


Abstract
In vivo mutational analysis of the yeast RPS28A ribosomal protein (rp-)gene promoter demonstrated that both the Abf1p binding site and the adjacent T-rich element are essential for efficient transcription. In vivo Mnase and DNaseI digestion showed that the RPS28A promoter contains a 50–60 bp long nucleosome-free region directly downstream from the Abf1p binding site, followed by an ordered array of nucleosomes. Mutating either the Abf1p binding site or the T-rich element has dramatic, but different, effects on the local chromatin structure. Failure to bind Abf1p appears to cause nucleosome positioning to become disorganized as concluded from the complete disappearance of Mnase hypersensitive sites. On the other hand, mutation of the T-rich element causes the downstream nucleosomal array to shift by ∼50 bp towards the Abf1p site, resulting in loss of the nucleosome-free region downstream of Abf1p. We conclude that Abf1p is a strong organizer of local chromatin structure that appears to act as a nucleosomal boundary factor requiring the downstream T-rich element to create a nucleosome-free region.
INTRODUCTION
Ribosomal protein (rp-)gene transcription in Saccharomyces cerevisiae is regulated by the global regulatory proteins (GRFs) Rap1p and Abf1p (1). The high transcriptional activity of rp-genes has been attributed in part to the presence of T-rich regions downstream of the GRF binding site(s) in the rp-gene promoters. T-rich elements appeared to have the capacity to activate transcription even by themselves, albeit with low efficiency. However, high transcriptional yields are reached when these elements function in synergism with a binding site for Rap1p, Abf1p or Reb1p (2,3). The latter factors are abundant, essential for cell viability and are involved in multiple cellular functions. In many instances a binding site for one GRF can be replaced by another GRF binding site and, furthermore, protein domains can be exchanged among GRFs without loss of function (2,4–6). Abf1p, the GRF that binds the promoters of several rp-genes (7), including RPS28A, functions in transcriptional regulation of a very diverse set of genes, but is also involved in silencing at HML and HMR, as well as in DNA replication. Finally, it binds in nearly all X-regions of telomeres (reviewed in 8). The mechanism by which Abf1p exerts its functions is still elusive. Several observations, however, suggest that it affects chromatin structure. De Winde et al. (9) reported that Abf1p is involved in the formation of a nuclease-hypersensitive region at the QCR8 promoter, while Venditti et al. (10) concluded that it acts as a nucleosomal boundary element at the autonomous replication sequence 1.
Although chromatin structure is dynamic in general, nucleosomes are often located at specific positions. The actual position of a nucleosome is determined by histone–DNA interactions, elements that function as a nucleosomal boundary, and/or by chromatin folding (11). Since the presence of nucleosomes is generally repressive to transcription, it has long been thought that T-stretches present in promoter regions act to exclude nucleosomes. However, stretches of As or Ts have been found in nucleosome-protected regions in vivo (12–14), and homopolymeric (dA:dT) elements (15) as well as naturally occurring promoter-proximal T-stretches can be incorporated into nucleosomes in vitro (16). Nevertheless, Iyer and Struhl (17) have found that introduction of a homopolymeric (dA:dT) element increases the occupancy of a neighbouring transcription factor binding site in vivo. Furthermore, it was observed that the stimulatory effect of such elements on transcription increases with length and that homopolymeric (dG:dC) elements which, like homopolymeric (dA:dT) elements, are structurally rigid DNA sequences, have a similar stimulatory effect on transcription. These and other observations led to the proposal that such structurally rigid elements increase the accessibility of neighbouring DNA, either by causing a local decrease in nucleosomal occupancy or by altering nucleosomal conformation.
Many naturally occurring T-rich elements, however, are not homopolymeric. These elements may harbour recognition site(s) for a specific DNA binding protein(s). A yeast protein that has been shown to specifically recognize T-rich elements is Dat1p. This protein binds any 11 bp sequence of which 10 bp are dA:dT (18). Unexpectedly, Dat1p appears to be able to affect transcription both negatively and positively. Genes that contain putative Dat1p-binding T-rich elements are 1.5–2.5-fold derepressed in a dat1 deletion strain (18) and the transcriptional activity of promoters containing homopolymeric dA:dT elements decreases when Dat1p is overexpressed (17). On the other hand, Moreira et al. (19) have shown that Dat1p functions as a transcriptional activator at the ILV1 promoter.
In order to obtain more insight into the role of the T-rich elements in transcriptional activation of yeast rp-genes we have studied the T-rich element that is present adjacent to the Abf1p binding site in the RPS28A rp-gene promoter. This element is located at the centre of the 294 bp intergenic region separating the coding regions of RPS28A and the oppositely transcribed GLN4 gene, which encodes glutamine tRNA synthetase (20). Our data indicate that the Abf1p binding site and the T-rich element act synergistically to create a nucleosome-free region downstream of the Abf1p-binding site followed by an ordered array of nucleosomes. This local (re-)organization of the chromatin may serve to increase the accessibility of downstream promoter sequences for additional protein factors.
MATERIALS AND METHODS
Yeast strains and growth conditions
The haploid strain BJ2168 (MATa, leu2, trp1, ura3-52, prc1-407, prb-1122, pep4-3) of Saccharomyces cerevisiae was used to analyse the GUS-reporter constructs (with respect to the Escherichia coli β-glucuronidase gene). The DAT1 (EWY-1002C-1) and dat1-Δ3 (BRY1004-3: EWY-1002C-1 with a null dat1 allele without a selectable marker) were kindly provided by Edward Winter (Thomas Jefferson University, USA) (21). Yeast cells were grown in minimal medium containing 2% (w/v) glucose, 0.67% (w/v) yeast nitrogen base and the appropriate supplements.
Synthetic oligonucleotides
Oligonucleotides were generated using a Millipore Expedite™ Nucleic Acid Synthesis System.
PGKfor, 5′-GGAATTCCAGATCTCCCATGTCTCTACTGG-3′;
PGKrev, 5′-TCCCGCGGGAGACTAGTCGCGGCCGCCTTA-AAATACGCTGAAACCCG-3′;
GUS-primer, 5′-CACCAACGCTGATCAATTCC-3′;
EV2, 5′-GCGCAGGGTTTTCCCAGTCACGAC-3′;
TS1for, 5′-GCCGTCTAGAGTGACCAGCATGGGCCCGGA-TAAATTTTTTTTTCTTGGTCG-3′;
TS2for, 5′-CGCGGCCGTCTAGAGTGACCACGCACTTT-TTTTTTTTTTTTTTTTTTCTTGGTCGTTGAACTAC-3′;
TS3for, 5′-GACCACGCACTTTTTTGATAAAGGGCCCAA-CGCCGGTCGTTGAACTACTTGG-3′;
TS4for, 5′-CACTTTTTTGATAAATTTTTTTTTCTTAAGG-GCCCTCTTACTTGGAATAAAGAAAATGAAATTTC-3′;
TS6for, 5′-CTAGAGTGACCACGCACAAAGCCGATAAA-GCCTCTCGAGTTGAGGGCC-3′;
Tcfor, 5′-CTAGAGTGACCACGCACCCCCCCGATAAACC-CCCCCCCCATGGGGGCC-3′.
The complementary strands of the oligonucleotides containing the sequences with which we wanted to replace the T-rich element were synthesized in such a way that, after isolation of a PCR-product or annealing of oligonucleotides, the resulting products contained flanking restriction sites or suitable overhangs for insertion in the appropriate restriction sites of the plasmids used (as described below).
Plasmids
The pRS306-based plasmids IRS3 and IRS* were obtained by replacing the RPL25 terminator region in the plasmids RS3 and RS* (22) for the PGK terminator, by ligation of EcoRI/SpeI-digested plasmid DNA with an EcoRI/SpeI-digested PCR product of the PGK terminator (primers PGKfor and PGKrev, template PUP23).
T-rich element mutant TS1 was generated as follows: with RS3 as a template and using 10 PCR cycles, two separate PCR products were generated with primer combinations TS1rev/EV2 and primers TS1for/GUS. These PCR products, containing the appropriate nucleotide replacements and flanking restriction sites, were digested with HindIII/ApaI or ApaI/NheI, respectively and were ligated in a triple-ligation reaction to the HindIII/NheI-digested RS3 plasmid. Subsequently the HindIII/NheI RPS28A promoter fragment was cloned into the HindIII/NheI fragment of plasmid IRS3, containing the GUS-reporter, PGK-terminator and pRS306 plasmid sequences. For ITS2, ITS3 and ITS4 the same strategy was used, with the appropriate primers (for TS2: XbaI was used instead of ApaI).
The ITS6 construct was made by triple-ligation of a double stranded TS6 oligonucleotide (annealed TS6for and TS6rev primers) with the 2.5 kb PstI/XbaI fragment and the 5.0 kb ApaI/PstI fragment of the ITS4 plasmid. Construct Tc was generated similarly. In this case the ds-oligo was ligated with a 2.4 kb ApaI/KpnI fragment and a 5.1 kb KpnI/XbaI fragment of the ITS4 plasmid.
All PCR-mediated cloning steps were verified by sequencing. All resulting integrative plasmids as described above were linearized with StuI and integrated at the URA3 locus. Single copy integrants were selected after Southern analysis.
ERS3, ERS* and ETS6 were constructed by subcloning of the 3.2 kb HindIII/KpnI fragments from IRS3, IRS* and ITS6 in the HindIII/KpnI-digested YEplac181 vector (23).
Northern analysis
Yeast cells were grown to OD 0.2–0.3. RNA was isolated according to the acid phenol procedure (Current Protocols; 24), electrophoretically separated on formaldehyde/agarose gels (24) and blotted onto Hybond-N. Membranes were hybridized at 65°C in 0.5 M Na2PO4 pH 7.2, 7% SDS, 1 mM EDTA, containing probes (Prime-a-Gene labeling system®; Promega) corresponding to the coding regions as indicated in the figure legends. mRNA levels are visualized by a PhosphorImager and quantified using ImageQuant™ (Molecular Dynamics).
Naked DNA and chromatin digestion
Cultures (800 ml; OD 0.2–0.3) were spheroplasted and permeabilized by nystatin (final volume of 8 ml) according to Venditti and Camilloni (25). For chromatin samples, after the 0 min time sample, either 250 U/4 ml Mnase (micrococcal endonuclease) or 180 U/4 ml DNaseI (both from Boehringer-Mannheim) was added to the nystatin-permeabilized spheroplasts, and 500 µl time samples (1, 2.5, 5, 9, 15 and 25 min) were added to 50 µl StopMnase (10% SDS, 0.1 M EDTA and 50 µg proteinase K). For isolation of undigested DNA, 800 µl StopMnase was added to ∼8 ml spheroplasts in Nystatine buffer. Samples were incubated at 56°C for 3 h or at room temperature overnight. To obtain protein-free DNA, samples were extracted twice with 25:24:1 phenol:chloroform:isoamyl alcohol (PCI) and once with 24:1 chloroform:isoamyl alcohol. DNA was treated with RNaseA, re-extracted and precipitated with NaAc/ethanol and dissolved in TE (10 mM Tris–Cl pH 8.0, 1 mM EDTA). For digestion of naked DNA, 300 µl bufferA (20 mM Tris pH 8.0, 150 mM NaCl, 5 mM KCl, 1 mM EDTA) and 15 µl 0.1 M CaCl2 was added to 40 µl DNA (16 µg/µl). DNA was digested with 0, 0.125, 0.5 and 1.25 U Mnase or DNaseI for 5 min at 37°C. After phenol extractions and precipitation, DNA was dissolved in TE. Proper digestion of chromatin and naked DNA samples was checked by running on a 1% agarose gel.
Indirect end-labeling
Chromosomal DNA (10 µg) was digested to completion with MluI and HindIII. For the XbaI-digested control 4 µg of chromosomal DNA was digested with MluI and XbaI and 6 µg of undigested chromosomal DNA was added before gel electrophoresis. As markers either PstI-digested λDNA or a 250 bp ladder (MRC Holland) were used. After addition of loading buffer, DNA samples were electrophoresed using a 1% agarose gel (20 cm) in 1× TBE containing 0.1 µg/ml ethidium bromide at 30–40 V for 15–18 h at room temperature. DNA was transferred to Hybond-N by the alkaline Southern blotting method. For hybridization conditions, see above. The 152 bp MluI-probe flanking the first MluI restriction site of the GUS-reporter gene was generated by PCR with primers GUS-3444for (5′-GGTGATTACCGACGAAAACG-3′) and GUS-3596rev (5′-TTACAGTCTTGCGCGACATG-3′).
Primer extension
PAGE-purified Primer GUS3028rev (5′-TTGGGGTTTCTACAGGACGGACCATGG-3′) was 5′ end labeled (26) and purified using Qiaquick (Qiagen). Conditions for primer extension on Mnase digests of episomal (multicopy) constructs, with five primer extension cycles, were according to Zhu and Thiele (12). Reactions were terminated on ice. DNA was extracted with PCI (equilibrated with 150 mM NaCl, 50 mM Tris–Cl pH 7.5, 1 mM EDTA), precipitated and electrophoretically fractionated on a 6% polyacrylamide–urea gel. Dried gels were exposed to a PhosphorImaging screen (Molecular Dynamics). For sequencing a cycle-sequencing kit (Amersham) was used with a 5′ end labeled GUS3028rev primer.
RESULTS
Both the Abf1p binding site and the T-rich element are essential for high level transcription of the RPS28A ribosomal protein gene
In order to determine the sequence requirements of the T-rich element in more detail we fused a 1 kb fragment containing the wild-type RPS28A promoter to the GUS reporter gene (construct RS3, Fig. 1A). Both the wild-type fusion construct and various mutant forms were integrated in single copy into the yeast genome. Figure 1B and C depicts the various mutations and their effect on transcription as determined by northern hybridization.
Figure 1.
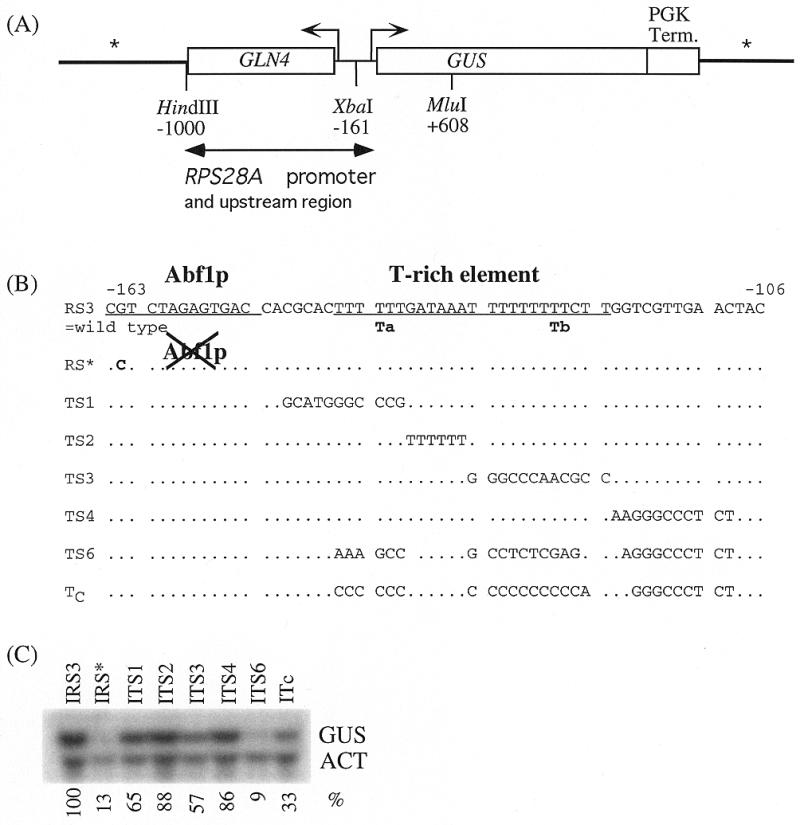
Effect of mutations in the Abf1p binding site and T-rich element of the RPS28A promoter on transcription of the GUS reporter gene [with respect to the E.coli β-glucuronidase gene (32)]. (A) Overview of the reporter used. Vector sequences, Integrative or Episomal, are denoted by an asterisk. (B) Sequences of the various mutants. (C) Northern analysis of the transcript levels of the integrated GUS-reporter constructs. Quantification of mRNA levels is indicated in percentages relative to the wild-type construct IRS3, using actin mRNA as loading control.
Introduction of a single point mutation into the Abf1p site (Integrated construct IRS*), which destroys Abf1p binding in vitro (T.M.Doorenbosch, unpublished results) reduced transcription to ∼10% of the wild-type level (Fig. 1C). This is consistent with results obtained in promoter reconstitution experiments previously performed in our laboratory (22). Clearly, binding of Abf1p is essential for optimal activation of transcription by the RPS28A promoter.
Interestingly, the T-rich element located 7 bp downstream of the Abf1p binding site is equally important for attaining high transcriptional yields. When both runs of thymidines present within this element were replaced by a randomized sequence (construct ITS6) transcription was reduced to the same extent as in the IRS* mutant (Fig. 1C). Substitution of either the first (construct ITS1) or second (construct ITS3) run of Ts resulted in a less dramatic, but still significant reduction to ∼65 and 55% of the wild-type level, respectively. Replacement of either the A-rich region that separates stretches Ta and Tb (construct ITS2) or sequences downstream of Tb (construct ITS4) did not significantly affect transcription.
These results indicate that optimal activity of the RPS28A promoter requires the presence of a functional Abf1p binding site as well as both homopolymeric T stretches (Ta and Tb) of the T-rich element. However, Ta and Tb can independently stimulate transcription to a significant extent in synergism with the Abf1p binding site.
Ta and Tb can be functionally replaced by other structurally rigid elements
Activation of transcription by T-rich elements is thought to take place via either of two different mechanisms. They may themselves contain a binding site for a transcriptional activator, notably Dat1p (19) or, particularly in the case of homopolymeric T-stretches, they may increase the accessibility of neighbouring protein binding sites by affecting local chromatin structure as a consequence of their structural rigidity. The latter explanation derives from the observation that homopolymeric (dT:dA) stretches can be functionally replaced by homopolymeric (dG:dC) or (dC:dG) stretches that are also structurally inflexible (17).
As can be seen in Figure 1B the sequence of the 3′ portion of the T-rich element of the RPS28A promoter conforms to the consensus Dat1p binding site proposed by Reardon et al. (18), with respect to any 11 bp homopolymeric (dT:dA) tract with one mismatch allowed. However, deleting the DAT1 gene did not result in decreased transcription of the RPS28A gene (Fig. 2). Rather, a slightly (∼20%) higher level of the RPS28A transcript was observed in the dat1 deletion strain. Therefore, we conclude that Dat1p does not function as a transcriptional activator of the RPS28A gene.
Figure 2.
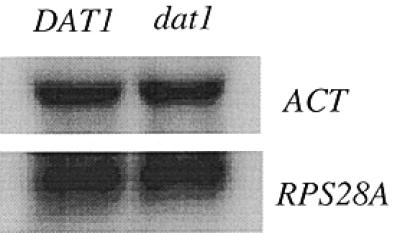
Effect of Dat1p on transcription of the RPS28A rp-gene. A dat1 deletion strain (see Materials and Methods) and the isogenic wild-type DAT1 strain were monitored for the expression of the RPS28A and RPL25 rp-genes. When the expression of RPS28A in the dat1 deletion strain was quantified from independent blots, an increase of 10–20% was found relative to the wild-type control. Actin mRNA was used as loading control.
In a further set of experiments we generated the ITc construct (Fig. 1B) in which both the Ta and Tb stretches of the RPS28A T-rich element were replaced by stretches of cytidines. As shown in Figure 1C the mutant promoter does stimulate transcription, albeit less efficiently than its wild-type counterpart (mutant ITS4, in which the replacements were made). This suggests that the T-rich element of the RPS28A promoter functions through a nucleosome destabilization mechanism similar to that proposed for homopolymeric (dA:dT) elements.
Loss of Abf1p binding results in loss of ordered nucleosome positioning
In order to investigate the effect of Abf1p binding on chromatin structure yeast cells carrying an integrated single copy of either the wild-type IRS3 or the mutant IRS* construct were spheroplasted, permeabilized with nystatin and subjected to treatment with Mnase. Figure 3 shows the results of indirect end-labeling experiments on the digested chromatin using a probe complementary to a sequence upstream of the MluI site in the coding region of the GUS gene (Fig. 1A). In the wild-type construct well-defined Mnase (hyper)sensitive sites as well as protected regions can be seen, while in the mutant construct, which does not bind Abf1p, no such sites are visible.
Figure 3.
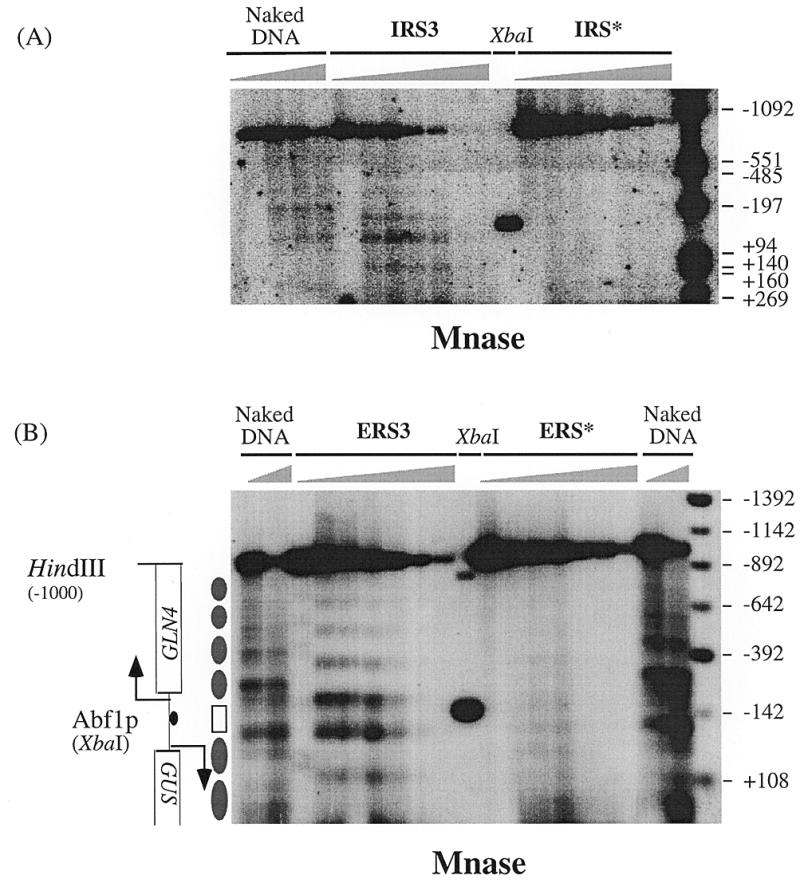
In vivo mapping of nucleosome positions on wild-type RPS28A promoters and promoters containing a mutated Abf1p site. (A) Spheroplasts prepared from single copy integrants of the reporter construct IRS3 (wild-type) and IRS* (unable to bind Abf1p) were permeabilized with nystatin and treated with Mnase. Purified DNA was digested with MluI and HindIII (Fig. 1A) and subjected to indirect end-labeling using a probe flanking the MluI site. The XbaI site corresponds to the position of the Abf1p site. As control, deproteinized DNA from the IRS3 integrant was digested with Mnase (Naked DNA). (B) As in (A), using cells carrying either multicopy plasmids ERS3 (wild-type) or ERS* (unable to bind Abf1p). Naked DNA digests on genomic DNA from both ERS3 and ERS* are included. Distances are from the translational start codon ATG. Protected regions of 160–170 bp are interpreted as positioned nucleosomes (closed elipses). The region between the two strongest Mnase hypersensitive sites (open box) spans 130 bp of the RPS28A–GLN4 intergenic region.
In order to increase the sensitivity of our analysis we repeated the experiment using multicopy vectors carrying the same constructs (Episomal constructs ERS3 and ERS*, respectively). Again for the wild-type promoter (construct ERS3) a clear pattern of Mnase-induced cuts was obtained which is identical to that observed for the integrated construct, while the ERS* construct fails to produce such a pattern. These data indicate a defined positioning of nucleosomes in the wild-type construct which depends on the presence of a functional Abf1p binding site.
From Figure 3 it is evident that binding of Abf1p induces two well-defined Mnase hypersensitive sites on either side of its binding site, as well as a regular pattern of sensitive sites and protected regions in the coding regions of both the GUS and the GLN4 gene. The two hypersensitive sites are separated by a region of ∼130 bp that appears to be protected against Mnase digestion (Fig. 3A and B). Although such a small region might reflect a positioned nucleosome, data presented below clearly demonstrate that this is not the case. In the coding regions of the GLN4 gene and the GUS reporter, on the other hand, the protected regions span a distance of 160–170 bp that corresponds to the length of DNA that is wrapped around a positioned nucleosome. Thus, nucleosomal arrays spread both downstream and upstream of the GLN4-RPS28A promoter regions.
Since our main interest concerns the RPS28A promoter region, the nucleosomal border of the nucleosome located downstream of Abf1p was mapped by primer extension with linear amplification (five cycles). As can be observed (ERS3 in Fig. 5), several hypersensitive bands were found which map between positions –49 and –83 upstream from the ATG. Identical hypersensitive sites were found with the integrated wild-type construct IRS3 (data not shown). We conclude that the first nucleosome downstream of the T-rich element can occupy several different translational positions. The ∼10 bp periodicity of the main hypersensitive bands (see Fig. 5) may suggest rotational phasing of the corresponding nucleosome.
Figure 5.
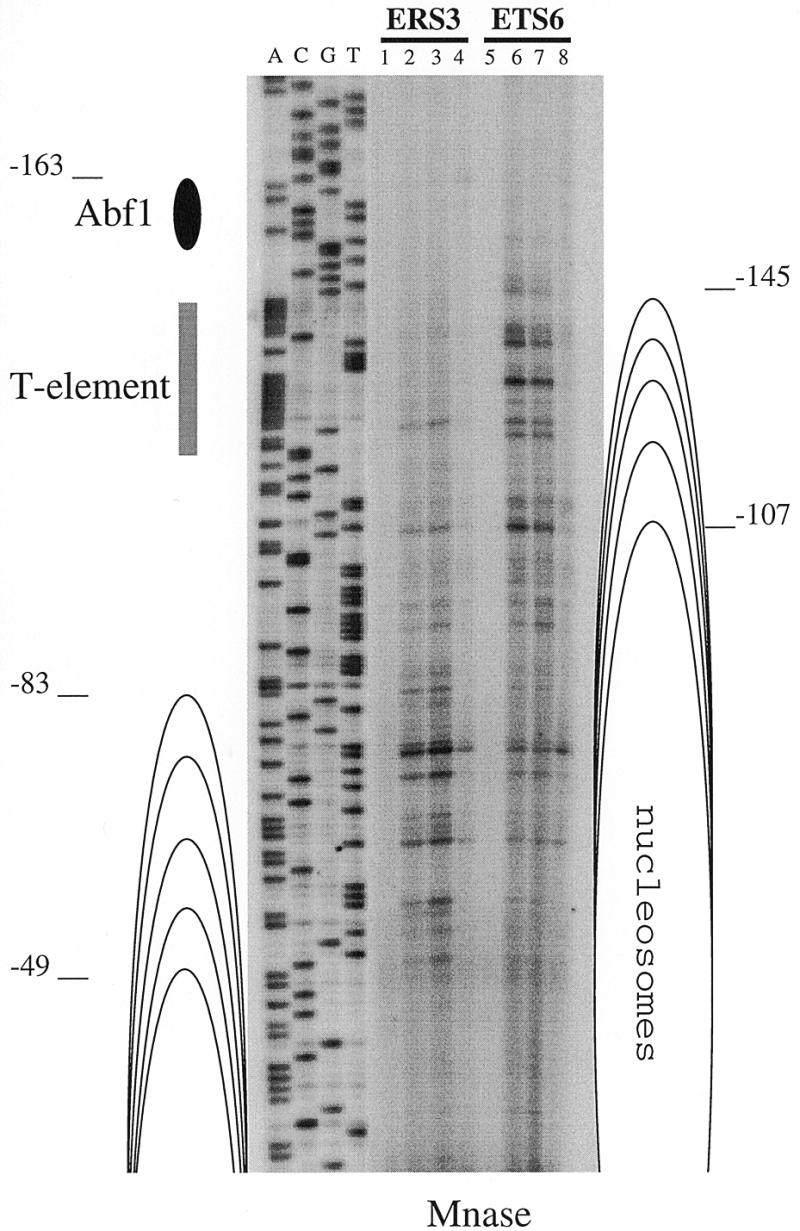
High resolution analysis of in vivo Mnase accessibility of the wild-type RPS28A promoter (ERS3) and a RPS28A promoter that contains a mutated T-rich element (ETS6). Primer GUS3028rev was used for unidirectional, linear amplification (five cycles) on DNA obtained after in vivo Mnase digests (Figs 3B and 4). Lane 1–3, chromatin DNA of ERS3 (time samples 0, 1 and 2.5 min); lane 4, naked DNA of ERS3; lane 5–7, chromatin DNA of ETS6 (time samples 0, 1 and 2.5 min); lane 8, naked DNA of ETS6. Sequencing reactions using the same primer are indicated by A, C, G and T. The position of the Abf1p binding site, T-rich element and the multiple translational positions of the mapped nucleosomes are indicated. Distances are from the translational start codon ATG.
In conclusion, binding of Abf1p is required to maintain a highly ordered nucleosome organization of the RPS28A and the GLN4 genes.
Generation of a nucleosome-free region depends upon the T-rich element
The possible role of the T-rich element in organizing the chromatin structure of the RPS28A promoter was investigated in the same manner as described above. The TS6 mutant, in which the T-rich element has been inactived (Fig. 1B), as well as its wild-type counterpart were introduced into yeast cells on a multicopy vector and subjected to digestion with either Mnase or DNaseI. Figure 4 shows the results of an indirect end-labeling experiment to detect the cleavage sites within the wild-type and mutant promoters (ERS3 and ETS6, respectively). Mnase and DNaseI sites in naked DNA do not differ between these constructs (Fig. 3 and data not shown). From these results it is clear that functional inactivation of the T-rich element notably changes the chromatin structure of the downstream region of the RPS28A promoter and the adjacent GUS reporter gene, while the upstream region and the GLN4 gene remain unaffected. In the ETS6 construct the lower one of the two hypersensitive sites is shifted to a position ∼50 bp closer to the Abf1p site. Since a similar shift has occurred in the pattern of protected and sensitive regions in the coding region of the GUS gene, it shows that the entire array of nucleosomes has shifted towards the Abf1p site. As a result of this nucleosomal rearrangement, the 130 bp protected region present in the wild-type ERS3 promoter has decreased to 85 bp in the T-rich element mutant ETS6 (open boxes in Fig. 4). Apparently this region is not protected by a positioned nucleosome and should therefore be considered as a nucleosome-free region.
Figure 4.
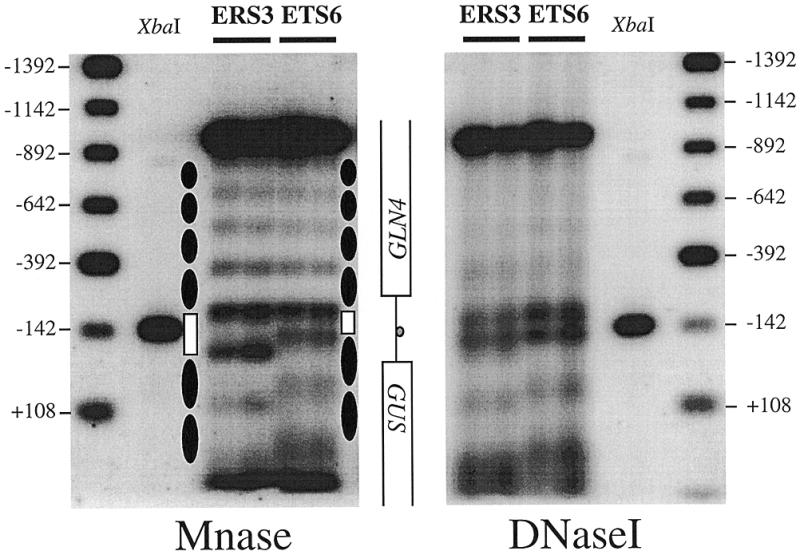
Comparison of in vivo nucleosome positions on the wild-type RPS28A promoter and a promoter containing a mutated T-rich element. Nystatin-permeabilized spheroplasts prepared from wild-type (ERS3) and mutant cells (ETS6) were treated with either Mnase (A) or DNaseI (B). Distances are from the translational start codon ATG. Protected regions of 160–170 bp are interpreted as positioned nucleosomes (closed elipses). The 130 bp Mnase-protected region that covers the Abf1p site (XbaI) in the wild-type promoter (ERS3) has decreased to 85 bp when the T-rich element is mutated (ERS6). Concomitantly, downstream nucleosomes are shifted towards the Abf1p binding site.
The upstream boundary of the nucleosomal array covering the region downstream of the Abf1p site in the ETS6 construct was mapped by primer extension. As shown in Figure 5, the hypersensitive sites that are found in the wild-type ETS3 construct, from –83 to –49, are protected in the ETS6 construct, and a new set of hypersensitive bands appears between positions –145 and –107. Hence, inactivation of the T-rich element repositions the first nucleosome downstream of the Abf1p site 50–60 bp closer to this site compared to the situation in the wild-type promoter.
We conclude that inactivation of the T-rich element causes the downstream nucleosomal array as a whole to shift by ∼50 bp towards the Abf1p site, resulting in loss of the nucleosome-free region downstream of Abf1p.
DISCUSSION
The results reported in this paper shed more light onto the mechanism by which the high efficiency of transcription of yeast rp-genes is attained. Using a fusion between the promoter of the RPS28A gene and the GUS reporter gene we have performed a mutational analysis that establishes a correlation between the transcriptional activation by the common elements within the rp-gene promoter and the effect of these elements on the local chromatin structure.
The data presented in Figure 1 demonstrate that the Abf1p binding site and the T-rich element of the RPS28A promoter are equally important for full transcriptional activity, confirming earlier conclusions (3,22). However, substitution of the individual segments of the bipartite T-rich element causes a much smaller reduction in transcriptional activity, showing that even a short T-stretch still has a substantial stimulatory capacity. The extent of the stimulatory activity shows an approximate correlation with the size of the segment; in mutant ITS1, in which the remaining portion of the T-stretch is 9 residues long, transcriptional activity is ∼65% of the wild-type level versus 55% in mutant ITS3 having a six residue long T-stretch. Changing the bipartite T-rich element into a 21 residue long homopolymeric T sequence (mutant ITS2), however, did not significantly increase transcription over the wild-type level. This contrasts with the results of Iyer and Struhl (17), who did observe an increase in transcriptional activity with increasing size of the homopolymeric T-stretch. Thus, the bipartite structure of the T-rich element in the RPS28A promoter might have functional significance.
Two observations prompted us to analyze the effect of the various mutations in the RPS28A promoter elements on the local chromatin structure. First, we found that deletion of the DAT1 gene, encoding the only protein so far known to influence transcription by binding specifically to T-rich elements (17–19), had very little effect on RPS28A promoter-driven transcription of the GUS reporter (Fig. 2). Secondly, as shown in Figure 1, replacement of the two (dA:dT) stretches of the T-rich element, which are thought to form a rigid DNA structure (27), by similarly rigid stretches of dC:dG (mutant ITc) suggests the requirement for structural rigidity of the DNA downstream of Abf1p in order to activate transcription. These two observations make it unlikely that the T-rich element functions as a recognition site for specific transcriptional activator proteins.
The chromatin structure of the wild-type promoter shows a well-defined organization consisting of a protected region, delineated by two hypersensitive sites, that spans the Abf1p binding site. Several lines of evidence indicate that this protected region of 130 bp does not correspond to a positioned nucleosome: (i) the relatively small size of the region; (ii) the finding that the protected region in DNaseI digests is much smaller, ∼90 bp (Fig. 4); and (iii) the 130 bp protected region present in the wild-type promoter (ERS3) has decreased to a region of ∼85 bp when the T-rich element is mutated (Figs 4 and 5). Hence, the 130 bp protected region should be considered as the result of two independent protections on either side of the Abf1p binding site. Analysis of the nucleosome organization of the IRS* and ERS* mutants, carrying a non-functional Abf1p binding site, revealed the complete loss of this highly ordered chromatin structure (Fig. 3). Thus, we conclude that after binding to its recognition site in the RPS28A promoter, Abf1p acts as a strong organizer of chromatin structure both upstream and downstream of the binding site.
Whereas the organization of nucleosomes into an ordered array is due to the binding of Abf1p, the creation of the nucleosome-free region adjacent to the Abf1p binding site depends upon the presence of the T-rich element. In the ETS6 mutant, which lacks a functional T-rich element (Fig. 1), the positioning of the nucleosomal array upstream of the Abf1p binding site, extending into the GLN4 gene, remains the same as in the wild-type. Downstream of the Abf1p site, however, the array of positioned nucleosomes has shifted towards the Abf1p binding site by ∼60 bp, thus completely obliterating the nucleosome-free region present in the wild-type RPS28A promoter. Apparently, Abf1p is not able to create a nucleosome-free region but merely acts as a nucleosomal boundary element.
High-resolution mapping (Fig. 5) shows that the nucleosome-free region extends 35–55 bp beyond the T-rich element. Since sequences from 18 bp downstream of the T-rich element are A-rich, having one A4 and two A3 stretches, the observed shift of the nucleosome beyond this region may be attributed to nucleosome exclusion properties of both the T-rich element and the A-rich region. Such nucleosome exclusion properties are known for stretches of As and Ts (see Introduction). However, the strong nucleosomal protection of both the core promoter and the A-rich region upon replacement of the T-rich element (Figs 3–5), suggests that the nucleosome exclusion properties of the A-rich region are very minor indeed. An alternative explanation for the extended nucleosome-free region at the RPS28A promoter may be that yet unidentified proteins bind downstream of the T-rich element and form a physical boundary for nucleosomes. Previous in vitro DNase footprinting (28) and methylation interference experiments (T.M.Doorenbosch, unpublished data) carried out in our laboratory, indicate the existence of a protein(s), called GDUF, that binds to the region of the RPS28A promoter downstream of the T-rich element between positions –108 and –87. Since the latter position is very close to the upstream boundary of the nucleosomal array at position –83, this factor(s) may form a physical boundary for nucleosomes in vivo. So far, however, both the identity of these proteins and their possible role in transcriptional activation remains unclear. Nevertheless, the observation that the nucleosome-free region is completely lost when the T-rich element is mutated consequently suggests that the T-rich element enables binding of these proteins to downstream sequences. This strongly correlates with the observations that homopolymeric (dA:dT) elements increase the accessibility of neighbouring binding sites in vivo (12,17). The idea that the T-rich element functions through a similar mechanism to these homopolymeric (dA:dT) elements is further supported by our finding that structurally rigid poly(dC:dG) can functionally replace the poly(dA:dT) sequences of the T-rich element (ITc in Fig. 1), suggesting that the T-rich element also functions by virtue of its intrinsic DNA structure and in this manner increases the accessibility of adjacent sequences to additional proteins.
Even though a T-stretch encompassing as few as six residues can still act in synergism with the Abf1p site to cause substantial transcriptional activation, the fully active T-rich element consists of two stretches of thymidines separated by an A-rich region (Fig. 1). This type of alternating sequence is expected to cause bending of the DNA (29,30), rather than form the ‘rod-like’ structure characteristic of homopolymeric sequences. Gel electrophoretic analysis of DNA fragments containing the various wild-type and mutant promoter sequences depicted in Figure 1 have indeed revealed differences in mobility that suggest differences in intrinsic DNA bending (data not shown). Abf1p binding, however, also causes the DNA to bend (31). Altering the phasing between these adjacently positioned DNA-bending elements by insertion of 5 or 10 bp between Abf1p binding site and T-rich element did not significantly affect transcription, neither did insertion of 5 or 10 bp downstream of these elements (data not shown). Apparently, the respective bends in the DNA are not interdependent and are not likely to be involved in, for instance, DNA looping. Alternatively, since any anomalous intrinsic DNA structure may affect the curved path of the DNA around a nucleosome, both the intrinsic DNA-bending properties and the structural rigidity of the thymidine stretches of the T-rich element may contribute in locally affecting DNA–nucleosome interactions, similar to the proposed nucleosome destabilizing action of structurally rigid sequences (17).
In conclusion, the data presented in this paper demonstrate that the Abf1p binding site and the T-rich element each play a distinct role in organizing the chromatin structure of an rp-gene promoter and the adjacent coding region. We favour a model in which Abf1p is able to organize chromatin by forming a physical boundary for nucleosomes and the T-rich element subsequently enables binding of other proteins to downstream sequences, creating a nucleosome-free region of ∼65 bp downstream of the Abf1p binding site.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Gerard Griffioen and Ruud Laan for their contribution and Dr Fritz Thoma (Switzerland) for his advice in the early stages of the experiments. Furthermore, we are endebted to Dr Dick Raué (this laboratory) for advice and critical reading of the manuscript and thank Dr Edward Winter (Thomas Jefferson University, USA) for providing the DAT1 and dat1-deletion strains. The work presented in this paper was financially supported by the Dutch Organization for Scientific Research (NWO).
REFERENCES
- 1.Planta R.J., Gonçalves,P.M. and Mager,W.H. (1995) Biochem. Cell Biol., 73, 825–834. [DOI] [PubMed] [Google Scholar]
- 2.Gonçalves P.M., Maurer,K., van Nieuw Amerongen,G., Bergkamp-Steffens,K., Mager,W.H. and Planta,R.J. (1996) Mol. Microbiol., 19, 535–543. [DOI] [PubMed] [Google Scholar]
- 3.Buchman A.R. and Kornberg,R.D. (1990) Mol. Cell. Biol., 10, 887–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kraakman L.S., Mager,W.H., Grootjans,J.J. and Planta,R.J. (1991) Biochim. Biophys. Acta, 1090, 204–210. [DOI] [PubMed] [Google Scholar]
- 5.Remacle J.E. and Holmberg,S. (1992) Mol. Cell. Biol., 12, 5516–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marahrens Y. and Stillman,B. (1992) Science, 255, 817–823. [DOI] [PubMed] [Google Scholar]
- 7.Gray W.M. and Fassler,J.S. (1993) Gene Expr., 3, 237–251. [PMC free article] [PubMed] [Google Scholar]
- 8.Planta R.J. (1997) Yeast, 13, 1505–1518. [DOI] [PubMed] [Google Scholar]
- 9.De Winde J.H., Van Leeuwen,H.C. and Grivell,L.A. (1993) Yeast, 9, 847–857. [DOI] [PubMed] [Google Scholar]
- 10.Venditti P., Costanzo,G., Negri,R. and Camilloni,G. (1994) Biochim. Biophys. Acta, 1219, 677–689. [DOI] [PubMed] [Google Scholar]
- 11.Thoma F. (1992) Biochim. Biophys. Acta, 1130, 1–19. [DOI] [PubMed] [Google Scholar]
- 12.Zhu Z. and Thiele,D.J. (1996) Cell, 87, 459–470. [DOI] [PubMed] [Google Scholar]
- 13.Verdone L., Camilloni,G., Di Mauro,E. and Caserta,M. (1996) Mol. Cell. Biol., 16, 1978–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rubbi L., Camilloni,G., Caserta,M., Di Mauro,E. and Venditti,S. (1997) Biochem. J., 328, 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Puhl H.L., Gudibande,S.R. and Behe,M.J. (1991) J. Mol. Biol., 222, 1149–1160. [DOI] [PubMed] [Google Scholar]
- 16.Losa R., Omari,S. and Thoma,F. (1990) Nucleic Acids Res., 18, 3495–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iyer V. and Struhl,K. (1995) EMBO J., 14, 2570–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reardon B.J., Gordon,D., Ballard,M.J. and Winter,E. (1995) Nucleic Acids Res., 23, 4900–4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moreira J.M., Remacle,J.E., Kielland-Brandt,M.C. and Holmberg,S. (1998) Mol. Gen. Genet., 258, 95–103. [DOI] [PubMed] [Google Scholar]
- 20.Ludmerer S.W. and Schimmel,P. (1985) J. Bacteriol., 163, 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winter E. and Varshavsky,A. (1989) EMBO J., 8, 1867–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gonçalves P.M., Griffioen,G., Minnee,R., Bosma,M., Kraakman,L.S., Mager,W.H. and Planta,R.J. (1995) Nucleic Acids Res., 23, 1475–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gietz R.D. and Sugino,A. (1988) Gene, 74, 527–534. [DOI] [PubMed] [Google Scholar]
- 24.Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1994) Current Protocols in Molecular Biology. John Wiley and Sons, Inc., New York.
- 25.Venditti S. and Camilloni,G. (1994) Mol. Gen. Genet., 242, 100–104. [DOI] [PubMed] [Google Scholar]
- 26.Venema J., Planta,R.J. and Raue,H.A. (1998) Methods Mol. Biol., 77, 257–270. [DOI] [PubMed] [Google Scholar]
- 27.Stanway C.A., Gibbs,J.M., Kearsey,S.E., Lopez,M.C. and Baker,H.V. (1994) Mol. Gen. Genet., 243, 207–214. [DOI] [PubMed] [Google Scholar]
- 28.Herruer M.H., Mager,W.H., Doorenbosch,T.M., Wessels,P.L., Wassenaar,T.M. and Planta,R.J. (1989) Nucleic Acids Res., 17, 7427–7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hagerman P.J. (1992) Biochim. Biophys. Acta, 1131, 125–132. [DOI] [PubMed] [Google Scholar]
- 30.Haran T.E., Kahn,J.D. and Crothers,D.M. (1994) J. Mol. Biol., 244, 135–143. [DOI] [PubMed] [Google Scholar]
- 31.McBroom L.D. and Sadowski,P.D. (1994) J. Biol. Chem., 269, 16461–16468. [PubMed] [Google Scholar]
- 32.Schmitz U.K., Lonsdale,D.M. and Jefferson,R.A. (1990) Curr. Genet., 17, 261–264. [DOI] [PubMed] [Google Scholar]