

Abstract
Spinal muscular atrophy (SMA) with respiratory distress type 1 (SMARD1) is an autosomal recessive motor neuron disease that is characterized by distal and proximal muscle weakness and diaphragmatic palsy that leads to respiratory distress. Without intervention, infants with the severe form of the disease die before 2 years of age. SMARD1 is caused by mutations in the IGHMBP2 gene that determine a deficiency in the encoded IGHMBP2 protein, which plays a critical role in motor neuron survival because of its functions in mRNA processing and maturation. Although it is rare, SMARD1 is the second most common motor neuron disease of infancy, and currently, treatment is primarily supportive. No effective therapy is available for this devastating disease, although multidisciplinary care has been an essential element of the improved quality of life and life span extension in these patients in recent years. The objectives of this review are to discuss the current understanding of SMARD1 through a summary of the presently known information regarding its clinical presentation and pathogenesis and to discuss emerging therapeutic approaches. Advances in clinical care management have significantly extended the lives of individuals affected by SMARD1 and research into the molecular mechanisms that lead to the disease has identified potential strategies for intervention that target the underlying causes of SMARD1. Gene therapy via gene replacement or gene correction provides the potential for transformative therapies to halt or possibly prevent neurodegenerative disease in SMARD1 patients. The recent approval of the first gene therapy approach for SMA associated with mutations in the SMN1 gene may be a turning point for the application of this strategy for SMARD1 and other genetic neurological diseases.
Keywords: Distal hereditary motor neuropathy type 6, SMARD1, Motor neuron disease, IGHMBP2, Gene therapy, Oligonucleotides
Introduction
Spinal muscular atrophy with respiratory distress type 1 (SMARD1, OMIM # 604320) is an early onset genetic degenerative motor neuron disease caused by autosomal recessive mutation in the IGHMBP2 gene, mainly characterized by progressive distal muscular atrophy and respiratory failure due to diaphragmatic palsy [35, 37]. It is also known as distal spinal muscular atrophy 1 (DMSA1) and distal hereditary motor neuropathy type 6 (dHMN6) [55]. Recessive mutations in IGHMBP2 cause a disease continuum with a neonatal onset and severe distal motor neuropathy with diaphragmatic weakness at one end (SMARD1) and a later onset of milder CMT2 at the other end (CMT2S). Both are thought to be due to a loss of IGHMBP2 function.
The first description of SMARD1 dates back to 1974, when Mellins et al. described two infants with a disease that resembled an atypical form of Werdnig–Hoffmann disease (SMA1) [69]. In 1989, Bertini et al. defined this disorder as a spinal muscular atrophy (SMA) variant mainly characterized by diaphragm involvement [7]. In 1996, Rudnik-Schoneborn et al. recognized SMARD1 as a separate disease from SMA [88]. More than 100 cases have been reported in the literature [101]. The actual prevalence of this disorder may be significantly higher since many studies have described diaphragmatic palsy in ~ 1% of patients with a diagnosis of early onset SMA [34].
Pitt and his group proposed diagnostic criteria for SMARD1 based on its clinical, histopathological and electromyographic features (Table 1) [81]. However, currently, a diagnosis of SMARD1 (due to IGHMBP2 mutations) can be made only with genetic testing, which is now relatively straightforward. In addition, NGS allows comprehensive genetic screening, especially for atypical cases in which IGHMBP2 is not the immediate candidate gene. Whole-genome sequencing can also identify variants in noncoding regions, allowing correct diagnosis in patients in which traditional analysis can identify only one variant [13].
Table 1.
Diagnostic criteria proposed by Pitt et al. to distinguish SMARD1 from other similar conditions and to facilitate the classification of the disease [81]
| Clinical criteria | Histopathological criteria | EMG criteria |
|---|---|---|
| Low birth weight (< 3rd percentile) | Reduced myelinated fiber diameter in sural nerve biopsiesa | Evidence of acute or chronic distal denervation |
| Onset of symptoms within the first 3 months of life | Slight evidence of progressive myelinated fiber degeneration in biopsies taken up to 3–4 months of age | Evidence of significant slowing (< 70% of LLN) in one or more motor a/o sensory nerves |
| Unilateral or bilateral diaphragmatic weakness | No evidence of regeneration or demyelination that can justify the reduction in fiber size | |
| Ventilator dependence within < 1 month of onset associated with an inability to wean | ||
| No evidence of other dysmorphology or other conditions |
LLN lower limit of normal range
aSince the thickness of the myelin sheath is appropriate for the axon size, its reduction in diameter originates from the axon, the size of which is similarly reduced
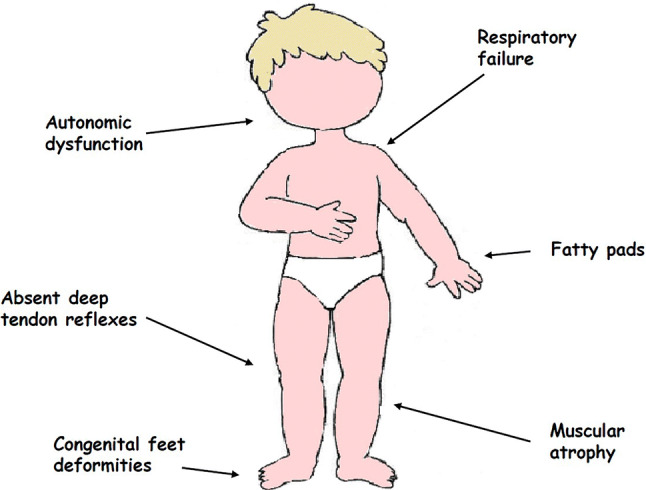
SMARD1 is clinically characterized by early onset respiratory involvement due to diaphragmatic palsy that is so severe that permanent ventilator support is required. This is the most common and pathognomonic sign that discriminates SMARD1 from other neuromuscular diseases, in particular from SMA1 5q, the most severe and common form of SMA [25, 29, 37] (Fig. 1). The first clinical manifestations of respiratory involvement in SMARD1 patients are inspiratory stridor, weak cry, recurrent bronchopneumonia and failure to swallow [34–37, 53, 88].
Fig. 1.
Clinical hallmarks of SMARD1
The second most typical clinical manifestation of SMARD1 is distal and progressive muscle paralysis caused by motor neuron death and neuropathic atrophy (Fig. 1). In contrast to the muscle involvement in SMA 5q patients, that in SMARD1 patients starts from the distal regions of the inferior limbs. Afterwards, the disease usually spreads to the distal superior limbs and eventually to the proximal regions of the four limbs and to the trunk muscles, and the progressive development of kyphoscoliosis occurs [37, 89].
Another frequent sign in these patients is muscular degeneration in the feet and hands that leads to adipose tissue accumulations in the phalanges, called fatty pads, and to foot deformities; these symptoms usually develop during intrauterine life and are present at birth, with finger contractures developing secondary to muscle atrophy [37, 89].
Consistent with these clinical manifestations, laboratory findings, such as neurogenic modifications by electromyography, decreased velocity of motor nerve conduction and absent motor response after maximal stimulation, are found in SMARD1 patients. Furthermore, muscle biopsies show neurogenic modifications, such as atrophy and hypertrophy of fibers [37]. The autonomic nervous system is often affected in SMARD1 patients, and in some rare cases, its involvement is prominent [76]. Sensory nerves are also affected and usually show axonal degeneration similar to that present in the biopsy specimens of SMA patients [37]. The natural history and long-term clinical outcomes of these patients are still not completely defined.
In 2012, Eckart and his group conducted a prospective and partially retrospective study that followed 11 genetically diagnosed SMARD1 patients aged 3 months–14 years for a mean observation period of 7.8 years. In general, disease progression showed a worsening trend in the first 2 years of life and then reached a plateau. Interestingly, biochemical analysis showed that a better prognosis was associated with residual IGHMBP2 enzymatic activity in vitro and with IGHMBP2 mutations that allow a certain degree of protein production [25]. Finally, hepatic and cardiac involvement have been reported in the literature in only a few SMARD1 patients [27]. Nevertheless, myocardial dysfunction is present in a mouse model of SMARD1 [64], and we cannot exclude late-onset cardiac involvement in these patients.
As previously mentioned, SMARD1 is an early onset disease that usually manifests during the first few years of life [25, 37]. Unfortunately, the rarity of the disease makes it difficult to conduct prenatal screening and almost all information about its prenatal features in the present literature is derived from retrospective studies. Indeed, sudden infant death syndrome is possible, and it is caused by multifactorial risks, including neuromuscular diseases similar to SMARD1.
In cases of unclear death in consanguineous families, it is important to investigate mutations causing lethal autosomal recessive disorders. In fact, another sign of suspicion for SMARD1 is a family history of recurrent infant deaths [58]. Next-generation sequencing approaches are useful for the identification of the genetic cause of recurrent deaths in consanguineous families.
Atypical presentations and juvenile cases complicate the diagnosis of SMARD1 and a high index of suspicion is needed to guide management and genetic counseling. Patients diagnosed with SMARD1 later in life have been described and these cases are designated juvenile SMARD1 [8, 25, 39, 40, 41, 54, 89]. Atypical forms have been reported [15, 45, 71, 76]. The patients were considered atypical because they did not exhibit prominent respiratory involvement but rather exhibited autonomic, neuropathic, bone, and multisystem involvement.
In 2014, Cottenie et al. identified compound heterozygous mutations (truncating and missense) in IGHMBP2 in a subgroup of CMT2 patients, which thus represents another genetic cause of CMT2 designated CMT2S. The mutations in IGHMBP2 identified to be involved in CMT2S were mainly loss-of-function nonsense mutations in the 5′ region of the gene that are associated with a truncating frameshift, missense, or homozygous frameshift mutation in the last exon and were predicted to be less aggressive than those involved in SMARD1 [19]. Overall, this finding suggests that SMARD1 and CMT2 are part of a clinical continuum that manifests differently according to the extent of protein reduction caused by different mutations.
Pathogenesis
The causes of SMARD1, as mentioned before, are homozygous or compound heterozygous mutations in the IGHMBP2 gene (IGHMBP2 [MIM 600502]; RefSeq: NM_002180.2) that cause a reduction in the level of the IGHMBP2 protein. The first evidence that SMA1 and SMARD1 are different diseases was reported by Rudnik-Schnoeborn et al. [88]; more than 200 SMA1 patients were analyzed and it was found that ~ 1% had diaphragmatic involvement and were negative for SMN1 gene mutations [88]. It was not until 1999 that Grohmann and her group identified the genetic cause of the disease. They studied a total of nine patients with a classic SMARD1 phenotype from three families from three different countries (Italy, Germany and Lebanon). The authors carried out linkage analysis in these patients and revealed associated markers on chromosome 11q13, which led to the identification of SMARD1 as a nosological entity separate from SMA1 [35]. Finally, in 2001, Grohmann et al. analyzed six more families to narrow the large cosegregating region on chromosome 11q that they had isolated in 1999, which led to the identification of the genetic mutations in the IGHMBP2 gene that cause SMARD1 [36].
IGHMBP2 protein
The IGHMBP2 gene is located on chromosome 11q13.2-q13.4. It is composed of 15 exons that encode a 993-amino-acid protein (the molecular mass of the protein is 109,149 Da) called immunoglobulin µ-binding protein 2 (Ighmbp2) or immunoglobulin S-µ-binding protein 2 (Sµbp-2).
The IGHMBP2 protein (OMIM*600502) consists of four domains, namely, an ATPase domain, an R3H domain that binds single-stranded nucleic acids, a DEXDc domain and an AN1-type zinc finger motif [38, 40, 41, 52]. Classification based on sequence homology places IGHMBP2 in the superfamily 1 (SF1) of helicases. In particular, IGHMBP2 shares high homology with UPf1, a protein that is involved in nonsense-mediated mRNA decay, and other similar members of the UPf1-like subfamily of SF1 (Fig. 2). The UPf1-like subfamily is characterized by the ability to unwind both DNA and RNA duplexes in the 3′–5′ direction [40, 41, 98]. In addition to UPf1, senataxin, a protein that has an important role in transcriptional regulation, is part of the UPf1-like subfamily [5, 14, 103].
Fig. 2.

Helicase domains of SETX, Upf1 and IGHMBP2 proteins. Upf1-like family of helicases contain two RecA domains (RecA1 and RecA2) with helicase motifs that bind nucleic acids and ATP, and two specific subdomains (1B and 1C) that modulate RNA binding
Heterozygous mutations in SETX cause a motor neuronopathy identified as ALS4, while several other recessive mutations are causative for ataxia with neuropathy called ataxia-oculomotor apraxia type 2 (AOA2 [MIM 606002]). The overlap in homology between IGHMBP2 and senataxin indicates that DNA/RNA helicase alteration may play a key role in the evolution of different types of neuropathy. The helicase domain of SETX also has homology with UPF1, which, similar to IGHMBP2, plays an important role in the production of mature mRNA [19]. Thus, a possible hypothesis is that mutations in IGHMBP2 might cause a dysfunction of protein helicase activity, which might determine the inability of neurons to generate error-free mature mRNA, thus inducing neuronal degeneration.
Despite these examples of homology that suggest the possible functions of IGHMBP2, the roles of this protein are still unclear. IGHMBP2 is highly expressed in the central and peripheral nervous systems during development [19]. After birth, IGHMBP2 expression levels increase in the cerebellar cortex but decrease slightly in other brain regions. IGHMBP2 seems to be expressed stably throughout adult life [19]. In adults, the highest IGHMBP2 expression levels are also in the cerebellum. IGHMBP2 is expressed ubiquitously in other body tissues and is expressed moderately in fibroblasts and lymphoblastoid cell lines. These findings highlight the relevance of the IGHMBP2 protein in peripheral nerves but suggest that, in other tissues such as the cerebellum, in which the protein is highly expressed, the protein has a less relevant function, as patients with IGHMBP2 mutations do not show signs of cerebellar dysfunction. In fact, while IGHMBP2 is ubiquitously expressed, when it is absent due to mutation, the predominant cells that degenerate are the α-motor neurons of the anterior horns of the spinal cord [40, 41]. In particular, it is not clear whether this cell-specific susceptibility to IGHMBP2 loss is due to a particular function of IGHMBP2 in motor neurons or whether the normal functions of IGHMBP2 are more fundamental to motor neurons than to other cell types. Nevertheless, IGHMBP2 is thought to be involved in many cellular processes, such as pre-mRNA processing, immunoglobulin class switching, the regulation of DNA replication and interactions with TATA binding protein [36, 62].
The structural analysis of IGHMBP2 shows that this protein is able to hydrolyze ATP to produce the energy necessary to accomplish its functions [40, 41]. In particular, IGHMBP2 acts as an ATP-dependent helicase for both DNA and RNA, which are bound by its R3H domain [52]. R3H is a polynucleotide-binding domain that can bind, due to specific sequence identification, both single-stranded DNA (ssDNA) and single-stranded RNA (ssRNA). R3H domains are found in more than 700 other proteins that present the same Arg-X-X-X-His sequence and are possibly associated with different domains, such as ring-type zinc finger nuclease domains, ATPase domains and DEAH helicase domains [52]. The terminal segment of the IGHMBP2 protein (from amino acids 638 to 786), in which the R3H domain is located, binds ssDNA sequences rich in 5′-phosphorylated guanines and acts as an anchor for the attachment of the entire protein and the execution of ATP-dependent helicase activity by the helicase domain [32, 52, 72]. The helicase domain is, in theory, able to bind RNA itself and exerts ATPase activity, while the R3H domain enhances these functions. Similarly, the R3H domain alone does not have sufficient affinity for RNA. The R3H domain and helicase domain strengthen each other in a cooperative manner, guaranteeing high-affinity binding to RNA. This is confirmed by evidence that a truncation in the N-terminal part of the R3H domain reduces the affinity for RNA and DNA [32, 52].
Genetics of SMARD1
Different mutations are causative of SMARD1. In particular, splice donor mutations, in-frame deletions, missense mutations, nonsense mutations and frameshift mutations have been reported [37].
In their first paper in 2001, Grohmann and her group studied IGHMBP2 and its connection with SMARD1 by analyzing six families and reported three recessive missense mutations, two nonsense mutations, one splice donor mutation and one frameshift deletion [36]. Later, nine other mutations, specifically seven missense mutations and two nonsense mutations, were identified in five SMARD1 patients by [67]. Interestingly, seven out of nine mutations were in highly conserved regions of IGHMBP2, which are most likely responsible for the enzymatic activity of the protein [67]. By 2010, 26 other mutations in IGHMBP2, specifically 14 missense mutations, 6 nonsense mutations, 4 frameshift mutations, 1 frameshift insertion and 1 in-frame deletion, had been reported in children with clinical phenotypes of SMARD1 [3, 37, 39, 114].
The majority of pathogenic mutations in IGHMBP2 that lead to SMARD1 clinical phenotypes are in its helicase domain, highlighting the importance of this domain for the function of the IGHMBP2 protein and suggesting that the disease is mainly caused by a biochemical loss of IGHMBP2 function, in particular its helicase or ATPase activity [19, 40, 41]. Indeed, seven out of nine typical missense mutations cause an alteration in ATPase binding/hydrolysis activity or a reduction in helicase motor stability [40, 41].
However, it is possible that other mechanisms besides biochemical functional alterations in IGHMBP2 contribute to the disease. In fact, Guenther and his group also reported mutations outside the helicase domain, such as the p.T491I mutation, which probably causes the disease by lowering the steady-state amount of the protein; the p.N583I and p.R603H mutations, which alter the nucleic acid binding ability of IGHMBP2; and the p.D565N mutation, which uncouples ATPase activity from RNA unwinding. Thus, different mechanisms, including altered ATPase activity, altered helicase activity and reduced steady-state levels of the protein, may be responsible for SMARD1 [40, 41].
As mentioned before, there is significant variability in the clinical presentation of SMARD1; at the same time, there are several reported IGHMBP2 mutations that may have different impacts on IGHMBP2 function. In the literature, it is difficult to find a clear genotype–phenotype correlation, although different authors have attempted to find a correlation between the phenotype and the protein level.
In 2009, Guenther et al. reported a case of two siblings with two compound heterozygous mutations in IGHMBP2 and a SMARD1 clinical phenotype. Interestingly, the two patients shared one mutation, while the other mutation was different. One of the two dissimilar mutations corresponded with a lower protein level and this was correlated with the more severe clinical phenotype [40, 41]. The hypothesis that a greater reduction in IGHMBP2 corresponds to a more severe SMARD1 clinical phenotype has been addressed in the literature. For instance, Litvinenko et al. reported in 2013 that a total absence of IGHMBP2 protein is correlated with more severe SMARD1 [63]. The same was suggested in 2014 by Cottenie and her group, who demonstrated that IGHMBP2 mutations are responsible for some cases of CMT2 and suggested that IGHMBP2 mutations that have a greater impact on IGHMBP2 protein levels are responsible for SMARD1, while milder mutations are responsible for CMT2, which is characterized by lower clinical severity [19]. However, few reports asserting the opposite are present in the literature; a case of two siblings with SMARD1 that carried the same mutation but exhibited a significantly different phenotype, which suggests the possible existence of disease modifiers in individuals, was reported [54]. In 2013, Stalpers and her group studied ten SMARD1 patients and showed that patients with homozygous mutations present with a more severe phenotype than that of patients with heterozygous compound mutations [101], suggesting that there is no precise genotype–phenotype correlation. Overall, while the type of mutation present is not completely predictive, a general correlation between a lower protein level and a more severe phenotype can be observed in the literature and this supports the hypothesis that a restoration of functional protein expression can be therapeutic.
Molecular pathogenesis
Although many hypotheses on the function of IGHMBP2 are driven by its structural analysis, the real role of IGHMBP2 in human cells and, in particular, the reason that it is so important in motor neurons and causes their death when its levels are reduced need to be further analyzed.
Subcellular localization and ribosome interaction: IGHMBP2 is a multifunctional protein
The first step to better understand the cellular roles of IGHMBP2 is to clarify its subcellular localization, especially in motor neurons. This was carried out by Guenther and his group in 2009. The authors reported that IGHMBP2 is predominantly localized to the cytoplasm, particularly the perinuclear cytoplasm, in motor neurons and spreads to growth cones and axons. This was demonstrated in mouse embryo-derived motor neurons, which are the first cells to degenerate in neuromuscular degeneration (nmd) mice, a mouse model of SMARD1 that harbors a spontaneous splice-site mutation in murine Ighmbp2 and presents as a neuromuscular disorder analogous to human SMARD1. The authors confirmed this localization, demonstrating a reduction in Ighmbp2 in nmd mice at these sites [40, 41].
Other research groups focused on IGHMBP2 function and found that IGHMBP2 physically interacts with ribosomes in the cytoplasm, suggesting a role for this protein in ribosomal biogenesis and translation [42]. This was also confirmed by Guenther’s group, who claimed IGHMBP2 to be a ribosomal-associated RNA ATP-dependent helicase that relies on the cytoplasmic localization of IGHMBP2 in association with ribosomes. Thus, IGHMBP2 may be responsible for mRNA translation. Specifically, IGHMBP2 is associated with the ribosomal 80S subunit but not with polysomes and it is probably involved in the first steps of translation, perhaps during its initiation [40, 41]. Regarding its role in ribosomal biogenesis, IGHMBP2 has been shown to have a close physical interaction with ABT1 and U3snoRNP, which are involved in the processing of the 5′ external transcribed sequence of 45S pre-rRNA [22]. However, Guenther previously reported that IGHMBP2 was shown to be involved in replication and pre-mRNA splicing and to be localized mainly to the nucleus, concluding that IGHMBP2 could be a multifunctional protein that plays a key role in motor neurons in different pathways [40, 41].
Genetic disease modifiers
Some hints on possible IGHMBP2 functions also come from the clinical aspects of SMARD1. Indeed, an increasing number of atypical presentations and slowly progressing and milder forms have been described in the literature, as previously mentioned. Since no definitive genotype–phenotype correlation has been described so far, a possible explanation of these clinical differences is the presence of genetic disease modifiers that are different for each patient and can affect disease onset and severity by influencing different biological pathways. Interestingly, a modifier locus that influences the clinical severity of the disease independent of Ighmbp2 protein levels was found on chromosome 13 in nmd mice [20]. The nmd mouse model presents with motor neuron degeneration, axonal loss and neurogenic muscular atrophy, all of which lead to death between 8 and 12 weeks [16]. The neuropathological phenotypes observed in nmd mice can be completely rescued by the expression of transgenic wild-type IGHMBP2 driven by a neuron-specific promoter [64], confirming that they are definitely caused by a reduction in Ighmbp2 levels in MNs. The expression of the genetic modifier on chromosome 13 mitigates both the neuropathological features and the clinical outcomes of the nmd mouse model [77].
Dysregulation of RNA metabolic pathways leads to the accumulation of toxic tRNA fragments
In 2000, Oda et al. demonstrated that this murine modifier locus contains five genes encoding 5 tRNATyrs and a gene encoding the activator of basal transcription 1 (ABT1), a protein that is fundamental for ribosome biogenesis and function [77]. The syntenic genomic area in humans also contains four tRNATyr genes and the activator of basal transcription 1 (ABT1) gene [19]. De Planell-Saguer et al. confirmed the relevance of these genetic elements, demonstrating a physical interaction between IGHMBP2 and tRNAs, particularly tRNATyr and ABT1 [22].
Moreover, IGHMBP2 has also been found to associate with other components of the translational machinery, such as the transcription factor IIIC-220 kDa (TFIIIC220), which is responsible for tRNA transcription, and with helicases Pontin and Reptin, which are involved in transcription and ribosome biogenesis [22, 33, 56, 94]. These findings strengthen the concept that the IGHMBP2 protein is involved in translational machinery and RNA metabolic pathways [22], which, together with findings reported by [40, 41] about the interactions of IGHMBP2 and 80S ribosomal subunits, add SMARD1 to the list of neurodegenerative diseases caused by the dysregulation of RNA metabolic pathways.
Indeed, RNA metabolism alterations are a common theme in neurological disorders. In particular, the observation that the inactivation of the RNA kinase CLP1, a component of the tRNA splicing endonuclease complex, results in the accumulation of toxic tRNA fragments and consequently the progressive loss of spinal MNs, muscle denervation and paralysis in a transgenic mouse model [46, 113] further highlights the possible involvement of tRNA dysregulation in MN diseases. Hanada and his group demonstrated that tRNA fragments derived from the aberrant processing of tyrosine pre-tRNA lead to an increase in oxidative stress that results in p53 activation and p53-mediated cell death. The relevance of tRNA metabolism impairment in neurodegenerative disorders has also been shown in humans, as patients with homozygous missense mutations in CLP1 show altered tRNA splicing and suffer from severe motor sensory neuropathy and cortical dysgenesis that leads to microcephaly [57, 93].
Several lines of evidence suggest a role for tRNA metabolism dysregulation in SMARD1 pathogenesis. IGHMBP2 has been found to be able to interact with tRNAs, such as tRNATyr, and with other molecules that are important for tRNA metabolism, such as TFIIIC220, Pontin and Reptin. Moreover, the modifier locus found in the nmd mouse model contains genes encoding the same tRNAs that interact with IGHMBP2. Thus, the absence of IGHMBP2 activity may determine the deficiency in tRNA–IGHMBP2 binding in SMARD1 patients, which leads to altered tRNA splicing, toxic fragment accumulation, and p53-mediated death. Conversely, the presence of the murine modifier locus in this model may correct the missing molecular pathways that encode important tRNAs. Many studies are needed to confirm these theories and fully elucidate the pathogenic mechanism of SMARD1 to determine an effective therapeutic approach.
A possible role in mRNA translation
The other main issue that may help us to fully understand the pathogenic mechanisms of SMARD1 concerns the selective vulnerability of MNs, as all of the previously discussed mechanisms should occur in all cells since IGHMBP2 is a ubiquitously expressed protein. Guenther et al. suggested that this might be attributed to the importance of IGHMBP2 to specific MNs for the initiation of translation of some mRNAs, such as those presenting an excessive number of secondary or tertiary structures. This hypothesis could also explain the involvement of cardiomyocytes and diaphragmatic cells in the murine nmd model [40, 41]. Inefficient DNA repair machinery or more fragile translational machinery, possibly due to the extension of the axons and dendrites of MNs, are other intriguing explanations, but these explanations may be incorrect or describe just a part of other mechanisms. Consistent with this notion, Ighmbp2 deficiency in nmd motor neurons was recently correlated with alterations in protein biosynthesis, especially that of the β-actin protein [104]. β-actin is an abundant cytoskeletal protein, and aberrations in its regulation have been previously observed in Smn (survival motor neuron)-deficient motor neurons, suggesting that it plays a crucial role in cell development and differentiation [50, 87]. Although Ighmbp2-deficient primary cultured motor neurons display a significant and selective reduction in β-actin protein levels in axonal growth cones, no abnormalities are detected in the amount of corresponding mRNA or total protein, indicating a translational delay [104]. These results may further indicate the involvement of IGHMBP2 in translation, although deeper studies are necessary to draw conclusions on its direct and indirect impacts.
IGHMBP2 deficiency may lead to R-loop accumulation
In addition to the processes reported, another molecular mechanism that may be involved in SMARD1 pathogenesis is R-loop dysregulation. R-loops are temporary three-stranded nucleic acid structures that form physiologically during transcription when a nascent RNA transcript hybridizes with the DNA template strand, leaving a single strand of displaced nontemplate DNA [26, 80, 92]. They are involved in different physiological processes, but their abnormal persistence, determined both by a deficiency in molecules responsible for their resolution and by the presence of stabilizing sequences in the nontemplate DNA strand, may cause DNA double-strand breaks (DSBs), genomic instability and eventually cell death [61, 79, 116].
Not by chance, they have recently been found to be increased or to abnormally persist in different human disorders, such as cancers and neurodegenerative diseases [80, 84]. In particular, different motor neuron disorders are linked to genetic mutations that induce R-loop formation and consequent motor neuron death. Among these disorders, ALS4, a juvenile form of amyotrophic lateral sclerosis (ALS), and ataxia with oculomotor apraxia type 2 (AOA2) are caused by mutations in senataxin (SETX), an RNA/DNA helicase that has been confirmed to be involved in R-loop resolution in different human genes [47, 80, 99]. Similarly, ALS seems to be closely connected to R-loops since C9ORF72 hexanucleotide repeat expansions (C9 HREs) have been demonstrated to stabilize R-loop formation [43]. Additionally, TDP43 and FUS are RNA binding proteins that may contribute to the regulation of the level of R-loops [91], as C9 HREs and TDP-43 and FUS mutations are some of the most frequent genetic alterations found in hereditary and sporadic forms of ALS. SMA may also be caused by R-loop dysregulation since the absence of SMN (which is encoded by the gene responsible for the disease) causes diffuse splicing alterations and abnormal R-loop accumulation, which induce p53 pathway activation and the expression of other markers of the DNA damage response [51].
Pathogenic R-loop pathways may also be present in SMARD1 and may represent a very important pathogenic mechanism and therapeutic target for the disease. Indeed, IGHMBP2 was initially described as belonging to R-loop-dependent class-switch recombination proteins and it has been described as having ATP-dependent 5′ to 3′ DNA/RNA helicase activity [32]. As previously mentioned, the majority of IGHMBP2 mutations observed in SMARD1 patients are located in the helicase domain, and importantly, IGHMBP2 has homology with SETX [40, 41, 62], which plays a key role in R-loop resolution pathways; this suggests a probable link between IGHMBP2 deficiency and R-loop accumulation, which eventually leads to MN death.
Other studies are needed to fully elucidate these pathways and understand their therapeutic potential.
SMARD1 preclinical disease models
SMARD1 is a disease that mainly affects the α-motor neurons of the spinal cord, which are not accessible for direct study. Thus, modeling this disease for pathogenetic and therapeutic preclinical studies is critical for the advancement of this field.
Induced pluripotent stem cells (iPSCs)
Stem cells are able to self-renew indefinitely and to differentiate into different cell types [83, 112]: [100], including motor neurons.
iPSCs are cells with a molecular profile and differentiation potential similar to that of embryonic stem cells (ESCs), but they can be generated from somatic cells (fibroblasts) by the expression of a combination of transcription factors usually involved in the maintenance of ESC self-renewal and totipotency [105, 106, 115]. iPSCs can be generated through viruses (retroviruses, lentiviruses, adenoviruses, and Sendai viruses), DNA (plasmids, episomal plasmids, transposons), and cell-penetrating peptides. Today, nonintegrating systems are preferred [96].
iPSCs present some undisputed advantages; they have human origins, are easily accessible (they can be derived from skin or blood cells), exhibit expandability in vitro, can differentiate into almost any human cell type, including neuronal cells, and thus can be used to model neurological disorders and solve the ethical concerns related to the use of human ESCs. Furthermore, since iPSCs can be derived from affected patients themselves, they represent a personalized disease model that may represent the future of precision medicine. In addition, iPSCs represent a potential source of personal stem cells for affected patients, opening unprecedented transplantation possibilities; they are isogenic, so no immunosuppressive therapy is needed, and they represent the first milestone for personalized medicine [96].
Cells derived from iPSCs usually have a relatively immature phenotype, so there is greater confidence that they represent a better model of early onset diseases than late-onset diseases, in which the accumulation of cellular stressors and aging-related processes may be key contributors to disease pathogenesis [102]. Therefore, iPSCs are better models for diseases such as SMARD1 and SMA1 than for disorders such as Alzheimer’s disease or ALS.
Furthermore, there is another important advantage of using iPSCs for disease modeling, which is the possibility of generating different disease-relevant cell types and studying the interactions among them [44, 73]. In SMARD1, for instance, the possibility of simultaneously generating MNs, glial cells and muscle cells and studying their interaction represents a major breakthrough for elucidating the disease mechanisms.
To date, iPSCs have been generated to model different human disorders, including motor neuron diseases such as ALS [23] and SMA [18, 24]. Apart from our group’s works, no iPSCs have been developed for SMARD1 [97].
We differentiated iPSCs into motor neurons and analyzed the processes that are relevant to SMARD1 pathogenesis, including cell survival, axonal elongation, and growth cone formation, showing that even though SMARD1 iPSC-derived motor neurons do not present developmental defects compared to wild-type cells, they autonomously degenerate in long-term cultures [97].
SMARD1 mouse model
B6.BKS Ighmbp2nmd-2J/J mice are commonly used to model SMARD1 [64]. The IGHMBP2 region is conserved in these mice, but it is localized to chromosome 19 instead of chromosome 11 [16]. Affected mice have a homozygous mutation (A-G) in intron 4 of the Ighmbp2 gene, which causes abnormal splicing in almost 80% of transcripts; only 20% of transcripts are full-length. Thus, Ighmbp2 protein levels are greatly reduced in nmd mice. Both [90] and [22] reported a direct correlation between a decrease in Ighmbp2 protein levels and motor neuron degeneration [22, 90]. Homozygous mice present a typical SMARD1 phenotype that usually becomes evident during the 2nd postnatal week. They present a loss of motor neurons and consequent progressive paralysis, which first involves the hindlimbs and then extends to the forelimbs, and respiratory involvement that, unlike that in human SMARD1 patients, is present only in late stages of the disease and possibly represents the cause of death [38]. Another difference between humans and mice is heart involvement, which is not present in SMARD1 patients but is important and occurs early in mice and leads to progressive dilated cardiomyopathy and congestive heart failure [64]. In 2004 and later in 2005, Maddatu and his group demonstrated that transgenic mice that selectively express the Ighmbp2 protein in the CNS or in cardiomyocytes develop disease hallmarks only in the CNS or heart, respectively, suggesting that the histological patterns related to the disease are dependent on defective Ighmbp2 expression and that the correction of the expression levels of this protein can reverse the disease phenotype in mice [64, 65]. Death usually occurs between 8 and 12 weeks of age, but this can vary [20, 22, 59, 64].
Histopathological studies have demonstrated a preferential involvement of lumbar motor neurons rather than thoracic or cervical neurons. Degenerating neurons can also be located in the sympathetic chain. The death of motor neurons, which is present by 10 days of life, occurs significantly earlier than the appearance of muscular atrophy and adipose tissue infiltration, which are late signs in affected mice and are more prominent in distal muscles than in proximal muscles. The overall pattern of affected muscles and motor neurons was thought to be random [16, 20, 36, 72] until recently, when Villalón et al. demonstrated that some motor units in symptomatic nmd mice appear to be more susceptible to degeneration than others. Neck muscles and the trunk muscles that are necessary for respiration and trunk movement have been described as resistant or slightly vulnerable to neuromuscular junction (NMJ) denervation. This is consistent with the clinical manifestation of nmd mice, which involves respiratory distress only in the latest stage of the disease. Except for the EDL and lumbricals, the distal appendicular muscles are vulnerable. All these results suggest that NMJ degeneration does not affect all muscles equally. Surprisingly, endplate fragmentation has been observed to occur completely independent of NMJ denervation. Consistent with this notion, compared to wild-type mice, nmd mice exhibit a reduction in internode length and nerve fiber diameter, which are both typical signs of demyelination/remyelination events. Overall, these results show the specific motor units that can be further analyzed to clarify the molecular pathways that underlie selective vulnerability in nmd mice.
Interestingly, these pathological changes can be significantly restored in neonatal nmd mice by gene therapy involving an injection of AAV9-IGHMBP2, demonstrating that this approach protects clinically relevant muscles from denervation and endplate fragmentation. This suggests that IGHMBP2 may play a relevant role in endplate maintenance [110].
Moreover, the existence of a disease-modifying gene that is able to delay disease onset and progression raises interesting questions regarding the possible use of disease modifiers for the determination of the life expectancy of nmd mice [20, 65].
SMARD1 therapy
To date, there are no approved therapies for SMARD1 that can modify progression and patient prognosis. The only clinical approaches for treating these patients involve using drugs and devices, such as antibiotics, mechanical ventilators and nutritional devices, to support vital functions and mitigate symptoms [25, 53]. Managing affected children thus falls on families and requires significant effort [82, 109].
Moreover, because SMARD1 is a very rare disease, only a few pharmacological treatments or therapeutic strategies have been tested, all at the preclinical stage. However, recent research advancements have led to the identification of interesting therapeutic strategies that may be effective in patients; these strategies are summarized in Fig. 2. Nevertheless, tolerance/adverse effect studies and randomized clinical trials are still needed to guarantee the safety and efficacy of these strategies and the path to a cure for SMARD1 remains long. For this reason, continued multidisciplinary care is fundamental to the management of patients.
Current achievements in preclinical studies
Neuroprotection via small molecules
Only a few pharmacological treatments have been tested for SMARD1, and until now, none have reached clinical trials (Fig. 2a). Insulin-like growth factor 1 (IGF1), a hormone with several functions, such as stimulating cell survival and neurotrophic factors, may be another interesting therapeutic molecule since it is able to increase the axonal growth and axonal sprouting of motor neurons in paralyzed and denervated muscles and diminish muscular atrophy [2, 11, 12, 49, 66, 74, 86]. Krieger et al. recently demonstrated that IGF1 is reduced in nmd mice, suggesting that it may cause some of the neuropathological hallmarks of SMARD1, such as motor neuron degeneration and muscular atrophy. When administered to nmd mice, IGF1 conjugated to polyethylene glycol (PEG-IGF1) was found to restore IGF1 serum levels and improve some of the core features of the disease without significantly increasing motor neuron survival, probably due to insufficient dose [59].
In 2005, a monoclonal antibody, Mab2256, an agonist of tyrosine kinase receptor C (TrkC), was tested in nmd mice by Ruiz et al. [90]; the researchers reported an initial beneficial effect on muscular strength and function, which was confirmed by electrophysiological studies, and a slowing of disease progression [48, 90]. However, the effects were only transient and did not prolong survival. The results of these studies were not positive, but considering the possible beneficial effects of IGF1, Mab2256 and other neurotrophic factors or molecules that activate neurotrophic pathways on different neurological disorders, these small molecules cannot be excluded as therapeutic strategies for SMARD1 and other neuropathies.
Cell transplantation improves the clinical outcomes of the nmd mouse model
Neural stem cell (NSC) transplantation represents a potential therapeutic strategy for ameliorating the neurological phenotype of SMARD1 through several different mechanisms, including neuroprotection and the replacement of different CNS cells [1, 28]. The positive effects of stem cell transplantation (neural and nonneural) have been demonstrated in earlier studies by our laboratory. Indeed, in 2006, our group demonstrated that the transplantation of ALDH high/side-scatter low (ALDHhiSSClo) NSCs obtained from mouse spinal cords into nmd mice improved the clinical outcomes of the mice [18]. The same therapeutic efficacy was achieved through the transplantation of more differentiated cells, such as precursors of motor neurons, into the spinal cords of nmd mice [17]. Later, we transplanted human iPSC-derived NSCs (Fig. 2d) and demonstrated that these cells not only correctly localized to the anterior horn of the mouse spinal cord but also differentiated into neuronal cells, particularly motor neurons, and improved the phenotype and life span of the mice [97].
Gene therapy with AAV9-IGHMBP2 rescues the disease phenotype in nmd mice
In autosomal recessive diseases, gene therapy is used to substitute mutated genes with healthy wild-type ones, allowing the correct expression of key proteins that are reduced and treating diseases at their origins. This is possible using viral vectors that carry a healthy copy of the gene that is mutated in the disease (Fig. 2e) [9, 30, 68, 85, 108]. In 2009, Foust et al. published a revolutionary study that showed that adeno-associated virus serotype 9 (AAV9) injected intravenously can cross the blood–brain barrier (BBB) and target neurons and astrocytes [31]. Since then, many other preclinical studies have been conducted on different neurological disorders and have confirmed the efficacy and safety of the AAV9 vector. Indeed, AAV9 is an interesting vector for targeting the CNS since it is nonimmunogenic and noninflammatory; it induces long-term expression (over 10 years in humans in clinical trials, which can reduce the number of administrations needed to maintain the effect to one or only a few), it does not integrate into the cell genome (and thus is not oncogenic), it can infect both dividing and nondividing cells, and it can cross the BBB and reach the cells affected by neurodegenerative disorders, even when administered in a noninvasive manner (Fig. 3).
Fig. 3.

a Neuroprotection via small molecules. PEG-IGF1 is a neurotrophic factor. Mab2256 is a monoclonal antibody with an agonistic effect on tyrosine kinase receptor C that has neuroprotective effect. b Enhancement of muscle functionality. Small molecules to enhance the muscle contractility (CK2127107) and mass of the muscles (myostatin activation inhibitor) can be considered. c mRNA modulation. ASO and small molecules can modulate mRNA splicing and transcription increasing the IGHMBP2 protein level. d Cell transplantation. Human induced pluripotent stem cells (iPSCs) can be generated from adult fibroblasts. The iPSCs are then differentiated into neural stem cells (NSCs) or GFP-positive MNs and transplanted into a SMARD1 mouse to improve the phenotype of the animal. e Gene therapy. This approach is based on the replacement of the defective gene using self-complementary adeno-associated viral vectors
The first clinical outcomes of AAV9 treatment in spinal muscular atrophy were so encouraging [70] that other SMA1 AAV9 trials were approved and are ongoing (http://www.clinicaltrial.gov). Indeed, on May 24, 2019, the FDA granted approval for this gene therapy approach for all SMA patients under 2 years of age.
Thus, success is encouraging for the application of this therapeutic technology for SMARD1 since SMARD1 is a monogenic disorder due to a loss of function, thus, the transfer of a functional copy of the defective gene could theoretically improve the phenotype and even reverse the disease.
Furthermore, in our laboratory, preclinical studies in a murine model confirmed the possibility of delivering the IGHMBP2 gene using an AAV9 vector and modifying murine clinical phenotypes using this therapeutic strategy [75]. We reported that the disease phenotype in a SMARD1 mouse model, the nmd mouse, was rescued after the therapeutic administration of an AAV9 construct carrying wild-type IGHMBP2 via systemic injection to replace the mutant gene. AAV9-IGHMBP2 transfer (5x1011 viral particles administered IV at P1) increased protein levels, restored motor function and neuromuscular physiology, increased life span (450% increase), and improved pathological features in the central nervous system, muscles, and heart. In this set of experiments, we also generated spinal motor neurons from SMARD1 patient-derived iPSCs to test the efficacy of gene transfer in a human model. Motor neuron survival, axonal length, and growth cone formation were restored in SMARD1 motor neurons that were genetically corrected by IGHMBP2 transferred via a viral vector due to an increase in protein expression levels. Thus, transferring wild-type IGHMBP2 can protect human motor neurons from SMARD1-induced degeneration.
An important aspect of the gene therapy approach is the route of delivery. To examine how delivery route can impact efficacy, a direct comparison of the IV and ICV delivery of AAV9-IGHMBP2 was performed [95]. A low dose of single-stranded AAV9-IGHMBP2 (1.25 × 1011) delivered either through ICV or IV injection was demonstrated to be sufficient to extend the life span and increase the weight of treated nmd mice compared to untreated nmd controls [95]. The authors concluded that while IV delivery of a low dose does not improve hindlimb phenotypes or motor function, the partial restoration of cardiac performance is sufficient to significantly increase the survival of nmd mice. As mentioned before, the therapeutic window considerably affects the effectiveness of a treatment. The study of gene therapy in nmd mice has demonstrated that this approach is highly effective when carried out in presymptomatic mice at birth. Unfortunately, the diagnosis of newborns is difficult, which narrows the opportunities for therapeutic intervention in SMARD1 patients. Therefore, the identification of treatment strategies that are also efficacious when administered during the symptomatic phase is crucial.
The feasibility of AAV-based technologies opens the possibility for novel therapeutic strategies that may prevent disease progression and neurodegeneration for both SMARD1 and other motor neuron diseases related to genetic disorders. However, at the present time, there are no ongoing trials for gene therapy in SMARD1 patients.
Emerging therapeutic options for SMARD1 treatment
Different therapeutic strategies employed for the treatment of other motor neuron diseases can also be exploited in SMARD1. Pharmacological and cellular approaches must be studied and evaluated in depth in vitro in SMARD1 cellular models and in vivo in nmd mouse models, but they surely represent a promising tool for the development of novel treatments.
mRNA modulation
In patients who harbor premature termination codons (PTCs) in the coding regions of IGHMBP2 mRNA, which lead to the incorrect termination of translation and the production of nonfunctional truncated proteins, the translational readthrough of PTCs induced by pharmaceutical compounds is a hopeful tool for the recovery of functional full-length protein expression and the amelioration of disease symptoms without directly affecting the genome or transcriptome of the patient. Stop codon readthrough therapy is available through the use of the drug ataluren (Translarna PTC Therapeutics), which is approved for Duchenne muscular dystrophy patients with stop codon mutations [10, 107]. This approach should be investigated in SMARD1 patients who carry a stop codon mutation [21].
For mutations that alter splicing and thus induce the inappropriate inclusion or skipping of an exon, oligonucleotide-based therapies that inhibit or activate specific splicing events can allow the expression of functional full-length IGHMBP2 protein, providing a therapeutic approach [78] (Fig. 2c). A similar therapeutic strategy was approved by the FDA/EMA as the first therapy for SMA.
An alternative to oligonucleotides is represented by small molecules that can modulate splicing, increasing the level of the protein. In the case of SMA, small molecules that promote the increase in SMN protein levels have been identified and risdiplam is now in phase II trials in Europe (NCT02913482 and NCT03032172). The positive feature of these small molecules is that they are orally available and thus can be easily administered with the advantage of increasing target protein levels in all tissues, including those outside the central nervous system. A similar approach for SMARD1 patients will require extensive in vitro screening and likely the design of a personalized therapy with N-of-1 trials, which appears difficult in the current state of the art but can be theoretically hypothesized in the future.
Gene correction with CRISPR/Cas9
In 2012, our group demonstrated that iPSCs obtained from SMA1 patients can be genetically corrected to generate healthy iPSCs, iPSC-derived NSCs and MNs with corrected phenotypes [18]. The same could be achieved with the iPSCs of SMARD1 patients and isogenic corrected cells could be used as a cell source for transplantation. Having demonstrated the validity of transplant therapy with iPSC-derived NSCs and motor neurons in nmd mice in the abovementioned studies, it is important to understand whether this protocol can be performed with autologous cells after genetic correction.
Viral vector-based gene therapy approaches certainly provide an advantageous system for correcting gene defects, although they have some limitations. In this sense, clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein-9 nuclease (Cas9) represent the newest and most advanced genome engineering system in the gene editing field. This sophisticated mechanism exploits the defense machinery that protects bacteria and archaea from bacteriophages in nature [4] and it is based on a cleavage/repair process at a specific genomic locus targeted by RNA-guided nucleases [6]. This specificity represents the greatest potential for gene therapy and allows higher efficiency and easier customization.
CRISPR/Cas9 technology has been recently employed in several fields of research, including neurodegenerative disorder modeling at different levels. Indeed, this powerful genome editing tool is used to generate genetically modified animals as well as customized cell lines with the aim of achieving more successful models that can help researchers better understand the physiopathology of diseases [60]. More specifically, in recent years, isogenic wild-type iPSC lines have been generated using CRISPR/Cas9 technology and a donor plasmid carrying wild-type coding sequences and homology arms as the repair template [111]. In this way, locus-specific gene correction can be achieved. In the same study, isogenic iPSCs were differentiated into motor neurons and compared with ALS cells to analyze relevant molecular differences.
In light of these results, because SMARD1 is a monogenic disorder, the application of CRISPR/Cas9 for gene correction in SMARD1 is promising. The mutated IGHMBP2 gene in SMARD1 cells could be substituted with the corresponding wild-type gene by homologous recombination to generate isogenic wild-type cell lines that represent the best-matched controls in molecular studies.
Conclusions
SMARD1 is a devastating motor neuron disease that causes infantile death within 2 years of life in most cases. The significant advances achieved by next-generation sequencing in recent years may contribute to an increased rate of diagnosis, particularly of atypical phenotypes such as extended survival or a lack of diaphragmatic involvement, thus increasing the need for a more effective therapy.
The pathomechanisms of SMARD1, especially the mechanisms that underlie the selective degeneration of some motor neuron subsets, remain unknown, although alterations in mRNA maturation and a disturbance in tRNAs due to a reduction in IGHMBP2 seem to be key elements. Indeed, little information on the causative gene (IGHMBP2) and the protein it encodes is available and further studies are needed to shed light on the molecular pathogenesis that results from its reduced expression. Expanding our current understanding of these issues will likely allow the identification of novel therapeutic targets and pharmacological approaches for SMARD1.
Despite the absence of a cure, different strategies, including pharmacological treatment, cell therapy and gene therapy, have been tested at the preclinical level with relatively satisfactory results.
Because SMARD1 is caused by a single gene mutation, it may be an appropriate candidate disease for gene therapy. Indeed, IGHMBP2 gene transfer results in the highly efficacious rescue of survival and pathological phenotypes of SMARD1 in mice. Two important issues that need to be addressed in this regard are the optimal delivery route (local into the cerebrospinal fluid by lumbar puncture or systemic by intravenous injection) and the therapeutic time window. In SMA, the rescue of SMN protein with therapies not only stops the disease but also partially reverses the phenotype in precociously treated patients. Whether a similar situation can occur in SMARD1 patients in the case of IGHMBP2 rescue in particular in helping with respiratory distress has to be demonstrated. Preclinical findings support the feasibility of AAV-based technologies as an efficacious therapeutic approach for the treatment of SMARD1 and the clinical translation of this strategy for the treatment of SMARD1 patients. The experience in SMA studies suggests the need for early treatment even at the presymptomatic stage. Performing newborn screening of IGHMBP2 with single gene analysis is not practical, but NGS screening approaches can overcome this issue in the future. With the development of potential therapies, there is a parallel need to delineate a more accurate natural history and outcome measures as well as for the identification of biomarkers to evaluate clinical stage and therapeutic response. Molecular and physiological biomarkers will help in the clinical management process of when and how to treat. The feasibility in the future of the design of small molecules and oligonucleotides tailored for a specific patient can be hypothesized, while approaches that are IGHMBP2 independent, such as neuroprotectants or muscle activators, that might be developed for SMA can also be quickly applied in SMARD1 if proven efficacious. Combinatorial therapies encompassing treatments to increase IGHMBP2 levels and treatments to support muscle function and motor neuron protection will be tailored to each patient. The success story of the approval of transformative, life-saving therapies for SMA has to be quickly learned and translated in the SMARD1 field for the benefit of the patients and their families.
Acknowledgements
We thank Association Centro Dino Ferrari for its support. This work was partially supported by Italian Ministry of Health to GPC and MN. This work was partially supported also by Cariplo Giovani Grant “Assessing the pathogenetic role of tRNA and rRNA deregulation in disease-specific human and mouse models to understand pathogenesis and identify molecular therapeutics targets for Spinal Muscular Atrophy with Respiratory Distress type 1 (SMARD1)” to Monica Nizzardo and RICERCA FINALIZZATA 2016 (PROJECT NUMBER: GR-2016-02362377) to Monica Nizzardo, Host Institution: IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Project Title: “Unravelling therapeutic window and central nervous system delivery of AAV9-IGHMBP2 gene therapy for SMARD1”. This work was partially supported by Italian Ministery of Health (Ricerca Corrente 2020) to GPC and NB. The image was generated using images from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License. http://smart.servier.com/
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Abati E, Bresolin N, Comi GP, Corti S. Preconditioning and cellular engineering to increase the survival of transplanted neural stem cells for motor neuron disease therapy. Mol Neurobiol. 2019;56(5):3356–3367. doi: 10.1007/s12035-018-1305-4. [DOI] [PubMed] [Google Scholar]
- 2.Allodi I, Comley L, Nichterwitz S, Nizzardo M, Simone C, Benitez JA, Cao M, Corti S, Hedlund E. Differential neuronal vulnerability identifies IGF-2 as a protective factor in ALS. Sci Rep. 2016;16(6):25960. doi: 10.1038/srep25960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.AlSaman A, Tomoum H. Infantile spinal muscular atrophy with respiratory distress type 1: a case report. J Child Neurol. 2010;25(6):764–769. doi: 10.1177/0883073809344121. [DOI] [PubMed] [Google Scholar]
- 4.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 5.Bhattacharya A, Czaplinski K, Trifillis P, He F, Jacobson A, Peltz SW. Characterization of the biochemical properties of the human Upf1 gene product that is involved in nonsense-mediated mRNA decay. RNA. 2000;6(9):1226–1235. doi: 10.1017/s1355838200000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet. 2011;45:273–297. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- 7.Bertini E, Gadisseux JL, Palmieri G, Ricci E, Di Capua M, Ferriere G, Lyon G. Distal infantile spinal muscular atrophy associated with paralysis of the diaphragm: a variant of infantile spinal muscular atrophy. Am J Med Genet. 1989;33(3):328–335. doi: 10.1002/ajmg.1320330309. [DOI] [PubMed] [Google Scholar]
- 8.Blaschek A, Gläser D, Kuhn M, Schroeder AS, Wimmer C, Heimkes B, Schön C, Müller-Felber W. Early infantile sensory-motor neuropathy with late onset respiratory distress. Neuromuscul Disord. 2014;24(3):269–271. doi: 10.1016/j.nmd.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 9.Braun R, Wang Z, Mack DL, Childers MK. Gene therapy for inherited muscle diseases: where genetics meets rehabilitation medicine. Am J Phys Med Rehabil. 2014;93(11 Suppl 3):S97–S107. doi: 10.1097/PHM.0000000000000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bushby K, Finkel R, Wong B, Barohn R, Campbell C, Comi GP, Connolly AM, Day JW, Flanigan KM, Goemans N, Jones KJ, Mercuri E, Quinlivan R, Renfroe JB, Russman B, Ryan MM, Tulinius M, Voit T, Moore SA, Lee Sweeney H, Abresch RT, Coleman KL, Eagle M, Florence J, Gappmaier E, Glanzman AM, Henricson E, Barth J, Elfring GL, Reha A, Spiegel RJ, O’donnell MW, Peltz SW, Mcdonald CM, PTC124-GD-007-DMD Study Group Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014;50(4):477–487. doi: 10.1002/mus.24332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caroni P, Grandes P. Nerve sprouting in innervated adult skeletal muscle induced by exposure to elevated levels of insulin-like growth factors. J Cell Biol. 1990;110(4):1307–1317. doi: 10.1083/jcb.110.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caroni P, Schneider C, Kiefer MC, Zapf J. Role of muscle insulin-like growth factors in nerve sprouting: suppression of terminal sprouting in paralyzed muscle by IGF-binding protein 4. J Cell Biol. 1994;125(4):893–902. doi: 10.1083/jcb.125.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cassini TA, Duncan L, Rives LC, Newman JH, Phillips JA, Koziura ME, Brault J, Hamid R, Cogan J, Undiagnosed Diseases Network Whole genome sequencing reveals novel IGHMBP2 variant leading to unique cryptic splice-site and Charcot-Marie-Tooth phenotype with early onset symptoms. Mol Genet Genom Med. 2019 doi: 10.1002/mgg3.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen YZ, Hashemi SH, Anderson SK, Huang Y, Moreira MC, Lynch DR, Glass IA, Chance PF, Bennett CL. Senataxin, the yeast Sen1p orthologue: characterization of a unique protein in which recessive mutations cause ataxia and dominant mutations cause motor neuron disease. Neurobiol Dis. 2006;23(1):97–108. doi: 10.1016/j.nbd.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 15.Chiu ATG, Chan SHS, Wu SP, Ting SH, Chung BHY, Chan AOK, Wong VCN. Spinal muscular atrophy with respiratory distress type 1—a child with atypical presentation. Child Neurol Open. 2018;5:2329048X18769811. doi: 10.1177/2329048x18769811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cook SA, Johnson KR, Bronson RT, Davisson MT. Neuromuscular degeneration (nmd): a mutation on mouse chromosome 19 that causes motor neuron degeneration. Mamm Genome. 1995;6(3):187–191. doi: 10.1007/BF00293010. [DOI] [PubMed] [Google Scholar]
- 17.Corti S, Nizzardo M, Nardini M, Donadoni C, Salani S, Del Bo R, Papadimitriou D, Locatelli F, Mezzina N, Gianni F, Bresolin N, Comi GP. Motoneuron transplantation rescues the phenotype of SMARD1 (Spinal Muscular Atrophy with Respiratory Distress type 1) J Neurosci. 2009;29(38):11761–11771. doi: 10.1523/JNEUROSCI.2734-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corti S, Nizzardo M, Simone C, Falcone M, Nardini M, Ronchi D, Donadoni C, Salani S, Riboldi G, Magri F, Menozzi G, Bonaglia C, Rizzo F, Bresolin N, Comi GP. Genetic correction of human induced pluripotent stem cells from patients with spinal muscular atrophy. Sci Transl Med. 2012;4(165ra162):162. doi: 10.1126/scitranslmed.3004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cottenie E, Kochanski A, Jordanova A, Bansagi B, Zimon M, Horga A, Jaunmuktane Z, Saveri P, Rasic VM, Baets J, Bartsakoulia M, Ploski R, Teterycz P, Nikolic M, Quinlivan R, Laura M, Sweeney MG, Taroni F, Lunn MP, Moroni I, Gonzalez M, Hanna MG, Bettencourt C, Chabrol E, Franke A, von Au K, Schilhabel M, Kabzińska D, Hausmanowa-Petrusewicz I, Brandner S, Lim SC, Song H, Choi BO, Horvath R, Chung KW, Zuchner S, Pareyson D, Harms M, Reilly MM, Houlden H. Truncating and missense mutations in IGHMBP2 cause Charcot-Marie Tooth disease type 2. Am J Hum Genet. 2014;95(5):590–601. doi: 10.1016/j.ajhg.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cox GA, Mahaffey CL, Frankel WN. Identification of the mouse neuromuscular degeneration gene and mapping of a second site suppressor allele. Neuron. 1998;21(6):1327–1337. doi: 10.1016/s0896-6273(00)80652-2. [DOI] [PubMed] [Google Scholar]
- 21.Dabrowski M, Bukowy-Bieryllo Z, Zietkiewicz E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol Med. 2018;24(1):25. doi: 10.1186/s10020-018-0024-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Planell-Saguer M, Schroeder DG, Rodicio MC, Cox GA, Mourelatos Z. Biochemical and genetic evidence for a role of IGHMBP2 in the translational machinery. Hum Mol Genet. 2009;18(12):2115–2126. doi: 10.1093/hmg/ddp134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, Wichterle H, Henderson CE, Eggan K. Induced pluripotent stem cells generated from patients with ALS can be different in motor neurons. Science. 2008;321(5893):1218–1221. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- 24.Ebert AD, Svendsen CN. Stem cell model of spinal muscular atrophy. Arch Neurol. 2010;67(6):665–669. doi: 10.1001/archneurol.2010.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eckart M, Guenther UP, Idkowiak J, Varon R, Grolle B, Boffi P, Van Maldergem L, Hübner C, Schuelke M, von Au K. The natural course of infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1) Pediatrics. 2012;129(1):e148–e156. doi: 10.1542/peds.2011-0544. [DOI] [PubMed] [Google Scholar]
- 26.Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, Padmanabhan A, Clay-Falcone D, McCluskey L, Elman L, Juhr D, Gruber PJ, Rüb U, Auburger G, Trojanowski JQ, Lee VM, Van Deerlin VM, Bonini NM, Gitler AD. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466(7310):1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fanos V, Cuccu A, Nemolato S, Marinelli V, Faa G. A new nonsense mutation of the IGHMBP2 gene responsible for the first case of SMARD1 in a Sardinian patient with giant cell hepatitis. Neuropediatrics. 2010;41(3):132–134. doi: 10.1055/s-0030-1262852. [DOI] [PubMed] [Google Scholar]
- 28.Faravelli I, Nizzardo M, Comi GP, Corti S. Spinal muscular atrophy—recent therapeutic advances for an old challenge. Nat Rev Neurol. 2015;11(6):351–359. doi: 10.1038/nrneurol.2015.77. [DOI] [PubMed] [Google Scholar]
- 29.Feldkötter M, Schwarzer V, Wirth R, Wienker TF, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002;70(2):358–368. doi: 10.1086/338627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foust KD, Kaspar BK. Over the barrier and through the blood: to CNS delivery we go. Cell Cycle. 2009;8(24):4017–4018. doi: 10.4161/cc.8.24.10245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27(1):59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fukita Y, Mizuta TR, Shirozu M, Ozawa K, Shimizu A, Honjo T. The human S mu bp-2, a DNA-binding protein specific to the single-stranded guanine-rich sequence related to the immunoglobulin mu chain switch region. J Biol Chem. 1993;268(23):17463–17470. [PubMed] [Google Scholar]
- 33.Geiduschek EP, Kassavetis GA. The RNA polymerase III transcription apparatus. J Mol Biol. 2001;310(1):1–26. doi: 10.1006/jmbi.2001.4732. [DOI] [PubMed] [Google Scholar]
- 34.Giannini A, Pinto AM, Rossetti G, Prandi E, Tiziano D, Brahe C, Nardocci N. Respiratory failure in infants due to spinal muscular atrophy with respiratory distress type 1. Intensive Care Med. 2006;32(11):1851–1855. doi: 10.1007/s00134-006-0346-8. [DOI] [PubMed] [Google Scholar]
- 35.Grohmann K, Wienker TF, Saar K, Rudnik-Schöneborn S, Stoltenburg-Didinger G, Rossi R, Novelli G, Nürnberg G, Pfeufer A, Wirth B, Reis A, Zerres K, Hübner C. Diaphragmatic spinal muscular atrophy with respiratory distress is heterogeneous, and one form Is linked to chromosome 11q13-q21. Am J Hum Genet. 1999;65(5):1459–1462. doi: 10.1086/302636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grohmann K, Schuelke M, Diers A, Hoffmann K, Lucke B, Adams C, Bertini E, Leonhardt-Horti H, Muntoni F, Ouvrier R, Pfeufer A, Rossi R, Van Maldergem L, Wilmshurst JM, Wienker TF, Sendtner M, Rudnik-Schöneborn S, Zerres K, Hübner C. Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat Genet. 2001;29(1):75–77. doi: 10.1038/ng703. [DOI] [PubMed] [Google Scholar]
- 37.Grohmann K, Varon R, Stolz P, Schuelke M, Janetzki C, Bertini E, Bushby K, Muntoni F, Ouvrier R, Van Maldergem L, Goemans NM, Lochmüller H, Eichholz S, Adams C, Bosch F, Grattan-Smith P, Navarro C, Neitzel H, Polster T, Topaloğlu H, Steglich C, Guenther UP, Zerres K, Rudnik-Schöneborn S, Hübner C. Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1) Ann Neurol. 2003;54(6):719–724. doi: 10.1002/ana.10755. [DOI] [PubMed] [Google Scholar]
- 38.Grohmann K, Rossoll W, Kobsar I, Holtmann B, Jablonka S, Wessig C, Stoltenburg-Didinger G, Fischer U, Hübner C, Martini R, Sendtner M. Characterization of Ighmbp2 in motor neurons and implications for the pathomechanism in a mouse model of human spinal muscular atrophy with respiratory distress type 1 (SMARD1) Hum Mol Genet. 2004;13(18):2031–2042. doi: 10.1093/hmg/ddh222. [DOI] [PubMed] [Google Scholar]
- 39.Guenther UP, Schuelke M, Bertini E, D’Amico A, Goemans N, Grohmann K, Hübner C, Varon R. Genomic rearrangements at the IGHMBP2 gene locus in two patients with SMARD1. Hum Genet. 2004;115(4):319–326. doi: 10.1007/s00439-004-1156-0. [DOI] [PubMed] [Google Scholar]
- 40.Guenther UP, Handoko L, Laggerbauer B, Jablonka S, Chari A, Alzheimer M, Ohmer J, Plöttner O, Gehring N, Sickmann A, von Au K, Schuelke M, Fischer U. IGHMBP2 is a ribosome-associated helicase inactive in the neuromuscular disorder distal SMA type 1 (DSMA1) Hum Mol Genet. 2009;18(7):1288–1300. doi: 10.1093/hmg/ddp028. [DOI] [PubMed] [Google Scholar]
- 41.Guenther UP, Handoko L, Varon R, Stephani U, Tsao CY, Mendell JR, Lützkendorf S, Hübner C, von Au K, Jablonka S, Dittmar G, Heinemann U, Schuetz A, Schuelke M. Clinical variability in distal spinal muscular atrophy type 1 (DSMA1): determination of steady-state IGHMBP2 protein levels in five patients with infantile and juvenile disease. J Mol Med (Berl) 2009;87(1):31–41. doi: 10.1007/s00109-008-0402-7. [DOI] [PubMed] [Google Scholar]
- 42.Hachiya Y, Arai H, Hayashi M, Kumada S, Furushima W, Ohtsuka E, Ito Y, Uchiyama A, Kurata K. Autonomic dysfunction in cases of spinal muscular atrophy type 1 with long survival. Brain Dev. 2005;27(8):574–578. doi: 10.1016/j.braindev.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 43.Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, Rothstein JD, Wang J. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507(7491):195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, Song S, Likhite S, Murtha MJ, Foust KD, Rao M, Eagle A, Kammesheidt A, Christensen A, Mendell JR, Burghes AH, Kaspar BK. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29(9):824–828. doi: 10.1038/nbt.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han C, Mai J, Tian T, He Y, Liao J, Wen F, Yi X, Yang Y. Patient with spinal muscular atrophy with respiratory distress type 1 presenting initially with hypertonia. Brain Dev. 2015;37(5):542–545. doi: 10.1016/j.braindev.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 46.Hanada T, Weitzer S, Mair B, Bernreuther C, Wainger BJ, Ichida J, Hanada R, Orthofer M, Cronin SJ, Komnenovic V, Minis A, Sato F, Mimata H, Yoshimura A, Tamir I, Rainer J, Kofler R, Yaron A, Eggan KC, Woolf CJ, Glatzel M, Herbst R, Martinez J, Penninger JM. CLP1 links tRNA metabolism to progressive motor-neuron loss. Nature. 2013;495(7442):474–480. doi: 10.1038/nature11923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hatchi E, Skourti-Stathaki K, Ventz S, Pinello L, Yen A, Kamieniarz-Gdula K, Dimitrov S, Pathania S, McKinney KM, Eaton ML, Kellis M, Hill SJ, Parmigiani G, Proudfoot NJ, Livingston DM. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol Cell. 2015;57(4):636–647. doi: 10.1016/j.molcel.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- 49.Hughes RA, Sendtner M, Thoenen H. Members of several gene families influence survival of rat motoneurons in vitro and in vivo. J Neurosci Res. 1993;36(6):663–671. doi: 10.1002/jnr.490360607. [DOI] [PubMed] [Google Scholar]
- 50.Jablonka S, Beck M, Lechner BD, Mayer C, Sendtner M. Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J Cell Biol. 2007;179(1):139–149. doi: 10.1083/jcb.200703187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jangi M, Fleet C, Cullen P, Gupta SV, Mekhoubad S, Chiao E, Allaire N, Bennett CF, Rigo F, Krainer AR, Hurt JA, Carulli JP, Staropoli JF. SMN deficiency in severe models of spinal muscular atrophy causes widespread intron retention and DNA damage. Proc Natl Acad Sci USA. 2017;114(12):E2347–E2356. doi: 10.1073/pnas.1613181114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jaudzems K, Jia X, Yagi H, Zhulenkovs D, Graham B, Otting G, Liepinsh E. Structural basis for 5′-end-specific recognition of single-stranded DNA by the R3H domain from human Sμbp-2. J Mol Biol. 2012;424(1–2):42–53. doi: 10.1016/j.jmb.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 53.Jędrzejowska M, Madej-Pilarczyk A, Fidziańska A, Mierzewska H, Pronicka E, Obersztyn E, Gos M, Pronicki M, Kmieć T, Migdał M, Mierzewska-Schmidt M, Walczak-Wojtkowska I, Konopka E, Hausmanowa-Petrusewicz I. Severe phenotypes of SMARD1 associated with novel mutations of the IGHMBP2 gene and nuclear degeneration of muscle and Schwann cells. Eur J Paediatr Neurol. 2014;18(2):183–192. doi: 10.1016/j.ejpn.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 54.Joseph S, Robb SA, Mohammed S, Lillis S, Simonds A, Manzur AY, Walter S, Wraige E. Interfamilial phenotypic heterogeneity in SMARD1. Neuromuscul Disord. 2009;19(3):193–195. doi: 10.1016/j.nmd.2008.11.013. [DOI] [PubMed] [Google Scholar]
- 55.Kaindl AM, Guenther UP, Rudnik-Schöneborn S, Varon R, Zerres K, Gressens P, Schuelke M, Hubner C, von Au K. Distal spinal-muscular atrophy 1 (DSMA1 or SMARD1) Arch Pediatr. 2008;15(10):1568–1572. doi: 10.1016/j.arcped.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 56.Kanemaki M, Makino Y, Yoshida T, Kishimoto T, Koga A, Yamamoto K, Yamamoto M, Moncollin V, Egly JM, Muramatsu M, Tamura T. Molecular cloning of a rat 49-kDa TBP-interacting protein (TIP49) that is highly homologous to the bacterial RuvB. Biochem Biophys Res Commun. 1997;235(1):64–68. doi: 10.1006/bbrc.1997.6729. [DOI] [PubMed] [Google Scholar]
- 57.Karaca E, Weitzer S, Pehlivan D, Shiraishi H, Gogakos T, Hanada T, Jhangiani SN, Wiszniewski W, Withers M, Campbell IM, Erdin S, Isikay S, Franco LM, Gonzaga-Jauregui C, Gambin T, Gelowani V, Hunter JV, Yesil G, Koparir E, Yilmaz S, Brown M, Briskin D, Hafner M, Morozov P, Farazi TA, Bernreuther C, Glatzel M, Trattnig S, Friske J, Kronnerwetter C, Bainbridge MN, Gezdirici A, Seven M, Muzny DM, Boerwinkle E, Ozen M, Baylor Hopkins Center for Mendelian Genomics. Clausen T, Tuschl T, Yuksel A, Hess A, Gibbs RA, Martinez J, Penninger JM, Lupski JR. Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell. 2014;157(3):636–650. doi: 10.1016/j.cell.2014.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim YA, Jin HY, Kim YM. Diagnostic Odyssey and application of targeted exome sequencing in the investigation of recurrent infant deaths in a Syrian consanguineous family: a case of spinal muscular atrophy with respiratory distress type 1. J Korean Med Sci. 2019;34(9):e54. doi: 10.3346/jkms.2019.34.e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krieger F, Elflein N, Saenger S, Wirthgen E, Rak K, Frantz S, Hoeflich A, Toyka KV, Metzger F, Jablonka S. Polyethylene glycol-coupled IGF1 delays motor function defects in a mouse model of spinal muscular atrophy with respiratory distress type 1. Brain. 2014;137(Pt 5):1374–1393. doi: 10.1093/brain/awu059. [DOI] [PubMed] [Google Scholar]
- 60.Kruminis-Kaszkiel E, Juranek J, Maksymowicz W, Wojtkiewicz J. CRISPR/Cas9 technology as an emerging tool for targeting amyotrophic lateral sclerosis (ALS) Int J Mol Sci. 2018;19(3):E906. doi: 10.3390/ijms19030906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li DK, Tisdale S, Lotti F, Pellizzoni L. SMN control of RNP assembly: from post-transcriptional gene regulation to motor neuron disease. Semin Cell Dev Biol. 2014;32:22–29. doi: 10.1016/j.semcdb.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lim SC, Bowler MW, Lai TF, Song H. The Ighmbp2 helicase structure reveals the molecular basis for disease-causing mutations in DMSA1. Nucleic Acids Res. 2012;40(21):11009–11022. doi: 10.1093/nar/gks792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Litvinenko I, Kirov AV, Georgieva R, Todorov T, Malinova Z, Mitev V, Todorova A. One novel and one recurrent mutation in IGHMBP2 gene, causing severe spinal muscular atrophy respiratory distress 1 with onset soon after birth. J Child Neurol. 2014;29(6):799–802. doi: 10.1177/0883073813477203. [DOI] [PubMed] [Google Scholar]
- 64.Maddatu TP, Garvey SM, Schroeder DG, Hampton TG, Cox GA. Transgenic rescue of neurogenic atrophy in the nmd mouse reveals a role for Ighmbp2 in dilated cardiomyopathy. Hum Mol Genet. 2004;13(11):1105–1115. doi: 10.1093/hmg/ddh129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maddatu TP, Garvey SM, Schroeder DG, Zhang W, Kim SY, Nicholson AI, Davis CJ, Cox GA. Dilated cardiomyopathy in the nmd mouse: transgenic rescue and QTLs that improve cardiac function and survival. Hum Mol Genet. 2005;14(21):3179–3189. doi: 10.1093/hmg/ddi349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martins KJ, Gehrig SM, Naim T, Saenger S, Baum D, Metzger F, Lynch GS. Intramuscular administration of PEGylated IGF-I improves skeletal muscle regeneration after myotoxic injury. Growth Horm IGF Res. 2013;23(4):128–133. doi: 10.1016/j.ghir.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 67.Maystadt I, Zarhrate M, Landrieu P, Boespflug-Tanguy O, Sukno S, Collignon P, Melki J, Verellen-Dumoulin C, Munnich A, Viollet L. Allelic heterogeneity of SMARD1 at the IGHMBP2 locus. Hum Mutat. 2004;23(5):525–526. doi: 10.1002/humu.9241. [DOI] [PubMed] [Google Scholar]
- 68.McCarty DM. Self-complementary AAV vectors; advances and applications. Mol Ther. 2008;16(10):1648–1656. doi: 10.1038/mt.2008.171. [DOI] [PubMed] [Google Scholar]
- 69.Mellins RB, Hays AP, Gold AP, Berdon WE, Bowdler JD. Respiratory distress as the initial manifestation of Werdnig-Hoffmann disease. Pediatrics. 1974;53(1):33–40. [PubMed] [Google Scholar]
- 70.Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, Lowes L, Alfano L, Berry K, Church K, Kissel JT, Nagendran S, L’Italien J, Sproule DM, Wells C, Cardenas JA, Heitzer MD, Kaspar A, Corcoran S, Braun L, Likhite S, Miranda C, Meyer K, Foust KD, Burghes AHM, Kaspar BK. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 71.Messina MF, Messina S, Gaeta M, Rodolico C, Salpietro Damiano AM, Lombardo F, Crisafulli G, De Luca F. Infantile spinal muscular atrophy with respiratory distress type I (SMARD 1): an atypical phenotype and review of the literature. Eur J Paediatr Neurol. 2012;16(1):90–94. doi: 10.1016/j.ejpn.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 72.Mizuta TR, Fukita Y, Miyoshi T, Shimizu A, Honjo T. Isolation of cDNA encoding a binding protein specific to 5′-phosphorylated single-stranded DNA with G-rich sequences. Nucleic Acids Res. 1993;21(8):1761–1766. doi: 10.1093/nar/21.8.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10(5):615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Neff NT, Prevette D, Houenou LJ, Lewis ME, Glicksman MA, Yin QW, Oppenheim RW. Insulin-like growth factors: putative muscle-derived trophic agents that promote motoneuron survival. J Neurobiol. 1993;24(12):1578–1588. doi: 10.1002/neu.480241203. [DOI] [PubMed] [Google Scholar]
- 75.Nizzardo M, Simone C, Rizzo F, Salani S, Dametti S, Rinchetti P, Del Bo R, Foust K, Kaspar BK, Bresolin N, Comi GP, Corti S. Gene therapy rescues disease phenotype in a spinal muscular atrophy with respiratory distress type 1 (SMARD1) mouse model. Sci Adv. 2015;1(2):e1500078. doi: 10.1126/sciadv.1500078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nomura T, Takenouchi T, Fukushima H, Shimozato S, Kosaki K, Takahashi T. Catastrophic autonomic crisis with cardiovascular collapse in spinal muscular atrophy with respiratory distress type 1. J Child Neurol. 2013;28(7):949–951. doi: 10.1177/0883073812453321. [DOI] [PubMed] [Google Scholar]
- 77.Oda T, Kayukawa K, Hagiwara H, Yudate HT, Masuho Y, Murakami Y, Tamura TA, Muramatsu MA. A novel TATA-binding protein-binding protein, ABT1, activates basal transcription and has a yeast homolog that is essential for growth. Mol Cell Biol. 2000;20(4):1407–1418. doi: 10.1128/mcb.20.4.1407-1418.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oren YS, Pranke IM, Kerem B, Sermet-Gaudelus I. The suppression of premature termination codons and the repair of splicing mutations in CFTR. Curr Opin Pharmacol. 2017;34:125–131. doi: 10.1016/j.coph.2017.09.017. [DOI] [PubMed] [Google Scholar]
- 79.Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, Meyer T, Cimprich KA. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell. 2009;35(2):228–239. doi: 10.1016/j.molcel.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Perego MGL, Taiana M, Bresolin N, Comi GP, Corti S. R-loops in motor neuron diseases. Mol Neurobiol. 2019;56(4):2579–2589. doi: 10.1007/s12035-018-1246-y. [DOI] [PubMed] [Google Scholar]
- 81.Pitt M, Houlden H, Jacobs J, Mok Q, Harding B, Reilly M, Surtees R. Severe infantile neuropathy with diaphragmatic weakness and its relationship to SMARD1. Brain. 2003;126(Pt 12):2682–2692. doi: 10.1093/brain/awg278. [DOI] [PubMed] [Google Scholar]
- 82.Porro F, Rinchetti P, Magri F, Riboldi G, Nizzardo M, Simone C, Zanetta C, Faravelli I, Corti S. The wide spectrum of clinical phenotypes of spinal muscular atrophy with respiratory distress type 1: a systematic review. J Neurol Sci. 2014;346(1–2):35–42. doi: 10.1016/j.jns.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 83.Raff M. Adult stem cell plasticity: fact or artifact? Annu Rev Cell Dev Biol. 2003;19(1):1–22. doi: 10.1146/annurev.cellbio.19.111301.143037. [DOI] [PubMed] [Google Scholar]
- 84.Richard P, Manley JL. R loops and links to human disease. J Mol Biol. 2017;429(21):3168–3180. doi: 10.1016/j.jmb.2016.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Robbins PD, Tahara H, Ghivizzani SC. Viral vectors for gene therapy. Trends Biotechnol. 1998;16(1):35–40. doi: 10.1016/S0167-7799(97)01137-2. [DOI] [PubMed] [Google Scholar]
- 86.Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, Yancopoulos GD, Glass DJ. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol. 2001;3(11):1009–1013. doi: 10.1038/ncb1101-1009. [DOI] [PubMed] [Google Scholar]
- 87.Rossoll W, Jablonka S, Andreassi C, Kröning AK, Karle K, Monani UR, Sendtner M. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol. 2003;163(4):801–812. doi: 10.1083/jcb.200304128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rudnik-Schöneborn S, Zerres K, Hahnen E, Meng G, Voit T, Hanefeld F, Wirth B. Apparent autosomal recessive inheritance in families with proximal spinal muscular atrophy affecting individuals in two generations. Am J Hum Genet. 1996;59(5):1163–1165. [PMC free article] [PubMed] [Google Scholar]
- 89.Rudnik-Schöneborn S, Stolz P, Varon R, Grohmann K, Schächtele M, Ketelsen UP, Stavrou D, Kurz H, Hübner C, Zerres K. Long-term observations of patients with infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1) Neuropediatrics. 2004;35(3):174–182. doi: 10.1055/s-2004-820994. [DOI] [PubMed] [Google Scholar]
- 90.Ruiz R, Lin J, Forgie A, Foletti D, Shelton D, Rosenthal A, Tabares L. Treatment with trkC agonist antibodies delays disease progression in neuromuscular degeneration (nmd) mice. Hum Mol Genet. 2005;14(13):1825–1837. doi: 10.1093/hmg/ddi189. [DOI] [PubMed] [Google Scholar]
- 91.Salvi JS, Mekhail K. R-loops highlight the nucleus in ALS. Nucleus. 2015;6(1):23–29. doi: 10.1080/19491034.2015.1004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Santos-Pereira JM, Aguilera A. R loops: new modulators of genome dynamics and function. Nat Rev Genet. 2015;16(10):583–597. doi: 10.1038/nrg3961. [DOI] [PubMed] [Google Scholar]
- 93.Schaffer AE, Eggens VR, Caglayan AO, Reuter MS, Scott E, Coufal NG, Silhavy JL, Xue Y, Kayserili H, Yasuno K, Rosti RO, Abdellateef M, Caglar C, Kasher PR, Cazemier JL, Weterman MA, Cantagrel V, Cai N, Zweier C, Altunoglu U, Satkin NB, Aktar F, Tuysuz B, Yalcinkaya C, Caksen H, Bilguvar K, Fu XD, Trotta CR, Gabriel S, Reis A, Gunel M, Baas F, Gleeson JG. CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell. 2014;157(3):651–663. doi: 10.1016/j.cell.2014.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schramm L, Hernandez N. Recruitment of RNA polymerase III to its target promoters. Genes Dev. 2002;16(20):2593–2620. doi: 10.1101/gad.1018902. [DOI] [PubMed] [Google Scholar]
- 95.Shababi M, Villalón E, Kaifer KA, DeMarco V, Lorson CL. A direct comparison of IV and ICV delivery methods for gene replacement therapy in a mouse model of SMARD1. Mol Ther Methods Clin Dev. 2018;17(10):348–360. doi: 10.1016/j.omtm.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov. 2017;16(2):115–130. doi: 10.1038/nrd.2016.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Simone C, Nizzardo M, Rizzo F, Ruggieri M, Riboldi G, Salani S, Bucchia M, Bresolin N, Comi GP, Corti S. iPSC-Derived neural stem cells act via kinase inhibition to exert neuroprotective effects in spinal muscular atrophy with respiratory distress type 1. Stem Cell Rep. 2014;3(2):297–311. doi: 10.1016/j.stemcr.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 99.Skourti-Stathaki K, Proudfoot NJ, Gromak N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell. 2011;42(6):794–805. doi: 10.1016/j.molcel.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Smith A. A glossary for stem-cell biology. Nature. 2006 doi: 10.1038/nature04954. [DOI] [Google Scholar]
- 101.Stalpers XL, Verrips A, Poll-The BT, Cobben JM, Snoeck IN, de Coo IF, Brooks A, Bulk S, Gooskens R, Fock A, Verschuuren-Bemelmans C, Sinke RJ, de Visser M, Lemmink HH. Clinical and mutational characteristics of spinal muscular atrophy with respiratory distress type 1 in The Netherlands. Neuromuscul Disord. 2013;23(6):461–468. doi: 10.1016/j.nmd.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 102.Studer L, Vera E, Cornacchia D. Programming and reprogramming cellular age in the era of induced pluripotency. Cell Stem Cell. 2015;16(6):591–600. doi: 10.1016/j.stem.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sun X, Perlick HA, Dietz HC, Maquat LE. A mutated human homologue to yeast Upf1 protein has a dominant-negative effect on the decay of nonsense-containing mRNAs in mammalian cells. Proc Natl Acad Sci USA. 1998;95(17):10009–10014. doi: 10.1073/pnas.95.17.10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Surrey V, Zöller C, Lork AA, Moradi M, Balk S, Dombert B, Saal-Bauernschubert L, Briese M, Appenzeller S, Fischer U, Jablonka S. Impaired local translation of β-actin mRNA in Ighmbp2-deficient motoneurons: implications for spinal muscular atrophy with respiratory distress (SMARD1) Neuroscience. 2018;21(386):24–40. doi: 10.1016/j.neuroscience.2018.06.019. [DOI] [PubMed] [Google Scholar]
- 105.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 106.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 107.Thangarajh M, Elfring GL, Trifillis P, McIntosh J, Peltz SW, Ataluren Phase 2b Study Group The relationship between deficit in digit span and genotype in nonsense mutation Duchenne muscular dystrophy. Neurology. 2018;91(13):e1215–e1219. doi: 10.1212/wnl.0000000000006245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Trono D. Lentiviral vectors: turning a deadly foe into a therapeutic agent. Gene Ther. 2000;7(1):20–23. doi: 10.1038/sj.gt.3301105. [DOI] [PubMed] [Google Scholar]
- 109.Vanoli F, Rinchetti P, Porro F, Parente V, Corti S. Clinical and molecular features and therapeutic perspectives of spinal muscular atrophy with respiratory distress type 1. J Cell Mol Med. 2015;19(9):2058–2066. doi: 10.1111/jcmm.12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Villalón E, Shababi M, Kline R, Lorson ZC, Florea KM, Lorson CL. Selective vulnerability in neuronal populations in nmd/SMARD1 mice. Hum Mol Genet. 2015;27(4):679–690. doi: 10.1093/hmg/ddx434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang L, Yi F, Fu L, Yang J, Wang S, Wang Z, Suzuki K, Sun L, Xu X, Yu Y, Qiao J, Belmonte JCI, Yang Z, Yuan Y, Qu J, Liu GH. CRISPR/Cas9-mediated targeted gene correction in amyotrophic lateral sclerosis patient iPSCs. Protein Cell. 2017;8(5):365–378. doi: 10.1007/s13238-017-0397-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Weiss S, Reynolds BA, Vescovi AL, Morshead C, Craig CG, van der Kooy D. Is there a neural stem cell in the mammalian forebrain? Trends Neurosci. 1996;19(9):387–393. doi: 10.1016/s0166-2236(96)10035-7. [DOI] [PubMed] [Google Scholar]
- 113.Weitzer S, Hanada T, Penninger JM, Martinez J. CLP1 as a novel player in linking tRNA splicing to neurodegenerative disorders. Wiley Interdiscip Rev RNA. 2015;6(1):47–63. doi: 10.1002/wrna.1255. [DOI] [PubMed] [Google Scholar]
- 114.Wong VC, Chung BH, Li S, Goh W, Lee SL. Mutation of gene in spinal muscular atrophy respiratory distress type I. Pediatr Neurol. 2006;34(6):474–477. doi: 10.1016/j.pediatrneurol.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 115.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 116.Zhao DY, Gish G, Braunschweig U, Li Y, Ni Z, Schmitges FW, Zhong G, Liu K, Li W, Moffat J, Vedadi M, Min J, Pawson TJ, Blencowe BJ, Greenblatt JF. SMN and symmetric arginine dimethylation of RNA polymerase II C-terminal domain control termination. Nature. 2016;529(7584):48–53. doi: 10.1038/nature16469. [DOI] [PubMed] [Google Scholar]