

Abstract
Redox homeostasis is an essential requirement of the biological systems for performing various normal cellular functions including cellular growth, differentiation, senescence, survival and aging in humans. The changes in the basal levels of reactive oxygen species (ROS) are detrimental to cells and often lead to several disease conditions including cardiovascular, neurological, diabetes and cancer. During the last two decades, substantial research has been done which clearly suggests that ROS are essential for the initiation, progression, angiogenesis as well as metastasis of cancer in several ways. During the last two decades, the potential of dysregulated ROS to enhance tumor formation through the activation of various oncogenic signaling pathways, DNA mutations, immune escape, tumor microenvironment, metastasis, angiogenesis and extension of telomere has been discovered. At present, surgery followed by chemotherapy and/or radiotherapy is the major therapeutic modality for treating patients with either early or advanced stages of cancer. However, the majority of patients relapse or did not respond to initial treatment. One of the reasons for recurrence/relapse is the altered levels of ROS in tumor cells as well as in cancer-initiating stem cells. One of the critical issues is targeting the intracellular/extracellular ROS for significant antitumor response and relapse-free survival. Indeed, a large number of FDA-approved anticancer drugs are efficient to eliminate cancer cells and drug resistance by increasing ROS production. Thus, the modulation of oxidative stress response might represent a potential approach to eradicate cancer in combination with FDA-approved chemotherapies, radiotherapies as well as immunotherapies.
Keywords: Reactive oxygen species (ROS), Mitochondrial ROS (mROS), Antioxidant system, Ferroptosis, Signaling pathways, Cancer stem cells (CSCs), Metastasis, Angiogenesis, Immune escape, Tumor microenvironment, ROS scavenger, Chemotherapy
Introduction
Reactive oxygen species (ROS) are characterized as oxygen-carrying molecules having reactive properties which consist of radicals including O2− (superoxide), HO• (hydroxyl) and non-radicals including H2O2 (hydrogen peroxide) [1–4]. These ROS molecules originate from oxygen which is utilized in several metabolic responses in the mitochondria and endoplasmic reticulum (ER) along with peroxisomes [5, 6]. Around 2% of the oxygen is utilized through mitochondria to generate O2−. Therefore, mitochondria are recognized as an utmost source of ROS [3, 6, 7]. The ER provides an oxidizing environment for proper folding of proteins by forming disulfide bonds and increasing ROS levels by oxidation of proteins [8]. Peroxisomes play a dual role: (a) scavenging of ROS through the catalytic degradation of H2O2 and (b) generation of ROS via β-oxidation of the fatty acids. ROS can be produced by either enzymatic and/or non-enzymatic mechanisms. The enzymatic mechanism involves NADPH oxidases (NOXs), endothelial nitric oxide synthase (eNOS), xanthine oxidase, arachidonic acid, lipoxygenase, enzymes of cytochrome P450 and cyclooxygenase. Non-enzymatic mechanism of ROS generation is through the mitochondrial respiratory chain [1, 2, 9, 10]. Therefore, coordination of ROS/redox homeostasis is pivotal for regulating the normal biological functions including cell growth, senescence, cell survival and aging. A controlled regulation of ROS inducer, as well as ROS scavenger pathways, is required because low/moderate levels ROS is important for proliferation, differentiation, migration, and survival, whereas excessive ROS levels are harmful [7] (Figs. 1 and 2). Alteration in the H2O2 or ROS has a potential effect on cellular functions, because of the fact that signaling pathways and transcription factors (TFs) related to cell division, stem cell differentiation and cellular stress networks are susceptible to the redox environment [11–16]. ROS can easily interact with DNA and other biomolecules. This can lead to DNA damage, incorporation of oncogenic mutations in the normal cells that results in genomic instability and cancer [16–19]. Cancer cells have increased aerobic glycolysis (Warburg effect) which is correlated with augmented ROS/oxidative stress [9]. The increased levels of ROS in cancer cells are because of alterations in key signaling pathways related to cellular metabolism. In the present review, we are focusing on the involvement of ROS as an important regulator of a variety of cellular processes including regulation of cellular homeostasis, various signaling pathways, telomerase, metastasis, angiogenesis, cancer stem cell, immune response and microbiome for the initiation, progression and treatment of human malignancies.
Fig. 1.
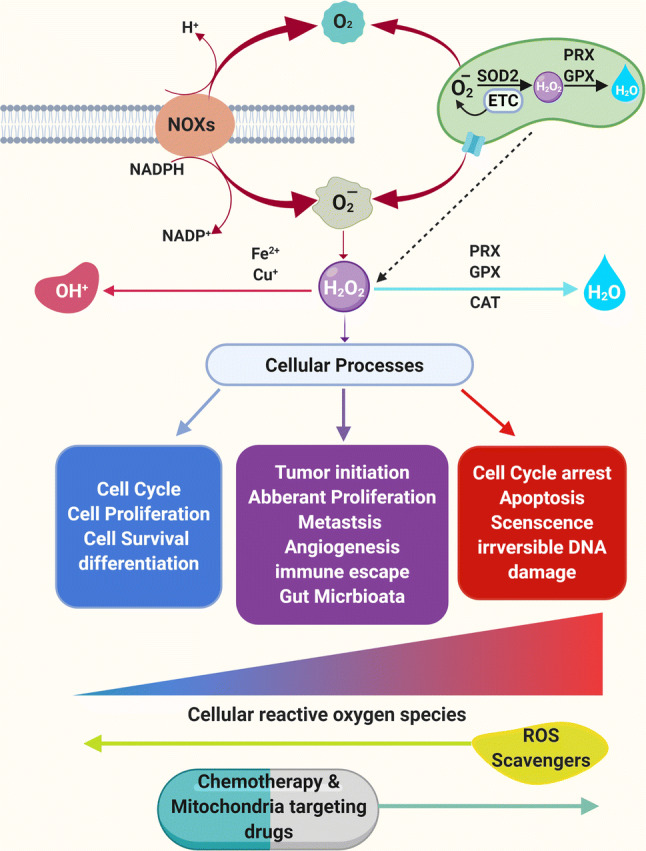
Formation and regulation of ROS and its effects on cellular functions. Mitochondria and NADPH oxidases are major sources of O2−, HO•, and H2O2 (ROS) formation. Superoxide dismutase (SOD1 or SOD2) can convert O2− into H2O2. H2O2 can be converted into H2O (water) by peroxiredoxin (PRX), glutathione peroxidase (GPX) and catalase (CAT) in mitochondria and cytosol. ROS are generated during normal cellular functioning and homeostasis is maintained by antioxidants expressed by the cells. Low ROS (green) is the basic need to maintain normal cellular proliferation, survival, and differentiation. Moderate to high ROS (tumor favoring ROS; light red) is the signal for the increased cellular proliferation, survival, tumor initiation, immune escape to genomic instability, metastasis, invasion and angiogenesis. Extremely high ROS produced by chemotherapeutic agents (dark red) is dangerous for the cells and leads to cell cycle arrest, apoptosis, senescence and unrepairable DNA damage
Fig. 2.

Maintenance of cellular homeostasis through inducers and scavengers of ROS. ROS can be produced by mitochondria, NADPH oxidases, hypoxia, metabolism, ER stress, cyclooxygenase and oncogenes including HRAS, FLT3-ITD, BCR-ABL, AKT, NF-kB, STAT3 and STAT5. On the other hand, ROS can be eliminated via activation of the dietary antioxidants, glutathione peroxidase, peroxiredoxin, catalase, NRF2, NADPH, SOD and tumor suppressor gnes including BRCA1, BRCA2, TP53, PTEN, FXOP3 and ATM
Regulation of ROS generation
ROS balance is maintained by several enzymes that neutralize toxic oxidants. Superoxide dismutases (SODs) are responsible for the conversion of O2− into H2O2. To avoid cellular damage, catalase (CAT), glutathione peroxidase (GPXs), and peroxiredoxins (PRXs) convert H2O2 into water and oxygen [20–22] (Fig. 1). There are six different types of PRXs that are localized in ER, cytosol, peroxisome, and mitochondria and this makes them ideal scavengers for ROS/H2O2 [21, 23]. PRXs function is to accept oxidants through active cysteine residue. These oxidized PRXs are then reduced via thioredoxin (TRX), as a result of which TRX gets oxidized and subsequently reduced by TRX reductase [14, 21]. In human cancers, deregulation of TRX metabolism has been found to be involved in drug resistance. Elevated levels of TRX have been noticed in different cancers including colorectal, pancreatic, lung, cervix, liver, and breast [24–29]. Glutathione (GSH) is a well-known antioxidant that functions as a scavenger for free radicals. GSH plays a critical role in multiple cellular processes such as cellular proliferation, division as well as differentiation. GSH is synthesized by glutamate-cysteine ligase (GCL) and GSH synthetase (GSS) [30]. The glutathione antioxidant system comprises GSH, glutathione reductase, GPX and glutathione S-transferases (GST). GSH guards the cells against oxidative stress by minimizing disulfide bond formation to the cysteine residues present on the cytoplasmic proteins. To perform the antioxidant function, GSH has been shown to be oxidized into GSSG. Glutathione peroxidases (GPX) act as a catalyst and accelerate the breakdown of hydroperoxides as well as H2O2 [47, 48]. GSH reductase has been shown to reduce GSSG and replenish the pool of GSH via the utilization of NADPH [49] (Fig. 3a). Generation of NADPH inside the cell is mostly controlled by cellular metabolism that includes glucose and glutamine metabolism, pentose phosphate pathway, conversion of pyruvate to malate by malic enzyme and conversion of isocitrate to α-ketoglutarate by isocitrate dehydrogenase (IDH) [1]. Under normal physiological conditions, GSH always occurs in its reduced form inside the cells due to the constitutive activity of glutathione reductase [50]. The reduced form of glutathione plays critical roles to control cellular levels of ROS. Moreover, mitochondrial GSH has been observed to react with ROS and protect from apoptosis. Modification of GSH metabolism has been observed in many tumors [31]. GSH dysregulation has been displayed to be involved in multidrug and radiation resistance. For example, an increase in GSH levels within tumor cells has been correlated with resistance to anthracyclines, platinum-based anticancer drugs, and alkylating agents. Another study showed that overutilization of cysteine for GSH synthesis can mediate tamoxifen resistance against breast cancer cells [32]. GSTs belongs to a class of detoxifying enzymes that accelerate the concurrence of GSH to a number of exogenous and endogenous electrophilic compounds for alimentation of cellular integrity, genomic stability by preventing DNA damage, oxidative stress [33, 34] (Fig. 3b). GSTs showed decreased hydroperoxides and 4-HNE, products of lipid peroxidation, to keep the oxidative stress under control [35]. GSTs have been reported to be robustly expressed in almost all human malignancies to modulate mitogen-activated protein kinase (MAPK) pathways [35]. Also, overexpression of GSTs has been correlated with tumor progression and drug resistance in human cancers [33, 35] (Table 1).
Fig. 3.
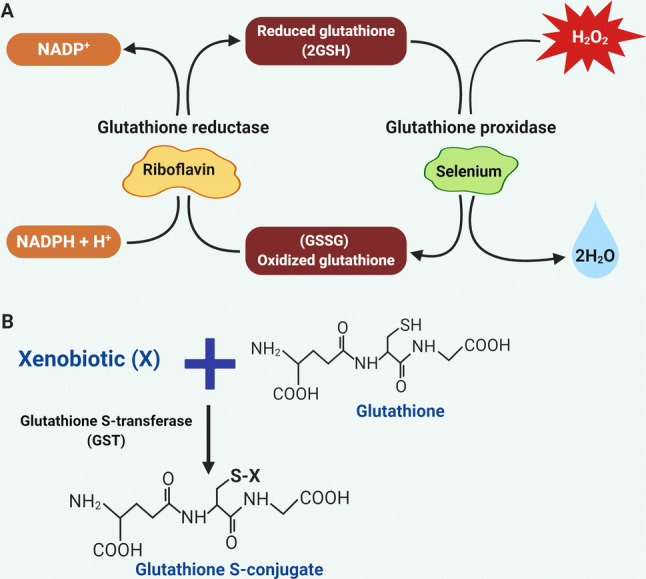
Glutathione antioxidant system. a Schematics for the reduction of hydrogen peroxide. Nicotinamide Adenine Dinucleotide Phosphate is essential for the regeneration of GSH via glutathione reductase. Hydrogen peroxide (H2O2) is reduced to water (H2O) via glutathione peroxidase. b Mechanism of the glutathione S transferases (GSTs). Glutathione conjugation with xenobiotic (X) is mainly catalyzed via GST to from glutathione S conjugate
Table 1.
Anticancer drugs or agents that directly or indirectly modulate reactive oxygen species in human malignancies
| Name of the drug or agent | Mechanism of the action or their target | Human malignancies | Status | References |
|---|---|---|---|---|
| Targeting antioxidant system, lipid ROS and ferroptosis | ||||
| Buthionine sulfoximine (BSO) | Block synthesis of GSH, induce lipid ROS | Ovarian cancer, breast cancer, melanoma, rhabdomyosarcoma | Approved | [51, 54, 55 177, 178, 182, 184] |
| Sulfasalazine (SSZ) | Inhibition of system Xc-, Induction of ferroptosis | Glioma, pancreatic carcinoma, lung carcinoma | Approved | [52, 54, 55, 194–196] |
| Artesunate (ART) | Induce ferroptosis through iron metabolism mediated lethal lipid ROS | Pancreatic carcinoma, ovarian cancer, lung carcinoma, head and neck cancer | Phase I/II | [51, 52, 54, 55, 192, 193] |
| Erastin | Inhibit VDAC2/VDAC3, block GSH synthesis, increase lipid peroxidation and lipid ROS | Fibrosarcoma, lung carcinoma, prostate cancer, osteosarcoma | Phase I/II/III | [51, 52, 54, 55, 184, 197] |
| Sorafenib | Inhibition of system Xc-, deplete GSH leading to accumulation of lipid ROS | Hepatocellular carcinoma | FDA Approved | [51, 54, 55, 191] |
| Cisplatin | Suppress GSH and GPX levels | Ovarian cancer, colon cancer | FDA Approved | [54, 200, 249, 250, 253, 254] |
| RSL-3 | Inhibit GPX4 and deplete GSH to induce ROS | Lung carcinoma, colon cancer | Clinical | [52, 54, 55] |
| ML-162 | Inhibit GPX4, enhances ROS production | Colon cancer, melanoma | [51, 54, 55, 202] | |
| ML-210 | Inhibit GPX4, increased ROS production | Lung carcinoma, colon cancer | [51, 54, 55, 202] | |
| FIN56 | Degrade GPX4 or inhibit the function GPX4 | Fibrosarcoma and transformed human fibroblast cells | [51, 53–55, 58, 59] | |
| FINO2 | Inhibit GPX4 | Fibrosarcoma, renal cell carcinoma | [51, 53, 54, 58] | |
| Lanperisone | Inhibition of system Xc-, enhance ROS production | Lung carcinoma, Kras-mutant mouse embryonic fibroblast | FDA Approved | [201, 202] |
| Artenimol | Promotes iron metabolism and ROS-mediated ferroptosis | Colon cancer, lung carcinoma | [51, 54, 55, 190] | |
| Salinomycin and Ionomycin | Iron-mediated ROS production | Breast cancer, colon cancer | FDA Approved | [202, 203] |
| Cotylenin A (CN-A) | Induces ferroptosis by increasing ROS | Pancreatic carcinoma | [54, 208] | |
| N-acetyl-l-cysteine (NAC) | Inhibit ROS production and ferroptosis via oxidative pathway | Fibrosarcoma, colon cancer, breast cancer | FDA Approved | [51, 55, 85, 182, 205, 206, 236] |
| Vitamin E | Inhibit ferroptosis via suppression of LOX | Knockout Gpx4 murine model | [54, 55, 205, 209] | |
| Ferrostatin | Inhibit ferroptosis via ROS generation from lipid peroxidation | Fibrosarcoma, murine embryonic fibroblast | [54, 56, 204, 205] | |
| Liproxstatin | Inhibit ferroptosis via ROS generation from lipid peroxidation | Murine hippocampal, fibrosarcoma | [54, 56, 204, 205] | |
| EUK-134 | SOD mimetic, inhibit H2O2 | Lung carcinoma, breast cancer | [1, 54, 180] | |
| NOV-002 | Modulate of intracellular GSSG/GSH ratio and increase oxidative stress, glutathione disulfide mimetic | Breast cancer, lung carcinoma | Phase I/II | [1, 7, 37, 180, 181] |
| DZNep (EZH2 inhibitor) | Silence thioredoxin and increases ROS | Acute myeloid leukemia | Phase I | [186] |
| All-trans-retinoic acid (ATRA) and arsenic trioxide (ATO) | Inhibit translocation of NRF2, enhance ROS | Leukemia, breast cancer, ovarian cancer | FDA Approved | [187, 188] |
| AEM1 | Repress transcriptional activation of NRF2 | Lung carcinoma | [189] | |
| Auranofin | Inhibitor of thioredoxin | Head and neck cancer, ovarian cancer, rhabdomyosarcoma | Phase I/II | [182–185] |
| Targeting mitochondrial and mitochondrial ROS | ||||
| Mitoquinone (MitoQ) | Mitochondrial respiratory chain complexes I, III, and IV to enhance ROS production | Renal cell carcinoma, Kras model of pancreatic carcinoma | [85, 157] | |
| MitoTEMPO | Activate SOD2 and inhibit mitochondrial superoxide | Renal cell carcinoma | [85, 157] | |
| Arsenic trioxide (AS2O3) | Enhance ROS production, inhibit the mitochondrial respiratory function | A promyelocytic leukemia, lung carcinoma, myeloma | FDA Approved | [217–220] |
| Paclitaxel | Increased mitochondria ROS that results in activation of STAT3 signaling | Lung Carcinoma, breast cancer | [117] | |
| Ivosidenib | Specific inhibitors for IDH1/2 mutant and target mROS for the anticancer effect | Acute myeloid Leukemia and Glioblastoma | FDA Approved | [215] |
| Enasidenib | Specific inhibitors for IDH1/2 mutant and target mROS for the anticancer effect | Acute myeloid Leukemia and Glioblastoma | FDA Approved | [215] |
| Disulfiram | Inhibit mitochondrial ALDH activity, activate the p38 pathway and ROS | Glioblastoma | FDA Approved | [146, 216] |
| 2-Deoxyglucose | Induce oxidative stress via accumulation of glutathione disulfide and NADP + /NADPH | Pancreatic carcinoma, Prostate cancer, cervical carcinoma | Phase I/II trials | [221–223] |
| Metformin | Mitochondrial complex I inhibitor, Inhibition of oxygen consumption, activate AMPK signaling | Hepatocellular carcinoma, murine cancer models (B16 for melanoma; MC38 for colon adenocarcinoma) | FDA Approved | [159–161] |
| Nutraceuticals | ||||
| Epigallocatechin-3-gallate (EGCG) | Modulation of ROS production, inhibition of NF-κB, regulation of MAPKs | Pancreatic carcinoma, colon cancer, breast cancer, lung carcinoma | Phase I/II | [227, 230] |
| Phenylethyl isothiocyanate (PEITC) | Deplete GPX and induce ROS | Bladder cancer, renal cell carcinoma, prostate cancer | In clinical trials | [179, 208] |
| Benzyl isothiocyanate (BITC) | Increase ROS production, activate JNK and p38 pathways | Pancreatic carcinoma, breast cancer, lung carcinoma | Phase I | [231, 234] |
| Vitamin A | Enhance ROS production | Ovarian cancer | [224] | |
| Vitamin C | Attenuated tumor growth in mutant Kras (G12D)/Apc murine models | Colorectal carcinoma, pancreatic carcinoma | In clinical trials | [225] |
| Vitamin D | Alteration in the ratio of GSSG and GSH, regulate thioredoxin-interacting protein | Endometrial cancer, breast cancer, colorectal carcinoma | In clinical trials | [226] |
| Bromelain | Downregulate CoA ligase 4, induce ROS in lipid membranes | KRAS mutant colon cancer | [207] | |
| Pancratistatin | Mitochondrial permeabilization increases ROS | Leukemia, colon cancer | Phase I | [239] |
| Aminoflavone | Enhance intracellular ROS by degenerating mitochondrial membrane potential | Pancreatic carcinoma, breast cancer, colorectal carcinoma | Phase II | [236] |
| Curcumin | Enhance intracellular ROS by increasing the potential of mitochondrial membrane | Almost all cancers | Phase II/III | [240, 241] |
| Nimbolide | Modulation of GSH/GSSG ratio leads to ROS production, inhibit STAT3 pathway | [242, 243] | ||
| β-Caryophyllene oxide | Suppress tumor growth and support apoptosis by suppressing ROS-mediated activation of MAPKs | Prostate cancer colon cancer Leukemia, lung carcinoma, multiple myeloma, Prostate cancer | [75, 244] | |
| Chemotherapeutic agents | ||||
| Doxorubicin, daunorubicin | Block DNA synthesis and topoisomerase II activity; inhibit complex I/II leading to an increase in the production of mitochondrial ROS | Acute myeloid leukemia, acute lymphocytic leukemia, breast cancer, chronic myelogenous leukemia, lymphoma, bladder cancer, Kaposi's sarcoma | FDA Approved | [245, 246] |
| Salvicine (SAL) | Inhibit topoisomerase II, GSH depletion trigger H2O2 production, DNA double-strand breaks | Gastric carcinoma, leukemia, cervical carcinoma | Phase I/II | [247, 248] |
| Carboplatin | Maintain very high levels of ROS to induce cell death | Breast cancer, ovarian cancer, lung carcinoma | FDA approved | [249, 250] |
| Oxaliplatin | Retain DACH by the formation of platinum–DNA adducts, block DNA replication | Colon carcinoma, Ovarian cancer, lung carcinoma | FDA approved | [249, 250] |
| Temozolomide (TMZ) | Inhibit autophagy, induces cell death via the accumulation of lipid ROS | Glioblastoma stem cells | FDA approved | [190, 197, 198] |
| PARP inhibitors (Olaparib, niraparib, rucaparib) | Inhibit the activity of PARP enzyme, enhance ROS mediated DNA damage | Breast cancer, ovarian cancer, pancreatic carcinoma, prostate carcinoma, lung carcinoma |
FDA approved European Medicines Agency |
[253–255] |
| 5-Fluorouracil (5-FU) | Inhibit thymidylate synthetase, block DNA and RNA synthesis, increase ROS | Colorectal carcinoma, breast cancer, pancreatic carcinoma | FDA approved | [251] |
| Vorinostat | Suppress SLC7A11, enhance ROS lead to DNA damage | FDA approved | [252] | |
Nuclear factor erythroid 2-related factor 2 (NRF2) is a well-known transcription factor. NRF2 is an important master regulator for maintaining redox balance while enhancing the expression of antioxidant proteins inside the cells [36, 37]. Under normal physiological conditions, NRF2 undergoes proteasomal degradation due to its ability to interact with Kelch-like ECH-associated protein 1(KEAP1), along with Cullin 3 (Cul3) E3 ubiquitin ligase [38, 39]. On the other hand, when there is an increase in the ROS levels during oxidative stress, KEAP1 gets oxidized and obstructs the binding of NRF2 to the KEAP1 degradation complex [40]. This leads to the stabilization of NRF2 in the cytoplasm and its translocation into the nucleus to drive the expression of several genes involved in antioxidants (PRXs, CAT GPXs), redox balance, detoxification, NADPH and GSH synthesis [1, 40–42] (Fig. 4). The constitutive activation of NRF2 has been observed in several human cancers including lung, breast, ovarian, skin, and prostate [41, 43–47]. Moreover, mutations in either KEAP1 or NRF2 and well-established oncogenes (KRAS, Myc) have been found to activate NRF2 [43, 46]. Deregulation in the NRF2–KEAP1 pathway has been reported in drug resistance, genomic instability, resistance to apoptosis, metastasis and metabolic reprogramming in several cancer cells [43, 46–49]. The depletion of NRF2 has displayed decreased tumor growth by enhancing oxidative stress-dependent cell death [40, 46, 47]. Therefore, therapeutic strategies that modulate TRX, PRX, GSH, GPX and NRF2 levels within tumor cells could increase the efficacy of anticancer therapies [50].
Fig. 4.

Role of the NRF2/KEAP1 antioxidant pathway for maintaining cellular homeostasis. Under normal physiological condition, NRF2 interact with KEAP1 to activate Cul3‐dependent ubiquitination and its degradation via the proteasome. Under stress or induced condition, NRF2 dissociates from KEAP1 and translocates into the nucleus. NRF2 forms a heterodimer with sMaf protein as well as to ARE to initiate the transcription of several downstream genes
Role of lipid ROS and ferroptosis in human malignancies
Regulated or programmed cell death is an important process and is required for several key biological processes including development and cellular homeostasis. Programmed cell death can be achieved either via apoptosis or non-apoptotic pathways, including ferroptosis [51–53]. Ferroptosis can be easily distinguished from other types of programmed cell death such as apoptosis, necrosis, and autophagy based on morphology and biochemical reaction [51–54]. Ferroptosis is a different class of cell death that relies on iron metabolism and lipid ROS [51, 52, 55]. Ferroptosis has shown to be initiated either with the depletion of cysteine or loss of glutathione peroxidase 4 (GPX4, an enzyme involved in lipid repair). The loss of GPX4 has been noticed with the accumulation of peroxides in the lipid membrane that leads to aggregation of destructive lipid ROS. The knockout of the Gpx4 gene in the murine model has been observed with increased lethal lipid ROS [56]. Also, the silencing of GPX4 in human cells has been found to induce the accumulation of lipid ROS and ferroptosis cell death [57]. Further, pharmacological inhibitors such as FIN56, FINO2, and RSL3 have reported to either degrade GPX4 or inhibit the function of GPX4 [51, 53–55, 58, 59]. Accumulation of fatal lipid ROS has been noticed with stimulation of polyunsaturated fatty acids (PUFAs) through long-chain fatty acid—CoA ligase 4 (ACSL4) and their addition within the membrane lysophospholipids [60]. However, several reports have proved beyond doubt that the peroxidation of PUFAs is catalyzed by lipoxygenases (LOXs) enzymes [61] (Fig. 5). Moreover, the suppression of system Xc− (erastin or RSL3) linked with indirect repression of GPX4 enzymatic activity [52]. System Xc− belongs to the cystine/glutamate antiporter system, which is associated with the import of extracellular cystine to replace intracellular glutamate [62]. Cysteine (reduced form of cystine) acts as a precursor for the synthesis of glutathione (GSH). GSH functions as a cofactor for GPX4 to catalyze the inhibition of lipid peroxides. The impairment of system Xc− using small molecules displayed an aggregation of lethal lipid peroxides and ROS that led to ferroptosis [57] (Fig. 5).
Fig. 5.

Mechanism and inducer of ferroptosis. Suppression of system Xc−/GPX4 activity caused ferroptosis to induce cell death. Elevation of lipid ROS results in the ferroptopsis
Role of ROS in the activated signaling pathways in human malignancies
Human malignancies are one of the major causes of deaths, more than tuberculosis, malaria and acquired immune deficiency syndrome around the world [63]. Cancer is a genetic and metabolic disorder that arises from internal factors (inherited mutations, translocations, abnormal activation of signaling pathways initiated by growth factors and hormones, immune conditions) and external factors (environment, infection, food, alcohol, tobacco, radiation) [63–67]. Both these factors can influence critical genes including proto-oncogenes, tumor suppressor genes, DNA repair, and cell cycle genes through the formation of cellular intermediates such as ROS [68]. The association between ROS and cellular transformation was unveiled by initial studies, where activating RAS mutations and growth factors (epidermal growth factor (EGF), insulin) pathways can enhance the intracellular levels of H2O2 to induce tumor growth [69–71]. Now, it is more evident through the laboratory experiments that ROS can lead to carcinogenesis, either by activation of several oncogenic pathways or through oncogenic mutations in the DNA. In this section, we focus on the most relevant signaling pathways such as MAPK/extracellular regulated kinase (ERK)/c-jun N-terminal kinase (JNK) pathway, PI3K/AKT/mTOR pathway, ROS in the NF-κB pathway, signal transducer and activator of transcription (STAT) signaling affected by ROS in cancers.
The MAPK family consisting of ERK1/2, JNK and p38 MAPKs pathways are intracellular signaling pathways required for cellular growth, differentiation and survival. ROS have been shown to oxidize and deactivate MAPK phosphatases, while activating the epidermal growth factor receptor (EGFR) and platelet-derived growth factor receptors (PDGFR) signaling in a ligand-independent fashion through the RAS and ERK pathways [72–78]. Several other studies have demonstrated that H2O2 is an important mediator for ligand-independent phosphorylation of receptor tyrosine kinases (RTKs) [79, 80]. For example, metabolism of estrogen in breast carcinoma results in the production of H2O2 which in turn activates ERK1/2 to increase cellular proliferation and survival. Mutant HRAS (G12V)-transformed NIH/3T3 fibroblast cells have been shown to generate a huge amount of O2− via RAC1 [81]. Moreover, ROS can activate HRAS, NRAS, and KRAS oncogenic switch through oxidation of the cysteine residue [82]. Weinberg and colleagues have observed that mitochondrial ROS (mROS) is essential for Kras-mediated tumorigenesis in murine lung carcinoma model via the ERK–MAPK signaling pathway [83]. Mitochondrial transcription factor A (TFAM) is important for the replication of mitochondrial DNA, and depletion of TFAM suppressed the growth of lung tumors in Kras murine models. Moreover, TFAM heterozygous knockout mice have elevated mROS levels and showed increased intestinal tumors in APC Min/+ murine model, suggesting the pivotal role of mROS in carcinogenesis [84]. Similarly, KRAS (G12D, G12V) mutation induces mROS and activates various signaling pathways in the acinar cells for the progression of pancreatic carcinoma [85, 86]. Inhibition of ROS using NAC and MitoQ showed a marked reduction in the initiation and progression of pre-cancerous lesions in Kras-driven murine models of pancreatic cancer [85]. On the contrary, activation of ERK1/2 signaling with exogenous H2O2 displayed apoptosis in human pancreatic carcinoma and glioma because of the extremely high level of ROS. Excessive levels of ROS have been found to be positively correlated with senescence, cell cycle arrest, and apoptosis through the ASK1/JNK/p38 signaling cascade [87]. ASK-1 (apoptosis signal-regulated kinase-1) and reduced TRX form a complex that results in the inactivation of ASK-1. During excessive stress, H2O2 has been shown to oxidize cysteine residues of TRX, leading to dissociation of ASK-1 for activation of JNK and p38 cascade leading to apoptosis [87–89]. Similarly, glutathione S-transferase P dissociates from JNK to facilitate JNK activation under elevated ROS/H2O2 [90] (Fig. 6). Higher levels of H2O2/ROS result in prolonged activation of the JNK/p38 that can prevent the proliferation of tumor cells [89–92].
Fig. 6.

ROS activate RAS and PI3/AKT signaling pathways. Growth factor receptor signaling can generate ROS through growth factors, NOXs and mitochondria. ROS can activate RAS/MAPK and PI3K/AKT/mTOR signaling cascade either though inactivation of phosphatases such as PTEN or PTP at cysteine residues or by direct oxidation of kinases. Other mechanisms by which ROS induce cellular signaling are through activation NF-kB signaling
PI3K/PTEN is another important signaling pathway in the tumorigenesis and metastasis where several key intermediates are highly sensitive to redox dysregulation [23, 93]. ROS (O2− and H2O2) can hyperactivate the PI3K/AKT/mTOR pathway through oxidation of the cysteine thiol group of various phosphatases (PTEN, PTP1B, PP2A), resulting in their inactivation [94–97]. Moreover, ROS can indirectly phosphorylate casein kinase II, which promotes degradation of PTEN protein via proteasomes. PTEN is mostly dysregulated in breast, glioblastomas, melanoma, endometrial and prostate cancers because of an increase in ROS (O2− and H2O2) production to favor tumor cell growth and survival [48, 98, 99]. H2O2 is generated during the binding of estrogen and growth factors (EGF, PDGF) to their respective receptors (Fig. 6). This has been displayed to activate the PI3K/AKT signaling in breast and ovarian carcinoma [100]. NRF2 protein binds to KEAP1 (E3 ubiquitin ligase) protein to maintain low levels of NRF2 protein in the cytosol under lower concentrations of cellular ROS, whereas high concentrations of ROS lead to oxidation at the cysteine residues of KEAP1, which allows cytosolic NRF2 to translocate into the nucleus to upregulate the expression of antioxidants. Also, activation of PI3K/AKT signaling is essential for the nuclear transportation of NRF2. PI3K inhibitors (LY294002, wortmannin) suppressed NRF2-dependent upregulation of antioxidant genes in neuroblastoma cells [101]. BRCA1 mutant breast tumors are deficient in DNA repair mechanisms and accumulate more ROS, leading to genetic modification. BKM120 treatment impedes estrogen-dependent activation of NRF2-mediated PI3K/AKT signaling, indicating that BRCA1-deficient tumors can be treated by elevating ROS levels [102].
NF-κB is a major TF which plays a critical role in inflammation, cellular proliferation, differentiation, and various immunological responses [103–107]. The NF-κB protein expression has been observed to be triggered via H2O2 [108]. For instance, treatment of breast carcinoma cells with IL-1β, TNFα, or sodium arsenite generates H2O2 and O2−, which in turn activate NF-κB and enhance cellular growth [109, 110]. Interestingly, knockdown of superoxide dismutase (SOD) showed an increase in the basal ROS levels and NF-κB activity in oral carcinoma. It has been reported that IKK-based NF-κB signaling is activated by increased cellular oxidative stress either by H2O2, rotenone-mediated O2− or by inhibition of the glutathione system. On the other hand, IKK-independent activation of NF-κB occurs through phosphorylation of IκBα at tyrosine residue in response to ROS which releases NF-κB [108] (Fig. 6). ROS have been found to activate NF-κB and NRF2 to support cancer cell survival by increasing the levels of antioxidants to escape cancer cell death in an ROS-dependent fashion [108, 111]. Mutant KRAS generates mROS and activates NF-κB through PKD1, which leads to the formation of precancerous lesions in the pancreas [85].
It has been well established that tumor undergoes metabolic reprogramming due to oxidative phosphorylation (OXPHOS) to control energy requirements. Particularly, tumors addicted to oncogene and drug resistanve have been noticed to rely on ROS/OXPHOS-mediated STAT3 signaling as an alternative mechanism for their survival. Several signaling pathways coincide with STAT3; therefore, translocation of STAT3 to the mitochondria can extend the connection across oncogene-mediated signaling pathways and cancer cell metabolism [112–116]. Radiotherapy treatment has been noticed with markedly lower ROS and elevated protein expression of phospho-STAT3, along with BCL2 in triple-negative breast cancer (TNBC) and radio-resistance. Uncoupling protein 2 (UCP-2) is responsible for reducing ROS levels. UCP2 is highly upregulated to maintain low mROS and resistance to paclitaxel in epithelial lung carcinoma (A549, H460). Paclitaxel resistance was reversed by the silencing of UCP-2 through the STAT3 pathway [117]. Further, niclosamide (STAT3 inhibitor) or STAT3 silencing sensitized the TNBC cells via induction of ROS and inhibition of BCL2 [118]. NOX4 is robustly expressed in NSCLC cells and helps in ROS-dependent IL-6 secretion, which eventually phosphorylates STAT3 (Y705). On the other hand, NOX4 knockdown proved that reduced H2O2 inhibited IL-6 dependent STAT3 activity. Also, exogenous IL-6 showed STAT3 activation via NOX4 (Fig. 7). This suggests a positive loop among NOX–ROS–IL-6 and STAT3 [119]. The STAT5 signaling pathway is activated in acute myeloid leukemia (AML) with FLT3/ITD. FLT3/ITD expression in AML has been noticed with increased H2O2 in a NOX-dependent manner [120]. FLT3 inhibitor (PKC412) and NOX inhibitors (DPI, VAS2870) have been shown to inhibits ROS production in FLT3/ITD expressing AML cells [121]. STAT5 expression has a positive link with BCR-ABL mutation in chronic myeloid leukemia (CML). STAT5 upregulation has been noticed with high ROS and more BCR-ABL mutation in CML cells. STAT5-induced ROS led to double-strand DNA breaks and witnessed by γH2AX [122].
Fig. 7.

ROS-dependent STAT3 pathway in metastasis and drug resistance. Growth factors, ionizing radiation, mitochondria and NOX4 result in the production of intracellular ROS. ROS activate cancer cells and cancer-associated fibroblast cells to secrete IL-6. IL-6 activates the STAT3 pathway and promotes tumor metastasis, resistance to chemotherapy and radiotherapy, and CSC self-renewal
ROS as an important regulator of telomerase
Human telomerase reverse transcriptase (hTERT) is localized in mitochondria and is important for mitochondrial function [123, 124]. hTERT is critical for respiratory chain function and to maintain low ROS [125–127]. In hepatocellular carcinoma, there is a marked increase in the ROS levels from early to late stage which is positively correlated with increased telomeres length. It has been observed that H2O2 extends telomeres by enhancing telomerase activity through AKT signaling in HCC, lung cancer and leukemias. Interestingly, there is a positive association between ROS levels, phosphorylation of AKT, length of telomere and prognosis in human cancers [128]. AKT inhibitors (perifosine, GSK690693, SH-6, and MK‐2206) displayed compromised telomerase activity as well as shortening of telomere length while decreasing ROS levels, viability, H2O2-mediated migration and invasion in human malignancies [129, 130]. Now, this is known that mitochondrial TERT can increase intracellular-reduced glutathione to escape ROS-mediated apoptosis [131, 132]. Translocation of hTERT from the nucleus to mitochondria results in multidrug resistance in cancers due to reduced ROS which provides protection to mtDNA. Elevated levels of H2O2 have been found to be associated with the shortening of the telomere [133, 134].
ROS is essential for metastasis, angiogenesis and cancer stem cell
Metastasis is the major cause of mortality and only limited number of cells can metastasize to distant organs [135]. Growing pieces of evidence witness the fact that higher levels of ROS are vital to facilitate and sustain the aggressive metastatic phenotype of cancer cells [136]. NOX-dependent ROS/NF-κB pathway accelerates migration and invasion of tumor cells by enhancing TGF-β1, uPA and MMP-9 expression [137]. Mutant TP53 was observed to enhance Nox4-dependent metastasis either through TGF-β1 or independent of TGF-β1 signaling. Treatment of colon carcinoma cells with H2O2 stimulated MMP-7 production in an AP1-dependent fashion. Also, ROS can lead to the overexpression of MMP1/2/9 to enhance metastasis. Other reports have displayed that activated integrin-Rac signaling can efficiently generate ROS which results in migration, invasion and epithelial to mesenchymal transition through MMP-3. Matsuno and colleagues found that ROS-activated Nrf2 leads to EMT and metastasis via Notch signaling. ROS can activate TGF-β1 through the TAK1 (TGF-β-activated kinase 1) pathway to metastasize the cancer cells to another organ. NRF2 and ATF4 are involved in antioxidant response by enhancing glutathione synthesis and heme oxygenase 1 to bypass oxidative stress, promoting survival during metastasis by blocking anoikis. Addition of either H2O2 or SOD in culture medium displayed EMT phenotype where TWIST1, vimentin and SLUG were upregulated and E-cadherin was downregulated in human malignant mesothelioma and pancreatic carcinoma cells, respectively [138]. It has been noticed that ROS stimulates tumor cells and stromal cells to secrete IL-6, which in turn activates STAT3 signaling and triggers EMT and drug-resistant phenotype by altering the protein expression of E-cadherin, N-cadherin, vimentin, and snail (Fig. 7). ROS activate NF-κB to maintain CSCs and cause resistance to chemotherapy and radiotherapy [119]. Also, the silencing of thioredoxin-like 2 (TXNL2) showed decreased mammosphere formation, metastasis, and tumor growth by inducing ROS levels and suppressing NF-kB activity in breast carcinoma [139].
In normoxia, HIF-1α is degraded due to hydroxylation of PHD2 and recognition through von Hippel–Lindau protein. H2O2 has been shown to contribute to metastasis and angiogenesis through the stabilization of HIF and activation of one-carbon metabolism as well as AMPK signaling networks to enhance NADPH production [140]. Hypoxia triggers mROS production which stabilizes HIF-1α subunit by forming a dimer along with HIF-1β to drive the expression of hypoxia-responsive genes to increase angiogenesis in tumor mass [141] (Fig. 8). AKT activation results in the formation of superoxide and H2O2, which turn on HIF-1 and induce VEGF expression [142]. Notably, H2O2 can promote angiogenesis via the Ang1 and p44/42 MAPK axis. Nox2-generated ROS induces the migration of endothelial cells to tumor mass to promote angiogenesis through several pathways such as PI3K/AKT, Src, and ERK [143]. Importantly, ROS have been noticed to regulate the expression of several TFs and remodeling proteins (p300, VEGF-A, HIF-1α, p53, and MMPs) essential for angiogenesis [144].
Fig. 8.

Mitochondrial ROS in hypoxia and angiogenesis. In oxygen-rich conditions, HIF-1α forms complex with VHL with the help of PHD2. This results in ubiquitination and proteasome-mediated degradation of the complex. On the other hand, mROS can cause the depletion of oxygen levels and inhibition of PHD2 activity resulting in HIF-1 α stabilization, by forming a dimer with HIF-1β. This dimer moves to the nucleus and results in transcriptional activation of VEGF, EPO
Cancer stem cells (CSCs) are correlated with clinical hallmark features such as resistance to therapy, tumor recurrence and metastasis [86, 145, 146]. CD44-positive leukemic stem cells (LSC) have lower ROS because of PKC-θ silencing by NOTCH1 [147]. The frequency of LSC in AML has been correlated with the expression of Gpx3 (ROS scavenger) to keep lower ROS [148]. In breast carcinoma, the Snail-G9a-DNMT1 complex pauses the promoter of E-cadherin and for promoter methylation of fructose-1,6-biphosphatase (FBP1). The silencing of FBP1 cuts down oxygen utilization as well as ROS due to compromised mitochondrial oxidative phosphorylation (OXPHOS). This increases CSC-like properties and tumorigenicity through β-catenin [149, 150]. On the contrary, CSCs are known to have high mROS, which helps them to alter the metabolic reprogramming through fatty acid β-oxidation and MAPK signaling, leading to transcriptional activation of EMT markers in several cancers [151, 152]. Several studies provided evidence that low ROS in CSCs helps them to overcome the effect of chemotherapeutic drugs. These suggested that low levels of ROS are needed to preserve LSC/CSCs.
Role of ROS in the immune response during tumor progression
The tumor microenvironment is composed of myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs) and tumor-associated macrophages (TAMs). MDSCs, Tregs cells and TAMs provide an immune-suppressive environment for tumor growth, metastasis, invasion and resistance to chemotherapeutics drugs. CD8+ T cells are crucial for anticancer immune response in the tumors. Nonetheless, the tumor microenvironment creates an immunosuppressive environment which eventually results in the suppression of CTL response, leading to cancer progression. High ROS have been noticed as one of the major factors for immunosuppression and inhibition for T cell activation and proliferation, while low ROS can bring the T cell back into action inside the tumor microenvironment. Complexes I and III of the mitochondrial electron transport chain (ETC) are excellent sources of mROS and T cell activation [153–155]. Tumor-infiltrating T cells can be activated by overexpression of PGC1α which is involved in the biogenesis of mitochondria and resumes anticancer activity [156]. ROS scavengers such as MitoQ and MitoTEMPO enhance CD8+ tumor-infiltrating lymphocyte activation in kidney tumors by activating SOD2 [157]. T cells expressing chimeric antigen receptor (CAR) and CAT have been shown to be correlated with decreased intracellular oxidative stress and an increased ability of T cells (CAR-CAT) to kill cancer cells [158]. CAR-CAT T cells showed better antitumor response than traditional CAR T cells even under extracellular oxidative stress [158]. Program Death receptor 1 (PD-1) is a negative regulator of the immune system, which is present on the surface of T cells. PD-1 can efficiently bind to either PD-L1 and/or PD-L2, which results in the recruitment of SHP2 and inhibits cytotoxic T-lymphocytes (T-CTLs) to mediate killing of cancer cells. It has been observed that T-CTLs extracted from murine treated with PD-L1 antibody have elevated O2− and cellular ROS. Further, exposure of these cells to tert-butyl hydroperoxide or a mitochondrial respiratory chain uncoupler showed a synergistic reduction in tumor growth. It has been observed that when HCC xenografts were treated with metformin, oxygen consumption was inhibited in murine tumors, leading to enhanced oxygen supply inside the tumor cells. This results in decreased levels of intratumor hypoxia by suppressing the expression of HIF-1α in HCC xenograft [159]. The combination of metformin with PD-1 blockade markedly enhanced intratumor T cell activation and proliferation, leading to tumor clearance through alleviation of tumor hypoxia [160]. This observation suggests that non-responders to PD-1 antibodies might have high mROS and less hypoxic microenvironment, which results in compromised CTL response. Several studies have observed that elevated ROS or oxidative stress led to immunosuppression inside the tumor microenvironment through Tregs. Furthermore, Tregs hinder the therapeutic ability of the PD-L1 antibody in murine cancer models. Kunisada and colleagues have evaluated that metformin (complex I inhibitor) decreased the number of tumor-infiltrating Tregs by reducing the differentiation ability of the naïve CD4+ T cells into Tregs via Foxp3 (the transcriptional regulator for metabolic reprogramming) [161]. Weinberg and group have demonstrated that mitochondrial complex III is needed for inhibiting Treg function [162]. It is clear from the above studies that more research is required to discover the key mechanisms of ROS involved in extracellular and tumor-infiltrating cells in modulating tumor immunity. MDSCs are immunosuppressive cells within the tumor microenvironment (TME). Tumor-induced MDSCs showed a block in T cell proliferation and support colorectal carcinoma cell growth through the production of ROS [163]. Interestingly, catalase (ROS inhibitors) rescued the activity of T cells by suppressing the negative effect of MDSCs [164]. On the contrary, high ROS inhibits T cell responses by suppressing the formation of TCR and MHC antigen complex [165]. TAMs are present within the TME and are important moderators of inflammation and carcinogenesis. ROS are involved in the activation of macrophage signaling. ROS generated from macrophages have been shown to induce Tregs [166]. Another study displayed that ROS promote an invasive phenotype in TAMs extracted from skin cancer (melanoma) through secretion of tumor necrosis factor α [167]. It has been observed that several key mitochondrial genes are highly expressed in TAMs obtained from melanomas, suggesting mROS is the major source of oxidative stress within TAMs. Now, it is very clear that ROS is not only involved in oxidative stress, but also important in immune modulation in human malignancies (Fig. 9).
Fig. 9.

Involvement of ROS in tumor microenvironment and immunosuppression. Myeloid-derived suppressor cells (MDSCs) are generated due to secretion of growth factors (GM-CSF, M-CSF, VEGF) and pro-inflammatory cytokines (IFN-ϒ, IL-1β, IL-4, TNFα by tumor cells. MDSC secrete ROS, nitric oxide (NO) and arginase (ARG) to inactivate T cell and TGFβ, and IL10 to activate regulatory T cells (Tregs). ROS convert M0 macrophages into TAMs and secrete immune-suppressive factors and cytokines to block NK and CTLs. Tumor cells and stromal cells express TGFβ, checkpoint ligands and FasL to cause T cell apoptosis. ROS help tumor cells to overexpress PDL1/2 and CTLA4 to inhibit CTLs. TGFβ stimulates NOXs within the Treg cells to trigger ROS production. Macrophage-induced ROS leads to the accumulation of Treg cells. MDSC produces a large amount of ROS to trigger Tregs and suppress T cells
Importance of ROS in the gut microbiome
It is universally accepted that host microbiota can support tumorigenesis via induction of pro-inflammatory toxins, signaling pathways or escape of antitumor immune functions. Interestingly, several host–microbiota have been associated with the generation of ROS, leading to tumorigenic state [168, 169]. Enterococcus faecalis have been shown to generate extracellular O2−, which is converted to H2O2 and can damage DNA in eukaryotes [170]. Bacteroides fragilis generate toxin, which is required for bacterial growth while maintaining polyamine catabolism. This is the major cause of ROS production, DNA damage and tumor initiation in the colon [171]. Several groups have shown that diverse species of bacteria can consume bile acid for their growth and generate ROS as a by-product which induces gastrointestinal cancers and DNA damage [172, 173]. On the contrary, damaged mucosal epithelium utilizes low redox/ROS signaling for repair [174]. It has been observed that host mROS decide diversity in the gut microbiome [175]. High-throughput sequencing of gut microbiota discovered mutations in different genes, leading to change in mitochondrial function and composition of the gut microbiota. Furthermore, modulation of ROS levels displayed higher diversity in the murine gut microbiota [175]. In a recent report, it has been noticed that melanoma patients, who respond well to immunotherapy, have displayed an increase in the diversity of gut microbiota [176]. These studies indicate that modulation of mROS could be used to increase the sensitivity of immunotherapies in cancer patients in clinics.
Role of ROS and ROS scavengers/antioxidants in cancer prevention and treatment
Several chemotherapeutic approaches are designed with the aim of increasing intracellular ROS levels to increase unrepairable damages which result in apoptosis of tumor cells. This is one of the promising approaches which can be easily achieved via chemotherapeutic drugs and radiotherapy depending on the origin of the tumor.
Drugs or agents affecting antioxidant system, lipid ROS and ferroptosis
GCL is an important rate-limiting enzyme in GSH synthesis. GSH metabolism has been displayed to enhance drug resistance by preventing cell death of the tumor cells. Buthionine sulfoximine (BSO) is a well-known inhibitor of de novo GSH synthesis and is clinically used for melanoma, ovarian and breast cancers [177, 178]. Phenylethyl isothiocyanate depletes GPX, and GSH has been reported with anticancer effect in preclinical ovarian cancer murine model [179]. EUK-134 (SOD mimetic) and NOV-002 (glutathione disulfide mimetic) are the antioxidants under clinical development for clinical practice in cancer and other diseases [180]. NOV-002 was injected in patients with HER2-negative breast carcinoma in combination with doxorubicin/cyclophosphamide/docetaxel and demonstrated a favorable antitumor activity with manageable side effect than adjuvant therapy [181]. Auranofin is a well-known inhibitor of thioredoxin and used as an antirheumatic drug in clinics. Importantly, the combination of auranofin with BSO showed enhanced sensitivity in head and neck cancer toward EGFR inhibitors and this effect was reversed in the treatment with NAC [182]. In another study, auranofin treatment of cisplatin-resistant ovarian cancer cells resulted in cytochrome c-mediated cell death via attenuation of TRX reductase [183]. BSO or erastin in combination with auranofin has displayed synergistic anticancer activity in rhabdomyosarcoma by increasing the ubiquitination of proteins [184]. Auranofin inhibited side population, expression of stem cell markers as well as the ability to initiate tumors in lung cancer xenograft model [185]. TRX interacting protein is one of the crucial targets of polycomb-repressive complex 2 and is silenced in AML. DZNep (EZH2 inhibitor) treatment restores the TRX-interacting protein expression, which in turn inhibits thioredoxin and increases ROS, leading the way to apoptosis in AML [186]. These data highlight the importance of thioredoxin metabolism in the survival of cancer cells [183]. Particularly, combination therapy using antioxidants with therapeutic drugs that strongly trigger apoptosis independent of oxidative stress may be effective. Combined treatment of all-trans-retinoic acid (ATRA) and ATO has been reported to prevent the translocation of NRF2 into the nucleus and displayed significant cell death in leukemia and breast cancer cells [187]. ATRA sensitizes the CSCs in ovarian cancer by inhibiting NRF2 and ALDH1 activity [188]. AEM1 showed promising anticancer activity in lung carcinoma by repressing transcriptional activation of NRF2 at ARE site in the nucleus [189]. However, the major challenge for suppressing NRF2 is specificity and toxicity. Sulfasalazine (SSZ), artesunate (ART), erastin, temozolomide (TMZ), sorafenib, BSO, lapatinib, altretamine, ML-162, RSL-3, ML-210, and ATRA are well-known inhibitors for induction of ferroptosism [51, 52, 54, 55, 190]. Sorafenib was initially discovered as an inducer of ferroptosis in hepatocellular cancer cells [191]. Mechanistically, sorafenib depletes GSH along with the accumulation of lipid ROS [191]. ART has been shown to induces ferroptosis in human cancer cells including pancreas, head and neck, and ovarian through iron metabolism-mediated ROS [192, 193]. SSZ induces ferroptosis in glioma cells (GBM), pancreatic carcinoma and lung carcinoma via inhibition of system Xc− [54, 194–196]. Erastin triggered ferroptosis in fibrosarcoma, lung, prostate, and osteosarcoma cells [197]. TMZ in combination with erastin can be a potential therapeutic agent in GBM [197]. TMZ inhibits autophagy in glioblastoma stem cells and induces cell death via the accumulation of lipid ROS [190, 198, 199]. Cisplatin exerts an anticancer effect in HCT116 (colon cancer) and A549 (lung cancer) cells through apoptosis via reduced GSH and GPX [200]. Lanperisone enhances the production of ROS to induce ferroptotic death in K-Ras-mutant mouse embryonic fibroblasts and lung cancer cells in the mouse model [201, 202]. Moreover, salinomycin and ionomycin are clinically approved antibiotics that promote ferroptosis in colon and breast cancer cells through iron metabolism-mediated ROS [202, 203]. Ferrostatin, liproxstatin and zileuton have been reported to suppress erastin and RSL3-induced ferroptosis in fibrosarcoma, murine hippocampal and murine embryonic fibroblasts [54, 56, 204, 205]. Several natural compounds including bromelain, baicalein, artenimol, artemisinin, cotylenin A (CN-A), N-acetyl-l-cysteine (NAC) and vitamins can control cell death via ferroptosis, lipid peroxidation and ROS production [52, 54–56, 190, 206–209].
Drugs or agents affecting mitochondria and mitochondrial ROS
IDH1/2 are mutated in blood cancers and brain tumors and result in the formation of 2-hydroxyglutarate (oncometabolite) [210–213]. In the Idh1 mutant knock-in murine model, there is a decrease in the intracellular ROS, leading to an increase in the NADP(+)/NADPH ratio and expression of Hif1α target gene in brain and hematopoietic cells [214]. The lower levels of ROS have been associated with metabolism and overexpression of BCL2 protein in leukemic stem cells in IDH1/2 mutant AML. Ivosidenib and enasidenib are specific inhibitors for IDH1/2 mutant and target mROS for the anticancer effect. These inhibitors showed promising antileukemic activity in patients with AML in clinical trials and are approved by the FDA for the treatment of elderly AML patients [215]. Disulfiram, an ALDH inhibitor in combination with copper (Cu), has been reported to inhibit cancer stem cells and tumor growth of GBM cells via suppression of mitochondrial ALDH activity and generation of ROS along with the activation of p38 pathway [216]. Disulfiram/Cu specifically eliminates leukemia-initiating cells by silencing of NRF2/NF-kB cascade and elevating ROS-dependent JNK pathway [146]. Arsenic trioxide (AS2O3) is one of the most successful FDA-approved therapies for leukemia, lung, and myeloma [217, 218]. AS2O3 exposure enhances ROS production and is sensed by PML to enhance nuclear body formation which eventually activates p53 to induces differentiation and cell death of leukemic cells [217, 219]. AS2O3 combined with ascorbic acid in phase 1 study and was found to be effective against patients with relapsed/refractory multiple myeloma [220]. Paclitaxel treatment revealed an elevated level of ROS through mitochondria which results in activation of STAT3 and JAK2 through phosphorylation in lung carcinoma cells, leading to BCL-2 mediated programmed cell death [117]. 2DG (2-deoxyglucose; glucose analog) has been shown to impede glucose metabolism that results in the accumulation of GSSG to induce oxidative stress. This was associated with radio-sensitization and marked apoptosis in a variety of cancers including pancreatic, prostate and cervical [221–223].
Nutraceuticals with antioxidant properties
Importantly, the intake of natural antioxidant-rich foods has been recommended as one of the best ways to protect against cancer. Several nutrients (vitamins A, C, and D, epigallocatechin-3-gallate (EGCG), genistein, curcumin, piperine, theanine, and choline) have strong antioxidant properties and have been found to control the expansion of cancer stem cells and tumorigenesis in pancreatic, ovarian, breast, colorectal and brain tumors. Wang and colleagues have performed a meta-analysis in a large cohort to find out the correlation between vitamin A and patients with ovarian cancer [224]. KRAS or BRAF mutations are the most recurrent mutations in colorectal carcinoma. It has been observed that high doses of vitamin C showed selective killing of colorectal cancer cells having either KRAS or BRAF mutations because of increased uptake of the dehydro-ascorbate (DHA, the oxidized form of vitamin C) through GLUT1 [225]. This led to the accumulation of ROS, inhibition of glyceraldehyde 3-phosphate dehydrogenase, energy crisis. Interestingly, vitamin C attenuated tumor growth in mutant Kras (G12D)/Apc murine models [225]. More recently, Grant has observed that vitamin D can lower the risk of colorectal and breast cancer, whereas it was the opposite in prostate cancer [226]. Yang and colleagues have reviewed the role, molecular mechanism and signaling pathways of EGCG in several murine cancer models as well as in human cancers [227]. To date, there is a limited therapeutic option for pancreatic cancer which includes gemcitabine in combination with trichostatin A, EGCG, benzyl isothiocyanate (BITC), and capsaicin [227–233]. The above drugs are known to increase intracellular ROS levels to promote apoptosis. BITC operates through ROS-dependent ERK/JNK/p38MAPK and G2/M arrest by reducing cyclin B1, Cdc2, and Cdc25C in pancreatic and other cancer [231, 234]. EGCG treatment suppressed the expression of the BCL-2, IAP, BCL-XL, and cIAP (antiapoptotic) and enhanced the expression of the BAD, FAS, and BAX pro-apoptotic [230]. Sulindac is the FDA-approved drug that enhances intracellular ROS levels in colorectal and lung cancer cells which makes them sensitive to H2O2-mediated apoptosis [235]. Aminoflavone induces cell death in breast cancer cells (MCF7, MDA-MB231), but is non-toxic in MCF-10A (non-malignant breast cells). Aminoflavone displayed a marked increase in intracellular ROS and was significantly correlated with the activation of caspase 3-mediated cell death. Further, inhibition of ROS production using NAC reverses the effect of amino flavone [236]. NAC treatment suppressed migration, invasion, and EMT through matrix metallopeptidase 3. Pancratistatin, IOA, thymoquinone, and Triphala induce apoptosis of breast carcinoma cells by enhancing intracellular ROS by increasing the potential of mitochondrial membrane [113, 237–239]. Curcumin is a well-known natural antioxidant that has been used as an anticancer agent in almost all human malignancies. Curcumin at lower concentrations has been correlated with reduced ROS production, while curcumin at higher concentrations displayed increased ROS levels in leukemia and solid tumors [240, 241]. Nimbolide has been found to induce oxidative stress, which caused delay in tumor growth in the transgenic prostate cancer model via STAT3 signaling [242, 243]. β-Caryophyllene oxide has been shown to suppress tumor growth and support apoptosis by suppressing ROS-mediated activation of MAPKs [75, 244].
Chemotherapeutic drugs or cytotoxic agents
Anthracyclines and topoisomerase inhibitors such as doxorubicin, adriamycin, daunorubicin, and epirubicin have been reported with anticancer activity in both solid and blood cancers, because these drugs can block DNA synthesis, topoisomerase II activity and complex I/II leading to increase in the production of mitochondrial ROS [245, 246]. Salvicine (SAL) is a known topoisomerase II poison that has been successful in clinical trials for cancer patients. SAL triggers H2O2 production, DNA double-strand breaks which induce G2M arrest and apoptosis in cervical carcinoma, leukemia and gastric carcinoma [247, 248]. Platinum-based drugs including cisplatin, carboplatin, oxaliplatin and other alkylating drugs are known for maintaining very high levels of ROS to induce cell death in several human malignancies [249, 250]. On the other hand, nucleotide analogs, antimetabolites, taxanes, and alkaloids treatments eliminate cancer cells by maintaining low ROS. The 5-fluorouracil (5-FU) is FDA approved for the treatment of patients with various malignancies. 5-FU sensitizes the tumors by producing mROS in a p53-dependent fashion [251]. Vorinostat displayed effective antitumor activity against BRAF and or MEK inhibitors resistant to melanoma in clinical trials. Treatment with vorinostat suppresses SLC7A11 which enhances ROS levels and induces DNA damage and cell death [252]. Under normal conditions, DNA damage is sensed and corrected either by DNA single-strand break repair (SSBR) mechanism or double-strand break (DSB) repair pathways [253]. PARP enzymes are essential for SSBR [253]. It is conceivable that loss of DNA damage repair due to PARP inhibitors can sensitize cancer cells to cisplatin- or carboplatin-induced oxidative stress [254, 255]. Interestingly, PARP inhibitors displayed synergy with cisplatin leading to increase in DNA damage as well as permeabilization of the mitochondrial membrane in lung carcinoma [253, 254]. More research is still required for a deeper and better understanding of clinical-grade ROS scavengers and inducers and will be beneficial for the treatment.
Conclusions
During the last five decades, our knowledge has greatly increased in context with the potential applicability of oxidative stress/ROS in normal physiological functions as well as in human malignancies. As we know, in the current scenario we use several toxic chemicals, preservatives, and plastics to process and preserve packed food items and color in food items, and have harmful practices such as excessive smoking and drinking. These are excellent sources of ROS right from birth and can lead to genomic instability, DNA mutations, activation of growth factor-mediated signaling, change in microbiota, metabolism and compromised immunity which ultimately lead to cancer and other diseases. Currently, with the advancement in novel technologies (DNA sequencing, metabolomics), we are starting to understand that even mutations in oncogenes and tumor suppressor genes induce oxidative stress/ROS. ROS are emerging as one of the key modulators of gut microbiota and tumor microenvironment. In future, modulation of ROS can be utilized to redefine or boost the immune response by releasing the immunosuppressive effect for better efficacy anticancer therapies. Moreover, this is very evident from many reports that ROS are involved in aberrant proliferation, tumorigenesis, angiogenesis, metastasis, and apoptosis through the activation of several signal transduction cascades including MAPK, PI3K, NF-kB, STAT3, HIF-1α, and ferroptosis. Importantly, in 2019, the Nobel Prize has been given for discovering the hypoxia-responsive pathway and how cell responds under varying oxygen levels by altering the transcription of HIF-1α regulated genes [140, 141]. Modulation of H2O2 through ROS scavengers in transformed cells has been shown to inhibit tumor growth and angiogenesis by blocking peroxide-dependent HIF-1α. On the other hand, several successful chemotherapeutic agents work by maintaining high ROS. Given the fact that ROS are critical for promoting tumorigenesis, ROS modulator or antioxidant has emerged as an alternative anticancer therapeutic and recently incorporated with chemotherapeutic drugs in clinical trials. Many studies were successful in reducing the tumor burden and provided proof of this concept in patients with late stages [256].
We must be a little careful, knowledgeable and considerable while using ROS modulator because ROS levels are crucial for the alimony of normal cells especially stem cells. ROS may be used as a biomarker for assessing the drug response where the aim of the chemotherapy drugs is to increase the ROS. One can think that ROS not only targets tumor cells, but also activate other cells in the tumor such as immune cells, macrophage, microbiota. This is what is required for a successful antitumor therapy and to overcome the drug resistance.
Acknowledgements
This work was supported and funded by the Department of Biotechnology (DBT), Government of India under its Ramalingaswami Fellowship (No. BT/RLF/Re-entry/24/2014) award to Dr. Manoj Garg and Early Career Research Award (ECRA) from Science & Engineering Research Board (SERB; ECR/2016/001519), Department of Science and Technology, Government of India. We acknowledge BioRender online software for illustration of figures.
Abbreviations
- 5-FU
5-Fluorouracil
- ABC
ATP-binding cassette
- AML
Adult acute myeloid leukemia
- AMPK 5′
AMP-activated protein kinase
- APAF1
Apoptosis protease-activating factor 1
- ATP
Adenosine triphosphate
- ART
Artesunate
- ASK-1
Apoptosis signal-regulated kinase 1
- BSO
Buthionine sulfoximine
- BCL-2
B cell lymphoma 2
- CAR
Chimeric antigen receptor
- CAT
Catalase
- CSCs
Cancer stem cells
- EGFR
Epidermal growth factor receptor
- EGF
Epidermal growth factor
- ER
Endoplasmic reticulum
- ERK
Extracellular regulated kinase
- ETC
Electron transport chain
- EMT
Epithelial–mesenchymal transition
- eNOS
Endothelial nitric oxide synthase
- GPX
Glutathione peroxidase
- GSH
Glutathione
- GCL
Glutamate-cysteine ligase
- GSS
GSH synthetase
- GSSG
GSH disulfide
- GPX4
Glutathione peroxidase 4
- H2O2
Hydrogen peroxide
- HCC
Hepatocellular carcinoma
- HER2
Human epidermal growth factor receptor 2
- HGF
Hepatocyte growth factor
- HIF-1
Hypoxia-inducible factor
- hTERT
Human telomerase reverse transcriptase
- IDH1
Isocitrate dehydrogenase 1
- IDH2
Isocitrate dehydrogenase 2
- IL-6
Interleukin 6
- JNK
C-Jun N-terminal kinase
- LDH
Lactate dehydrogenase
- LSC
Leukemic stem cells
- MAPK
Mitogen-activated protein kinase
- MDSC
Myeloid-derived suppressor cell
- mROS
Mitochondrial reactive oxygen species
- mtDNA
Mitochondrial DNA
- NADH
Nicotinamide adenine dinucleotide
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NO
Nitrogen oxide
- NOS
Nitric oxide synthase
- NOX
NADPH oxidase
- NRF2
Nuclear factor erythroid 2-related factor 2
- O2•
Superoxide
- OH•
Hydroxy radical
- OXPHOS
Oxidative phosphorylation
- PRX
Peroxiredoxins
- PDAC
Pancreatic ductal adenocarcinoma
- PD-1
Programmed death protein 1
- PD-L1
Programmed death ligand 1
- PD-L2
Programmed death ligand 2
- PDGFR
Platelet-derived growth factor receptors
- PDGF
Platelet-derived growth factor receptors
- PI3K
Phosphoinositide 3-kinases
- PML
Promyelocytic leukemia
- PTEN
Phosphatase and tensin homolog
- RTK
Receptor tyrosine kinase
- RNS
Reactive nitrogen species
- ROS
Reactive oxygen species
- SAL
Salvicine
- SOD
Superoxide dismutase
- SSZ
Sulfasalazine
- STAT3
Signal transducer and activator of transcription 3
- TF
Transcription factor
- Treg
Regulatory T cells
- TAM
Tumor-associated macrophages
- TFAM
Mitochondrial transcription factor A
- TMZ
Temozolomide
- TNBC
Triple-negative breast cancer
- UCP-2
Uncoupling protein 2
Author Contributions
MG conceived the idea and designed the format of the manuscript. MG, AK, and GS wrote the manuscript and presented the concepts in the manuscript. MG and AK created the figures and the tables. MG, AK, and GS revised the manuscript and agreed to the published version of the manuscript.
Compliance with ethical standards
Conflict of interest
All the authors have read the manuscript and have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Gautam Sethi, Email: phcgs@nus.edu.sg.
Manoj Garg, Email: mgarg@amity.edu, Email: nuscsimg@gmail.com.
References
- 1.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 2.D'Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 3.Handy DE, Loscalzo J. Redox regulation of mitochondrial function. Antioxid Redox Signal. 2012;16:1323–1367. doi: 10.1089/ars.2011.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cross CE, Halliwell B, Borish ET, Pryor WA, Ames BN, Saul RL, McCord JM, Harman D. Oxygen radicals and human disease. Ann Intern Med. 1987;107:526–545. doi: 10.7326/0003-4819-107-4-526. [DOI] [PubMed] [Google Scholar]
- 5.Brewer TF, Garcia FJ, Onak CS, Carroll KS, Chang CJ. Chemical approaches to discovery and study of sources and targets of hydrogen peroxide redox signaling through NADPH oxidase proteins. Annu Rev Biochem. 2015;84:765–790. doi: 10.1146/annurev-biochem-060614-034018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glasauer A, Chandel NS. Ros. Curr Biol. 2013;23:R100–102. doi: 10.1016/j.cub.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 7.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012;48:158–167. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 9.Hart PC, Mao M, de Abreu AL, Ansenberger-Fricano K, Ekoue DN, Ganini D, Kajdacsy-Balla A, Diamond AM, Minshall RD, Consolaro ME, et al. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat Commun. 2015;6:6053. doi: 10.1038/ncomms7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vignais PV. The superoxide-generating NADPH oxidase: structural aspects and activation mechanism. Cell Mol Life Sci. 2002;59:1428–1459. doi: 10.1007/s00018-002-8520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rhee SG. Cell signalling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 12.Chong SJF, Lai JXH, Eu JQ, Bellot GL, Pervaiz S. Reactive oxygen species and oncoprotein signalling—a dangerous liaison. Antioxid Redox Signal. 2018;29:1553–1588. doi: 10.1089/ars.2017.7441. [DOI] [PubMed] [Google Scholar]
- 13.Pervaiz S. Redox dichotomy in cell fate decision: evasive mechanism or Achilles heel? Antioxid Redox Signal. 2018;29:1191–1195. doi: 10.1089/ars.2018.7586. [DOI] [PubMed] [Google Scholar]
- 14.Reczek CR, Chandel NS. ROS-dependent signal transduction. Curr Opin Cell Biol. 2015;33:8–13. doi: 10.1016/j.ceb.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weinberg F, Chandel NS. Reactive oxygen species-dependent signaling regulates cancer. Cell Mol Life Sci. 2009;66:3663–3673. doi: 10.1007/s00018-009-0099-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles' heel? Nat Rev Cancer. 2014;14:709–721. doi: 10.1038/nrc3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srinivas US, Tan BWQ, Vellayappan BA, Jeyasekharan AD. ROS and the DNA damage response in cancer. Redox Biol. 2019;25:101084. doi: 10.1016/j.redox.2018.101084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Somyajit K, Gupta R, Sedlackova H, Neelsen KJ, Ochs F, Rask MB, Choudhary C, Lukas J. Redox-sensitive alteration of replisome architecture safeguards genome integrity. Science. 2017;358:797–802. doi: 10.1126/science.aao3172. [DOI] [PubMed] [Google Scholar]
- 19.Sallmyr A, Fan J, Rassool FV. Genomic instability in myeloid malignancies: increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008;270:1–9. doi: 10.1016/j.canlet.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 20.Rhee SG, Yang KS, Kang SW, Woo HA, Chang TS. Controlled elimination of intracellular H(2)O(2): regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid Redox Signal. 2005;7:619–626. doi: 10.1089/ars.2005.7.619. [DOI] [PubMed] [Google Scholar]
- 21.Rhee SG, Woo HA, Kil IS, Bae SH. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J Biol Chem. 2012;287:4403–4410. doi: 10.1074/jbc.R111.283432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chelikani P, Fita I, Loewen PC. Diversity of structures and properties among catalases. Cell Mol Life Sci. 2004;61:192–208. doi: 10.1007/s00018-003-3206-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24:R453–462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raffel J, Bhattacharyya AK, Gallegos A, Cui H, Einspahr JG, Alberts DS, Powis G. Increased expression of thioredoxin-1 in human colorectal cancer is associated with decreased patient survival. J Lab Clin Med. 2003;142:46–51. doi: 10.1016/S0022-2143(03)00068-4. [DOI] [PubMed] [Google Scholar]
- 25.Han H, Bearss DJ, Browne LW, Calaluce R, Nagle RB, Von Hoff DD. Identification of differentially expressed genes in pancreatic cancer cells using cDNA microarray. Cancer Res. 2002;62:2890–2896. [PubMed] [Google Scholar]
- 26.Kim HJ, Chae HZ, Kim YJ, Kim YH, Hwangs TS, Park EM, Park YM. Preferential elevation of Prx I and Trx expression in lung cancer cells following hypoxia and in human lung cancer tissues. Cell Biol Toxicol. 2003;19:285–298. doi: 10.1023/b:cbto.0000004952.07979.3d. [DOI] [PubMed] [Google Scholar]
- 27.Hedley D, Pintilie M, Woo J, Nicklee T, Morrison A, Birle D, Fyles A, Milosevic M, Hill R. Up-regulation of the redox mediators thioredoxin and apurinic/apyrimidinic excision (APE)/Ref-1 in hypoxic microregions of invasive cervical carcinomas, mapped using multispectral, wide-field fluorescence image analysis. Am J Pathol. 2004;164:557–565. doi: 10.1016/S0002-9440(10)63145-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi JH, Kim TN, Kim S, Baek SH, Kim JH, Lee SR, Kim JR. Overexpression of mitochondrial thioredoxin reductase and peroxiredoxin III in hepatocellular carcinomas. Anticancer Res. 2002;22:3331–3335. [PubMed] [Google Scholar]
- 29.Cha MK, Suh KH, Kim IH. Overexpression of peroxiredoxin I and thioredoxin1 in human breast carcinoma. J Exp Clin Cancer Res. 2009;28:93. doi: 10.1186/1756-9966-28-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015;27:211–222. doi: 10.1016/j.ccell.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 31.Desideri E, Ciccarone F, Ciriolo MR. Targeting glutathione metabolism: partner in crime in anticancer therapy. Nutrients. 2019;11:1926. doi: 10.3390/nu11081926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ryu CS, Kwak HC, Lee JY, Oh SJ, Phuong NT, Kang KW, Kim SK. Elevation of cysteine consumption in tamoxifen-resistant MCF-7 cells. Biochem Pharmacol. 2013;85:197–206. doi: 10.1016/j.bcp.2012.10.021. [DOI] [PubMed] [Google Scholar]
- 33.Chatterjee A, Gupta S. The multifaceted role of glutathione S-transferases in cancer. Cancer Lett. 2018;433:33–42. doi: 10.1016/j.canlet.2018.06.028. [DOI] [PubMed] [Google Scholar]
- 34.Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 35.Sharma R, Yang Y, Sharma A, Awasthi S, Awasthi YC. Antioxidant role of glutathione S-transferases: protection against oxidant toxicity and regulation of stress-mediated apoptosis. Antioxid Redox Signal. 2004;6:289–300. doi: 10.1089/152308604322899350. [DOI] [PubMed] [Google Scholar]
- 36.Tonelli C, Chio IIC, Tuveson DA. Transcriptional regulation by Nrf2. Antioxid Redox Signal. 2018;29:1727–1745. doi: 10.1089/ars.2017.7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chandel NS, Tuveson DA. The promise and perils of antioxidants for cancer patients. N Engl J Med. 2014;371:177–178. doi: 10.1056/NEJMcibr1405701. [DOI] [PubMed] [Google Scholar]
- 38.Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 39.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179–2191. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu KC, Cui JY, Klaassen CD. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol Sci. 2011;123:590–600. doi: 10.1093/toxsci/kfr183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kerins MJ, Ooi A. A catalogue of somatic NRF2 gain-of-function mutations in cancer. Sci Rep. 2018;8:12846. doi: 10.1038/s41598-018-31281-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang P, Singh A, Yegnasubramanian S, Esopi D, Kombairaju P, Bodas M, Wu H, Bova SG, Biswal S. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol Cancer Ther. 2010;9:336–346. doi: 10.1158/1535-7163.MCT-09-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Almeida M, Soares M, Ramalhinho AC, Moutinho JF, Breitenfeld L, Pereira L. The prognostic value of NRF2 in breast cancer patients: a systematic review with meta-analysis. Breast Cancer Res Treat. 2020;179:523–532. doi: 10.1007/s10549-019-05494-4. [DOI] [PubMed] [Google Scholar]
- 46.Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34:21–43. doi: 10.1016/j.ccell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Satoh H, Moriguchi T, Takai J, Ebina M, Yamamoto M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013;73:4158–4168. doi: 10.1158/0008-5472.CAN-12-4499. [DOI] [PubMed] [Google Scholar]
- 48.Garg M, Okamoto R, Nagata Y, Kanojia D, Venkatesan S, Anand MT, Braunstein GD, Said JW, Doan NB, Ho Q, et al. Establishment and characterization of novel human primary and metastatic anaplastic thyroid cancer cell lines and their genomic evolution over a year as a primagraft. J Clin Endocrinol Metab. 2015;100:725–735. doi: 10.1210/jc.2014-2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X, Zheng W, Wondrak GT, et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29:1235–1243. doi: 10.1093/carcin/bgn095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benhar M, Shytaj IL, Stamler JS, Savarino A. Dual targeting of the thioredoxin and glutathione systems in cancer and HIV. J Clin Invest. 2016;126:1630–1639. doi: 10.1172/JCI85339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dixon SJ. Ferroptosis: bug or feature? Immunol Rev. 2017;277:150–157. doi: 10.1111/imr.12533. [DOI] [PubMed] [Google Scholar]
- 52.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, Heindel DW, Zuckerman DS, Bos PH, Reznik E, et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol. 2018;14:507–515. doi: 10.1038/s41589-018-0031-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–379. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stockwell BR. A powerful cell-protection system prevents cell death by ferroptosis. Nature. 2019;575:597–598. doi: 10.1038/d41586-019-03145-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–1191. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234–245. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ, Stockwell BR. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12:497–503. doi: 10.1038/nchembio.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Muller T, Dewitz C, Schmitz J, Schroder AS, Brasen JH, Stockwell BR, Murphy JM, Kunzendorf U, Krautwald S. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol Life Sci. 2017;74:3631–3645. doi: 10.1007/s00018-017-2547-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bridges RJ, Natale NR, Patel SA. System xc(–) cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol. 2012;165:20–34. doi: 10.1111/j.1476-5381.2011.01480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 64.Garg M, Braunstein G, Koeffler HP. LAMC2 as a therapeutic target for cancers. Expert Opin Ther Targets. 2014;18:979–982. doi: 10.1517/14728222.2014.934814. [DOI] [PubMed] [Google Scholar]
- 65.Bishayee A, Sethi G. Bioactive natural products in cancer prevention and therapy: progress and promise. Semin Cancer Biol. 2016;40–41:1–3. doi: 10.1016/j.semcancer.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 66.Shanmugam MK, Lee JH, Chai EZ, Kanchi MM, Kar S, Arfuso F, Dharmarajan A, Kumar AP, Ramar PS, Looi CY, et al. Cancer prevention and therapy through the modulation of transcription factors by bioactive natural compounds. Semin Cancer Biol. 2016;40–41:35–47. doi: 10.1016/j.semcancer.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 67.Shanmugam MK, Warrier S, Kumar AP, Sethi G, Arfuso F. Potential role of natural compounds as anti-angiogenic agents in cancer. Curr Vasc Pharmacol. 2017;15:503–519. doi: 10.2174/1570161115666170713094319. [DOI] [PubMed] [Google Scholar]
- 68.Purohit V, Simeone DM, Lyssiotis CA. Metabolic regulation of redox balance in cancer. Cancers (Basel) 2019;11:955. doi: 10.3390/cancers11070955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oberley LW. Free radicals and diabetes. Free Radic Biol Med. 1988;5:113–124. doi: 10.1016/0891-5849(88)90036-6. [DOI] [PubMed] [Google Scholar]
- 70.Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- 71.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 72.Seth D, Rudolph J. Redox regulation of MAP kinase phosphatase 3. Biochemistry. 2006;45:8476–8487. doi: 10.1021/bi060157p. [DOI] [PubMed] [Google Scholar]
- 73.Son Y, Cheong YK, Kim NH, Chung HT, Kang DG, Pae HO. Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? J Signal Transduct. 2011;2011:792639. doi: 10.1155/2011/792639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sidhanth C, Manasa P, Krishnapriya S, Sneha S, Bindhya S, Nagare RP, Garg M, Ganesan TS. A systematic understanding of signaling by ErbB2 in cancer using phosphoproteomics. Biochem Cell Biol. 2018;96:295–305. doi: 10.1139/bcb-2017-0020. [DOI] [PubMed] [Google Scholar]
- 75.Park KR, Nam D, Yun HM, Lee SG, Jang HJ, Sethi G, Cho SK, Ahn KS. beta-Caryophyllene oxide inhibits growth and induces apoptosis through the suppression of PI3K/AKT/mTOR/S6K1 pathways and ROS-mediated MAPKs activation. Cancer Lett. 2011;312:178–188. doi: 10.1016/j.canlet.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 76.Lee JH, Kim C, Lee SG, Yang WM, Um JY, Sethi G, Ahn KS. Ophiopogonin D modulates multiple oncogenic signaling pathways, leading to suppression of proliferation and chemosensitization of human lung cancer cells. Phytomedicine. 2018;40:165–175. doi: 10.1016/j.phymed.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 77.Aggarwal V, Tuli HS, Varol A, Thakral F, Yerer MB, Sak K, Varol M, Jain A, Khan MA, Sethi G. Role of reactive oxygen species in cancer progression: molecular mechanisms and recent advancements. Biomolecules. 2019;9:735. doi: 10.3390/biom9110735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rana R, Jagadish N, Garg M, Mishra D, Dahiya N, Chaurasiya D, Suri A. Small interference RNA-mediated knockdown of sperm associated antigen 9 having structural homology with c-Jun N-terminal kinase-interacting protein. Biochem Biophys Res Commun. 2006;340:158–164. doi: 10.1016/j.bbrc.2005.11.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meves A, Stock SN, Beyerle A, Pittelkow MR, Peus D. H(2)O(2) mediates oxidative stress-induced epidermal growth factor receptor phosphorylation. Toxicol Lett. 2001;122:205–214. doi: 10.1016/s0378-4274(01)00359-9. [DOI] [PubMed] [Google Scholar]
- 80.Kanojia D, Garg M, Martinez J, Luty SB, Doan NB, Said JW, Forscher C, Tyner JW, Koeffler HP. Kinase profiling of liposarcomas using RNAi and drug screening assays identified druggable targets. J Hematol Oncol. 2017;10:173. doi: 10.1186/s13045-017-0540-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, Sundaresan M, Finkel T, Goldschmidt-Clermont PJ. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275:1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 82.Messina S, De Simone G, Ascenzi P. Cysteine-based regulation of redox-sensitive Ras small GTPases. Redox Biol. 2019;26:101282. doi: 10.1016/j.redox.2019.101282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Woo DK, Green PD, Santos JH, D'Souza AD, Walther Z, Martin WD, Christian BE, Chandel NS, Shadel GS. Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APC(Min/+) mice. Am J Pathol. 2012;180:24–31. doi: 10.1016/j.ajpath.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liou GY, Doppler H, DelGiorno KE, Zhang L, Leitges M, Crawford HC, Murphy MP, Storz P. Mutant KRas-induced Mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 2016;14:2325–2336. doi: 10.1016/j.celrep.2016.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shi X, Zhang Y, Zheng J, Pan J. Reactive oxygen species in cancer stem cells. Antioxid Redox Signal. 2012;16:1215–1228. doi: 10.1089/ars.2012.4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 88.Moon DO, Kim MO, Choi YH, Hyun JW, Chang WY, Kim GY. Butein induces G(2)/M phase arrest and apoptosis in human hepatoma cancer cells through ROS generation. Cancer Lett. 2010;288:204–213. doi: 10.1016/j.canlet.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 89.Lee S, Kim SM, Lee RT. Thioredoxin and thioredoxin target proteins: from molecular mechanisms to functional significance. Antioxid Redox Signal. 2013;18:1165–1207. doi: 10.1089/ars.2011.4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yin Z, Ivanov VN, Habelhah H, Tew K, Ronai Z. Glutathione S-transferase p elicits protection against H2O2-induced cell death via coordinated regulation of stress kinases. Cancer Res. 2000;60:4053–4057. [PubMed] [Google Scholar]
- 91.Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007;11:191–205. doi: 10.1016/j.ccr.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 92.Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 93.Koundouros N, Poulogiannis G. Phosphoinositide 3-kinase/Akt signaling and redox metabolism in cancer. Front Oncol. 2018;8:160. doi: 10.3389/fonc.2018.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 95.Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- 96.Baek SH, Ko JH, Lee JH, Kim C, Lee H, Nam D, Lee J, Lee SG, Yang WM, Um JY, et al. Ginkgolic acid inhibits invasion and migration and TGF-beta-induced EMT of lung cancer cells through PI3K/Akt/mTOR inactivation. J Cell Physiol. 2017;232:346–354. doi: 10.1002/jcp.25426. [DOI] [PubMed] [Google Scholar]
- 97.Siveen KS, Ahn KS, Ong TH, Shanmugam MK, Li F, Yap WN, Kumar AP, Fong CW, Tergaonkar V, Hui KM, Sethi G. Y-tocotrienol inhibits angiogenesis-dependent growth of human hepatocellular carcinoma through abrogation of AKT/mTOR pathway in an orthotopic mouse model. Oncotarget. 2014;5:1897–1911. doi: 10.18632/oncotarget.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wu H, Goel V, Haluska FG. PTEN signaling pathways in melanoma. Oncogene. 2003;22:3113–3122. doi: 10.1038/sj.onc.1206451. [DOI] [PubMed] [Google Scholar]
- 99.Behrend L, Henderson G, Zwacka RM. Reactive oxygen species in oncogenic transformation. Biochem Soc Trans. 2003;31:1441–1444. doi: 10.1042/bst0311441. [DOI] [PubMed] [Google Scholar]
- 100.Ding LW, Sun QY, Lin DC, Chien W, Hattori N, Dong XM, Gery S, Garg M, Doan NB, Said JW, et al. LNK (SH2B3): paradoxical effects in ovarian cancer. Oncogene. 2015;34:1463–1474. doi: 10.1038/onc.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nakaso K, Yano H, Fukuhara Y, Takeshima T, Wada-Isoe K, Nakashima K. PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin in human neuroblastoma cells. FEBS Lett. 2003;546:181–184. doi: 10.1016/s0014-5793(03)00517-9. [DOI] [PubMed] [Google Scholar]
- 102.Gorrini C, Gang BP, Bassi C, Wakeham A, Baniasadi SP, Hao Z, Li WY, Cescon DW, Li YT, Molyneux S, et al. Estrogen controls the survival of BRCA1-deficient cells via a PI3K-NRF2-regulated pathway. Proc Natl Acad Sci U S A. 2014;111:4472–4477. doi: 10.1073/pnas.1324136111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shin EM, Hay HS, Lee MH, Goh JN, Tan TZ, Sen YP, Lim SW, Yousef EM, Ong HT, Thike AA, et al. DEAD-box helicase DP103 defines metastatic potential of human breast cancers. J Clin Invest. 2014;124:3807–3824. doi: 10.1172/JCI73451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Manu KA, Shanmugam MK, Ramachandran L, Li F, Siveen KS, Chinnathambi A, Zayed ME, Alharbi SA, Arfuso F, Kumar AP, et al. Isorhamnetin augments the anti-tumor effect of capecitabine through the negative regulation of NF-kappaB signaling cascade in gastric cancer. Cancer Lett. 2015;363:28–36. doi: 10.1016/j.canlet.2015.03.033. [DOI] [PubMed] [Google Scholar]
- 105.Li F, Zhang J, Arfuso F, Chinnathambi A, Zayed ME, Alharbi SA, Kumar AP, Ahn KS, Sethi G. NF-kappaB in cancer therapy. Arch Toxicol. 2015;89:711–731. doi: 10.1007/s00204-015-1470-4. [DOI] [PubMed] [Google Scholar]
- 106.Manu KA, Shanmugam MK, Ramachandran L, Li F, Fong CW, Kumar AP, Tan P, Sethi G. First evidence that gamma-tocotrienol inhibits the growth of human gastric cancer and chemosensitizes it to capecitabine in a xenograft mouse model through the modulation of NF-kappaB pathway. Clin Cancer Res. 2012;18:2220–2229. doi: 10.1158/1078-0432.CCR-11-2470. [DOI] [PubMed] [Google Scholar]
- 107.Manu KA, Shanmugam MK, Rajendran P, Li F, Ramachandran L, Hay HS, Kannaiyan R, Swamy SN, Vali S, Kapoor S, et al. Plumbagin inhibits invasion and migration of breast and gastric cancer cells by downregulating the expression of chemokine receptor CXCR4. Mol Cancer. 2011;10:107. doi: 10.1186/1476-4598-10-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ruiz-Ramos R, Lopez-Carrillo L, Rios-Perez AD, De Vizcaya-Ruiz A, Cebrian ME. Sodium arsenite induces ROS generation, DNA oxidative damage, HO-1 and c-Myc proteins, NF-kappaB activation and cell proliferation in human breast cancer MCF-7 cells. Mutat Res. 2009;674:109–115. doi: 10.1016/j.mrgentox.2008.09.021. [DOI] [PubMed] [Google Scholar]
- 110.Li Q, Engelhardt JF. Interleukin-1beta induction of NFkappaB is partially regulated by H2O2-mediated activation of NFkappaB-inducing kinase. J Biol Chem. 2006;281:1495–1505. doi: 10.1074/jbc.M511153200. [DOI] [PubMed] [Google Scholar]
- 111.Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul. 2006;46:113–140. doi: 10.1016/j.advenzreg.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 112.Lee M, Hirpara JL, Eu JQ, Sethi G, Wang L, Goh BC, Wong AL. Targeting STAT3 and oxidative phosphorylation in oncogene-addicted tumors. Redox Biol. 2018;25:101073. doi: 10.1016/j.redox.2018.101073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kim C, Lee SG, Yang WM, Arfuso F, Um JY, Kumar AP, Bian J, Sethi G, Ahn KS. Formononetin-induced oxidative stress abrogates the activation of STAT3/5 signaling axis and suppresses the tumor growth in multiple myeloma preclinical model. Cancer Lett. 2018;431:123–141. doi: 10.1016/j.canlet.2018.05.038. [DOI] [PubMed] [Google Scholar]
- 114.Sethi G, Chatterjee S, Rajendran P, Li F, Shanmugam MK, Wong KF, Kumar AP, Senapati P, Behera AK, Hui KM, et al. Inhibition of STAT3 dimerization and acetylation by garcinol suppresses the growth of human hepatocellular carcinoma in vitro and in vivo. Mol Cancer. 2014;13:66. doi: 10.1186/1476-4598-13-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Arora L, Kumar AP, Arfuso F, Chng WJ, Sethi G. The role of signal transducer and activator of transcription 3 (STAT3) and its targeted inhibition in hematological malignancies. Cancers (Basel) 2018;10:327. doi: 10.3390/cancers10090327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rajendran P, Li F, Shanmugam MK, Vali S, Abbasi T, Kapoor S, Ahn KS, Kumar AP, Sethi G. Honokiol inhibits signal transducer and activator of transcription-3 signaling, proliferation, and survival of hepatocellular carcinoma cells via the protein tyrosine phosphatase SHP-1. J Cell Physiol. 2012;227:2184–2195. doi: 10.1002/jcp.22954. [DOI] [PubMed] [Google Scholar]
- 117.Su WP, Lo YC, Yan JJ, Liao IC, Tsai PJ, Wang HC, Yeh HH, Lin CC, Chen HH, Lai WW, Su WC. Mitochondrial uncoupling protein 2 regulates the effects of paclitaxel on Stat3 activation and cellular survival in lung cancer cells. Carcinogenesis. 2012;33:2065–2075. doi: 10.1093/carcin/bgs253. [DOI] [PubMed] [Google Scholar]
- 118.Lu L, Dong J, Wang L, Xia Q, Zhang D, Kim H, Yin T, Fan S, Shen Q. Activation of STAT3 and Bcl-2 and reduction of reactive oxygen species (ROS) promote radioresistance in breast cancer and overcome of radioresistance with niclosamide. Oncogene. 2018;37:5292–5304. doi: 10.1038/s41388-018-0340-y. [DOI] [PubMed] [Google Scholar]
- 119.Li J, Lan T, Zhang C, Zeng C, Hou J, Yang Z, Zhang M, Liu J, Liu B. Reciprocal activation between IL-6/STAT3 and NOX4/Akt signalings promotes proliferation and survival of non-small cell lung cancer cells. Oncotarget. 2015;6:1031–1048. doi: 10.18632/oncotarget.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stanicka J, Russell EG, Woolley JF, Cotter TG. NADPH oxidase-generated hydrogen peroxide induces DNA damage in mutant FLT3-expressing leukemia cells. J Biol Chem. 2015;290:9348–9361. doi: 10.1074/jbc.M113.510495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Woolley JF, Naughton R, Stanicka J, Gough DR, Bhatt L, Dickinson BC, Chang CJ, Cotter TG. H2O2 production downstream of FLT3 is mediated by p22phox in the endoplasmic reticulum and is required for STAT5 signalling. PLoS ONE. 2012;7:e34050. doi: 10.1371/journal.pone.0034050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Warsch W, Grundschober E, Berger A, Gille L, Cerny-Reiterer S, Tigan AS, Hoelbl-Kovacic A, Valent P, Moriggl R, Sexl V. STAT5 triggers BCR-ABL1 mutation by mediating ROS production in chronic myeloid leukaemia. Oncotarget. 2012;3:1669–1687. doi: 10.18632/oncotarget.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Santos JH, Meyer JN, Van Houten B. Mitochondrial localization of telomerase as a determinant for hydrogen peroxide-induced mitochondrial DNA damage and apoptosis. Hum Mol Genet. 2006;15:1757–1768. doi: 10.1093/hmg/ddl098. [DOI] [PubMed] [Google Scholar]
- 124.Li Y, Cheng HS, Chng WJ, Tergaonkar V. Activation of mutant TERT promoter by RAS-ERK signaling is a key step in malignant progression of BRAF-mutant human melanomas. Proc Natl Acad Sci U S A. 2016;113:14402–14407. doi: 10.1073/pnas.1611106113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sharma NK, Reyes A, Green P, Caron MJ, Bonini MG, Gordon DM, Holt IJ, Santos JH. Human telomerase acts as a hTR-independent reverse transcriptase in mitochondria. Nucleic Acids Res. 2012;40:712–725. doi: 10.1093/nar/gkr758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Khattar E, Kumar P, Liu CY, Akincilar SC, Raju A, Lakshmanan M, Maury JJ, Qiang Y, Li S, Tan EY, et al. Telomerase reverse transcriptase promotes cancer cell proliferation by augmenting tRNA expression. J Clin Invest. 2016;126:4045–4060. doi: 10.1172/JCI86042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Akincilar SC, Khattar E, Boon PL, Unal B, Fullwood MJ, Tergaonkar V. Long-range chromatin interactions drive mutant TERT promoter activation. Cancer Discov. 2016;6:1276–1291. doi: 10.1158/2159-8290.CD-16-0177. [DOI] [PubMed] [Google Scholar]
- 128.Akincilar SC, Low KC, Liu CY, Yan TD, Oji A, Ikawa M, Li S, Tergaonkar V. Quantitative assessment of telomerase components in cancer cell lines. FEBS Lett. 2015;589:974–984. doi: 10.1016/j.febslet.2015.02.035. [DOI] [PubMed] [Google Scholar]
- 129.Ko E, Seo HW, Jung G. Telomere length and reactive oxygen species levels are positively associated with a high risk of mortality and recurrence in hepatocellular carcinoma. Hepatology. 2018;67:1378–1391. doi: 10.1002/hep.29604. [DOI] [PubMed] [Google Scholar]
- 130.Lim SO, Gu JM, Kim MS, Kim HS, Park YN, Park CK, Cho JW, Park YM, Jung G. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: methylation of the E-cadherin promoter. Gastroenterology. 2008;135:2128–2140. doi: 10.1053/j.gastro.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 131.Low KC, Tergaonkar V. Telomerase: central regulator of all of the hallmarks of cancer. Trends Biochem Sci. 2013;38:426–434. doi: 10.1016/j.tibs.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 132.Lipinska N, Romaniuk A, Paszel-Jaworska A, Toton E, Kopczynski P, Rubis B. Telomerase and drug resistance in cancer. Cell Mol Life Sci. 2017;74:4121–4132. doi: 10.1007/s00018-017-2573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ziech D, Franco R, Pappa A, Panayiotidis MI. Reactive oxygen species (ROS)–induced genetic and epigenetic alterations in human carcinogenesis. Mutat Res. 2011;711:167–173. doi: 10.1016/j.mrfmmm.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 134.von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002;27:339–344. doi: 10.1016/s0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- 135.Peiris-Pages M, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Metastasis and oxidative stress: are antioxidants a metabolic driver of progression? Cell Metab. 2015;22:956–958. doi: 10.1016/j.cmet.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 136.Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, Morrison SJ. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186–191. doi: 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tobar N, Villar V, Santibanez JF. ROS-NFkappaB mediates TGF-beta1-induced expression of urokinase-type plasminogen activator, matrix metalloproteinase-9 and cell invasion. Mol Cell Biochem. 2010;340:195–202. doi: 10.1007/s11010-010-0418-5. [DOI] [PubMed] [Google Scholar]
- 138.Kim MC, Cui FJ, Kim Y. Hydrogen peroxide promotes epithelial to mesenchymal transition and stemness in human malignant mesothelioma cells. Asian Pac J Cancer Prev. 2013;14:3625–3630. doi: 10.7314/apjcp.2013.14.6.3625. [DOI] [PubMed] [Google Scholar]
- 139.Qu Y, Wang J, Ray PS, Guo H, Huang J, Shin-Sim M, Bukoye BA, Liu B, Lee AV, Lin X, et al. Thioredoxin-like 2 regulates human cancer cell growth and metastasis via redox homeostasis and NF-kappaB signaling. J Clin Invest. 2011;121:212–225. doi: 10.1172/JCI43144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Prabhakar NR, Semenza GL. Oxygen sensing and homeostasis. Physiology (Bethesda) 2015;30:340–348. doi: 10.1152/physiol.00022.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Pugh CW, Ratcliffe PJ. New horizons in hypoxia signaling pathways. Exp Cell Res. 2017;356:116–121. doi: 10.1016/j.yexcr.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liu LZ, Hu XW, Xia C, He J, Zhou Q, Shi X, Fang J, Jiang BH. Reactive oxygen species regulate epidermal growth factor-induced vascular endothelial growth factor and hypoxia-inducible factor-1alpha expression through activation of AKT and P70S6K1 in human ovarian cancer cells. Free Radic Biol Med. 2006;41:1521–1533. doi: 10.1016/j.freeradbiomed.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 143.Govindarajan B, Sligh JE, Vincent BJ, Li M, Canter JA, Nickoloff BJ, Rodenburg RJ, Smeitink JA, Oberley L, Zhang Y, et al. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J Clin Invest. 2007;117:719–729. doi: 10.1172/JCI30102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer. 2008;8:425–437. doi: 10.1038/nrc2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer. 2016;114:1305–1312. doi: 10.1038/bjc.2016.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Xu B, Wang S, Li R, Chen K, He L, Deng M, Kannappan V, Zha J, Dong H, Wang W. Disulfiram/copper selectively eradicates AML leukemia stem cells in vitro and in vivo by simultaneous induction of ROS-JNK and inhibition of NF-kappaB and Nrf2. Cell Death Dis. 2017;8:e2797. doi: 10.1038/cddis.2017.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Giambra V, Jenkins CR, Wang H, Lam SH, Shevchuk OO, Nemirovsky O, Wai C, Gusscott S, Chiang MY, Aster JC, et al. NOTCH1 promotes T cell leukemia-initiating activity by RUNX-mediated regulation of PKC-theta and reactive oxygen species. Nat Med. 2012;18:1693–1698. doi: 10.1038/nm.2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Herault O, Hope KJ, Deneault E, Mayotte N, Chagraoui J, Wilhelm BT, Cellot S, Sauvageau M, Andrade-Navarro MA, Hebert J, Sauvageau G. A role for GPx3 in activity of normal and leukemia stem cells. J Exp Med. 2012;209:895–901. doi: 10.1084/jem.20102386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, Lin Y, Yao J, Shi J, Kang T, et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. 2013;23:316–331. doi: 10.1016/j.ccr.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hirpara J, Eu JQ, Tan JKM, Wong AL, Clement MV, Kong LR, Ohi N, Tsunoda T, Qu J, Goh BC, Pervaiz S. Metabolic reprogramming of oncogene-addicted cancer cells to OXPHOS as a mechanism of drug resistance. Redox Biol. 2018;25:101076. doi: 10.1016/j.redox.2018.101076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wang C, Shao L, Pan C, Ye J, Ding Z, Wu J, Du Q, Ren Y, Zhu C. Elevated level of mitochondrial reactive oxygen species via fatty acid beta-oxidation in cancer stem cells promotes cancer metastasis by inducing epithelial-mesenchymal transition. Stem Cell Res Ther. 2019;10:175. doi: 10.1186/s13287-019-1265-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Pervaiz S. Cell signaling and fate through the redox lens. Redox Biol. 2019;25:101298. doi: 10.1016/j.redox.2019.101298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang CR, Schumacker PT, Licht JD, Perlman H, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 2013;38:225–236. doi: 10.1016/j.immuni.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kaminski M, Kiessling M, Suss D, Krammer PH, Gulow K. Novel role for mitochondria: protein kinase Ctheta-dependent oxidative signaling organelles in activation-induced T-cell death. Mol Cell Biol. 2007;27:3625–3639. doi: 10.1128/MCB.02295-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Warrier VU, Makandar AI, Garg M, Sethi G, Kant R, Pal JK, Yuba E. Engineering anti-cancer nanovaccine based on antigen cross-presentation. Biosci Rep. 2019;39:BSR20193220. doi: 10.1042/BSR20193220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, Delgoffe GM. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity. 2016;45:701–703. doi: 10.1016/j.immuni.2016.08.009. [DOI] [PubMed] [Google Scholar]
- 157.Siska PJ, Beckermann KE, Mason FM, Andrejeva G, Greenplate AR, Sendor AB, Chiang YJ, Corona AL, Gemta LF, Vincent BG, et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight. 2017;2:e93411. doi: 10.1172/jci.insight.93411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ligtenberg MA, Mougiakakos D, Mukhopadhyay M, Witt K, Lladser A, Chmielewski M, Riet T, Abken H, Kiessling R. Coexpressed catalase protects chimeric antigen receptor-redirected T cells as well as bystander cells from oxidative stress-induced loss of antitumor activity. J Immunol. 2016;196:759–766. doi: 10.4049/jimmunol.1401710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhou X, Chen J, Yi G, Deng M, Liu H, Liang M, Shi B, Fu X, Chen Y, Chen L, et al. Metformin suppresses hypoxia-induced stabilization of HIF-1alpha through reprogramming of oxygen metabolism in hepatocellular carcinoma. Oncotarget. 2016;7:873–884. doi: 10.18632/oncotarget.6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol Res. 2017;5:9–16. doi: 10.1158/2326-6066.CIR-16-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kunisada Y, Eikawa S, Tomonobu N, Domae S, Uehara T, Hori S, Furusawa Y, Hase K, Sasaki A, Udono H. Attenuation of CD4(+)CD25(+) regulatory T cells in the tumor microenvironment by metformin, a type 2 diabetes drug. EBioMedicine. 2017;25:154–164. doi: 10.1016/j.ebiom.2017.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martinez-Reyes I, Gao P, Helmin KA, Abdala-Valencia H, Sena LA, et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature. 2019;565:495–499. doi: 10.1038/s41586-018-0846-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.OuYang LY, Wu XJ, Ye SB, Zhang RX, Li ZL, Liao W, Pan ZZ, Zheng LM, Zhang XS, Wang Z, et al. Tumor-induced myeloid-derived suppressor cells promote tumor progression through oxidative metabolism in human colorectal cancer. J Transl Med. 2015;13:47. doi: 10.1186/s12967-015-0410-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wei J, Zhang M, Zhou J. Myeloid-derived suppressor cells in major depression patients suppress T cell responses through the production of reactive oxygen species. Psychiatry Res. 2015;228:695–701. doi: 10.1016/j.psychres.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 165.Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. 2015;42:406–417. doi: 10.1016/j.immuni.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kraaij MD, Savage ND, van der Kooij SW, Koekkoek K, Wang J, van den Berg JM, Ottenhoff TH, Kuijpers TW, Holmdahl R, van Kooten C, Gelderman KA. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc Natl Acad Sci U S A. 2010;107:17686–17691. doi: 10.1073/pnas.1012016107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lin X, Zheng W, Liu J, Zhang Y, Qin H, Wu H, Xue B, Lu Y, Shen P. Oxidative stress in malignant melanoma enhances tumor necrosis factor-alpha secretion of tumor-associated macrophages that promote cancer cell invasion. Antioxid Redox Signal. 2013;19:1337–1355. doi: 10.1089/ars.2012.4617. [DOI] [PubMed] [Google Scholar]
- 168.Garrett WS. Cancer and the microbiota. Science. 2015;348:80–86. doi: 10.1126/science.aaa4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Weinberg F, Ramnath N, Nagrath D. Reactive oxygen species in the tumor microenvironment: an overview. Cancers (Basel) 2019;11:1191. doi: 10.3390/cancers11081191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Huycke MM, Abrams V, Moore DR. Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis. 2002;23:529–536. doi: 10.1093/carcin/23.3.529. [DOI] [PubMed] [Google Scholar]
- 171.Goodwin AC, Destefano Shields CE, Wu S, Huso DL, Wu X, Murray-Stewart TR, Hacker-Prietz A, Rabizadeh S, Woster PM, Sears CL, Casero RA., Jr Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc Natl Acad Sci U S A. 2011;108:15354–15359. doi: 10.1073/pnas.1010203108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bernstein H, Bernstein C, Payne CM, Dvorak K. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J Gastroenterol. 2009;15:3329–3340. doi: 10.3748/wjg.15.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature. 2012;487:104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jones RM, Mercante JW, Neish AS. Reactive oxygen production induced by the gut microbiota: pharmacotherapeutic implications. Curr Med Chem. 2012;19:1519–1529. doi: 10.2174/092986712799828283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Yardeni T, Tanes CE, Bittinger K, Mattei LM, Schaefer PM, Singh LN, Wu GD, Murdock DG, Wallace DC. Host mitochondria influence gut microbiome diversity: A role for ROS. Sci Signal. 2019;12:eaaw3159. doi: 10.1126/scisignal.aaw3159. [DOI] [PubMed] [Google Scholar]
- 176.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103. doi: 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.O'Dwyer PJ, Hamilton TC, LaCreta FP, Gallo JM, Kilpatrick D, Halbherr T, Brennan J, Bookman MA, Hoffman J, Young RC, et al. Phase I trial of buthionine sulfoximine in combination with melphalan in patients with cancer. J Clin Oncol. 1996;14:249–256. doi: 10.1200/JCO.1996.14.1.249. [DOI] [PubMed] [Google Scholar]
- 178.Lewis-Wambi JS, Kim HR, Wambi C, Patel R, Pyle JR, Klein-Szanto AJ, Jordan VC. Buthionine sulfoximine sensitizes antihormone-resistant human breast cancer cells to estrogen-induced apoptosis. Breast Cancer Res. 2008;10:R104. doi: 10.1186/bcr2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Gupta P, Wright SE, Kim SH, Srivastava SK. Phenethyl isothiocyanate: a comprehensive review of anti-cancer mechanisms. Biochim Biophys Acta. 2014;1846:405–424. doi: 10.1016/j.bbcan.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Townsend DM, Tew KD. Pharmacology of a mimetic of glutathione disulfide, NOV-002. Biomed Pharmacother. 2009;63:75–78. doi: 10.1016/j.biopha.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Montero AJ, Diaz-Montero CM, Deutsch YE, Hurley J, Koniaris LG, Rumboldt T, Yasir S, Jorda M, Garret-Mayer E, Avisar E, et al. Phase 2 study of neoadjuvant treatment with NOV-002 in combination with doxorubicin and cyclophosphamide followed by docetaxel in patients with HER-2 negative clinical stage II–IIIc breast cancer. Breast Cancer Res Treat. 2012;132:215–223. doi: 10.1007/s10549-011-1889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sobhakumari A, Love-Homan L, Fletcher EV, Martin SM, Parsons AD, Spitz DR, Knudson CM, Simons AL. Susceptibility of human head and neck cancer cells to combined inhibition of glutathione and thioredoxin metabolism. PLoS ONE. 2012;7:e48175. doi: 10.1371/journal.pone.0048175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Marzano C, Gandin V, Folda A, Scutari G, Bindoli A, Rigobello MP. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic Biol Med. 2007;42:872–881. doi: 10.1016/j.freeradbiomed.2006.12.021. [DOI] [PubMed] [Google Scholar]
- 184.Habermann KJ, Grunewald L, van Wijk S, Fulda S. Targeting redox homeostasis in rhabdomyosarcoma cells: GSH-depleting agents enhance auranofin-induced cell death. Cell Death Dis. 2017;8:e3067. doi: 10.1038/cddis.2017.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hou GX, Liu PP, Zhang S, Yang M, Liao J, Yang J, Hu Y, Jiang WQ, Wen S, Huang P. Elimination of stem-like cancer cell side-population by auranofin through modulation of ROS and glycolysis. Cell Death Dis. 2018;9:89. doi: 10.1038/s41419-017-0159-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhou J, Bi C, Cheong LL, Mahara S, Liu SC, Tay KG, Koh TL, Yu Q, Chng WJ. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood. 2011;118:2830–2839. doi: 10.1182/blood-2010-07-294827. [DOI] [PubMed] [Google Scholar]
- 187.Valenzuela M, Glorieux C, Stockis J, Sid B, Sandoval JM, Felipe KB, Kviecinski MR, Verrax J, Buc Calderon P. Retinoic acid synergizes ATO-mediated cytotoxicity by precluding Nrf2 activity in AML cells. Br J Cancer. 2014;111:874–882. doi: 10.1038/bjc.2014.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kim D, Choi BH, Ryoo IG, Kwak MK. High NRF2 level mediates cancer stem cell-like properties of aldehyde dehydrogenase (ALDH)-high ovarian cancer cells: inhibitory role of all-trans retinoic acid in ALDH/NRF2 signaling. Cell Death Dis. 2018;9:896. doi: 10.1038/s41419-018-0903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Bollong MJ, Yun H, Sherwood L, Woods AK, Lairson LL, Schultz PG. A small molecule inhibits deregulated NRF2 transcriptional activity in cancer. ACS Chem Biol. 2015;10:2193–2198. doi: 10.1021/acschembio.5b00448. [DOI] [PubMed] [Google Scholar]
- 190.Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, Li B. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12:34. doi: 10.1186/s13045-019-0720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lachaier E, Louandre C, Godin C, Saidak Z, Baert M, Diouf M, Chauffert B, Galmiche A. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014;34:6417–6422. [PubMed] [Google Scholar]
- 192.Eling N, Reuter L, Hazin J, Hamacher-Brady A, Brady NR. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2:517–532. doi: 10.18632/oncoscience.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017;11:254–262. doi: 10.1016/j.redox.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Patel D, Kharkar PS, Gandhi NS, Kaur E, Dutt S, Nandave M. Novel analogs of sulfasalazine as system xc(–) antiporter inhibitors: Insights from the molecular modeling studies. Drug Dev Res. 2019;80:758–777. doi: 10.1002/ddr.21557. [DOI] [PubMed] [Google Scholar]
- 195.Lo M, Ling V, Low C, Wang YZ, Gout PW. Potential use of the anti-inflammatory drug, sulfasalazine, for targeted therapy of pancreatic cancer. Curr Oncol. 2010;17:9–16. doi: 10.3747/co.v17i3.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Guan J, Lo M, Dockery P, Mahon S, Karp CM, Buckley AR, Lam S, Gout PW, Wang YZ. The xc- cystine/glutamate antiporter as a potential therapeutic target for small-cell lung cancer: use of sulfasalazine. Cancer Chemother Pharmacol. 2009;64:463–472. doi: 10.1007/s00280-008-0894-4. [DOI] [PubMed] [Google Scholar]
- 197.Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, Stockwell BR. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523. doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sehm T, Fan Z, Ghoochani A, Rauh M, Engelhorn T, Minakaki G, Dorfler A, Klucken J, Buchfelder M, Eyupoglu IY, Savaskan N. Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. Oncotarget. 2016;7:36021–36033. doi: 10.18632/oncotarget.8651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Buccarelli M, Marconi M, Pacioni S, De Pascalis I, D'Alessandris QG, Martini M, Ascione B, Malorni W, Larocca LM, Pallini R, et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018;9:841. doi: 10.1038/s41419-018-0864-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, Dai X, Li Z, Wu G. Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res Treat. 2018;50:445–460. doi: 10.4143/crt.2016.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Shaw AT, Winslow MM, Magendantz M, Ouyang C, Dowdle J, Subramanian A, Lewis TA, Maglathin RL, Tolliday N, Jacks T. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc Natl Acad Sci U S A. 2011;108:8773–8778. doi: 10.1073/pnas.1105941108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Mai TT, Hamai A, Hienzsch A, Caneque T, Muller S, Wicinski J, Cabaud O, Leroy C, David A, Acevedo V, et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat Chem. 2017;9:1025–1033. doi: 10.1038/nchem.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Klose J, Trefz S, Wagner T, Steffen L, Preissendorfer Charrier A, Radhakrishnan P, Volz C, Schmidt T, Ulrich A, Dieter SM, et al. Salinomycin: anti-tumor activity in a pre-clinical colorectal cancer model. PLoS ONE. 2019;14:e0211916. doi: 10.1371/journal.pone.0211916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, Pratt DA. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci. 2017;3:232–243. doi: 10.1021/acscentsci.7b00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci. 2016;73:2195–2209. doi: 10.1007/s00018-016-2194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ye J, Zhang R, Wu F, Zhai L, Wang K, Xiao M, Xie T, Sui X. Non-apoptotic cell death in malignant tumor cells and natural compounds. Cancer Lett. 2018;420:210–227. doi: 10.1016/j.canlet.2018.01.061. [DOI] [PubMed] [Google Scholar]
- 207.Park S, Oh J, Kim M, Jin EJ. Bromelain effectively suppresses Kras-mutant colorectal cancer by stimulating ferroptosis. Anim Cells Syst (Seoul) 2018;22:334–340. doi: 10.1080/19768354.2018.1512521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kasukabe T, Honma Y, Okabe-Kado J, Higuchi Y, Kato N, Kumakura S. Combined treatment with cotylenin A and phenethyl isothiocyanate induces strong antitumor activity mainly through the induction of ferroptotic cell death in human pancreatic cancer cells. Oncol Rep. 2016;36:968–976. doi: 10.3892/or.2016.4867. [DOI] [PubMed] [Google Scholar]
- 209.Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Garg M, Nagata Y, Kanojia D, Mayakonda A, Yoshida K, Haridas Keloth S, Zang ZJ, Okuno Y, Shiraishi Y, Chiba K, et al. Profiling of somatic mutations in acute myeloid leukemia with FLT3-ITD at diagnosis and relapse. Blood. 2015;126:2491–2501. doi: 10.1182/blood-2015-05-646240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Barthel FP, Johnson KC, Varn FS, Moskalik AD, Tanner G, Kocakavuk E, Anderson KJ, Abiola O, Aldape K, Alfaro KD, et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature. 2019;576:112–120. doi: 10.1038/s41586-019-1775-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Chien W, Sun QY, Ding LW, Mayakonda A, Takao S, Liu L, Lim SL, Tan KT, Garg M, Varela ADSM, et al. Diagnosis and relapse: cytogenetically normal acute myelogenous leukemia without FLT3-ITD or MLL-PTD. Leukemia. 2017;31:762–766. doi: 10.1038/leu.2016.343. [DOI] [PubMed] [Google Scholar]
- 213.Sun QY, Ding LW, Tan KT, Chien W, Mayakonda A, Lin DC, Loh XY, Xiao JF, Meggendorfer M, Alpermann T, et al. Ordering of mutations in acute myeloid leukemia with partial tandem duplication of MLL (MLL-PTD) Leukemia. 2017;31:1–10. doi: 10.1038/leu.2016.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio II, Cairns RA, McCracken S, Wakeham A, Haight J, et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012;26:2038–2049. doi: 10.1101/gad.198200.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, Stone RM, DeAngelo DJ, Levine RL, Flinn IW, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130:722–731. doi: 10.1182/blood-2017-04-779405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Liu P, Brown S, Goktug T, Channathodiyil P, Kannappan V, Hugnot JP, Guichet PO, Bian X, Armesilla AL, Darling JL, Wang W. Cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancer-stem-like cells. Br J Cancer. 2012;107:1488–1497. doi: 10.1038/bjc.2012.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Jeanne M, Lallemand-Breitenbach V, Ferhi O, Koken M, Le Bras M, Duffort S, Peres L, Berthier C, Soilihi H, Raught B, de The H. PML/RARA oxidation and arsenic binding initiate the antileukemia response of As2O3. Cancer Cell. 2010;18:88–98. doi: 10.1016/j.ccr.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 218.Huang W, Zeng YC. A candidate for lung cancer treatment: arsenic trioxide. Clin Transl Oncol. 2019;21:1115–1126. doi: 10.1007/s12094-019-02054-6. [DOI] [PubMed] [Google Scholar]
- 219.Miller WH, Jr, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002;62:3893–3903. [PubMed] [Google Scholar]
- 220.Bahlis NJ, McCafferty-Grad J, Jordan-McMurry I, Neil J, Reis I, Kharfan-Dabaja M, Eckman J, Goodman M, Fernandez HF, Boise LH, Lee KP. Feasibility and correlates of arsenic trioxide combined with ascorbic acid-mediated depletion of intracellular glutathione for the treatment of relapsed/refractory multiple myeloma. Clin Cancer Res. 2002;8:3658–3668. [PubMed] [Google Scholar]
- 221.Coleman MC, Asbury CR, Daniels D, Du J, Aykin-Burns N, Smith BJ, Li L, Spitz DR, Cullen JJ. 2-Deoxy-d-glucose causes cytotoxicity, oxidative stress, and radiosensitization in pancreatic cancer. Free Radic Biol Med. 2008;44:322–331. doi: 10.1016/j.freeradbiomed.2007.08.032. [DOI] [PubMed] [Google Scholar]
- 222.Lin X, Zhang F, Bradbury CM, Kaushal A, Li L, Spitz DR, Aft RL, Gius D. 2-Deoxy-d-glucose-induced cytotoxicity and radiosensitization in tumor cells is mediated via disruptions in thiol metabolism. Cancer Res. 2003;63:3413–3417. [PubMed] [Google Scholar]
- 223.Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, Le Marchand-Brustel Y, Giorgetti-Peraldi S, Cormont M, Bertolotto C, et al. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010;70:2465–2475. doi: 10.1158/0008-5472.CAN-09-2782. [DOI] [PubMed] [Google Scholar]
- 224.Wang Q, He C. Dietary vitamin A intake and the risk of ovarian cancer: a meta-analysis. Biosci Rep. 2020;40:BSR20193979. doi: 10.1042/BSR20193979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K, Roper J, Chio II, Giannopoulou EG, Rago C, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015;350:1391–1396. doi: 10.1126/science.aaa5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Grant WB. Review of recent advances in understanding the role of vitamin D in reducing cancer risk: breast, colorectal, prostate, and overall cancer. Anticancer Res. 2020;40:491–499. doi: 10.21873/anticanres.13977. [DOI] [PubMed] [Google Scholar]
- 227.Yang CS, Wang X, Lu G, Picinich SC. Cancer prevention by tea: animal studies, molecular mechanisms and human relevance. Nat Rev Cancer. 2009;9:429–439. doi: 10.1038/nrc2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Chien W, Sudo M, Ding LW, Sun QY, Wuensche P, Lee KL, Hattori N, Garg M, Xu L, Zheng Y, et al. Functional genome-wide screening identifies targets and pathways sensitizing pancreatic cancer cells to dasatinib. J Cancer. 2018;9:4762–4773. doi: 10.7150/jca.25138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Gahr S, Ocker M, Ganslmayer M, Zopf S, Okamoto K, Hartl A, Leitner S, Hahn EG, Herold C. The combination of the histone-deacetylase inhibitor trichostatin A and gemcitabine induces inhibition of proliferation and increased apoptosis in pancreatic carcinoma cells. Int J Oncol. 2007;31:567–576. [PubMed] [Google Scholar]
- 230.Shankar S, Ganapathy S, Hingorani SR, Srivastava RK. EGCG inhibits growth, invasion, angiogenesis and metastasis of pancreatic cancer. Front Biosci. 2008;13:440–452. doi: 10.2741/2691. [DOI] [PubMed] [Google Scholar]
- 231.Srivastava SK, Singh SV. Cell cycle arrest, apoptosis induction and inhibition of nuclear factor kappa B activation in anti-proliferative activity of benzyl isothiocyanate against human pancreatic cancer cells. Carcinogenesis. 2004;25:1701–1709. doi: 10.1093/carcin/bgh179. [DOI] [PubMed] [Google Scholar]
- 232.Chien W, Ding LW, Sun QY, Torres-Fernandez LA, Tan SZ, Xiao J, Lim SL, Garg M, Lee KL, Kitajima S, et al. Selective inhibition of unfolded protein response induces apoptosis in pancreatic cancer cells. Oncotarget. 2014;5:4881–4894. doi: 10.18632/oncotarget.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Jagadish N, Rana R, Mishra D, Garg M, Chaurasiya D, Hasegawa A, Koyama K, Suri A. Immunogenicity and contraceptive potential of recombinant human sperm associated antigen (SPAG9) J Reprod Immunol. 2005;67:69–76. doi: 10.1016/j.jri.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 234.Rao CV. Benzyl isothiocyanate: double trouble for breast cancer cells. Cancer Prev Res (Phila) 2013;6:760–763. doi: 10.1158/1940-6207.CAPR-13-0242. [DOI] [PubMed] [Google Scholar]
- 235.Ayyanathan K, Kesaraju S, Dawson-Scully K, Weissbach H. Combination of sulindac and dichloroacetate kills cancer cells via oxidative damage. PLoS ONE. 2012;7:e39949. doi: 10.1371/journal.pone.0039949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.McLean L, Soto U, Agama K, Francis J, Jimenez R, Pommier Y, Sowers L, Brantley E. Aminoflavone induces oxidative DNA damage and reactive oxidative species-mediated apoptosis in breast cancer cells. Int J Cancer. 2008;122:1665–1674. doi: 10.1002/ijc.23244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Woo CC, Hsu A, Kumar AP, Sethi G, Tan KH. Thymoquinone inhibits tumor growth and induces apoptosis in a breast cancer xenograft mouse model: the role of p38 MAPK and ROS. PLoS ONE. 2013;8:e75356. doi: 10.1371/journal.pone.0075356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Wang H, Sim MK, Loke WK, Chinnathambi A, Alharbi SA, Tang FR, Sethi G. Potential protective effects of ursolic acid against gamma irradiation-induced damage are mediated through the modulation of diverse inflammatory mediators. Front Pharmacol. 2017;8:352. doi: 10.3389/fphar.2017.00352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.McLachlan A, Kekre N, McNulty J, Pandey S. Pancratistatin: a natural anti-cancer compound that targets mitochondria specifically in cancer cells to induce apoptosis. Apoptosis. 2005;10:619–630. doi: 10.1007/s10495-005-1896-x. [DOI] [PubMed] [Google Scholar]
- 240.Chen J, Wanming D, Zhang D, Liu Q, Kang J. Water-soluble antioxidants improve the antioxidant and anticancer activity of low concentrations of curcumin in human leukemia cells. Pharmazie. 2005;60:57–61. [PubMed] [Google Scholar]
- 241.Shanmugam MK, Rane G, Kanchi MM, Arfuso F, Chinnathambi A, Zayed ME, Alharbi SA, Tan BK, Kumar AP, Sethi G. The multifaceted role of curcumin in cancer prevention and treatment. Molecules. 2015;20:2728–2769. doi: 10.3390/molecules20022728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Zhang J, Ahn KS, Kim C, Shanmugam MK, Siveen KS, Arfuso F, Samym RP, Deivasigamanim A, Lim LH, Wang L, et al. Nimbolide-induced oxidative stress abrogates STAT3 signaling cascade and inhibits tumor growth in transgenic adenocarcinoma of mouse prostate model. Antioxid Redox Signal. 2016;24:575–589. doi: 10.1089/ars.2015.6418. [DOI] [PubMed] [Google Scholar]
- 243.Kim SM, Kim C, Bae H, Lee JH, Baek SH, Nam D, Chung WS, Shim BS, Lee SG, Kim SH, et al. 6-Shogaol exerts anti-proliferative and pro-apoptotic effects through the modulation of STAT3 and MAPKs signaling pathways. Mol Carcinog. 2015;54:1132–1146. doi: 10.1002/mc.22184. [DOI] [PubMed] [Google Scholar]
- 244.Kim C, Cho SK, Kim KD, Nam D, Chung WS, Jang HJ, Lee SG, Shim BS, Sethi G, Ahn KS. Beta-Caryophyllene oxide potentiates TNFalpha-induced apoptosis and inhibits invasion through down-modulation of NF-kappaB-regulated gene products. Apoptosis. 2014;19:708–718. doi: 10.1007/s10495-013-0957-9. [DOI] [PubMed] [Google Scholar]
- 245.Young RC, Ozols RF, Myers CE. The anthracycline antineoplastic drugs. N Engl J Med. 1981;305:139–153. doi: 10.1056/NEJM198107163050305. [DOI] [PubMed] [Google Scholar]
- 246.Garg M, Kanojia D, Mayakonda A, Ganesan TS, Sadhanandhan B, Suresh S, Nagare RP, Said JW, Doan NB, et al. Selinexor (KPT-330) has antitumor activity against anaplastic thyroid carcinoma in vitro and in vivo and enhances sensitivity to doxorubicin. Sci Rep. 2017;7:9749. doi: 10.1038/s41598-017-10325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Cai YJ, Lu JJ, Zhu H, Xie H, Huang M, Lin LP, Zhang XW, Ding J. Salvicine triggers DNA double-strand breaks and apoptosis by GSH-depletion-driven H2O2 generation and topoisomerase II inhibition. Free Radic Biol Med. 2008;45:627–635. doi: 10.1016/j.freeradbiomed.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 248.Qing C, Jiang C, Zhang JS, Ding J. Induction of apoptosis in human leukemia K-562 and gastric carcinoma SGC-7901 cells by salvicine, a novel anticancer compound. Anticancer Drugs. 2001;12:51–56. doi: 10.1097/00001813-200101000-00007. [DOI] [PubMed] [Google Scholar]
- 249.Mikula-Pietrasik J, Witucka A, Pakula M, Uruski P, Begier-Krasinska B, Niklas A, Tykarski A, Ksiazek K. Comprehensive review on how platinum- and taxane-based chemotherapy of ovarian cancer affects biology of normal cells. Cell Mol Life Sci. 2019;76:681–697. doi: 10.1007/s00018-018-2954-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Yang H, Villani RM, Wang H, Simpson MJ, Roberts MS, Tang M, Liang X. The role of cellular reactive oxygen species in cancer chemotherapy. J Exp Clin Cancer Res. 2018;37:266. doi: 10.1186/s13046-018-0909-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 252.Wang L, Leite de Oliveira R, Huijberts S, Bosdriesz E, Pencheva N, Brunen D, Bosma A, Song JY, Zevenhoven J, Los-de Vries GT, et al. An acquired vulnerability of drug-resistant melanoma with therapeutic potential. Cell. 2018;173:1413–1425.e1414. doi: 10.1016/j.cell.2018.04.012. [DOI] [PubMed] [Google Scholar]
- 253.Franzese E, Centonze S, Diana A, Carlino F, Guerrera LP, Di Napoli M, De Vita F, Pignata S, Ciardiello F, Orditura M. PARP inhibitors in ovarian cancer. Cancer Treat Rev. 2019;73:1–9. doi: 10.1016/j.ctrv.2018.12.002. [DOI] [PubMed] [Google Scholar]
- 254.Michels J, Vitale I, Galluzzi L, Adam J, Olaussen KA, Kepp O, Senovilla L, Talhaoui I, Guegan J, Enot DP, et al. Cisplatin resistance associated with PARP hyperactivation. Cancer Res. 2013;73:2271–2280. doi: 10.1158/0008-5472.CAN-12-3000. [DOI] [PubMed] [Google Scholar]
- 255.Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105:17079–17084. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Goodman M, Bostick RM, Kucuk O, Jones DP. Clinical trials of antioxidants as cancer prevention agents: past, present, and future. Free Radic Biol Med. 2011;51:1068–1084. doi: 10.1016/j.freeradbiomed.2011.05.018. [DOI] [PubMed] [Google Scholar]