

Abstract
The chemotherapeutic drug epirubicin increases the exosomal export of miR-503 in endothelial cells. To understand the mechanisms behind this process, we transfected endothelial cells with miR-503 carrying a biotin tag. Then, we pulled-down the proteins interacting with miR-503 and studied their role in microRNA exosomal export. A total of four different binding partners were identified by mass spectrometry and validated by western blotting and negative controls, among them ANXA2 and hnRNPA2B1. Using knock-down systems combined with pull-down analysis, we determined that epirubicin mediates the export of miR-503 by disrupting the interaction between hnRNPA2B1 and miR-503. Then, both ANXA2 and miR-503 are sorted into exosomes while hnRNPA2B1 is relocated into the nucleus. The combination of these processes culminates in the increased export of miR-503. These results suggest, for the first time, that RNA-binding proteins can negatively regulate the exosomal sorting of microRNAs.
Electronic supplementary material
The online version of this article (10.1007/s00018-019-03425-6) contains supplementary material, which is available to authorized users.
Keywords: Exosomes, EVs, MicroRNAs, Exosomal export, RNA-binding proteins
Introduction
Exosomes are small bilipidic vesicles ranging in size between 30 and 150 nm [1] produced in the endosomal compartment of virtually all cells. These vesicles mediate extracellular communication through the exchange of information via receptor signaling and/or by absorption, via either endocytic processes or pinocytosis/phagocytosis [2]. The content of exosomes is dictated by the parent cell type, as well as by the status and the environment of the cells at the time of production [3]. Even though exosomes may carry different cargo (coding and non-coding RNA [4], DNA [5], lipids [6] and proteins [7]), multiple studies have shown that the loading of specific RNAs (in disease) can promote phenotypic changes in the recipient cells [8].
MicroRNAs are small non-coding RNAs involved in the negative regulation of gene expression. Although changes in the exosomal RNA profiles often reflect changes in the parental cells, cellular stimulus can also modify the encapsulation of specific microRNAs. An example of this mechanism is the anti-tumoral microRNA miR-503. Our group previously reported that breast cancer patients receiving the neoadjuvant epirubicin had increased circulating levels of miR-503. We then demonstrated that this chemotherapeutic drug could promote, in endothelial cells, the production of exosomes with anti-tumoral properties loaded with miR-503 [9]. In conclusion, we highlighted an exosome-dependent transfer of microRNAs from endothelial to tumor cells that contributes to the anti-tumoral effect of epirubicin. Our data suggested the presence of an underlying mechanism leading to the selective export of miR-503 in endothelial cells. To date, several studies have focused on motif-based RNA-binding protein (RBP) recognition to explain exosomal microRNA export, but the mechanisms behind this process still remain mostly unknown. In this study, combining microRNA pull-down techniques along with proteomic assays and knock-down studies, we discovered that epirubicin promotes the exosomal export of miR-503 by destabilizing the interaction between this microRNA and hnRNPA2B1. Process that culminates in the increased exosomal encapsulation of miR-503.
Methods
Cells and culture conditions
Human umbilical vein endothelial cells were isolated as previously described by [10]). HUVECs were amplified in flasks coated with gelatin (0.2%) in endothelial cell growth media-2 (EGM2) (Lonza, Germany) lacking heparin and supplemented with 5% donor bovine serum (DBS) at 37 °C and 5% CO2. Human microvascular endothelial cells (HMVECs) (Lonza, Germany) were cultured in EGM2 lacking heparin and supplemented with 5% fetal bovine serum (FBS) at 37 °C and 5% CO2. All exosomes experiments were performed in exosome-depleted media prepared using serum that had been centrifuged at 110,000g for 16 h at 4 °C to reduce exosomal contamination (Beckman Coulter Optima L-90K, SW32 Rotor). Cells were stored in liquid nitrogen for further use and used for experimental procedures from passage 6 (P6) to P10. The concentration of epirubicin used in all experiments was 1 µg/ml in complete EGM2. The levels used for this test were decided after calculating the circulating concentration of this drug in patients [9].
Exosome purification and characterization
HUVECs and HMVECs were cultured in heparin-free EGM2 supplemented with full 5% DBS (v/v) or exosome-depleted DBS. Cells were incubated at 37 °C and 5% CO2 up to P10 at a seeding density of 1.8 × 106 cells per 175 cm2 flask. 3 days later, the supernatant was recovered, and exosomes were purified by sequential ultracentrifugation. The media were first centrifuged at 2000g for 20 min at 4 °C to remove unattached cells, followed by a second round of centrifugation at 12,000g (45 min at 4 °C) to remove cell debris and larger vesicles. The supernatant was then collected and passed through a 0.2 μm filter and ultracentrifuged at 110,000g for 2 h at 4 °C to pellet the exosomes (Beckman Coulter Opitima L-90K, SW32 rotor). The pellet was washed with PBS to remove any possible coprecipitated protein complexes, and with a final round of centrifugation at 110,000g for 2 h at 4 °C, the pellet was recovered and stored in PBS at − 80 °C. Exosomes were characterized by dynamic light scattering (DLS) for vesicle size and by western blotting for protein composition. In brief, the protein characterization of cells and exosomes was performed following the MISEV 2018 guidelines [11] using the following antibodies: CD63 (#10628D, Invitrogen), CD9 (#sc20048, Santa Cruz), CD81 (#10630D, Invitrogen), SYN (#ab133267, Abcam), and cytochrome c (#556433, BD Pharmingen) for immune detection.
Electron microscopy of whole-mounted immuno-labelled exosomes
Isolated exosomes were placed on Formvar/carbon coated nickel grids for 1 h, washed three times with PBS and fixed with 2% paraformaldehyde for 10 min. After three washes, grids were then incubated for 2 h with the following antibodies: anti-CD63 (#H5C6, BD Phamingen) or anti-CD105 (#M3527, DAKO). Exosomes were then washed five times and incubated with a 10 nm-gold labeled secondary antibody. They were washed five more times and post-fixed with 2.5% glutaraldehyde for 10 min. Samples were contrasted using 2.5% uranyl acetate for 10 min followed by four washes and an incubation of 10 min in lead citrate. Grids were finally washed four times in deionized water and examined with a JEOL JEM-1400 transmission electron microscope at 80 kV.
Design of the synthetic microRNA
The microRNA mimics miR-503-biotin and cel-miR-67-biotin are double-stranded RNAs designed following the publications by Orom and Lund [12] and Betancur et al. [13]. In summary, the mature microRNA strand was modified by the addition of a biotin to the 3′ –OH and a phosphorylation at the 5′ end. The carrier strand (miR-503-reverse or cel-miR-67 reverse) was the complementary RNA sequence, which was also phosphorylated at the 5′ end and carried a two bases 3′ overhang with mutations near the 3′ end to thermodynamically destabilize the strand and induce faster degradation. Supplementary Fig S1 shows the sequence and modifications induced in the microRNA duplexes. Oligonucleotides were purchased from Eurogentec.
Preparation of protein lysates and western blotting
HUVECs were washed with PBS 1 × and RIPA buffer was added (50 mM Tris pH 7.5, 150 mM NaCl, 10 mM CaCl2, 0.5% NP40, 0.25% sodium deoxycholate and 0.1% SDS) at 75 µl/106 cells. The plates/flasks were then scratched and the cellular lysate was centrifuged at 10,000g and 4 °C for 15 min. The cleared supernatant was then recovered and the pellet (cellular debris) discarded. The quantification of cellular lysates was performed using the BCA Protein Assay Kit (Pierce) following the manufacturer’s instructions. For exosomes, the lysis of the samples was performed using exosome lysis buffer (10% Triton, 1% SDS) and the quantification of exosomal protein was performed with BCA Protein Assay Kit (Pierce) incubating the samples at 60 °C for 60 min. Prior gel loading, samples were denatured by boiling at 95 °C for 7 min in 1 × loading buffer (40% Glycerol, 240 mM Tris/HCl pH 6.8, 8% SDS, 0.025% Bromophenol Blue) without 5% β-mercaptoethanol for the detection of tetraspanins (CD9, CD63, and CD81) and with the reducing agent for all the other proteins. Equal amounts of protein lysates (10 µg) were electrophoresed on a 12% SDS–polyacrylamide gels and transferred to a polyvinylidene fluoride membrane using a wet transfer system. The blots were then blocked with either 5% BSA (in the case of hnRNPA2B1) or commercial powdered milk at 8% for 1 h. Blots were then incubated overnight at 4 °C with the primary antibody [ANXA2 (#8235, Cell Signaling), FN1 (#610077, BD Phamingen), HNRNPA2B1 (#Ab6102, Abcam), TSP1 (#MA5-13398, Thermo Fisher), VIM (#MO72529, DAKO), VINC (#Ab129002, Abcam), and SYN (#Ab133267, Abcam)]. After three washes of 10 min with TBS/0.1% Tween-20 (TBST), the membranes were incubated for 1 h at room temperature with an HRP-conjugated secondary antibody (anti-rabbit (#7074S, Cell Signaling), anti-mouse (#7076S, Cell Signaling) and washed twice with TBST and once with TBS. The blots were then incubated with enhanced chemiluminescence (ECL) substrate (Pierce Biotechnology) and exposed to films. All films were scanned and the intensity of the bands was quantified using ImageJ.
MiR-503 and cel-miR-67 pull-down
To detect the proteins associated with miR-503, HUVECs or HMVECs were transfected with 10 nM miR-503-biotin or cel-miR-67-biotin duplexes at the time of seeding. For this purpose, 7.5 µl of DharmaFECT 4 (Dharmacon) were mixed with 2242.5 µl of serum-free EBM2 (Lonza) and incubated at room temperature for 10 min. After this, a second solution containing 2235 µl of EBM2 and 15 µl of 10 µM miR-503-biotin/reverse was prepared. Both solutions were mixed and incubated at room temperature for 20 more minutes. In parallel, 1.8 × 106 cells were seeded in 10.5 ml of full EGM2 (supplemented with 5% DBS) in a 145 cm2 round dish previously coated with gelatine 0.2%. The transfectant-miRNA solution was then added to the cells to achieve a final microRNA concentration of 10 nM in 15 ml. The next day, the media were replaced with 20 ml of fully supplemented EGM2 and 2 days later we proceeded to pull down the microRNA and its putative partners. Forty plates were used for the identification of the miR-503-biotin partners by mass spectrometry (see supplementary methods for detailed protocol), and ten plates were used for the validation of the results.
The protocol used in the pull down followed the methodology described by Rambout X [14] with some modifications. Two crosslinking steps were performed. First, after washing twice with ice-cold PBS, 5 ml of a 1 mM 3,3-dithiobis(sulfosuccinimidylpropionate) (DTSSP) solution was added to the plates and incubated for 2 h at 4 °C to induce the crosslinking between proteins. The supernatant was removed and the cells were incubated with 20 mM Tris–HCl pH 7.6 for 30 min to stop the crosslinking reaction. Then, the plates were washed twice with ice-cold PBS. In a second crosslinking step, the formation of covalent bonds between proteins and RNA was induced via UV irradiation. For that purpose, the cells were set on an ice tray and UV-irradiated (0.4 J/cm2 of 365-nm UV light with a Stratalinker 2400). Then, 2 ml of pull-down lysis buffer 1 (20 mM Tris–HCl pH 7.5, 0.5% SDS, 1 mM EDTA), 200 U/ml RNAse inhibitor (Roche, Sigma-Aldrich), 1 × Halt Protease and phosphatase inhibitor cocktail (Thermo Scientific) and 1 pill/10 ml of cOmplete Protease Inhibitor (Roche, Sigma-Aldrich) was added per plate and the cells scratched. The lysate was recovered and passed through a needle (22G) 2–3 times to reduce viscosity. At this point, an aliquot of the lysate (1%) was recovered and stored at − 20 °C as input. Then, 25 µl (per plate) of magnetic streptavidin-coated beads (New England BioLabs) was added to the lysate and the mix was incubated in soft agitation for 2 h at 4 °C. The beads were then separated using a magnet for 30 min and washed with pull-down washing buffer 1 (20 mM Tris–HCl pH 7.5, 500 mM LiCl, 0.1% SDS, 1 mM EDTA). The beads were then washed again with washing buffer 2 (20 mM Tris–HCl pH 7.5, 500 mM LiCl, 1 mM EDTA), separated for 20 min and washed one last time with washing buffer 3 (20 mM Tris–HCl pH 7.5, 200 mM LiCl, 1 mM EDTA) followed by magnetic separation for 15 min. After all the washes, the beads were resuspended again in denaturing 4 × loading buffer and boiled for 10 min at 97 °C. Next, the different pulled-down components were separated by electrophoresis on a 12% SDS–polyacrylamide gel until the loading buffer left the gel. The protocol used for the identification of the putative partners of miR-503 by mass spectrometry can be found in the Supplementary Methods.
Immunoprecipitation assays and qPCR
To detect the affinity of the identified proteins with miR-503, HUVECs or HMVECs were transfected with a synthetic miR-503 (10 nM) (identical to the native mature miR-503) following the same protocol used for miR-503-biotin transfection. Twenty-four hours after removing the media and 48 h after transfection, cells were washed twice with ice-cold PBS and incubated with 1 mM dithiobis (succinimidyl propionate) DSP (ThermoFisher) in soft agitation for 2 h at 4 °C. Then, the reaction was stopped by removing the supernatant, adding 20 mM Tris HCl and incubating at room temperature for additional 30 min. After washing twice with ice-cold PBS, the plates were set on an ice tray and irradiated with UV light to induce RNA–protein crosslinking (0.4 J/cm2 of 365-nm UV light with a Stratalinker 2400). Then, 25 µl of RIPA buffer was added to the plates and these were scratched using a scraper. The lysate was then recovered into an Eppendorf tube and incubated on ice for 15 min. Then, the samples were centrifuged, and the pellet (cellular debris) discarded. A preclearing of the samples (to reduce non-specific binding of peptides to the beads), was performed by adding 50 µl of protein A agarose beads (per plate) (Sigma-Aldrich, 11719408001) and incubating the sample for 2 h at 4 °C with rotation. After that, the samples were centrifuged at 2800 rpm and 4 °C for 3 min to pellet the beads. The supernatant was recovered and 5 µl of antibody was added per plate used. As a negative control, immunoglobulin G (IgG) was used instead of the specific antibody during immunoprecipitation. The samples were left to incubate and rotate overnight at 4 °C. The following day, 50 µl of protein A agarose beads (per plate) was added, and the samples were incubated again for 2 h more at 4 °C in rotation. Then, the beads were recovered by centrifugation (3 min at 2800g and 4 °C) and washed three times with IP Lysis Buffer (50 nM Hepes pH 7.5; 150 nM NaCl; 1% Triton X-100) (lacking protease inhibitors) for 1 h. After the final wash, the beads were resuspended in 100 µl of PBS and RNA extraction was performed using miRNEasy kit (Qiagen) following the manufacturer instructions. The antibodies used for immunoprecipitation were ANXA2 (#610069, BD Pharmingen), FN1 (#610077, BD Pharmingen), HNRNPA2B1 (#Ab6102, Abcam), TSP1 (#MA5-13398, Thermo Fisher), VIM (#MO72529, DAKO), and IgG control (#31903, Invitrogen). To identify unspecific interactions, HUVECs were transfected with 10 nM of cel-miR-67 and the same protocol was followed.
Small interfering RNA assays
Small interfering RNAs (siRNAs) were used to knock down the levels of expression of anxa2, hnrnpA2B1, tbsh1, and vim. A scramble siRNA (a siRNA without targets in the human genome) was used as a control in all experiments at the same concentration used for the knock down of the other proteins. Oligonucleotides were purchased from Eurogentec and sequences are shown in the supplementary material. The transfection protocol was as follows per mL of final volume to transfect: 700 µl of a solution of EGM2 and cells was combined with 300 µl of a solution containing the transfectant and siRNA in EBM2. To prepare this mixture, 0.5 µl of DharmaFECT 4 (Dharmacon) was mixed with 149.5 µl of serum-free EBM2 (for each milliliter) and incubated for 10 min at room temperature. Posteriorly, a solution containing 149 µl of serum-free EBM2 and 1 µl of siRNA at a concentration of 20 µM was mixed with the DharmaFECT dilution and incubated again at room temperature for another 20 min. Finally, the cells (700 µl) were seeded in a plate or flask previously coated with gelatine, and the siRNA-DharmaFECT solution was added. The following day, the supernatant was removed, and full media (EGM2) was added to the cells. To validate the efficiency of protein knock-down, 100,000 HUVECs were seeded in a 6-well plate and transfected with the siRNAs. The plates/flasks were then scratched, and the cells were either lysed with RIPA buffer for further protein analysis or mixed with TRIzol for RNA extraction. The time points used for the validation were 24, 48, and 72 h for RNA and 48, 72, and 96 h for proteins. The rationale behind the 24 h delay (for protein level assessment) was to allow the normal levels of the protein in the cell to naturally decrease and thus more reliably observe the effect of the gene knock-down at the protein level. For the production of exosomes after protein knock-down and treatment with epirubicin, 1.8 × 106 cells [for the non-treated conditions (NT)] and 5.5 × 106 cells (for cells treated with epirubicin) were seeded in each T175 flask and transfected to a final volume of 18 ml following the protocol mentioned above. Then, the next day, the supernatant was fully recovered and replaced with 20 ml of full EGM2 supplemented (or not) with 1 µg/ml epirubicin. The next day, the entire supernatant was discarded, and 20 ml of exosome-depleted (exofree) EGM2 was added. Four T175 flasks were used per condition. The cells were incubated in these conditions for 72 h, after which the exosomes were purified. The levels of miR-503 in the cells after the knock-down of the genes were assessed as 24 after the addition of the exosome-depleted media.
Quantitative analysis of gene and microRNA expression by RT-qPCR
The purification of RNA from cellular sources was performed using the miRNeasy kit (Qiagen) following the manufacturer’s instructions. The RNA was resuspended in RNAse-free water and quantified by Nanodrop (ThermoFisher). The same kit was used to extract RNA from exosomes although a modification step was added: five volumes of TRIzol were added to the exosomes and the volumes of chloroform and ethanol were adjusted accordingly. RNA was suspended in RNAse-free water and quantified with the Quant-it Ribogreen RNA assay kit (R11490, ThermoFisher) on black 96-well plates. The emitted fluorescence was assessed using a spectrophotometer (2030 Multilabel Reader VICTORTM X3 from Perkin Elmer) at 592 nm (emission range of fluorescein).
Two different approaches were used to measure the levels of different genes depending on their type. The variation in coding genes (gapdh, anxa2, hnRNPA2B1, thbs1, vim) were assessed by qRT-PCR. For that purpose, the synthesis of cDNA was performed starting from 500 ng of RNA using the iScript Kit (BioRad) following the manufacturer’s instructions. The levels of mRNA in the samples were then assessed by qPCR using a SYBR system (Takyon, Eurogentec) and detected with a thermocycler (Applied Biosystems 7900HT, Applied Biosystems). gapdh was used as housekeeping gene to normalized variations in the levels of input RNA. For each experiment performed, two negative controls were used: a sample lacking retrotranscriptase enzyme (RT–) and one lacking primers. Only experiments with undetected gene levels for these two controls were considered. The sequences used for the design of the probes are in the Supplementary Methods.
For the detection of cellular non-coding RNAs (RNU44, RNU48, miR-503, let-7d, miR-16 miR-210, and cel-miR-67), 3.33 ng of RNA were retrotranscribed using the TaqMan Reverse Transcription Kit (Applied Biosystem) with the TaqMan microRNA Assay (Applied Biosystems). The detection of the levels of these genes was performed using 2.2 µl of the cDNA product, 1.7 µl of TaqMan microRNA assay reagent (Applied Biosystem) and a dilution of TaqMan Universal Master Mix (Applied Biosystem) (16.5 µl of enzyme diluted with 12.6 µl of RNAse free water). The mix was prepared prior to plating and 10 µl of solution was added per well. The average levels of RNU44 and RNU48 were used to normalize RNA input in the cells. In the case of exosomal RNA detection, 1.67 µl of RNA extract were used per reaction and the normalization was performed against the average levels of let-7d and miR-16. Since this system of detection does not amplify DNA products, only the negative control lacking primers was prepared per experiment. In all cases, the level of the genes was assessed using the ΔΔCt method [15].
Immunofluorescence assays
HUVECs were seeded in coverslips at a density of 2.5 × 104 cells/well and treated (or not) with epirubicin, from 24 to 72 h. Cells were then washed three times with PBS, fixed for 10 minutes with 4% paraformaldehyde (PFA) and then permeabilized with 70% ethanol overnight. After permeabilization, the cells were washed again three times with PBS and blocked with 5% BSA for 30 min at room temperature. Then, the cells were incubated with anti-ANXA2 (1:100; #8235, Cell Signaling), anti-hnRNPA2B1 (1:100; #Ab6102, Abcam), anti-VIM (1:100; #MO72529, DAKO), and anti-TSP1 (1:100; # MA5-13398, Thermo Fisher) for 1 h at room temperature. After incubation, cells were washed three times with PBS, and incubated an additional 1 h with Alexa488-conjugated goat anti-mouse IgG (1:300; Thermo Fisher Scientific) and DAPI (1:500; Thermo Fisher Scientific). Then, cells were washed three times with PBS and once with distilled water. Finally, the coverslips were mounted on slides using Prolong (Thermo Fisher Scientific) and visualized under a confocal microscope (Leica SP5, Leica Microsystems, Wetzlar, Germany). The fluorescence intensity of hnRNPA2B1 staining in HUVEC cells was assessed using the plugin “Intensity Ratio Nuclei-Cytoplasm” on ImageJ-FIJI.
Subcellular fractionation
Endothelial cells were treated (or not) with epirubicin (1 µg/ml; Sigma-Aldrich, Saint-Louis, MO, USA) for 24 h. At 24, 48, and 72 h after starting treatment, cells were trypsinized and pelleted by centrifugation at 1000g for 5 min at 4 °C. Later, the cells were washed twice with cold PBS and permeabilized with 0.5% PBS-Tween for 10 min. Then, the cells were resuspended in buffer A (1 M Hepes pH 7.9; 1 M KCl; 1 M MgCl2; 1 M sucrose; 10% glycerol; 1 M, dithiothreitol 0.01‰; Triton X-100 protease and phosphatase inhibitors (Sigma-Aldrich, Saint-Louis, MO, USA); diethylpyrocarbonate-(DEPC) water) and incubated on ice for 5 min. The lysates were then centrifuged at 1300g for 4 min at 4 °C. The supernatant (S1) was recovered and centrifuged at 20,000g for 15 min at 4 °C. The cytosolic fraction (second supernatant, S2) was recovered and stored at − 20 °C. The pellet (P1) was washed with buffer A twice, and resuspended in buffer B (EDTA 100 mM pH 8; EGTA 50 mM pH 8; Dithiothréitol 1 M; protease and phosphatase inhibitors (Sigma-Aldrich, Saint-Louis, MO, USA); DEPC-water). The solution was then incubated on ice for 30 min followed by centrifugation at 1700g for 4 min (4 °C). The nuclear fraction present in the supernatant (S3), was recovered and stored at − 20 °C. The different fractions were characterized by western blotting using the nuclear marker Histone H3 (#ab1791, Abcam) and the cytoplasmic marker GAPDH (#ab8245; Abcam). The levels of hnRNPA2B1 were detected by western blotting in equimolar quantities of protein (10 µg) from the cytoplasmic and nuclear fractions and quantified using ImageJ.
Statistical analysis
All experiments were performed a minimum of three times unless otherwise stated. The plotted values represent the mean of the biological replicates ± the standard error of the mean (SEM) or the standard deviation (SD); the technique used is specified for each case in the figure legend. The statistical significance of the results was assessed using an unpaired t test.
Results
Characterization of exosomes and identification of miR-503 binding partners
Extracellular vesicles (EVs) were purified using ultracentrifugation and characterized by western blotting detecting the following exosomal (CD63, CD9, CD81, and syntenin) and cellular markers (mitochondrial cytochrome C) (Fig. 1a). Dynamic light scattering revealed vesicles with an average size of 100 nm (Fig. 1b), suggesting an enrichment of exosomes in our preparation. Electronic microscopy analysis confirms the size as well as the presence of the tetraspanin CD63 and the endothelial marker CD105 (Fig. 1c).
Fig. 1.

Exosome characterization and identification of miR-503 binding proteins: a exosome characterization of HUVEC lysates and exosomes (10 µg) against the EV markers: CD9, CD63, CD81, syntenin (SYN), and the cellular marker cytochrome C (Cyt C). b Dynamic light scattering analysis of exosomal preparations. c Electron microscopy images of HUVEC exosomes labeled with anti-CD63 and anti-CD105, scale bars = 100 nm. d Cells were treated with epirubicin for 24 h. Cell lysates were prepared 24 h after treatment and exosomes were collected 72 h after removing the chemotherapeutic drug. Cellular and exosomal levels of miR-503 were evaluated by qPCR. Data show mean ± SEM (n = 3). **p < 0.01 vs. respective control. e Schematic representation of the protocol used to identify the MEC proteins. HUVECs (30 × 106 cells) were transfected with miR-503-biotin (10 nM). The following day, the cells were crosslinked with DTSSP and UV. HUVECs lysates were incubated with streptavidin beads. Both input (IN) (cellular lysate) and pull-down fractions (PD) were separated by SDS-PAGE. Isolated proteins were identified by mass spectrometry and f validated by western blotting against pull-down (PD) and input (IN) (1% of cell lysate) fractions using vinculin as loading control. Cel-miR-67 was used as a negative control for the pull-down
As previously shown [9], epirubicin increases the export of miR-503 into exosomes in human umbilical vein endothelial cells (HUVECs) (Fig. 1d). To identify the binding partners of miR-503 we transfected HUVECs with a synthetic miR-503-biotin. We then determined if the biotinylation of miR-503 interfered with the mechanism of export triggered by epirubicin by assessing the levels of miR-503 in exosomes after treatment (Supplementary Fig. S2a). We found little to no effect linked to the presence of a biotin tag in the selective export of miR-503 in response to epirubicin, ranging around the twofold increase in native conditions (Fig. 1d) and after miR-503 biotin transfection (Fig S2a).
To identify the miR-503 binding partners, we transfected HUVECs with the biotin-microRNA construct and followed a double crosslinking strategy (Fig. 1e): first, by inducing the formation of RNA–protein bonds and, second, by stabilizing protein–protein interactions. Following this methodology, we pulled down the microRNA and identified its binding protein partners by mass spectrometry. The efficiency of the transfection of miR-503-Biotin was assessed by qPCR on cellular lysates (Fig S2b).
A total of nine different proteins were identified by mass spectrometry: propionyl-CoA carboxylase alpha subunit (PCCA), pyruvate carboxylase (PC), heparan sulfate proteoglycan 2 (Perlecan), fibronectin 1 (FN1), thrombospondin-1 (TSP1), β-actin (ACTB), vimentin (VIM), annexin A2 (ANXA2), and heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNPA2B1). A brief description of their main roles in the cell is summarized in Supplementary Table 1. Both PCCA and PC are binders of endogenous biotin, hence their identification [16]; they were not considered for further analysis. Perlecan, TSP1 and FN1 are part of the extracellular matrix [17–19], while ACTB and VIM are main components of the cytoskeleton and regulate motility, cellular stability, and cellular division [20, 21]. ANXA2, via its involvement in cellular transduction, can modulate cellular growth and various signaling processes. In addition, it was also recently discovered that ANXA2 can bind to mRNAs [22–24] and mediate the exosomal export of microRNAs. The most well-known function of hnRNPA2B1 is shuttling mRNA from the nucleus to the cytoplasm and regulating post-transcriptional gene expression [25]. Interestingly, hnRNPA2B1 can also bind microRNAs with a specific sequence and promote their export into exosomes [26].
The microRNA-protein association between miR-503 and its putative partners was then validated by western blotting with the proteins of interest in the biotinylated-miR-503 pull-down fraction. Figure 1f shows that, ACTB and Perlecan were not found in association with miR-503-botin while FN1, TSP1, hnRNPA2B1 and, to a lesser extent, VIM and ANXA2, were enriched in the pull-down fraction.
To mitigate bias associated with the unspecific detection of proteins, we transfected HUVECs with a c. elegans microRNA carrying a biotin tag (cel-miR-67-biotin). The efficiency of the transfection is shown in Figure S2c and the effect of Epi on the exosomal export of cel-miR-67 in Fig S2d. Then, we pulled down the microRNA and detected by western blot, the proteins previously validated. Our results (Fig. 1f) pointed to FN1 as a non-specific contaminant of our pull-down and was excluded from further analysis.
The putative complex formed by miR-503 in combination with ANXA2, hnRNPA2B1, TSP1, and VIM will be referred to in the following sections as the MicroRNA Exporting Complex or MEC.
Epirubicin does not regulate the expression of the MEC components
To study whether epirubicin regulates the expression of some of the components of the MEC in HUVECs, we assessed the mRNA and protein abundance in cells after treatment. As shown in Fig. 2a, epirubicin reduces the expression of thbs1 and hnRNPA2B1. Interestingly, the regulation is only significant 24 h after treating the cells with epirubicin. At protein level (Fig. 2b, c), we observe that epirubicin has little effect on the protein abundance of the MEC components: while changes in the abundance of ANXA2 and TSP1 follow a similar trend to the changes observed at RNA level, the effect is too modest to be significant. These results suggest that epirubicin does not affect the export of miR-503 by regulating the abundance of the MEC components.
Fig. 2.
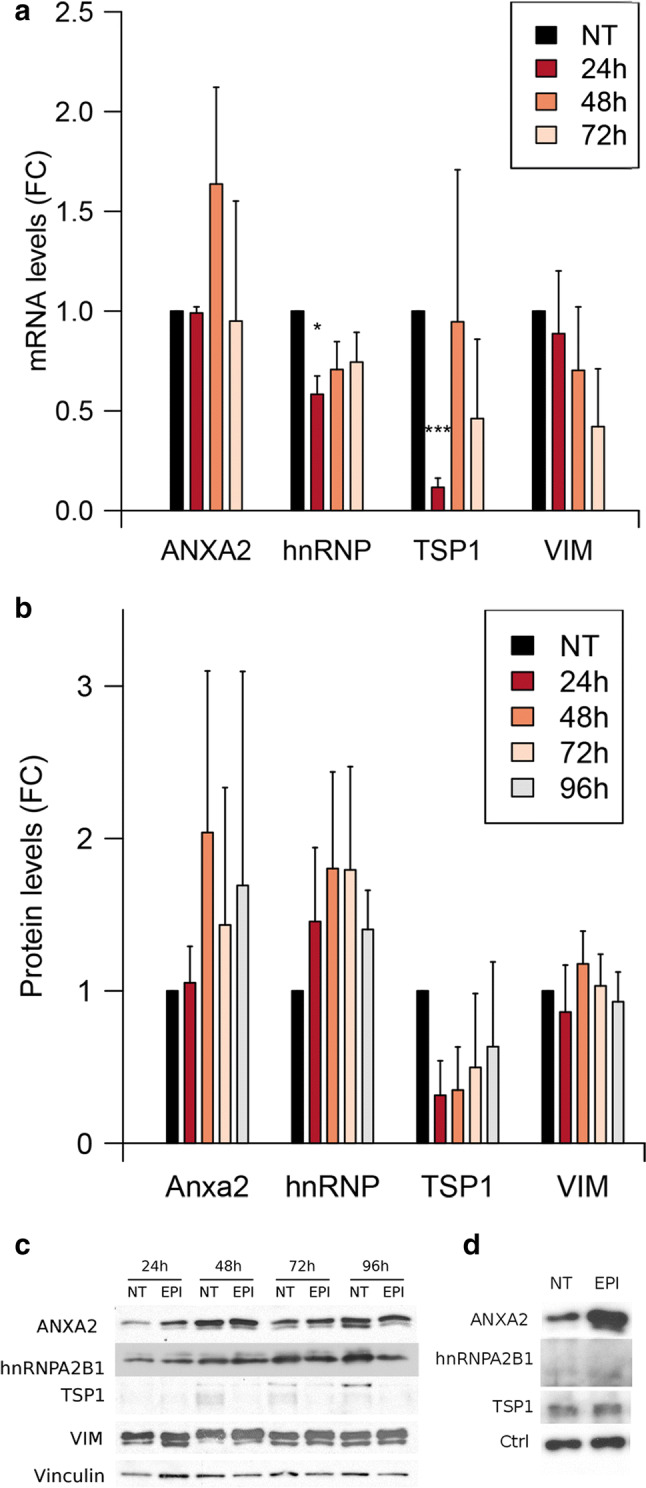
Epirubicin regulates the mRNA levels of TSP1 and hnRNPA2B1. HUVECs were treated with epirubicin for 24 h and the levels of a RNA and b proteins were assessed, respectively, by qPCR at 24, 48, and 72 h after treatment and western blotting at 24, 48, 72, and 96 h after treatment. Data represents fold change against non-treated cells. Plots show mean and SEM from three independent experiments. *p < 0.05, ***p < 0.001. c Representative western blot. d HUVECs were treated with epirubicin for 24 h and exosomes were produced for additional 72 h. Exosomes were purified by ultracentrifugation and the levels of proteins were determined by western blotting (10 µg/lane). Representative example of N = 3
The exosomal export of ANXA2 is regulated by epirubicin
To determine if miR-503 is co-exported with all or some of the components of the MEC, we treated HUVECs with epirubicin and then collected the exosomes produced during the following 72 h. With the exception of VIM, all MEC proteins were detected in exosomes (Fig. 2d). Interestingly, only ANXA2 showed a strong enrichment (average 8-fold) in exosomes after the treatment with epirubicin. Taken together, these results suggest that the MEC is, at least, partially destabilized upon epirubicin treatment.
ANXA2 and miR-503 present the most stable interaction among the MEC components
To study the composition of the MEC, we decided to pull down miR-503 prior any crosslinking and after only inducing the formation of protein–RNA bonds with UV crosslinking. Theoretically, the strongest interaction between the components of the MEC and the microRNA should be maintained in non-crosslinked conditions while the weakest interactions would only be detected when both UV and chemical crosslinkings were performed. Our results (Fig. 3a) suggest that in the absence of crosslinking, ANXA2 is the most abundant MEC protein present in the pull-down. To a much lesser extent, VIM can also be detected in non-crosslinked conditions. Interestingly, both TSP1 and hnRNPA2B1 can only be detected when both crosslinkings are performed.
Fig. 3.

Epirubicin disrupts the interaction between miR-503 and VIM and hnRNPA2B1. a HUVECs (30·106 cells) were transfected with miR-503-biotin (10 nM). The following day, the cells were crosslinked with DTSSP and UV (UV + CHL), only UV (UV) or not subjected to any crosslinking (No CL). HUVECs lysates were incubated with streptavidin beads. Both input (IN) (cellular lysate) and pull-down fractions (PD) were separated by SDS-PAGE and revealed by western blotting using indicated proteins. Input (IN) = 1% of cell lysate. Vinculin was used as loading control. b, c HUVECs were transfected with miR-503 (10 nM). 48 h later, immunoprecipitation assays were performed using the indicated antibodies or an IgG control and the levels of miR-503 were evaluated by qPCR. Plots show b fold change of miR-503 in the immunoprecipitated (IP) vs input fractions (IN) in non-treated cells and c fold change of immunoprecipitated miR-503 in epirubicin-treated (EPI) vs non-treated cells (NT). Plots represent mean and SEM from three independent experiments,*p < 0.05; **p < 0.01, ***p < 0.001. IN = 1% of the cellular lysate before immunoprecipitation
Epirubicin disrupts the interaction between miR-503 and hnRNPA2B1 and VIM
We then assessed if the interaction between miR-503 and the MEC components was affected by epirubicin treatment. For that purpose, the components of the MEC were immunoprecipitated and the levels of miR-503 determined by qPCR. As shown in Fig. 3b, in untreated conditions, miR-503 was coprecipitated with all MEC components, although the levels bound to ANXA2 were minimal. Interestingly, hnRNPA2B1 showed very high affinity for miR-503 (10, 36, and 79-fold change when compared to the cellular lysate).To study if the interaction between the components of the MEC and miR-503 was affected by the chemotherapeutic drug, we treated endothelial cells with epirubicin and measured the levels of miR-503 in the immunoprecipitates before and after treatment (Fig. 3c). In the cases of VIM, and hnRNPA2B1 a significant reduction of miR-503 in the immunoprecipitates was observed after treatment (p = 0.025 and p = 0.00003, respectively). Strikingly, the opposite effect was observed for ANXA2 (~ 2.3-fold increase, p = 0.019). In addition, when using the transfection of cel-miR-67 as a negative control (Fig S3a and S3b), we observed that epirubicin does not have any effect on the interaction between these proteins and the exogenous RNA. In combination, these results suggest that epirubicin modulates the interaction between miR-503 and some components of the MEC (ANXA2, VIM, and hnRNPA2B1).
Epirubicin treatment promotes the relocation of hnRNPA2B1 into the nucleus
Aiming to assess whether the changes in affinity between MEC components and miR-503 were associated to fluctuations in protein distribution, we decided to study the effect of epirubicin on protein localization. Confocal analysis revealed that, in untreated conditions, hnRNPA2B1 is mainly located in the nucleus and in some cytoplasmic granules (Fig. 4a, b). Interestingly, epirubicin treatment promotes the relocation of hnRNPA2B1 into the nucleus 24 and 48 h after treatment. This migration was not observed for any of the other MEC components (Fig S4a). The relocation of hnRNPA2B1 was confirmed by western blotting analysis of subcellular fractions (validation of the technique in Supplementary Fig. S4b) showing an increase in the ratio between cytoplasmic and nuclear abundance at 24 and 48 h that is partially reverted at 72 h (Fig. 4c, d).
Fig. 4.

hnRNPA2B1 re-localizes into the nucleus after epirubicin treatment. a Confocal images of hnRNPA2B1 (green) and DAPI (blue) of HUVECs treated with epirubicin for 24 h. Pictures taken at the indicated times after starting the treatment. b Quantification of the intensity in the cytoplasm and nucleus were performed using ImageJ on epifluorescence images. c Nuclear and cytoplasmic fractions from HUVECs treated with epirubicin and analysed by western blotting for hnRNPA2B1 localization. d Abundance ratio of hnRNPA2B1 between nucleus and cytoplasm in subcellular fractions. All plots show results from three independent experiments. *p < 0.05. Validation of subcellular fractionation method in Supplementary Figure S4
ANXA2 and hnRNPA2B1 are key mediators of the exosomal export of miR-503
To study the role of the MEC components in the export of miR-503, we used a dual approach. First, we studied if the abundance of the components of the MEC could determine the fate of miR-503 or miR-210, a microRNA-control present in the cell at similar concentrations than miR-503. For that purpose, we knocked down the putative partners of the microRNA (validation of siRNA knock-down Figure S5) and assessed the exosomal export of the microRNAs. Our results (Fig. 5a) suggest that the knock-down of thbs1 reduces significantly the export of miR-503, although the level by which is reduced is minimal (fold change 0.8). Interestingly, both the knock-down of ANXA2 and hnRNPA2B1 increased the export of miR-503. While lower levels of ANXA2 induce a 1.5-fold increase, slightly below the increase observed upon epirubicin treatment in non-transfected conditions, the knock-down of hnRNPA2B1 reproduced the effect of the chemotherapeutic drug to the same levels (ranging in two- to three-fold change increase). No significant differences were observed for the knock-down of vim in untreated conditions and none of the MEC protein knock-downs affected significantly the export of miR-210 (Fig. 5b).
Fig. 5.

ANXA2, hnRNPA2B1, TSP1 and VIM are necessary for the effect of epirubicin on the exosomal export of miR-503. HUVECs were transfected with siRNA for the indicated protein or control siRNA (siScr) (20 nM). Then, cells were treated with epirubicin for 24 h (or not) and exosomes were produced for the following 72 h. The levels of miR-503 (a, c, e) and miR-210 (b, d, f) in purified cells and exosomes were then assessed via qPCR. Data show fold change of miR-503 (a) and miR-210 (b) in exosomes from siRNA-transfected cells vs Scramble RNA-transfected (siScr) cells in non-treated conditions. Fold change of miR-503 and mR-210 respectively in exosomes (c, d) and cells (e, f) after siRNA transfection and epirubicin treatment normalized to untreated conditions. Plots show mean and SEM of three independent experiments. (*p < 0.05; **p < 0.01, ***p < 0.001)
Second, we studied whether the knock-down of the MEC proteins could affect the incorporation of miR-503 into exosomes triggered by the treatment with epirubicin. For that purpose, cells transfected with siRNA against the components of the MEC (or siScramble) were treated with epirubicin and the levels of exosomal miR-503 were assessed. Our results show that, the knock-down of any of the components of the MEC reduced the exosomal encapsulation of miR-503 upon epirubicin treatment (Fig. 5c). Interestingly, the knock-down of both ANXA2 and hnRNPA2B1 had the most dramatic effect on the exosomal encapsulation of miR-503. While no effect was observed in untreated conditions, epirubicin seemed to reduce the exosomal export of miR-210 in most conditions, although when compared to siScr, none of the knock-downs had any effect (Fig. 5d).
Finally, we determined whether the changes observed in the exosomal microRNA profiles were a reflection of changes in the cellular levels of miR-503, or the consequence of a specific export mechanism (Fig. 5e). The knock-down of any of the components of the MEC failed to increase the cellular production of miR-503. Only when hnRNPA2B1 was knocked down we observed a modest, but significant reduction in the cellular levels of miR-503. In the case of miR-210 (Fig. 5f), any treatment of the cells with either siRNA or epirubicin induced the production of reduced levels of this microRNA.
The mecanism of exosomal export of miR-503 is conserved in microvascular cells
To validate our findings, we confirmed our results in human microvascular endothelial cells (HMVECs), presenting a closer phenotype to cells located in the tumor microenvironment. First, we assessed whether epirubicin also induced the exosomal export of miR-503. For that purpose, we isolated HMVEC exosomes from epirubicin-treated or untreated cells. Our findings suggest (Fig. 6a) that the treatment with the chemotherapeutic drug also induces miR-503 sorting into exosomes. Then, we determined if miR-503 also interacted with the same MEC partners in HMVEC. Our results showed (Fig. 6b) that, after pulling down miR-503-biotin, we could detect two of the previously identified MEC components (ANXA2 and hnRNPA2B1) while TSP1 and VIM were not detected.
Fig. 6.

ANXA2 and hnRNPA2B1 are necessary for the effect of epirubicin on the exosomal export of miR-503 in microvascular endothelial cells. HMVECs were treated with epirubicin for 24 h. Cell lysates were prepared 24 h after treatment and exosomes were collected 72 h after removing the chemotherapeutic drug. Cellular and exosomal levels of miR-503 were evaluated by qPCR. Plot represents mean and SEM (n = 3). b HMVECs were transfected with miR-503-biotin (10 nM). The following day, the cells were crosslinked with DTSSP and UV. HMECs lysates were incubated with streptavidin beads. Both input (IN) (cellular lysate) and pull-down fractions (PD) were separated by SDS-PAGE. Previously identified components of the MEC complex were validated by western blotting against pull-down (PD) and input (IN) (1% of cell lysate) fractions using vinculin as loading control. c HMVECs were transfected with miR-503 (10 nM). 48 h later, immunoprecipitation assays were performed using the indicated antibodies and the levels of miR-503 were evaluated by qPCR. Plot shows fold change of miR-503 in the immunoprecipitated (IP) vs input fractions (IN) in non-treated cells and (d) fold change of immunoprecipitated miR-503 in epirubicin-treated (EPI) vs non-treated cells (NT). Plots represent mean and SEM from three independent experiments, *p < 0.05, **p < 0.01, ***p < 0.001. IN = 1% of the cellular lysate before immunoprecipitation
To study if epirubicin also affects the interaction between miR-503 and some components of the MEC in HMVECs, we treated these cells with epirubicin and then immunoprecipitated the proteins of interest. Our results show that, both in untreated conditions (Fig. 6c) and after treatment (Fig. 6d), the interaction between miR-503 and the MEC components follows the same trends observed for HUVECs: before treatment, the levels of ANXA2 binding miR-503 are low while the opposite is observed for hnRNPA2B1 (Fig. 6c). After treatment, these interactions are reversed and increased miR-503 is found in the immunoprecipitated ANXA2 while less microRNA is associated to hnRNPA2B1 (Fig. 6d).
Discussion
In previous work we showed that the treatment with the chemotherapeutic agent epirubicin induces the over export of the anti-tumoral miRNA-503 in endothelial cells. These results suggested the presence of a specific mechanism behind the sorting of this microRNA. Unfortunately, little is known about the machinery regulating the exosomal export of microRNAs. Only a few studies have reported the role of RNA-binding proteins in promoting the export of some subclasses of microRNAs (hnRNPA2B1 [26], Syncrip [27], MVP [28], MEX3C [29] and Y-Box [30]). Surprisingly, in this study we show that the exosomal sorting of miR-503 is negatively regulated by its binding to hnRNPA2B1. This is the first evidence showing that RBPs can bind and prevent the export of miRNAs into exosomes.
The proteomic analysis of the binding partners of miR-503 revealed nine potential proteins possibly involved in exosomal miR-503 export. Leaving aside the two proteins identified as natural biotin binders, one protein identified as a non-specific contaminant and those not validated by western blotting, the identified partners of miR-503 were TSP1, VIM, ANXA2, and hnRNPA2B1.
While no relationship has been established between TSP1 and the binding or export of RNAs, thrombospondin-1 has often been studied in the extracellular compartment. This protein is underrepresented in exosomes derived from nasopharyngeal carcinoma cells [31] and in EVs derived from cells undergoing epithelial to mesenchymal transition (EMT) [32]. Despite this, most of the publications studying circulating TSP1 have focused on its role in angiogenesis [33, 34] and as a marker of EMT [35]. Our results propose an ambiguous role for TSP1 in the MEC: the knock-down of this protein suggests that TSP1 has a modest, but significant effect in the exosomal export of miR-503 independently of the treatment with epirubicin. However, no significant differences were observed when the interaction between TSP1 and miR-503 was studied. These results suggest that TSP1 may be involved in the interaction between secondary partners of the MEC and the microRNA export machinery, but not directly binding the microRNA and regulating its export.
Our study suggests that VIM could be involved in the export of miR-503: in cells with reduced VIM abundance, epirubicin fails to promote the exosomal export of miR-503. Given that in untreated conditions the knock-down of this protein does not have any significant effect, it is possible that VIM regulates the localization of the MEC before the release of miR-503 for exosomal encapsulation. This hypothesis is supported by two factors: first, this protein was mostly pulled down after inducing the formation of protein–protein bonds. Second, by the physiological role of VIM in the cell: vimentin regulates the reorganization of the cellular cytoskeleton and the rearrangement of extracellular adhesion molecules [36]. Several publications have also found that VIM can be encapsulated into exosomes and that the levels at which it can be found often correlate with aggressiveness in the parent cells [37]. Unfortunately, in the cellular model used in this study, we could not detect this protein in the extracellular fraction. In addition, VIM was recently found to bind RNAs: VIM can stabilize collagen mRNA by binding to a stem loop region found at the 5′UTR [38]. After this study, it has been repeatedly showed that VIM can stabilize several other mRNAs following the same mechanism: alkaline phosphatase mRNA [39], mu-opioid receptor [40], and eIF2α [41]. Even though our experiments failed to prove direct binding between VIM and miR-503, we show that epirubicin can disrupt the interaction between these partners and propose a role for VIM during microRNA exosomal export.
ANXA2 is one of the top 20 most common proteins found in exosomes [42] and mediates EV uptake via the immobilization of the vesicles to the surface of the recipient cells [43]. At the cellular level, ANXA2 has been associated with exo- and endocytosis, as well as with the trafficking of membranous bodies, lipid raft formation, and signal transduction (reviewed in Ref. [44]). Moreover, ANXA2 can induce EMT by increasing the migration and invasion capacities of cancer cells [45], reducing apoptosis and mediating multi-drug resistance [46]. At the extracellular level, ANXA2 can induce pro-angiogenic processes and metastatic phenotypic switch by promoting cellular motility [47–49]. Regarding RNA, some studies have found that ANXA2 can bind some mRNAs such as c-myc [50] and “infectious bronchitis virus pseudoknot” RNA [51]. Additionally, another recently published study shows that some proteins of the same calcium-biding signaling family regulate the export of exosomal microRNAs. Although promising, these results showed that ANXA2 only participates in the encapsulation of six specific microRNAs without shared motifs [52] (none of them is miR-503). According to our results, ANXA2 has an affinity for miR-503 stable enough that it is pulled down without any sort of crosslinking. These results, in combination with previous evidence showing that ANXA2 can bind RNA suggest that ANXA2 is the main protein in the MEC directly interacting with miR-503. Although the treatment with epirubicin only increases modestly but not significantly the levels of cellular ANXA2, its exosomal export is increased by eightfold. Even though the relative quantity of ANXA2 binding to miR-503 under untreated conditions is very low, likely due to the high abundance of this protein in the cell, the treatment with epirubicin strongly increases the pull-down of miR-503-bound ANXA2 (2.5-fold). This switch could indicate that either epirubicin induces the binding of ANXA2 to the microRNA promoting its exosomal export or that epitopes unavailable for IP are revealed upon treatment. Since in untreated conditions, ANXA2 already shows a strong interaction with miR-503, our results support the latter hypothesis. At the same time, given that reduced levels of ANXA2 promote the export of miR-503 in untreated conditions but impair the export induced by epirubicin, our results point to a crucial role for ANXA2 in the microRNA export mechanism triggered by chemotherapy.
The second component of the MEC, key in the export of miR-503 into exosomes is hnRNPA2B1. The known roles of hnRNPA2B1 include DNA replication and repair, RNA nuclear export, pre-mRNA splicing, mRNA stability [25], pri-miRNA processing and mediation of splicing events [53], among others. In addition, hnRNPA2B1 can also bind to lncRNAs and regulates the expression of some genes at post-transcriptional level [54]. Several studies have also focused on the role of this protein in disease. In hepatocellular carcinoma, hnRNPA2B1 can act as an oncogene via the control of alternative splicing processes [55] and induction of EMT [56]. Moreover, hnRNAP2B1 has been found to be a circulating biomarker of lung cancer [57] and a dual mediator of the development of breast cancer. First, hnRNPA2B1 is associated with the loss of breast cancer susceptibility gene 1 (brca1) [58] and, second, by being a regulator of the STAT3-ERK1/2-signaling pathway [59]. hnRNPA2B1 has also been linked to the efficiency of chemotherapy in vitro: Inhibition of hnRNPA2B1 expression improved chemosensitivity to gemcitabine, 5-fluorouracil (5-FU), and oxaliplatin in pancreatic cancer cell lines [60].
Regarding exosomal RNA loading, hnRNPA2B1 was the first RBP known to regulate the export of microRNAs. In their study, Villarroya et al. discovered that this protein binds to microRNAs with a specific motif and promotes their exosomal export upon sumoylation [26]. Interestingly, miR-503 does not have the “exo-motif” described in this study, likely suggesting that the involvement of hnRNPA2B1 in the export of this microRNA does not follow the same mechanism. Moreover, unlike in their study, we did not observe any differences in the sumoylation of hnRNPA2B1 between cells and exosomes, likely due to differences in the cellular model used. Another recent study showed that hnRNPA2B1 can bind the host gene of miR-503 [61]. Their results suggest that the binding region of this RBP falls outside the coordinates of the mature miR-503, thus supporting the hypothesis that hnRNPA2B1 does not directly bind to miR-503. Our data showed that hnRNPA2B1 can only be identified along with miR-503 when both protein–protein and protein–UV crosslinking is induced. These results propose the hypothesis that hnRNPA2B1 does not bind directly to miR-503, but to other components of the MEC. Our findings also point to hnRNPA2B1 having high affinity for miR-503 (avg 40-fold) and that, upon epirubicin treatment, this affinity is strongly reduced. At the same time, we have shown that epirubicin induces the relocation of cytoplasmic hnRNPA2B1 towards the nucleus. Interestingly, reduced levels of hnRNPA2B1 also promote the export of exosomal miR-503 to the same levels observed by the treatment with epirubicin alone (~ 2.5-fold).
The combination of these results suggests that epirubicin induces the increased exosomal export of miR-503 by disrupting the interaction between hnRNPA2B1 and miR-503/ANXA2. Then, hnRNPA2B1 is relocated into the nucleus and a fraction of the initial MEC, composed by ANXA2 binding to miR-503, is sorted into exosomes. Our findings suggest that ANXA2 mediates the interaction between miR-503 and hnRNPA2B1. These results are supported by the evidence showing that when the levels of ANXA2 are reduced (by siRNA), epirubicin cannot promote the over export of miR-503 because hnRNPA2B1 is no longer associated to this microRNA. The validation of our results in HMVECs proves the conservation of a miR-503/ANXA2/hnRNPA2B1 axis across different subtypes of endothelial cells thus supporting the role of both RBPs in mediating the export of microRNAs. Given that hnRNPA2B1 mediates repair processes at sites of DNA double-strand break (DBS) hotspots [62], we hypothesize that the relocation of hnRNPA2B1 towards the nucleus upon epirubicin treatment could respond to a recycling mechanism initiated by the DNA destabilization triggered by the chemotherapeutic drug. Whether this relocation is the cause of the MEC destabilization or its consequence, remains unknown. Although several independent studies [26, 27] have found a series of RBPs that can bind and promote the export of microRNAs, this study is the first one to propose a mechanism by which proteins can promote cellular retention and inhibit the exosomal export of a specific microRNA.
Supplementary methods
The parameters used for the mass spectrometry analysis, the quantification of immunofluorescence, the primer sequences, and the conditions used for the detection of the potential components of the MEC are all explained in detail in the Supplementary Methods.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig S1: Design of miR-503-biotin and cel-miR-67-biotin duplexes. In black is shown the natural miR-503 and cel-miR-67 sequences and the modifications are indicated in colors: orange for the biotin addition, blue for the 3' tailing, light purple for the phosphorylations and in bold for the mismatch included to de-stabilize the carrier (reverse) strand (TIF 746 kb)
Fig S2: Biotinylation of miR-503 does not prevent the exosomal export of miR-503 triggered by Epirubicin and transfection validations. (a) Cells were transfected with miR-503-biotin. After 24h, cells were treated with Epirubicin and the export of miR-503 was assessed by RT-qPCR in exosomes from treated (green) and untreated (grey) cells. (b) The efficiency of the transfection of the synthetic RNAs was assessed by RT-qPCR in mock and (b) miR-503-bio or (c) cel-miR-67-bio transfected cells. (d) Cells were transfected with cel-miR-67. After 24h, cells were treated with Epirubicin and the exosomal and cellular abundance of cell-miR-67 was assessed by RT-qPCR in treated (green) and untreated (grey) conditions. Data shows mean and SEM of three independent experiments (TIF 821 kb)
Fig S3: cel-miR-67 is not immunoprecipitated with ANXA1 and hnRNPA2B1. HUVECs were transfected with cel-miR-67 (10 nM). 48h later, immunoprecipitation assays were performed using the indicated antibodies or an IgG control (IgG). Then the levels of cel-miR-67 were evaluated by qPCR. Plots show (a) fold change of cel-miR-67 in the immunoprecipitated (IP) vs input fractions (IN) in non-treated conditions and (b) fold change of immunoprecipitated cel-miR-67 in Epirubicin-treated (EPI) vs non-treated cells (NT). Plots represent mean and SEM from three independent experiments,* p<0.05; ** p<0.01, ***p<0.001. IN= 1% of the cellular lysate before immunoprecipitation (TIF 14120 kb)
Fig S4: ANXA2, Thrombspondin-1 (TSP-1) and Vimentin (VIM ) do not re-localize into the nucleus after Epirubicin treatment. (a) Confocal images of ANXA2 (top panels), TSP1 (middle panel) and VIM (lower panels) in green and DAPI in blue of HUVECs treated with Epirubicin for 24h. Pictures taken at the indicated times after starting the treatment. (b) Western blotting for GAPDH (cytoplasm marker) and Histone H3 (nucleus marker) in different subcellular fractions (TIF 17473 kb)
Fig S5: Validation of the knock down of the components of ExoMiR complex. Cells were transfected with siRNAs against the indicated genes or control siRNA (siScr). The levels of (a) mRNA were assessed at 24, 48 and 72h, and the levels of (b) protein were assessed with a 24h delay by Western Blotting. (* p<0.05; ** p<0.01, ***p<0.001) (TIF 14393 kb)
Acknowledgements
We thank the technology platform support staff at the GIGA Research Center. We thank Marie-Alice Meuwis for her support in designing and interpreting the mass spectrometry data. We thank Nicolas Bovy and Marc Thiry for the generation of the electronic microscopy data. This study was supported by the University of Liège (ULiege), the Fonds National de la Recherche Scientifique (FNRS), Télévie and the Fonds Léon Frédéricq. The authors declare that they have no competing interests.
Abbreviations
- Epi
Epirubicin
- EVs
Extracellular vesicles
- HUVECs
Human umbilical vein endothelial cells
- HMVECs
Human microvascular endothelial cells
- ANXA2
Annexin A2
- hnRNPA2B1
Heteronuclear ribonucleoprotein A2/B1
- TSP1/thbs1
Thrombospondin 1
- SYN
Syntenin
- VIM
Vimentin
- Vinculin
Vinculin
- MEC
MicroRNA exporting complex
Author contributions
JPB designed, supervised, conducted and performed experiments and statistical analyses, interpreted the data, and wrote the manuscript. AB performed the experiments with HMVECs, immunofluorescence, fractionation experiments, and exosome characterization. ML performed experiments and provided technical backup. FD participated in data analysis and provided scientific suggestions. IS conceived and designed the study, coordinated the experiments, and wrote the manuscript. All authors read and approved the final manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Jennifer Pérez-Boza and Amandine Boeckx contributed equally to the work.
References
- 1.van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018 doi: 10.1038/nrm.2017.125. [DOI] [PubMed] [Google Scholar]
- 2.McKelvey KJ, Powell KL, Ashton AW, et al. Exosomes: mechanisms of uptake. J Circ Biomark. 2015 doi: 10.5772/61186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dreyer F, Baur A. Biogenesis and functions of exosomes and extracellular vesicles. Methods Mol Biol. 2016;1448:201–216. doi: 10.1007/978-1-4939-3753-0_15. [DOI] [PubMed] [Google Scholar]
- 4.Pérez-Boza J, Lion M, Struman I. Exploring the RNA landscape of endothelial exosomes. RNA. 2018;24:423–435. doi: 10.1261/rna.064352.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalluri R, LeBleu VS (2017) Discovery of double-stranded genomic DNA in circulating exosomes. Cold Spring Harb Symp Quant Biol LXXXI:030932. 10.1101/sqb.2016.81.030932 [DOI] [PubMed]
- 6.Record M, Carayon K, Poirot M, Silvente-Poirot S. Exosomes as new vesicular lipid transporters involved in cell-cell communication and various pathophysiologies. Biochim Biophys Acta. 2014;1841:108–120. doi: 10.1016/j.bbalip.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 7.Beach A, Zhang H-G, Ratajczak MZ, Kakar SS. Exosomes: an overview of biogenesis, composition and role in ovarian cancer. J Ovarian Res. 2014;7:14. doi: 10.1186/1757-2215-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Q, Shao Y, Zhang X, et al. Plasma long noncoding RNA protected by exosomes as a potential stable biomarker for gastric cancer. Tumour Biol J Int Soc Oncodevelopmental Biol Med. 2015;36:2007–2012. doi: 10.1007/s13277-014-2807-y. [DOI] [PubMed] [Google Scholar]
- 9.Bovy N, Blomme B, Frères P et al (2015) Endothelial exosomes contribute to the antitumor response during breast cancer neoadjuvant chemotherapy via microRNA transfer. Oncotarget 6:10253–10266. 10.18632/oncotarget.3520 [DOI] [PMC free article] [PubMed]
- 10.Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Investig. 1973;52:2745–2756. doi: 10.1172/JCI107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Théry C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2019;8:1535750. doi: 10.1080/20013078.2018.1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ørom UA, Lund AH. Isolation of microRNA targets using biotinylated synthetic microRNAs. Methods. 2007;43:162–165. doi: 10.1016/j.ymeth.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 13.Betancur JG, Yoda M, Tomari Y. miRNA-like duplexes as RNAi triggers with improved specificity. Front Genet. 2012;3:2008–2013. doi: 10.3389/fgene.2012.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rambout X, Detiffe C, Bruyr J, et al. The transcription factor ERG recruits CCR4-NOT to control mRNA decay and mitotic progression. Nat Struct Mol Biol. 2016;23:663–672. doi: 10.1038/NSMB.3243. [DOI] [PubMed] [Google Scholar]
- 15.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 16.Tong L. Structure and function of biotin-dependent carboxylases. Cell Mol Life Sci C. 2013;70:863–891. doi: 10.1007/s00018-012-1096-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neill T, Jones HR, Crane-Smith Z, et al. Decorin evokes rapid secretion of thrombospondin-1 in basal breast carcinoma cells via inhibition of RhoA/ROCK1. FEBS J. 2013;280:2353–2368. doi: 10.1111/febs.12148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hellewell AL, Gong X, Schärich K, et al. Modulation of the extracellular matrix patterning of thrombospondins by actin dynamics and thrombospondin oligomer state. Biosci Rep. 2015 doi: 10.1042/BSR20140168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Viana LS, Affonso RJ, Silva SRM, et al. Relationship between the expression of the extracellular matrix genes SPARC, SPP1, FN1, ITGA5 and ITGAV and clinicopathological parameters of tumor progression and colorectal cancer dissemination. Oncology. 2013;84:81–91. doi: 10.1159/000343436. [DOI] [PubMed] [Google Scholar]
- 20.Dave JM, Bayless KJ (2014) Vimentin as an integral regulator of cell adhesion and endothelial sprouting. Microcirc (New York, NY 1994) 21:333–344. 10.1111/micc.12111 [DOI] [PubMed]
- 21.Dominguez R, Holmes KC. Actin structure and function. Annu Rev Biophys. 2011;40:169–186. doi: 10.1146/annurev-biophys-042910-155359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aukrust I, Hollås H, Strand E, et al. The mRNA-binding site of annexin A2 resides in helices C–D of its domain IV. J Mol Biol. 2007;368:1367–1378. doi: 10.1016/j.jmb.2007.02.094. [DOI] [PubMed] [Google Scholar]
- 23.Vedeler A, Hollås H, Grindheim AK, Raddum AM. Multiple roles of annexin A2 in post-transcriptional regulation of gene expression. Curr Protein Pept Sci. 2012;13:401–412. doi: 10.2174/138920312801619402. [DOI] [PubMed] [Google Scholar]
- 24.Luo M, Hajjar KA. Annexin A2 system in human biology: cell surface and beyond. Semin Thromb Hemost. 2013;39:338–346. doi: 10.1055/s-0033-1334143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He Y, Smith R. Nuclear functions of heterogeneous nuclear ribonucleoproteins A/B. Cell Mol Life Sci. 2009;66:1239–1256. doi: 10.1007/s00018-008-8532-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Villarroya-Beltri C, Gutiérrez-Vázquez C, Sánchez-Cabo F, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. 2013;4:2980. doi: 10.1038/ncomms3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santangelo L, Giurato G, Cicchini C, et al. The RNA-binding protein SYNCRIP is a component of the hepatocyte exosomal machinery controlling microRNA sorting. Cell Rep. 2016;17:799–808. doi: 10.1016/j.celrep.2016.09.031. [DOI] [PubMed] [Google Scholar]
- 28.Teng Y, Ren Y, Hu X, et al. MVP-mediated exosomal sorting of miR-193a promotes colon cancer progression. Nat Commun. 2017;8:1–16. doi: 10.1038/ncomms14448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu P, Li H, Li N, et al. MEX3C interacts with adaptor-related protein complex 2 and involves in miR-451a exosomal sorting. PLoS ONE. 2017;12:1–25. doi: 10.1371/journal.pone.0185992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shurtleff M, Karfilis KV, Temoche-Diaz M, et al. Y-box protein 1 is required to sort microRNAs into exosomes in cells and in a cell-free reaction. Elife. 2016;5:e19276. doi: 10.1101/040238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan Y-K, Zhang H, Liu P, et al. Proteomic analysis of exosomes from nasopharyngeal carcinoma cell identifies intercellular transfer of angiogenic proteins. Int J Cancer. 2015;137:1830–1841. doi: 10.1002/ijc.29562. [DOI] [PubMed] [Google Scholar]
- 32.Tauro BJ, Mathias RA, Greening DW, et al. Oncogenic H-ras reprograms Madin–Darby canine kidney (MDCK) cell-derived exosomal proteins following epithelial-mesenchymal transition. Mol Cell Proteomics MCP. 2013;12:2148–2159. doi: 10.1074/mcp.M112.027086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dudek AZ, Mahaseth H. Circulating angiogenic cytokines in patients with advanced non-small cell lung cancer: correlation with treatment response and survival. Cancer Investig. 2005;23:193–200. doi: 10.1081/CNV-200055949. [DOI] [PubMed] [Google Scholar]
- 34.Gonzalez FJ, Rueda A, Sevilla I, et al. Shift in the balance between circulating thrombospondin-1 and vascular endothelial growth factor in cancer patients: relationship to platelet a-granule content and primary activation. Int J Biol Markers. 2004;19:221–228. doi: 10.5301/JBM.2008.1959. [DOI] [PubMed] [Google Scholar]
- 35.Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell. 2009;15:195–206. doi: 10.1016/j.ccr.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 36.Liu C-Y, Lin H-H, Tang M-J, Wang Y-K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget. 2015;6:15966–15983. doi: 10.18632/oncotarget.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeppesen DK, Nawrocki A, Jensen SG, et al. Quantitative proteomics of fractionated membrane and lumen exosome proteins from isogenic metastatic and nonmetastatic bladder cancer cells reveal differential expression of EMT factors. Proteomics. 2014;14:699–712. doi: 10.1002/pmic.201300452. [DOI] [PubMed] [Google Scholar]
- 38.Challa AA, Stefanovic B. A novel role of vimentin filaments: binding and stabilization of collagen mRNAs. Mol Cell Biol. 2011;31:3773–3789. doi: 10.1128/MCB.05263-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmidt Y, Biniossek M, Stark GB, et al. Osteoblastic alkaline phosphatase mRNA is stabilized by binding to vimentin intermediary filaments. Biol Chem. 2015;396:253–260. doi: 10.1515/hsz-2014-0274. [DOI] [PubMed] [Google Scholar]
- 40.Song KY, Choi HS, Law P-Y, et al. Vimentin interacts with the 5’-untranslated region of mouse mu opioid receptor (MOR) and is required for post-transcriptional regulation. RNA Biol. 2013;10:256–266. doi: 10.4161/rna.23022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chatterjee S, Panda AC, Berwal SK, et al. Vimentin is a component of a complex that binds to the 5′-UTR of human heme-regulated eIF2α kinase mRNA and regulates its translation. FEBS Lett. 2013;587:474–480. doi: 10.1016/j.febslet.2013.01.013. [DOI] [PubMed] [Google Scholar]
- 42.Mathivanan S, Fahner CJ, Reid GE, Simpson RJ. ExoCarta 2012: database of exosomal proteins, RNA and lipids. Nucleic Acids Res. 2012;40:D1241–D1244. doi: 10.1093/nar/gkr828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koumangoye RB, Sakwe AM, Goodwin JS, et al. Detachment of breast tumor cells induces rapid secretion of exosomes which subsequently mediate cellular adhesion and spreading. PLoS ONE. 2011;6:e24234. doi: 10.1371/journal.pone.0024234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang C-Y, Lin C-F. Annexin A2: its molecular regulation and cellular expression in cancer development. Dis Markers. 2014 doi: 10.1155/2014/308976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang T, Yuan J, Zhang J et al (2015) Anxa2 binds to STAT3 and promotes epithelial to mesenchymal transition in breast cancer cells. Oncotarget 6:30975–30992. 10.18632/oncotarget.5199 [DOI] [PMC free article] [PubMed]
- 46.Zhang F, Zhang H, Wang Z, et al. P-glycoprotein associates with Anxa2 and promotes invasion in multidrug resistant breast cancer cells. Biochem Pharmacol. 2014;87:292–302. doi: 10.1016/j.bcp.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 47.Maji S, Chaudhary P, Akopova I, et al. Exosomal annexin A2 promotes angiogenesis and breast cancer metastasis. Mol Cancer Res MCR. 2016 doi: 10.1158/1541-7786.MCR-16-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ling Q, Jacovina AT, Deora A, et al. Annexin II regulates fibrin homeostasis and neoangiogenesis in vivo. J Clin Investig. 2004;113:38–48. doi: 10.1172/JCI19684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharma MR, Rothman V, Tuszynski GP, Sharma MC. Antibody-directed targeting of angiostatin’s receptor annexin II inhibits Lewis Lung Carcinoma tumor growth via blocking of plasminogen activation: possible biochemical mechanism of angiostatin’s action. Exp Mol Pathol. 2006;81:136–145. doi: 10.1016/j.yexmp.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 50.Filipenko NR, MacLeod TJ, Yoon C-S, Waisman DM. Annexin A2 is a novel RNA-binding protein. J Biol Chem. 2004;279:8723–8731. doi: 10.1074/jbc.M311951200. [DOI] [PubMed] [Google Scholar]
- 51.Kwak H, Park MW, Jeong S. Annexin A2 Binds RNA and reduces the frameshifting efficiency of infectious bronchitis virus. PLoS ONE. 2011;6:e24067. doi: 10.1371/journal.pone.0024067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hagiwara K, Katsuda T, Gailhouste L, et al. Commitment of Annexin A2 in recruitment of microRNAs into extracellular vesicles. FEBS Lett. 2015;589:4071–4078. doi: 10.1016/j.febslet.2015.11.036. [DOI] [PubMed] [Google Scholar]
- 53.Alarcón CR, Goodarzi H, Lee H, et al. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell. 2015;162:1299–1308. doi: 10.1016/j.cell.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lan X, Yan J, Ren J, et al (2016) A novel long noncoding RNA Lnc-HC binds hnRNPA2B1 to regulate expressions of Cyp7a1 and Abca1 in hepatocytic cholesterol metabolism: hepatology, Vol. XX, No. X, 2015 Lan et al. Hepatology 64:58–72. 10.1002/hep.28391 [DOI] [PubMed]
- 55.Shilo A, Ben Hur V, Denichenko P, et al. Splicing factor hnRNP A2 activates the Ras-MAPK-ERK pathway by controlling A-Raf splicing in hepatocellular carcinoma development. RNA. 2014;20:505–515. doi: 10.1261/rna.042259.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou Z-J, Dai Z, Zhou S-L, et al. HNRNPAB induces epithelial-mesenchymal transition and promotes metastasis of hepatocellular carcinoma by transcriptionally activating SNAIL. Cancer Res. 2014;74:2750–2762. doi: 10.1158/0008-5472.CAN-13-2509. [DOI] [PubMed] [Google Scholar]
- 57.Dowling P, Pollard D, Larkin A, et al. Abnormal levels of heterogeneous nuclear ribonucleoprotein A2B1 (hnRNPA2B1) in tumour tissue and blood samples from patients diagnosed with lung cancer. Mol Biosyst. 2015;11:743–752. doi: 10.1039/c4mb00384e. [DOI] [PubMed] [Google Scholar]
- 58.Santarosa M, Del Col L, Viel A, et al. BRCA1 modulates the expression of hnRNPA2B1 and KHSRP. Cell Cycle. 2010;9:4666–4673. doi: 10.4161/cc.9.23.14022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hu Y, Sun Z, Deng J, et al. Splicing factor hnRNPA2B1 contributes to tumorigenic potential of breast cancer cells through STAT3 and ERK1/2 signaling pathway. Tumour Biol J Int Soc Oncodevelopmental Biol Med. 2017;39:1010428317694318. doi: 10.1177/1010428317694318. [DOI] [PubMed] [Google Scholar]
- 60.Gu W-J, Liu H-L. Induction of pancreatic cancer cell apoptosis, invasion, migration, and enhancement of chemotherapy sensitivity of gemcitabine, 5-FU, and oxaliplatin by hnRNP A2/B1 siRNA. Anticancer Drugs. 2013;24:1. doi: 10.1097/CAD.0b013e3283608bc5. [DOI] [PubMed] [Google Scholar]
- 61.Wang H, Liang L, Dong Q, et al. Long noncoding RNA miR503HG, a prognostic indicator, inhibits tumor metastasis by regulating the HNRNPA2B1/NF-κB pathway in hepatocellular carcinoma. Theranostics. 2018;8:2814–2829. doi: 10.7150/thno.23012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tchurikov NA, Kretova OV, Fedoseeva DM, et al. DNA double-strand breaks coupled with PARP1 and HNRNPA2B1 binding sites flank coordinately expressed domains in human chromosomes. PLoS Genet. 2013;9:e1003429. doi: 10.1371/journal.pgen.1003429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1: Design of miR-503-biotin and cel-miR-67-biotin duplexes. In black is shown the natural miR-503 and cel-miR-67 sequences and the modifications are indicated in colors: orange for the biotin addition, blue for the 3' tailing, light purple for the phosphorylations and in bold for the mismatch included to de-stabilize the carrier (reverse) strand (TIF 746 kb)
Fig S2: Biotinylation of miR-503 does not prevent the exosomal export of miR-503 triggered by Epirubicin and transfection validations. (a) Cells were transfected with miR-503-biotin. After 24h, cells were treated with Epirubicin and the export of miR-503 was assessed by RT-qPCR in exosomes from treated (green) and untreated (grey) cells. (b) The efficiency of the transfection of the synthetic RNAs was assessed by RT-qPCR in mock and (b) miR-503-bio or (c) cel-miR-67-bio transfected cells. (d) Cells were transfected with cel-miR-67. After 24h, cells were treated with Epirubicin and the exosomal and cellular abundance of cell-miR-67 was assessed by RT-qPCR in treated (green) and untreated (grey) conditions. Data shows mean and SEM of three independent experiments (TIF 821 kb)
Fig S3: cel-miR-67 is not immunoprecipitated with ANXA1 and hnRNPA2B1. HUVECs were transfected with cel-miR-67 (10 nM). 48h later, immunoprecipitation assays were performed using the indicated antibodies or an IgG control (IgG). Then the levels of cel-miR-67 were evaluated by qPCR. Plots show (a) fold change of cel-miR-67 in the immunoprecipitated (IP) vs input fractions (IN) in non-treated conditions and (b) fold change of immunoprecipitated cel-miR-67 in Epirubicin-treated (EPI) vs non-treated cells (NT). Plots represent mean and SEM from three independent experiments,* p<0.05; ** p<0.01, ***p<0.001. IN= 1% of the cellular lysate before immunoprecipitation (TIF 14120 kb)
Fig S4: ANXA2, Thrombspondin-1 (TSP-1) and Vimentin (VIM ) do not re-localize into the nucleus after Epirubicin treatment. (a) Confocal images of ANXA2 (top panels), TSP1 (middle panel) and VIM (lower panels) in green and DAPI in blue of HUVECs treated with Epirubicin for 24h. Pictures taken at the indicated times after starting the treatment. (b) Western blotting for GAPDH (cytoplasm marker) and Histone H3 (nucleus marker) in different subcellular fractions (TIF 17473 kb)
Fig S5: Validation of the knock down of the components of ExoMiR complex. Cells were transfected with siRNAs against the indicated genes or control siRNA (siScr). The levels of (a) mRNA were assessed at 24, 48 and 72h, and the levels of (b) protein were assessed with a 24h delay by Western Blotting. (* p<0.05; ** p<0.01, ***p<0.001) (TIF 14393 kb)