

Abstract
Prior studies have established the important role of extracellular signal-regulated kinase 1/2 (ERK1/2) as a mediator of acute kidney injury (AKI). We demonstrated rapid ERK1/2 activation induced renal dysfunction following ischemia/reperfusion (IR)-induced AKI and downregulated the mitochondrial biogenesis (MB) regulator, peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) in mice. In this study, ERK1/2 regulation of cellular nicotinamide adenine dinucleotide (NAD) and PGC-1α were explored. Inhibition of ERK1/2 activation during AKI in mice using the MEK1/2 inhibitor, trametinib, attenuated renal cortical oxidized NAD (NAD+) depletion. The rate-limiting NAD biosynthesis salvage enzyme, NAMPT, decreased following AKI, and this decrease was prevented by ERK1/2 inhibition. The microRNA miR34a decreased with the inhibition of ERK1/2, leading to increased NAMPT protein. Mice treated with a miR34a mimic prevented increases in NAMPT protein in the renal cortex in the presence of ERK1/2 inhibition. In addition, ERK1/2 activation increased acetylated PGC-1α, the less active form, whereas inhibition of ERK1/2 activation prevented an increase in acetylated PGC-1α after AKI through SIRT1 and NAD+ attenuation. These results implicate IR-induced ERK1/2 activation as an important contributor to the downregulation of both PGC-1α and NAD+ pathways that ultimately decrease cellular metabolism and renal function. Inhibition of ERK1/2 activation prior to the initiation of IR injury attenuated decreases in PGC-1α and NAD+ and prevented kidney dysfunction.
Electronic supplementary material
The online version of this article (10.1007/s00018-019-03391-z) contains supplementary material, which is available to authorized users.
Keywords: ERK1/2, Nicotinamide adenine dinucleotide, Kidney, Mitochondrial biogenesis, Cellular metabolism, Ischemia–reperfusion, miR-34a
Introduction
Extracellular regulated kinases 1/2 (ERK1/2) are protein-serine/threonine kinases that are activated through phosphorylation by the upstream mitogen-activated protein kinase kinase 1/2 (MEK1/2) [1, 2]. This signaling pathway is initiated by various extracellular stimuli and intracellular signaling that ultimately targets several physiological and pathological pathways [3, 4]. Many reviews have covered the cellular processes that ERK1/2 is known to participate, such as proliferation, differentiation, cell survival, migration, death, and metabolism [2, 5–9]. Because ERK1/2 regulates a broad array of processes, and many of these processes are seemingly opposite in nature, context is very important in ERK1/2 signaling.
We previously demonstrated rapid ERK1/2 activation following renal ischemia–reperfusion (IR) injury downregulates mitochondrial biogenesis (MB) by decreasing PGC-1α, the master regulator of MB, and the downstream targets of MB expression in renal cortical epithelial cells, as well as kidney injury markers [10, 11]. This is especially relevant as epithelial cells of the proximal tubules have a high mitochondrial content to produce ATP that drives various transport processes. This is not unique to the epithelial cells of the kidney, as the epithelium of the intestine, lung (alveolar type II cell), and the liver (hepatocytes) require proper functioning mitochondria for their energy demands [12–15]. To meet the energy demands, renal proximal tubules rely on mitochondrial oxidative phosphorylation exclusively and any mitochondrial dysfunction can disrupt the energy balance resulting in cellular injury/death and prevent recovery from injury [16, 17].
PGC-1α is a transcriptional coactivator involved in the regulation of cellular and mitochondrial metabolism through a number of pathways that include MB, mitochondrial dynamics, and mitophagy [18–20]. PGC-1α is enriched in tissues with high mitochondrial content and energy demands, such as the heart, skeletal muscle, and kidney [21–23]. Specifically, PGC-1α is highly expressed in the renal cortical epithelial cells and has been demonstrated to play a vital protective role in multiple diverse renal diseases, including sepsis, IR injury, diabetes, and fibrosis [20].
Using the potent and specific MEK1/2 inhibitor, trametinib, we previously blocked ERK1/2 phosphorylation in the kidney of naive mice and observed increases in PGC-1α and downstream targets of PGC-1α, including nuclear-encoded mitochondrial transcription factor A (TFAM) and nuclear respiratory factor-1 (NRF1), and the mitochondrial-encoded electron transport chain protein cytochrome c oxidase I (COX1) [10]. Trametinib pretreatment prevented ERK1/2 activation due to IR injury and attenuated IR-induced PGC-1α mRNA and protein loss, ultimately preventing a rise in serum creatinine, a marker of renal dysfunction (Fig. 1a). Similarly, ERK1/2 inhibition also prevented the downregulation of renal PGC-1α in a lipopolysaccharide (LPS)-induced sepsis model in mice and attenuated kidney dysfunction [24].
Fig. 1.

ERK1/2 inhibition-induced PGC-1α and miR34a regulation and the connection to the NAD+ salvage pathway through the enzyme, NAMPT. Illustrative connections between trametinib pretreatment and inhibition of ERK1/2 phosphorylation and the NAD salvage pathway. a By inhibiting ERK1/2 phosphorylation before renal IR AKI renal dysfunction is prevented. Trametinib treatment mediated mitochondrial biogenesis through an increase in PGC-1α non-acetylated protein and ameliorated NAD+ and the rate-limiting NAD salvage enzyme, NAMPT, loss following IR by decreasing miR34a. miR34a did not change due to IR alone. b Illustration of NAD generation from both the de novo pathway and the salvage pathway
Overexpression of PGC-1α in cultures of primary rabbit renal proximal tubule epithelial cells (RPTC) promoted mitochondrial and cellular recovery following oxidant injury [25]. Similar results were obtained in RPTC using a pharmacological compound, formoterol, which acts through the β2-adrenergic receptor to increase PGC-1α expression leading to an increase in maximal oxygen consumption rate and MB. In vivo PGC-1α upregulation using formoterol increased mitochondrial electron transport chain proteins and improved renal recovery following IR injury [26, 27].
NAD+ is a vital coenzyme used in cellular energy metabolism, redox signaling, and contributes to overall cellular health [28, 29]. NAD+ is often depleted during injury or disease, and restoration or prevention of this depletion averts worsening injury and often promotes recovery [30]. The kidney is highly susceptible to NAD+ depletion as it has the lowest NAD+ half-life at ~ 30 min, yet is also among the highest NAD+ containing organs [31].
Tran et al. reported a connection between PGC-1α and nicotinamide adenine dinucleotide (NAD) metabolism [32, 33]. Utilizing both PGC-1α knockout and overexpressing transgenic mice, the group determined that PGC-1α is required for less severe AKI, including improved renal recovery and increased survival. Additionally, the group found that PGC-1α-mediated AKI protection was largely due to the prevention of NAD+ depletion through upregulation of the de novo NAD+ pathway, not the salvage pathway (Fig. 1b). Exogenous nicotinamide (NAM) administered to PGC-1α KO mice improved renal function after injury by increasing the NAD+ pool [32].
The importance of NAD+ in the kidney was also confirmed by Guan et al. by preventing NAD+ depletion and renal dysfunction in both cisplatin and IR injury-induced AKI in 3- and 20-month-old mice by administering nicotinamide mononucleotide (NMN), a NAD+ precursor [34]. NMN also restored NAD+ and sirtuin 1 (SIRT1) levels [34]. Age-associated AKI microarray data pointed to MAPK signaling as an important contributor following cisplatin injury, possibly connecting MAPK signaling and NAD. However, less is known about the interactions between ERK1/2 and NAD+ metabolism in the proximal tubules and kidney. Considering the important roles of ERK1/2, PGC-1α, and NAD+ physiologically and pathologically, and our previous reports of ERK1/2 regulation on renal PGC-1α, we hypothesized early ERK1/2 activation following IR-induced AKI regulates NAD+ metabolism.
Results
ERK1/2 inhibition prevents IR-induced downregulation of PGC-1α and downstream targets and attenuates kidney dysfunction
The effect of ERK1/2 activation and inhibition on PGC-1α signaling in the renal cortex 24 h after renal IR injury was examined. We previously demonstrated the potent and selective MEK1/2 inhibitor trametinib blocks ERK1/2 activation in this model [10] and confirmed ERK1/2 phosphorylation increased in the IR group and was blocked by trametinib, without a change in total ERK1/2 (Fig. 2A). PGC-1α mRNA and protein decreased ~ 70% and ~ 55%, respectively, 24 h after IR injury (Fig. 2B, C). Pretreatment with trametinib 1 h before IR injury partially prevented this decrease in PGC-1α mRNA and protein at 24 h (Fig. 2B, C). MB proteins involved in PGC-1α signaling and the mitochondrial electron transport chain such as nuclear-encoded NDUFS1 and NDUFB8, and the mitochondrial-encoded COX1 decreased after IR injury and were completely attenuated by trametinib pretreatment (Fig. 2A, B). Specific downstream transcriptional targets of PGC-1α, including TFAM, superoxide dismutase 2 (SOD2), and forkhead box protein O1 (FOXO1) mRNA, that were previously demonstrated to be regulated by ERK1/2 [10] were reduced ~ 70% after 24 h and was partially attenuated by trametinib (Fig. 2C). In addition, the renal dysfunction marker, serum creatinine, increased ~ tenfold after IR and trametinib completely prevented the increase in serum creatinine, and the loss of renal function in response to IR injury (Fig. 2D). PGC-1α signaling has been shown to play a critical role in MB, and preventing and restoring renal function in settings of AKI [10, 26, 32, 35]. MB proteins and mRNA were consistently decreased 24 h after injury and trametinib pretreatment partially or completely prevented the decreases, ultimately, preventing a rise in serum creatinine.
Fig. 2.

ERK1/2 inhibition prevents IR-induced downregulation of PGC-1α and downstream targets and attenuates kidney dysfunction. A Representative immunoblot of PGC-1α, NDUFS1, NDUFB8, COX1, pERK1/2, and tERK1/2 following IR AKI. p phosphorylated, t total. B Densitometry analysis of PGC-1α, NDUFS1, NDUFB8, and COX1 proteins following 24 h IR. C mRNA expression of PGC-1α, TFAM, SOD2, and FOXO1 following IR AKI. D Serum creatinine was assessed at 24 h after IR AKI. Data are represented as mean ± S.E.M., n ≥ 4. Different superscripts indicate statistically significant differences (P < 0.05); ‘a’ means not statistically different from sham, ‘b’ means statistically different from sham, ‘c’ means statistically different from sham and IR
ERK1/2 inhibition attenuates PGC-1α acetylation increases following 24 h AKI
Because PGC-1α acetylation status alters its activity, particularly following an acute injury [36–39], we used acetylated-lysine immunoprecipitation to look at PGC-1α acetylation. In the IR group, PGC-1α was more acetylated, corresponding to less activity, compared to the sham group (Fig. 3A). However, in the trametinib + IR group there was less acetylated PGC-1α compared to the IR group and not different than the sham group (Fig. 3A). PGC-1α is a major target of deacetylation [40, 41] for the NAD-dependent deacetylase, SIRT1. SIRT1 deacetylation of PGC-1α has been reported to play a critical role during AKI [36, 37]. After IR injury SIRT1 protein decreased and this decrease was prevented in the trametinib + IR group (Fig. 3B). SIRT1 mRNA was downregulated at 24 h after IR injury and was not altered in the trametinib + IR group (Fig. 3B). These results reveal that trametinib pretreatment preserves PGC-1α in a more active state by preventing PGC-1α acetylation following 24 h IR injury and may be mediated by the prevention of SIRT1 protein loss.
Fig. 3.

ERK1/2 inhibition attenuates PGC-1α acetylation increases following 24 h AKI. A Representative immunoblot of PGC-1α following IP of acetylated lysine. B Representative immunoblot of SIRT1, and analysis of SIRT1 protein and mRNA. Data are represented as mean ± S.E.M., n ≥ 4. Different superscripts indicate statistically significant differences (P < 0.05); ‘a’ means not statistically different from sham, ‘b’ means statistically different from sham
ERK1/2 inhibition attenuates NAD+ loss after 24 h IR injury
As SIRT1 enzymatic activity is dependent on the availability of cellular NAD+, we examined the effect ERK1/2 activation and inhibition has on NAD+ metabolism in the renal cortex following renal IR injury [42]. At 3 and 24 h after renal IR injury trametinib pretreatment blocked ERK1/2 phosphorylation (Fig. 2A; Fig. 4A). NAD+ and NADH did not change at 3 h following IR injury in any group; however, the ratio trended up in the trametinib + IR group (Fig. 4B–D). At 24 h, NAD+ and NADH decreased ~ 50% with no change in the NAD+:NADH ratio in response to IR injury alone (Fig. 4E–G). Pretreatment with trametinib 1 h before IR injury partially prevented the decrease of NAD+ without a change in NADH loss, resulting in a marked increase in the NAD+:NADH ratio (Fig. 4E–G). These results reveal that ERK1/2 plays a role in NAD+ metabolism during IR injury and inhibiting ERK1/2 attenuates the NAD+ loss.
Fig. 4.

ERK1/2 inhibition attenuates NAD+ loss after 24 h IR injury. A Representative immunoblot of pERK1/2 and tERK1/2 after IR AKI at 3 h. p phosphorylated, t total. B, C, E, F NAD + and NADH measurement after IR AKI at 3 and 24 h in the renal cortex. D, G Ratio of NAD + :NADH analyzed after IR AKI at 3 and 24 h. Data are represented as mean ± S.E.M., n ≥ 5. Different superscripts indicate statistically significant differences (P < 0.05); ‘a’ means not statistically different from sham, ‘b’ means statistically different from sham
NAMPT protein decreases following 24 h IR and trametinib decreases miR34a and prevents IR-induced NAMPT loss
To elucidate the mechanism of ERK1/2-mediated NAD+ and NADH loss in renal IR injury, protein and mRNA levels of the enzymes responsible for NAD+ biosynthesis were measured (Fig. 1). NAMPT protein was measured 3 and 24 h after IR injury and no change was observed at 3 h (Fig. 5A); however, at 24 h NAMPT protein was decreased 50% compared to sham mice (Fig. 5B, C). Trametinib pretreatment increased NAMPT protein compared to the IR group at 3 h (Fig. 5A) and at 24 h prevented the NAMPT protein loss observed in the IR group (Fig. 5B, C).
Fig. 5.

NAMPT protein decreases following 24 h IR and trametinib decreases miR34a and prevents IR-induced NAMPT loss. A Representative immunoblot and densitometry of NAMPT after IR AKI at 3 h. Note: β-actin is the same β-actin from the membrane as Fig. 4A. B Representative immunoblot of NAMPT, NMNAT1,2,3 after IR AKI at 24 h. C Densitometry analysis of NAMPT and NMNAT1,2,3 after IR AKI at 24 h. D mRNA expression measured for NAD biosynthetic pathway enzymes. E NAMPT activity measured at 24 h after IR AKI. F, G miR34a expression measured after IR AKI at 3 and 24 h. Data are represented as mean ± S.E.M., n ≥ 5. Different superscripts indicate statistically significant differences (P < 0.05); ‘a’ means not statistically different from sham, ‘b’ means statistically different from sham. c’ means statistically different from IR
Protein levels for the three enzymes responsible for conversion of NMN to NAD + revealed that only NMNAT1 was decreased in the IR group and trametinib prevented this decrease (Fig. 5B, C). NMNAT2 and NMNAT3 proteins were not changed in the IR group nor trametinib + IR group (Fig. 5B, C). The activity of NAMPT following immunoprecipitation was performed and there were no observed differences between any groups indicating no posttranslational modifications that modify activity (Fig. 5E). These results reveal that ERK1/2 inhibition increases NAMPT early after IR injury and prevents NAMPT and NMNAT1 protein loss 24 h after IR injury. As both NAMPT and NMNAT1 contribute to NAD synthesis, and NAMPT is the rate-limiting enzyme in the salvage pathway (Fig. 1) [43], and the salvage pathway has been shown to be responsible for up to 99% of the total NAD synthesis [31], ERK1/2 mediated NAD + loss is the result of NAMPT and NMNAT1 protein loss.
To determine if the decrease in NAMPT and NMNAT1 proteins was the result of decreased transcription, mRNA of the respective proteins were measured. Both vehicle and trametinib-treated IR groups exhibited ~ 60% reduction in NAMPT and NMNAT1 mRNA at 24 h (Fig. 5D). Thus, ERK1/2 inhibition did not restore NAMPT and NMNAT1 mRNA yet restored protein (Fig. 5C), suggesting that ERK1/2 regulates these enzymes independent of transcription. Other NAD+ salvage enzymes, NMNAT3 and NRK1, and the important NAD de novo enzyme, QPRT, also exhibited marked decreases in mRNA levels in the IR and trametinib + IR groups [44] (Fig. 5D). These results demonstrate that following IR injury the synthesis of key NAD+ metabolizing enzyme mRNA are substantially decreased.
The microRNA, miR34a, can directly bind to the 3′-UTR of NAMPT mRNA and prevent NAMPT translation, thus regulating NAMPT protein expression, and are inversely correlated [45]. Because NAMPT protein was increased at 3 h and attenuated at 24 h in the trametinib + IR group (Fig. 5A, B), miR34a was measured in these groups. At 3 h miR34a levels decreased in the trametinib + IR group and were not altered in the 3 h IR group (Fig. 5F). At 24 h following IR injury, miR34a levels remained low in the trametinib + IR group compared to the sham and IR groups (Fig. 5G). These results correlate ERK1/2 inhibition with decreased miR34a levels and increased NAMPT protein.
ERK1/2 physiologically regulates miR34a and ERK1/2 inhibition decreases miR-34a, leading to increases in NAMPT protein
To examine if ERK1/2 also regulates miR34a and NAMPT protein at a physiological level we administered trametinib to naïve mice. Following a 4-h trametinib treatment, ERK1/2 phosphorylation was completely inhibited in the renal cortex (Fig. 6A). NAMPT protein increased 44% in the trametinib group compared to the control group, without influencing NAMPT mRNA (Fig. 6A, B). The expression of renal miR34a decreased by 40% in the trametinib-treated group, indicating the rise in NAMPT protein following ERK1/2 inhibition is miR34a-mediated (Fig. 6B). Interestingly, NAD+ and NADH were not altered in response to the increased NAMPT protein (Fig. 6C). We also observed increased NAMPT protein in trametinib-treated primary rabbit renal tubule epithelial cells along with a concomitant increase in NAD+ content (Fig. S1, 2). These data reveal that ERK1/2 affects NAMPT and miR34a under physiological conditions and does so without changing NAD+ and NADH content in mouse renal cortex.
Fig. 6.

ERK1/2 physiologically regulates miR34a and ERK1/2 inhibition decreases miR-34a, leading to increases in NAMPT Protein. A Representative immunoblot of pERK1/2, tERK1/2, and NAMPT protein 4 h after trametinib administration. p phosphorylated, t total. B Densitometry analysis of NAMPT protein, and NAMPT mRNA and miR34a expression measured at 4 h after trametinib treatment. C NAD + and NADH content in renal cortex measured at 4 h after trametinib treatment. D miR34a expression after 34a mimic treatment with and without trametinib in mice. E Representative immunoblot of NAMPT protein after 34a mimic treatment in the renal cortex. F Densitometry analysis of NAMPT protein after 34a mimic treatment. Data are represented as mean ± S.E.M., n ≥ 5. Different superscripts indicate statistically significant differences (P < 0.05); ‘a’ means not statistically different from sham, ‘b’ means statistically different from sham. c’ means statistically different from IR
To further investigate whether ERK1/2 inhibition-induced increase in NAMPT protein is miR34a dependent, we utilized a miR34a mimic to prevent trametinib-induced decrease of miR34a. Naïve mice were treated with a mirVana miR34a mimic mixed with in vivo-jetPEI via retro-orbital injection to prevent trametinib treatment from decreasing miR34a [46]. The miR34a mimic increased miR34a levels in both the 34a mimic group and in the 34a mimic + trametinib group at least twofold compared to vehicle (Fig. 6D). Increasing miR34a levels in the cortex prevented trametinib-induced NAMPT protein upregulation and minimally decreased NAMPT protein in both the miR34a mimic group and the 34a + trametinib group (Fig. 6E, F). In summary, ERK1/2 phosphorylation inhibition decreases miR34a, which leads to an increase in NAMPT protein. This ERK1/2/miR34a/NAMPT pathway is prevented by increasing miR34a levels in the kidney exogenously using a miR34a mimic. Trametinib was unable to increase NAMPT protein with miR34a levels elevated.
Trametinib-induced prevention of serum creatinine elevation following IR is dependent on NAMPT
Because ERK1/2 inhibition increased NAMPT protein in a miR34a-dependent pathway we sought to clarify the role that NAMPT protein contributes to trametinib-induced prevention of serum creatinine elevation in IR-induced AKI. We utilized the NAMPT inhibitor, FK866, [47, 48] and mice were pretreated before IR surgery with either vehicle or FK866 20 mg/kg [49]. Mice pretreated with trametinib were protected from IR-induced serum creatinine increases compared to the Veh + IR group (Fig. 7). In contrast, mice pretreated with both FK866 and trametinib had the same increase in serum creatinine as the Veh + IR and FK866 + IR groups and reversed the protective effect of trametinib (Fig. 7). Mice pretreated with FK866 and subjected to IR injury had a similar increase in serum creatinine as in the Veh + IR group. These results provide evidence that ERK1/2 inhibition by trametinib pretreatment protects against IR-induced serum creatinine elevation after 24 h by maintaining NAMPT protein expression and ameliorating NAD+ loss following IR AKI.
Fig. 7.
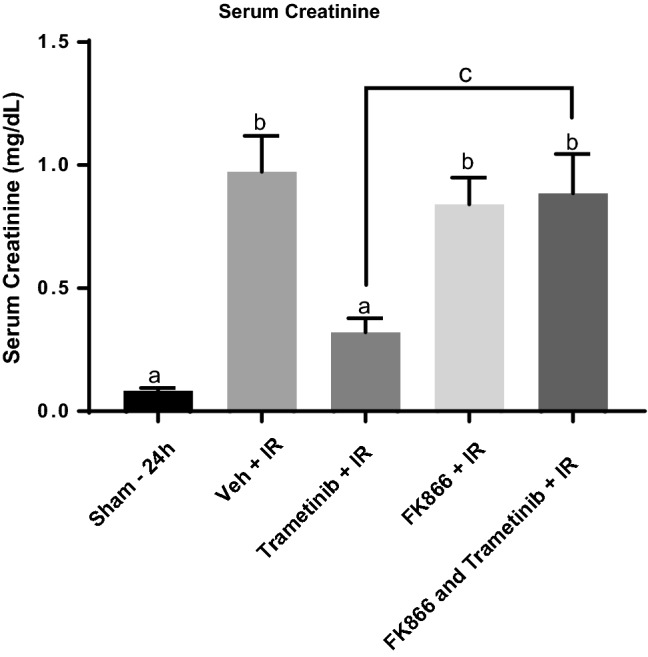
Trametinib-induced prevention of serum creatinine elevation following IR is dependent on NAMPT. Serum creatinine measurements following pretreatment with trametinib, FK866, and a combination of FK866 and trametinib after IR AKI at 24 h. Data are represented as mean ± S.E.M., n ≥ 4. Different superscripts indicate statistically significant differences (P < 0.05); ‘a’ means not statistically different from sham, ‘b’ means statistically different from sham. c’ means trametinib + IR and FK866 + trametinib + IR are statistically different from each other
Discussion
We have previously studied the role of ERK1/2 in PGC-1α signaling under physiological and pathological conditions and determined that early activation of ERK1/2 following renal IR injury was responsible for the initial decrease in PGC-1α mRNA expression, and correlated with kidney dysfunction [10]. Here, we have expanded our studies to understand the connection between ERK1/2 and NAD+ regulation under physiological and renal IR conditions, and in context with PGC-1α signaling. We determined that inhibition of ERK1/2 using trametinib, a MEK1/2 inhibitor, decreased miR34a, the microRNA regulator of both SIRT1 and NAMPT protein expression (Fig. 1). A decrease in miR34a in naïve mice led to an increase in NAMPT protein and attenuated the loss of NAMPT and SIRT1 proteins after renal IR injury. By preventing NAMPT loss at the protein level, NAD+ loss was attenuated following IR injury. Likewise, by mediating the SIRT1 protein and NAD+ loss PGC-1α acetylation was decreased likely through activation of SIRT1. Furthermore, this trametinib-induced miR34a/NAMPT regulation was prevented by pretreatment with a miR34a mimic, indicating increased NAMPT protein expression by ERK1/2 inhibition was dependent on miR34a.
Similar to our miR34a results, Choi SE et al., observed NAMPT protein regulation by miR34a in the liver of obese mice [45]. Hepatic NAMPT protein was lower in obese mice compared to controls, and was caused by an increase in miR34a. SIRT1 was also shown to correspond with miR34a levels, as miR34a can bind to and repress NAMPT and SIRT1 mRNA translation by binding to the 3′UTR of both [45, 50, 51]. ERK1/2 regulation of miR34a has not been well characterized; however, a few studies demonstrated that ERK1/2 phosphorylation is reduced as a result of a rise in miR34a expression [52, 53]. In liver cancer, active ERK1/2 was shown to phosphorylate exportin-5, which led to a decrease in pre-miRNA export from the nucleus and a global reduction in miRNA [54]. However, a regulatory connection between a decrease in phosphorylated ERK1/2 and a decrease in miR34a expression, as reported in this study, has not been elucidated.
Because NAMPT protein increased at 4 h after trametinib administration without any change in NAMPT mRNA, we conclude ERK1/2 inhibition rapidly decreases miR34a in the renal cortex and subsequently, induces an increase in NAMPT protein. Because NAMPT is the rate-limiting enzyme in the NAD-+ salvage pathway, and proximal tubules have been shown to have a high abundance of NAMPT [43, 55, 56], any prevention of the loss of this enzyme has the potential to directly impact the NAD-+ content and severity of the injury. Using primary culture, we identified renal tubule epithelial cells as being responsive to trametinib treatment, similar to our physiological studies, by inhibiting ERK1/2 phosphorylation and increasing NAMPT and PGC-1α protein as well as NAD+ content (Fig. S1–3). As both Tran et al. and Guan et al. demonstrated within the kidney, NAD+ levels can directly influence AKI susceptibility [32, 34]. However, this is not a kidney-specific phenomenon, as NAD+ boosting or pathway upregulation has been shown to have positive health outcomes relating to multiple organs, including the liver, heart, pancreas, skeletal muscle, brain, and nervous system [57–64].
NAD+ and NADH were not altered when NAMPT protein increased at 4 h possibly demonstrating the salvage pathway maintains only a fixed NAD+ level physiologically without the addition of NAD precursors. However, during an acute renal injury, we found that preventing NAMPT protein loss led to an attenuation of the NAD+ decline at 24 h following IR injury. In addition, using the NAMPT inhibitor, FK866, we found trametinib treatment did not prevent an increase in serum creatinine if given in combination with FK866. This indicates the trametinib-induced prevention of IR AKI is NAMPT dependent.
Trametinib administration prevented ERK1/2 phosphorylation in uninjured and IR-injured renal cortical tissue. PGC-1α expression decreased following IR injury at 24 h, which trametinib pretreatment attenuated. PGC-1α acetylation increased following IR injury in mice, which decreases PGC-1α transcriptional co-activator activity [40, 65]. Our group has previously demonstrated the SIRT1 activator SRT1720, in an AKI model, increased the deacetylation status of PGC-1α allowing for more effective recruitment of various transcription factors that stimulate the expression of downstream target genes [36, 40]. Here, we demonstrate the same concept using trametinib, and observed an increase in deacetylated PGC-1α in the trametinib mice following IR injury and an increase in MB associated proteins, including NDUFS1, NDUFB8, and COX1. Furthermore, trametinib pretreatment prevented the rise in serum creatinine at 24 h post IR injury observed in the vehicle-treated group.
Because trametinib altered the acetylation status of PGC-1α and increased PGC-1α downstream targets of MB, SIRT1 and the necessary coenzyme for SIRT1 enzymatic activity, NAD+, were further studied. The increase in deacetylated PGC-1α due to trametinib administration was discovered to likely stem from the attenuation of the decrease in SIRT1 protein and NAD+ content following IR injury. The availability of NAD+ is directly linked to the activity of SIRT1; therefore, having more SIRT1 and NAD+ available in the cortex in the trametinib group prevented an increase in PGC-1α acetylation following IR injury [41, 42, 66].
An increase of PGC-1α in a renal tubule inducible PGC-1α transgenic mouse model upregulated the enzymes of the de novo NAD biosynthetic pathway [32]. The opposite was found as a decrease in PGC-1α suppressed the enzymes of the NAD biosynthetic pathway, as observed in both Pgc1α−/− uninjured kidneys and WT IR kidneys [32]. We observed similar results in our studies where PGC-1α is downregulated and NAD content is decreased after renal IR injury [10]. Better renal functional outcomes were observed in the PGC-1α transgenic mice compared to controls, and worse outcomes were observed in the Pgc1α−/− mice, linking PGC-1α and NAD to AKI severity. Here, a decrease in PGC-1α protein and PGC-1α acetylation prevented renal dysfunction as measured by serum creatinine in addition to amelioration of the AKI-induced NAD+ loss.
These experimental results have identified three important findings: (1) ERK1/2 inhibition before IR AKI attenuates the downregulation of PGC-1α and certain MB targets out to 24 h, (2) renal ERK1/2 inhibition decreases miR34a levels acutely leading to SIRT1 and NAMPT protein increases, and (3) NAMPT protein is vital for ERK1/2 inhibition-induced reduction in IR AKI severity (Figs. 1a, 7). In addition, trametinib pretreatment prevented ERK1/2 phosphorylation and attenuated kidney dysfunction as measured by serum creatinine at 24 h following IR-induced AKI. An accumulation of factors likely attributes to the trametinib-induced prevention of renal AKI dysfunction, which includes attenuation of PGC-1α, SIRT1, and NAMPT protein reductions, PGC-1α acetylation, and NAD+ loss.
Methods
In vitro studies
Female New Zealand White rabbits (2 kg) were purchased from Charles River (Oakwood, MI/Canada). Renal proximal tubule cells (RPTC) were isolated using the iron oxide perfusion method and grown in 35-mm tissue culture dishes under improved conditions similar to what is observed in vivo [67]. The culture medium was a 1:1 mixture of Dulbecco’s modified Eagle’s medium/F-12 (without glucose, phenol red, or sodium pyruvate) supplemented with 15 mM HEPES buffer, 2.5 mM l-glutamine, 1 μM pyridoxine HCl, 15 mM sodium bicarbonate, and 6 mM lactate. Hydrocortisone (50 nM), selenium (5 ng/ml), human transferrin (5 ug/ml), bovine insulin (10 nM), and l-ascorbic acid-2-phosphate (50 μM) were added to fresh culture medium. Confluent RPTC were used for all experiments. RPTC monolayers were treated with various compounds or vehicle (DMSO).
Naïve and AKI studies
Trametinib (GSK1120212, [68] was purchased from Selleckchem Chemicals (Houston, TX). Eight- to nine-week-old male C57BL/6 mice (20–25 g) were acquired from Charles River Laboratories (Frederick, MD) and administered an injection of trametinib (1 mg/kg) or vehicle control (NEOBEE M-5 – Fisher Scientific) intraperitoneally (ip). Four hours after injection, kidneys were collected and flash frozen in liquid nitrogen for further analysis.
For IRI-induced AKI, mice were assigned to three groups: (1) Sham, (2) IR + Vehicle, (3) IR + trametinib. Vehicle or trametinib were administered ip 1 h before surgery. In some experiments (Fig. 7), mice were pretreated 48 and 24 h before IR surgery with vehicle, FK866 (20 mg/kg) (Selleckchem) [S2799], or NMN (500 mg/kg) (Sigma-Aldrich) [N3501] by ip injection [49]. IRI mice were subjected to ischemia/reperfusion surgery by bilateral renal pedicle clamping for 18.5 min as described previously [69]. Briefly, the renal artery and vein were isolated and blood flow was occluded with a vascular clamp while maintaining a constant body temperature of 36.5 ~ 37.5 °C. Sham mice were treated the same as IRI mice, except the renal pedicles were not clamped. Mice were killed at 3 or 24 h after surgery, and blood and kidneys (flash frozen in liquid nitrogen) were collected for analysis. All studies were conducted in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Animal use was approved by the Institutional Animal Care and Use Committee at both the Medical University of South Carolina and the University of Arizona.
Retro-orbital injection of microRNA 34a mimic
Polyplus transfection (Illkirch, France) delivery reagent, in vivo-jetPEI, was used for delivering microRNA 34a mimic to the kidney. Preparation of delivery reagent was carried out according to manufacturer’s instructions. A 5% glucose solution mixed with in vivo-jetPEI was added to a 5% glucose solution containing mirVana miRNA 34a mimic. Combined solutions were vortexed and allowed to incubate at RT for 15–20 min. Retro-orbital injection was performed using a solution containing 40 μg of miR34a mimic and a N/P ratio of eight. An ip injection of vehicle control (NEOBEE M-5) or trametinib (1 mg/kg) was administered 2 h later. Mice were euthanized 6 h following retro-orbital injection. Mice were assigned to three groups: (1) jetPEI (retro-orbital) + Vehicle (ip), (2) miR34a mimic + Vehicle, (3) miR34a mimic + trametinib.
Serum creatinine measurement
Serum creatinine (SCr) was determined using the Creatinine Enzymatic Reagent Assay kit (Pointe Scientific, Canton, MI) [c7548-120] based on the manufacturer’s directions. All values are expressed as serum creatinine concentration in milligrams per deciliter.
RT-qPCR analysis of mRNA and microRNA expression
Total RNA was isolated from renal cortical tissue with TRIzol reagent (Life Technologies). The iScript Advanced cDNA Synthesis Kit (Bio-Rad) was used according to the manufacturer’s protocol. The generated cDNA was used with the SsoAdvanced Universal SYBR Green Supermix reagent (Bio-Rad). The relative mRNA expression of all genes was determined by the 2− ΔΔCt method, and mouse actin RNA was used as a reference for normalization (see Table 1 for Primer pairs). For microRNA the MystiCq microRNA cDNA synthesis kit was used according to the manufacturer’s instructions (Sigma-Aldrich) [MIRRT]. MystiCq SYBR green (Sigma-Aldrich) [MIRRM00] and universal PCR primer (Sigma-Aldrich) [MIRUP] were used for RT-qPCR. The relative microRNA expression was determined by the 2−ΔΔCt method, and the mouse small nucleolar RNA, SNORD48 was used as a reference small non-coding RNA for normalization (Sigma-Aldrich) [MIRCP00007].
Table 1.
Mouse primer sequence pairs with forward (F) and reverse (R) primers identified
| Gene | F/R | Mouse—Primer Sequences |
|---|---|---|
| Actin | F | 5’-GGGATGTTTGCTCCAACCAA |
| R | 5’-GCGCTTTTGACTCAGGATTTAA | |
| FOXO1 | F | 5’-CGGAAAATCACCCCGGAGAA |
| R | 5’-TACACCAGGGAATGCACGTC | |
| NAMPT | F | 5’-GTGACTTAAGCAACGGAGCG |
| R | 5’-CTTTGCTTGTGTTGGGTGGG | |
| NMNAT1 | F | 5’-GTGGAGACTGTGAAGGTGCTC |
| R | 5’-GTGAGCTTTGTGGGTAACTGC | |
| NMNAT3 | F | 5’-GGTGTGGAGGTGTGTGACAGC |
| R | 5’-GCCATGGCCACTCGGTGATGG | |
| NMRK1 | F | 5’-CTTGAAGCTTGCTCTGCGAC |
| R | 5’-GTGTCGTCTTCCCTCCGTTT | |
| PGC-1α | F | 5’-AGGAAATCCGAGCGGAGCTGA |
| R | 5’-GCAAGAAGGCGACACATCGAA | |
| QPRT | F | 5’-TGGAAAGGGCAGTGCTGAA |
| R | 5’-AGGCACTGGGGTATCTCCTT | |
| SIRT1 | F | 5’-AATCCAGTCATTAAACGGTCTACAA |
| R | 5’-TAGGACCATTACTGCCAGAGGA | |
| SOD2 | F | 5’-ACACATTAACGCGCAGATCA |
| R | 5’-AGCCTCCAGCAACTCTCCTT | |
| TFAM | F | 5’-GCTGATGGGTATGGAGAAG |
| R | 5’-GAGCCGAATCATCCTTTGC |
Immunoblot analysis
Protein was extracted from renal cortex using RIPA assay buffer (50 mM Tris–HCl, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, pH 7.4). Protease inhibitor cocktail (1:100), 1 mM sodium fluoride, and 1 mM sodium orthovanadate (Sigma-Aldrich) and nicotinamide and 4-phenylbutyrate were added fresh before each extraction.
Equal protein quantities (10–60 μg) were loaded onto 4–15% SDS-PAGE gels, resolved by gel electrophoresis, and transferred onto nitrocellulose membranes (Bio-Rad). Membranes were blocked in 5% bovine serum albumin or 5% milk in TBST and incubated overnight with primary antibody at 4 °C with gentle agitation. Primary antibodies used in these studies included phospho-ERK1/2 (1:1000) [#4370], total ERK1/2 (1:1000) [#4695]. β-actin (1:2000; Santa Cruz Biotechnology, Dallas, TX) [sc-47778].
Membranes were incubated with the appropriate horseradish peroxidase conjugated secondary antibody before visualization using enhanced chemiluminescence (Thermo Scientific) and the GE ImageQuant LAS4000 (GE Life Sciences). Optical density was determined using the ImageJ software from NIH and Image Studio Lite from LI-COR.
Immunoprecipitation
Following protein extraction, protein quantification was performed using Pierce 660 nm Assay (ThermoFisher Scientific) [22660]. Equal amounts of protein from every group were utilized according to the instructions from Dynabeads Protein G Immunoprecipitation Kit (ThermoFisher Scientific). Immunoprecipitation was performed using acetylated-lysine antibody (1:100; Cell Signaling) [#9441]. PGC-1α antibody (1:1000; abcam) [ab54481] was used for immunoblotting pull-down and input. Veriblot (1:400; abcam) [ab131366] was used for detection of immunoblotted target protein before visualization using enhanced chemiluminescence (Thermo Scientific) and the GE ImageQuant LAS4000 (GE Life Sciences).
NAD+ and NADH measurement
Quantification of NAD+ and NADH was carried out using the NAD/NADH Assay Kit (abcam) [#ab65348] according to the manufacturer’s instructions. NAD+ and NADH values were normalized to kidney cortex wet weight.
NAMPT activity assay
To quantify the enzymatic activity of NAMPT in kidney cortex, protein extracts were assayed according to the manufacturer’s instructions using the Cyclex NAMPT Colorimetric Assay Kit (MBL International) [#CY-1251].
Statistical analysis
All data are shown as mean ± S.E.M. When comparing two experimental groups, an unpaired, two-tailed t test or Mann–Whitney U was used to determine statistical differences. A one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was performed for comparisons of multiple groups. P < 0.05 was considered statistically significant. All statistical tests were performed using GraphPad Prism software (GraphPad Software, San Diego, CA).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This study was funded by National Institutes of Health National Institute of General Medical Sciences [Grant R01GM084147] award to R.G.S.; the Biomedical Laboratory Research and Development Program of the Department of Veterans Affairs [Grant BX000851] awarded to R.G.S.; and the Ruth L. Kirschstein National Research Service Award Individual Predoctoral Fellowship through the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant F31DK105782] awarded to J.B.C.
Author contributions
Participated in research design: JBC, RGS. Conducted experiments: JBC. Performed data analysis: JBC. Wrote or contributed to the writing of the manuscript: JBC, RGS.
Compliance with ethical standards
Conflict of interests
The authors declare that they have no conflict of interest.
Ethical standards
All studies were conducted in accordance with the recommendations set forth in the Guide for the Care and Use of Laboratory Animals by the National Institutes of Health. Animal use was approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina and the University of Arizona.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Forrester SJ, et al. Epidermal growth factor receptor transactivation: mechanisms, pathophysiology, and potential therapies in the cardiovascular system. Annu Rev Pharmacol Toxicol. 2016;56:627–653. doi: 10.1146/annurev-pharmtox-070115-095427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eblen ST. Extracellular-regulated kinases: signaling from Ras to ERK substrates to control biological outcomes. Adv Cancer Res. 2018;138:99–142. doi: 10.1016/bs.acr.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daub H, et al. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379(6565):557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 4.Luttrell LM, et al. Gbetagamma subunits mediate Src-dependent phosphorylation of the epidermal growth factor receptor. A scaffold for G protein-coupled receptor-mediated Ras activation. J Biol Chem. 1997;272(7):4637–4644. doi: 10.1074/jbc.272.7.4637. [DOI] [PubMed] [Google Scholar]
- 5.Wainstein E, Seger R. The dynamic subcellular localization of ERK: mechanisms of translocation and role in various organelles. Curr Opin Cell Biol. 2016;39:15–20. doi: 10.1016/j.ceb.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Roskoski R., Jr ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012;66(2):105–143. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 7.Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death–apoptosis, autophagy and senescence. FEBS J. 2010;277(1):2–21. doi: 10.1111/j.1742-4658.2009.07366.x. [DOI] [PubMed] [Google Scholar]
- 8.Keshet Y, Seger R. The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol Biol. 2010;661:3–38. doi: 10.1007/978-1-60761-795-2_1. [DOI] [PubMed] [Google Scholar]
- 9.Jain R, et al. ERK activation pathways downstream of GPCRs. Int Rev Cell Mol Biol. 2018;338:79–109. doi: 10.1016/bs.ircmb.2018.02.003. [DOI] [PubMed] [Google Scholar]
- 10.Collier JB, et al. Rapid renal regulation of peroxisome proliferator-activated receptor gamma coactivator-1alpha by extracellular signal-regulated kinase 1/2 in physiological and pathological conditions. J Biol Chem. 2016;291(52):26850–26859. doi: 10.1074/jbc.M116.754762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collier JB, Schnellmann RG. Extracellular signal-regulated kinase 1/2 regulates mouse kidney injury molecule-1 expression physiologically and following ischemic and septic renal injury. J Pharmacol Exp Ther. 2017;363(3):419–427. doi: 10.1124/jpet.117.244152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berger E, et al. Mitochondrial function controls intestinal epithelial stemness and proliferation. Nat Commun. 2016;7:13171. doi: 10.1038/ncomms13171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel AS, et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS One. 2015;10(3):e0121246. doi: 10.1371/journal.pone.0121246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lottes RG, et al. Lactate as substrate for mitochondrial respiration in alveolar epithelial type II cells. Am J Physiol Lung Cell Mol Physiol. 2015;308(9):L953–L961. doi: 10.1152/ajplung.00335.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rui L. Energy metabolism in the liver. Compr Physiol. 2014;4(1):177–197. doi: 10.1002/cphy.c130024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zuk A, Bonventre JV. Acute kidney injury. Annu Rev Med. 2016;67:293–307. doi: 10.1146/annurev-med-050214-013407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall AM, et al. Multiphoton imaging reveals differences in mitochondrial function between nephron segments. J Am Soc Nephrol. 2009;20(6):1293–1302. doi: 10.1681/ASN.2008070759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vainshtein A, et al. Role of PGC-1alpha during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am J Physiol Cell Physiol. 2015;308(9):C710–C719. doi: 10.1152/ajpcell.00380.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Z, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98(1):115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 20.Lynch MR, Tran MT, Parikh SM. PGC1alpha in the kidney. Am J Physiol Renal Physiol. 2018;314(1):F1–F8. doi: 10.1152/ajprenal.00263.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88(2):611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- 22.Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24(1):78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 23.Puigserver P, et al. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92(6):829–839. doi: 10.1016/S0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 24.Smith JA, et al. Suppression of mitochondrial biogenesis through toll-like receptor 4-dependent mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signaling in endotoxin-induced acute kidney injury. J Pharmacol Exp Ther. 2015;352(2):346–357. doi: 10.1124/jpet.114.221085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rasbach KA, Schnellmann RG. PGC-1alpha over-expression promotes recovery from mitochondrial dysfunction and cell injury. Biochem Biophys Res Commun. 2007;355(3):734–739. doi: 10.1016/j.bbrc.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 26.Jesinkey SR, et al. Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. J Am Soc Nephrol. 2014;25(6):1157–1162. doi: 10.1681/ASN.2013090952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cameron RB, Beeson CC, Schnellmann RG. Structural and pharmacological basis for the induction of mitochondrial biogenesis by formoterol but not clenbuterol. Sci Rep. 2017;7(1):10578. doi: 10.1038/s41598-017-11030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katsyuba E, Auwerx J. Modulating NAD(+) metabolism, from bench to bedside. EMBO J. 2017;36(18):2670–2683. doi: 10.15252/embj.201797135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Preyat N, Leo O. Complex role of nicotinamide adenine dinucleotide in the regulation of programmed cell death pathways. Biochem Pharmacol. 2016;101:13–26. doi: 10.1016/j.bcp.2015.08.110. [DOI] [PubMed] [Google Scholar]
- 30.Hershberger KA, Martin AS, Hirschey MD. Role of NAD(+) and mitochondrial sirtuins in cardiac and renal diseases. Nat Rev Nephrol. 2017;13(4):213–225. doi: 10.1038/nrneph.2017.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mori V, et al. Metabolic profiling of alternative NAD biosynthetic routes in mouse tissues. PLoS One. 2014;9(11):e113939. doi: 10.1371/journal.pone.0113939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tran MT, et al. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature. 2016;531(7595):528–532. doi: 10.1038/nature17184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allison SJ. Acute kidney injury: improved fuel metabolism protects against AKI. Nat Rev Nephrol. 2016;12(5):255. doi: 10.1038/nrneph.2016.45. [DOI] [PubMed] [Google Scholar]
- 34.Guan Y, et al. Nicotinamide mononucleotide, an NAD(+) precursor, rescues age-associated susceptibility to AKI in a sirtuin 1-dependent manner. J Am Soc Nephrol. 2017;28(8):2337–2352. doi: 10.1681/ASN.2016040385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tran M, et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest. 2011;121(10):4003–4014. doi: 10.1172/JCI58662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Funk JA, Schnellmann RG. Accelerated recovery of renal mitochondrial and tubule homeostasis with SIRT1/PGC-1alpha activation following ischemia-reperfusion injury. Toxicol Appl Pharmacol. 2013;273(2):345–354. doi: 10.1016/j.taap.2013.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Funk JA, Odejinmi S, Schnellmann RG. SRT1720 induces mitochondrial biogenesis and rescues mitochondrial function after oxidant injury in renal proximal tubule cells. J Pharmacol Exp Ther. 2010;333(2):593–601. doi: 10.1124/jpet.109.161992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim SB, et al. Acetylation of PGC1alpha by histone deacetylase 1 downregulation is implicated in radiation-induced senescence of brain endothelial cells. J Gerontol A Biol Sci Med Sci. 2018;74(6):787–793. doi: 10.1093/gerona/gly167. [DOI] [PubMed] [Google Scholar]
- 39.Jeninga EH, Schoonjans K, Auwerx J. Reversible acetylation of PGC-1: connecting energy sensors and effectors to guarantee metabolic flexibility. Oncogene. 2010;29(33):4617–4624. doi: 10.1038/onc.2010.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gerhart-Hines Z, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007;26(7):1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morigi M, Perico L, Benigni A. Sirtuins in renal health and disease. J Am Soc Nephrol. 2018;29(7):1799–1809. doi: 10.1681/ASN.2017111218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Canto C, Auwerx J. Targeting sirtuin 1 to improve metabolism: all you need is NAD(+)? Pharmacol Rev. 2012;64(1):166–187. doi: 10.1124/pr.110.003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279(49):50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- 44.Ishidoh K, et al. Quinolinate phosphoribosyl transferase, a key enzyme in de novo NAD(+) synthesis, suppresses spontaneous cell death by inhibiting overproduction of active-caspase-3. Biochim Biophys Acta. 2010;1803(5):527–533. doi: 10.1016/j.bbamcr.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 45.Choi SE, et al. Elevated microRNA-34a in obesity reduces NAD + levels and SIRT1 activity by directly targeting NAMPT. Aging Cell. 2013;12(6):1062–1072. doi: 10.1111/acel.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonnet ME, et al. Systemic delivery of sticky siRNAs targeting the cell cycle for lung tumor metastasis inhibition. J Control Release. 2013;170(2):183–190. doi: 10.1016/j.jconrel.2013.05.015. [DOI] [PubMed] [Google Scholar]
- 47.Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003;63(21):7436–7442. [PubMed] [Google Scholar]
- 48.Yoshino J, Baur JA, Imai SI. NAD(+) intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab. 2018;27(3):513–528. doi: 10.1016/j.cmet.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshino J, et al. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14(4):528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. 2008;105(36):13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee J, Kemper JK. Controlling SIRT1 expression by microRNAs in health and metabolic disease. Aging (Albany NY) 2010;2(8):527–534. doi: 10.18632/aging.100184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang Y, et al. The epigenetically-regulated miR-34a targeting c-SRC suppresses RAF/MEK/ERK signaling pathway in K-562 cells. Leuk Res. 2017;55:91–96. doi: 10.1016/j.leukres.2017.01.020. [DOI] [PubMed] [Google Scholar]
- 53.Zarone MR, et al. Evidence of novel miR-34a-based therapeutic approaches for multiple myeloma treatment. Sci Rep. 2017;7(1):17949. doi: 10.1038/s41598-017-18186-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun HL, et al. ERK activation globally downregulates miRNAs through phosphorylating Exportin-5. Cancer Cell. 2016;30(5):723–736. doi: 10.1016/j.ccell.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wakino S, Hasegawa K, Itoh H. Sirtuin and metabolic kidney disease. Kidney Int. 2015;88(4):691–698. doi: 10.1038/ki.2015.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hasegawa K, et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med. 2013;19(11):1496–1504. doi: 10.1038/nm.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mouchiroud L, et al. The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013;154(2):430–441. doi: 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamamoto T, et al. Nicotinamide mononucleotide, an intermediate of NAD + synthesis, protects the heart from ischemia and reperfusion. PLoS One. 2014;9(6):e98972. doi: 10.1371/journal.pone.0098972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Horton JL, et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight. 2016;2(1):e84897. doi: 10.1172/jci.insight.84897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Revollo JR, et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007;6(5):363–375. doi: 10.1016/j.cmet.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang H, et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352(6292):1436–1443. doi: 10.1126/science.aaf2693. [DOI] [PubMed] [Google Scholar]
- 62.Hou Y, et al. NAD(+) supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc Natl Acad Sci USA. 2018;115(8):E1876–E1885. doi: 10.1073/pnas.1718819115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Di Stefano M, et al. NMN deamidase delays wallerian degeneration and rescues axonal defects caused by NMNAT2 deficiency in vivo. Curr Biol. 2017;27(6):784–794. doi: 10.1016/j.cub.2017.01.070. [DOI] [PubMed] [Google Scholar]
- 64.Rajman L, Chwalek K, Sinclair DA. Therapeutic potential of NAD-boosting molecules: the in vivo evidence. Cell Metab. 2018;27(3):529–547. doi: 10.1016/j.cmet.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rohas LM, et al. A fundamental system of cellular energy homeostasis regulated by PGC-1alpha. Proc Natl Acad Sci USA. 2007;104(19):7933–7938. doi: 10.1073/pnas.0702683104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kong L, et al. Sirtuin 1: a target for kidney diseases. Mol Med. 2015;21:87–97. doi: 10.2119/molmed.2014.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nowak G, Schnellmann RG. L-ascorbic acid regulates growth and metabolism of renal cells: improvements in cell culture. Am J Physiol. 1996;271(6 Pt 1):C2072–C2080. doi: 10.1152/ajpcell.1996.271.6.C2072. [DOI] [PubMed] [Google Scholar]
- 68.Gilmartin AG, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17(5):989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 69.Funk JA, Schnellmann RG. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am J Physiol Renal Physiol. 2012;302(7):F853–F864. doi: 10.1152/ajprenal.00035.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.