] [PubMed] [Google Scholar]
- 40.Heim A, Brehm C, Stille-Siegener M, et al. Cultured human myocardial fibroblasts of pediatric origin: natural human interferon-alpha is more effective than recombinant interferon-alpha 2a in carrier-state coxsackievirus B3 replication. J Mol Cell Cardiol. 1995;27:2199–2208. doi: 10.1016/s0022-2828(95)91515-x. [DOI] [PubMed] [Google Scholar]
- 41.Pinkert S, Klingel K, Lindig V, et al. Virus-host coevolution in a persistently coxsackievirus B3-infected cardiomyocyte cell line. J Virol. 2011;85:13409–13419. doi: 10.1128/JVI.00621-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richardson SJ, Willcox A, Bone AJ, et al. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia. 2009;52:1143–1151. doi: 10.1007/s00125-009-1276-0. [DOI] [PubMed] [Google Scholar]
- 43.Pujol-Borrell R, Todd I, Doshi M, et al. Differential expression and regulation of MHC products in the endocrine and exocrine cells of the human pancreas. Clin Exp Immunol. 1986;65:128–139. [PMC free article] [PubMed] [Google Scholar]
- 44.Deitz SB, Dodd DA, Cooper S, et al. MHC I-dependent antigen presentation is inhibited by poliovirus protein 3A. Proc Natl Acad Sci USA. 2000;97:13790–13795. doi: 10.1073/pnas.250483097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moffat K, Howell G, Knox C, et al. Effects of foot-and-mouth disease virus nonstructural proteins on the structure and function of the early secretory pathway: 2BC but not 3A blocks endoplasmic reticulum-to-Golgi transport. J Virol. 2005;79:4382–4395. doi: 10.1128/JVI.79.7.4382-4395.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kirkegaard K, Taylor MP, Jackson WT. Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nat Rev Microbiol. 2004;2:301–314. doi: 10.1038/nrmicro865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Jong AS, Visch H-J, de Mattia F, et al. The coxsackievirus 2B protein increases efflux of ions from the endoplasmic reticulum and Golgi, thereby inhibiting protein trafficking through the Golgi. J Biol Chem. 2006;281:14144–14150. doi: 10.1074/jbc.M511766200. [DOI] [PubMed] [Google Scholar]
- 48.Cornell CT, Kiosses WB, Harkins S, Whitton JL. Inhibition of protein trafficking by coxsackievirus b3: multiple viral proteins target a single organelle. J Virol. 2006;80:6637–6647. doi: 10.1128/JVI.02572-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- 50.Li ZM, Liu ZC, Guan ZZ, et al. Inhibition of DNA primase and induction of apoptosis by 3,3′-diethyl-9-methylthia-carbocyanine iodide in hepatocellular carcinoma BEL-7402 cells. World J Gastroenterol. 2004;10:514–520. doi: 10.3748/wjg.v10.i4.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vives-Pi M, Rodríguez-Fernández S, Pujol-Autonell I. How apoptotic β-cells direct immune response to tolerance or to autoimmune diabetes: a review. Apoptosis. 2015;20:263–272. doi: 10.1007/s10495-015-1090-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mathis D, Vence L, Benoist C. Beta-cell death during progression to diabetes. Nature. 2001;414:792–798. doi: 10.1038/414792a. [DOI] [PubMed] [Google Scholar]
- 53.O’Brien BA, Geng X, Orteu CH, et al. A deficiency in the in vivo clearance of apoptotic cells is a feature of the NOD mouse. J Autoimmun. 2006;26:104–115. doi: 10.1016/j.jaut.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 54.Eizirik DL, Grieco FA. On the immense variety and complexity of circumstances conditioning pancreatic-cell apoptosis in type 1 diabetes. Diabetes. 2012;61:1661–1663. doi: 10.2337/db12-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilson RG, Anderson J, Shenton BK, et al. Natural killer cells in insulin dependent diabetes mellitus. Br Med J. 1986;293:244. doi: 10.1136/bmj.293.6541.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hussain MJ, Alviggi L, Millward BA, et al. Evidence that the reduced number of natural killer cells in type 1 (insulin-dependent) diabetes may be genetically determined. Diabetologia. 1987;30:907–911. doi: 10.1007/BF00295872. [DOI] [PubMed] [Google Scholar]
- 57.Negishi K, Waldeck N, Chandy G, et al. Natural killer cell and islet killer cell activities in type 1 (insulin-dependent) diabetes. Diabetologia. 1986;29:352–357. doi: 10.1007/BF00903343. [DOI] [PubMed] [Google Scholar]
- 58.Lorini R, Moretta A, Valtorta A, et al. Cytotoxic activity in children with insulin-dependent diabetes mellitus. Diabetes Res Clin Pract. 1994;23:37–42. doi: 10.1016/0168-8227(94)90125-2. [DOI] [PubMed] [Google Scholar]
- 59.Qin H, Lee IF, Panagiotopoulos C, et al. Natural killer cells from children with type 1 diabetes have defects in NKG2D-dependent function and signaling. Diabetes. 2011;60:857–866. doi: 10.2337/db09-1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rodacki M, Svoren B, Butty V, et al. Altered natural killer cells in type 1 diabetic patients. Diabetes. 2007;56:177–185. doi: 10.2337/db06-0493. [DOI] [PubMed] [Google Scholar]
- 61.Hofmann P, Schmidtke M, Stelzner A, Gemsa D. Suppression of proinflammatory cytokines and induction of IL-10 in human monocytes after coxsackievirus B3 infection. J Med Virol. 2001;64:487–498. doi: 10.1002/jmv.1076. [DOI] [PubMed] [Google Scholar]
- 62.Ylipaasto P, Klingel K, Lindberg AM, et al. Enterovirus infection in human pancreatic islet cells, islet tropism in vivo and receptor involvement in cultured islet beta cells. Diabetologia. 2004;47:225–239. doi: 10.1007/s00125-003-1297-z. [DOI] [PubMed] [Google Scholar]
- 63.Schulte BM, Bakkers J, Lanke KHW, et al. Detection of enterovirus RNA in peripheral blood mononuclear cells of type 1 diabetic patients beyond the stage of acute infection. Viral Immunol. 2010;23:99–104. doi: 10.1089/vim.2009.0072. [DOI] [PubMed] [Google Scholar]
- 64.Willcox A, Richardson SJ, Bone AJ, et al. Immunohistochemical analysis of the relationship between islet cell proliferation and the production of the enteroviral capsid protein, VP1, in the islets of patients with recent-onset type 1 diabetes. Diabetologia. 2011;54:2417–2420. doi: 10.1007/s00125-011-2192-7. [DOI] [PubMed] [Google Scholar]
- 65.Lima JF, Oliveira LMS, Pereira NZ, et al. Polyfunctional natural killer cells with a low activation profile in response to Toll-like receptor 3 activation in HIV-1-exposed seronegative subjects. Sci Rep. 2017;7:524. doi: 10.1038/s41598-017-00637-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Flodström M, Maday A, Balakrishna D, et al. Target cell defense prevents the development of diabetes after viral infection. Nat Immunol. 2002;3:373–382. doi: 10.1038/ni771. [DOI] [PubMed] [Google Scholar]