

Abstract
Hereditary fructose intolerance (HFI) is a rare inborn disease characterized by a deficiency in aldolase B, which catalyzes the cleavage of fructose 1,6-bisphosphate and fructose 1-phosphate (Fru 1P) to triose molecules. In patients with HFI, ingestion of fructose results in accumulation of Fru 1P and depletion of ATP, which are believed to cause symptoms, such as nausea, vomiting, hypoglycemia, and liver and kidney failure. These sequelae can be prevented by a fructose-restricted diet. Recent studies in aldolase B-deficient mice and HFI patients have provided more insight into the pathogenesis of HFI, in particular the liver phenotype. Both aldolase B-deficient mice (fed a very low fructose diet) and HFI patients (treated with a fructose-restricted diet) displayed greater intrahepatic fat content when compared to controls. The liver phenotype in aldolase B-deficient mice was prevented by reduction in intrahepatic Fru 1P concentrations by crossing these mice with mice deficient for ketohexokinase, the enzyme that catalyzes the synthesis of Fru 1P. These new findings not only provide a potential novel treatment for HFI, but lend insight into the pathogenesis of fructose-induced non-alcoholic fatty liver disease (NAFLD), which has raised to epidemic proportions in Western society. This narrative review summarizes the most recent advances in the pathogenesis of HFI and discusses the implications for the understanding and treatment of fructose-induced NAFLD.
Keywords: Hereditary fructose intolerance, Glucokinase regulatory protein, Ketohexokinase, Fructose, De novo lipogenesis, Non-alcoholic fatty liver disease
Introduction
Hereditary fructose intolerance (HFI; OMIM 22960), an inborn error of fructose metabolism, was first reported in 1956 by Chambers and Pratt [1]. A 24-year-old woman was admitted for evaluation of faintness, abdominal pain, and nausea upon fruit or sugar ingestion. The physicians subjected her to systematic, single-blinded exposure to a variety of oral sugars. Administration of solely fructose and sucrose, not glucose, galactose or lactose, provoked symptoms of nausea in a dose-dependent manner. Based on these findings, the patient was diagnosed with ‘idiosyncrasy to fructose’ [1]. Six years later, Hers and Joassin identified the enzymatic defect of HFI in two liver biopsy specimens as a ‘functional deficiency of fructose-1-aldolase activity,’ i.e., aldolase B [2].
Recent experimental and clinical studies have provided more insight into the pathogenesis of HFI, in particular its liver phenotype. In the present narrative review, we will give an overview of these studies and subsequently elaborate on the implications, not only for the treatment of HFI, but also for the current epidemic of fructose overconsumption.
Background
Clinical manifestations
The first symptoms of HFI appear when a neonate is exposed to fructose-containing infant formulas [3] or when fructose-containing foods, such as fruits and vegetables, are introduced to young infants [4, 5]. Signs of acute intoxication are vomiting, abdominal pain, lactic acidosis, hyperuricemia, hypoglycemia, and acute liver failure. Persistent fructose ingestion can lead to failure to thrive, liver disease (i.e., hepatic steatosis, fibrosis, and cirrhosis), signs of proximal renal tubular dysfunction (i.e., Fanconi syndrome), and eventually death. These sequelae can be prevented when treated with a fructose-restricted diet. Further, since fructose can also be synthesized endogenously from sorbitol (via the polyol pathway, Fig. 1), HFI patients additionally should avoid sorbitol-containing food products and high levels of high-glycemic foods [4, 5]. When adhering to these dietary restrictions, the prognosis of HFI appears excellent, although little is known about the long-term pathology of adults with HFI [6–9].
Fig. 1.
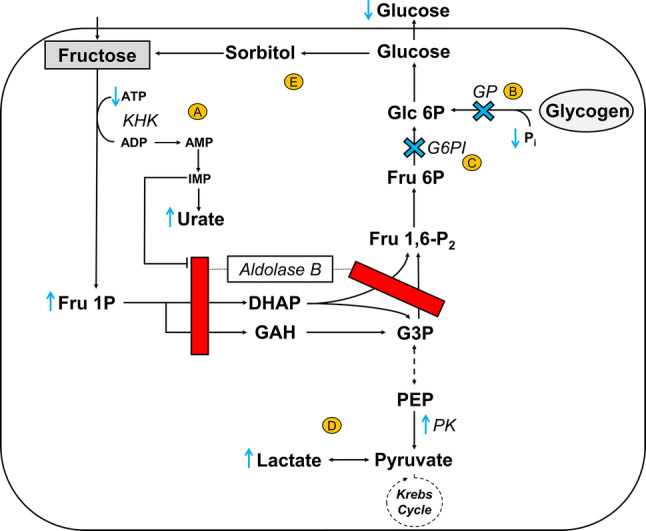
Metabolic consequences of aldolase B deficiency in the liver after an oral fructose load. In physiological states, fructose is rapidly phosphorylated by KHK and subsequently converted by aldolase B to trioses (DHAP and GAH) that enter the glycolytic/gluconeogenic pathways. Aldolase B also catalyzes the conversion of Fru 1,6-P2 to triose phosphates (DHAP and G3P). In aldolase B deficiency, the catabolism of Fru 1P is impaired, and the metabolism of Fru 1,6-P2 is blocked (red bar). Accumulation of Fru 1P has several acute downstream effects denoted in yellow circled letters as follows: (1) depletion of intracellular inorganic phosphate (Pi) and ATP, and consequently formation of IMP and urate (A); (2) impairment of glycogenolysis (by inhibition of GP and loss of Pi) (B) and gluconeogenesis (by inhibition of G6PI) (C), resulting in hypoglycemia; and (3) stimulation of PK activity that—in combination with an impaired gluconeogenesis—promotes hyperlactatemia (D). Further, fructose, which can be produced endogenously from sorbitol (via the polyol pathway), may contribute to the accumulation of Fru 1P (E). Blue cross indicates blocked pathway as a consequence of Fru 1P accumulation. Dashed arrow indicates multiple intermediate enzymatic steps that have not been visualized for simplicity purposes. ADP adenosine diphosphate, AMP adenosine monophosphate, ATP adenosine triphosphate, DHAP dihydroxyacetone phosphate, Fru 6P fructose 6-phosphate, Fru 1P fructose 1-phosphate, Fru 1,6-P2 fructose 1,6-biphosphate, G3P glyceraldehyde 3-phosphate, Glc 6P glucose 6-phosphate, G6PI glucose-6-phosphate isomerase, GAH glyceraldehyde, GP glycogen phosphorylase, IMP inosine monophosphate, KHK ketohexokinase, PEP phosphoenolpyruvate, Pi inorganic phosphate, PK pyruvate kinase
Genetics and epidemiology
The human gene for aldolase B (ALDOB) has been mapped to chromosome 9q22.3 [10, 11]. At present, over 40 causative mutations of the ALDOB gene have been documented, of which c.448G > C (p.A149P), c.524C > A (p.A174D), c.357delAAAC (∆4E4), and c.1005C > G (p.N334 K) account for 59% and 86% of HFI mutations in North Americans and Europeans, respectively [12–17]. Based on the carrier frequency of the most common mutations in neonates, it has been estimated that the incidence of HFI is 1:18,000–20,000 in live births [18, 19].
Metabolic derangements
The metabolic derangements of aldolase B deficiency have been the scope of previous, high-quality review papers [9, 20, 21]. Briefly, fructose-1,6-bisphosphate aldolase (aldolase; EC 4.1.2.13) is responsible for the reversible conversion of fructose 1,6-bisphosphate (Fru 1,6-P2) or fructose 1-phosphate (Fru 1P) to the triose phosphate dihydroxyacetone phosphate (DHAP) and either glyceraldehyde 3-phosphate (G3P) or glyceraldehyde, respectively, which are intermediates of the glycolytic/gluconeogenic pathway (Fig. 1) [22]. At least three aldolase isozymes (A, B, and C) have been described which differ in tissue expression and activity for the substrates Fru 1,6-P2 and Fru 1P. Aldolase B is expressed in the liver, kidney, and small intestine and has activity for both Fru 1,6-P2 and Fru 1P. This is in contrast to both aldolase A (predominantly expressed in skeletal muscle) and aldolase C (predominantly expressed in brain and smooth muscle) which have the highest efficiencies for Fru 1,6-P2 as a substrate [23, 24], although aldolase C may perform fructose metabolism in the brain [25].
Liver biopsies of HFI patients show substantially reduced Fru 1P aldolase activity (0–15%), but preserved Fru 1,6-P2 aldolase activity (5–30%) leading to a marked increase in the ratio of Fru 1,6-P2 to Fru 1P activities, which was used as a diagnostic before the introduction of genetic testing [26]. This remains the only definitive diagnostic test as so many HFI-causing mutations remain unknown or variants found by DNA testing have unknown consequences [15]. The relatively preserved Fru 1,6-P2 aldolase activity could theoretically be explained by residual aldolase A activity in the liver that compensates for the defect in aldolase B activity for the substrate Fru 1,6-P2, but not for Fru 1P [20], or, alternatively, aldolase A activity in erythrocytes, which are also present in liver lysates.
As a consequence of the catalytic deficiency of aldolase B, a fructose load in HFI patients results in the rapid accumulation of Fru 1P and, hence, intracellular inorganic phosphate (Pi) and adenosine triphosphate (ATP) depletion [27, 28]. Reduced Pi concentrations lead to an increased rate of degradation of adenosine 5'-monophosphate (AMP) [29]. As a result, adenosine deaminase and xanthine oxidase activities are increased and inosine monophosphate (IMP) and urate are rapidly formed (Fig. 1) [29]. The specific inhibition of aldolase B by the increased IMP further accentuates the increase in Fru 1P [28].
High levels of intrahepatic Fru 1P—in combination with the loss of Pi—inhibit glycogenolysis by impairment of glycogen phosphorylase (GP) [30–33]. This is also illustrated by the failure of exogenous glucagon to correct for the fructose-induced hypoglycemia in HFI patients [34, 35]. Further, high levels of Fru 1P impair gluconeogenesis by competitive inhibition of glucose-6-phosphate isomerase (G6PI) [36, 37]. The rate of gluconeogenesis may also depend on the intrahepatic concentration of ATP [38], which is low in case of HFI following fructose ingestion. The impaired gluconeogenesis is evidenced by the inability of dihydroxyacetone administration (which enters the gluconeogenic pathway) to prevent fructose-induced hypoglycemia in HFI patients [35]. In conclusion, fructose-induced, impaired glycogenolysis and gluconeogenesis both result in a decreased hepatic glucose production and, consequently, the rapid development of hypoglycemia. Of note, in the absence of fructose, gluconeogenesis is not impaired in HFI [39].
In addition, an impaired gluconeogenesis together with Fru-1P-induced activation of pyruvate kinase (PK) promotes accumulation of lactate and, consequently, hyperlactatemia [40, 41] (Fig. 1). Notably, these metabolic defects do not only occur after oral intake of fructose, but also upon sorbitol consumption [4, 42]. This is due to the oxidation of sorbitol to fructose via the polyol pathway (Fig. 1). This pathway of endogenous fructose production can be activated through dehydration and hyperosmolarity as well as high-glycemic foods [43–46].
Recent advances from animal studies
The phenotype of aldolase B knockout mice resembles the human HFI phenotype
Recent work has demonstrated that aldolase B knockout (ALDOB-KO) mice exhibit similar metabolic features as HFI patients [47, 48]. In these mice, chronic exposure to fructose resulted in growth retardation and death [47, 48]. An acute, oral fructose load caused a rise in serum liver enzymes and intestinal injury, characterized by the destruction of apical villi and the presence of apoptotic cells in the duodenum and jejunum [48]. In addition, ALDOB-KO mice exposed to an oral fructose load showed decreased hepatic ATP and phosphate levels, and elevated serum urate concentrations [48]. Finally, oral fructose provoked severe hypoglycemia in a dose-dependent fashion [48]. Exploration of the gluconeogenic pathway by a pyruvate tolerance test revealed a reduced, but not absent ability for gluconeogenesis [48]. This is remarkable given the absence of aldolase B, which not only affects fructolysis but also glycolysis/gluconeogenesis (Fig. 1). Furthermore, there was no residual aldolase A or C expression in the liver (Lanaspa, personal communication) and suggests that gluconeogenesis occurs in other tissues [49]. Some key enzymes of gluconeogenesis (i.e., phosphoenolpyruvate carboxykinase and glucose-6-phosphatase) were found to be upregulated in the livers of ALDOB-KO mice [48].
Although Fru-1P-mediated impairment of glycogenolysis was not specifically studied, the ALDOB-KO mice were characterized by an increased hepatic glycogen content after an oral fructose load [48]. Of interest, glycogen synthase activity—determined by the ratio of phosphorylated to total glycogen synthase—was increased [48], suggesting an enhanced glycogenesis. Of additional interest, the increased hepatic glycogen content and decreased serum glucose and insulin were also observed in ALDOB-KO mice that were not exposed to an acute oral fructose load [48]. This chronic feature could be due to the endogenous fructose production via the polyol pathway [4, 42] or, alternatively, an increased hepatic glucose uptake (see below).
Aldolase B knockout mice are characterized by an increased intrahepatic triglyceride content
In addition to the above-described metabolic features, ALDOB-KO mice chronically exposed to small amounts of fructose in the chow (~ 0.3%) displayed an increased amount of hepatic triglycerides, hepatic inflammation—characterized by the presence of apoptotic and necrotic cells, and diffuse macrophage infiltration—and signs of periportal fibrosis [47, 48]. Hepatic expression of enzymes involved in de novo lipogenesis (DNL), i.e., ATP-citrate lyase (ACL), acetyl-CoA carboxylase (ACC), fatty acid synthase (FAS), was greater in ALDOB-KO mice, suggesting that this pathway accounts, at least in part, for the increased hepatic triglycerides levels [48]. In addition, cytosolic glucokinase (GCK) was more abundant in ALDOB-KO mice when compared to wild-type mice [48].
GCK converts glucose to glucose 6-phosphate (Glc 6P) in the liver, pancreas, and pituitary and is the first step in glycolysis. Thanks to its unique kinetic properties, GCK is a major regulator of hepatic glucose uptake and pancreatic insulin secretion [50]. In the post-absorptive state, hepatic GCK is bound to glucokinase regulatory protein (GKRP), a liver-specific protein. The GKRP-GCK complex resides in the nucleus and thus inactivates GCK [51, 52]. In the postprandial state, a rise in intracellular glucose facilitates the dissociation of GCK from GKRP and migration of GCK to the cytosolic space where it facilitates phosphorylation and, hence, storage of glucose. Of interest, Fru 1P is a very potent disruptor of the GKRP-GCK complex. Experimental studies have shown that only trace amounts of Fru 1P are required to dissociate GCK from GKRP [52–57]. Notably, intrahepatic Fru 1P concentrations in ALDOB-KO mice were also elevated after chronic exposure to only small amounts of fructose in the chow [48]. From these studies, it can be speculated that accumulation of Fru 1P in ALDOB-KO mice chronically fed small amounts of fructose induces dissociation of the GKRP-GCK complex, which would explain the greater cytosolic GCK activity in ALDOB-KO mice. Consequently, hepatic glucose uptake is stimulated, thereby contributing to the reduced serum glucose and insulin levels in these mice. The metabolic fate of the glucose taken up by the liver can be several fold, among others an enhanced storage of glycogen and fat. Although the latter requires glycolysis (which appears to be blocked in case of aldolase B deficiency) and subsequent DNL, the pentose phosphate pathway (PPP)—a metabolic pathway that parallels glycolysis—may serve as an alternative pathway to convert Glc 6P to G3P (Fig. 2). Of interest, a previous experimental study has shown that the PPP increases in parallel to DNL in rat fatty livers [58].
Fig. 2.
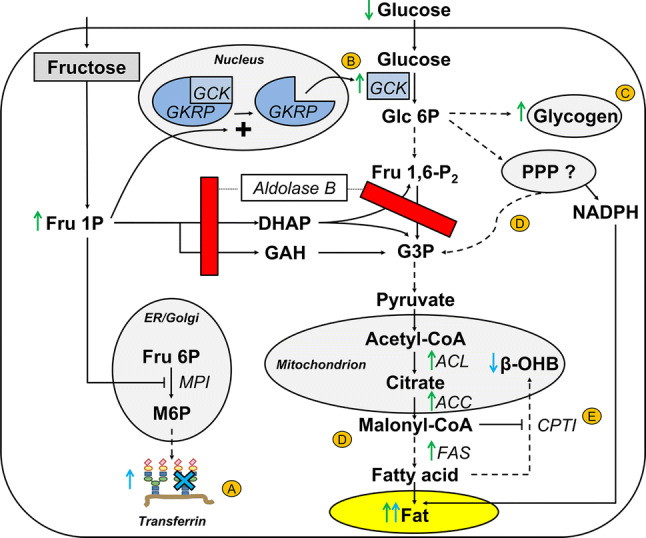
Hypothesized pathogenesis of hepatic fat accumulation in aldolase B deficiency. Accumulation of Fru 1P has several chronic downstream effects leading to fat accumulation denoted in yellow circled letters. ALDOB-KO mice fed a low-fructose diet (~ 0.3%) display increased hepatic Fru 1P concentrations. This also seems to be the case in adult HFI patients treated with a fructose-restricted diet, as can be deduced from an abundancy of circulating hypoglycosylated transferrin. Hepatic Fru 1P inhibits glycosylation of transferrin by impairment of MPI (A). Catalytic amounts of Fru 1P dissociate GCK from GKRP in the nucleus, which allows migration of GCK toward the cytosolic space where it converts glucose to Glc 6P and, as a consequence, facilitates hepatic glucose uptake (B). The metabolic fates of an increased hepatic glucose uptake can be: (1) storage as glycogen (C) and (2) storage as fat via DNL with carbons and electrons derived from possibly the pentose phosphate pathway (PPP) (D). Malonyl-CoA, an intermediate of DNL, inhibits fatty acid beta-oxidation (and formation of β-OHB) through impairment of the mitochondrial fatty acid transporter CPTI (E). Of note, alternative mechanisms may contribute to the development of hepatic fat accumulation in aldolase B deficiency as well, such as Fru 1P-induced formation of urate and activation of ChREBP, which both stimulate DNL (see text). Green arrows indicate observations in ALDOB-KO mice. Blue arrows and blue cross indicate observations in HFI patients. Dashed arrow indicates multiple intermediate enzymatic steps that have not been visualized for simplicity purposes. ACC acetyl-CoA carboxylase, ACL ATP-citrate lyase, ALDOB aldolase B, β-OHB beta-hydroxybutyrate, CPTI carnitine palmitoyltransferase I, DHAP dihydroxyacetone phosphate, ER endoplasmic reticulum, Fru 6P fructose 6-phosphate, FAS fatty acid synthase, Fru 1P fructose 1-phosphate, Fru 1,6-P2 fructose 1,6-biphosphate, G3P glyceraldehyde 3-phosphate, Glc 6P glucose 6-phosphate, GAH glyceraldehyde, GCK glucokinase, GKRP glucokinase regulatory protein, M6P mannose 6-phosphate, MPI mannose-6-phosphate isomerase, NADPH nicotinamide adenine dinucleotide phosphate, PPP pentose phosphate pathway
There are other biologically plausible mechanisms that could explain the upregulated DNL pathway leading to hepatic fat accumulation in ALDOB-KO mice. First, experimental studies have shown that activation of the AMP-deaminase pathway and formation of urate (Fig. 1) induces mitochondrial dysfunction, which results in downregulation of fatty acid oxidation and stimulation of DNL [59]. Second, carbohydrate-responsive element-binding protein (ChREBP) is activated upon intracellular phosphate depletion and stimulates expression of glucose-6-phosphatase and DNL genes [60, 61], all in accordance with the observations in ALDOB-KO mice [48].
Inhibition of ketohexokinase protects ALDOB-KO mice from metabolic derangements
The importance of Fru 1P in the pathogenesis of the metabolic derangements as observed in ALDOB-KO mice was unequivocally demonstrated by inhibition of ketohexokinase (KHK), the enzyme that catalyzes the first step in fructose metabolism: the phosphorylation of fructose to yield Fru 1P. In most mammals, including humans, KHK exists as two isoforms, A and C [62]. KHK-C has high affinity for fructose and is abundant in the liver, intestine, and kidney. In contrast, KHK-A has much lower affinity for fructose and is more ubiquitously expressed [63]. Nearly, all of the aforementioned metabolic abnormalities in ALDOB-KO mice ameliorated when they were crossed with KHK knockout (KHK-KO) mice, i.e., both KHK-A and KHK-C [48]. Further, similar results were observed after treatment with osthole, a natural KHK inhibitor [64]. Fructose-loaded ALDOB-KO mice treated with osthole were protected from intrahepatic ATP depletion, hyperuricemia, rise in liver enzymes, and severe hypoglycemia [48]. In addition, osthole treatment resulted in a decrease in the GCK cytosol/nucleus ratio, indicative of more GCK bound to GKRP in the nucleus [48].
Importantly, ALDOB-KO mice were not protected from the above-mentioned metabolic abnormalities when crossed with KHK-A specific knockout mice [48]. In fact, the mice possessing only KHK-C resulted in an exacerbated phenotype [48]. This observation is likely explained by the fact that inhibition of KHK-A results in reduced metabolism of fructose in peripheral tissues and, hence, a greater supply to the liver, which is detrimental in case of aldolase B deficiency. These findings suggest that inhibition KHK-C may serve as a therapeutic target that could make the fructose-restricted diet redundant in HFI patients.
Recent advances in humans
Patients with HFI are characterized by an increased intrahepatic triglyceride content
Until recently, only anecdotal reports suggested that hepatic fat accumulation persists in HFI patients, despite a fructose-restricted diet [6]. A recent cross-sectional observational study including 16 genetically diagnosed HFI patients reported a high prevalence of fatty liver, as assessed by ultrasound or hepatic magnetic resonance imaging [65]. This issue was recently more structurally addressed in 15 adult HFI patients who were on a lifelong fructose-restricted diet, ranging from 0.3 to 7.0 grams of fructose per day (the average fructose intake of American adults ranges from 32 to 75 grams per day [66]). Magnetic resonance imaging spectroscopy of the liver revealed that intrahepatic triglyceride (IHTG) content was higher in HFI patients in comparison with 15 healthy age-, sex-, and BMI-matched individuals [67]. Although liver stiffness, a non-invasive marker of liver fibrosis, was not significantly different between both groups, one HFI patient displayed a liver stiffness measurement compatible with liver fibrosis stage 3 or higher. Metabolic profiling revealed that HFI patients were more glucose intolerant, as reflected by higher plasma glucose excursions during a standard 75-gram oral glucose tolerance test [67].
Further investigations to delineate the underlying mechanism that leads to an increased IHTG content in HFI patients were limited due to the noninvasive nature of human studies. Nevertheless, the use of liver-specific plasma biomarkers allowed some insight. First, hypoglycosylated transferrin, a liver-specific protein, was more abundant in HFI patients, which is in line with previous studies [68, 69]. Experimental studies have shown that Fru 1P inhibits mannose-6-phosphate isomerase (MPI) activity, one of the first enzymes involved in the glycosylation process (Fig. 2) [70]. The higher levels of hypoglycosylated transferrin (yet within the normal range) therefore suggest that intrahepatic Fru 1P concentrations are higher in HFI patients than in controls, even on a fructose-restricted diet. This may be explained by the minute levels of ingested fructose (blocked by aldolase B) or, alternatively, by endogenous fructose production via the polyol pathway. Despite the suggestion of higher intrahepatic Fru 1P levels in HFI patients on a fructose-restricted diet, plasma uric acid concentrations were not different between both groups [67]. This finding is consistent with observations in ALDOB-KO mice, which only displayed increased plasma uric acid levels after an oral fructose load [48].
Second, plasma beta-hydroxybutyrate levels, a liver-specific biomarker of beta-oxidation, were significantly lower in HFI patients compared to healthy individuals [67]. Notably, DNL and beta-oxidation are reciprocally regulated. Malonyl-CoA, a precursor of de novo synthesized fatty acids, inhibits the activity of the long-chain fatty acid transporter carnitine palmitoyltransferase I (CPTI). Consequently, the transport of long-chain fatty acids over the mitochondrial membrane is hampered and beta-oxidation is impaired [71]. It can therefore be concluded that the biomarker patterns in HFI patients are similar to the in-depth phenotyping of the ALDOB-KO mice (as illustrated in Fig. 2).
Variants in the GKRP gene show phenotypic similarities with ALDOB-KO mice and HFI patients
Unfortunately, it is not possible to non-invasively measure the GKRP-GCK interaction as a potential explanation for the increased IHTG content in HFI patients, since this would require liver biopsies. Nevertheless, genetic epidemiology is a valuable tool in predicting the metabolic consequences of increased GKRP-GCK disruption in humans [72]. Rs1260326 and rs789004 are common variants in the GKRP gene (GCKR), which are in strong linkage disequilibrium. The former is a functional variant that encodes a GKRP protein that dissociates from GCK more easily [73], comparable to the effect of Fru 1P on the GKRP-GCK complex. The previously reported associations of these common gene variants with cardiometabolic traits in the general population show some striking similarities with the metabolic abnormalities observed in ALDOB-KO mice and HFI patients (Table 1). First, variants in GCKR have been associated with reduced beta-hydroxybutyrate levels, pronounced DNL, and a greater IHTG content [74–77]. Further, these variants have been associated with lower fasting insulin concentrations [78, 79] and higher 2-h post-glucose load glucose levels [80], the former in agreement with ALDOB-KO mice [48] and the latter with HFI patients [67]. Of note, despite the consistently reported association between GCKR variants and increased plasma triglycerides [78, 79], HFI patients were characterized by normal plasma triglycerides levels [67]. This discrepancy may be explained by the fact that HFI patients were (relatively) metabolically healthy, i.e., non-(abdominally) obese [67]. We previously reported that GCKR interacts with metabolic health on plasma triglycerides, i.e., the unhealthier the greater the effect on plasma triglycerides levels [81]. Finally, a recent meta-analysis suggested that the common variants in GCKR protect against chronic kidney disease, but predisposes to cardiovascular disease (CVD) [82]. These relevant clinical endpoints have not been addressed in HFI patients chronically treated with a fructose-restricted diet and therefore deserve further study.
Table 1.
Cardiometabolic features in ALDOB-KO mice, HFI patients, and human carriers of common variants in the GCKR gene
| ALDOB-KO micea | HFI patientsb | GCKRc | References | |
|---|---|---|---|---|
| Intrahepatic triglycerides | ↑ | ↑ | ↑ | [48, 65, 67, 105] |
| Serum AST/ALTd | ↑ | ↑ | ↑ | [48, 65, 106] |
| DNLe | ↑ | ? | ↑ | [48, 76] |
| Serum beta-hydroxybutyrate | ? | ↓ | ↓ | [67, 74] |
| Intrahepatic glycogen | ↑ | ? | ? | [48] |
| Serum glucose | ↔ | ↔ | ↓ | [48, 67, 78–80] |
| Serum glucose, 2 h post-glucose load | ? | ↑ | ↑ | [48, 67, 80] |
| Serum insulin | ↓ | ↔ | ↓ | [48, 67, 79, 80] |
| Serum urate | ↔ | ↔ | ↑ | [48, 67, 107, 108] |
| Serum triglycerides | ? | ↔ | ↑ | [65, 67, 78, 79, 81] |
| eGFRf | ? | ? | ↑ | [82] |
| Coronary artery disease | ? | ? | ↑ | [82] |
Arrows indicate the direction of association, not the effect size
aObservations in ALDOB-KO mice fed a low-fructose diet (~ 0.3%)
bObservations in adult HFI patients chronically treated with a fructose-restricted diet
cCommon variants in rs1260326 and rs780084, which encode a GKRP protein that binds glucokinase less effectively
dAspartate transaminase (AST) and alanine transaminase (ALT)
eDe novo lipogenesis (DNL) is assessed by hepatic expression of key enzymes (ALDOB-KO mice) and stable isotopes (GCKR)
feGFR: estimated glomerular filtration
Implications for the current epidemic of fructose overconsumption
Since the industrial revolution, the intake of fructose in the USA has risen dramatically [66]. Fructose—which has a sweeter taste than glucose—is often added as a sweetener (e.g., as high fructose corn syrup) to processed foods. Given the parallel increase in fructose consumption and the current obesity epidemic and its sequelae (dyslipidemia, type 2 diabetes mellitus [T2DM], gout, and CVD) in Western society, fructose has been implicated as a major contributing factor [83–86].
Non-alcoholic fatty liver disease (NAFLD), a histological spectrum ranging from simple steatosis to steatohepatitis, fibrosis, and cirrhosis, is another frequently encountered phenomenon in obese individuals [87]. NAFLD may not only progress to end-stage liver failure and hepatocellular carcinoma, and it has also been associated with new-onset T2DM and CVD [88, 89]. The pathogenesis of NAFLD involves a complex interaction between genetic factors and unhealthy lifestyle habits [90].
Experimental studies in rodents and humans have unequivocally demonstrated that fructose overfeeding leads to an increased hepatic fat content [91–95] and many symptoms of the metabolic syndrome [96]. The mechanism by which fructose causes hepatic fat accumulation can be directly by serving as a substrate for DNL. Further, fructose can also indirectly enhance DNL via the hitherto mentioned mechanisms: (1) Fru 1P-induced disruption of the GKRP-GCK complex, which facilitates hepatic glucose uptake and consequently DNL (Fig. 2); (2) Fru 1P-induced ATP depletion and urate formation, which stimulates DNL [27–29]; and (3) Fru 1P-induced intracellular phosphate depletion, which activates ChREBP, a transcription factor with multiple downstream effects, among other stimulation of DNL [60, 61]. Of note, these processes have been observed in humans with normal aldolase B function [97–99].
The recent studies in ALDOB-KO mice and HFI patients suggest that the direct lipogenic effects of fructose do not necessarily play a role in the pathogenesis of fructose-induced NAFLD [48, 67]. Moreover, they suggest that the accumulation of intermediates of fructolysis, i.e., Fru 1P, is a key element in the pathogenesis of fructose-induced NAFLD.
From these findings, it can also be deduced that inhibition of Fru 1P formation by blocking upstream KHK activity may be a novel therapeutic modality, not only for HFI, but also for fructose-induced NAFLD. Indeed, the fatty liver phenotype in fructose-fed mice improved after treatment with liver-specific small interfering RNA (siRNA) targeting KHK expression [100]. Further, previous experimental studies have demonstrated that fructose-fed KHK-KO mice were protected from hepatic fat accumulation and other metabolic abnormalities, such as obesity and hyperinsulinemia, when compared to wild-type mice [101–103]. Again, analogous to the observations in ALDOB-KO [48], specific knockout of KHK-A resulted in an exacerbation of the metabolic abnormalities, including increased hepatic fat accumulation [102, 103]. Of interest, in humans, a loss of KHK results in essential fructosuria (OMIM #229800) [39]. This benign condition is not known to provoke any clinical symptoms [39] and, hence, emphasizes the therapeutic potential of KHK inhibition.
Future perspectives
The recent studies in aldolase B-deficient mice and HFI patients have contributed to our understanding of the pathogenesis of HFI and fructose-induced NAFLD [48, 67]. There are, however, several issues that deserve further study.
First, experimental studies are warranted to establish the exact roles (and their relative contributions) of the GKRP-GCK complex, urate, and ChREBP as potential mediators in the pathogenesis of hepatic fat accumulation in aldolase B deficiency. Furthermore, although the recent studies have convincingly identified Fru 1P as the key driver behind hepatic fat accumulation in aldolase B deficiency, the exact contribution of endogenous fructose production (via the polyol pathway) to the accumulation of intrahepatic Fru 1P remains to be elucidated. Future studies are warranted to determine to what extent gluconeogenesis and glycolysis are functional in aldolase B-deficient livers, and which alternative pathways (e.g., PPP) are involved. Long-term follow-up of a large cohort of HFI patients is needed to study whether these patients are protected from chronic kidney disease and predisposed to CVD, similar to individuals carrying common variants in GCKR [82]. Finally, clinical studies are required to demonstrate whether KHK inhibition will: a) replace the fructose-restricted diet as a treatment for HFI and b) be efficacious in the treatment of fructose-induced NAFLD in the general population. Interestingly, Huard et al. [104] recently reported the discovery of a small molecule that selectively inhibits KHK activity in vitro and in vivo more effectively than osthole.
Concluding remarks
HFI is a rare inborn error of fructose metabolism. Recent studies in ALDOB-KO mice and HFI patients have proposed a prominent role for Fru 1P in the pathogenesis of hepatic fat accumulation, and suggest that an increased dissociation of GCK from GKRP is involved. These findings have therapeutic implications for not only HFI, but also for fructose-induced NAFLD in the general population. These studies clearly demonstrate that fructose-induced NAFLD can benefit from the insight gained from rare inborn errors of metabolism, and vice versa.
Acknowledgements
The study in HFI patients was subsidized by the Netherlands Heart Foundation (#2015T042) and Stofwisselkracht. MB received a personal grant from the Diabetes Foundation (#2017.82.004). DT was supported by Colorado Research Partners LLC and National Institutes of Health (R01DK108859).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Dean R. Tolan, Email: tolan@bu.edu
Martijn C.G.J. Brouwers, Email: mcgj.brouwers@mumc.nl
References
- 1.Chambers RA, Pratt RT. Idiosyncrasy to fructose. Lancet. 1956;271(6938):340. doi: 10.1016/s0140-6736(56)92196-1. [DOI] [PubMed] [Google Scholar]
- 2.Herrs H, Joassin G. Anomalie de l’aldolase hepatique dans l’intolerance au fructose. Enzymol Biol Clin. 1961;1:4–14. [PubMed] [Google Scholar]
- 3.Li H, Byers HM, Diaz-Kuan A, Vos MB, Hall PL, Tortorelli S, Singh R, Wallenstein MB, Allain M, Dimmock DP, Farrell RM, McCandless S, Gambello MJ. Acute liver failure in neonates with undiagnosed hereditary fructose intolerance due to exposure from widely available infant formulas. Mol Genet Metab. 2018;123(4):428–432. doi: 10.1016/j.ymgme.2018.02.016. [DOI] [PubMed] [Google Scholar]
- 4.Sanchez-Lozada LG, Andres-Hernando A, Garcia-Arroyo FE, Cicerchi C, Li N, Kuwabara M, Roncal-Jimenez CA, Johnson RJ, Lanaspa MA. Uric acid activates aldose reductase and the polyol pathway for endogenous fructose and fat production causing development of fatty liver in rats. J Biol Chem. 2019 doi: 10.1074/jbc.RA118.006158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan LJ. Redox imbalance stress in diabetes mellitus: role of the polyol pathway. Animal Model Exp Med. 2018;1(1):7–13. doi: 10.1002/ame2.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Odievre M, Gentil C, Gautier M, Alagille D. Hereditary fructose intolerance in childhood. Diagnosis, management, and course in 55 patients. Am J Dis Child. 1978;132(6):605–608. doi: 10.1001/archpedi.1978.02120310069014. [DOI] [PubMed] [Google Scholar]
- 7.Baerlocher K, Gitzelmann R, Steinmann B, Gitzelmann-Cumarasamy N. Hereditary fructose intolerance in early childhood: a major diagnostic challenge. Survey of 20 symptomatic cases. Helv Paediatr Acta. 1978;33(6):465–487. [PubMed] [Google Scholar]
- 8.Mock DM, Perman JA, Thaler M, Morris RC., Jr Chronic fructose intoxication after infancy in children with hereditary fructose intolerance. A cause of growth retardation. N Engl J Med. 1983;309(13):764–770. doi: 10.1056/nejm198309293091305. [DOI] [PubMed] [Google Scholar]
- 9.Ali M, Rellos P, Cox TM. Hereditary fructose intolerance. J Med Genet. 1998;35(5):353–365. doi: 10.1136/jmg.35.5.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lebo RV, Tolan DR, Bruce BD, Cheung MC, Kan YW. Spot-blot analysis of sorted chromosomes assigns a fructose intolerance disease locus to chromosome 9. Cytometry. 1985;6(5):478–483. doi: 10.1002/cyto.990060513. [DOI] [PubMed] [Google Scholar]
- 11.Lench NJ, Telford EA, Andersen SE, Moynihan TP, Robinson PA, Markham AF. An EST and STS-based YAC contig map of human chromosome 9q22.3. Genomics. 1996;38(2):199–205. doi: 10.1006/geno.1996.0616. [DOI] [PubMed] [Google Scholar]
- 12.Cross NC, de Franchis R, Sebastio G, Dazzo C, Tolan DR, Gregori C, Odievre M, Vidailhet M, Romano V, Mascali G, et al. Molecular analysis of aldolase B genes in hereditary fructose intolerance. Lancet. 1990;335(8685):306–309. doi: 10.1016/0140-6736(90)90603-3. [DOI] [PubMed] [Google Scholar]
- 13.Tolan DR, Brooks CC. Molecular analysis of common aldolase B alleles for hereditary fructose intolerance in North Americans. Biochem Med Metab Biol. 1992;48(1):19–25. doi: 10.1016/0885-4505(92)90043-x. [DOI] [PubMed] [Google Scholar]
- 14.Cross NC, Stojanov LM, Cox TM. A new aldolase B variant, N334 K, is a common cause of hereditary fructose intolerance in Yugoslavia. Nucleic Acids Res. 1990;18(7):1925. doi: 10.1093/nar/18.7.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coffee EM, Yerkes L, Ewen EP, Zee T, Tolan DR. Increased prevalence of mutant null alleles that cause hereditary fructose intolerance in the American population. J Inherit Metab Dis. 2010;33(1):33–42. doi: 10.1007/s10545-009-9008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dazzo C, Tolan DR. Molecular evidence for compound heterozygosity in hereditary fructose intolerance. Am J Hum Genet. 1990;46(6):1194–1199. [PMC free article] [PubMed] [Google Scholar]
- 17.Cross NC, Tolan DR, Cox TM. Catalytic deficiency of human aldolase B in hereditary fructose intolerance caused by a common missense mutation. Cell. 1988;53(6):881–885. doi: 10.1016/s0092-8674(88)90349-2. [DOI] [PubMed] [Google Scholar]
- 18.James CL, Rellos P, Ali M, Heeley AF, Cox TM. Neonatal screening for hereditary fructose intolerance: frequency of the most common mutant aldolase B allele (A149P) in the British population. J Med Genet. 1996;33(10):837–841. doi: 10.1136/jmg.33.10.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gitzelmann R, Baerlocher K. Vorteile und Nachteile der Fruktose in der Nahrung. Pädiat Fortbildk Praxis. 1973;37:40–55. [Google Scholar]
- 20.Cox TM. Aldolase B and fructose intolerance. FASEB J. 1994;8(1):62–71. doi: 10.1096/fasebj.8.1.8299892. [DOI] [PubMed] [Google Scholar]
- 21.Tolan DR. Molecular basis of hereditary fructose intolerance: mutations and polymorphisms in the human aldolase B gene. Hum Mutat. 1995;6(3):210–218. doi: 10.1002/humu.1380060303. [DOI] [PubMed] [Google Scholar]
- 22.Hers H. Le métabolisme du fructose. Bruxelles: Editions Arscia; 1957. [Google Scholar]
- 23.Penhoet EE, Rutter WJ. Catalytic and immunochemical properties of homomeric and heteromeric combinations of aldolase subunits. J Biol Chem. 1971;246(2):318–323. [PubMed] [Google Scholar]
- 24.Penhoet E, Rajkumar T, Rutter WJ. Multiple forms of fructose diphosphate aldolase in mammalian tissues. Proc Natl Acad Sci USA. 1966;56(4):1275–1282. doi: 10.1073/pnas.56.4.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oppelt SA, Zhang W, Tolan DR. Specific regions of the brain are capable of fructose metabolism. Brain Res. 2017;1657:312–322. doi: 10.1016/j.brainres.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steinmann B, Gitzelmann R. The diagnosis of hereditary fructose intolerance. Helv Paediatr Acta. 1981;36(4):297–316. [PubMed] [Google Scholar]
- 27.Oberhaensli RD, Rajagopalan B, Taylor DJ, Radda GK, Collins JE, Leonard JV, Schwarz H, Herschkowitz N. Study of hereditary fructose intolerance by use of 31P magnetic resonance spectroscopy. Lancet. 1987;2(8565):931–934. doi: 10.1016/s0140-6736(87)91419-x. [DOI] [PubMed] [Google Scholar]
- 28.Woods HF, Eggleston LV, Krebs HA. The cause of hepatic accumulation of fructose 1-phosphate on fructose loading. Biochem J. 1970;119(3):501–510. doi: 10.1042/bj1190501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van den Berghe G, Bronfman M, Vanneste R, Hers HG. The mechanism of adenosine triphosphate depletion in the liver after a load of fructose. A kinetic study of liver adenylate deaminase. Biochem J. 1977;162(3):601–609. doi: 10.1042/bj1620601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaufmann U, Froesch ER. Inhibition of phosphorylase-a by fructose-1-phosphate, alpha-glycerophosphate and fructose-1,6-diphosphate: explanation for fructose-induced hypoglycaemia in hereditary fructose intolerance and fructose-1,6-diphosphatase deficiency. Eur J Clin Invest. 1973;3(5):407–413. doi: 10.1111/j.1365-2362.1973.tb02208.x. [DOI] [PubMed] [Google Scholar]
- 31.Thurston JH, Jones EM, Hauhart RE. Decrease and inhibition of liver glycogen phosphorylase after fructose. An experimental model for the study of hereditary fructose intolerance. Diabetes. 1974;23(7):597–604. doi: 10.2337/diab.23.7.597. [DOI] [PubMed] [Google Scholar]
- 32.Van Den Berghe G, Hue L, Hers HG. Effect of administration of the fructose on the glycogenolytic action of glucagon. An investigation of the pathogeny of hereditary fructose intolerance. Biochem J. 1973;134(2):637–645. doi: 10.1042/bj1340637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van den Berghe G. Biochemical aspects of hereditary fructose intolerance. In: Hommes FA, van den Berg CJ, editors. Normal and pathological development of energy metabolism. London: Academic Press; 1975. pp. 221–228. [Google Scholar]
- 34.Dubois RLH, Malaisse-Lagaw F, Toppet M. Etude clinique et anatomo-pathologique de deux cas d’intolerance congenitale au fructose. Pediatrie. 1965;20:5–14. [Google Scholar]
- 35.Rossier AMG, Colin J, Job J-C, Brault A, Beauvais P, Lemerle J. Intolérance congénitale au fructose. deux cas damiliaux avec étude biochimique in vitro. Arch Fr Pediatr. 1966;23:533. [Google Scholar]
- 36.Zalitis J, Oliver IT. Inhibition of glucose phosphate isomerase by metabolic intermediates of fructose. Biochem J. 1967;102(3):753–759. doi: 10.1042/bj1020753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Froesch E, Prader A, Wolf H, Labhart A. Die hereditare Fructoseintoleranz. Helvetica Paediatrica Acta. 1959;14:99–112. [PubMed] [Google Scholar]
- 38.Wilkening J, Nowack J, Decker K. The dependence of glucose formation from lactate on the adenosine triphosphate content in the isolated perfused rat liver. Biochim Biophys Acta. 1975;392(2):299–309. doi: 10.1016/0304-4165(75)90011-2. [DOI] [PubMed] [Google Scholar]
- 39.Gitzelmann R, Steinmann B, Gvd Berghe. Disorders of Fructose Metabolism. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. New York: McGraw-Hill; 1995. pp. 905–934. [Google Scholar]
- 40.Woods HF, Alberti KG. Dangers of intravenous fructose. Lancet. 1972;2(7791):1354–1357. doi: 10.1016/s0140-6736(72)92791-2. [DOI] [PubMed] [Google Scholar]
- 41.Bergstrom J, Hultman E, Roch-Norlund AE. Lactic acid accumulation in connection with fructose infusion. Acta Med Scand. 1968;184(5):359–364. doi: 10.1111/j.0954-6820.1968.tb02471.x. [DOI] [PubMed] [Google Scholar]
- 42.Lanaspa MA, Ishimoto T, Li N, Cicerchi C, Orlicky DJ, Ruzycki P, Rivard C, Inaba S, Roncal-Jimenez CA, Bales ES, Diggle CP, Asipu A, Petrash JM, Kosugi T, Maruyama S, Sanchez-Lozada LG, McManaman JL, Bonthron DT, Sautin YY, Johnson RJ. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat Commun. 2013;4:2434. doi: 10.1038/ncomms3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson RJ, Rodriguez-Iturbe B, Roncal-Jimenez C, Lanaspa MA, Ishimoto T, Nakagawa T, Correa-Rotter R, Wesseling C, Bankir L, Sanchez-Lozada LG. Hyperosmolarity drives hypertension and CKD–water and salt revisited. Nat Rev Nephrol. 2014;10(7):415–420. doi: 10.1038/nrneph.2014.76. [DOI] [PubMed] [Google Scholar]
- 44.Roncal Jimenez CA, Ishimoto T, Lanaspa MA, Rivard CJ, Nakagawa T, Ejaz AA, Cicerchi C, Inaba S, Le M, Miyazaki M, Glaser J, Correa-Rotter R, Gonzalez MA, Aragon A, Wesseling C, Sanchez-Lozada LG, Johnson RJ. Fructokinase activity mediates dehydration-induced renal injury. Kidney Int. 2014;86(2):294–302. doi: 10.1038/ki.2013.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sward P, Rippe B. Acute and sustained actions of hyperglycaemia on endothelial and glomerular barrier permeability. Acta Physiol (Oxf) 2012;204(3):294–307. doi: 10.1111/j.1748-1716.2011.02343.x. [DOI] [PubMed] [Google Scholar]
- 46.Lyons PA, Gould S, Wise PH, Palmer TN. Activation of erythrocyte aldose reductase in man in response to glycaemic challenge. Diabetes Res Clin Pract. 1991;14(1):9–13. doi: 10.1016/0168-8227(91)90047-h. [DOI] [PubMed] [Google Scholar]
- 47.Oppelt SA, Sennott EM, Tolan DR. Aldolase-B knockout in mice phenocopies hereditary fructose intolerance in humans. Mol Genet Metab. 2015;114(3):445–450. doi: 10.1016/j.ymgme.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 48.Lanaspa MA, Andres-Hernando A, Orlicky DJ, Cicerchi C, Jang C, Li N, Milagres T, Kuwabara M, Wempe MF, Rabinowitz JD, Johnson RJ, Tolan DR. Ketohexokinase C blockade ameliorates fructose-induced metabolic dysfunction in fructose-sensitive mice. J Clin Invest. 2018;128(6):2226–2238. doi: 10.1172/JCI94427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coffee E, Tolan D. Gluconeogenesis. In: Lee B, Scaglia F, editors. Inborn errors of metabolism: from neonatal screening to metabolic pathways. New York: Oxford University Press; 2014. p. 384. [Google Scholar]
- 50.Agius L. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem J. 2008;414(1):1–18. doi: 10.1042/BJ20080595. [DOI] [PubMed] [Google Scholar]
- 51.Agius L, Peak M, Van Schaftingen E. The regulatory protein of glucokinase binds to the hepatocyte matrix, but, unlike glucokinase, does not translocate during substrate stimulation. Biochem J. 1995;309(Pt 3):711–713. doi: 10.1042/bj3090711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Schaftingen E, Veiga-da-Cunha M, Niculescu L. The regulatory protein of glucokinase. Biochem Soc Trans. 1997;25(1):136–140. doi: 10.1042/bst0250136. [DOI] [PubMed] [Google Scholar]
- 53.van Schaftingen E, Vandercammen A, Detheux M, Davies DR. The regulatory protein of liver glucokinase. Adv Enzyme Regul. 1992;32:133–148. doi: 10.1016/0065-2571(92)90013-p. [DOI] [PubMed] [Google Scholar]
- 54.Vandercammen A, Van Schaftingen E. Species and tissue distribution of the regulatory protein of glucokinase. Biochem J. 1993;294(Pt 2):551–556. doi: 10.1042/bj2940551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Veiga-Da-Cunha M, Detheux M, Watelet N, Van Schaftingen E. Cloning and expression of a Xenopus liver cDNA encoding a fructose-phosphate-insensitive regulatory protein of glucokinase. Eur J Biochem. 1994;225(1):43–51. doi: 10.1111/j.1432-1033.1994.00043.x. [DOI] [PubMed] [Google Scholar]
- 56.Beck T, Miller BG. Structural basis for regulation of human glucokinase by glucokinase regulatory protein. Biochemistry. 2013;52(36):6232–6239. doi: 10.1021/bi400838t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choi JM, Seo MH, Kyeong HH, Kim E, Kim HS. Molecular basis for the role of glucokinase regulatory protein as the allosteric switch for glucokinase. Proc Natl Acad Sci USA. 2013;110(25):10171–10176. doi: 10.1073/pnas.1300457110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jin ES, Lee MH, Murphy RE, Malloy CR. Pentose phosphate pathway activity parallels lipogenesis but not antioxidant processes in rat liver. Am J Physiol Endocrinol Metab. 2018;314(6):E543–E551. doi: 10.1152/ajpendo.00342.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lanaspa MA, Sanchez-Lozada LG, Choi YJ, Cicerchi C, Kanbay M, Roncal-Jimenez CA, Ishimoto T, Li N, Marek G, Duranay M, Schreiner G, Rodriguez-Iturbe B, Nakagawa T, Kang DH, Sautin YY, Johnson RJ. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J Biol Chem. 2012;287(48):40732–40744. doi: 10.1074/jbc.M112.399899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim MS, Krawczyk SA, Doridot L, Fowler AJ, Wang JX, Trauger SA, Noh HL, Kang HJ, Meissen JK, Blatnik M, Kim JK, Lai M, Herman MA. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J Clin Invest. 2016;126(11):4372–4386. doi: 10.1172/JCI81993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hannou SA, Haslam DE, McKeown NM, Herman MA. Fructose metabolism and metabolic disease. J Clin Invest. 2018;128(2):545–555. doi: 10.1172/JCI96702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hayward BE, Bonthron DT. Structure and alternative splicing of the ketohexokinase gene. Eur J Biochem. 1998;257(1):85–91. doi: 10.1046/j.1432-1327.1998.2570085.x. [DOI] [PubMed] [Google Scholar]
- 63.Diggle CP, Shires M, Leitch D, Brooke D, Carr IM, Markham AF, Hayward BE, Asipu A, Bonthron DT. Ketohexokinase: expression and localization of the principal fructose-metabolizing enzyme. J Histochem Cytochem. 2009;57(8):763–774. doi: 10.1369/jhc.2009.953190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Le MT, Lanaspa MA, Cicerchi CM, Rana J, Scholten JD, Hunter BL, Rivard CJ, Randolph RK, Johnson RJ. Bioactivity-guided identification of botanical inhibitors of ketohexokinase. PLoS One. 2016;11(6):e0157458. doi: 10.1371/journal.pone.0157458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aldamiz-Echevarria L, de Las Heras J, Couce ML, Alcalde C, Vitoria I, Bueno M, Blasco-Alonso J, Concepcion Garcia M, Ruiz M, Suarez R, Andrade F, Villate O. Non-alcoholic fatty liver in hereditary fructose intolerance. Clin Nutr. 2019 doi: 10.1016/j.clnu.2019.02.019. [DOI] [PubMed] [Google Scholar]
- 66.Marriott BP, Cole N, Lee E. National estimates of dietary fructose intake increased from 1977 to 2004 in the United States. J Nutr. 2009;139(6):1228S–1235S. doi: 10.3945/jn.108.098277. [DOI] [PubMed] [Google Scholar]
- 67.Simons N, Debray FG, Schaper NC, Kooi ME, Feskens EJM, Hollak CEM, Lindeboom L, Koek GH, Bons JAP, Lefeber DJ, Hodson L, Schalkwijk CG, Stehouwer CDA, Cassiman D, Brouwers M. Patients with aldolase B deficiency are characterized by an increased intrahepatic triglyceride content. J Clin Endocrinol Metab. 2019 doi: 10.1210/jc.2018-02795. [DOI] [PubMed] [Google Scholar]
- 68.Adamowicz M, Ploski R, Rokicki D, Morava E, Gizewska M, Mierzewska H, Pollak A, Lefeber DJ, Wevers RA, Pronicka E. Transferrin hypoglycosylation in hereditary fructose intolerance: using the clues and avoiding the pitfalls. J Inherit Metab Dis. 2007;30(3):407. doi: 10.1007/s10545-007-0569-z. [DOI] [PubMed] [Google Scholar]
- 69.Quintana E, Sturiale L, Montero R, Andrade F, Fernandez C, Couce ML, Barone R, Aldamiz-Echevarria L, Ribes A, Artuch R, Briones P. Secondary disorders of glycosylation in inborn errors of fructose metabolism. J Inherit Metab Dis. 2009;32(Suppl 1):S273–S278. doi: 10.1007/s10545-009-1219-4. [DOI] [PubMed] [Google Scholar]
- 70.Jaeken J, Pirard M, Adamowicz M, Pronicka E, van Schaftingen E. Inhibition of phosphomannose isomerase by fructose 1-phosphate: an explanation for defective N-glycosylation in hereditary fructose intolerance. Pediatr Res. 1996;40(5):764–766. doi: 10.1203/00006450-199611000-00017. [DOI] [PubMed] [Google Scholar]
- 71.Foster DW. Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. J Clin Invest. 2012;122(6):1958–1959. doi: 10.1172/JCI63967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brouwers M, Jacobs C, Bast A, Stehouwer CDA, Schaper NC. Modulation of glucokinase regulatory protein: a double-edged sword? Trends Mol Med. 2015;21(10):583–594. doi: 10.1016/j.molmed.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 73.Beer NL, Tribble ND, McCulloch LJ, Roos C, Johnson PR, Orho-Melander M, Gloyn AL. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum Mol Genet. 2009;18(21):4081–4088. doi: 10.1093/hmg/ddp357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mahendran Y, Vangipurapu J, Cederberg H, Stancakova A, Pihlajamaki J, Soininen P, Kangas AJ, Paananen J, Civelek M, Saleem NK, Pajukanta P, Lusis AJ, Bonnycastle LL, Morken MA, Collins FS, Mohlke KL, Boehnke M, Ala-Korpela M, Kuusisto J, Laakso M. Association of ketone body levels with hyperglycemia and type 2 diabetes in 9,398 Finnish men. Diabetes. 2013;62(10):3618–3626. doi: 10.2337/db12-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Santoro N, Zhang CK, Zhao H, Pakstis AJ, Kim G, Kursawe R, Dykas DJ, Bale AE, Giannini C, Pierpont B, Shaw MM, Groop L, Caprio S. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology. 2012;55(3):781–789. doi: 10.1002/hep.24806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Santoro N, Caprio S, Pierpont B, Name MV, Savoye M, Parks EJ. Hepatic de novo lipogenesis in obese youth is modulated by a common variant in the GCKR gene. J Clin Endocrinol Metab. 2015 doi: 10.1210/jc.2015-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, Gudnason V, Eiriksdottir G, Garcia ME, Launer LJ, Nalls MA, Clark JM, Mitchell BD, Shuldiner AR, Butler JL, Tomas M, Hoffmann U, Hwang SJ, Massaro JM, O’Donnell CJ, Sahani DV, Salomaa V, Schadt EE, Schwartz SM, Siscovick DS, Voight BF, Carr JJ, Feitosa MF, Harris TB, Fox CS, Smith AV, Kao WH, Hirschhorn JN, Borecki IB. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7(3):e1001324. doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Orho-Melander M, Melander O, Guiducci C, Perez-Martinez P, Corella D, Roos C, Tewhey R, Rieder MJ, Hall J, Abecasis G, Tai ES, Welch C, Arnett DK, Lyssenko V, Lindholm E, Saxena R, de Bakker PI, Burtt N, Voight BF, Hirschhorn JN, Tucker KL, Hedner T, Tuomi T, Isomaa B, Eriksson KF, Taskinen MR, Wahlstrand B, Hughes TE, Parnell LD, Lai CQ, Berglund G, Peltonen L, Vartiainen E, Jousilahti P, Havulinna AS, Salomaa V, Nilsson P, Groop L, Altshuler D, Ordovas JM, Kathiresan S. Common missense variant in the glucokinase regulatory protein gene is associated with increased plasma triglyceride and C-reactive protein but lower fasting glucose concentrations. Diabetes. 2008;57(11):3112–3121. doi: 10.2337/db08-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vaxillaire M, Cavalcanti-Proenca C, Dechaume A, Tichet J, Marre M, Balkau B, Froguel P, Group DS. The common P446L polymorphism in GCKR inversely modulates fasting glucose and triglyceride levels and reduces type 2 diabetes risk in the DESIR prospective general French population. Diabetes. 2008;57(8):2253–2257. doi: 10.2337/db07-1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bi M, Kao WH, Boerwinkle E, Hoogeveen RC, Rasmussen-Torvik LJ, Astor BC, North KE, Coresh J, Kottgen A. Association of rs780094 in GCKR with metabolic traits and incident diabetes and cardiovascular disease: the ARIC Study. PLoS One. 2010;5(7):e11690. doi: 10.1371/journal.pone.0011690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Simons N, Dekker JM, van Greevenbroek MM, Nijpels G, t Hart LM, van der Kallen CJ, Schalkwijk CG, Schaper NC, Stehouwer CD, Brouwers MC. A common gene variant in glucokinase regulatory protein interacts with glucose metabolism on diabetic dyslipidemia: the combined CODAM and Hoorn studies. Diabetes Care. 2016;39(10):1811–1817. doi: 10.2337/dc16-0153. [DOI] [PubMed] [Google Scholar]
- 82.Simons P, Simons N, Stehouwer CDA, Schalkwijk CG, Schaper NC, Brouwers M. Association of common gene variants in glucokinase regulatory protein with cardiorenal disease: a systematic review and meta-analysis. PLoS One. 2018;13(10):e0206174. doi: 10.1371/journal.pone.0206174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bray GA, Nielsen SJ, Popkin BM. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am J Clin Nutr. 2004;79(4):537–543. doi: 10.1093/ajcn/79.4.537. [DOI] [PubMed] [Google Scholar]
- 84.Bray GA. Fructose: should we worry? Int J Obes (Lond) 2008;32(Suppl 7):S127–S131. doi: 10.1038/ijo.2008.248. [DOI] [PubMed] [Google Scholar]
- 85.Stanhope KL, Havel PJ. Fructose consumption: potential mechanisms for its effects to increase visceral adiposity and induce dyslipidemia and insulin resistance. Curr Opin Lipidol. 2008;19(1):16–24. doi: 10.1097/MOL.0b013e3282f2b24a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lustig RH. Fructose: metabolic, hedonic, and societal parallels with ethanol. J Am Diet Assoc. 2010;110(9):1307–1321. doi: 10.1016/j.jada.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 87.Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: mayo clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55(7):434–438. [PubMed] [Google Scholar]
- 88.Targher G, Byrne CD, Lonardo A, Zoppini G, Barbui C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: a meta-analysis. J Hepatol. 2016;65(3):589–600. doi: 10.1016/j.jhep.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 89.Mantovani A, Byrne CD, Bonora E, Targher G. Nonalcoholic fatty liver disease and risk of incident type 2 diabetes: a meta-analysis. Diabetes Care. 2018;41(2):372–382. doi: 10.2337/dc17-1902. [DOI] [PubMed] [Google Scholar]
- 90.Stender S, Kozlitina J, Nordestgaard BG, Tybjaerg-Hansen A, Hobbs HH, Cohen JC. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet. 2017;49(6):842–847. doi: 10.1038/ng.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang L, Perdomo G, Kim DH, Qu S, Ringquist S, Trucco M, Dong HH. Proteomic analysis of fructose-induced fatty liver in hamsters. Metabolism. 2008;57(8):1115–1124. doi: 10.1016/j.metabol.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mock K, Lateef S, Benedito VA, Tou JC. High-fructose corn syrup-55 consumption alters hepatic lipid metabolism and promotes triglyceride accumulation. J Nutr Biochem. 2017;39:32–39. doi: 10.1016/j.jnutbio.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 93.Schultz A, Barbosa-da-Silva S, Aguila MB, Mandarim-de-Lacerda CA. Differences and similarities in hepatic lipogenesis, gluconeogenesis and oxidative imbalance in mice fed diets rich in fructose or sucrose. Food Funct. 2015;6(5):1684–1691. doi: 10.1039/c5fo00251f. [DOI] [PubMed] [Google Scholar]
- 94.Ackerman Z, Oron-Herman M, Grozovski M, Rosenthal T, Pappo O, Link G, Sela BA. Fructose-induced fatty liver disease: hepatic effects of blood pressure and plasma triglyceride reduction. Hypertension. 2005;45(5):1012–1018. doi: 10.1161/01.HYP.0000164570.20420.67. [DOI] [PubMed] [Google Scholar]
- 95.Chiu S, Sievenpiper JL, de Souza RJ, Cozma AI, Mirrahimi A, Carleton AJ, Ha V, Di Buono M, Jenkins AL, Leiter LA, Wolever TM, Don-Wauchope AC, Beyene J, Kendall CW, Jenkins DJ. Effect of fructose on markers of non-alcoholic fatty liver disease (NAFLD): a systematic review and meta-analysis of controlled feeding trials. Eur J Clin Nutr. 2014;68(4):416–423. doi: 10.1038/ejcn.2014.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Perez-Pozo SE, Schold J, Nakagawa T, Sanchez-Lozada LG, Johnson RJ, Lillo JL. Excessive fructose intake induces the features of metabolic syndrome in healthy adult men: role of uric acid in the hypertensive response. Int J Obes (Lond) 2010;34(3):454–461. doi: 10.1038/ijo.2009.259. [DOI] [PubMed] [Google Scholar]
- 97.Fox IH, Kelley WN. Studies on the mechanism of fructose-induced hyperuricemia in man. Metabolism. 1972;21(8):713–721. doi: 10.1016/0026-0495(72)90120-5. [DOI] [PubMed] [Google Scholar]
- 98.Hurtado del Pozo C, Vesperinas-Garcia G, Rubio MA, Corripio-Sanchez R, Torres-Garcia AJ, Obregon MJ, Calvo RM. ChREBP expression in the liver, adipose tissue and differentiated preadipocytes in human obesity. Biochim Biophys Acta. 2011;1811(12):1194–1200. doi: 10.1016/j.bbalip.2011.07.016. [DOI] [PubMed] [Google Scholar]
- 99.Petersen KF, Laurent D, Yu C, Cline GW, Shulman GI. Stimulating effects of low-dose fructose on insulin-stimulated hepatic glycogen synthesis in humans. Diabetes. 2001;50(6):1263–1268. doi: 10.2337/diabetes.50.6.1263. [DOI] [PubMed] [Google Scholar]
- 100.Softic S, Gupta MK, Wang GX, Fujisaka S, O’Neill BT, Rao TN, Willoughby J, Harbison C, Fitzgerald K, Ilkayeva O, Newgard CB, Cohen DE, Kahn CR. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest. 2017;127(11):4059–4074. doi: 10.1172/JCI94585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Miller C, Yang X, Lu K, Cao J, Herath K, Rosahl TW, Askew R, Pavlovic G, Zhou G, Li C, Akiyama TE. Ketohexokinase knockout mice, a model for essential fructosuria, exhibit altered fructose metabolism and are protected from diet-induced metabolic defects. Am J Physiol Endocrinol Metab. 2018 doi: 10.1152/ajpendo.00027.2018. [DOI] [PubMed] [Google Scholar]
- 102.Ishimoto T, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA, Orlicky DJ, Cicerchi C, McMahan RH, Abdelmalek MF, Rosen HR, Jackman MR, MacLean PS, Diggle CP, Asipu A, Inaba S, Kosugi T, Sato W, Maruyama S, Sanchez-Lozada LG, Sautin YY, Hill JO, Bonthron DT, Johnson RJ. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology. 2013;58(5):1632–1643. doi: 10.1002/hep.26594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ishimoto T, Lanaspa MA, Le MT, Garcia GE, Diggle CP, Maclean PS, Jackman MR, Asipu A, Roncal-Jimenez CA, Kosugi T, Rivard CJ, Maruyama S, Rodriguez-Iturbe B, Sanchez-Lozada LG, Bonthron DT, Sautin YY, Johnson RJ. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc Natl Acad Sci USA. 2012;109(11):4320–4325. doi: 10.1073/pnas.1119908109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Huard K, Ahn K, Amor P, Beebe DA, Borzilleri KA, Chrunyk BA, Coffey SB, Cong Y, Conn EL, Culp JS, Dowling MS, Gorgoglione MF, Gutierrez JA, Knafels JD, Lachapelle EA, Pandit J, Parris KD, Perez S, Pfefferkorn JA, Price DA, Raymer B, Ross TT, Shavnya A, Smith AC, Subashi TA, Tesz GJ, Thuma BA, Tu M, Weaver JD, Weng Y, Withka JM, Xing G, Magee TV. Discovery of fragment-derived small molecules for in vivo inhibition of ketohexokinase (KHK) J Med Chem. 2017;60(18):7835–7849. doi: 10.1021/acs.jmedchem.7b00947. [DOI] [PubMed] [Google Scholar]
- 105.Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, Gudnason V, Eiriksdottir G, Garcia ME, Launer LJ, Nalls MA, Clark JM, Mitchell BD, Shuldiner AR, Butler JL, Tomas M, Hoffmann U, Hwang SJ, Massaro JM, O’Donnell CJ, Sahani DV, Salomaa V, Schadt EE, Schwartz SM, Siscovick DS, Nash CRN, Consortium G, Investigators M, Voight BF, Carr JJ, Feitosa MF, Harris TB, Fox CS, Smith AV, Kao WH, Hirschhorn JN, Borecki IB, Consortium G. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;73:e1001324. doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Seedorf U, Sen-Chowdhry S, Siminovitch KA, Smit JH, Spector TD, Tan W, Teslovich TM, Tukiainen T, Uitterlinden AG, Van der Klauw MM, Vasan RS, Wallace C, Wallaschofski H, Wichmann HE, Willemsen G, Wurtz P, Xu C, Yerges-Armstrong LM, Alcohol Genome-wide Association C. Diabetes Genetics R. Meta-analyses S. Genetic Investigation of Anthropometric Traits C. Global Lipids Genetics C. Genetics of Liver Disease C. International Consortium for Blood P. Meta-analyses of G. Insulin-Related Traits C. Abecasis GR, Ahmadi KR, Boomsma DI, Caulfield M, Cookson WO, van Duijn CM, Froguel P, Matsuda K, McCarthy MI, Meisinger C, Mooser V, Pietilainen KH, Schumann G, Snieder H, Sternberg MJ, Stolk RP, Thomas HC, Thorsteinsdottir U, Uda M, Waeber G, Wareham NJ, Waterworth DM, Watkins H, Whitfield JB, Witteman JC, Wolffenbuttel BH, Fox CS, Ala-Korpela M, Stefansson K, Vollenweider P, Volzke H, Schadt EE, Scott J, Jarvelin MR, Elliott P, Kooner JS. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet. 2011;43(11):1131–1138. doi: 10.1038/ng.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.van der Harst P, Bakker SJ, de Boer RA, Wolffenbuttel BH, Johnson T, Caulfield MJ, Navis G. Replication of the five novel loci for uric acid concentrations and potential mediating mechanisms. Hum Mol Genet. 2010;19(2):387–395. doi: 10.1093/hmg/ddp489. [DOI] [PubMed] [Google Scholar]
- 108.Kolz M, Johnson T, Sanna S, Teumer A, Vitart V, Perola M, Mangino M, Albrecht E, Wallace C, Farrall M, Johansson A, Nyholt DR, Aulchenko Y, Beckmann JS, Bergmann S, Bochud M, Brown M, Campbell H, Consortium E, Connell J, Dominiczak A, Homuth G, Lamina C, McCarthy MI, Consortium E, Meitinger T, Mooser V, Munroe P, Nauck M, Peden J, Prokisch H, Salo P, Salomaa V, Samani NJ, Schlessinger D, Uda M, Volker U, Waeber G, Waterworth D, Wang-Sattler R, Wright AF, Adamski J, Whitfield JB, Gyllensten U, Wilson JF, Rudan I, Pramstaller P, Watkins H, Consortium P, Doering A, Wichmann HE, Study K, Spector TD, Peltonen L, Volzke H, Nagaraja R, Vollenweider P, Caulfield M, Wtccc Illig T, Gieger C. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet. 2009;5(6):e1000504. doi: 10.1371/journal.pgen.1000504. [DOI] [PMC free article] [PubMed] [Google Scholar]