

Abstract
Alzheimer’s disease (AD) is a leading cause of age-related dementia worldwide. Despite more than a century of intensive research, we are not anywhere near the discovery of a cure for this disease or a way to prevent its progression. Among the various molecular mechanisms proposed for the description of the pathogenesis and progression of AD, the amyloid cascade hypothesis, according to which accumulation of a product of amyloid precursor protein (APP) cleavage, amyloid β (Aβ) peptide, induces pathological changes in the brain observed in AD, occupies a unique niche. Although multiple proteins have been implicated in this amyloid cascade signaling pathway, their structure–function relationships are mostly unexplored. However, it is known that two major proteins related to AD pathology, Aβ peptide, and microtubule-associated protein tau belong to the category of intrinsically disordered proteins (IDPs), which are the functionally important proteins characterized by a lack of fixed, ordered three-dimensional structure. IDPs and intrinsically disordered protein regions (IDPRs) play numerous vital roles in various cellular processes, such as signaling, cell cycle regulation, macromolecular recognition, and promiscuous binding. However, the deregulation and misfolding of IDPs may lead to disturbed signaling, interactions, and disease pathogenesis. Often, molecular recognition-related IDPs/IDPRs undergo disorder-to-order transition upon binding to their biological partners and contain specific disorder-based binding motifs, known as molecular recognition features (MoRFs). Knowing the intrinsic disorder status and disorder-based functionality of proteins associated with amyloid cascade signaling pathway may help to untangle the mechanisms of AD pathogenesis and help identify therapeutic targets. In this paper, we have used multiple computational tools to evaluate the presence of intrinsic disorder and MoRFs in 27 proteins potentially relevant to the amyloid cascade signaling pathway. Among these, BIN1, APP, APOE, PICALM, PSEN1 and CD33 were found to be highly disordered. Furthermore, their disorder-based binding regions and associated short linear motifs have also been identified. These findings represent important foundation for the future research, and experimental characterization of disordered regions in these proteins is required to better understand their roles in AD pathogenesis.
Electronic supplementary material
The online version of this article (10.1007/s00018-019-03414-9) contains supplementary material, which is available to authorized users.
Keywords: Alzheimer’s disease, Amyloid cascade signaling, Intrinsically disordered proteins, Amyloid-beta, Molecular recognition features
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder, which is a leading cause of aging-related dementia worldwide. The first clinical description of this disease was published by Dr. Alois Alzheimer, wherein he described the case of a 50-year-old female patient with “peculiar severe disease process of the cerebral cortex” [1]. He observed two distinct pathologies, neurofibrillary tangles and neuritic plaques in the brain tissue of the patient when he performed an autopsy [2]. In 1984, Glenner and Wong showed that neuritic plaques are mainly composed of a 4.2 kDa peptide which was 40–42 amino acid long [3]. They speculated that this peptide was formed after cleavage of a larger precursor. This prediction was verified in 1987, when the amyloid precursor protein (APP) was identified and the short peptide derived from its processing is now known as the amyloid β (Aβ) peptide [4–7].
According to the 2018 World Alzheimer’s report, currently there are 50 million cases of dementia reported worldwide. This number is estimated to rise to 82 million by 2030 and to 152 million by 2050 as the current population ages. Even now, more than 110 years after the AD discovery, there is no clinically accepted treatment for complete cure of this disease and prevention of its progression (https://www.alz.co.uk/research/WorldAlzheimerReport 2018.pdf). Currently, there are only five FDA-approved drugs for treatment of AD [8]. These include cholinesterase inhibitors, N-methyl-d-aspartate (NMDA) receptor antagonist or a combination of both. However, these drugs provide only temporary symptomatic relief to the patients and have severe side effects [9].
Even though the detailed pathogenesis of the disease is still unclear, over the past century many hypotheses to explain its origin have been developed. These include Aβ cascade hypothesis, tau hypothesis, inflammation hypothesis, cholinergic and oxidative stress hypothesis, etc. [9]. The Aβ cascade hypothesis, which was proposed by Hardy and Higgins in 1992 [10], is the most widely accepted and best-defined hypothesis for AD pathogenesis. It postulates that the Aβ deposition is the primary and sole event for the AD initiation that leads to formation of senile plaques, neurofibrillary tangles (NFT), nerve cell death and finally clinical dementia [10]. APP processing through the amyloid cascade generates Aβ peptide and promotes its aggregation that accelerates downstream deleterious events, such as tau hyperphosphorylation, oxidative stress, mitochondrial dysfunction, synaptotoxicity, and neurotoxicity [11, 12].
AD is a highly heritable disease (76% heritability) characterized by a complex genetic profile of susceptibility [13]. Genome-wide association studies (GWAS) have reported more than 45 genes/loci potentially associated with the risk of AD development [14]. APP digestion and Aβ production are at the heart of AD pathogenesis [14, 15]. Many of these proteins are involved in pathological events of AD, such as APP processing, Aβ production, tau toxicity, aggregation, degradation, and clearance of Aβ [14]. Here, we report intrinsic disorder analysis and structural and functional characterization of 27 proteins involved in the amyloid cascade signaling pathway (see Fig. 1). A brief description of involvement of these proteins in AD pathogenesis is provided below.
Fig. 1.

Schematic representation of amyloid cascade signaling and AD-associated risk genes. Cleavage of APP occurs by three enzymes (α, β and γ-secretase). ADAM 9, ADAM 10, and ADAM 17 are associated with APP processing through α-secretase that follows the non-amyloidogenic pathway. Cleavage by β-secretase and γ-secretase complex follows amyloidogenic pathway that leads to formation of Aβ peptide. γ-secretase complex, a multi-subunit protease consists of PSEN1, PSEN2, APH1, PEN2, and NCSTN. SORL1 and BIN1 regulate APP processing and control Aβ production. Further, Aβ clearance occurs via vascular system where APOE, ABCA7, CLU, PICALM, and A2M are involved in Aβ clearance via BBB. LRP1 mediates brain Aβ clearance. CD33 and TREM2 are associated with Aβ degradation via microglia. Aβ degradation via enzymes occurs by Aβ degrading enzymes such as NEP, IDE, and ECE2. Aβ degradation also occurs by plasmin where PLAT, PLAU, and PLG play an important role. The troubles in APP processing, Aβ clearance and Aβ degradation leads to formation of Aβ oligomers and fibrils. Further, Aβ oligomers and fibrils stimulate formation of NFTs and senile plaques, the pathogenic hallmark of AD. Senile plaques and NFTs deposit into synapses and damage nerve cells that leads to loss of memory, cognition and further AD. All reported proteins are represented in red color
The α-secretase enzyme is crucial for the normal processing of APP. Proteins belonging to the ADAM (disintegrin and metalloprotease domain-containing protein) family, namely, ADAM9 [16], ADAM10 [17], and ADAM17 [18] act as α-secretases for APP [19] and catalyze the non-amyloidogenic digestion of APP, thus preventing generation of Aβ [20]. BACE 1, which is also known as β-secretase, is crucial for the initiation of Aβ generation [21, 22]. The γ-secretase complex is made up of five subunits, namely, PSEN1, PSEN2, APH1A, PEN2, and nicastrin. All these subunits are essential for full proteolytic activity of this machine, as each of them contributes differently to Aβ production [23, 24]. Autosomal dominant mutations in PSEN1, PSEN2, and APP lead to early onset familial Alzheimer’s disease (FAD) [12, 25]. Apolipoprotein E (APOE) isoforms play a key role in Aβ deposition and neuritic plaque formation [26]. Aβ clearance is conducted by Aβ-degrading enzymes, such as neprilysin, insulin-degrading enzyme (IDE), and plasminogen (PLG). However, the levels of IDE, neprilysin, and PLG were found to be reduced in AD [27, 28].
ATP-binding cassette transporter A7 (ABCA7) is important for the normal processing of APP and its loss of function leads to AD pathology [29]. Bridging integrator 1(BIN1), a member of amphiphysin family, is involved in endocytosis and the endosomal sorting of membrane proteins that regulate intracellular BACE1 trafficking and Aβ formation [30]. Phosphatidylinositol-binding clathrin assembly protein (PICALM) modulates autophagy for the degradation and clearance of Aβ and tau [31, 32]. Microglial phagocytosis is linked to a cluster of differentiation 33 (CD33) protein that acts as a strong risk factor for late-onset Alzheimer’s disease (LOAD). Increased expression of CD33 leads to impairment of microglia-mediated clearance of Aβ that further generates senile plaques in the brain [33, 34]. Clusterin (CLU), also known as apolipoprotein J (APOJ), is a ubiquitous multifunctional chaperone molecule which binds to the Aβ peptide and prevents its fibrillation [35, 36]. Its expression is increased in AD and it has now been identified as a risk factor for AD pathology [37].
Low-density lipoprotein receptor-related protein 1 (LRP1) is an endocytic receptor involved in Aβ clearance through the blood–brain barrier (BBB) and its impaired function is associated with AD pathogenesis [38, 39]. Alpha 2 macroglobulin (A2M) is a serum pan-protease inhibitor involved in the degradation and clearance of Aβ [40]. Endothelin-converting enzyme-2 (ECE2) cleaves big endothelin and produces endothelin-1. The ECE2 deficiency is associated with increased Aβ peptide level which supports the statement that ECE2 degrades Aβ peptide [41]. Triggering receptor expressed on myeloid cells 2 (TREM2) proteins expressed specifically in microglia. Although there is a conflict on whether TREM2 is beneficial or harmful, there are many reports about its significant role in AD pathogenesis [42].
Many proteins need to have stable, unique 3D structures to perform their function and these are known as structured or globular proteins. Apart from these, the proteomes of all the organisms studied so far also contain proteins that lack well-defined 3D structures but are still functional. These proteins are referred to as intrinsically disordered proteins (IDPs) [43]. Such disordered proteins are often involved in regulation, signaling and control processes. Their structural flexibility and plasticity enable them to interact with a large number of binding partners [44–47]. Upon binding to specific partners, some of these proteins might undergo a transition from disordered state to ordered state, in which the entire protein or a region of the protein adopts a well-structured conformation and is now ready to perform its function [44, 48–52]. In fact, IDPs/IDPRs are able to interact with various partners via multiple binding scenarios [53–56], form static, semi-static, and dynamic or fuzzy complexes, with the degree of such binding-induced folding being different in various systems, thereby forming complexes with broad structural and functional heterogeneity [57, 58].
Proteins play extremely crucial roles in the maintenance of life and hence their dysfunction can lead to occurrence of disease pathologies [44]. IDPs and IDPRs, proteins due to their specific structural and functional properties, are prone to misfold, which can lead to misidentification, misregulation, and missignaling and thus development of diseases [59, 60]. The misfolded peptides also tend to aggregate and gain toxic functions [59, 60]. IDPs are considered as potential and promising drug targets, since they are linked to the pathogenesis of numerous human diseases [61, 62]. In fact, many proteins associated with neurodegenerative diseases and various amyloidoses are intrinsically disordered or contain long IDPRs [63–66]. The conformational variation of IDPs into misfolding and aggregation is the major factor for occurrence of various diseases [67]. The disordered proteins associated with aggregation and disease pathologies include Aβ, tau and α-synuclein in neurodegenerative diseases, p53 and c-Myc in cancer, and amylin in diabetes [67]. Structural flexibility of IDPs is essential for their functions in various cellular processes. However, highly soluble disordered tau protein forms highly insoluble filamentous lesions in AD [68]. Since IDPs are very common in various proteomes and disorder content varies from viruses to higher organisms [47, 53, 56, 69–75], they must be protected from aggregation, as they contain few hydrophobic amino acids that can stick together and lead to pathogenesis [76]. Therefore, IDP analysis of the proteins involved in the amyloid cascade signaling pathway which leads to Aβ formation is crucial. As an essential step in that direction, we extensively analyzed these proteins for the presence of disordered regions. We have also performed MoRF analysis to identify binding sites on the proteins. The identified disordered regions may have a significant role in the pathogenesis of AD.
It is important to explore the potential role of intrinsic disorder and MoRFs in protein–protein interactions of amyloid cascade-related proteins. Using STRING database, we have obtained a protein–protein interaction network of these proteins. Also, we have used eukaryotic linear motif resource to identify short linear motifs in these protein sequences. Further characterization of the disordered regions can help identify new drug targets for AD as well as other diseases where these proteins are significantly involved in AD pathogenesis. Moreover, due to presence of long disordered regions in many of the proteins analyzed, in the near future, it is vital to study the cross-seeding with each other and with Aβ/tau peptides.
Materials and methods
Identification of proteins involved in the amyloid cascade signaling
The proteins involved in the amyloid cascade signaling pathway were identified in three different review articles published in 2005 [77], 2012 [78], and 2016 [79]. The 27 proteins that were identified and their role in AD are summarized in Table 1 and Fig. 1. The reviewed protein sequences of all proteins in Table 1 have been retrieved in FASTA format from the UniProt database [80].
Table 1.
Human risk genes/proteins susceptible to Alzheimer’s disease
| Sr. no. | Human gene/protein | Length (amino acids) | Functional relevance to AD | Relevent pathogenic event in AD | UniProt ID |
|---|---|---|---|---|---|
| 1 | BIN1 | 593 | APP processing, control Aβ production | Aβ generation and/Aβ aggregation | O00499 |
| 2 | APOE | 317 | Aβ clearance | Aβ aggregation, Aβ clearance | P02649 |
| 3 | APP | 770 | Synapse formation, neural plasticity | Aβ42/Aβ40 ratio, Aβ production and Aβ aggregation | P05067 |
| 4 | PICALM | 652 | Modulates autophagy for Aβ and tau clearance | Aβ- and Tau-mediated toxicity | Q13492 |
| 5 | CD33 | 364 | Microglia-mediated Aβ clearance | CD 33 expression, microglia-mediated Aβ clearance | P20138 |
| 6 | PSEN1 | 467 | α-Secretase complex, APP processing | Aβ42/Aβ40 ratio and Aβ aggregation | P49768 |
| 7 | ADAM 17 | 824 | APP processing by α-secretase | Aβ production | P78536 |
| 8 | Clusterin | 449 | Aβ clearance, prevent Aβ fibrillization | CLU expression, Aβ clearance | P10909 |
| 9 | PSEN2 | 448 | APP processing, α-secretase complex | Aβ42/Aβ40 ratio and Aβ aggregation | P49810 |
| 10 | PLG | 810 | Aβ degradation (via plasmin) | PLG activity, Aβ clearance | P00747 |
| 11 | ADAM 9 | 819 | APP processing by α-secretase | Aβ generation and/Aβ aggregation | Q13443 |
| 12 | ADAM 10 | 748 | Catalyzes non-amyloidogenic α-secretase-mediated cleavage of APP | Aβ production | O14672 |
| 13 | ABCA7 | 2,146 | APP processing, Aβ clearance | Aβ aggregation, Aβ clearance | Q8IZY2 |
| 14 | LRP1 | 4,544 | Aβ export/clearance through blood via BBB | Impaired LRP1 function, Aβ clearance | Q07954 |
| 15 | TREM2 | 230 | Modulates microglial function in response to Aβ and Tau | Dysfunction in TREM2 impairs brain Aβ metabolism | Q9NZC2 |
| 16 | ECE2 | 811 | Aβ degradation | ECE2 deficiency, Aβ degradation | P0DPD6 |
| 17 | SORL1 | 2,214 | Aβ metabolism | Aβ aggregation, Aβ clearance | Q92673 |
| 18 | A2M | 1,474 | Aβ export/clearance | Aβ clearance from the brain | P01023 |
| 19 | BACE1 | 501 | APP processing | Aβ production and Aβ aggregation | P56817 |
| 20 | Neprilysin | 750 | Aβ degradation | Neprilysin level, Aβ degradation | P08473 |
| 21 | Nicastrin | 709 | APP processing, a subunit of γ-secretase complex | Aβ generation and/Aβ aggregation | Q92542 |
| 22 | PEN2 | 101 | APP processing, a subunit of γ-secretase complex | Aβ production and/Aβ aggregation | Q9NZ42 |
| 23 | IDE | 1,019 | Aβ/AICD degradation | IDE level in brain, Aβ degradation | P14735 |
| 24 | PLAU | 431 | Aβ degradation (Via plasmin) | Aβ clearance, Aβ aggregation | P00749 |
| 25 | PLAT | 559 | Aβ degradation (via plasmin) | Aβ clearance, Aβ aggregation | P00750 |
| 26 | APH1A | 265 | APP processing, a subunit of γ-secretase complex | Aβ generation and/Aβ aggregation | Q96BI3 |
| 27 | APH1B | 257 | Intramembrane proteolysis of APP | Aβ production | Q8WW43 |
Evaluation of intrinsic disorder propensity of proteins related to the amyloid cascade hypothesis
To evaluate the predisposition of these proteins to disorder, computational tools available at the DisProt [81–83], a database of protein disorder have been used. DisProt includes proteins and protein regions with the experimentally validated intrinsic disorder and which has links to several computational tools commonly used for predicting intrinsic disorder in query proteins. In this study, several members of the Predictor of Natural Disordered Regions (PONDR) family have been used, namely PONDR® VSL2 [84], PONDR® VL3 [85], PONDR® VLXT [86], and PONDR® FIT [87].The various PONDRs are distinguished by the training sets and machine learning models used to develop them [48, 88]. Further, two forms of the IUPred tool have also been used [89] for prediction of long and short IDPRs in query proteins. Residues with disorder score above the 0.5 threshold value are considered as intrinsically disordered, whereas residues with the predicted disorder scores between 0.2 and 0.5 are considered as flexible. An overall predicted percent of intrinsic disorder (PPID) was calculated for each protein from outputs of six predictors. MobiDB [90], another database of protein disorder, was also used for analyzing the given proteins, and the mean PPID value from MobiDB was compared to the six predictors that were used.
Using a combined CH-CDF analysis approach to predict disorder
The charge-hydropathy (CH) plot [91] and the cumulative distribution function (CDF) are binary predictors that determine whether the query protein is mostly ordered or mostly disordered [92]. These tools are a part of PONDR web page (https://www.pondr.com/). When one combines the result from these two tools into a CH-CDF analysis, one analyzes proteins based on their charge, hydropathy, and PONDR score to distinguish between highly ordered and highly disordered proteins [93, 94].
Identification of molecular recognition features (MoRFs) in proteins involved in the amyloid cascade hypothesis.
In this study, MoRF regions in protein sequences have been identified using D2P2 and ANCHOR. D2P2 is a database of disordered protein predictions. But along with disorder, it also reports disordered regions that undergo disorder-to-order transition upon binding to their interacting partners [95]. These regions are known as MoRFs. ANCHOR [96], which is a web server associated with IUPred is also used for identifying these regions. The protein residues with an ANCHOR score above the threshold value of 0.5 are considered MoRF regions.
Generation of protein–protein interaction network
To obtain a comprehensive view of all protein–protein associations involving proteins of the amyloid cascade signaling, Search Tool for Retrieval of Interacting Genes (STRING) database has been used [97]. This database integrates all the information on protein–protein interactions that is publically available, complements it with computational predictions and returns an overview of all possible interactions of query protein(s) with other proteins [97].
Identification of motifs
The eukaryotic linear motif (ELM) resource was used to identify short linear motifs (SLiMs), which are often found in IDPRs. The ELM server performs globular domain, structural and context filtering on these predicted SLiMs and the motifs retained after filtering are annotated [98]. The retained motifs that are associated with the predicted MoRF residues have been described here. This paper mentions all six types of annotations for the SLiMs that are described by the ELM server [98]. These include motifs that act as proteolytic cleavage sites (ELM_CLV); degron motifs that play a role in polyubiquitylation and targeting of proteins to proteasomal degradation (ELM_DEG); docking motifs (ELM_DOC); ligand-binding motifs (ELM_LIG); sites for post-translational modifications (ELM_MOD) and motifs for targeting to subcellular compartments (ELM_TRG) [98].
Mapping of IDPRs and MoRFs on available structures
Known structures of proteins from amyloid cascade signaling with best resolution have been obtained from PDB. The IDPRs and MoRF residues (from D2P2) have been mapped on these structures using Maestro which is an interface for all Schrödinger software.
Results and discussion
Global analysis of the disorder predisposition of proteins associated with the amyloid cascade hypothesis
Table 2 represents the predicted percentage of intrinsic disorder (PPID) for each of the 27 proteins involved in the amyloid cascade signaling in AD. Figure 2a shows the 2D-disorder plot presenting the PPIDPONDR®FIT vs. PPIDmean plot to illustrate the peculiarities of disorder predisposition of 27 proteins from the amyloid cascade pathway. According to the overall levels of intrinsic disorder, proteins can be classified as highly ordered (PPID < 10%), moderately disordered (10% ≤ PPID < 30%) and highly disordered (PPID ≥ 30%) [99]. From this PPID-based classification and data shown in Table 2 and Fig. 2a, we can conclude that the proteins BIN1, APOE, PICALM, APP, PSEN1, and CD33 are highly disordered; APH1A, APH1B, PLAT, PLAU, IDE, neprilysin, PEN2, and nicastrin are highly ordered, whereas the remaining proteins in this dataset are moderately disordered. For more detailed computational analyses (see below), we selected six highly disordered proteins (APP, APOE, BIN1, PICALM, PSEN1, and CD33), six moderately disordered proteins (PSEN2, ADAM17, PEN2, PLG, clusterin, and BACE1), and three highly ordered proteins (APH1A, APH1B, and nicastrin) from this set of 27 proteins.
Table 2.
Predicted percentage of intrinsic disorder (PPID) in proteins involved in the amyloid cascade signaling
| Protein Name | PPID VSL2 | PPID VL3 | PPID VLXT | PPID FIT | PPID IUPRED long | PPID IUPRED short | PPID mean | Disorder content (MobiDB) |
|---|---|---|---|---|---|---|---|---|
| BIN1 | 63.91 | 59.53 | 53.63 | 47.05 | 53.12 | 37.44 | 52.45 | 28 |
| APOE | 74.76 | 63.09 | 61.20 | 45.11 | 15.77 | 6.31 | 44.37 | 0 |
| APP | 47.53 | 44.55 | 44.81 | 30.13 | 37.01 | 28.18 | 38.70 | 12 |
| PICALM | 59.20 | 49.23 | 34.36 | 36.20 | 25.46 | 13.96 | 36.40 | 3 |
| CD33 | 42.31 | 25.27 | 32.42 | 27.75 | 32.14 | 26.37 | 31.04 | 21 |
| PSEN1 | 33.40 | 28.48 | 31.05 | 32.12 | 23.98 | 27.84 | 29.48 | 18 |
| ADAM17 | 52.79 | 43.81 | 26.46 | 16.87 | 15.17 | 16.02 | 28.52 | 11 |
| Clusterin | 59.69 | 43.65 | 27.17 | 20.94 | 11.36 | 6.24 | 28.17 | 0 |
| PSEN2 | 30.58 | 28.79 | 32.14 | 22.54 | 15.85 | 25.00 | 25.82 | 16 |
| PLG | 50.86 | 36.91 | 25.06 | 11.48 | 11.85 | 14.32 | 25.08 | 5 |
| ADAM9 | 39.56 | 33.82 | 20.27 | 18.68 | 10.62 | 13.43 | 22.73 | 9 |
| ADAM10 | 44.52 | 34.09 | 16.31 | 11.63 | 12.17 | 12.30 | 21.84 | 6 |
| ABCA7 | 30.71 | 25.91 | 30.57 | 14.40 | 10.25 | 11.79 | 20.60 | 4 |
| LRP1 | 44.32 | 37.21 | 14.39 | 6.38 | 2.88 | 1.87 | 17.84 | 0 |
| TREM2 | 24.35 | 17.39 | 28.26 | 16.52 | 5.65 | 10.87 | 17.17 | 0 |
| ECE2 | 25.40 | 12.95 | 25.89 | 11.96 | 9.37 | 8.01 | 15.60 | No entry |
| SORL1 | 30.08 | 21.00 | 12.47 | 5.56 | 1.31 | 2.44 | 12.14 | 0 |
| A2M | 25.58 | 14.59 | 18.11 | 4.21 | 3.39 | 3.46 | 11.56 | 0 |
| BACE1 | 13.37 | 8.98 | 15.77 | 11.18 | 5.79 | 5.79 | 10.15 | 4 |
| Neprilysin | 18.53 | 5.07 | 19.20 | 7.07 | 1.87 | 3.20 | 9.16 | 3 |
| Nicastrin | 17.63 | 9.03 | 18.76 | 5.08 | 2.96 | 0.28 | 8.96 | 0 |
| PEN2 | 15.84 | 0.00 | 7.92 | 20.79 | 0.00 | 6.93 | 8.58 | 0 |
| IDE | 16.39 | 5.79 | 18.06 | 3.43 | 2.94 | 3.43 | 8.34 | 0 |
| PLAU | 14.85 | 7.42 | 9.51 | 7.19 | 0.46 | 2.55 | 7.00 | 0 |
| PLAT | 17.79 | 7.65 | 9.07 | 2.31 | 0.89 | 1.60 | 6.55 | 0 |
| APH1A | 9.43 | 5.28 | 7.55 | 6.79 | 0.00 | 3.02 | 5.35 | 0 |
| APH1B | 10.51 | 0.00 | 6.23 | 7.78 | 0.00 | 1.17 | 4.28 | 0 |
Fig. 2.

Evaluation of the overall disorder status of 27 human prpoteins from the amyloid cascade signaling pathway. a 2D disorder plot showing presenting the PPIDPONDR®FIT vs. PPIDmean dependence. b CH-CDF plot. In this plot, the coordinates of each spot are calculated as a distance of the corresponding protein in the CH-plot from the boundary (Y-coordinate) and an average distance of the respective CDF curve from the CDF boundary (X-coordinate)
To gain further insight into the nature of disorder in proteins associated with the amyloid cascade, we used CH-CDF analysis [94, 100, 101], which is based on a combined utilization of two binary disorder classifiers (i.e., predictors that evaluate the predisposition of a given protein to be ordered or disordered as a whole), charge-hydropathy (CH) plot [91, 92] and cumulative distribution function (CDF) plot [92, 101]. In the resulting CH-CDF plot, the coordinates of each spot are calculated as a distance of the corresponding protein in the CH-plot from the boundary (Y-coordinate) and an average distance of the respective CDF curve from the CDF boundary (X-coordinate) [94, 100, 101]. The primary difference between the binary predictors is that the CH-plot is a linear classifier that takes into account only two parameters of the particular sequence (charge and hydropathy), whereas CDF analysis is dependent on the output of the PONDR® predictor, a nonlinear classifier, which was trained to distinguish order and disorder based on a significantly larger feature space. According to these methodological differences, CH-plot analysis is predisposed to discriminate proteins with a substantial amount of extended disorder (random coils and pre-molten globules) from proteins with compact conformations (molten globule-like and ordered globular proteins). On the other hand, CDF analysis discriminates all disordered conformations, including native coils, native pre-molten globules, and native molten globules, from ordered globular proteins. Therefore, this computational discrepancy provides a useful tool for discrimination of proteins with extended disorder from molten globules and hybrid proteins containing noticeable (comparable) levels of ordered and disordered regions. Here, positive and negative Y values in the CH-CDF plot correspond to proteins predicted within CH-plot analysis to be extended or compact, respectively. On the other hand, positive and negative X values are attributed to proteins predicted within the CDF analysis to be ordered or intrinsically disordered, respectively. Therefore, the CDF-CH phase space can be separated on four quadrants that correspond to the following expectations: Q1, ordered proteins; Q2, proteins predicted to be disordered by CDFs, but compact by CH-plots (i.e., putative native molten globules or hybrid proteins); Q3, proteins predicted to be disordered by both methods (i.e., proteins with extended disorder); and Q4, proteins predicted to be disordered by CH-plots, but ordered by CDF [94, 100, 101]. Figure 2b represents the results of this analysis for the proteins associated with the amyloid cascade hypothesis and shows that these proteins are located within the two quadrants within the CH-CDF phase space, Q1 and Q2. Based on this analysis, one can conclude that ten proteins APOE, BIN1, APP, PICALM, PSEN1, PSEN2, TREM2, CD33, ADAM17, and ABCA7 are expected to be mostly disordered, whereas the remaining proteins associated with the amyloid cascade hypothesis are predicted as mostly ordered.
IDPs and IDPRs might contain certain sub-regions that tend to become ordered upon binding to appropriate partners. These sites are known as molecular recognition features (MoRFs), which represent short regions with increased order propensity embedded within longer IDPRs and which can be identified using various computational tools, including ANCHOR [96]. The D2P2database also identifies these potential binding sites located in disordered regions. Table 3 shows the MoRF-containing residues of each protein of the amyloid cascade signaling pathway predicted by ANCHOR and D2P2. This table also has a record of the percentage of MoRF residues in each protein. The protein BIN1 shows the highest percentage of MoRF residues. No MoRF residues were predicted in APH1A, APH1B, PEN2, nicastrin, SORL1, clusterin, PLAT, PLAU, neprilysin, A2M, and TREM2. This could be because most of these proteins have a low disorder score (PPID mean score < 20%), except clusterin, which has a mean PPID score of 28.2%, but shows no MoRF regions.
Table 3.
Identification of motifs and MoRF regions present in the proteins involved in the amyloid cascade hypothesis
| Protein | Retained (R) and Total (T) SLiMs | SLiMs | Instances | ANCHOR | D2P2 | Percentage of MoRF | |
|---|---|---|---|---|---|---|---|
| ANCHOR | D2P2 | ||||||
| BIN1 |
R T |
45 68 |
127 232 |
259–343 (85) 350–526 (177) 535–566 (32) |
67–72 (6) 234–246 (13) 260–286 (27) 304–336 (33) 353–402 (50) 411–452 (42) 456–475 (20) 478–503 (26) 513–528 (16) 541–552 (12) |
49.58 | 41.32 |
| APOE |
R T |
17 44 |
24 111 |
278–283 (6) | 0 | 1.89 | |
| APP |
R T |
53 65 |
139 240 |
200–289 (90) 354–439 (86) 611–649 (39) |
181–190 (10) 205–243 (39) 251–275 (25) 283–291 (9) 301–322 (22) 336–346 (11) 391–396 (6) 426–437 (12) 471–479 (9) 491–497 (7) 545–550 (6) 606–626 (21) |
27.9 | 22.99 |
| PICALM |
R T |
51 70 |
160 269 |
334–377 (44) 389–404 (16) 533–552 (20) 562–566 (5) 577–592 (16) |
308–314 (7) 335–344 (10) 366–378 (13) 392–397 (6) 506–514 (9) 540–553 (14) |
15.49 | 9.05 |
| CD33 |
R T |
21 52 |
35 143 |
324–364 (41) | 334–348 (15) | 11.26 | 4.12 |
| PSEN1 |
R T |
23 67 |
29 192 |
1–31 (31) 38–53 (16) 327–349 (22) |
1–26 (26) | 14.78 | 5.57 |
| ADAM17 |
R T |
61 72 |
178 298 |
735–815 (81) |
225–236 (12) 433–439 (7) 745–761 (17) 774–815 (42) |
9.83 | 9.47 |
| Clusterin |
R T |
0 57 |
0 201 |
0 | 0 | ||
| PSEN2 |
R T |
26 62 |
42 202 |
1–28 (28) 39–49 (11) |
1–25 (25) | 8.71 | 5.58 |
| PLG |
R T |
50 76 |
157 296 |
463–470 (8) | 0 | 0.99 | |
| ADAM9 |
R T |
58 80 |
120 283 |
759–786 (28) 798–819 (22) |
767–781 (15) 799–819 (21) |
6.11 | 4.40 |
| ADAM10 |
R T |
54 81 |
133 277 |
719–748 (30) |
313–318 (6) 415–420 (6) 741–748 (8) |
3.83 | 2.67 |
| ABCA7 |
R T |
78 95 |
469 839 |
1176–1182 (7) 1326–1337 (12) 1353–1359 (7) 2117–2147 (30) |
124–131 (8) 156–164 (9) 195–201 (7) 1142–1147 (6) 1166–1173 (8) 1327–1333 (7) 1352–1360 (9) 1386–1397 (12) 2067–2072 (6) 2092–2102 (11) 2133–2146 (14) |
2.61 | 4.52 |
| LRP1 |
R T |
75 112 |
277 1395 |
4508–4515 (8) 4525–4544 (20) |
3965–3970 (6) 4505–4513 (9) |
0.62 | 0.33 |
| TREM2 |
R T |
13 44 |
15 86 |
0 | 0 | ||
| ECE2 |
R T |
73 81 |
246 300 |
1–53 (53) |
1–22 (22) 718–724 (7) |
6.54 | 3.58 |
| SORL1 |
R T |
42 100 |
88 808 |
0 | 0 | ||
| A2M |
R T |
65 91 |
280 653 |
1148–1155 (8) 1209–1218 (10) |
0 | 1.22 | |
| BACE1 |
R T |
44 63 |
80 198 |
57–67 (11) | 2.20 | 0 | |
| Neprilysin |
R T |
54 77 |
104 252 |
0 | 0 | ||
| Nicastrin |
R T |
56 67 |
236 312 |
0 | 0 | ||
| PEN2 |
R T |
15 29 |
18 41 |
0 | 0 | ||
| IDE |
R T |
66 80 |
184 335 |
70–77 (8) 108–115 (8) |
0 | 1.57 | |
| PLAU |
R T |
41 64 |
74 146 |
No result found | 0 | No result | |
| PLAT |
R T |
40 61 |
85 194 |
0 | 0 | ||
| APH1A |
R T |
16 43 |
21 122 |
0 | 0 | ||
| APH1B |
R T |
9 41 |
11 110 |
0 | 0 | ||
R retained, T total SLiMs
The direct interactions of misfolded proteins are less studied, yet are a very important field, since such interactions may be involved in the development of different pathological conditions. The cross-seeding between misfolded proteins, such as Aβ/tau [102], Aβ/α-synuclein [103], tau/α-synuclein [104] was reported in neurodegenerative diseases, such as AD and PD. Furthermore, studies in transgenic mouse model by Clinton et al. have shown that Aβ, tau, and α-synuclein interact in vivo, promote each other’s aggregation, and contribute to the cognitive failure [105]. The prion protein (PrP) affects the biological activity of Aβ42 via direct interactions [106]. The monomers of Aβ and some of its interacting proteins are disordered that provides template for interaction with each other [107]. Here in our analysis, Fig. 3 shows the interaction network of all 27 proteins involved in the amyloid cascade signaling. This interaction network was obtained using STRING resource, which is a consortium of information from experimental annotated and predicted protein–protein interactions (PPIs). Figure 3a shows that these proteins are highly interconnected, generating a rather dense PPI network. In fact, 27 nodes (proteins) in this PPI network have 171 edges (connections), which is significantly higher (p value < 10–16) than the expected number of edges (9) for a random set of proteins of similar size, drawn from the genome. The average node degree (i.e., is the number of connections it node has to other nodes) of this network is 12.7, and it has an average local clustering coefficient (which defines how close its neighbors are to being a complete clique; local clustering coefficient is equal to 1, if every neighbor connected to a given node Ni is also connected to every other node within the neighborhood, and it is equal to 0 if no node that is connected to a given node Ni connects to any other node that is connected to Ni) of 0.744. In addition to Aβ metabolic process, Aβ formation, regulation of Aβ formation, amyloid precursor protein catabolic process, regulation of Aβ clearance, positive regulation of amyloid fibril formation, and Aβ clearance, negative regulation of Aβ formation, positive regulation of Aβ formation the major GO (gene ontology) biological processes assigned to this network include membrane protein proteolysis, membrane protein ectodomain proteolysis, Notch receptor processing, membrane protein intracellular domain proteolysis, and regulation of tau protein kinase activity, to name a few. Among the GO molecular functions associated with this pathway are Aβ binding, endopeptidase activity, lipoprotein particle receptor binding, metalloendopeptidase activity, hydrolase activity, apolipoprotein binding, and tau protein binding.
Fig. 3.

Analysis of interactivity of human proteins involved in the amyloid cascade pathway. a Interactivity of 27 proteins mentioned in Table 1 was analyzed using the STRING resource. The medium confidence score of 0.4 was used to ensure inclusion of all these proteins in a single PPI network. b STRING-generated PPI network centered at 27 human proteins involved in the amyloid cascade pathway. The highest confidence score of 0.9 was used here. The network includes 27 query proteins (red circles) and 500 first shell interactors (white circles)
Next, we extended PPI network to include a first shell of interactors of these 27 proteins involved in the amyloid cascade signaling. The corresponding STRING-generated a PPI network is shown in Fig. 3b. To generate the most reliable network, we used the highest confidence of 0.9 as a minimum required interaction score. Note that due to the computational limitations, STRING allows analysis of the first 500 interactors only. Therefore, the resulting network shown in Fig. 3b includes 527 proteins connected by 13,313 interactions instead of the expected 4634 interactions for a randomly selected set of comparable size. The average node degree of this PPI network centered at 27 proteins involved in the amyloid cascade signaling is 50.5, and it has an average local clustering coefficient of 0.732. This analysis showed that the proteins from the amyloid cascade signaling pathway are not only highly connected themselves, but are also engaged in a very dense and highly developed network between proteins engaged in a wide spectrum of biological processes. In fact, top 10 GO-annotated biological processes ascribed to this network include vesicle-mediated transport, regulation of transport, localization, regulation of localization, establishment of localization, response to stimulus, regulation of response to stimulus, regulation of multicellular organismal process, cell-surface receptor signaling pathway, and endocytosis. Among top GO-annotated molecular functions of this network are receptor regulator activity, receptor ligand activity, molecular function regulator, signaling receptor activity, transmembrane signaling receptor activity, peptidase inhibitor activity, endopeptidase inhibitor activity, hormone activity, growth factor activity, peptidase regulator activity, protein binding, and binding of different partners, such as signaling receptor, G protein-coupled receptor, protein-containing complexes, cell adhesion molecule, peptide, lipoprotein particle receptor, enzyme, amide, clathrin, identical protein, integrin, amyloid-beta, lipid binding, and calcium ion, to name a few.
Intrinsic disorder propensity of APP
Amyloid precursor protein (APP; UniProt ID: P05067) is a single-pass transmembrane protein with a large N-terminal extracellular domain and a small C-terminal intracellular domain. Eleven isoforms of the protein ranging in length from 305 to 770 residues exist due to alternative splicing. Out of these, three most common isoforms include 695-amino-acid-long isoform found in CNS, and ubiquitously expressed 751- and 770-amino-acid-long isoforms [4]. APP belongs to the APP family of proteins, which also include APP-like protein 1 and 2 (APLP1, APLP2) and APP-like (APPL in Drosophila) [108]. The intracellular C-terminal region of APP acts as a transcriptional regulator. It houses the YENPTY domain, which is 100% conserved and is responsible for the intracellular sorting of APP. It regulates clathrin-coated pit internalization of the protein. Mutations in this domain alter endocytosis of the protein, thus diminishing production of Aβ peptide. Crystal structures of truncated APP protein are available, some of them with high resolution are discussed here. A 1.40 Å resolution structure of residues 18–190 (PDB ID: 4PWQ) which houses the E1 domain, 0.85 Å resolution structure of residues 133–189 (PDB ID: 2FMA), 2.31 Å resolution structure of residues 370–710 (PDB ID: 5BUO) which contain the E2 domain and 2 Å resolution structure of residues 739–770 (PDB ID: 3DXE) are available. Thus, 65.07% of the protein structure is known.
Although the exact physiological function of APP is not known, based on the single, double, and triple knockout studies in mice performed for determining its function, it has been concluded that APP and other members of its family play an essential role in neuritic outgrowth and synaptic pruning [4]. In single knockouts, subtle phenotypic changes were observed, which indicated that members of the APP family compensate for the loss of one another, while in double and triple knockouts, the mice died soon after birth. APP also plays a role in neuronal migration in embryogenesis; however, its role in mature CNS is still not understood [108].
Post-translation, APP sorting occurs in the endoplasmic reticulum (ER) and Golgi complex. It is then delivered to the axon and transported to the synaptic terminals by fast axonal transport. APP from the trans-Golgi network (TGN) can be either transported to cell surface or can be packed in endosomal compartments. Transport along both these paths is mediated by clathrin-coated vesicles. APP must pass through the cell surface for its processing. But at a time, a very little amount of APP is present on the cell surface which helps us conclude that the process is very rapid. APP driven to the cell surface first undergoes proteolysis by α-secretase and then by γ-secretase. However, this process does not generate Aβ peptide. Some of the APP from cell surface gets repacked into clathrin-coated endosomal compartments containing BACE1 and γ-secretase. Following cleavage of APP by these enzymes, Aβ peptide is produced. Why APP undergoes either one of these two processes is still unclear. The Aβ peptide produced is then dumped in extracellular space or degraded in lysosomes [4]. Abnormally high Aβ accumulation in the brain causes formation of neuritic plaques, which is one of the hallmarks of Alzheimer’s disease (AD). The reasons for Aβ accumulation could be either lowered clearance of the peptide through CSF or an alteration in the APP processing [4].
Best characterized APP protein-binding partners are X11 (adapter protein X11-α orAβ A4 precursor protein-binding family A member 1) and Fe65 (Aβ A4 precursor protein-binding family B member 1). These proteins are highly expressed in the brain and interact with all members of APP family. Studies in cell culture have provided evidence that these proteins couple APP to the neuronal sorting protein-related receptor SorLA/LR11 in the trans-Golgi network, thereby preventing it from interacting with BACE1. When mouse X11 or Fe65 knockouts were crossed with the APP overexpressing mice, progeny showed a significant increase in accumulation of Aβ peptide. When X11 or Fe65 were overexpressed, diminished levels of Aβ accumulation in the brain were observed [4].
Figure 4d represents a per-residue disorder profile generated for human APP. Figure 4a–c show the PDB structures of the residues present in the corresponding white regions of the graph below them. APP is characterized by a mean PPID score of 38.7% (Table 2), which is much higher than the PPID of 12% predicted by MobiDB. Note that, as a rule, the PPID scores generated for proteins by MobiDB are noticeably lower than the corresponding values derived using other computational tools. The circular dichroism (CD) spectroscopy analysis by Botelho et al. on extracellular 595-residue-long domain of APP (sAPPα695) showed α-helical secondary structure [109]. Furthermore, the C-terminal 105-residue-long region of APP was characterized as α-helical [110], which has been shown ordered in our analysis. Studies also reported that the transmembrane domain of APP (residues 700–723) has α-helical structure [111], which is consistent with the outcomes of our analyses, where the transmembrane region has a predicted disorder score below 0.5, which corresponds to the mostly ordered structure of a protein. Additionally, previous results based on CD studies of peptides derived from APP have also been found consistent with the results of our analysis. The 316–346 peptide of APP showed α-helix secondary structure, which is ordered in our analysis. On the other hand, APP peptides 347–381, 382–415, and 416–447 were characterized by CD spectra typical for the disordered polypeptides [112]. In our analysis, these three regions also showed high disordered propensity.
Fig. 4.

Evaluation of intrinsic disorder propensity of APP a 1.40 Å resolution structure of the E1 domain of APP (PDB ID: 4PWQ). b 2.31 Å resolution structure of APP residues 370–710 (PDB ID: 5BUO) c 2 Å resolution structure of APP residues 739–770 (PDB ID: 3DXE). The proteins interacting with APP are shown in faded salmon color. The IDP regions (violet color), MoRF regions (green color) and MoRF regions in IDPRs (red color) are mapped on the protein structures (olive color). d Disorder profile generated by PONDR® VSL2 (grey line), PONDR® VL3 (red line), PONDR® VLXT (blue line), PONDR® FIT (magenta line), IUPRED long (purple line) and IUPRED short (orange line). The cyan line represents the mean disorder propensity calculated using the disorder scores from all six predictors. The light magenta shadow region represents the PONDR® FIT error distribution and the light cyan region represents the mean error distribution. The area of the graph marked in grey color either represents the residues which are missing in the PDB structures or the residues for which no PDB structure is available. e Evaluation of MoRF sites and PTMs in APP by D2P2. The D2P2 profile presents information related to the protein propensity for intrinsic disorder, while also commenting on their disorder-related functionality. At the top of plot, there is a side-by-side comparison of seven separate disorder predictors (Espritz-D, Espritz-X, Espritz-N, IUPred-L, IUPred-S, PV2, PrDOS, VSL2b, and VLXT), bars indicate positions of intrinsically disordered regions predicted by each tool. The middle of the plot comments on predicted domains associated with protein function. At the bottom of the plot, there is information related to coherence between predictors, labeled as ‘Predicted Disorder Agreement’ presenting the level of agreement between all of the disorder predictors, which is shown as color intensity in an aligned gradient bar below the stack of predictions. The green segments represent disorder that is not found within a predicted SCOP domain. The blue segments are where the disorder predictions intersect the SCOP domain prediction. Below the disorder agreement line disorder-based binding region (i.e., disordered regions that fold upon interaction with binding partners and known as molecular recognition features, MoRFs) predicted by ANCHOR are displayed as yellow blocks with zigzag infill. Finally, the bottom of the plot shows positions of various PTMs within the query proteins. These are shown by differently colored circles
The protein is predicted to harbor three large MoRF regions by ANCHOR, which constitute 27.9% of the protein and 12 small MoRF regions by D2P2 which make up almost 23% of the protein. Despite the huge variation in the number of predicted regions by both the software, not much variation is seen in the predicted percentage of MoRF regions present in the protein. According to the prediction from D2P2, which is represented in Fig. 4e, APP has 15 glycosylation sites out of which 12 are present in IDPR, 11 phosphorylation sites out of which 2 are located in IDPR, 2 ubiquitylation sites out of which 1 is in the IDPR,1 SUMOylation site and 2 acetylation sites. Importantly, alternatively spliced isoforms of APP are often generated by altering its IDPRs. For example, the shortest isoform APP305 lacks C-terminal domain (residues 306–770), which is predicted to contain a high level of intrinsic disorder (its PPID exceeds 50%). Residues 637–654 (PPID = 100%) are missing in isoforms L-APP677, L-APP696, and L-APP733. Residues 290–364 (PPID = 34%) are not present in the isoforms APP639, APP695, and L-APP677. Isoforms L-APP696 and APP714 are missing residues 290–345 (PPID = 14%), residues 346–364 (PPID = 100%) are not present in isoforms APP751, L-APP733, and isoform-11, whereas residues 19–74 (PPID = 20%) are missing in the APP639 isoform. As a result, the alternative splicing-generated proteoforms of APP are characterized by noticeable variability of their intrinsic disorder predispositions, with their PONDR® VSL2-based PPIDs (PPIDPONDR®VSL2 scores) being 47.53%, 41.64%, 46.68%, 48.92%, 48.13%, 50.28%, 43.79%, 45.94%, 45.48%, 51.33%, and 46.38% for the canonical (APP770), APP305, L-APP677, APP695, L-APP695, APP714, L-APP733, APP751, L-APP752, APP639, and isoform-11, respectively.
ELM server predicted that 53 SLiMs and their 139 instances in APP are retained after filtering (Table 3). 14 motif instances are embedded in D2P2-predicted MoRF regions, 20 motif instances are overlapped and 3 motif instances are in their close proximity (Supplementary Table 1). The annotations of these motifs from ELM server are grouped into six categories as described in methods (Supplementary Table 1).
In line with the presence multiple disorder-based binding sites, STRING-based analysis of the APP interactivity using the highest confidence of 0.9 as a minimum required interaction score generated a protein–protein interaction (PPI) network that includes at least 501 nodes connected by 34,459 edges, which significantly exceeds the number of 7508 edges expected for a random set of proteins of similar size, drawn from the genome (see Supplementary Figure S1A). This network is characterized by the average node degree of 138, an average clustering coefficient of 0.9, and PPI enrichment p value < 10–16. Most common molecular functions in this APP-centered PPI network are G protein-coupled receptor activity, transmembrane signaling receptor activity, signaling receptor activity, molecular transducer activity, and G protein-coupled peptide receptor activity. Top five GO-annotated biological processes associated with this network include G protein-coupled receptor signaling pathway, signal transduction, signaling, cell communication, and response to stimulus.
Intrinsic disorder propensity of protein ADAM17
Alpha disintegrin and metalloproteinase (ADAM, UniProt ID: P78536) enzymes are cell-surface proteins which belong to the adamalysin family. Due to their adhesive and proteolytic properties, they play a crucial role in cell adhesion and proteolytic cleavage of cell-surface molecules. This makes them important for mediating cell signaling events which decide the fate, proliferation and growth of cells. As ADAMs contribute greatly to physiological and pathophysiological processes, they are regarded as potential therapeutic targets in various diseases. The human genome consists of 41 ADAMs, out of which 21 have been described in literature and 13 are proteolytically active. ADAM 17 is the most well-studied ADAM enzyme [113].
ADAM17 was discovered in 1997 as an enzyme, which acts on membrane-bound tumor necrotic factor-alpha (TNF-α) precursor and converts it to its soluble form. It is an 824-amino-acid-long protein and is made up of a signal sequence (residues 1–17), prodomain (residues 18–214), metalloenzyme or catalytic domain (residues 215–473), disintegrin domain (residues 474–572), cysteine-rich domain (residues 603–671), transmembrane domain (residues 672–694) and a cytoplasmic tail (residues 695–824). Its gene is located on chromosome 2p25 and is expressed mainly in brain, heart, kidney and skeletal muscle. Varied expression patterns are observed at the embryonic level and in an adult. ADAM 17 shows very little sequence similarity to any other ADAM enzyme. According to BLAST, ADAM 17 is closest to ADAM10, and they have only 30% sequence homology [113]. A 1.80 Å resolution structure of the catalytic domain of ADAM17 (PDB ID: 3EWJ) has been obtained by XRD [114] and the residues 215–217, 357–360 and 476–485 are missing in the structure, thus only 30.8% of the protein structure is known.
ADAM17 functions as a proteolytic enzyme which cleaves the ectodomains of various transmembrane proteins like APP, EGFR, and TNFα. ADAM17 and ADAM10 act as the α-secretase and cleave APP to release the soluble non-amyloidogenic fragment APPsα. The γ-secretase further acts as the released fragment and a soluble extracellular fragment is formed which does not contribute to amyloid plaques. Lack of α-secretase activity allows β-secretase to act on APP which results in formation of an amyloidogenic β peptide fragment which after action of γ-secretase leads to formation of Aβ peptide which aggregates to form amyloid plaques. Thus, a better understanding of ADAM17 and ADAM10 is required to use them as a therapeutic target in AD, as they can reduce Aβ peptide formation [115].
ADAM17 also acts as a sheddase in leucocyte activation and cleaves molecules like integrins and selectins. Patients affected with chronic inflammations like arthritis have elevated levels of ADAM17. As the enzyme acts as a sheddase and helps release growth factors necessary for tumor growth and progression, it has been implicated in carcinogeneses like ovarian cancer and breast cancer. ADAM17 is vital for heart development but also contributes to various heart conditions. Elevated levels of ADAM17 are observed in patients with myocarditis, advanced congestive heart failure, and myocardial infarction. Experimental evidence suggests that ADAM17-based TNFα inhibition could be used to treat non-obese insulin-resistant diabetes [116]. Elevated levels of ADAM17 have been correlated with increased EGFR production in several kidney diseases like polycystic kidney disease [117].
Figure 5b shows the intrinsic disorder profile of ADAM17. Figure 5a represents the PDB structure of the residues presented in the corresponding white region of the graph below. ADAM17 shows a mean PPID score of 28.52% (Table 2), which is much higher than 11% predicted by MobiDB. The protein is predicted by ANCHOR to harbor a long MoRF region, which constitutes 9.8% of the protein and 4 short MoRFs identified by D2P2, which make up 9.5% of the protein. According to the prediction from D2P2, which is represented in Fig. 5c, ADAM17 has 12 phosphorylation sites out of which 6 are located in IDPR and 6 ubiquitylation sites out of which 3 are in the IDPR region. A canonical form of ADAM17 has PPIDPONDR®VSL2 scores of 52.79%, whereas this value decreases to 45.39% in alternatively spliced isoform II that misses a large portion of a highly disordered C-terminal domain (residues 695–824, PPID = 92.3%). The CD spectroscopy studies by Dusterhoft et al. of the conserved ADAM17 dynamic interaction sequence (CANDIS) (residues 643–666) showed spectra characteristic for the α-helical polypeptide [118], which is consistent with our analysis, where this region was predicted to have an ordered structure.
Fig. 5.

Evaluation of intrinsic disorder propensity of ADAM17 a 1.80 Å resolution structure of the catalytic domain of ADAM17 (PDB ID: 3EWJ). The IDP regions (violet color) are mapped on the protein structure (olive color). b Disorder profile generated by PONDR® VSL2 (grey line), PONDR® VL3 (red line), PONDR® VLXT (blue line), PONDR® FIT (magenta line), IUPRED long (purple line) and IUPRED short (orange line). The cyan line represents the mean disorder propensity calculated using the disorder scores from all six predictors. The light magenta shadow region represents the PONDR® FIT error distribution and the light cyan region represents the mean error distribution. The grey shaded area of the graph represents residues which are either missing in the PDB structure or the residues for which no structure is available. c Evaluation of MoRF sites and PTMs in ADAM17 by D2P2
ELM server predicts that ADAM17 has retained 61 SLiMs and 178 instances after filtering (Table 3). Thirteen of these instances are embedded in, ten instances overlap with and one instance is in close proximity with the MoRF residues predicted by D2P2 (Supplementary Table 1).
STRING-based analysis of the ADAM17 interactvity using the highest confidence of 0.9 as a minimum required interaction score generated a protein–protein interaction (PPI) network that includes 39 nodes connected by 217 edges, which significantly exceeds the number of 76 edges expected for a set of proteins of similar size randomly drawn from the genome (see Supplementary Figure S1B). This network is characterized by the average node degree of 11.1, an average clustering coefficient of 0.857, and PPI enrichment p value < 10–16. Most common molecular functions in this network are signaling receptor binding, phosphatidylinositol-4,5-bisphosphate 3-kinase activity, growth factor activity, enzyme binding, and receptor ligand activity, whereas its most common biological processes include cell-surface receptor signaling pathway, positive regulation of signal transduction, regulation of response to stimulus, ERBB2 signaling pathway, and enzyme-linked receptor protein signaling pathway.
Intrinsic disorder propensity of protein BACE1
β-Secretase 1 (BACE1, UniProt ID: P56817) is a transmembrane aspartic protease, which is also known as Asp-2 and memapsin-2. It is co-localized within the trans-Golgi network and endosomal system along with amyloid precursor protein (APP). When coupled with γ-secretase, it cleaves APP, and Aβ peptide is produced. BACE1 is recycled by re-internalizing from cell surface into endosomal compartments which later bring it back to the cell surface along with APP. A 1.46 Å resolution structure of the full-length BACE1 was obtained by X-ray crystallography [119] and is available in PDB (PDB ID: 6EJ2). The residues 1–57, 219–228, 376–377 and 446–501, are missing in the structure, indicating the structural coverage of 75.3% for this protein.
With time, Aβ accumulation gives rise to neuritic plaques which is one of the hallmarks of Alzheimer’s disease (AD). Aβ accumulation also initiates hyperphosphorylation of tau protein which later forms neurofibrillary tangles, another hallmark of AD. As BACE1 has a crucial role to play in the generation of toxic Aβ peptide, it is considered a major target for AD therapy. Many research groups are working on designing an inhibitor for BACE1 to block the production of Aβ peptide [120, 121]. The active site of the enzyme is covered by a flexible flap region made up of antiparallel β-hairpin structure.
BACE1 is indispensable for Aβ generation. Elevated levels of Aβ peptide in AD patients disturb the reactive oxygen species (ROS) balance and thus, can interfere with the pre- and post-synaptic functions (Ca2+ channels, Acetylcholine receptors). Also, BACE1-mediated cleavage of APP can give rise to a 99 amino-acid-long C-terminal fragment (βAPPc), which is known to impair synaptic function. Thus, BACE1 represents a very good therapeutic target for AD [120].
In mouse AD models, when the BACE1 gene is deleted, hippocampal-dependent memory deficits that occur due to Aβ accumulation can be reversed [122]. But, BACE1 also plays a role in normal synaptic transmission and plasticity in certain regions of the hippocampus. BACE1 null mice show altered hippocampal pre-synaptic plasticity, changes in paired-pulse facilitation (PPF) ratio and post-synaptic modifications in AMPA receptors. Normal physiological concentration of Aβ peptide is essential to facilitate synaptic plasticity (in pM range). BACE1 deficiency causes reduction in levels of Aβ, thus causing synaptic deficits [120].
The protein has a mean PPID score of 10.2%, while MobiDB predicts 4% disorder (Table 2). It has been reported that the secondary structure analysis of BACE1 using CD spectra has shown a noticeable α-helical structure [123], which is also in agreement with our analysis, which characterized BACE1 as a mostly ordered protein. According to ANCHOR, BACE1 has one MoRF region which makes up 2.2% of the protein, while D2P2 identifies no MoRF region (Table 3). Figure 6b represents the disorder profile of BACE1. Figure 6a shows the crystal structure of the protein corresponding to residues presented in the white regions of the graph below. Although according to D2P2, BACE1 has no PTMs, UniProt entry of this protein shows that there are multiple acetylation sites (residues 126, 275, 279, 285, 299, 300, and 377), four glycosylation sites (residues 153, 172, 223, and 354), a phosphorylation site (residue 498), and four palmitoylation sites (residues 474, 478, 482, and 485). Although BACE1 is a moderately disordered protein, it has six isoforms generated by alternative splicing. The PPIDPONDR®VSL2 scores for these isoforms are 13.37% (canonical isoform or isoform A, or BACE-1A, or BAC-501), 14.08% [isoform B, or BACE-1B, or BACE-I-476 with missing residues 190–214 (PPID = 0%)], 16.63% [isoform C, or BACE-1C, or BACE-I-457 with missing residues 146–189 (PPID = 0%)], 15.51% [isoform D, or BACE-1D, or BACE-I-432 with missing residues 146–189 (PPID = 0%) and 190–214 (PPID = 0%)], 3.99% [isoform 5 with missing residues 21–120 (PPID = 49%) and residues 1–20 changed from MAQALPWLLLWMGAGVLPAH (PPID = 10%) to MVPFIYLQAHFTLCS GWSST (PPID = 0%)], and 4.26% [isoform 6 with missing residues 21–120 (PPID = 49%) and 190–214 (PPID = 0%) and with residues 1–20 changed from MAQALPWLLLWMGAGVLPAH (PPID = 10%) to MVPFIYLQAHFTLCSGWSST (PPID = 0%)].
Fig. 6.

Evaluation of intrinsic disorder propensity of BACE1 a 1.46 Å resolution structure of full-length BACE1 (PDB ID: 6EJ2). The IDP regions (violet color) are mapped on the protein structure (olive color). b Disorder profile generated by PONDR® VSL2 (grey line), PONDR® VL3 (red line), PONDR® VLXT (blue line), PONDR® FIT (magenta line), IUPRED long (purple line) and IUPRED short (orange line). The cyan line denotes the mean disorder propensity calculated using the disorder scores from all six predictors. The light magenta shadow region represents the PONDR® FIT error distribution and the light cyan region represents the mean error distribution. The grey shaded area of the graph represents residues which are either missing in the PDB structure or the residues for which no structure is available. c Evaluation of MoRF sites and PTMs in BACE1 by D2P2
ELM server predicts that after globular domain filtering, structural filtering and context filtering, BACE1 has retained 44 SLiMs and 80 instances (Table 3). But as the protein has no predicted MoRF residues by D2P2, none of the retained motifs are associated with MoRFs. STRING-based analysis of the BACE1 interactvity using high confidence of 0.7 generated a PPI network containing 31 nodes with 94 edges, which significantly exceeds the number of 36 edges expected for a set of proteins of similar size randomly drawn from the genome (see Supplementary Figure S1C). This network is characterized by the average node degree of 6.06, an average clustering coefficient of 0.837, and PPI enrichment p value < 1.33 × 10–15. Most common molecular functions in this network are endopeptidase activity, ADP-ribosylation factor binding, peptide binding, amyloid-beta binding, and aspartic-type endopeptidase activity, and top five biological processes associated with the BACE1-centered PPI network include amyloid-beta metabolic process, membrane protein ectodomain proteolysis, Notch receptor processing, membrane protein intracellular domain proteolysis, and Notch receptor processing, ligand-dependent.
Intrinsic disorder propensity of the proteins of the γ-secretase complex
γ-Secretase is an acidic proteolytic enzyme which catalyzes the final step of APP processing giving rise to Aβ peptide and its C-terminal APP intracellular domain (AICD) [124, 125]. γ-Secretase is a multiprotein complex made up of five subunits: presinilin-1(PSEN1), presenilin-2 (PSEN2), APH1A (presenilin-stabilization factor), nicastrin, and PEN2 [4]. A 2.60 Å resolution structure of all subunits of γ-secretase except PSEN2 (Fig. 7) cross-linked with APP has been determined by Cryo-electron microscopy [126] and is available on PDB (PDB ID:6IYC). γ-Secretase has been an appealing target to design a cure for AD, as it is of utmost importance in the final step of Aβ production from APP. However, blocking this enzyme can have other side effects, such as impairment of NOTCH signaling.
Fig. 7.
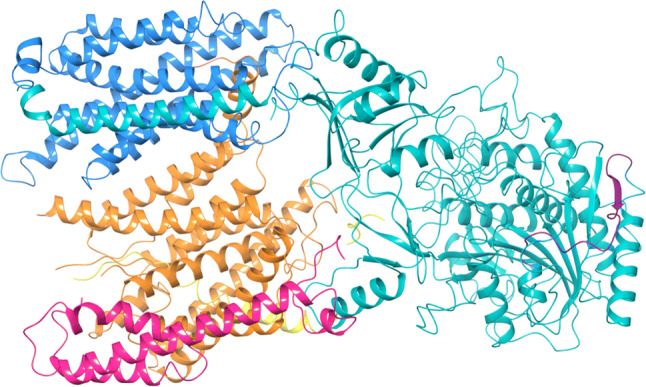
PDB structure of γ-secretase (PDB ID: 6IYC): structure of all subunits of γ-secretase complex except PSEN2 cross-linked with APP (yellow color) was obtained by cryo-electron microscopy (2.60 Å resolution). The structure includes subunits nicastrin (teal color), PSEN1 (orange color), APH1A (azure color) and PEN2 (pink color). The IDP regions (violet color) have been mapped on the structure
Presenilin 1 (PSEN1)
PSEN1 and PSEN2 form the catalytic domain of γ-secretase [124]. PSEN1 is a 467-amino-acid-long protein and its mutations are the most common cause of familial Alzheimer’s disease (FAD). They cause early onset of FAD compared to APP mutations [127]. Two hypotheses have been proposed to show the link between FAD and PSEN1 mutations.
Amyloid hypothesis
PSEN1 mutations increase the pathogenesis of AD by increasing the production of Aβ42 peptide in plasma. Similar observations have been recorded in cell lines as well as transfected mice. PSEN1 mutations enhance APP processing leading to excessive production of Aβ peptide, thus, FAD pathogenesis is triggered. To fix the inconsistencies with this model, it was further revised. Now, the amyloid hypothesis states that PSEN1 with pathogenic mutations increase the Aβ42/Aβ40 ratio by relative increase in Aβ42 production [127].
Presenilin1 (PSEN) hypothesis
Earlier findings suggest that presenilin plays an essential role in memory, learning and neuronal survival during aging in cerebral cortex of adult mice. Knock-in studies show the role of PSEN1 in synaptic plasticity [128]. PSEN hypothesis suggests that loss of these essential functions due to PSEN1 mutations triggers neurodegeneration and dementia in FAD [127].
A recent study by Sun et. al. published in 2016 challenges the amyloid hypothesis and supports the PSEN hypothesis by suggesting that PSEN1 mutations suppress γ-secretase activity and Aβ production [128]. They collected 138 known PSEN1 mutations and used them to reconstitute γ-secretase in vitro with APH1A. Contrary to amyloid hypothesis, 75% mutations led to decreased production of Aβ40 and Aβ42 compared to WT of γ-secretase. The vast majority of these mutations show increased Aβ42/Aβ40 ratio. This was because levels of Aβ40 are decreased more than Aβ42 levels. Thus, they increase the pathogenesis of FAD [128].
The role of PSEN1 in mitophagy processes has been studied in cell lines and in transgenic mice. PSEN1 affects mRNA and protein levels of PINK1. Using inhibitors of γ-secretase or PSEN1 mutations can completely abolish PINK1 activity. It has been shown that APP mediates PSEN1-induced γ-secretase-mediated effect on PINK1. PARK2–PINK1 interplay drives homeostasis and regulates mitochondrial dynamics [124].
The available structure of PSEN1 in γ-secretase complex structure has residues 1–72 and residues 292–375 missing. Thus, the deduced structure of PSEN1 covers 66.8% of its sequence. Figure 8a is a disorder propensity graph of PSEN1. Mean PPID score of PSEN1 (UniProt ID: P49768) is 29.5% (Table 2) and that predicted by MobiDB is only 18%. According to ANCHOR, the protein has three MoRF regions which constitute 14.8% of the protein and D2P2 shows a single MoRF region of 5.6% (Table 3). According to the CD spectroscopic analysis by Yang et al., PSEN1 showed α-helical spectra [129], and our analysis predicted PSEN1 as moderately disordered protein. D2P2 also shows that the protein has 16 phosphorylation sites out of which 13 lie in IDPR and 2 ubiquitylation sites out of which 1 lies in IDPR (Fig. 8b). In addition to the canonical form (I-467), PSEN1 has six isoforms generated by alternative splicing (isoforms I-463, I-374, minilin, and isoforms 5, 6, and 7) with the corresponding PPIDPONDR®VSL2 scores of 33.40%, 32.83%, 39.04%, 42.39%, 39.68%, 23.46%, and 35.94%.
Fig. 8.

Evaluation of intrinsic disorder propensity of PSEN1: a Disorder profile generated by PONDR® VSL2 (grey line), PONDR® VL3 (red line), PONDR® VLXT (blue line), PONDR® FIT (magenta line), IUPRED long (purple line) and IUPRED short (orange line). The cyan line represents the mean disorder propensity calculated using the disorder scores from all six predictors. The light magenta shadow region represents the PONDR® FIT error distribution and the light cyan region represents the mean error distribution. The area of the graph marked in grey color represents the residues which are missing in the PDB structure. b Evaluation of MoRF sites and PTMs in PSEN1 by D2P2
ELM server predicts that PSEN1 has 23 SLiMs and 29 instances which are retained after filtering (Table 3). Among these, MoRF residues predicted by D2P2 contain one motif which is in close proximity, two motifs that overlap and three motifs that are embedded in them (Supplementary Table 1).
STRING-based analysis of the PSEN1 interactvity using the highest confidence level of 0.9 generated a PPI network containing 95 nodes with 1746 edges (see Supplementary Figure S1D), which significantly exceeds the number of 170 edges expected for a set of proteins of similar size randomly drawn from the genome. This network is characterized by the average node degree of 36.8, an average clustering coefficient of 0.929, and PPI enrichment p value < 10–16. Most common molecular functions in this network are transmembrane receptor protein tyrosine kinase activity, ephrin receptor activity, protein tyrosine kinase activity, axon guidance receptor activity, and protein dimerization activity, whereas among top biological processes associated with the PSEN1-centered PPI network are neutrophil degranulation, myeloid leukocyte activation, cell activation involved in immune response, leukocyte activation involved in immune response, and regulated exocytosis.
Presenilin 2 (PSEN2)
In 1995, PSEN2 was initially reported as a gene responsible for AD. PSEN2 gene is localized on chromosome 1q42.13 and has 12 exons of which exon 1 and exon2 contain the untranslated regions. Two promoter elements P1 and P2 are located in exon 1 and exon 2. P1 is a housekeeping promoter and its activity depends on stimulating protein-1 (Sp-1) binding site. P2 is induced by Egr-1 which represses activity of P1 [130]. PSEN2 is a transmembrane protein made up of 448 amino acids and has molecular weight 55 kDa which spans the lipid bilayer 9 times. It shows 67% homology with PSEN1 and their hydrophobic regions are highly conserved. Two aspartyl residues D263 and D366 are found in the adjacent transmembrane regions of PSEN2 (TM-VI and TM-VII) that are involved in the formation of active site of γ-secretase complex.
Two isoforms of PSEN2 have been normally observed and the difference between them is that isoform-2 lacks part of the sequence from residue 263 to 296, which are present in isoform-1. This protein is localized in ER and Golgi network of neurons. An aberrant splice variant of PSEN2 lacks exon-5, which leads to the insertion of five residues ‘-SSMAG-’ into the variant chain and introduces a premature stop codon in exon-6. Aggregation of this variant is observed in hippocampus and cerebral cortex of people suffering from sporadic AD. This variant has been found in frontal lobe of patients with bipolar disorder and in patients with schizophrenia. In cell culture, under hypoxic conditions, this variant of PSEN2 was found to be upregulated. Furthermore, it influences tau conformation in neuroblastoma cells [130].
PSEN-2 mutations have been rarely reported and mainly detected in European and African populations. Patients with early onset of Alzheimer’s disease (EOAD), i.e., below 65 years of age show autosomal dominant forms of mutations in APP, PSEN1 or PSEN2. More than 200 mutations of PSEN1 and only 38 mutations for PSEN2 have been reported. PSEN2 mutations are a cause of familial EOAD, because they enhance Aβ production. PSEN2 mutations alter the intracellular Ca2+ signaling which further results in Aβ accumulation to form neuronal plaques and lead to neuronal cell death. PSEN2 mutations might increase γ-secretase activity. This was inferred from studies on cell lines and in mouse model that show increased production of Aβ42. Vito et al. [131] and Wolozin et al. [132] in separate studies published in 1996 have shown that PSEN2 is also involved in apoptosis. PSEN2 mutations can promote apoptosis and Bcl-2 can down-regulate pro-apoptotic activities induced by PSEN2.
No PDB structure of PSEN2 is available as of yet. The protein has a mean PPID of 25.8% while MobiDB predicts 16% disorder (Table 2). Similar to PSEN1, the CD spectra of PSEN2 indicated the presence of noticeable α-helical secondary structure [129], and also in our analysis, it was observed that PSEN2 has a few disordered regions. According to ANCHOR, PSEN2 (UniProt ID: P49810) has two MoRF regions which constitute 8.7% of the protein and D2P2 prediction shows a single MoRF region which makes up 5.6% of the protein (Table 3). The graph in Fig. 9a shows the disorder propensity of residues of the protein PSEN2. According to D2P2, PSEN2 has 11 phosphorylation sites, out of which 10 lie in the IDPRs, 1 acetylation site and 1 ubiquitylation site.
Fig. 9.

Evaluation of intrinsic disorder propensity of PSEN2: a Disorder profile generated by PONDR® VSL2 (grey line), PONDR® VL3 (red line), PONDR® VLXT (blue line), PONDR® FIT (magenta line), IUPRED long (purple line) and IUPRED short (orange line). The cyan line represents the mean disorder propensity calculated using the disorder scores from all six predictors. The light magenta shadow region represents the PONDR® FIT error distribution and the light cyan region represents the mean error distribution. b Evaluation of MoRF sites and PTMs in PSEN2 by D2P2
According to the ELM server predictions, PSEN2 has 26 SLiMs and 42 instances that are retained after filtering (Table 3). Out of these, ten SLiMs are embedded in, seven SLiMs overlap with and one SLiM is in close proximity with MoRF residues predicted by D2P2 (Supplementary Table 1).
STRING-based analysis of the PSEN2 interactivity using the highest confidence level of 0.9 generated a PPI network containing 26 nodes with 174 edges (see Supplementary Figure S1E), which significantly exceeds the number of 29 edges expected for a set of proteins of similar size randomly drawn from the genome. This network is characterized by the average node degree of 13.4, an average clustering coefficient of 0.86, and PPI enrichment p value < 10–16. Most common molecular functions in this network are ephrin receptor activity, axon guidance receptor activity, signaling receptor activity, Notch binding, and catalytic activity, acting on a protein, whereas the top five biological processes associated with the PSEN2-centered PPI include Notch signaling pathway, cell-surface receptor signaling pathway, membrane protein intracellular domain proteolysis, Notch receptor processing, and signal transduction.
Nicastrin
Nicastrin (UniProt ID: Q92542), which is a 709-amino-acid-long single-pass type I membrane protein, is the largest component of the γ-secretase complex. The extracellular domain of nicastrin is essential for substrate recognition of γ-secretase. Integral membrane proteins need to shed their ectodomain prior to cleavage by γ-secretase. The size of the shed ectodomain determines whether a protein becomes a substrate for γ-secretase or not. Bulky ectodomains, i.e., 200–300 residues long prevent cleavage of proteins by γ-secretase, while proteins with smaller ectodomain, i.e., 50 residues long are efficiently cleaved [133].
Nicastrin functions as a γ-secretase substrate receptor but has no role in cleavage [134]. After removal of N-terminal ectodomain by action of sheddases, newly generated short N-termini are recognized by nicastrin. Nicastrin then positions the bound substrates in the lipid bilayer to facilitate their cleavage by the catalytic presenilin subunit in the γ-secretase complex [133, 134].
Crystal structure of nicastrin from Dictyostelium purpureum with the resolution of 1.95 Å has been reported. Nicastrin protein is made up of 2 lobes: a large lobe, which contains the substrate recognition site, and a small lobe, which is connected to the large lobe by a hydrophobic pivot. The substrate-binding pocket is shielded by a lid from the small lobe, which blocks substrate entry [135].
The available structure of nicastrin within the γ-secretase complex has two regions of missing electron density, residues 1–33 and 701–709. Thus, structural information is available for 94.1% of nicastrin sequence. Figure 10a represents disorder propensity of nicastrin. Nicastrin shows a mean PPID score of 9.0% (Table 2) and 0% is predicted by MobiDB. In a previous study reported by Yu et al., nicastrin was shown to have far-UV CD spectra typical for the ordered proteins [136], which is consistent with our analysis showing that nicastrin is expected to be a highly ordered protein. According to the predictions by ELM server, nicastrin has 56 SLiMs and 236 instances retained after filtering (Table 3). Both ANCHOR and D2P2 predict that the protein has no MoRF regions. Hence, none of the predicted motifs are associated with MoRF regions. According to the prediction from D2P2 (Fig. 10b), nicastrin has nine phosphorylation sites out of which four are in IDPR, three acetylation sites, two ubiquitylation sites, and one monomethylation site. In addition to the canonical form, nicastrin has an alternatively spliced isoform, which is missing the mostly disordered N-terminal tail (residues 1–20, PPID = 75%).
Fig. 10.

Evaluation of intrinsic disorder propensity of nicastrin: a Disorder profile generated by PONDR® VSL2 (grey line), PONDR® VL3 (red line), PONDR® VLXT (blue line), PONDR® FIT (magenta line), IUPRED long (purple line) and IUPRED short (orange line). The cyan line represents the mean disorder propensity calculated using the disorder scores from all six predictors. The light magenta shadow region represents the PONDR® FIT error distribution and the light cyan region represents the mean error distribution. The area of the graph marked in grey color represents the residues which are missing in the PDB structure of γ-secretase b Evaluation of MoRF sites and PTMs in nicastrin by D2P2
STRING-based analysis of the nicastrin interactivity using the highest confidence level of 0.9 generated a PPI network containing 92 nodes with 1755 edges (see Supplementary Figure S1F), which significantly exceeds the number of 165 edges expected for a set of proteins of similar size randomly drawn from the genome. This network is characterized by the average node degree of 38.2, an average clustering coefficient of 0.924, and PPI enrichment p value < 10–16. Most common biological processes associated with the nicastrin-centered PPI include neutrophil degranulation, myeloid leukocyte activation, cell activation involved in immune response, leukocyte activation involved in immune response, and leukocyte activation, and top five molecular functions in this network are Notch binding, ephrin receptor activity, axon guidance receptor activity, aspartic-type endopeptidase activity, and calcium ion binding.
γ-Secretase subunits APH1
γ-Secretase subunit APH1A also known as presenilin-stabilization factor (UniProt ID: Q96BI3) is a 265-amino-acid-long subunit of the γ-secretase complex. Exact function of APH1A is not known but it is thought to be responsible for assembling of PSEN1 and PSEN2 to form the catalytic domain of the enzyme [137]. APH1B (UniProt ID: Q8WW43) is 257 amino acid long and is a presenilin-stabilization factor-like protein. In humans, two genes APH1A and APH1B encode for the protein APH1. In mice, Aph1B duplicates and gives rise to Aph1C. Gene knockout studies in mice show the following results.
Deletion of Aph1A (Aph1A −/−) is lethal to embryos and generates defective phenotype, such as vascular reorganization defects in yolk sac, neuronal tube malformations, and mild somitogenesis defects were observed. Defects in vasculogenesis were also observed in cases of PSEN 1−/−, PSEN2 −/− and Nct −/−. The other two phenotypes were observed for the first time in Aph1A deletion. This deletion leads to 70% decrease in γ-secretase activity [138].
Deletion of Aph1B or Aph1C or both (Aph1B −/− or Aph1C −/− or AphBC −/−): These mice survive into adulthood and no gross abnormality in fertility, survival or anatomy were observed. Although a mild but significant reduction in APP processing was observed in certain regions of adult brain that manifested in form of APP-CTF accumulation. Mice are even less affected than PSEN2 mutations [138]. (AphBC −/− in mice is equivalent to Aph1B −/− in humans).
APH1B and APH1C show 58% sequence homology in their amino acid sequences and 96.3% similarity at the nucleotide level. They are both clustered on chromosome 9. In a recent study, specific inactivation of APH1B subunit in murine AD model led to an improvement in AD-related phenotypic features without any Notch-related side effects [139].
In the available structure of APH1A in the γ-secretase complex, residues 1 and 245–265 are missing. Thus, the available structure covers 91.7% of the APH1A sequence. Figure 11a, b show an intrinsic disorder profile of APH1A and APH1B, respectively. APH1A shows a mean PPID score of 5.4%, while that of APH1B is 4.28% (Table 2). MobiDB predicts 0% disorder in both APH1A and APH1B. APH1A has retained 16 SLiMs and 21 instances, while APH1B has retained 9 SLiMs and 11 instances after filtering the SLiMs predicted by ELM server (Table 3). Both ANCHOR and D2P2 predict that APH1A and APH1B have no MoRF regions. Thus, none of these retained motifs are associated with MoRFs. According to the prediction from D2P2 (Fig. 11a1, b1), APH1A has three phosphorylation sites and two ubiquitylation sites and APH1B has two ubiquitylation sites. Three alternatively spliced isoforms of human APH1A are characterized by PPIDPONDR®VSL2 scores of 9.43%, 9.72%, and 5.13%. Here, isoform 2 is different from the canonical form by missing C-terminal residues 248–265 (PPID = 39%), whereas in the isoform 3, residues 39–120 changed to a shorter sequence RCSALPTTSCLI.
Fig. 11.

Evaluation of intrinsic disorder propensity of APH1: a, b Disorder profile generated by PONDR® VSL2 (grey line), PONDR® VL3 (red line), PONDR® VLXT(blue line), PONDR® FIT (magenta line), IUPRED long (purple line)and IUPRED short (orange line) of APH1A and APH1B, respectively. The cyan line represents the mean disorder propensity calculated using the disorder scores from all six predictors. The light magenta shadow region represents the PONDR® FIT error distribution and the light cyan region represents the mean error distribution. The area of the graph marked in grey color denotes the residues for which no PDB structure is available. a1, b1 Evaluation of MoRF sites and PTMs in APH1A and APH1B, respectively, by D2P2
STRING-based analysis of the APH1A interactivity using the highest confidence level of 0.9 generated a PPI network containing 37 nodes with 252 edges (see Supplementary Fig. 1G), which significantly exceeds the number of 45 edges expected for a set of proteins of similar size randomly drawn from the genome. This network is characterized by the average node degree of 13.6, an average clustering coefficient of 0.854, and PPI enrichment p value < 10–16. Most common biological processes associated with the APH1A-centered PPI include Notch signaling pathway, cell-surface receptor signaling pathway, membrane protein intracellular domain proteolysis, Notch receptor processing, and signal transduction, and among the molecular functions associated with network are Notch binding, ephrin receptor activity, axon guidance receptor activity, signaling receptor activity, and calcium ion binding. Similarly, a APH1B-centered PPI network generated by STRING using the highest confidence level of 0.9 included 35 nodes with 242 edges (see Supplementary Figure S1H), which significantly exceeds the number of edges (40) expected for a set of proteins of similar size randomly drawn from the genome. This network is characterized by the average node degree of 13.8, an average clustering coefficient of 0.855, and PPI enrichment p value < 10–16. Most common biological processes and molecular functions here were identical to those associated with the APH1A-centered PPI network.
γ-Secretase subunit PEN2
PEN2, or presenilin enhancer protein-2 (UniProt ID: Q9NZ42), is 101 amino acid long and is the smallest subunit of the human γ-secretase complex. It consists of two hydrophobic domains. It has been proposed that the protein shows a hairpin topology with its loop domain facing towards intracellular side of the membrane [140].
The role of this subunit in the catalytic activity of the enzyme is not well understood. But RNAi-mediated down-regulation studies suggest its importance in endoproteolysis of presenilin and thus in the activation of γ-secretase complex [140]. Ahn et al. have demonstrated in an in vitro system the importance of PSEN1 and PEN2 combination for proteolysis and activation of γ-secretase [141]. The C-terminus of PEN2 harbors a conserved sequence motif, DYSLF, which is crucial for γ-secretase assembly and stabilization of the presenilin fragments after endoproteolysis [140].
The structure of PEN2 in γ-secretase complex has only one missing residue (101). Thus, the structure is available for 99.0% residues of PEN2. The mean disorder propensity for full-length PEN2 protein is 8.6% and 0% as predicted by MobiDB (Table 2, Fig. 12a). ELM server predictions show that after filtering PEN2 retains 15 SLiMs and 18 instances. Both ANCHOR and D2P2 predict that the protein has no MoRF regions (Table 3). Hence, none of the retained SLiMs are associated with MoRFs. According to the prediction from D2P2 (Fig. 12b), PEN2 has only one ubiquitylation site.
Fig. 12.

Evaluation of intrinsic disorder propensity of PEN2: a Disorder profile generated by PONDR® VSL2 (grey line), PONDR® VL3 (red line), PONDR® VLXT (blue line), PONDR® FIT (magenta line), IUPRED long (purple line) and IUPRED short (orange line). The cyan line represents the mean disorder propensity calculated using the disorder scores from all six predictors. The light magenta shadow region represents the PONDR® FIT error distribution and the light cyan region represents the mean error distribution. The area of the graph marked in grey color represents residue 1 of PEN2 which is missing in the PDB structure of γ-secretase. b Evaluation of MoRF sites and PTMs in PEN2 by D2P2
In the PPI network generated for PEN2 by STRING using the highest confidence level of 0.9, there are 36 nodes with 246 edges (see Supplementary Figure S1I), which significantly exceeds the number of 41 edges expected for a set of proteins of similar size randomly drawn from the genome. This network is characterized by the average node degree of 13.7, an average clustering coefficient of 0.858, and PPI enrichment p value < 10–16. Most common biological processes and molecular functions of this network were identical to those associated with the APH1A-centered PPI network (see above).
Intrinsic disorder propensity of protein APOE
APOE is the predominant apolipoprotein of the HDL (high-density lipoprotein) complex in brain. It belongs to the group of transmembrane proteins of the low-density lipoprotein receptor (LDLR) family. The gene for APOE is located on chromosome 19q.13 [4]. It has three isoforms formed by alternative splicing: APOE2, APOE3, and APOE4. Sequences of these isoforms differ in only two residues at the positions 112 and 158. Human APOE is a lipoprotein, which is synthesized in a form of 317-residue-long precursors containing N-terminal signal peptide. APOE is a major genetic risk factor for LOAD and is strongly associated with increased Aβ accumulation in the brain [4]. Interestingly, the lipid-free apoE form has been identified with Aβ peptide in amyloid plaques of AD patients [142].
APOE (UniProt ID: P02649) is made up of three structural domains: N-terminal domain which contains the receptor-binding region, interaction domain and a C-terminal domain which contains the lipid-binding region. Structure of the N-terminal domain was obtained by X-ray diffraction and the C-terminal domain structure was modeled by structure prediction and CD spectroscopy. Later, the NMR structure for full-length APOE3 (PDB ID: 2L7B) was obtained which suggested a different domain–domain interaction than the previously available models [143].
In APOE3 and APOE4, Arg 158 forms a salt bridge with Asp154 of the low-density lipoprotein receptor (LDLR) region, while, in APOE2, Arg150 and Asp154 form an alternative salt bridge in the LDLR region. In APOE4, Arg112 orients the side chain of Arg61 in an aqueous environment so that it can interact with Glu255 from C-terminal domain. Such domain interactions occur to a lesser extent in E2 and E3, as they contain a Cys instead of Arg at 112 resulting in a different Arg61 conformation. The residue at 112 affects the movement of Arg114 and thus the ionization of His140 resulting in difference in charge distribution and structural difference in APOE3 and APOE4 [144].
APOE has a major role in lipid recycling. In CNS, it takes up lipids generated from neuronal degeneration and redistributes them to other cells. These lipids are utilized by neurons for remyelination of new axons, membrane repair, and proliferation. APOE also modulates glutamate receptor function and synaptic plasticity.
The APOE4 isoform is a major risk factor for AD, while APOE2 protects against AD. In transgenic mice, APOE4 increased Aβ accumulation in the brain, while APOE2 and APOE3 diminished Aβ levels. The mechanism underlying the pathogenic nature of APOE4 is still not clearly understood. ApoE is also known to aggregate at different rates on the isoform-dependent manner (apoE4 > apoE3 > apoE2) [145]. Further reports demonstrated that ApoE4 accelerates early seeding of Aβ and facilitates Aβ amyloid formation during the early stage of aggregation [146]. Recent findings reported that the amyloidogenic regions in APOE are essential binding sites for oligomeric Αβ [147]. Individuals heterozygous for APOE4, i.e., carrying a single copy of APOE4 are at threefold more risk of AD and the risk curve for the disease shows a shift to 5 years earlier, while people who are homozygous for APOE4 are 15-fold more susceptible and show a shift in the risk curve by 10 years earlier. People who are heterozygous for APOE2 show a shift in the risk curve of 5 years later and homozygotes of APOE2 show a shift of 10 years later. Heterozygotes carrying E3 and E4 with a higher E4/E3 ratio are at greater risk of AD [4, 144].
In AD, APOE receptors mediate endocytosis of ligands and recycle them back to the cell surface. Cytosolic adaptors that bind APOE are able to interact with NPxY motif of APP and thus play a role in APP trafficking and Aβ production. Effects of APOE on APP trafficking and Aβ production has been investigated through cell culture studies and is isoform dependent [144]. Deposition of Aβ by APOE is isoform dependent and follows the trend, APOE4 > > APOE3 > APOE2. APOE-based clearance of Aβ is isoform dependent (APOE2 > > APOE3 > APOE4). The exact molecular mechanism and cellular process involved are still not clear. Apart from APP trafficking, Aβ production, deposition, and clearance, APOE might also play a role in tauopathies. APOE may promote tau hyperphosphorylation and lead to formation of neurofibrillary tangles of tau protein.
Like APP, proteolytic enzymes can also cleave APOE and produce toxic fragments. APOE4 is more susceptible to cleavage than APOE3 and APOE2. The N-terminal fragment formed after cleavage plays a role in neurotoxicity. It includes GTPase, CREB activation, Ca2+dysregulation and activation of AKT pathway. The C-terminal fragment produces AD like pathology and stimulates Tau phosphorylation. It forms NFT inclusion bodies both in vitro and in vivo.
APOE modulates inflammatory and immune response in an isoform-dependent manner (APOE4 > APOE3 > APOE2). E3 suppresses neuroinflammation while APOE activates a proinflammatory response. In APOE mice, genotype-dependent differences in secretion of cytokines are observed. In E4/E4 transgenic mice, a dramatic increase in proinflammatory cytokines is observed. APOE isoforms differently modulate innate immunity components through multiple signaling pathways and cause neuroinflammation [144].
APOE is a modulator of cholesterol transport and lipids between neurons as well as glial cells. In cell culture studies, it has been observed that E4 is less efficient than E2 and E3 in the transport of cholesterol in neurons. Aβ degradation is mediated by cholesterol efflux function of APOE. APOE lowers cholesterol levels in cells and promotes Aβ trafficking to lysosomes for degradation. APOE-mediated cholesterol efflux is also isoform dependent (E2 > E3 > E4) [144]. APOE4 carriers have structural and developmental alterations in their brain that provide a foothold for neuropathological changes associated with subsequent course of AD [144].
In a previous analysis of the APOE secondary structure, it has been shown that the full-length proteins of three APOE isoforms, APOE2, APOE3, and APOE4 have α-helical structure [148], while other reports on short peptides (residues 202–223, 223–244, 245–266, and 268–289) derived from APOE have been found disordered [149]. In our computational analysis of the per-residue intrinsic disorder predisposition, the full-length APOE protein has shown to have the mean PPID score of 44.4% (Table 2), which coincide with these experimental observations. According to ANCHOR, APOE has no MoRF regions (Table 3), while D2P2 predicts a short MoRF region of six residues which constitutes only 1.9% of the protein. The graph in Fig. 13b shows the disorder propensity of this protein. Figure 13a represents an NMR structure of APOE3. According to D2P2 (Fig. 13c), APOE has six glycosylation sites, one acetylation site and six glycosylation sites and all these PTM sites lie in IDPRs.
Fig. 13.

Evaluation of intrinsic disorder propensity of APOE a NMR structure of APOE3 which is an isoform of APOE (PDB ID: 2L7B). The IDP regions (violet color) are mapped on the protein structure (olive color). b Disorder profile generated by PONDR® VSL2 (grey line), PONDR® VL3 (red line), PONDR® VLXT (blue line), PONDR® FIT (magenta line), IUPRED long (purple line) and IUPRED short (orange line). The cyan line represents the mean disorder propensity calculated using the disorder scores from all six predictors. The light magenta shadow region represents the PONDR® FIT error distribution and the light cyan region denotes the mean error distribution. c Evaluation of MoRF sites and PTMs in APOE by D2P2
According to the prediction by ELM server, APOE has retained 17 SLiMs and 24 instances after filtering (Table 3). On comparing with the MoRF residues predicted by D2P2, it was observed that one SLiM is embedded in and five SLiMs overlap with the MoRF regions (Supplementary Table 1).
STRING-generated PPI network for APOE using the highest confidence level of 0.9 is a dense net of 147 nodes connected by 6,152 edges (see Supplementary Figure S1J), which significantly exceeds the number of edges (407) expected for a random set of proteins of similar size. This network is characterized by the average node degree of 83.3, an average clustering coefficient of 0.937, and PPI enrichment p value < 10–16. Most common biological processes associated with the APOE-centered PPI include post-translational protein modification, organonitrogen compound metabolic process, protein metabolic process, cellular protein modification process, and cellular protein metabolic process, and top five molecular functions of this network are lipoprotein particle binding, lipoprotein particle receptor binding, signaling receptor binding, apolipoprotein binding, and glycosaminoglycan binding.
Intrinsic disorder propensity of the proteins of BIN1
The bridging integrator 1 (BIN1, UniProt ID: O00499) is also known as amphiphysin 2. According to the Alzgene database, BIN1 has been identified as the most important genetic susceptibility locus after APOE in LOAD. The BIN1 gene is located on chromosome 2q14.3, which codes for 20 exons. BIN1 shows a diverse array of splice variants, which vary in inclusion of exons 4, 6a, 10, 12, and 13, and which show different tissue distributions. The canonical form of BIN1 protein is made up of 593 amino acids, but its full-length structure has not been determined yet [150].
Earlier, BIN1 was identified as a tumor suppressor, as it contains a MYC-interacting domain along with an SH3-domain at the C-terminal region and a BAR domain at the N-terminal region. A 1.99 Å resolution structure of the N-terminal residues 1–272, which houses the BAR domain, has been determined by X-ray crystallography and is available on PDB (PDB ID: 2FIC) [151]. An NMR structure of residues 301–593 (PDB ID: 1MV3) is also available [152].
BIN1 plays a significant role in the regulation of endocytosis, trafficking, inflammation, calcium homeostasis, and apoptosis. Very little is known about the role of BIN1 in AD pathogenesis. It has been speculated that the endocytosis and trafficking role of BIN1 could affect APP processing [150]. Other studies have suggested that BIN1 affects AD risk by modulating the tau-related pathology. A recent study has demonstrated a physical interaction between BIN1 and tau protein which strongly suggests that elevated levels of BIN1 with increased risk of AD [153]. Also, BIN1 knockout studies show significant suppression of tau neurotoxicity. BIN1 is emerging to be the new therapeutic target for AD, but more work needs to be done to pinpoint the exact role of BIN1 in the Tau pathway [150].
Figure 14a, b represents the PDB structures of the residues present in the corresponding white regions of the graph below. BIN1 shows a mean PPID score of 52.5% (Table 2, Fig. 14c), which is much higher than 28% predicted by MobiDB. The protein is predicted to harbor three long MoRF regions by ANCHOR, which constitute 49.6% of the protein and ten short MoRF regions by D2P2, which make up 41.3% of the protein. Despite the large variation in number of predicted regions by both the software, not much variation is seen in the predicted percentage of MoRF regions present in the protein. According to D2P2, BIN1 has 12 phosphorylation sites out of which 9 are located in IDPR and 1 acetylation site in the IDPR (Fig. 14c). Alternative splicing generates 11 isoforms of this protein, mostly by affecting disorder-containing regions. The PPIDPONDR®VSL2 scores for various isoforms of BIN1 are 63.91% (canonical or IIA isoform), 65.25% [isoform IIb with missing residues 174–204 (PPID = 6.5%) and 378–421 (PPID = 100%)], 57.71% [isoform IIC1 with missing residues 335–421 (PPID = 100%)], 62.11% [isoform IIC2 with missing residues 174–204 (PPID = 6.5%) and 335–421 (PPID = 100%)], 61.09% [isoform IID with missing residues 335–377 (PPID = 100%)], 62.66% [isoform II2 with missing residues 174–204 (PPID = 6.5%) and 378–457 (PPID = 100%)], 59.00% [isoform II3 with missing residues 174–204 (PPID = 6.5%) and 335–457 (PPID = 100%)], 60.35% [isoform BIN1 with missing residues 174–204 (PPID = 6.5%) and 335–457 (PPID = 100%) and with residue 285 being changed from P to PRKKSKLFSRLRRKKN], 55.75% [isoform BIN1-10-13 with missing residues 174–204 (PPID = 6.5%) and 335–487 (PPID = 100%)], 57.31% [isoform BIN1-13 with missing residues 174–204 (PPID = 6.5%) and 335–487 (PPID = 100%) and with residue 285 being changed from P to PRKKSKLFSRLRRKKN], and 63.78% [isoform BIN1 + 12Awith missing residues 174–204 (PPID = 6.5%) and 378–457 (PPID = 100%) and with residue 285 being changed from P to PRKKSKLFSRLRRKKN].
Fig. 14.

Evaluation of intrinsic disorder propensity of BIN1 a 1.99 Å resolution structure of the BAR domain of BIN1 (PDB ID: 2FIC). b NMR structure of residues 301–593 of BIN1 (PDB ID: 1MV3). The IDP regions (violet color), MoRF regions (green color) and MoRF regions in IDPRs (red color) are mapped on the protein structures (olive color). b Disorder profile generated by PONDR® VSL2 (grey line), PONDR® VL3 (red line), PONDR® VLXT (blue line), PONDR® FIT (magenta line), IUPRED long (purple line) and IUPRED short (orange line). The cyan line represents the mean disorder propensity calculated using the disorder scores from all six predictors. The light magenta shadow region represents the PONDR® FIT error distribution and the light cyan region represents the mean error distribution. The grey shaded area of the graph represents residues which are either missing in the PDB structures or the residues for which no structure is available. c Evaluation of MoRF sites and PTMs in BIN 1 by D2P2
According to the ELM server, BIN1 has retained 45 SLiMs and 127 instances (Table 3) after filtering. Out of these, 78 instances are embedded in, 21 instances overlap with and 11 instances are in close proximity to the MoRF regions predicted by D2P2 (Supplementary Table 1).
STRING-generated interactome of BIN1 using the highest confidence level of 0.9 represents a densely linked network of 125 nodes connected by 6833 edges (see Supplementary Figure S1K), significantly exceeding the number of edges (593) expected for a random protein set of similar size. This network is characterized by the average node degree of 109, an average clustering coefficient of 0.985, and PPI enrichment p value < 10–16. The most common biological processes include membrane organization, endocytosis, vesicle-mediated transport, establishment of localization, and transport, whereas the most common molecular functions associated with the APOE-centered PPI network are clathrin binding, lipid binding, protein binding, phospholipid binding, clathrin adaptor activity, and cargo receptor activity.
Intrinsic disorder propensity of clusterin
Clusterin (UniProt ID: P10909) is a 449-amino-acid-long chaperone protein which belongs to the clusterin family [154]. It is also known as ApoJ. Six isoforms of this protein exist, which are produced by alternative splicing. Clusterin has two nuclear localization motifs (residues 78–81 and 443–447) and no PDB structure of this protein is available.
Clusterin is involved in multiple physiological processes like lipid metabolism, cell apoptosis and Aβ peptide binding. Interaction studies of clusterin and Aβ show that clusterin is involved in Aβ plaque formation and modulates neurotoxicity generated by Aβ in AD pathogenesis. GWAS studies have identified clusterin as the third strongest genetic risk marker for LOAD. Multiple studies have shown that clusterin is associated with metabolic syndromes and inflammatory markers associated with AD risk. High level of clusterin is detected in plasma of patients with mild cognitive impairment (MCI) and AD [154].
Previous reports based on CD spectroscopy revealed that the full-length clusterin is characterized by α-helical secondary structure [155]. According to our analyses, clusterin is characterized as partially disordered with mean PPID score of 31% and 21% in MobiDB (Table 2). According to ANCHOR and D2P2, clusterin has no MoRF regions (Table 3). Figure 15a shows the per-residue disorder propensity of this protein. According to D2P2 (Fig. 15b), clusterin has ten phosphorylation sites out of which six lie in the IDPR, one acetylation site in IDPR, two ubiquitylation sites out of which one lies in IDPR and one glycosylation site which also lies in IDPR. The canonical form of clusterin (also known as CLU35) has a PPIDPONDR®VSL2 score of 59.99%. CLU34 isoform is generated by replacing N-terminal M residue with a highly disordered 52-residue-long peptide (PPID = 71.1%), leading to an increase in PPIDPONDR®VSL2 to 60.28%. Another isoform is generated by the removal of 175 N-terminal residues from canonical isoform that changes PPIDPONDR®VSL2 to 67.52%. According to the results from ELM server, none of the predicted motifs are retained after filtering (Table 3).
Fig. 15.

Evaluation of intrinsic disorder propensity of clusterin: a Disorder profile generated by PONDR® VSL2 (grey line), PONDR® VL3 (red line), PONDR® VLXT (blue line), PONDR® FIT (magenta line), IUPRED long (purple line) and IUPRED short (orange line). The cyan line represents the mean disorder propensity calculated using the disorder scores from all six predictors. The light magenta shadow region denotes the PONDR® FIT error distribution and the light cyan region represents the mean error distribution. b Evaluation of MoRF sites and PTMs in clusterin by D2P2
STRING-generated PPI network centered at clusterin using the highest confidence level of 0.9 includes 77 nodes connected by 2156 edges (see Supplementary Figure S1L), which significantly exceeds the number of 147 edges expected for a random set of proteins of similar size. This network is characterized by the average node degree of 56, an average clustering coefficient of 0.968, and PPI enrichment p value < 10–16. Most common biological processes associated with the clusterin-centered PPI network include platelet degranulation, regulated exocytosis, vesicle-mediated transport, localization, and regulation of response to external stimulus, whereas its most common molecular functions are signaling receptor binding, growth factor activity, molecular function regulator, receptor ligand activity, and peptidase regulator activity.
Intrinsic disorder propensity of protein PICALM
Phosphatidylinositol-binding clathrin assembly protein (PICALM, UniProt ID: Q13492) is a 652-amino-acid-long protein encoded by gene located on chromosome 11q14. In the CNS, PICALM is present in neurons, astrocytes, and oligodendrocytes. Recent immunolabelling studies have established that it mainly exists in the endothelial cells of vascular walls. In glial cells and neurons, weak immunolabelling was observed [156]. There is also experimental evidence suggesting that PICALM co-localizes with APP in neurons. Initially, this protein was known to be involved in acute lymphoblastic and acute myeloid leukemias. Hence, it is also known as clathrin assembly lymphoid myeloid leukemia protein. GWAS tell us that PICALM is a risk gene for LOAD [157]. Five different isoforms of the protein are produced through alternative splicing. No PDB structure is available for PICALM.
This protein potentially plays a role in growth, hematopoiesis, iron metabolism, and clathrin-mediated endocytosis (CME). CME of APP is a very crucial step for generation of Aβ peptide, therefore, PICALM is important for APP trafficking and development of AD-related pathogenesis [13]. The exact molecular mechanism by which PICALM acts in AD is still not elucidated. In cell culture models of AD, PICALM is reported to influence Aβ metabolism and increase amyloid loads [158].
Role of PICALM in AD can be Aβ-dependent and Aβ-independent. The latter suggests its importance in tauopathy, synaptic dysfunction, disorganized lipid metabolism, disorganized lipid metabolism, and disrupted iron homeostasis. The expression of PICALM in endothelial cells suggests that it could be affecting clearance of Aβ from the blood–brain barrier. It has been observed that inhibiting PICALM expression can reduce APP internalization and Aβ production. PICALM degradation by caspases can also block endocytosis. Thus, figuring out the exact mechanism by which PICALM affects AD pathogenesis is important and will fuel our understanding of AD mechanism [13].
The protein has a mean PPID score of 36.4%, while MobiDB predicts only 3% disorder (Table 2). According to ANCHOR, PICALM has five MoRF regions which make up 15.5% of the protein and D2P2 predicts it to have six MoRF regions that constitute 9.1% (Table 3). Figure 16a represents the intrinsic disorder profile of PICALM. According to D2P2 (Fig. 16b), PICALM has 26 phosphorylation sites out of which 15 lie in the IDPR, 1 acetylation site, 5 ubiquitylation sites out of which 3 lie in IDPR and 1 di-methylation site, which lies within the IDPR. There are five isoforms of human PICALM generated by alternative splicing. The PPIDPONDR®VSL2 scores for these isoforms are 59.20% (canonical isoform), 57.91% [isoform 2 with missing residues 594–613 (PPID = 100%)], 55.41% [isoform 3 with missing residues 420–469 (PPID = 100%) and residue 593 changed to a MNGMHFPQY peptide], 56.62% [isoform 4 with missing residues 1–51 (PPID = 56.9%) and 420–469 (PPID = 100%)], and 58.76% [isoform 5 with missing residues 420–426 (PPID = 100%)].
Fig. 16.

Evaluation of intrinsic disorder propensity of PICALM: a Disorder profile generated by PONDR® VSL2 (grey line), PONDR® VL3 (red line), PONDR® VLXT (blue line), PONDR® FIT (magenta line), IUPRED long (purple line) and IUPRED short (orange line). The cyan line represents the mean disorder propensity calculated using the disorder scores from all six predictors. The light magenta shadow region represents the PONDR® FIT error distribution and the light cyan region represents the mean error distribution. b Evaluation of MoRF sites and PTMs in PICALM by D2P2
ELM server predictions show that PICALM has 51 SLiMs and 160 instances which are retained after filtering. Out of these, 8 instances are embedded in, 17 instances overlap with and 8 instances are in close proximity to the MoRF regions predicted by D2P2 (Supplementary Table 1).
PPI network generated for PICALM by STRING using the highest confidence level of 0.9 includes 156 nodes connected by 8,010 edges (see Supplementary Figure S1M), which significantly exceeds the number of 736 edges expected for a random set of proteins of similar size. This network is characterized by the average node degree of 103, an average clustering coefficient of 0.935, and PPI enrichment p value < 10–16. Most common biological processes associated with the PICALM-centered PPI network include membrane organization, vesicle-mediated transport, endocytosis, transport, and establishment of localization, and top ten molecular functions of this network are clathrin binding, lipid binding, protein binding, phospholipid binding, clathrin adaptor activity, SNARE binding, enzyme binding, low-density lipoprotein particle receptor binding, syntaxin binding, and protein binding, bridging.
Intrinsic disorder propensity of protein CD33
CD33 (UniProt ID: P20138) is a type I transmembrane protein and is the shortest member of the sialic acid-binding Ig-like family (SIGLECs). This protein is a myeloid cell receptor which is expressed exclusively by microglia and macrophages in the brain. The CD33 gene is located on chromosome 19q13.33 and is made up of seven coding exons. CD33 is a 67 kDa, 364-residue-long protein, with its N-terminal being located in the extracellular space and C-terminal in the cytosol [33]. It consists of an 18-residue-long signal peptide, a canonical Ig-like V-type domain (residues 19–135), which is the functional domain of CD33 as it contains the sialic acid-binding site, C2-type Ig repeat structural domain (residues 145–228), transmembrane domain (residues 260–282), a membrane-proximal immunotyrosine inhibitory motif (ITIM) (residues 338–343), and a membrane distal ITAM-like motif (residues 356–361). A 1.75 Å resolution crystal structure containing the functional domain of CD33 (residues 18–143) is available in PDB (PDB ID: 6D4U). In this structure, the residues 17–19 and 143 are missing. Thus, the structure is currently known only for 33.79% of the protein sequence.
The exact function of CD33 and its role in AD has not been fully understood. As CD33 is expressed only on immune cells, it could have an important role in modulating the function of immune cells. This protein is also known to be involved in processes like adhesion in immune cells and cancer cells, growth and survival of immune cells and induction of apoptosis [33].
According to GWAS, CD33 is one of the top risk factors for AD. Microglia-based Aβ clearance is dependent on surface expression CD33 levels are high in patients with AD. It is responsible for microglial activation and blocks clearance of Aβ from the brain. CD33 −/− mouse model shows lower Aβ levels and reduced amyloid plaque burden in the brain [159]. Thus, reduced CD33 expression can allow better Aβ clearance by microglia and thus can protect against AD. This makes CD33 a good therapeutic target for AD.
Figure 17b shows a graph of per-residue disorder propensity of CD33 protein. Figure 17a represents a crystal structure of the Ig-like V-type domain. CD33 shows a mean PPID score of 31.0% (Table 2) and 21% disorder is predicted by MobiDB. The protein is predicted to harbor one MoRF region by both ANCHOR and D2P2. But the MoRF region predicted by ANCHOR is longer, as it constitutes 11.3% of the protein, and the region predicted by D2P2 only makes up 4.1% of the protein. According to the results of the D2P2 analysis represented in Fig. 17c, CD33 has eight phosphorylation sites out of which six are located in IDPR and two ubiquitylation sites in the IDPR. The canonical form of CD33 is characterized by the presence of long disordered C-terminal region (residues 290–364) and has a PPIDPONDR®VSL2 score of 42.31%. Alternative splicing generates two isoforms by (a) changing the C-terminal residues 309–364 to VR dipeptide to make an isoform 3 with a PPIDPONDR®VSL2 score of 32.58% or (b) removing residues 13–139 (PPID = 40.9%) to produce an isoform CD33m with a PPIDPONDR®VSL2 score of 43.04%.
Fig. 17.

Evaluation of intrinsic disorder propensity of CD33 a 2.24 Å resolution structure of CD33 (PDB ID: 5IHB). The IDPRs (violet color) are mapped on the protein structure (olive color). b Disorder profile generated by PONDR® VSL2 (grey line), PONDR® VL3 (red line), PONDR® VLXT (blue line), PONDR® FIT (magenta line), IUPRED long (purple line) and IUPRED short (orange line). The cyan line represents the mean disorder propensity calculated using the disorder scores from all six predictors. The light magenta shadow region represents the PONDR® FIT error distribution and the light cyan region represents the mean error distribution. The grey shaded area of the graph represents residues which are either missing in the PDB structure or the residues for which no structure is available. The protein is shown in olive color and the IDPRs (violet color) are mapped on the protein structure. c Evaluation of MoRF sites and PTMs in CD33 by D2P2
ELM server predicts that after filtering, the protein CD33 has 21 SLiMs and 35 instances (Table 3). Two of these retained SLiMs are embedded in and one SLiM overlaps with the MoRF regions predicted by D2P2. These motifs were classified based on the type of annotation and they include one ELM_LIG which is a phosphopeptide motif, one ELM_MOD which is a GSK3 phosphorylation recognition site and one ELM_TRG which is a tyrosine-based sorting signal (Supplementary Table 1).
STRING-generated PPI network centered at CD33 using the highest confidence level of 0.9 includes 114 nodes connected by 5,324 edges (see Supplementary Figure S1N), which significantly exceeds the number of 298 edges expected for a random set of proteins of similar size. This network is characterized by the average node degree of 93.4, an average clustering coefficient of 0.931, and PPI enrichment p value < 10–16. Most common biological processes associated with the CD33-centered PPI network include neutrophil degranulation, leukocyte migration, regulation of localization, regulation of immune system process, and regulation of immune response, whereas its most common molecular functions are carbohydrate binding, transporter activity, signaling receptor activity, transmembrane transporter activity, and ion transmembrane transporter activity.
Intrinsic disorder propensity of protein SORL1
SORL1 (UniProt ID: Q92673) is a 2,214-amino-acid-long type I transmembrane protein. It belongs to both low-density lipoprotein receptor family and vacuolar protein sorting 10 (VPS10) domain receptor family. The gene for this protein is located on 11q23.2–11q24.2. It is a 250 kDa membrane protein and has 7 distinct domains which include a NH2 terminal (domain I), a sequence homologous to VPS10 (domain II), 5 tandem LDLR repeats (domain III), cluster of 11 complement type repeats (domain IV), 6 motifs related to FN type III found in certain neural adhesion proteins (domain V), a putative membrane-spanning region (domain VI) and a COOH terminus (domain VII). SORL1 is predominantly expressed in hippocampal neurons, nuclei of brain cells and Purkinje fibers. In these cells, SORL1 is mainly localized to the TGN and early endosomes. SORL1 plays a role in cellular transport, intracellular sorting and cell signaling. It also shows chaperone-like activity. A 2.35 Å resolution structure (PDB ID: 3WSX) of residues 29–753 [160] and a 1.70 resolution structure (PDB ID: 3G2S) of residues 2202–2214 [161] are available in PDB.
The protein SORL1 is significantly reduced in brains of patients with LOAD and this is positively co-related with Aβ accumulation. Normally, SORL1 interacts with cytosolic adaptors and mediates the anterograde and retrograde movement of APP in the cell between TGN and early endosomes. It prevents the delivery of APP to endocytic compartments and thus prevents formation of Aβ. SORL1 also steers formed Aβ towards lysosomes where it undergoes degradation. In SORL1 knockout mice, enhanced production of Aβ peptide and its deposition are observed. Also, in cell culture studies, overexpression of SORL1 resulted in reduced amyloidogenic processing of APP. These results indicate that SORL1 could be a good therapeutic target for treatment of AD.
SORL1 has a mean PPID score of 12.14%, while MobiDB predicts 21% disorder (Table 2). According to ANCHOR and D2P2, SORL1 has no MoRF residues (Table 3). Figure 18c represents the disorder profile of SORL1. Figure 18a, b shows the crystal structures of the protein corresponding to residues presented in the white regions of the graph below. According to D2P2 (Fig. 18d), SORL1 has 22 phosphorylation sites, out of which 3 are located in the IDPRs and 1 glycosylation site. After filtering of ELM server predictions, 42 SLiMs and their 88 instances in SORL1 are retained (Table 3). As D2P2 does not predict presence of any MoRF residues in SORL1, none of these instances are associated with MoRF regions.
Fig. 18.

Evaluation of intrinsic disorder propensity of SORL1 a 2.35 Å resolution structure of SORL1 residues 29–753 (PDB ID: 3WSX). b 1.70 Å resolution structure of SORL1 residues 2202–2214 (PDB ID: 3G2S). The IDP regions (violet color) are mapped on the protein structure (olive color). The protein interacting with SORL1 is shown in faded salmon color. c Disorder profile generated by PONDR® VSL2 (grey line), PONDR® VL3 (red line), PONDR® VLXT (blue line), PONDR® FIT (magenta line), IUPRED long (purple line) and IUPRED short (orange line). The cyan line represents the mean disorder propensity calculated using the disorder scores from all six predictors. The light magenta shadow region represents the PONDR® FIT error distribution and the light cyan region represents the mean error distribution. The grey shaded area of the graph represents residues which are either missing in the PDB structures or the residues for which no structure is available. d Evaluation of MoRF sites and PTMs in SORL1 by D2P2
STRING-generated PPI network centered at SORL1 using the high confidence level of 0.7 includes 22 nodes connected by 66 edges (see Supplementary Figure S1O), which significantly exceeds the number of 23 edges expected for a random set of proteins of similar size. This network is characterized by the average node degree of 6, an average clustering coefficient of 0.8, and PPI enrichment p value < 4.05 × 10–13. Most common biological processes associated with this SORL1-centered PPI network include regulation of amyloid-beta formation, negative regulation of amyloid precursor protein catabolic process, negative regulation of amyloid-beta formation, positive regulation of amyloid fibril formation, and positive regulation of protein catabolic process, whereas most common molecular functions of this network are low-density lipoprotein particle receptor binding, ADP-ribosylation factor binding, peptide binding, amyloid-beta binding, and tau protein binding.
Intrinsic disorder propensity of protein PLG
Plasmin is a serine protease which is formed when tissue plasminogen activator (tPA) or urokinase PLG activator (uPA) cleaves its precursor, Plasminogen (PLG, UniProt ID: P00747) [162]. The PLG gene is localized on chromosome 6q27 and gives rise to an 810-residue-long protein. The protein is made up of a PAN domain, five kringle domains in tandem, and a peptidase S1 domain. A 2.45 Å resolution structure of full-length PLG (PDB ID: 4DUR) has been obtained by X-ray crystallography and the residues 1–21, 354–363, 457–476 and 708–710 are missing in the structure [163]. Therefore, the structure is currently known for 93.33% of the protein sequence.
Plasmin primarily plays a role in degradation and clearing of fibrin clots through hydrolysis and the process is known as fibrinolysis. Plasmin also plays a role in the innate immune system by acting as a chemoattractant for macrophages, thus increasing inflammation. Plasmin also has a proteolytic function and modifies extracellular matrix proteins either directly or by activating other proteins, growth factors complement and other proteins [162, 164]. Thus, apart from fibrinolysis, plasmin has been implicated in other cellular processes like wound healing, host defense, cell adhesion, cell migration, and cell signaling.
Plasminogen and its activators are responsible for promoting the development of autoimmune inflammatory arthritis [165, 166]. In a recent study, it was established that plasmin and plasminogen are responsible for regulating disease onset in autoimmune neuroinflammatory disease [164]. In a mouse model study of AD, it was found that conditional depletion of PLG from peripheral blood and not from the brain protects from Aβ deposition and generation of a neuroinflammatory response [162].
The protein has a PPIDPONDR®VSL2 score of 50.86% and a mean PPID score of 25.1%, while MobiDB predicts 5% of disordered residues in PLG (Table 2). According to ANCHOR, PLG has no MoRF regions, while D2P2 identifies an 8-residue-long MoRF (Table 3). The graph in Fig. 19b shows the disorder propensity of protein residues predicted by six different predictors and their mean. Figure 19a above the graph is the PDB structure of the residues present in the corresponding white region of the graph below. The missing residues in the PDB structure and the residues for which no structure is available are marked in grey. According to D2P2 (Fig. 19c), PLG has 14 phosphorylation sites. Currently, there are no experimentally validated isoforms of PLG generated by alternative splicing.
Fig. 19.

Evaluation of intrinsic disorder propensity of PLG a 2.45 Å resolution structure of full-length PLG (PDB ID: 4DUR). The IDPRs (violet color) and MoRF regions (green color) are mapped on the protein structure (olive color). b Disorder profile generated by PONDR® VSL2 (grey line), PONDR® VL3 (red line), PONDR® VLXT (blue line), PONDR® FIT (magenta line), IUPRED long (purple line) and IUPRED short (orange line). The cyan line represents the mean disorder propensity calculated using the disorder scores from all six predictors. The light magenta shadow region represents the PONDR® FIT error distribution and the light cyan region represents the mean error distribution. The grey shaded area of the graph represents residues which are either missing in the PDB structure or the residues for which no structure is available. c Evaluation of MoRF sites and PTMs in PLG by D2P2
PPI network generated for PLG by STRING using the highest confidence level of 0.9 includes 100 nodes connected by 2325 edges (see Supplementary Figure S1P), which significantly exceeds the number of edges (211) expected for a random set of proteins of similar size. This network is characterized by the average node degree of 46.5, an average clustering coefficient of 0.858, and PPI enrichment p value < 10–16. Most common biological processes associated with the PLG-centered PPI network include platelet degranulation, regulated exocytosis, secretion, vesicle-mediated transport, and extracellular matrix organization, and top five molecular functions of this network are signaling receptor binding, growth factor activity, molecular function regulator, endopeptidase inhibitor activity, and peptidase regulator activity.
Intrinsic disorder propensity of ADAM9, ADAM10, ABCA7, IDE, neprilysin, LRP1, A2M, ECE2, PLAU, PLAT, and TREM
We also conducted computational analysis of the intrinsic disorder predisposition and functionality of IDPRs in the remaining 11 proteins from the amyloid cascade signaling pathway. Results of these analyses are summarized in Supplementary Figures S2–S12, Tables 2 and 3, and Supplementary Table S1. This analysis revealed that all these proteins contain variable levels of intrinsic disorder and have multiple potentially disorder-based functions. All of them are engaged in multiple interactions with different partners, typically generating densely populated PPI networks with specific molecular functions and involvement in specific biological processes. It is likely that intrinsic disorder is at least in part responsible for the multifunctionality of these proteins.
Concluding remarks
Numerous biological studies have revealed that proteins involved in amyloid cascade signaling play vital roles in APP processing, Aβ production, Aβ aggregation, Aβ clearance, and nerve cell death. All these processes are critically involved in Alzheimer’s disease pathogenesis. This study is the first systematic analysis of functional intrinsic disorder in 27 proteins from the amyloid cascade signaling pathway. We have elucidated the importance of IDPRs in the functionality, protein–protein interactions, and PTMs of these proteins. Our data indicate that these proteins are characterized by structural polymorphism defined by their high disorder contents, presence of multiple PTM sites, and existence of numerous isoforms generated by alternative splicing. These features (intrinsic disorder, PTMs, and alternative splicing) were considered as means for generation of various proteoforms (i.e., a set of structurally and functionally distinct protein molecules encoded by a single gene [167, 168]), which constitute a foundation for the “protein structure–function continuum" model, where a given protein exists as a dynamic conformational ensemble containing multiple proteoforms [conformational/basic (i.e., proteoforms existing due to the presence of intrinsic disorder), inducible/modified (i.e., proteoforms induced by PTMs or originating from alternative splicing events), and functioning (i.e., proteoforms generated as a result of protein function)] characterized by a broad spectrum of structural features and possessing various functional potentials [169–171]. As per the analysis performed in this study, all proteins involved in the amyloid cascade signaling pathway contain noticeable levels of disorder. These observations align with the common knowledge that many disease-associated proteins are IDPs or hybrid proteins containing ordered domains and functional IDPRs [59, 60]. Such global association of intrinsic disorder with the pathogenesis of various diseases is linked to the facts that IDPs/IDPRs are commonly involved in recognition, regulation, and cell signaling, and therefore their deregulation and misfolding can lead to misidentification, misinteractions, and missignaling and, therefore, can be pathogenic [59, 66, 172–175].
According to GWAS studies, genes BIN1, APOE, Clusterin, PICALM, and CD33 are high-risk factors for LOAD. Our analysis revealed that proteins coded by these genes have shown a large amount of intrinsic disorder. APP (precursor of Aβ peptide), ADAM17 (which shows α-secretase activity), and PSEN1 and PSEN2 (subunits of γ-secretase complex) are major players in APP processing which are also found to be highly disordered. Furthermore, the nature of disorder in these proteins was also confirmed using CH-CDF analysis. Additionally, as SLiMs are functionally important for protein–protein interactions and post-translational modifications, our investigation shows a high abundance of motifs that are associated with MoRFs (disordered based protein–protein interaction sites) in BIN1 followed by APP, PICALM, PSEN1 and CD33. The exact role of these disorder regions in the pathogenesis and progression of AD needs to be determined experimentally. Amyloid forming proteins adopt extremely flexible structures in solution and act as promiscuous binding partners for other proteins. For example, Aβ interacts with some disordered proteins associated with various amyloid diseases, and these interactions mutually modulate toxicity of parties engaged in interaction. Therefore, in vitro and in vivo studies on the cross-amyloid interaction between Aβ and other highly disordered proteins of amyloid cascade signaling may help in identifying novel drug targets for Alzheimer’s disease.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
RG and KG are supported by the DBT project (BT/PR16871/NER/95/329/201). BG is grateful to DBT-IUSSTF sponsored Khorana scholarship 2019. PK would like to thank DBT for funding (BT/IN/IC-Impacts/21/DS/2016-2017). VU and RG are thankful to MHRD-SPARC (SPARC/2018-2019/P37/SL).
Abbreviations
- AD
Alzheimer’s disease
- AICD
APP intracellular domain
- APP
Amyloid precursor protein
- Aβ
Amyloid-beta
- CD
Cumulative distribution function
- CH
Charge-hydropathy
- CME
Clathrin-mediated endocytosis
- CSF
Cerebrospinal fluid
- D2P2
Database of disordered protein predictions
- ELM
Eukaryotic linear motifs
- FAD
Familial Alzheimer’s disease
- GWAS
Genome-wide association studies
- HDL
High-density lipoprotein
- IDP
Intrinsically disordered proteins
- IDPRs
Intrinsically disordered protein regions
- ITAM
Immunotyrosine inhibitory motif
- LDLR
Low-density lipoprotein receptor
- LOAD
Late onset of Alzheimer’s disease
- MCI
Mild cognitive impairment
- MoRFs
Molecular recognition features
- NFT
Neurofibrillary tangles
- NMDA
N-Methyl-d-aspartate
- PONDR
Predictor of natural disordered regions
- PPID
Predicted percentage of intrinsic disorder
- PTM
Post-translational modification
- SIGLECs
Sialic acid-binding Ig-like family
- SLiM
Short linear motifs
- STRING
Search tool for retrieval of interacting genes
- TGN
Trans-Golgi network
- TNF
Tumor necrotic factor
- tPA
Tissue plasminogen activator
- uPA
Urokinase PLG activator
Author contributions
RG and VNU conception, design and study supervision; KG, PG, BG, BX, VNU, and RG produced and analyzed data; KG, BG, VNU, and RG wrote and edited the manuscript.
Compliance with ethical standards
Conflict of interest
All authors declare that there are no conflicts.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Vladimir N. Uversky, Email: vuversky@usf.edu
Rajanish Giri, Email: rajanishgiri@iitmandi.ac.in.
References
- 1.Alzheimer A. Über einen eigenartigen schweren Erkrankungsprozeß der Hirnrinde. Neurologisches Centralblatt. 1906;23:1129–1136. [Google Scholar]
- 2.Hippius H, Neundorfer G. The discovery of Alzheimer's disease. Dialogues Clin Neurosci. 2003;5(1):101–108. doi: 10.31887/DCNS.2003.5.1/hhippius. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120(3):885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 4.O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robakis NK, Ramakrishna N, Wolfe G, Wisniewski HM. Molecular cloning and characterization of a cDNA encoding the cerebrovascular and the neuritic plaque amyloid peptides. Proc Natl Acad Sci USA. 1987;84(12):4190–4194. doi: 10.1073/pnas.84.12.4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldgaber D, Lerman MI, McBride OW, Saffiotti U, Gajdusek DC. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer's disease. Science. 1987;235(4791):877–880. doi: 10.1126/science.3810169. [DOI] [PubMed] [Google Scholar]
- 7.Tanzi RE, Gusella JF, Watkins PC, Bruns GA, St George-Hyslop P, Van Keuren ML, Patterson D, Pagan S, Kurnit DM, Neve RL. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science. 1987;235(4791):880–884. doi: 10.1126/science.2949367. [DOI] [PubMed] [Google Scholar]
- 8.Goyal D, Kaur A, Goyal B. Benzofuran and indole: promising scaffolds for drug development in Alzheimer's disease. ChemMedChem. 2018;13(13):1275–1299. doi: 10.1002/cmdc.201800156. [DOI] [PubMed] [Google Scholar]
- 9.Du X, Wang X, Geng M. Alzheimer's disease hypothesis and related therapies. Transl Neurodegener. 2018;7:2. doi: 10.1186/s40035-018-0107-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 11.Mohamed T, Shakeri A, Rao PP. Amyloid cascade in Alzheimer's disease: recent advances in medicinal chemistry. Eur J Med Chem. 2016;113:258–272. doi: 10.1016/j.ejmech.2016.02.049. [DOI] [PubMed] [Google Scholar]
- 12.Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10(9):698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- 13.Xu W, Tan L, Yu JT. The role of PICALM in Alzheimer's disease. Mol Neurobiol. 2015;52(1):399–413. doi: 10.1007/s12035-014-8878-3. [DOI] [PubMed] [Google Scholar]
- 14.Dourlen P, Kilinc D, Malmanche N, Chapuis J, Lambert JC. The new genetic landscape of Alzheimer's disease: from amyloid cascade to genetically driven synaptic failure hypothesis? Acta Neuropathol. 2019;138(2):221–236. doi: 10.1007/s00401-019-02004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gadhave K, Bolshette N, Ahire A, Pardeshi R, Thakur K, Trandafir C, Istrate A, Ahmed S, Lahkar M, Muresanu DF, Balea M. The ubiquitin proteasomal system: a potential target for the management of Alzheimer's disease. J Cell Mol Med. 2016;20(7):1392–1407. doi: 10.1111/jcmm.12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moss ML, Powell G, Miller MA, Edwards L, Qi B, Sang QX, De Strooper B, Tesseur I, Lichtenthaler SF, Taverna M, Zhong JL, Dingwall C, Ferdous T, Schlomann U, Zhou P, Griffith LG, Lauffenburger DA, Petrovich R, Bartsch JW. ADAM9 inhibition increases membrane activity of ADAM10 and controls alpha-secretase processing of amyloid precursor protein. J Biol Chem. 2011;286(47):40443–40451. doi: 10.1074/jbc.M111.280495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seegar TCM, Killingsworth LB, Saha N, Meyer PA, Patra D, Zimmerman B, Janes PW, Rubinstein E, Nikolov DB, Skiniotis G, Kruse AC, Blacklow SC. Structural basis for regulated proteolysis by the alpha-secretase ADAM10. Cell. 2017;171(7):1638 e1637–1648 e1637. doi: 10.1016/j.cell.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hartl D, May P, Gu W, Mayhaus M, Pichler S, Spaniol C, Glaab E, Bobbili DR, Antony P, Koegelsberger S, Kurz A, Grimmer T, Morgan K, Vardarajan BN, Reitz C, Hardy J, Bras J, Guerreiro R, Balling R, Schneider JG, Riemenschneider M. A rare loss-of-function variant of ADAM17 is associated with late-onset familial Alzheimer disease. Mol Psychiatry. 2018 doi: 10.1038/s41380-018-0091-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deuss M, Reiss K, Hartmann D. Part-time alpha-secretases: the functional biology of ADAM 9, 10 and 17. Curr Alzheimer Res. 2008;5(2):187–201. doi: 10.2174/156720508783954686. [DOI] [PubMed] [Google Scholar]
- 20.Asai M, Hattori C, Szabo B, Sasagawa N, Maruyama K, Tanuma S, Ishiura S. Putative function of ADAM9, ADAM10, and ADAM17 as APP alpha-secretase. Biochem Biophys Res Commun. 2003;301(1):231–235. doi: 10.1016/s0006-291x(02)02999-6. [DOI] [PubMed] [Google Scholar]
- 21.Yan R, Vassar R. Targeting the beta secretase BACE1 for Alzheimer's disease therapy. Lancet Neurol. 2014;13(3):319–329. doi: 10.1016/S1474-4422(13)70276-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pardeshi R, Bolshette N, Gadhave K, Ahire A, Ahmed S, Cassano T, Gupta VB, Lahkar M. Insulin signaling: an opportunistic target to minify the risk of Alzheimer's disease. Psychoneuroendocrinology. 2017;83:159–171. doi: 10.1016/j.psyneuen.2017.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Serneels L, Dejaegere T, Craessaerts K, Horre K, Jorissen E, Tousseyn T, Hebert S, Coolen M, Martens G, Zwijsen A, Annaert W, Hartmann D, De Strooper B. Differential contribution of the three Aph1 genes to gamma-secretase activity in vivo. Proc Natl Acad Sci USA. 2005;102(5):1719–1724. doi: 10.1073/pnas.0408901102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Strooper B. Aph-1, Pen-2, and nicastrin with presenilin generate an active gamma-Secretase complex. Neuron. 2003;38(1):9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 25.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2(8):864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 26.Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2000;97(6):2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barker R, Love S, Kehoe PG. Plasminogen and plasmin in Alzheimer's disease. Brain Res. 2010;1355:7–15. doi: 10.1016/j.brainres.2010.08.025. [DOI] [PubMed] [Google Scholar]
- 28.Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S. Abeta-degrading enzymes in Alzheimer's disease. Brain Pathol. 2008;18(2):240–252. doi: 10.1111/j.1750-3639.2008.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Satoh K, Abe-Dohmae S, Yokoyama S, St George-Hyslop P, Fraser PE. ATP-binding cassette transporter A7 (ABCA7) loss of function alters Alzheimer amyloid processing. J Biol Chem. 2015;290(40):24152–24165. doi: 10.1074/jbc.M115.655076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyagawa T, Ebinuma I, Morohashi Y, Hori Y, Young Chang M, Hattori H, Maehara T, Yokoshima S, Fukuyama T, Tsuji S, Iwatsubo T, Prendergast GC, Tomita T. BIN1 regulates BACE1 intracellular trafficking and amyloid-beta production. Hum Mol Genet. 2016;25(14):2948–2958. doi: 10.1093/hmg/ddw146. [DOI] [PubMed] [Google Scholar]
- 31.Moreau K, Fleming A, Imarisio S, Lopez Ramirez A, Mercer JL, Jimenez-Sanchez M, Bento CF, Puri C, Zavodszky E, Siddiqi F, Lavau CP, Betton M, O'Kane CJ, Wechsler DS, Rubinsztein DC. PICALM modulates autophagy activity and tau accumulation. Nat Commun. 2014;5:4998. doi: 10.1038/ncomms5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tian Y, Chang JC, Fan EY, Flajolet M, Greengard P. Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer's APP-CTF for terminal degradation via autophagy. Proc Natl Acad Sci USA. 2013;110(42):17071–17076. doi: 10.1073/pnas.1315110110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao L. CD33 in Alzheimer's disease—biology, pathogenesis, and therapeutics: a mini-review. Gerontology. 2019;65(4):323–331. doi: 10.1159/000492596. [DOI] [PubMed] [Google Scholar]
- 34.Jiang T, Yu JT, Hu N, Tan MS, Zhu XC, Tan L. CD33 in Alzheimer's disease. Mol Neurobiol. 2014;49(1):529–535. doi: 10.1007/s12035-013-8536-1. [DOI] [PubMed] [Google Scholar]
- 35.Wu ZC, Yu JT, Li Y, Tan L. Clusterin in Alzheimer's disease. Adv Clin Chem. 2012;56:155–173. doi: 10.1016/B978-0-12-394317-0.00011-X. [DOI] [PubMed] [Google Scholar]
- 36.Nuutinen T, Suuronen T, Kauppinen A, Salminen A. Clusterin: a forgotten player in Alzheimer's disease. Brain Res Rev. 2009;61(2):89–104. doi: 10.1016/j.brainresrev.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 37.Narayan P, Orte A, Clarke RW, Bolognesi B, Hook S, Ganzinger KA, Meehan S, Wilson MR, Dobson CM, Klenerman D. The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-beta(1–40) peptide. Nat Struct Mol Biol. 2011;19(1):79–83. doi: 10.1038/nsmb.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Storck SE, Pietrzik CU. Endothelial LRP1—a potential target for the treatment of Alzheimer's disease : theme: drug discovery, development and delivery in Alzheimer's disease guest editor: Davide Brambilla. Pharm Res. 2017;34(12):2637–2651. doi: 10.1007/s11095-017-2267-3. [DOI] [PubMed] [Google Scholar]
- 39.Shinohara M, Tachibana M, Kanekiyo T, Bu G. Role of LRP1 in the pathogenesis of Alzheimer's disease: evidence from clinical and preclinical studies. J Lipid Res. 2017;58(7):1267–1281. doi: 10.1194/jlr.R075796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blacker D, Wilcox MA, Laird NM, Rodes L, Horvath SM, Go RC, Perry R, Watson B, Jr, Bassett SS, McInnis MG, Albert MS, Hyman BT, Tanzi RE. Alpha-2 macroglobulin is genetically associated with Alzheimer disease. Nat Genet. 1998;19(4):357–360. doi: 10.1038/1243. [DOI] [PubMed] [Google Scholar]
- 41.Palmer JC, Baig S, Kehoe PG, Love S. Endothelin-converting enzyme-2 is increased in Alzheimer's disease and up-regulated by Abeta. Am J Pathol. 2009;175(1):262–270. doi: 10.2353/ajpath.2009.081054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deming Y, Li Z, Benitez BA, Cruchaga C. Triggering receptor expressed on myeloid cells 2 (TREM2): a potential therapeutic target for Alzheimer disease? Expert Opin Ther Targets. 2018;22(7):587–598. doi: 10.1080/14728222.2018.1486823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, Fuxreiter M, Gough J, Gsponer J, Jones DT, Kim PM, Kriwacki RW, Oldfield CJ, Pappu RV, Tompa P, Uversky VN, Wright PE, Babu MM. Classification of intrinsically disordered regions and proteins. Chem Rev. 2014;114(13):6589–6631. doi: 10.1021/cr400525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Habchi J, Tompa P, Longhi S, Uversky VN. Introducing protein intrinsic disorder. Chem Rev. 2014;114(13):6561–6588. doi: 10.1021/cr400514h. [DOI] [PubMed] [Google Scholar]
- 45.Mishra PM, Uversky VN, Giri R. Molecular recognition features in Zika virus proteome. J Mol Biol. 2018;430(16):2372–2388. doi: 10.1016/j.jmb.2017.10.018. [DOI] [PubMed] [Google Scholar]
- 46.Toto A, Camilloni C, Giri R, Brunori M, Vendruscolo M, Gianni S. Molecular recognition by templated folding of an intrinsically disordered protein. Sci Rep. 2016;6:21994. doi: 10.1038/srep21994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gianni S, Morrone A, Giri R, Brunori M. A folding-after-binding mechanism describes the recognition between the transactivation domain of c-Myb and the KIX domain of the CREB-binding protein. Biochem Biophys Res Commun. 2012;428(2):205–209. doi: 10.1016/j.bbrc.2012.09.112. [DOI] [PubMed] [Google Scholar]
- 48.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41(21):6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 49.Dunker AK, Brown CJ, Obradovic Z. Identification and functions of usefully disordered proteins. Adv Protein Chem. 2002;62:25–49. doi: 10.1016/S0065-3233(02)62004-2. [DOI] [PubMed] [Google Scholar]
- 50.Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. 2009;19(1):31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Toto A, Giri R, Brunori M, Gianni S. The mechanism of binding of the KIX domain to the mixed lineage leukemia protein and its allosteric role in the recognition of c-Myb. Protein Sci. 2014;23(7):962–969. doi: 10.1002/pro.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Giri R, Morrone A, Toto A, Brunori M, Gianni S. Structure of the transition state for the binding of c-Myb and KIX highlights an unexpected order for a disordered system. Proc Natl Acad Sci USA. 2013;110(37):14942–14947. doi: 10.1073/pnas.1307337110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. Intrinsically disordered protein. J Mol Graph Model. 2001;19(1):26–59. doi: 10.1016/S1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 54.Oldfield CJ, Dunker AK. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu Rev Biochem. 2014;83:553–584. doi: 10.1146/annurev-biochem-072711-164947. [DOI] [PubMed] [Google Scholar]
- 55.Uversky VN. Unusual biophysics of intrinsically disordered proteins. Biochim Biophys Acta. 2013;1834(5):932–951. doi: 10.1016/j.bbapap.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 56.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 2010;1804(6):1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uversky VN. Multitude of binding modes attainable by intrinsically disordered proteins: a portrait gallery of disorder-based complexes. Chem Soc Rev. 2011;40(3):1623–1634. doi: 10.1039/c0cs00057d. [DOI] [PubMed] [Google Scholar]
- 58.Uversky VN. Intrinsic disorder-based protein interactions and their modulators. Curr Pharm Des. 2013;19(23):4191–4213. doi: 10.2174/1381612811319230005. [DOI] [PubMed] [Google Scholar]
- 59.Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–246. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 60.Uversky VN, Dave V, Iakoucheva LM, Malaney P, Metallo SJ, Pathak RR, Joerger AC. Pathological unfoldomics of uncontrolled chaos: intrinsically disordered proteins and human diseases. Chem Rev. 2014;114(13):6844–6879. doi: 10.1021/cr400713r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kumar D, Sharma N, Giri R. Therapeutic interventions of cancers using intrinsically disordered proteins as drug targets: c-Myc as model system. Cancer Inform. 2017;16:1176935117699408. doi: 10.1177/1176935117699408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Uversky VN. Intrinsically disordered proteins and novel strategies for drug discovery. Expert Opin Drug Discov. 2012;7(6):475–488. doi: 10.1517/17460441.2012.686489. [DOI] [PubMed] [Google Scholar]
- 63.Uversky VN. Intrinsic disorder in proteins associated with neurodegenerative diseases. Front Biosci (Landmark Ed) 2009;14:5188–5238. doi: 10.2741/3594. [DOI] [PubMed] [Google Scholar]
- 64.Uversky VN. The roles of intrinsic disorder-based liquid-liquid phase transitions in the "Dr. Jekyll-Mr. Hyde" behavior of proteins involved in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Autophagy. 2017;13(12):2115–2162. doi: 10.1080/15548627.2017.1384889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Uversky VN. The triple power of D(3): protein intrinsic disorder in degenerative diseases. Front Biosci (Landmark Ed) 2014;19:181–258. doi: 10.2741/4204. [DOI] [PubMed] [Google Scholar]
- 66.Uversky VN. Targeting intrinsically disordered proteins in neurodegenerative and protein dysfunction diseases: another illustration of the D(2) concept. Expert Rev Proteom. 2010;7(4):543–564. doi: 10.1586/epr.10.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martinelli AHS, Lopes FC, John EBO, Carlini CR, Ligabue-Braun R. Modulation of disordered proteins with a focus on neurodegenerative diseases and other pathologies. Int J Mol Sci. 2019 doi: 10.3390/ijms20061322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Skrabana R, Skrabanova M, Csokova N, Sevcik J, Novak M. Intrinsically disordered tau protein in Alzheimer's tangles: a coincidence or a rule? Bratisl Lek Listy. 2006;107(9–10):354–358. [PubMed] [Google Scholar]
- 69.Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform. 2000;11:161–171. [PubMed] [Google Scholar]
- 70.Uversky VN. The mysterious unfoldome: structureless, underappreciated, yet vital part of any given proteome. J Biomed Biotechnol. 2010;2010:568068. doi: 10.1155/2010/568068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337(3):635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 72.Xue B, Dunker AK, Uversky VN. Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. J Biomol Struct Dyn. 2012;30(2):137–149. doi: 10.1080/07391102.2012.675145. [DOI] [PubMed] [Google Scholar]
- 73.Peng Z, Yan J, Fan X, Mizianty MJ, Xue B, Wang K, Hu G, Uversky VN, Kurgan L. Exceptionally abundant exceptions: comprehensive characterization of intrinsic disorder in all domains of life. Cell Mol Life Sci. 2015;72(1):137–151. doi: 10.1007/s00018-014-1661-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Singh A, Kumar A, Yadav R, Uversky VN, Giri R. Deciphering the dark proteome of Chikungunya virus. Sci Rep. 2018;8(1):5822. doi: 10.1038/s41598-018-23969-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Giri R, Kumar D, Sharma N, Uversky VN. Intrinsically disordered side of the Zika virus proteome. Front Cell Infect Microbiol. 2016;6:144. doi: 10.3389/fcimb.2016.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chouard T. Structural biology: breaking the protein rules. Nature. 2011;471(7337):151–153. doi: 10.1038/471151a. [DOI] [PubMed] [Google Scholar]
- 77.Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120(4):545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 78.Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012 doi: 10.1101/cshperspect.a006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rosenberg RN, Lambracht-Washington D, Yu G, Xia W. Genomics of Alzheimer disease: a review. JAMA Neurol. 2016;73(7):867–874. doi: 10.1001/jamaneurol.2016.0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O'Donovan C, Phan I, Pilbout S, Schneider M. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31(1):365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vucetic S, Obradovic Z, Vacic V, Radivojac P, Peng K, Iakoucheva LM, Cortese MS, Lawson JD, Brown CJ, Sikes JG, Newton CD, Dunker AK. DisProt: a database of protein disorder. Bioinformatics. 2005;21(1):137–140. doi: 10.1093/bioinformatics/bth476. [DOI] [PubMed] [Google Scholar]
- 82.Piovesan D, Tabaro F, Micetic I, Necci M, Quaglia F, Oldfield CJ, Aspromonte MC, Davey NE, Davidovic R, Dosztanyi Z, Elofsson A, Gasparini A, Hatos A, Kajava AV, Kalmar L, Leonardi E, Lazar T, Macedo-Ribeiro S, Macossay-Castillo M, Meszaros A, Minervini G, Murvai N, Pujols J, Roche DB, Salladini E, Schad E, Schramm A, Szabo B, Tantos A, Tonello F, Tsirigos KD, Veljkovic N, Ventura S, Vranken W, Warholm P, Uversky VN, Dunker AK, Longhi S, Tompa P, Tosatto SC. DisProt 7.0: a major update of the database of disordered proteins. Nucleic Acids Res. 2017;45(D1):D219–D227. doi: 10.1093/nar/gkw1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sickmeier M, Hamilton JA, LeGall T, Vacic V, Cortese MS, Tantos A, Szabo B, Tompa P, Chen J, Uversky VN, Obradovic Z, Dunker AK. DisProt: the database of disordered proteins. Nucleic Acids Res. 2007;35(Database issue):D786–D793. doi: 10.1093/nar/gkl893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Peng K, Radivojac P, Vucetic S, Dunker AK, Obradovic Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006;7:208. doi: 10.1186/1471-2105-7-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK, Obradovic Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. J Bioinform Comput Biol. 2005;3(1):35–60. doi: 10.1142/S0219720005000886. [DOI] [PubMed] [Google Scholar]
- 86.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42(1):38–48. doi: 10.1002/1097-0134(20010101)42:1<38::AID-PROT50>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 87.Xue B, Dunbrack RL, Williams RW, Dunker AK. Uversky VN (2010) PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim Biophys Acta. 1804;4:996–1010. doi: 10.1016/j.bbapap.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.He B, Wang K, Liu Y, Xue B, Uversky VN, Dunker AK. Predicting intrinsic disorder in proteins: an overview. Cell Res. 2009;19(8):929–949. doi: 10.1038/cr.2009.87. [DOI] [PubMed] [Google Scholar]
- 89.Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21(16):3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- 90.Piovesan D, Tabaro F, Paladin L, Necci M, Micetic I, Camilloni C, Davey N, Dosztanyi Z, Meszaros B, Monzon AM, Parisi G, Schad E, Sormanni P, Tompa P, Vendruscolo M, Vranken WF, Tosatto SCE. MobiDB 3.0: more annotations for intrinsic disorder, conformational diversity and interactions in proteins. Nucleic Acids Res. 2018;46(D1):D471–D476. doi: 10.1093/nar/gkx1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Uversky VN, Gillespie JR, Fink AL. Why are "natively unfolded" proteins unstructured under physiologic conditions? Proteins. 2000;41(3):415–427. doi: 10.1002/1097-0134(20001115)41:3<415::AID-PROT130>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 92.Oldfield CJ, Cheng Y, Cortese MS, Brown CJ, Uversky VN, Dunker AK. Comparing and combining predictors of mostly disordered proteins. Biochemistry. 2005;44(6):1989–2000. doi: 10.1021/bi047993o. [DOI] [PubMed] [Google Scholar]
- 93.Huang F, Oldfield CJ, Xue B, Hsu WL, Meng J, Liu X, Shen L, Romero P, Uversky VN, Dunker A. Improving protein order-disorder classification using charge-hydropathy plots. BMC Bioinform. 2014;15(Suppl 17):S4. doi: 10.1186/1471-2105-15-S17-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Huang F, Oldfield C, Meng J, Hsu WL, Xue B, Uversky VN, Romero P, Dunker AK (2012) Subclassifying disordered proteins by the CH-CDF plot method. Pac Symp Biocomput 128–139 [PubMed]
- 95.Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztanyi Z, Uversky VN, Obradovic Z, Kurgan L, Dunker AK, Gough J. D(2)P(2): database of disordered protein predictions. Nucleic Acids Res. 2013;41(Database issue):D508–D516. doi: 10.1093/nar/gks1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dosztanyi Z, Meszaros B, Simon I. ANCHOR: web server for predicting protein binding regions in disordered proteins. Bioinformatics. 2009;25(20):2745–2746. doi: 10.1093/bioinformatics/btp518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Mering CV. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gouw M, Samano-Sanchez H, Van Roey K, Diella F, Gibson TJ, Dinkel H. Exploring short linear motifs using the ELM database and tools. Curr Protoc Bioinform. 2017;58:8 22 21–28 22 35. doi: 10.1002/cpbi.26. [DOI] [PubMed] [Google Scholar]
- 99.Rajagopalan K, Mooney SM, Parekh N, Getzenberg RH, Kulkarni P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J Cell Biochem. 2011;112(11):3256–3267. doi: 10.1002/jcb.23252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mohan A, Sullivan WJ, Jr, Radivojac P, Dunker AK, Uversky VN. Intrinsic disorder in pathogenic and non-pathogenic microbes: discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol Biosyst. 2008;4(4):328–340. doi: 10.1039/b719168e. [DOI] [PubMed] [Google Scholar]
- 101.Xue B, Oldfield CJ, Dunker AK, Uversky VN. CDF it all: consensus prediction of intrinsically disordered proteins based on various cumulative distribution functions. FEBS Lett. 2009;583(9):1469–1474. doi: 10.1016/j.febslet.2009.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293(5534):1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 103.Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, Mucke L. beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer's disease and Parkinson's disease. Proc Natl Acad Sci USA. 2001;98(21):12245–12250. doi: 10.1073/pnas.211412398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300(5619):636–640. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- 105.Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic Interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci. 2010;30(21):7281–7289. doi: 10.1523/JNEUROSCI.0490-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zou WQ, Xiao X, Yuan J, Puoti G, Fujioka H, Wang X, Richardson S, Zhou X, Zou R, Li S, Zhu X, McGeer PL, McGeehan J, Kneale G, Rincon-Limas DE, Fernandez-Funez P, Lee HG, Smith MA, Petersen RB, Guo JP. Amyloid-beta42 interacts mainly with insoluble prion protein in the Alzheimer brain. J Biol Chem. 2011;286(17):15095–15105. doi: 10.1074/jbc.M110.199356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Luo J, Warmlander SK, Graslund A, Abrahams JP. Cross-interactions between the Alzheimer disease amyloid-beta peptide and other amyloid proteins: a further aspect of the amyloid cascade hypothesis. J Biol Chem. 2016;291(32):16485–16493. doi: 10.1074/jbc.R116.714576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.van der Kant R, Goldstein LS. Cellular functions of the amyloid precursor protein from development to dementia. Dev Cell. 2015;32(4):502–515. doi: 10.1016/j.devcel.2015.01.022. [DOI] [PubMed] [Google Scholar]
- 109.Botelho MG, Gralle M, Oliveira CL, Torriani I, Ferreira ST. Folding and stability of the extracellular domain of the human amyloid precursor protein. J Biol Chem. 2003;278(36):34259–34267. doi: 10.1074/jbc.M303189200. [DOI] [PubMed] [Google Scholar]
- 110.Kim HS, Park CH, Cha SH, Lee JH, Lee S, Kim Y, Rah JC, Jeong SJ, Suh YH. Carboxyl-terminal fragment of Alzheimer's APP destabilizes calcium homeostasis and renders neuronal cells vulnerable to excitotoxicity. FASEB J. 2000;14(11):1508–1517. doi: 10.1096/fj.14.11.1508. [DOI] [PubMed] [Google Scholar]
- 111.Gorman PM, Kim S, Guo M, Melnyk RA, McLaurin J, Fraser PE, Bowie JU, Chakrabartty A. Dimerization of the transmembrane domain of amyloid precursor proteins and familial Alzheimer's disease mutants. BMC Neurosci. 2008;9:17. doi: 10.1186/1471-2202-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mok SS, Sberna G, Heffernan D, Cappai R, Galatis D, Clarris HJ, Sawyer WH, Beyreuther K, Masters CL, Small DH. Expression and analysis of heparin-binding regions of the amyloid precursor protein of Alzheimer's disease. FEBS Lett. 1997;415(3):303–307. doi: 10.1016/s0014-5793(97)01146-0. [DOI] [PubMed] [Google Scholar]
- 113.Gooz M. ADAM-17: the enzyme that does it all. Crit Rev Biochem Mol Biol. 2010;45(2):146–169. doi: 10.3109/10409231003628015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ingram RN, Orth P, Strickland CL, Le HV, Madison V, Beyer BM. Stabilization of the autoproteolysis of TNF-alpha converting enzyme (TACE) results in a novel crystal form suitable for structure-based drug design studies. Protein Eng Des Sel. 2006;19(4):155–161. doi: 10.1093/protein/gzj014. [DOI] [PubMed] [Google Scholar]
- 115.Qian M, Shen X, Wang H. The distinct role of ADAM17 in APP proteolysis and microglial activation related to Alzheimer's disease. Cell Mol Neurobiol. 2016;36(4):471–482. doi: 10.1007/s10571-015-0232-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Togashi N, Ura N, Higashiura K, Murakami H, Shimamoto K. Effect of TNF-alpha-converting enzyme inhibitor on insulin resistance in fructose-fed rats. Hypertension. 2002;39(2 Pt 2):578–580. doi: 10.1161/hy0202.103290. [DOI] [PubMed] [Google Scholar]
- 117.Richards WG, Sweeney WE, Yoder BK, Wilkinson JE, Woychik RP, Avner ED. Epidermal growth factor receptor activity mediates renal cyst formation in polycystic kidney disease. J Clin Investig. 1998;101(5):935–939. doi: 10.1172/JCI2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dusterhoft S, Hobel K, Oldefest M, Lokau J, Waetzig GH, Chalaris A, Garbers C, Scheller J, Rose-John S, Lorenzen I, Grotzinger J. A disintegrin and metalloprotease 17 dynamic interaction sequence, the sweet tooth for the human interleukin 6 receptor. J Biol Chem. 2014;289(23):16336–16348. doi: 10.1074/jbc.M114.557322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Johansson P, Kaspersson K, Gurrell IK, Back E, Eketjall S, Scott CW, Cebers G, Thorne P, McKenzie MJ, Beaton H, Davey P, Kolmodin K, Holenz J, Duggan ME, Budd Haeberlein S, Burli RW. Toward beta-secretase-1 inhibitors with improved isoform selectivity. J Med Chem. 2018;61(8):3491–3502. doi: 10.1021/acs.jmedchem.7b01716. [DOI] [PubMed] [Google Scholar]
- 120.Das B, Yan R. Role of BACE1 in Alzheimer's synaptic function. Transl Neurodegener. 2017;6:23. doi: 10.1186/s40035-017-0093-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shimizu H, Tosaki A, Kaneko K, Hisano T, Sakurai T, Nukina N. Crystal structure of an active form of BACE1, an enzyme responsible for amyloid beta protein production. Mol Cell Biol. 2008;28(11):3663–3671. doi: 10.1128/MCB.02185-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hu X, Das B, Hou H, He W, Yan R. BACE1 deletion in the adult mouse reverses preformed amyloid deposition and improves cognitive functions. J Exp Med. 2018;215(3):927–940. doi: 10.1084/jem.20171831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.De Simone A, Mancini F, Real Fernandez F, Rovero P, Bertucci C, Andrisano V. Surface plasmon resonance, fluorescence, and circular dichroism studies for the characterization of the binding of BACE-1 inhibitors. Anal Bioanal Chem. 2013;405(2–3):827–835. doi: 10.1007/s00216-012-6312-0. [DOI] [PubMed] [Google Scholar]
- 124.Checler F, Goiran T, Alves da Costa C. Presenilins at the crossroad of a functional interplay between PARK2/PARKIN and PINK1 to control mitophagy: implication for neurodegenerative diseases. Autophagy. 2017;13(11):2004–2005. doi: 10.1080/15548627.2017.1363950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pardeshi R, Bolshette N, Gadhave K, Arfeen M, Ahmed S, Jamwal R, Hammock BD, Lahkar M, Goswami SK. Docosahexaenoic acid increases the potency of soluble epoxide hydrolase inhibitor in alleviating streptozotocin-induced Alzheimer's disease-like complications of diabetes. Front Pharmacol. 2019;10:288. doi: 10.3389/fphar.2019.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhou R, Yang G, Guo X, Zhou Q, Lei J, Shi Y. Recognition of the amyloid precursor protein by human gamma-secretase. Science. 2019 doi: 10.1126/science.aaw0930. [DOI] [PubMed] [Google Scholar]
- 127.Kelleher RJ, 3rd, Shen J. Presenilin-1 mutations and Alzheimer's disease. Proc Natl Acad Sci USA. 2017;114(4):629–631. doi: 10.1073/pnas.1619574114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sun L, Zhou R, Yang G, Shi Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Abeta42 and Abeta40 peptides by gamma-secretase. Proc Natl Acad Sci USA. 2017;114(4):E476–E485. doi: 10.1073/pnas.1618657114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yang G, Yu K, Kaitatzi CS, Singh A, Labahn J. Influence of solubilization and AD-mutations on stability and structure of human presenilins. Sci Rep. 2017;7(1):17970. doi: 10.1038/s41598-017-18313-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cai Y, An SS, Kim S. Mutations in presenilin 2 and its implications in Alzheimer's disease and other dementia-associated disorders. Clin Interv Aging. 2015;10:1163–1172. doi: 10.2147/CIA.S85808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Vito P, Wolozin B, Ganjei JK, Iwasaki K, Lacana E, D'Adamio L. Requirement of the familial Alzheimer's disease gene PS2 for apoptosis. Opposing effect of ALG-3. J Biol Chem. 1996;271(49):31025–31028. doi: 10.1074/jbc.271.49.31025. [DOI] [PubMed] [Google Scholar]
- 132.Wolozin B, Iwasaki K, Vito P, Ganjei JK, Lacana E, Sunderland T, Zhao B, Kusiak JW, Wasco W, D'Adamio L. Participation of presenilin 2 in apoptosis: enhanced basal activity conferred by an Alzheimer mutation. Science. 1996;274(5293):1710–1713. doi: 10.1126/science.274.5293.1710. [DOI] [PubMed] [Google Scholar]
- 133.De Strooper B. Nicastrin: gatekeeper of the gamma-secretase complex. Cell. 2005;122(3):318–320. doi: 10.1016/j.cell.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 134.Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CE, 3rd, Sudhof T, Yu G. Nicastrin functions as a gamma-secretase-substrate receptor. Cell. 2005;122(3):435–447. doi: 10.1016/j.cell.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 135.Xie T, Yan C, Zhou R, Zhao Y, Sun L, Yang G, Lu P, Ma D, Shi Y. Crystal structure of the gamma-secretase component nicastrin. Proc Natl Acad Sci USA. 2014;111(37):13349–13354. doi: 10.1073/pnas.1414837111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yu K, Yang G, Labahn J. High-efficient production and biophysical characterisation of nicastrin and its interaction with APPC100. Sci Rep. 2017;7:44297. doi: 10.1038/srep44297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pardossi-Piquard R, Yang SP, Kanemoto S, Gu Y, Chen F, Bohm C, Sevalle J, Li T, Wong PC, Checler F, Schmitt-Ulms G, St George-Hyslop P, Fraser PE. APH1 polar transmembrane residues regulate the assembly and activity of presenilin complexes. J Biol Chem. 2009;284(24):16298–16307. doi: 10.1074/jbc.M109.000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Du M, Fan X, Hanada T, Gao H, Lutchman M, Brandsma JL, Chishti AH, Chen JJ. Association of cottontail rabbit papillomavirus E6 oncoproteins with the hDlg/SAP97 tumor suppressor. J Cell Biochem. 2005;94(5):1038–1045. doi: 10.1002/jcb.20383. [DOI] [PubMed] [Google Scholar]
- 139.Serneels L, Van Biervliet J, Craessaerts K, Dejaegere T, Horre K, Van Houtvin T, Esselmann H, Paul S, Schafer MK, Berezovska O, Hyman BT, Sprangers B, Sciot R, Moons L, Jucker M, Yang Z, May PC, Karran E, Wiltfang J, D'Hooge R, De Strooper B. gamma-Secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer's disease. Science. 2009;324(5927):639–642. doi: 10.1126/science.1171176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bammens L, Chavez-Gutierrez L, Tolia A, Zwijsen A, De Strooper B. Functional and topological analysis of Pen-2, the fourth subunit of the gamma-secretase complex. J Biol Chem. 2011;286(14):12271–12282. doi: 10.1074/jbc.M110.216978. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 141.Ahn K, Shelton CC, Tian Y, Zhang X, Gilchrist ML, Sisodia SS, Li YM. Activation and intrinsic gamma-secretase activity of presenilin 1. Proc Natl Acad Sci USA. 2010;107(50):21435–21440. doi: 10.1073/pnas.1013246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kanekiyo T, Xu H, Bu G. ApoE and Abeta in Alzheimer's disease: accidental encounters or partners? Neuron. 2014;81(4):740–754. doi: 10.1016/j.neuron.2014.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chen J, Li Q, Wang J. Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc Natl Acad Sci USA. 2011;108(36):14813–14818. doi: 10.1073/pnas.1106420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yu JT, Tan L, Hardy J. Apolipoprotein E in Alzheimer's disease: an update. Annu Rev Neurosci. 2014;37:79–100. doi: 10.1146/annurev-neuro-071013-014300. [DOI] [PubMed] [Google Scholar]
- 145.Hatters DM, Zhong N, Rutenber E, Weisgraber KH. Amino-terminal domain stability mediates apolipoprotein E aggregation into neurotoxic fibrils. J Mol Biol. 2006;361(5):932–944. doi: 10.1016/j.jmb.2006.06.080. [DOI] [PubMed] [Google Scholar]
- 146.Liu CC, Zhao N, Fu Y, Wang N, Linares C, Tsai CW, Bu G. ApoE4 accelerates early seeding of amyloid pathology. Neuron. 2017;96(5):1024 e1023–1032 e1023. doi: 10.1016/j.neuron.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tsiolaki PL, Katsafana AD, Baltoumas FA, Louros NN, Iconomidou VA. Hidden aggregation hot-spots on human apolipoprotein E: a structural study. Int J Mol Sci. 2019 doi: 10.3390/ijms20092274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Raulin AC, Kraft L, Al-Hilaly YK, Xue WF, McGeehan JE, Atack JR, Serpell L. The molecular basis for apolipoprotein E4 as the major risk factor for late-onset Alzheimer's disease. J Mol Biol. 2019;431(12):2248–2265. doi: 10.1016/j.jmb.2019.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pande AH, Tripathy RK, Nankar SA. Membrane surface charge modulates lipoprotein complex forming capability of peptides derived from the C-terminal domain of apolipoprotein E. Biochim Biophys Acta. 2009;1788(6):1366–1376. doi: 10.1016/j.bbamem.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 150.Tan MS, Yu JT, Tan L. Bridging integrator 1 (BIN1): form, function, and Alzheimer's disease. Trends Mol Med. 2013;19(10):594–603. doi: 10.1016/j.molmed.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 151.Casal E, Federici L, Zhang W, Fernandez-Recio J, Priego EM, Miguel RN, DuHadaway JB, Prendergast GC, Luisi BF, Laue ED. The crystal structure of the BAR domain from human Bin1/amphiphysin II and its implications for molecular recognition. Biochemistry. 2006;45(43):12917–12928. doi: 10.1021/bi060717k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Pineda-Lucena A, Ho CS, Mao DY, Sheng Y, Laister RC, Muhandiram R, Lu Y, Seet BT, Katz S, Szyperski T, Penn LZ, Arrowsmith CH. A structure-based model of the c-Myc/Bin1 protein interaction shows alternative splicing of Bin1 and c-Myc phosphorylation are key binding determinants. J Mol Biol. 2005;351(1):182–194. doi: 10.1016/j.jmb.2005.05.046. [DOI] [PubMed] [Google Scholar]
- 153.Chapuis J, Hansmannel F, Gistelinck M, Mounier A, Van Cauwenberghe C, Kolen KV, Geller F, Sottejeau Y, Harold D, Dourlen P, Grenier-Boley B, Kamatani Y, Delepine B, Demiautte F, Zelenika D, Zommer N, Hamdane M, Bellenguez C, Dartigues JF, Hauw JJ, Letronne F, Ayral AM, Sleegers K, Schellens A, Broeck LV, Engelborghs S, De Deyn PP, Vandenberghe R, O'Donovan M, Owen M, Epelbaum J, Mercken M, Karran E, Bantscheff M, Drewes G, Joberty G, Campion D, Octave JN, Berr C, Lathrop M, Callaerts P, Mann D, Williams J, Buee L, Dewachter I, Van Broeckhoven C, Amouyel P, Moechars D, Dermaut B, Lambert JC, Consortium G. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry. 2013;18(11):1225–1234. doi: 10.1038/mp.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Beeg M, Stravalaci M, Romeo M, Carra AD, Cagnotto A, Rossi A, Diomede L, Salmona M, Gobbi M. Clusterin binds to Abeta1-42 oligomers with high affinity and interferes with peptide aggregation by inhibiting primary and secondary nucleation. J Biol Chem. 2016;291(13):6958–6966. doi: 10.1074/jbc.M115.689539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Matukumalli SR, Tangirala R, Rao CM. Clusterin: full-length protein and one of its chains show opposing effects on cellular lipid accumulation. Sci Rep. 2017;7:41235. doi: 10.1038/srep41235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Baig S, Joseph SA, Tayler H, Abraham R, Owen MJ, Williams J, Kehoe PG, Love S. Distribution and expression of picalm in Alzheimer disease. J Neuropathol Exp Neurol. 2010;69(10):1071–1077. doi: 10.1097/NEN.0b013e3181f52e01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, Heun R, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O'Donovan M, Owen MJ, Williams J. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41(10):1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Xiao Q, Gil SC, Yan P, Wang Y, Han S, Gonzales E, Perez R, Cirrito JR, Lee JM. Role of phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia (PICALM) in intracellular amyloid precursor protein (APP) processing and amyloid plaque pathogenesis. J Biol Chem. 2012;287(25):21279–21289. doi: 10.1074/jbc.M111.338376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78(4):631–643. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kitago Y, Nagae M, Nakata Z, Yagi-Utsumi M, Takagi-Niidome S, Mihara E, Nogi T, Kato K, Takagi J. Structural basis for amyloidogenic peptide recognition by sorLA. Nat Struct Mol Biol. 2015;22(3):199–206. doi: 10.1038/nsmb.2954. [DOI] [PubMed] [Google Scholar]
- 161.Cramer JF, Gustafsen C, Behrens MA, Oliveira CL, Pedersen JS, Madsen P, Petersen CM, Thirup SS. GGA autoinhibition revisited. Traffic. 2010;11(2):259–273. doi: 10.1111/j.1600-0854.2009.01017.x. [DOI] [PubMed] [Google Scholar]
- 162.Baker SK, Chen ZL, Norris EH, Revenko AS, MacLeod AR, Strickland S. Blood-derived plasminogen drives brain inflammation and plaque deposition in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2018;115(41):E9687–E9696. doi: 10.1073/pnas.1811172115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Law RH, Caradoc-Davies T, Cowieson N, Horvath AJ, Quek AJ, Encarnacao JA, Steer D, Cowan A, Zhang Q, Lu BG, Pike RN, Smith AI, Coughlin PB, Whisstock JC. The X-ray crystal structure of full-length human plasminogen. Cell Rep. 2012;1(3):185–190. doi: 10.1016/j.celrep.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 164.Shaw MA, Gao Z, McElhinney KE, Thornton S, Flick MJ, Lane A, Degen JL, Ryu JK, Akassoglou K, Mullins ES. Plasminogen deficiency delays the onset and protects from demyelination and paralysis in autoimmune neuroinflammatory disease. J Neurosci. 2017;37(14):3776–3788. doi: 10.1523/JNEUROSCI.2932-15.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cook AD, De Nardo CM, Braine EL, Turner AL, Vlahos R, Way KJ, Beckman SK, Lenzo JC, Hamilton JA. Urokinase-type plasminogen activator and arthritis progression: role in systemic disease with immune complex involvement. Arthritis Res Ther. 2010;12(2):R37. doi: 10.1186/ar2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Raghu H, Jone A, Cruz C, Rewerts CL, Frederick MD, Thornton S, Degen JL, Flick MJ. Plasminogen is a joint-specific positive or negative determinant of arthritis pathogenesis in mice. Arthritis Rheumatol. 2014;66(6):1504–1516. doi: 10.1002/art.38402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Uversky VN. p53 Proteoforms and intrinsic disorder: an illustration of the protein structure-function continuum concept. Int J Mol Sci. 2016;17(11):1874. doi: 10.3390/ijms17111874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Smith LM, Kelleher NL, Consortium for Top Down P Proteoform: a single term describing protein complexity. Nat Methods. 2013;10(3):186–187. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Uversky VN. Protein intrinsic disorder and structure-function continuum. Prog Mol Biol Transl Sci. 2019;166:1–17. doi: 10.1016/bs.pmbts.2019.05.003. [DOI] [PubMed] [Google Scholar]
- 170.Fonin AV, Darling AL, Kuznetsova IM, Turoverov KK, Uversky VN. Multi-functionality of proteins involved in GPCR and G protein signaling: making sense of structure-function continuum with intrinsic disorder-based proteoforms. Cell Mol Life Sci. 2019 doi: 10.1007/s00018-019-03276-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Uversky VN. p53 Proteoforms and intrinsic disorder: an illustration of the protein structure-function continuum concept. Int J Mol Sci. 2016 doi: 10.3390/ijms17111874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Midic U, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Unfoldomics of human genetic diseases: illustrative examples of ordered and intrinsically disordered members of the human diseasome. Protein Pept Lett. 2009;16(12):1533–1547. doi: 10.2174/092986609789839377. [DOI] [PubMed] [Google Scholar]
- 173.Uversky VN. Intrinsic disorder in proteins associated with neurodegenerative diseases. Front Biosci. 2009;14:5188–5238. doi: 10.2741/3594. [DOI] [PubMed] [Google Scholar]
- 174.Uversky VN, Oldfield CJ, Midic U, Xie H, Xue B, Vucetic S, Iakoucheva LM, Obradovic Z, Dunker AK. Unfoldomics of human diseases: linking protein intrinsic disorder with diseases. BMC Genom. 2009;10(Suppl 1):S7. doi: 10.1186/1471-2164-10-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Uversky VN. Wrecked regulation of intrinsically disordered proteins in diseases: pathogenicity of deregulated regulators. Front Mol Biosci. 2014;1:6. doi: 10.3389/fmolb.2014.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.