

Abstract
The water channel aquaporin 2 (AQP2) is responsible for water reabsorption by kidney collecting duct cells. A substitution of amino acid leucine 137 to proline in AQP2 (AQP2-L137P) causes Nephrogenic Diabetes Insipidus (NDI). This study aimed to determine the cell biological consequences of this mutation on AQP2 function. Studies were performed in HEK293 and MDCK type I cells, transfected with wildtype (WT) AQP2 or an AQP2-L137P mutant. AQP2-L137P was predominantly detected as a high-mannose form of AQP2, whereas AQP2-WT was observed in both non-glycosylated and complex glycosylated forms. In contrast to AQP2-WT, the AQP2-L137P mutant did not accumulate on the apical plasma membrane following stimulation with forskolin. Ubiquitylation of AQP2-L137P was different from AQP2-WT, with predominance of non-distinct protein bands at various molecular weights. The AQP2-L137P mutant displayed reduced half-life compared to AQP2-WT. Treatment of cells with chloroquine increased abundance of AQP2-WT, but not AQP2-L137P. In contrast, treatment with MG132 increased abundance of AQP2-L137P but not AQP2-WT. Xenopus oocytes injected with AQP2-WT had increased osmotic water permeability when compared to AQP2-L137P, which correlated with lack of the mutant form in the plasma membrane. From the localization of the mutation and nature of the substitution it is likely that AQP2-L137P causes protein misfolding, which may be responsible for the observed functional defects. The data suggest that the L137P mutation results in altered AQP2 protein maturation, increased AQP2 degradation via the proteasomal pathway and limited plasma membrane expression. These combined mechanisms are likely responsible for the phenotype observed in this class of NDI patients.
Keywords: Aquaporin 2, Water channel, Nephrogenic diabetes insipidus, Mutation, Xenopus oocytes
Introduction
The ability of the kidney to concentrate urine is an essential mechanism in maintaining normal body water homeostasis. Minor increases in plasma osmolality cause release of the antidiuretic hormone arginine vasopressin (AVP) from the pituitary gland [1]. Upon binding of AVP to the AVP type II receptor (V2R) in the principal cells of the kidney collecting duct, a variety of signaling mechanisms are initiated resulting in accumulation of the water channel aquaporin 2 (AQP2) in the apical plasma membrane. This increases the water permeability of the collecting duct epithelium, facilitating water reabsorption to the hyperosmotic interstitium [2].
Functionally, AQP2 exists as tetramer, with each monomer having six transmembrane spanning membrane domains that fold to create a water pore. AQP2 function, in particular apical plasma membrane accumulation of AQP2 following AVP stimulation, is tightly controlled by various post-translational modifications in the intracellular C-terminus of AQP2. AVP increases phosphorylation of AQP2 at ser256, ser264, and ser269 (threonine in human), but decreases phosphorylation of ser261 [3]. Phosphorylation of ser256 and ser269 is important for the AVP-mediated accumulation of AQP2 in the apical plasma membrane [4, 5]. AQP2 is also modified by K63-linked ubiquitylation at lys270, which promotes AQP2 internalization from the plasma membrane [6], but also AQP2 degradation via lysosomal and proteasomal pathways [6, 7].
The disease congenital nephrogenic diabetes insipidus (NDI) is characterized by the decreased ability of the kidney to respond to AVP and concentrate urine. This renders the affected person prone to hypernatremic dehydration if access to water is limited [8]. Hereditary mutations in the AQP2 gene are responsible for approximately 10% of congenital NDI cases [8]. Currently, 65 different mutations of the AQP2 gene have been reported to cause NDI [9]. The majority of these mutations are of recessive inheritance and generally caused by amino-acid substitutions in the transmembrane domains or connecting loops of the monomer resulting in AQP2 misfolding and intracellular retention [10–12]. In rare cases NDI can also be autosomal dominant. These cases are caused by AQP2 mutations residing in the carboxyl terminus of AQP2 resulting in misrouting of the protein to the Golgi complex, late endosomes or lysosomes [11, 13, 14]. A structural basis for further classifying how these mutations may alter AQP2 pore function, tetrameric assembly, monomer folding, or phosphorylation has recently been suggested [15].
We have previously identified an autosomal recessive form of NDI resulting from substitution of AQP2 leu137 with proline (AQP2-L137P) [16]. This amino acid substitution in the fourth membrane spanning region of AQP2 [15] results in a severe form of NDI, with the male patient harboring this mutation suffering from severe fatigue, polydipsia, prominent hyposthenuria, and more than 20 L of daily voiding. The aim of this study was to determine the cell biological consequences of this mutation on AQP2 function as a basis for this patient’s NDI, and whether the uncovered biology fits with existing observations for mutations in autosomal recessive NDI.
Materials and methods
Generation of constructs, transfection and maintenance of cell lines
Human AQP2 was cloned from human kidney cDNA and subcloned into the pcDNA5/FRT vector (Invitrogen). The mutant form of hAQP2 (AQP2-L137P) was generated by replacing Leu137 with Proline at amino acid 137 using site-directed mutagenesis. All plasmid inserts were checked by DNA sequencing. All cells were kept in a 5% CO2 incubator at 37 °C. For transient transfections, HEK293 cells were grown and maintained in DMEM high glucose with 10% donor bovine serum. Cells were seeded in 12 well plates and when 80% confluent, transfection was performed using Lipofectamine 2000 (Invitrogen) in accordance with manufacturer’s instructions. Cells were used for experiments the following day. For stable transfections, the MDCK type I cell line, MDCK-FRT pcDNA6/TR6 [17], was maintained in complete DMEM (DMEMGlutaMAX) containing 10% donor bovine serum and 5 µg/ml blasticidin-HCL and transfected as previously described [17]. Stable AQP2-expressing cells were selected using 5 µg/ul blasticidin and 500 µg/ml Hygromycin B.
Cycloheximide studies and immunoprecipitation studies
Stably transfected MDCK cells were seeded in 24-well plastic plates and grown to confluence in complete DMEM. Cells were rinsed in prewarmed pure media and incubated in 50 µM cycloheximide in pure media for various periods before lysis in Laemmli sample buffer containing 15 mg/ml DTT. The lysates were sonicated and heated to 65 °C for 15 min prior to immunoblotting. To determine half-lives, band densities for AQP2 were analyzed using Image Studio Lite Version 4 and data fitted with nonlinear regression and one-phase exponential decay using GraphPad Prism 6 software. For immunoprecipitation studies, cells were incubated for 3 h with 100 µM of the lysosomal inhibitor chloroquine (Sigma-Aldrich) or 10 µM of the proteasomal inhibitor Mg132 (Sigma-Aldrich) prior to AQP2 immunoprecipitation as described previously [18].
De-glycosylation of proteins
Cells were grown to confluence in plastic wells, rinsed in PBS and harvested in dissection buffer (300 mM sucrose, 25 mM imidazole, 1 mM EDTA, pH 7.2) containing protease inhibitors Leupeptin (1 mg/ml) and Pefa-block (0.1 mg/ml) (Boehringer Mannheim). Samples were deglycosylated using PNGase F or endoglycosidase H (EndoH) (New England BioLabs) in accordance to manufacturer’s instructions. Samples were prepared for SDS-PAGE by addition of Laemmli sample buffer with DTT as above.
Surface biotinylation
MDCK cells were seeded in 6-well filter plates and grown in complete DMEM until confluent. Cells were washed in pure media and treated with 25 µM forskolin or DMSO for 30 min before surface biotinylation as previously described [19].
Immunoblotting
Standard procedures were used for SDS-PAGE and ECL-detection. Densitometry for assessment of signal intensity was performed using Image Studio Lite software (Qiagen).
Immunocytochemistry and confocal microscopy
Performed as described in detail earlier [5]. A Leica TCS SL confocal microscope and a HCX PL APO 40×/1.25–0.75 Oil C9 lens were used for imaging.
Antibodies
Primary antibodies used were goat anti-AQP2 (C17, Santa Cruz, sc-9882), anti-AQP2 H7661 [20], anti-AQP2 9398 [21], rabbit anti-Actin (Sigma-Aldrich, A2066), and rabbit anti-Proteasome 20S (abcam, ab3325). For immunoprecipitation of AQP2 anti-AQP2 9398 was used and the mouse anti-ubiquitin antibody was from Cell Signaling (P4D1, 3936). In oocyte experiments, a rabbit anti-AQP2 was used (ab78230, Abcam).
Oocyte volume measurement experiments and isolation of oocyte membranes
AQP2-WT and AQP2-L137P were subcloned to the pT7Ts vector, linearized with SalI endonuclease and capped RNAs synthesized using in vitro transcription with T7 RNA polymerase (mMessage mMachine; Ambion, Austin, TX). Stage V and stage VI Xenopus laevis oocytes (Ecocyte Bioscience, Germany) were stored at 18 °C in Barth’s solution (88 mM NaCl, 3 mM KCl, 0.82 mM MgSO4, 0.4 mM CaCl2, 0.33 mM Ca(NO3)2 and 5 mM HEPES, pH 7.6) supplemented with donor horse serum (5%), sodium pyruvate (2.5 mM), and gentamycin (25 μg/ml). Oocytes were injected with water, WT (1 ng) or mutated (10 ng) AQP2 cRNAs, and further incubated for 24–72-h in Barth’s solution at 18 °C prior to the related experiments. Water flux measurements were performed similar to as previously described [22–24]. Briefly, 24 h after the injection, oocyte vitelline membranes were removed and volume measurements were performed at 18 °C after bathing solution was switched from 200 to 2 mOsM. The oocyte volume change was calculated from the cross-sectional areas of the images taken at 0 and 60 s, using the formula (A/A0) = V/V0. (A: surface area observed after swelling, A0: initial surface area, V: volume after swelling, V0: initial volume). The WT-AQP2 change was set as 100% and the relative volume change of the mutant was determined relative to this [25]. Total and plasma membranes from injected oocytes were isolated according to previous studies [24, 26]. Total membranes were isolated from 5 oocytes, whereas plasma membranes were isolated from 40 oocytes. Membrane equivalents of 5 oocytes (total membranes) and 40 oocytes (plasma membranes) were assessed by immunoblotting and standard procedures.
Visualization of AQP2 structure
To visualize the potential impact of the L137P mutation, the amino acid substitution was examined using the Pymol software [27] and the structure of AQP2 (PDB ID is 4NEF) [28].
Statistics
Data are expressed as mean ± SEM. Statistical analysis was performed using GraphPad Prism7 software. Differences between groups were analyzed as appropriate using unpaired t test or one-way ANOVA and Tukey’s multiple comparison tests. Differences were considered significant at p values < 0.05. The type of analysis is highlighted in the figure legends.
Results
AQP2-L137P exists as a high-mannose modified protein
Immunoblots of HEK293 cells transiently transfected with AQP2-WT demonstrated that AQP2 was expressed as a non-glycosylated form of approximately 25 kDa, a weaker protein form of approximately 27 kDa, and a smear centered at 37 kDa representing complex glycosylated AQP2 (Fig. 1a). In contrast, AQP2-L137P was detected only as 25 and 27 kDa forms, suggesting different glycosylation to AQP2-WT. In MDCK cells stably expressing either AQP2-WT or AQP2-L137P, a similar pattern was observed, with a pronounced higher molecular weight form of AQP2-L137P at approximately 27 kDa being dominant (Fig. 1b). In the MDCK lines, immunocytochemistry of AQP2 and confocal microscopy demonstrated that under basal conditions AQP2-WT was distributed in a punctate and distinct pattern, whereas AQP2-L137P staining was more dispersed (Fig. 1c).
Fig. 1.
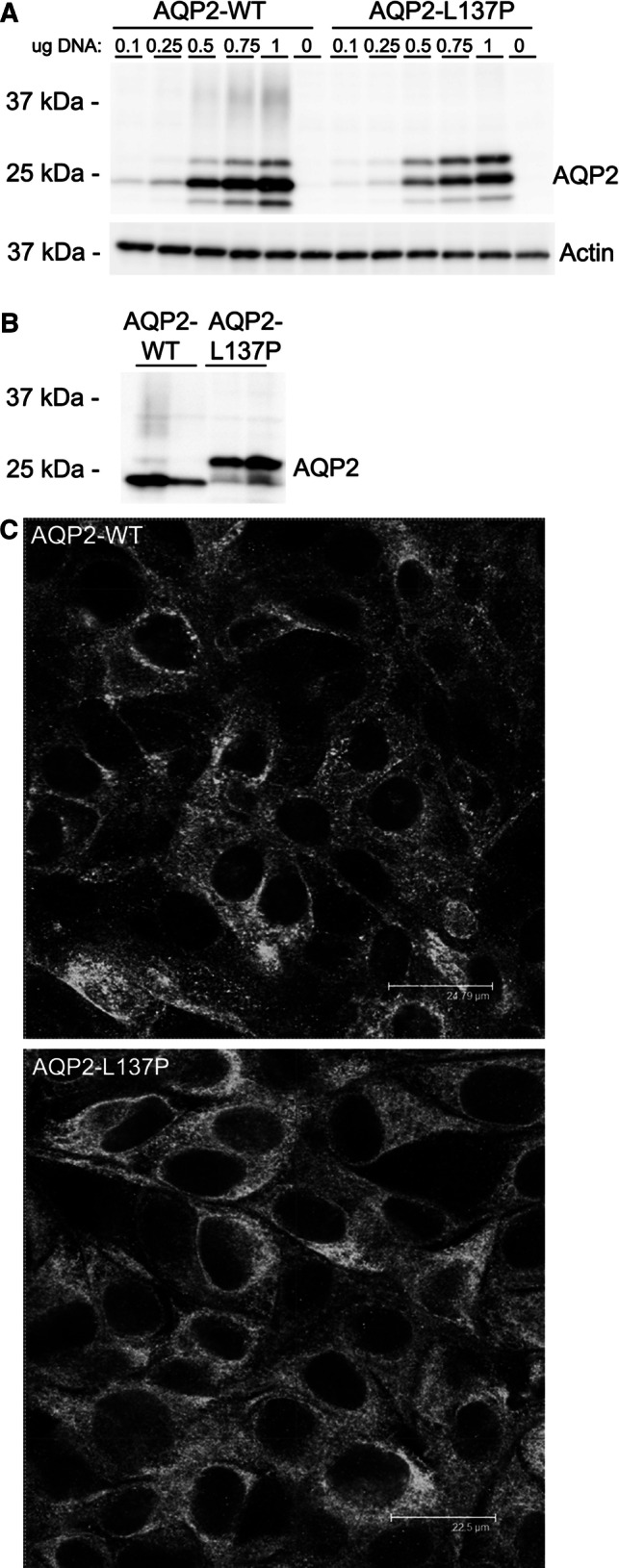
Compared to AQP2-WT, the AQP2-L137P mutant has a different molecular mass and subcellular distribution when expressed in kidney epithelial cell lines. a Immunoblotting of AQP2 on cell lysates isolated from HEK293 cells transiently transfected with AQP2-WT and AQP2-L137P constructs. Various amounts of plasmid DNA were used for transfection. AQP2-WT is mainly detected as a ~ 25 kDa band, but AQP2-L137P is distributed evenly between ~ 25 kDa and 27 kDa forms. Actin is used as a loading control. b Immunblotting of cell lysates from MDCK cells stably expressing AQP2-WT or AQP2-L137P revealed a similar tendency for the mutant protein to exist as a higher molecular weight form compared to WT-AQP2. Upon longer exposure a complex glycosylated smear could be observed for the AQP2-WT-expressing cells. Each lane represents a lysate from a different stable cell line. c Confocal images of AQP2 distribution in MDCK cells stably expressing AQP2-WT or AQP2-L137P
Mutant forms of AQP2 are often core-glycosylated (high-mannose), indicative of ER retention [29, 30]. To examine if this was similar for AQP2-L137P, lysates from either transiently transfected HEK293 cells or stable AQP2-expressing MDCK cells were deglycosylated. Treatment of HEK293 cell lysates with PNGase F (cleaving all types of asparginine-bound N-glycans) shifted the observed 27 kDa form of AQP2-L137P to 25 kDa, demonstrating that this is a glycosylated form of AQP2 (Fig. 2a). Treatment of similar lysates with endoglycosidase H (EndoH), cleaving high-mannose and some hybrid oligosaccharides of N-linked glycoproteins, resulted in a similar Mw shift, suggesting that AQP2-L137P exists in a high-mannose form (Fig. 2a). Similar results were obtained using PNGase F and MDCK cell lysates (Fig. 2b).
Fig. 2.
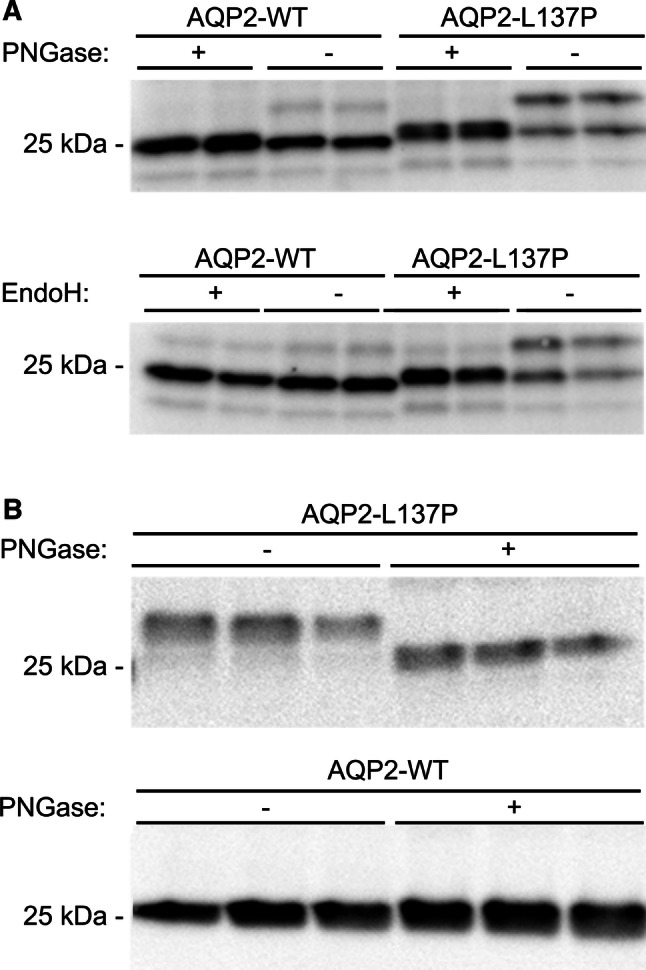
The AQP2-L137P mutant exists as an immature high-mannose form when expressed in kidney epithelial cells. a Cell lysates isolated from HEK293 cells transiently transfected with AQP2-WT and AQP2-L137P constructs were deglycosylated using PNGase F or EndoH and immunoblotted for AQP2. PNGase treatment shifted higher molecular weight bands of AQP2-WT and AQP2-L137P to a similar size, whereas EndoH only shifted the higher molecular weight bands of AQP2-L137P indicating it is a high-mannose form. b Immunblotting of PNGase-treated cell lysates from MDCK cells stably expressing AQP2-WT or AQP2-L137P revealed a similar size shift in the mutant cells. Each lane represents a sample obtained from a different batch of cells
In MDCK cells AQP2-L137P has a reduced half-life relative to AQP2-WT
High-mannose glycosylation can be indicative of the mutant protein being misfolded and trapped in the ER. As misfolded proteins are often degraded quicker and some mutant forms of AQP2 have decreased half-life [29], the half-life of AQP2-WT and AQP2-L137P in MDCK cells was examined using a cycloheximide chase experiment. Immunoblotting of a representative experiment is shown in Fig. 3a. Quantification of AQP2 levels from several experiments (Fig. 3b) was plotted using a one-phase exponential decay, which demonstrated that AQP2-WT had a significantly longer half-life (7.05 ± 1.12 h) compared to AQP2-L137P (2.08 ± 0.29 h) (p = 0.012).
Fig. 3.
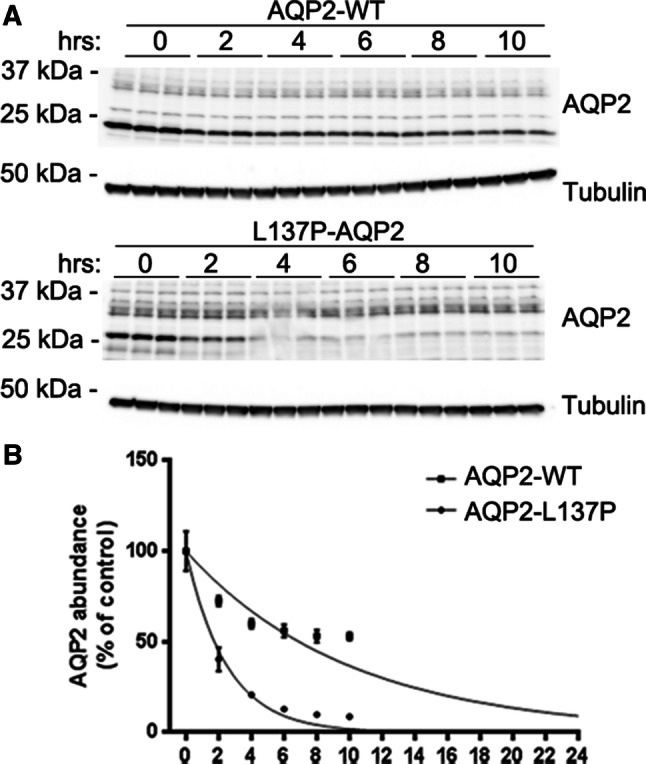
The AQP2-L137P mutant has a reduced protein half-life when expressed in MDCK cells. a Representative immunoblotting of cell lysates from MDCK cells stably expressing AQP2-WT or AQP2-L137P following cycloheximide treatment for various time points. Tubulin is used as a loading control. b Following quantification of band densities, data were fitted using a one-phase exponential decay to calculate half-life. A representative curve is shown. A total of four experiments were performed with three wells per condition for each experiment to calculate half-life. AQP2-WT half-life = 7.05 ± 1.12 h, AQP2-L137P half-life = 2.08 ± 0.29 h, p = 0.012
AQP2-L137P does not traffic to the apical plasma membrane following forskolin stimulation of MDCK cells
AQP2-expressing MDCK cells grown on semi-permeable supports were stimulated with forskolin, to increase intracellular cAMP levels, and surface proteins biotinylated from the apical side. In AQP2-WT cells, biotinylated levels of AQP2 relative to total cellular AQP2 levels increased following forskolin stimulation (Fig. 4). In contrast, no increase in surface levels of AQP2-L137P was observed under similar conditions. The small amount of AQP2-L137P observed at the apical plasma membrane in control conditions suggests that in the overexpression system some misfolded AQP2 is routed to the plasma membrane.
Fig. 4.

In MDCK cells the AQP2-L137P mutant does not traffic to the apical plasma membrane following forskolin stimulation. a AQP2-expressing MDCK cells grown on semi-permeable supports were stimulated with forskolin (for), to increase intracellular cAMP levels, and surface proteins biotinylated from the apical side. Representative immunoblotting from one of four experiments is shown. To assess the purity of the biotin isolation, biotin was omitted from some samples (− b) and immunoblotting with proteasome 20S (P20S) was used to determine if biotin passed the cell membrane. b Quantification of accumulated data from four independent biotinylation experiments. The biotinylated (surface) pool of AQP2 was normalized to total cellular AQP2 levels for each individual sample followed by normalization to first control within either AQP2-WT group or AQP2-L137P group. Each group was analysed by unpaired t test to determine significant difference between control and forskolin treatment. A significant effect of forskolin on surface AQP2 levels was observed in AQP2-WT-expressing cells that was not apparent in AQP2-L137P-expressing cells. *p < 0.05
Xenopus laevis oocytes expressing AQP2-L137P display reduced osmotic water permeability
In contrast to WT-AQP2-expressing oocytes, the relative volume change of oocytes expressing the AQP2-L137P mutant was similar to water-injected controls when oocytes were subjected to a hypo-osmotic challenge (Fig. 5a). To determine the basis of the reduced water permeability, the quantity of AQP2 in plasma membranes isolated from a similar batch of oocytes was assessed (Fig. 5b). In total membrane fractions, AQP2 was observed as a 29 kDa form in AQP2-WT-expressing oocytes, but similar to epithelial cell lines, as 29 kDa and 32 kDa forms in AQP2-L137P expressing oocytes. In WT-AQP2-expressing oocytes, AQP2 was detected in the plasma membrane fraction, whereas no plasma membrane associated AQP2 could be observed in the AQP2-L137P-expressing oocytes. Together, these data indicate that the reduced water permeability of AQP2-L137P-expressing oocytes could be due to misfolding of AQP2 and reduced membrane targeting.
Fig. 5.
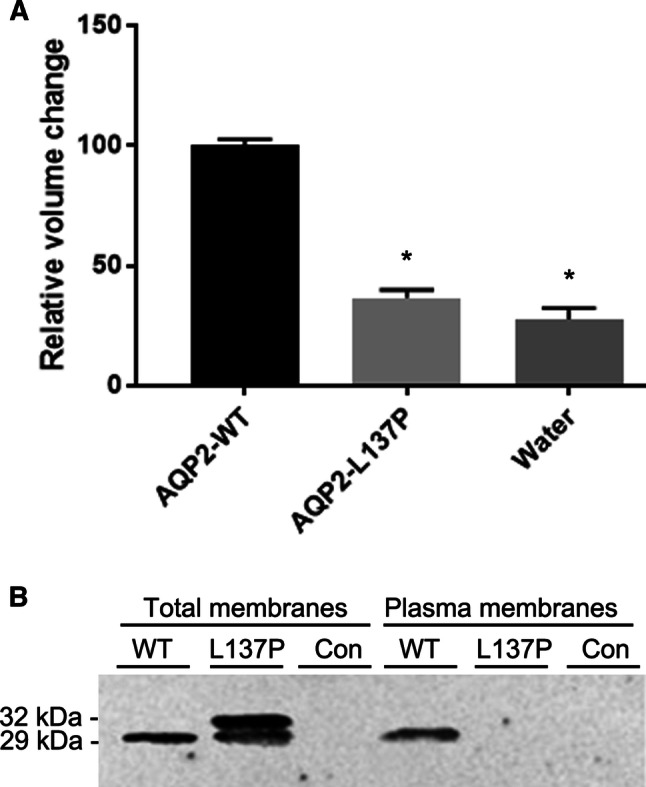
Xenopus laevis oocytes expressing AQP2-L137P display reduced osmotic water permeability relative to AQP2-WT. a Hypo-osmotic induced volume changes of oocytes expressing AQP2-L137P relative to AQP2-WT-injected oocytes. The water permeability of mutant expressing X. leavis oocytes was similar to water-injected oocytes and significantly lower than AQP2-WT-expressing oocytes. Data were analyzed using one-way ANOVA and a Turkey’s multiple comparison test. *p < 0.05. b Immunoblotting of total membranes and isolated plasma membranes from oocytes injected with either AQP2-L137P or AQP2-WT. AQP2 was not detected in the plasma membrane isolated from AQP2-L137P injected oocytes
Altered ubiquitylation of AQP2-L137P in MDCK cells
AQP2 is ubiquitylated at K270, which both mediates internalization from the plasma membrane [6, 18], but is also involved in both lysosomal and proteasomal degradation of AQP2 [7]. The decreased half-life and core glycosylated (high-mannose) form of AQP2-L137P indicative of ER retention suggests an altered rate of degradation, potentially via the ERAD system. To examine this, we examined the ubiquitylation pattern of AQP2 in the stable AQP2 expressing MDCK cells (Fig. 6a, b). The banding pattern of ubiquitylated WT-AQP2 was similar to that observed previously [18], with distinct protein moieties in the range of approximately 32–50 kDa. Although these distinct patterns were also observed in AQP2-L137P (considering the higher Mw of the core protein), the mutant also displayed a highly smeared ubiquitylated AQP2 pattern. After longer exposure (not shown), bands of smaller molecular weight were detected indicative of degraded AQP2. Attempts to detect specific K63- and K48-linked ubiquitylation of AQP2 using site-specific antibodies were not successful (not shown).
Fig. 6.
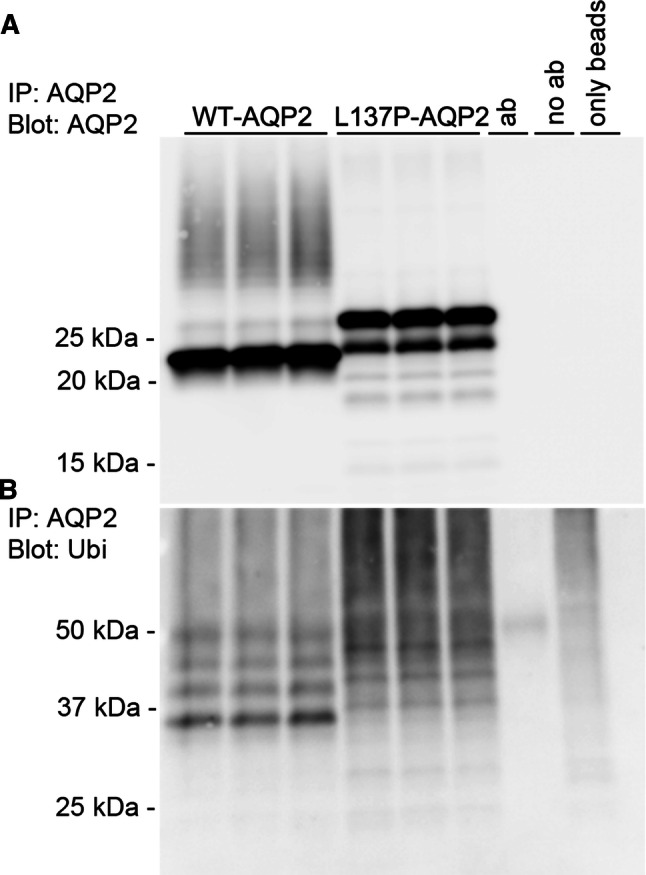
In MDCK cells the AQP2-L137P mutant has altered ubiquitylation. AQP2 was immunoprecipitated from cell lysates isolated from MDCK cells stably expressing AQP2-WT or AQP2-L137P. Each lane represents a sample from an individual well. Controls included performing the immunoprecipitation with antibody alone (ab), with lysate alone (no ab) or with no lysate or antibody added to the column (only beads). The experiment was repeated twice with similar results. a Immunoblotting of AQP2 demonstrates the higher molecular mass of the AQP2-L137P mutant. b Immunoblotting the same samples for ubiquitin highlights a highly smeared ubiquitylated AQP2 pattern for the mutant
AQP2-L137P is degraded predominantly via the proteasome
The previous data indicate that the mutated form of AQP2 was degraded via an alternative pathway to AQP2-WT. To confirm this, the AQP2-expressing MDCK cell lines were treated for 3 h with the proteasomal inhibitor Mg132 or the lysosomal inhibitor chloroquine and the levels of AQP2 assessed by immunoblotting. Chloroquine treatment resulted in increased abundance of only AQP2-WT, suggesting that normally AQP2 is predominantly degraded via the lysosomal pathway [6]. In contrast, blocking proteasomal degradation with Mg132 was associated with a build-up of ubiquitylated AQP2-L137P and ultimately increased AQP2-L137P levels (Fig. 7).
Fig. 7.
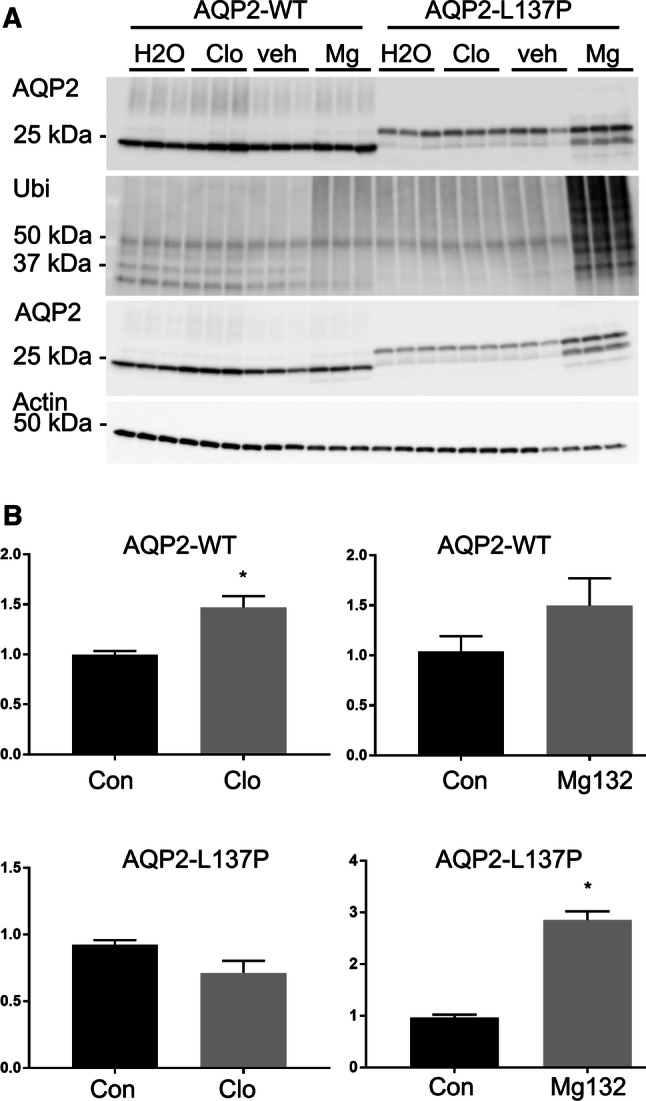
In MDCK cells the AQP2-L137P mutant is degraded predominantly by the proteasomal pathway. AQP2-expressing MDCK cells were treated for 3 h with the proteasomal inhibitor Mg132 (Mg) or the lysosomal inhibitor chloroquine (Clo). DMSO (veh) and water (H2O) represent solvent controls for the Mg132 and chloroquine, respectively. a Representative immunoblots of one of the experiments. The top two panels are AQP2-immunoprecipitated samples and the two lower panels are total cell lysates. Ubiquitylated levels of AQP2-L137P were greatly increased following Mg132 treatment. Actin is used as a loading control for the total lysates. b Quantification of accumulated data. The total abundance of AQP2-WT, but not AQP2-L137P, was significantly increased upon inhibition of the lysosomal degradation pathway using chloroquine. In contrast, inhibition of proteasomal degradation using Mg132 resulted in a significant increase in AQP2-L137P levels, but not AQP2-WT. Data were examined using an unpaired student t test *p < 0.05 relative to control group for each cell line and condition
Structural visualization of AQP2-L137P suggests destabilization of helical structure within the fourth membrane-spanning domain
Introduction of a proline residue in an alpha-helix will disturb its structure due to the restricted main chain conformation caused by the proline ring structure. As visualised in Fig. 8a, L137 is located in the middle of the fourth membrane spanning alpha-helix of the AQP2 monomer. As such, the proline substitution at L137 will cause a destabilization of the alpha-helix and may in turn disturb the folding of the entire membrane spanning domain of AQP2. Furthermore, the fourth membrane spanning helix is also involved in inter-subunit interactions. Thus, the L137P mutation could also limit formation of the functional AQP2 tetramer (Fig. 8b).
Fig. 8.

Structure and localization of the Leu137 localisation within the AQP2 monomer and tetramer (PDB = 4NEF) were examined using Pymol software. a AQP2-WT monomeric structure. The leucine residue at position 137 is shown in red and is localized within the middle of the fourth alpha-helix highlighted in green. b Tetramer structure of AQP2 shows that the fourth alpha-helix containing L137 is forming part of the interface between monomers
Discussion
The aim of this study was to determine the cell biological consequences of an L137P mutation in AQP2 that causes a severe form of NDI [16]. The majority of the studied NDI causing AQP2 mutations are suggested to result in misfolding of AQP2 monomers or defects in tetramer assembly [15]. Within the ER, AQP2 is likely assembled to a homotetramer and modified by addition of high-mannose sugar moieties. During passage through the Golgi, the high-mannose sugars are cleaved and complex glycosylation of AQP2 occurs at Asn123, possibly on one or two of the monomers [31]. In a number of AQP2 mutations resulting in recessive NDI, disruption of this natural maturation process is highlighted by AQP2 being detected as only non-glycosylated or high-mannose forms [12, 32]. The major observation from our study is that AQP2-L137P mainly exists as such as a high-mannose form, indicative of ER retention. In alignment with this, AQP2-L137P does not undergo regulated trafficking to the plasma membrane in MDCK cells or in Xenopus oocytes.
The proline substitution (L137P) likely causes changed conformation of the individual AQP2 monomers at the fourth membrane spanning domain (helix-4), which may result in AQP2 misfolding. In line with this, it has been hypothesized that another NDI-causing mutation, a L28P substitution within helix-1, interferes with alpha-helical structure and folding of the AQP2 monomer [15]. Additionally, it is plausible that proline substitution within helix-4 interferes with formation of tetramers due to its close proximity to residues thought to be important for tetramer formation, including F136, L139, and L143 [15]. Misfolded ER-retained proteins or unassembled subunits of multimeric proteins are usually ubiquitylated and subjected to ER-associated degradation (ERAD) via the proteasome [33, 34]. The altered ubiquitylation pattern of AQP2-L137P, in association with the increased abundance of the mutant following inhibition of the proteasome, suggests that AQP2-L137P is subjected to ERAD. In contrast, our results using chloroquine indicate that the majority of AQP2-WT is degraded via the lysosomal system. In line with these observations, we previously determined that AQP2-WT can be degraded by both lysosomal or proteasomal pathways and that the E3 ubiquitin ligase C-terminus of Hsc70 Interacting Protein (CHIP) plays a role in both processes [7]. Future studies examining if enhanced CHIP interaction occurs with the AQP2-L137P mutant to promote its ubiquitylation and facilitate proteasomal degradation may be informative.
Analysis of aquaporins using the Xenopus oocyte expression system is well established and the functional implications of many NDI-causing AQP2 mutations have been examined using this approach [12, 35]. Importantly, single-channel water permeability of some NDI-causing AQP2 mutants have been examined in this way [36]. Although in general the water permeability of these mutants is less than AQP2-WT, even small increases in osmotic water permeability of cell membranes would limit the water loss in NDI. Thus, rescuing these AQP2 mutants from intracellular retention and delivering them to the plasma membrane where they can act as functional water channels has the potential for future treatment of NDI [37]. Unfortunately, L137P-AQP2 did not reach the oocyte plasma membrane and thus its osmotic water permeability could not be assessed. Therefore, whether this mutant constitutes a functional channel and thus membrane rescue could be theoretically used as treatment strategy is unclear [10].
In conclusion, our data suggest that the novel AQP2 mutation L137P most likely results in channel misfolding, which underlies both the increase in proteasomal degradation and the intracellular retention of AQP2. Ultimately, this limits AQP2 trafficking to the plasma membrane and is the basis for the patients’ NDI.
Acknowledgments
Christian Westberg, Helle Høyer and Tina Drejer are thanked for technical assistance. Christian Brix Folsted Andersen and Kristian Stødkilde-Jørgensen are thanked for discussion and insights into Pymol Software. Nuhan Puralı, Berk Saglam and Bora Ergin are thanked for help with oocyte experiments.
Funding
This research was funded by The Scientific and Technological Research Council of Turkey (Project number: 115S499). Emel Saglar Ozer was supported by an EMBO Short Term Fellowship at the Department of Biomedicine, Aarhus University, Aarhus, Denmark (ASFT No: 583-2014). Further funding is provided by the Danish Medical Research Council, The Novo Nordisk Foundation and the Lundbeck Foundation.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Emel Saglar Ozer and Hanne B. Moeller contributed equally to this work.
Contributor Information
Emel Saglar Ozer, Email: esaglar@hacettepe.edu.tr.
Hanne B. Moeller, Email: HBMO@biomed.au.dk
References
- 1.Bourque CW. Central mechanisms of osmosensation and systemic osmoregulation. Nat Rev Neurosci. 2008;9(7):519–531. doi: 10.1038/nrn2400. [DOI] [PubMed] [Google Scholar]
- 2.Jung HJ, Kwon TH. Molecular mechanisms regulating aquaporin-2 in kidney collecting duct. Am J Physiol Renal Physiol. 2016;311(6):F1318–F1328. doi: 10.1152/ajprenal.00485.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffert JD, et al. Quantitative phosphoproteomics of vasopressin-sensitive renal cells: regulation of aquaporin-2 phosphorylation at two sites. Proc Natl Acad Sci USA. 2006;103(18):7159–7164. doi: 10.1073/pnas.0600895103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fushimi K, Sasaki S, Marumo F. Phosphorylation of serine 256 is required for cAMP-dependent regulatory exocytosis of the aquaporin-2 water channel. J Biol Chem. 1997;272(23):14800–14804. doi: 10.1074/jbc.272.23.14800. [DOI] [PubMed] [Google Scholar]
- 5.Hoffert JD, et al. Vasopressin-stimulated increase in phosphorylation at Ser269 potentiates plasma membrane retention of aquaporin-2. J Biol Chem. 2008;283(36):24617–24627. doi: 10.1074/jbc.M803074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamsteeg EJ, et al. Short-chain ubiquitination mediates the regulated endocytosis of the aquaporin-2 water channel. Proc Natl Acad Sci USA. 2006;103(48):18344–18349. doi: 10.1073/pnas.0604073103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu Q, et al. CHIP regulates aquaporin-2 quality control and body water homeostasis. J Am Soc Nephrol. 2018;29(3):936–948. doi: 10.1681/ASN.2017050526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bockenhauer D, Bichet DG. Pathophysiology, diagnosis and management of nephrogenic diabetes insipidus. Nat Rev Nephrol. 2015;11(10):576. doi: 10.1038/nrneph.2015.89. [DOI] [PubMed] [Google Scholar]
- 9.Milano S, et al. Hereditary nephrogenic diabetes insipidus: pathophysiology and possible treatment. An update. Int J Mol Sci. 2017;18(11):2385. doi: 10.3390/ijms18112385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moeller HB, Rittig S, Fenton RA. Nephrogenic diabetes insipidus: essential insights into the molecular background and potential therapies for treatment. Endocr Rev. 2013;34(2):278–301. doi: 10.1210/er.2012-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bichet DG, et al. Aquaporin-2: new mutations responsible for autosomal-recessive nephrogenic diabetes insipidus-update and epidemiology. Clin Kidney J. 2012;5(3):195–202. doi: 10.1093/ckj/sfs029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marr N, et al. Cell-biologic and functional analyses of five new aquaporin-2 missense mutations that cause recessive nephrogenic diabetes insipidus. J Am Soc Nephrol. 2002;13(9):2267–2277. doi: 10.1097/01.ASN.0000027355.41663.14. [DOI] [PubMed] [Google Scholar]
- 13.Mulders SM, et al. An aquaporin-2 water channel mutant which causes autosomal dominant nephrogenic diabetes insipidus is retained in the Golgi complex. J Clin Invest. 1998;102(1):57–66. doi: 10.1172/JCI2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marr N, et al. Heteroligomerization of an aquaporin-2 mutant with wild-type aquaporin-2 and their misrouting to late endosomes/lysosomes explains dominant nephrogenic diabetes insipidus. Hum Mol Genet. 2002;11(7):779–789. doi: 10.1093/hmg/11.7.779. [DOI] [PubMed] [Google Scholar]
- 15.Calvanese L, et al. Structural basis for mutations of human aquaporins associated to genetic diseases. Int J Mol Sci. 2018;19(6):1577. doi: 10.3390/ijms19061577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duzenli D, et al. Mutations in the AVPR2, AVP-NPII, and AQP2 genes in Turkish patients with diabetes insipidus. Endocrine. 2012;42(3):664–669. doi: 10.1007/s12020-012-9704-1. [DOI] [PubMed] [Google Scholar]
- 17.Rosenbaek LL, et al. Phosphorylation decreases ubiquitylation of the thiazide-sensitive cotransporter NCC and subsequent clathrin-mediated endocytosis. J Biol Chem. 2014;289(19):13347–13361. doi: 10.1074/jbc.M113.543710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moeller HB, et al. Phosphorylation and ubiquitylation are opposing processes that regulate endocytosis of the water channel aquaporin-2. J Cell Sci. 2014;127(Pt 14):3174–3183. doi: 10.1242/jcs.150680. [DOI] [PubMed] [Google Scholar]
- 19.Moeller HB, et al. Phosphorylation of aquaporin-2 regulates its endocytosis and protein–protein interactions. Proc Natl Acad Sci USA. 2010;107(1):424–429. doi: 10.1073/pnas.0910683107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poulsen SB, et al. Long-term vasopressin-V2-receptor stimulation induces regulation of aquaporin 4 protein in renal inner medulla and cortex of Brattleboro rats. Nephrol Dial Transplant. 2013;28(8):2058–2065. doi: 10.1093/ndt/gft088. [DOI] [PubMed] [Google Scholar]
- 21.Moeller HB, et al. Regulation of the water channel Aquaporin-2 via 14-3-3theta and -zeta. J Biol Chem. 2016;291(5):2469–2484. doi: 10.1074/jbc.M115.691121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeuthen T, et al. Water transport by the Na +/glucose cotransporter under isotonic conditions. Biol Cell. 1997;89(5–6):307–312. doi: 10.1016/s0248-4900(97)83383-7. [DOI] [PubMed] [Google Scholar]
- 23.Duquette PP, Bissonnette P, Lapointe JY. Local osmotic gradients drive the water flux associated with Na(+)/glucose cotransport. Proc Natl Acad Sci USA. 2001;98(7):3796–3801. doi: 10.1073/pnas.071245198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guyon C, et al. Characterization of D150E and G196D aquaporin-2 mutations responsible for nephrogenic diabetes insipidus: importance of a mild phenotype. Am J Physiol Renal Physiol. 2009;297(2):F489–F498. doi: 10.1152/ajprenal.90589.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El Tarazi A, et al. Functional recovery of AQP2 recessive mutations through hetero-oligomerization with wild-type counterpart. Sci Rep. 2016;6:33298. doi: 10.1038/srep33298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leduc-Nadeau A, et al. Elaboration of a novel technique for purification of plasma membranes from Xenopus laevis oocytes. Am J Physiol Cell Physiol. 2007;292(3):C1132–C1136. doi: 10.1152/ajpcell.00136.2006. [DOI] [PubMed] [Google Scholar]
- 27.Janson G, et al. PyMod 2.0: improvements in protein sequence-structure analysis and homology modeling within PyMOL. Bioinformatics. 2017;33(3):444–446. doi: 10.1093/bioinformatics/btw638. [DOI] [PubMed] [Google Scholar]
- 28.Frick A, et al. X-ray structure of human aquaporin 2 and its implications for nephrogenic diabetes insipidus and trafficking. Proc Natl Acad Sci USA. 2014;111(17):6305–6310. doi: 10.1073/pnas.1321406111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirano K, et al. The proteasome is involved in the degradation of different aquaporin-2 mutants causing nephrogenic diabetes insipidus. Am J Pathol. 2003;163(1):111–120. doi: 10.1016/S0002-9440(10)63635-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deen PM, et al. Water channels encoded by mutant aquaporin-2 genes in nephrogenic diabetes insipidus are impaired in their cellular routing. J Clin Invest. 1995;95(5):2291–2296. doi: 10.1172/JCI117920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baumgarten R, et al. Glycosylation is not essential for vasopressin-dependent routing of aquaporin-2 in transfected Madin–Darby canine kidney cells. J Am Soc Nephrol. 1998;9(9):1553–1559. doi: 10.1681/ASN.V991553. [DOI] [PubMed] [Google Scholar]
- 32.Mulders SM, et al. New mutations in the AQP2 gene in nephrogenic diabetes insipidus resulting in functional but misrouted water channels. J Am Soc Nephrol. 1997;8(2):242–248. doi: 10.1681/ASN.V82242. [DOI] [PubMed] [Google Scholar]
- 33.Preston GM, Brodsky JL. The evolving role of ubiquitin modification in endoplasmic reticulum-associated degradation. Biochem J. 2017;474(4):445–469. doi: 10.1042/BCJ20160582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feige MJ, Hendershot LM. Disulfide bonds in ER protein folding and homeostasis. Curr Opin Cell Biol. 2011;23(2):167–175. doi: 10.1016/j.ceb.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moeller HB, et al. Role of multiple phosphorylation sites in the COOH-terminal tail of aquaporin-2 for water transport: evidence against channel gating. Am J Physiol Renal Physiol. 2009;296(3):F649–F657. doi: 10.1152/ajprenal.90682.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tamarappoo BK, Verkman AS. Defective aquaporin-2 trafficking in nephrogenic diabetes insipidus and correction by chemical chaperones. J Clin Invest. 1998;101(10):2257–2267. doi: 10.1172/JCI2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang B, Zhao D, Verkman AS. Hsp90 inhibitor partially corrects nephrogenic diabetes insipidus in a conditional knock-in mouse model of aquaporin-2 mutation. FASEB J. 2009;23(2):503–512. doi: 10.1096/fj.08-118422. [DOI] [PMC free article] [PubMed] [Google Scholar]