

Abstract
Age-related macular degeneration (AMD) is a complex eye disease underlined by the death of photoreceptors and degeneration of retinal pigment epithelium (RPE) and choriocapillaris (CC). The mechanism(s) responsible for massive and progressive retinal degeneration is not completely known. Senescence, a state of permanent inhibition of cell growth, may be induced by many factors important for AMD pathogenesis and results in senescence-associated secretory phenotype (SASP) that releases growth factors, cytokines, chemokines, proteases and other molecules inducing inflammation and other AMD-related effects. These effects can be induced in the affected cell and neighboring cells, leading to progression of AMD phenotype. Senescent cells also release reactive oxygen species that increase SASP propagation. Many other pathways of senescence-related AMD pathogenesis, including autophagy, the cGAS–STING signaling, degeneration of CC by membrane attack complex, can be considered. A2E, a fluorophore present in lipofuscin, amyloid-beta peptide and humanin, a mitochondria-derived peptide, may link AMD with senescence. Further studies on senescence in AMD pathogenesis to check the possibility of opening a perspective of the use of drugs killing senescent cells (senolytics) and terminating SASP bystander effects (senostatics) might be beneficial for AMD that at present is an incurable disease.
Keywords: Age-related macular degeneration (AMD), Senescence, Stress-induced premature senescence, Oxidative stress, Inflammaging, A2E, Amyloid-beta, DNA damage
Introduction
Age-related macular degeneration (AMD) is a complex eye disease affecting the macula, a small, specialized structure in the middle of the retina responsible for central and fine vision [1]. AMD leads to vision loss due to dead or non-functional photoreceptors and the underlying retinal pigment epithelium (RPE). The RPE consists of polarized epithelial cells whose apical part is in contact with photoreceptor outer segments (POSs), while their basal side adheres to Bruch’s membrane (BrMb) that separates the RPE from choriocapillaris (CC) [2]. RPE cells phagocytose used POSs, absorb light and heat, release vascular endothelial growth factor (VEGF) and fulfill other functions essential for the maintenance of photoreceptors and choriocapillaris [3]. They also play an important role in the maintaining of the blood–retina barrier [4].
Clinically, AMD is divided into two forms: dry (atrophic, non-exudative) that can progress to geographic atrophy (GA) and wet (exudative) AMD characterized by choroidal neovascularization (CNV).
The exact mechanism of AMD pathogenesis in not known, but it appears that the disease-initiating events occur in a location depending on the type of AMD. The formation of large confluent drusen and hyperpigmentation that may be underlined by RPE dysfunction are initial insults in dry AMD that may progress to GA with drusen resorption and hypopigmentation induced by RPE loss [5] (Fig. 1). Dysfunction and eventual loss of photoreceptors and CC are rather secondary to RPE loss. In wet AMD, the initial insult to the photoreceptor/RPE/BrMb/CC complex is the loss of choroidal vasculature that may be underlined by a reduction in blood supply induced by stenosis of large vessels. That environment is a proinflammatory medium that accumulates proinflammatory molecules during AMD progression [6]. RPE remains intact, but becomes hypoxic and starts producing angiogenic substances, including VEGF that stimulate the formation of new vessels from CC (CNV). These events result in photoreceptors death mainly due to lack of nutrients. Both dry and wet AMD are characterized by dysfunction and/or death of all components of the photoreceptor/RPE/BrMb/CC complex, as they are mutually and functionally integrated. However, in its early phase, dry AMD may progress to either GA or wet AMD, but the exact mechanism of the dry AMD–wet AMD transformation is unknown. Therefore, the RPE plays an important role in the pathogenesis of either form of AMD. As damage, dysfunction and loss of retinal structures can support AMD induction and progression, the question about the ability of the retina to regenerate is justified.
Fig. 1.

Dry and wet AMD. The normal retina contains photoreceptors (not presented here) that are in contact with the retinal pigment epithelium (RPE)/Bruch’s membrane (BrMb)/choriocapillaris (CC) complex, which is underlined by large choroidal blood vessels (not shown). In dry age-related macular degeneration (AMD), RPE cells are progressively lost with disease progression. Some of the RPE cells in dry AMD may be damaged and some CC are lost (broken line). In wet AMD, CC are almost completely lost, become hypoxic and produce hypoxia-inducible growth factors, including vascular endothelial growth factor (VEGF, multi-color small objects) that induce formation of choroidal neovascularization (CNV) presented as a large purple vessel, penetrating BrMb and RPE. In advanced stage of wet AMD, almost all RPE cells are damaged, but they still reside on the top of CNV vessels [5]
Regenerative capability of the retina
Retinal cells death largely occurs in the final stage of AMD, but clinical symptoms associated with vision loss can be observed earlier when the cells become dysfunctional and degenerated [5]. Therefore, death of photoreceptors and RPE cells may not be casual for AMD, but rather results from its progression to the final stage. The process of RPE degeneration is irreversible and the only way to stop or slow down AMD progression is to replace degenerated cells with their normal counterparts.
So far, no adult stem cells have been identified in the mammalian retina in contrast to fish and amphibians, and to a lower extent to birds [7]. However, Salero et al. showed that subpopulation of human RPE cells in vitro could be activated to retinal pigment epithelium stem cells (RPESCs) that are able to self-renew and can produce both neural and mesenchymal progenitors [8]. As mesenchymal fates can be observed in some human pathologies, including proliferative vitroretinopathy, one can speculate that some of the RPE cells may self-renew in vivo and produce their progenitors [9-11]. These speculations need verification and provoke the question whether such RPESCs could replace degenerated RPE cells.
Over 30 years ago, Burke and Soref had shown that cells isolated from the human macula, located in the central retina, displayed a lower proliferative potential than their peripheral counterparts [12]. RPE cells in the central retina do not proliferate due to spatial restraints, but cells from peripheral regions of the retina are more relaxed and can proliferate so they may replace their damaged/dysfunctional complements. These cells must find their way to degenerated sites and must retain intrinsic capability to proliferate. RPE cells isolated from human eyes restore the ability to proliferate despite decades of dormancy.
The replacement of damaged RPE cells with their normal counterparts is possible when the latter are able to proliferate, but if they are senescent, they cannot do so (Fig. 2). On the other hand, senescence may also promote tissue repair [13-15]. Therefore, regenerative potential can be modulated by senescence-inducing factors, including oxidative stress, a major risk factor in AMD pathogenesis. Moreover, stress-induced premature senescence (SIPS) can directly induce AMD-like phenotype in RPE cells [16]. Therefore, senescence of RPE cells may play an important, if not the main, mechanistic role in AMD pathogenesis. Senescence of RPE as a key factor in AMD pathogenesis was hypothesized by Matsunaga et al. and developed by Kozlowski [17-19].
Fig. 2.
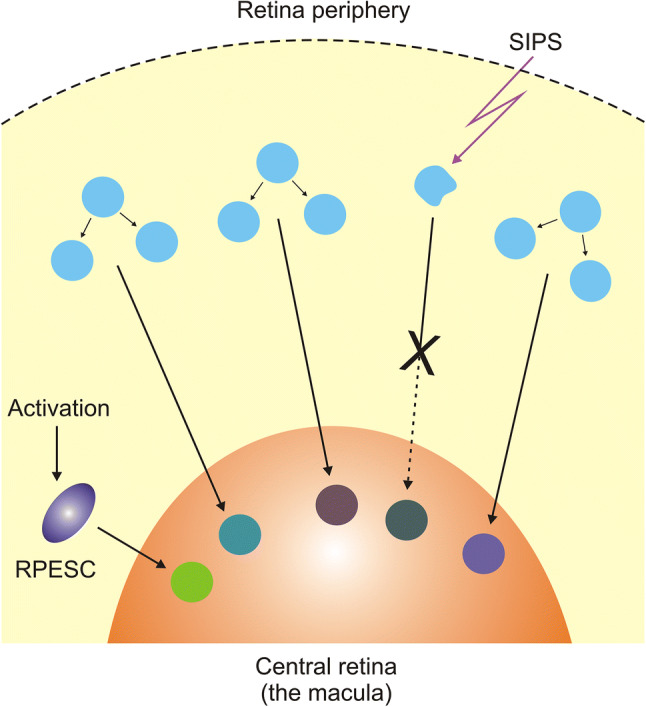
Regeneration of macular cells. Retinal pigment epithelium (RPE) cells in the normal macula are dormant and do not proliferate due to spatial constraints. Degenerated or dead RPE cells from the macula can be replaced by RPE cells from the periphery of the retina where they are more relaxed and can proliferate. RPE stem cells (RPESCs) can self-renew in vitro upon activation and it is hypothesized that they could do so in vivo. If the cells are exposed to stress, they may not be able to proliferate as they may be senescent. SIPS - stress-induced premature senescence
Cellular senescence
Cellular senescence (further: senescence) was identified in cultured human fibroblasts as a permanent exit from the cell cycle resulted from their replicative exhaustion [20]. Nowadays, cellular senescence is understood as a cellular response to stress originating from various sources, including telomere erosion and other DNA damage, mitochondrial dysfunction, oncogenic transformation and others [21]. In general, it is accepted that senescent cells are transiently present in early life and are mostly beneficial for the development, regeneration and homeostasis, but their accumulation in advanced age may be detrimental for the organism [22].
Senescence is a tumor-suppressing mechanism, as it stops proliferation of cells with potentially oncogenic damage [23]. Senescence has been linked to aging, but this link is not completely clear with several possible pathways [21]. Another aspect of cellular senescence is its involvement in wound healing or more general, tissue regeneration [24].
Senescence can be induced by a plethora of stress factors, including DNA damage from various sources, genotoxic factors, shortening of telomeres beyond critical length, oxidative stress, mitochondrial dysfunction and others. These factors may induce the activation of two tumor suppressor pathways p53/p21 and p16INK4a/Rb resulting in irreversible cell-cycle arrest (Fig. 3). Many senescent cells express p16INK4a that is transcribed from the CDK2A (cyclin-dependent kinase inhibitor 2A) gene, a tumor suppressor whose expression increases with age and correlates with the activity of the basic senescence marker, senescence-associated beta galactosidase (SA-β-gal) [25]. However, in contrary to p16INK4a/Rb, p53/21 not always induces irreversible cell growth arrest [26]. Cell-cycle arrest is induced by p21 and p16INK4a that are cyclin-dependent kinase inhibitors.
Fig. 3.
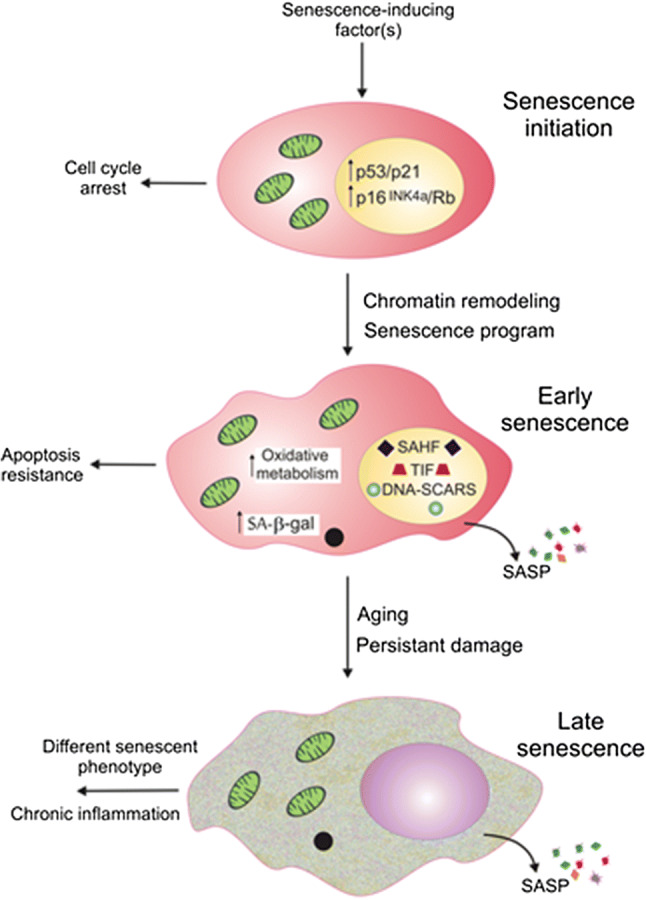
Three general phases of cellular senescence. Senescence can be initiated by various factors that lead to the activation of the p53/p21 and p16INK4a/Rb pathways, resulting in an irreversible cell cycle arrest. Early stages of senescence are associated with chromatin remodeling, forming DNA segments with chromatin alterations reinforcing senescence (DNA-SCARS) and telomere-induced dysfunctional foci (TIF) as well as senescence-associated heterochromatin foci (SAHF). Senescent cells start to release various molecules, including chemokines, cytokines, growth factors and others, determining senescence-associated secretory phenotype (SASP). Senescent cells increase mitochondrial metabolism and become resistant to apoptosis. Further changes associated with aging and long-lasting cellular damage result in variation in senescent phenotype and chronic inflammation
In early senescence, there are changes associated with chromatin remodeling, including the formation of DNA segments with chromatin alterations reinforcing senescence (DNA-SCARS) and telomere dysfunction-induced loci (TIFs) [27]. DNA-SCARS contains DNA fragments with associated proteins of DNA damage response (DDR), including phosphorylated ATM (ataxia telangiectasia mutated) and ATR (ataxia telangiectasia mutated and Rad3 related) [27]. DNA-SCARS can also contain dysfunctional telomeres or foci induced by such telomeres. Senescent cells are characterized by increased size, enhanced mitochondrial oxidative metabolism and SA-β-gal activity. They release various biomolecules, including chemokines, cytokines, proteases, growth factors and extracellular vesicles that can act in both autocrine and paracrine fashions, collectively determining senescence-associated secretory phenotype (SASP). Further changes occurring in late senescence result in different senescent phenotype and low level of chronic inflammation. Paracrine activity of SASP may result in changes in neighboring structures and cells (bystander effect), including senescence induction (paracrine senescence, senescence-induced senescence) [28]. In cells that do not express p16INK4a, inactivation of p53 may result in reinitiation of the cell cycle without SASP termination that together may lead to induction cancer phenotype in neighboring cells [29].
Oxidative stress is of particular significance in senescence, as it may induce premature senescence and overproduction of ROS may lead to DNA damage and permanent DDR that are essential for senescent phenotype. As showed by Passos et al., DDR triggered a dynamic feedback loop that locked the cell in the state of senescence [30]. That loop resulted in a long-term activation of the checkpoint gene CDKN1A, encoding p21, mitochondrial dysfunction and ROS production through the GADD45–MAPK14 (p38MAPK)–GRB2–TGFBR2–TGFβ signaling.
This is a simplified general presentation of cellular senescence and some further details will be provided in the context of AMD pathogenesis. However, it should be taken into account, that no senescence marker entirely specific to this phenomenon has been identified, some senescent cells are hardly distinguishable from quiescent or terminally differentiated cells and some can resume cell growth (see [31] for review).
Here we develop the idea that senescence may directly and indirectly lead to RPE degeneration, as it can evoke degenerative changes in RPE cells and suppress the replacement of degenerated RPE cells with their normal counterparts. Accumulation of senescent cells may be casual in aging and aging-related pathologies and selective killing of senescent cells extended life span of mice [32, 33]. However, elimination of senescent RPE cells from retina periphery may make some more space for other, originally more inner cells that could replace damaged cells from the central region of the retina, but it would result in RPE atrophy, typical for dry AMD.
Senescence-associated secretory phenotype—a key player in AMD pathophysiology
Senescence-associated secretory phenotype represents a highly plastic phenotype and can be a key player in senescence-relevant AMD pathophysiology [18, 31].
SASP-derived factors initiate or potentiate low-grade inflammatory process, which is of special relevance to aging and age-associated diseases [34]. It was shown that senescent endothelial cells (ECs) secrete IL-1β that might upregulate p21/p53 and enhance SASP [35]. Priming and activation of the NLR family pyrin domain-containing 3 (NLRP3) inflammasome in RPE cells plays a role in AMD pathogenesis [6]. IL-1β secretion, in turn, was mediated by NLRP3 activation and ROS overproduction. Furthermore, activation of NLRP3 was promoted by its association with TXNIP (thioredoxin-interacting protein) mediated by ROS. TXNIP regulates AKT-mediated cellular senescence by a direct interaction in glucose-mediated metabolic stress [36].
Aging and low-grade inflammation are linked by several molecular pathways. The inflammasome may be triggered by age-related alterations in redox balance, increase in the population of age-related senescent cells, SASP and non-efficient autophagy [34]. It is important that SASP-related release of inflammatory cytokines can be triggered by persistent DDR signaling [37].
Mechanistic target of rapamycin complex 1 (mTORC1) is the main signaling link between cellular growth and autophagy [38]. On the other hand, mTORC1 was shown to be involved in the control of SASP through the regulation of the translation of IL-1α and MAP kinase-activated protein kinase 2 (MAPKAPK2) [39, 40]. However, the interplay between cellular senescence and autophagy is not completely clear. It was shown that senescence induction activated autophagy, but other data suggest that inhibition of autophagy can favor cell senescence and that autophagy is required for senescence [41-43]. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1α), a major protein of mitochondrial biogenesis, was shown to regulate lysosomal activity of transcription factor E-box binding (TFEB) protein in RPE cells to improve autophagy flux and remove cellular damage [44, 45]. PGC-1α-deficient mice showed some abnormalities in their RPE associated with accelerated senescence. Moreover, downregulation of PGC-1α enhanced senescence induced by hydrogen peroxide. GATA4, a member of GATA transcription factors, has been identified as a key regulator of cellular senescence [46, 47]. GATA4 was shown to regulate DDR and SASP promoting chronic inflammation. Therefore, GATA4 may be a candidate to link senescence with autophagy in AMD pathogenesis. However, no experimental data supporting this hypothesis are available.
Besides soluble signaling cytokines and growth factors, senescent cells also secrete proteases as their SASP-derived products (reviewed in [25]). Matrix metalloproteinases (MMPs) may play a role in AMD pathogenesis (reviewed in [48]). The MMP pathway is impaired with aging resulting in a decreased pool of active MMPs for matrix degradation and consequently accumulation of denaturated collagen in various cellular structures, including BrMb [49]. Hussain et al. observed that total levels of active MMP-2 and MMP-9 were reduced in AMD donors [50]. They also noted an increased level of high-molecular-mass gelatinases containing homo- and heteropolymers of pro-MMP-2 and pro-MMP-9. These changes in the MMP pathway might result in impaired matrix degradation of BrMb and consequently disturbed transport of trophic substances to RPE cells and photoreceptors. These and other observations support the potential of the MMP-based, laser- and drug-mediated therapeutic interventions in AMD [51, 52].
Cao et al. showed that senescent human fetal RPE cells secreted higher levels of MMP-9 and IL-8 than their non-senescent counterparts [53]. Senescence was induced by the amyloid-beta (Aβ) peptide, which is a component of drusen observed in AMD patients, so it can be considered as a secondary risk factor in AMD [1]. Moreover, Aβ is implicated in the activation of the complement cascade, another pathway in AMD pathogenesis [54]. More details on the involvement of Aβ in AMD pathogenesis will be provided in next sections. MMP-9 released by senescent RPE cells might lyse tight junction proteins of the outer blood–retinal barrier resulting in its breakdown. MMP-9 might also increase activity of concomitantly released IL-8, which recruits and activates immune cells, such as NK cells and neutrophils that may increase chronic inflammation in the retina. In another work, Cao et al. showed a profile of cytokines that was specific for aging in human primary RPE cell cultures and RPE cells isolated from donor eyes, supporting the pivotal role of aging in AMD pathogenesis [55]. Moreover, these authors showed that RPE cells challenged by oxidative stress upregulated the angiogenic growth factor and VEGF. Taken together, advanced age and oxidative stress may contribute to AMD pathogenesis through the modulation of the cytokine profile to support pathological changes observed in AMD retinas. The important role of cytokines in senescence-related AMD pathogenesis was supported by Chaum et al., who showed that cytokines were an essential component of the network regulating p53 in RPE cells [56].
In summary, SASP results in the secretion of different kinds of molecules that can change the phenotype of the SASP cell and neighboring cells. These changes may correspond to changes observed in RPE cells affected by AMD (Fig. 4).
Fig. 4.

Senescence-associated secretory phenotype (SASP) may contribute to age-related macular degeneration (AMD) in several pathways. SASP results in release of cytokines, chemokines, growth factor and other molecules that may change the phenotype of the source cells and neighboring cells. These changes may be in line with pathways of AMD pathogenesis. BRB blood–retina barrier, BrMb Bruch’s membrane
Senescence in AMD pathogenesis—in vivo and in vitro studies
Zhu et al. showed that bone morphogenetic protein-4 (BMP4) was highly expressed in the RPE and adjacent extracellular matrix of dry AMD patients [57]. In a concomitant in vitro study these authors showed that sublethal oxidative stress upregulated BMP4 in RPE cells and BMP4 induced senescence in these cells through the p53-p21Cip1/WAF1/Rb pathway. Furthermore, they suggested that BMP4 played a mediator role in oxidative stress-induced senesce via the Smad and p38 signaling pathways to increase and activate p21 and decrease phosphorylated Rb. Therefore, BMP4 may be considered as an important element of dry AMD pathogenesis related to oxidative stress-induced senescence and so it can be also considered as a potential therapeutic target in this disease.
Senescence-prone mouse strain 8 (SAMP8) is characterized by an early onset and rapid progression of senescence and is used to model several human diseases, including AMD [58]. Using this strain, Feng et al. confirmed the important role of Aβ in senescence of retinal cells [59].They observed an age-dependent increase in drusen-like lesions in fundus of SAMP8 mice and degenerative changes in their RPE, including the presence of large vacuoles, thickened BrMb and loss of basal infolding, all known to be features of AMD. These changes were associated with increased deposits of Aβ in POS layer. Increased levels of proinflammatory IL-6 and IL-8 were also observed. Therefore, Aβ deposition and senescence may increase inflammation and play a role in AMD development.
Jadeja et al. evaluated the age-dependent expression of nicotinamide adenine dinucleotide (NAD+) biosynthetic genes and levels of NAD+ in mouse RPE [60]. They observed a decline in the expression of NAD+ with age that was correlated with decreased expression of nicotinamide phosphoribosyltransferase (NAMPT) and increased expression of senescence markers, including p16INK4a, p21 and β-gal as well as a decreased expression and activity of SIRT1. The use of FK866, which is a NAMPT inhibitor, simulated age-related decline in NAD+ level and increased senescence in ARPE-19 and mouse primary RPE cells. These results were confirmed in vivo in mice that were sub-retinally injected with FK866. Moreover, FK866 induced expression of inflammatory cytokines and ROS production in RPE, which is not surprised as FK866 evoked senescence. An in vitro treatment of RPE cells with nicotinamide mononucleotide preserved NAD+ and prevented senescence. Therefore, NAD+ may be molecular element of senescence-related AMD.
Senescence-accelerated OXYS rats are a model of accelerated aging and several neurodegenerative diseases, including AMD [61]. Kozvehnikova et al. showed that OXYS rats had increased levels of autophagic proteins LC3A/B, ATG7 and ATG12–ATG5 conjugate in the retina during manifestation of AMD-like retinopathy in 3-month-old animals and decreased levels of ATG7 and ATG12–ATG5 in 18-month-old rats [62]. An accumulation of Aβ in RPE and choroid of OXYS rats was observed to correlate with autophagic markers. These results indicate the important role of autophagy in AMD pathogenesis and point at the senescence–autophagy interplay to underline this role.
Idobenone, a synthestic analogue of the Q10 coenzyme, protected ARPE-19 cells against senescence induced by hydrogen peroxide [63]. Idobenone reduced the activity of SA-βgal, extent of intracellular ROS and DNA fragmentation. It was suggested that these effects were associated with reduction of the BAX/Bcl-2 ratio by idobenone.
Lutein is known to have beneficial effect on vision and it was shown that it protected ARPE-19 cells against premature senescence induced by hydrogen peroxide [64]. These studies showed that lutein upregulated heme oxygenase1, NAD(P)H quinone dehydrogenase 1, sirtuin 1 (SIRT1) and SIRT3.
Dvashi et al. observed a high expression of TGF-β-activated kinase 1 (TAK1) in ARPE-19 cells, but inhibition of TAK1 resulted in a decreased ratio of proliferation, cell-cycle arrest at G0/G1, and increased activity of SA-β-gal expression that are associated with cellular senescence [65]. When the cells with inhibited TAK1 were challenged with hydrogen peroxide, induction of senescence, decreased expression of apoptotic proteins and secretion of factors inducing hypertrophy and fibrotic changes were observed. Moreover, ARPE-19 cells with inhibited TAK1 exposed to oxidative stress or both secreted increased levels of MMP-9 as compared with cells in normal conditions. In conclusion, Dashi et al. stated that TAK1 was essential for maintaining normal functions of RPE cells, in particular in response to oxidative stress and its aberrant expression might induce senescence and changes in the retina typical for dry AMD. Therefore, TAK1 may be an important element of senescence-related pathogenesis of AMD.
As mentioned previously, human RPE contains a subpopulation of cells that can be activated in vitro to retinal epithelium stem cells (RPESCs) [8]. In other work from the same group, Lazzarini et al. showed that RPESCs could undergo replicative senescence, significantly affecting their proliferation and differentiation capability [66]. These cells acquired SASP that may create inflammatory environment to progress AMD. Therefore, these data support the hypothesis that senescence may be the key event in AMD pathogenesis, as it eliminates regenerative capacity of both “loose” retinal cells from retina periphery as well as, at present putative in vivo, RPESCs.
Sun et al. observed a decrease in the expression of small ubiquitin-like modifier (SUMO) enzymes, and global protein sumoylation in the aging retina of C57/B6 mice and in oxidative stress-induced premature senescence in primary RPE cells and ARPE-19 cell line [67]. Inhibition of sumoylation decreased oxidative stress-induced senescence as indicated by reduced expression of p21 and p53 and decreased cell-cycle arrest at G0/G1. In addition, inhibition of the SUMO E1 enzyme inhibited expression of proinflammatory cytokines and chemokines in premature senescent RPE cells, but it did not change DNA damage induced during senescence. Therefore, sumoylation may be of critical importance in senescence-related AMD pathogenesis.
Mechanisms of the involvement of senescence in AMD pathogenesis
Oxidative stress
Although ROS are not included in SASP, senescent cells trigger downstream signaling pathways that induce ROS production and release [30]. These ROS may contribute to oxidative stress in the host cell and neighboring cells. Oxidative stress, a recognized factor in AMD pathogenesis, may induce SIPS, so it has a special position in senescence-related AMD pathogenesis.
Marazita et al. showed that cigarette smoke extract and hydrogen peroxide induced senescence, SASP and increased levels of ROS in ARPE-19 cells [16]. An increased level of 8-hydroxydeoxyguanosine-immunoreactive (8-OHdG) DNA lesions and phosphorylated histone 2AX-immunoreactive (p-H2AX) nuclear foci were also observed. Co-treatment with N-acetylcysteine, a ROS scavenger, decreased expression of senescence markers, including SA-β-gal, p16INK4a and p21. Moreover, these authors showed downregulation of complement factor H as well as upregulation of IL-6, IL-8 and VEGF. VEGF is essential for the development of CNV and acquiring wet AMD phenotype. Therefore, oxidative stress can induce premature senescence in RPE cells. Inflammation is a consequence of SASP of RPE cells, but it is not known whether DNA damage and upregulation of VEGF are senescence-related or are directly induced by increased ROS. Furthermore, that work shows a closer link between AMD and cigarette smoking, which is considered as a risk factor for AMD.
It was shown that glutathione depletion in human ARPE-19 cells decreased cell viability and increased both soluble and lipid ROS, but it did not affect mitochondria-derived ROS or mitochondrial mass [68]. Decreased expression of ferroptotic modulator of glutathione peroxidase 4 (GPX4) was observed in that study. Increased expression of LC3 and autophagic flux suggested enhancement of autophagy. Senescence was evidenced by increased cells positive for SA-β-gal, increased SAHF and cell-cycle arrest at G0/G1. Further experiments with autophagy inhibitors/activators suggested that glutathione depletion induced ferroptosis, autophagy and SIPS and autophagy was activated in both ferroptosis and SIPS. These results could be considered in a broader context of detrimental effects of oxidative stress in RPE cells and their consequence for AMD pathogenesis.
DNA damage response
Several works show that DNA damage response (DDR) may be compromised in AMD (reviewed in [69]). Inefficient DNA repair may result in persistent nuclear and mitochondrial DNA (mtDNA) damage. Accumulation of damage to mtDNA may be linked with aging and therefore can play a role in AMD pathogenesis as aging is the main risk factor in AMD [70]. This accumulation is mainly due to increased ratio of replication errors and decreased efficacy of mtDNA repair. Several results suggest an increased susceptibility of mtDNA to damage in AMD (reviewed in [71]). However, the most important question in this context is whether such increased mtDNA damage is specific for AMD per se or may follow from advanced age, usually typical for AMD patients? To address this, problem Karunadharma et al. used human donor eyes from an eye bank [72]. They observed an increased mtDNA damage in the macula region of AMD donor eyes as compared with age-matched controls. Moreover, DNA damage in mitochondria of AMD donors was approximately eight times more pronounced than in nuclear DNA. These observations led to the conclusion that damage to mtDNA is an important element of AMD pathogenesis. That conclusion was strongly supported by Ferrington et al. who showed a positive correlation between mtDNA damage and a specific nuclear genetic variant of complement factor H that was associated with an increased risk of AMD [73]. Is that associated with senescence or death of retinal cells?
Laberge et al. showed that damage to mtDNA induced apoptosis in senescent cells [74]. However, the immediate question is whether this is not mtDNA damage that is primarily responsible for senescence and apoptosis? In other words, maybe a certain extent of mtDNA damage induces senescence and other, a higher extent—apoptosis? Although the work of Laberge et al. was specific for ganciclovir C9H5N13O4 (GCV), a nucleoside analogue used to target viruses and herpes simplex virus thymidine kinase (HSVtk), it presents a general idea that senescence can be induced by a factor evoking damage to nuclear DNA and then mtDNA can be targeted to induce apoptosis. GCV can kill dividing cells by induction of DNA double-strand breaks (DSBs) dependent on replication [75]. However, not all cells are killed at once and some of them become senescent. Therefore, they do not divide and consequently are resistant to replication-dependent DSBs. In general, this reflects the general property of senescent cells—their resistance to apoptosis. However, GCV might induce mtDNA damage or inhibit mtDNA synthesis, both resulting in apoptosis of non-dividing cells. It is known that senescent cells increase their mitochondrial mass, so they may be more prone to mitochondrial toxicity than non-senescent cells [76].
DNA damage, including telomere erosion beyond critical level, belongs to the most important senescence-inducing stimuli as the essential senescence event, the activation of the p53/p21 pathway, is induced by DDR [77]. DNA damage must not be too high to elicit a senescence response and a low level of DDR signaling, including p53/p21, may persist after DNA damage induction. Elevated levels of ROS in senescent cells may also trigger other signaling pathways, including the p16INK4a/Rb pathway responsible for irreversibly growth arrest. It is important that rapid transient signaling may rather result in cell death, not senescence and persistent DDR signaling, as developing slowly in cell culture after induction of DDR, is required for SASP to develop [27]. DNA-SCARS and TIF are of special importance for the development of SASP, as they contain activated DDR proteins needed for persistent DDR signaling [27].
Humanin
Humanin (HN) is a 21–24 aa peptide encoded by an open reading frame within the mitochondrial MT-RNR2 gene encoding 16S rRNA discovered in surviving neurons in patients with Alzheimer’s disease [78]. Several other HN-like encoding loci have been identified in both mitochondrial and nuclear DNA [79, 80]. Humanin displays a protective effect against oxidative stress and endoplasmic reticulum stress in RPE (reviewed in [81]).
Nashine et al. constructed transmitochondrial ARPE-19 cybrids through the replacement of original mitochondria in ARPE-19 cells by mitochondria derived from either AMD patients or age-matched controls [82]. In AMD cybrids, humanin G protected mitochondria, downregulated pro-apoptosis genes, and increased the protection against Aβ-induced damage. Therefore, HN may be involved in senescence-related AMD pathogenesis, as Aβ was reported to induce senescence in RPE cells [1]. On the other hand, HN was shown to induce chaperone-mediated autophagy and autophagy is an important mechanism in AMD pathogenesis [83, 84].
The protective action of HN against senescence in RPE cells was directly demonstrated by Sreekumar et al. who showed that HN decreased the number of SA-β-gal cells induced in RPE cells isolated from human fetal eyes and exposed to hydrogen peroxide [85]. Humanin co-treatment with H2O2 reduced the expression of p16INK4a and ApoJ transcript, another senescence marker, as compared with H2O2-only treatment.
Membrane attack complex-related degeneration of choriocapillaris
Dry form of AMD is featured by degeneration of CC that is involved in RPE hypoxia and atrophy [86-89]. Dry AMD is also characterized by the activation of complement in the sub-RPE space, resulting in the deposition of membrane attack complex (MAC) on the CC [6, 90]. MAC induces cell lysis and pores in cellular membrane, so it can be involved in CC degeneration occurring in dry AMD. However, MAC is common in CC of young, healthy eyes [90]. Therefore, there may be an age-related factor that induces or fuels MAC-related degenerative changes in CC. To address this problem, Cabrera et al. cultured monkey chorioretinal endothelial cells (ECs) to achieve replicative senescence [91]. They showed that senescence of ECs was associated with their increased stiffness that correlated with Rho GTP-ase activity in these cells and their susceptibility to MAC-induced injury. Therefore, aging in general and senescence in particular may be implicated in the MAC-related CC degeneration, playing a role in dry AMD pathogenesis. More specifically, senescence-associated stiffening of choroidal EC is a determinant of CC atrophy, typical for dry AMD.
The cGAS–STING pathway
Cyclic GMP–AMP synthase (cGAS) is the major sensor of cytosolic DNA regulating de novo transcriptional immune response [92, 93]. cGAS interacts with sugar-phosphate backbone of double-stranded DNA (dsDNA) recognizing a DNA molecule in a sequence-independent manner [94]. Binding dsDNA results in the catalysis of cyclic GMP–AMP (cGAMP), which is a second messenger for signal transduction and promotes stimulator of interferon genes (STING) that is a endoplasmic reticulum transmembrane protein [95]. These events lead to the activation and translocation of interferon-regulatory factor 3 (IRF3) and NF-κB that in concert in other factors induce the expression of interferons and other cytokines and chemokines that are present in SASP. Therefore, the cGAS–STING pathway may be important in the adaptation response to DNA damage-induced senescence [96].
As the cGAS–STING pathway is important in SASP acquiring and SASP may play a role in senescence-related AMD pathogenesis, it is justified to ask about the role of the cGAS–STING pathway in AMD. This problem was addressed by Wu et al. who hypothesized that the cGAS–STING pathway is a sensor of DNA damage associated with senescence and trigger of inflammation in early AMD [97]. They suggested that targeting this pathway might have a therapeutic significance in AMD as small-molecule inhibitors covalently binding STING were reported [98]. However, the cGAS–STING pathway is not the only way to induce SASP evoked by DNA damage. Second, senescence-related DNA damage is detected by DDR detectors, so how exactly cGAS–STING recognize DNA damage that does not result in DNA fragmentation and release to the cytosol? These questions should be addressed by the studies related to the potential of cGAS–STING in AMD therapy. However, the role of the cGAS–STING pathway in AMD pathogenesis may be prospectively considered, as it is essential for senescence and is proposed as a therapeutic target in cancer [99].
Lipofuscin and A2E
Continuous degradation of used POS in phagocytosis by RPE cells is essential for normal functioning of the retina and vision maintenance. Its impairment leads to accumulation of lipid–protein aggregates that are derived from oxidized unsaturated fatty acids that may play a role in AMD pathogenesis [100].
These yellow-brownish aggregates called lipofuscin contain oxidized cross-linked proteins, lipids with a tiny amount of saccharides and metals [101] (Fig. 5). Lipofuscin accumulation in RPE cells increases with age and results from impaired ability of these cells to remove cellular waste, first of all protein debris [102]. Boyer et al. showed that lipofuscin was derived from the free 11-cis-retinal that was continuously supplied to the rod for rhodopsin regeneration and POS renewal [103]. Iron is a main metal component of lipofuscin and it is a major source of oxidants [104]. Dunaief was the first who showed that iron homeostasis could be impaired in AMD [105-107]. These results were confirmed in later studies (reviewed in [108-111]). Moreover, it was shown that lipofuscin-mediated formation of oxidant with the involvement of iron occurred in senescent fibroblasts, independently of mitochondria and was toxic for these cells [104]. Therefore, lipofuscin cytotoxicity can be related to senescence through a mechanism involving redox reaction(s). Furthermore, the formation of lipofuscin was observed when mitochondrial fission was inhibited and the Lon protease, responsible for a selective degradation of aberrant mitochondrial proteins, was downregulated [112]. Impaired mitochondria feature senescence is an important element in AMD pathogenesis (reviewed in [71, 113]).
Fig. 5.
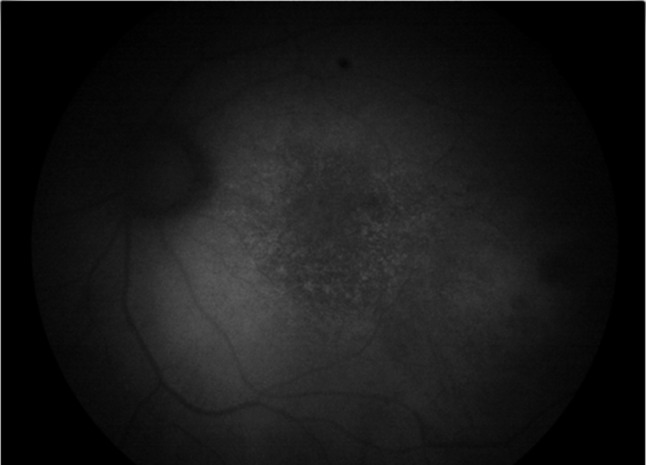
Lipofuscin accumulation in age-related macular degeneration (AMD) retina. Fundus autofluorescence image from an AMD macula shows increased accumulation of lipofuscin granules with increased autofluorescence signal (courtesy of Professor Kai Kaarniranta of University of Eastern Finland, Finland)
N-Retinylidene-N-retinylethanolamine (A2E) is a by-product of the visual cycle and the main fluorophore present in lipofuscin whose accumulation is associated with AMD development, but a causal relationship between them has not been established [114]. However, it was shown that A2E is the main component of lipofuscin that induced DNA damage to RPE cells [115]. A2E treatment of human RPE cells derived from induced pluripotent stem cells obtained from normal individuals revealed a novel senescence-related RPE protein, high-mobility group box 1 (HMGB1) [116]. HGMB1 upregulated caveolin-1, another senescence-related protein, so it was concluded that both HMGB1 and caveolin-1 might be important in senescence-related AMD pathogenesis. A2E accumulates in aging RPE cells and upregulates pro-inflammatory molecules and VEGF that are elements of SASP [117]. A2E can also be formed on the dimerization of a phosphate ester of retinaldehyde, a vitamin A aldehyde, on phosphatidylethanolamine in the retinal disc membrane, contributing to senescence and retinal degeneration in AMD [118].
As mentioned, lipofuscin may contribute to AMD pathogenesis by the interaction with mitochondria. To clarify this issue, Alaimo et al. showed that light-irradiated ARPE-19 cells displayed decreased viability and increased ROS production [119]. Moreover, using ARPE-19 cell loaded with A2E, they showed that blue light-induced mitochondrial fragmentation correlated with downregulation of mitochondrial-shaping proteins OPA1, DRP1 and OMA1. These results show a specific pathway of the involvement of A2E-mediated mitochondrial quality control in AMD pathogenesis.
Somatic cells shorten their telomeres with each cellular division that leads to crisis and resulting replicative senescence unless they escape from the crisis. Activation of telomerase, an enzyme capable of extending telomeres may be essential for such an escape and stability and maintenance of telomeres. Telomerase is a cellular reverse transcriptase, an RNA-dependent DNA polymerase that adds telomeric DNA to telomeres due to its inner RNA template [120]. Telomere shortening results from the inability of eukaryotic DNA polymerase to complete replication of linear chromosomes—some bases at the very 3′ end of each template strand are not copied (the end-replication problem) [121]. Telomere loss and SA-β-gal activation were observed in a human RPE cell line [19]. In humans, normal telomeres have a long 3′ single-stranded DNA overhang containing a 6-nucleotide sequence motif, 5′-TTAGGG-3′, that is tandemly repeated many times [122]. As it is a G-rich stretch, it may fold onto itself to form G-quadruplexes and various four-stranded DNA structures [123].
Wang et al. reasoned that since telomeres are G-rich structures, they might be targeted by A2E to induce DNA damage, as this fluorophore was shown to induce 8-oxo-guanine (8-oxoG), a hallmark of oxidative damage to DNA and oxidative stress marker [115, 124, 125]. Furthermore, these authors provided rationale that photosensitization of A2E could result in the senescence of RPE cells through the induction of DNA damage in telomeric structures. Indeed, they showed that photosensitization of A2E induced DNA damage and senescence of ARPE-19 cells. DNA damage, observed as co-localization of phosphorylated H2AX histone with a subunit of sheltering, a telomere protein, resulted in telomere loss. Ectopic expression of the catalytic subunit of telomerase decreased senescence in RPE cells suggesting that senescence was underlined by telomere loss/dysfunction. The NF-κB pathway was identified as the main mechanistic route in A2E photosensitization-related senescence and SASP. The use of N-acetyl cysteine (NAC), a free radical scavenger, partly ameliorated effects induced by A2E photosensitization. Therefore, Wang et al. presented a comprehensive mechanism of senescence-related AMD pathogenesis mediated by a RPE damaging factor that is not necessarily a recognized AMD risk factor (Fig. 6).
Fig. 6.
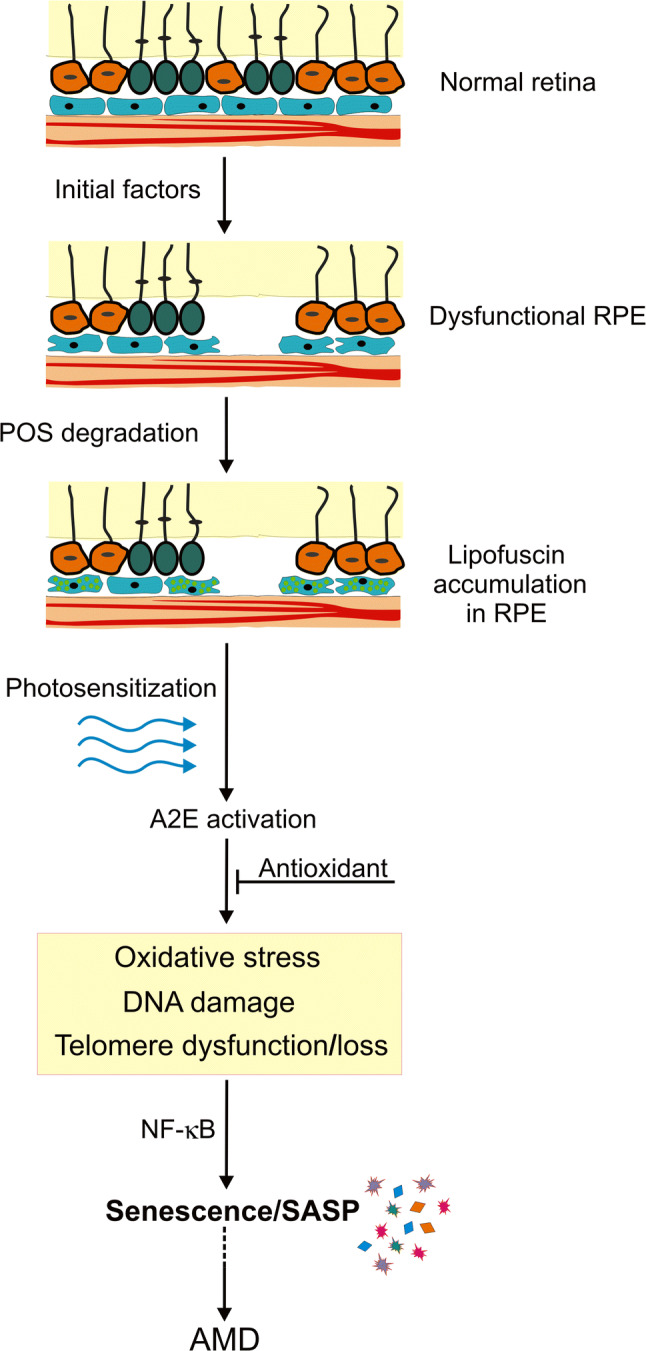
N-Retinylidene-N-retinylethanolamine (A2E) photosensitization-mediated senescence in the pathogenesis of age-related macular degeneration (AMD). A retinal pigment epithelium (RPE) damaging factor may impair the ability of RPE cells to phagocytose photoreceptor outer segments (POS) resulting in lipofuscin accumulation (light green spots) and AMD-like phenotype. Lipofuscin contains A2E that, upon photosensitization, may induce oxidative stress, DNA damage and telomere dysfunction/loss that leads to senescence and nuclear factor kappa B subunit 1 (NF-κB)-mediated senescence-associated secretory phenotype (SASP) that may result in damaging neighboring normal RPE cells and expansion of the AMD phenotype. Activated A2E may be neutralized by antioxidants
The amyloid-beta peptide
Feng et al. detected activation of autophagy in Aβ1-42-treated RPE cells [126]. They observed the generation of autophagic vehicles, altered pattern of LC3 expression, increased LC3-II and decreased p62 expression. The authors suggested that autophagy can be a protective mechanism of RPE against detrimental changes induced by Aβ.
The potential role of the Aβ peptide in senescence-related AMD pathogenesis was confirmed in several in vitro studies [126-129]. To address this issue in vivo, Liu et al. studied the effect of Aβ on senescence of RPE cells and senescence-associated inflammation in C57BL/6 mice that received subretinal injections with Aβ1-42 [130]. These mice showed a decreased electroretinographic response and severe degenerative changes in the eye, including disruption of the junction between inner and outer segments, vacuolation and thickening of BrMb. These mice also showed an increased fundus autofluorescence and p16INK4a expression. Moreover, an upregulation of the IL-6 and IL-8 genes was observed. The authors concluded that the subretinal injection of Aβ evoked AMD-like pathology in mice and suggested that senescence could be a mechanistic link between inflammation and retinal degeneration. Both in vitro and in vivo studies as well as clinical observations suggest that Aβ can be an important element of AMD progression, as it is present in AMD drusen, can induce senescence in RPE cells that can display SASP and release inflammatory cytokines that may stimulate AMD phenotype in the same cell and neighboring cells (Fig. 7). This would be a senescence- and inflammation-based feedback- or vicious cycle-like mechanism that would increase AMD phenotype in a single induced cell and extend it gradually on an increased number of retinal cells, which leads to AMD progression. Autophagy can inhibit these changes, which may partly support autophagy decline as an important factor of AMD pathogenesis, but the mechanism of protective role of autophagy in AMD is likely multi-pathway and requires further studies [84, 131]. Moreover, autophagy may not always be protective and contribute to the death of RPE cells [131-133].
Fig. 7.
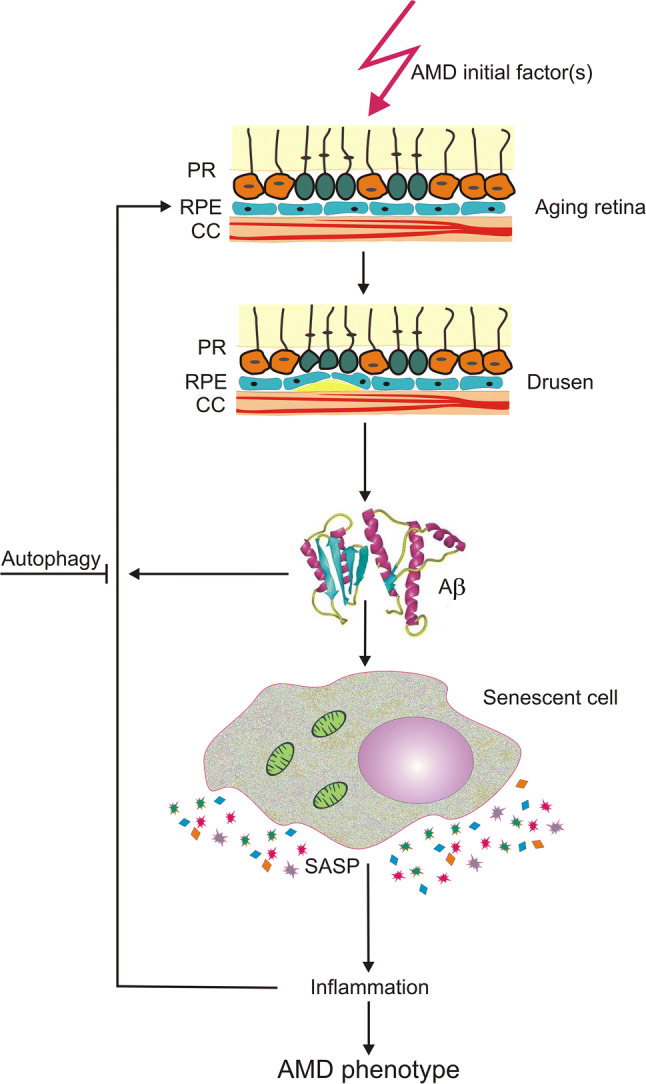
Amyloid-beta (Aβ) peptide in senescence-mediated pathogenesis of age-related macular degeneration (AMD). A factor of AMD pathogenesis may initiate drusen (yellow) formation between choriocapilaris (CC) and retinal pigment epithelium (RPE) cells. Amyloid-beta (Aβ) peptide is a component of drusen and can induce senescence in the affected cell, which acquires senescence-associated secretory phenotype (SASP), releasing inflammatory cytokines and inducing local inflammation that contribute to the expansion of AMD phenotype in the same cell and neighboring cells. Autophagy may be a protective mechanism against these detrimental changes in the retina. PR photoreceptors, Bruch’s membrane is not presented
Conclusions and perspectives
Degeneration of the retina is the main clinical hallmark of AMD. It is underlined by death of photoreceptors and various degenerative changes in RPE and choroid. In the final stage of AMD death of RPE cells and CC ECs can be observed, but its mechanism is not completely clear and RPE cell death is not required to express AMD phenotype [132]. We and others have postulated that persistent and progressive degeneration of retinal cells in AMD results from their stress-induced premature senescence (SIPS). A SIPS factor may induce SASP in a single cell resulting in the release of various molecules that may enhance SASP in the host cell (autocrine) and induce it in neighboring cells (paracrine). This lead to a cascade of events resulting in the progression of AMD phenotype expressed by an increased number of degenerated retinal cells. Degenerated retina has a limited capability to regenerate. RPE cells in the central retina, including the macula, are quiescent and cannot proliferate due to spatial restrictions. As functional and localized adult stem cells have not been recognized in the human retina, the only way to regenerate degenerated cells from central regions in the retina is to replace them with progenitor of their peripheral counterparts that are more spatially relaxed and likely able to proliferate. However, if these proliferation-capable cells are affected by SIPS, they will not be able to produce progeny to replace degenerated macular cells. Nonetheless, induction and expansion of SIPS seem crucial in AMD pathogenesis.
Several mechanisms may underline senescence-related pathogenesis of AMD. Aging is the most important factor of AMD pathogenesis and although the link between premature aging and SIPS is not completely clear, there are reports showing a correlation between senescence and aging in acquiring of AMD-like phenotype [97]. SASP is characterized by the release of pro-inflammatory molecules that create a local, low-grade inflammation. However, that “local” becomes “global” with the progression of senescence and SASP in neighboring cells. Chronic inflammation is a well-recognized factor associated with AMD [6]. Proteases released by SASP cells, in particular MMP-2 and MMP-9, may disturb the balance in matrix degradation of BrMb and consequently transport of trophic substances to RPE cells and photoreceptors. Moreover, MMP-9 released by senescent RPE cells may degrade tight junction proteins of the outer blood–retinal barrier resulting in its breakdown. MMP-9 may also enhance the activity of SASP IL-8, which recruits and activates immune cells that increase chronic inflammation in the retina. Autophagy impairment plays a role in AMD pathogenesis and mTORC1, the main protein relating autophagy with cell growth, was shown to control SASP through the regulation of IL-1α and MAPKAPK2 [39, 40, 84]. Although the relationships between senescence and autophagy as well as autophagy and AMD are not completely clear, GATA4 may link these phenomena, as it is regulated by autophagy and influence senescence and DDR. Therefore, further research on senescence-related AMD might include GATA4. The Aβ peptide is another candidate as it induces autophagy in RPE cells [126]. Potential role of Aβ in senescence-related AMD pathogenesis was confirmed in several in vitro and in vivo studies.
SIPS can be induced by ROS, but senescent cell releases ROS creating a local oxidative stress. However, these extracellular ROS may induce SIPS in neighboring cells that can release ROS leading to a cascade of event similar to SASP expansion and resulting in changing local into global oxidative stress. This leads to a persistent DDR that supports cellular senescence [30].
A2E, a fluorophore present in lipofuscin, can be involved in senescence induction, providing a link between AMD initiating factor and senescence [125]. A2E may play a special role in senescence-related AMD, as it is a DNA-damaging factor and may shorten telomeres beyond critical length. Therefore, A2E links SIPS with replicative senescence.
Drugs that kill senescent cells (senolytics) and compounds inhibiting bystander effect resulting from SASP (senostatics) are widely considered in cancer therapy (reviewed in [134]). Recently, a senolytic intervention proved to be promising in the first human, open-label study on idiopathic pulmonary fibrosis, a progressive, fatal cellular senescence- and age-associated disease [135]. Therefore, studies on the possibility of the use of senolytics in AMD, which is principle an incurable disease, are justified.
Acknowledgements
The author thanks Ms. Monika Kicinska for help in figures preparation. This work was supported by National Science Centre, Poland grant number 2017/27/B/NZ3/00872.
Abbreviations
- 8-oxoG
8-Oxo-guanine
- Aβ
Amyloid-beta
- AMD
Age-related macular degeneration
- ATM
Ataxia telangiectasia mutated
- ATR
Ataxia telangiectasia mutated and Rad3 related
- BMP4
Bone morphogenetic protein-4
- BrMb
Bruch’s membrane
- CC
Choriocapillaris
- CDK2A
Cyclin-dependent kinase inhibitor 2A
- cGAMP
Cyclic GMP–AMP
- cGAS
Cyclic GMP–AMP synthase
- CNV
Choroidal neovascularization
- DDR
DNA damage response
- DNA-SCARS
DNA segments with chromatin alterations reinforcing senescence
- DSB
DNA double-strand break
- GA
Geographic atrophy
- GCV
Ganciclovir C9H5N13O4
- HN
Humanin
- HSVtk
Herpes simplex virus thymidine kinase
- MAC
Membrane attack complex
- MAPKAPK2
MAP kinase-activated protein kinase 2
- MMP
Matrix metalloproteinases
- mtDNA
Mitochondrial DNA
- mTORC1
Mechanistic target of rapamycin complex 1
- NAMPT
Nicotinamide phosphoribosyltransferase
- NF-κB
Nuclear factor kappa B subunit 1
- NLRP3
NLR family pyrin domain-containing 3
- PGC-1α
Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha
- POS
Photoreceptor outer segment
- Rb
Retinoblastoma
- RPE
Retinal pigment epithelium
- RPESC
Retinal pigment epithelium stem cell
- SA-β-gal
Senescence-associated beta galactosidase
- SAHF
Senescence-associated heterochromatin foci
- SAMP8
Senescence-prone mouse strain 8
- SASP
Senescence-associated secretory phenotype
- SIRT
Sirtuin
- STING
Stimulator of interferon genes
- TAK1
TGF-β-activated kinase 1
- TIF
Telomere dysfunction-induced loci
- VEGF
Vascular endothelial growth factor
Compliance with ethical standards
Conflict of interest statement
The author does not declare any conflict of interest associated with this paper.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ratnayaka JA, Serpell LC, Lotery AJ. Dementia of the eye: the role of amyloid beta in retinal degeneration. Eye (Lond) 2015;29(8):1013–1026. doi: 10.1038/eye.2015.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hogan MJ. Role of the retinal pigment epithelium in macular disease. Trans Am Acad Ophthalmol Otolaryngol. 1972;76(1):64–80. [PubMed] [Google Scholar]
- 3.Saint-Geniez M, Kurihara T, Sekiyama E, Maldonado AE, Amore PA. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc Natl Acad Sci. 2009;106(44):18751. doi: 10.1073/pnas.0905010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005;85(3):845–881. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- 5.Bhutto I, Lutty G. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch's membrane/choriocapillaris complex. Mol Aspects Med. 2012;33(4):295–317. doi: 10.1016/j.mam.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kauppinen A, Paterno JJ, Blasiak J, Salminen A, Kaarniranta K. Inflammation and its role in age-related macular degeneration. Cell Mol Life Sci. 2016;73(9):1765–1786. doi: 10.1007/s00018-016-2147-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eymann J, Salomies L, Macri S, Di-Poi N. Variations in the proliferative activity of the peripheral retina correlate with postnatal ocular growth in squamate reptiles. J Comp Neurol. 2019;527(14):2356–2370. doi: 10.1002/cne.24677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salero E, Blenkinsop TA, Corneo B, Harris A, Rabin D, Stern JH, Temple S. Adult human RPE can be activated into a multipotent stem cell that produces mesenchymal derivatives. Cell Stem Cell. 2012;10(1):88–95. doi: 10.1016/j.stem.2011.11.018. [DOI] [PubMed] [Google Scholar]
- 9.Kampik A, Kenyon KR, Michels RG, Green WR, de la Cruz ZC. Epiretinal and vitreous membranes: comparative study of 56 cases. 1981. Retina (Philadelphia, Pa) 2005;25(5 Suppl):1445–1454. doi: 10.1097/00006982-200507001-00010. [DOI] [PubMed] [Google Scholar]
- 10.Newsome DA, Rodrigues MM, Machemer R. Human massive periretinal proliferation. In vitro characteristics of cellular components. Arch Ophthalmol (Chicago, Ill) 1981;99(5):873–880. doi: 10.1001/archopht.1981.03930010873017. [DOI] [PubMed] [Google Scholar]
- 11.Vemuganti GK, Honavar SG, Jalali S. Intraocular osseous metaplasia. A clinico-pathological study. Indian J Ophthalmol. 2002;50(3):183–188. [PubMed] [Google Scholar]
- 12.Burke JM, Soref C. Topographical variation in growth in cultured bovine retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1988;29(12):1784–1788. [PubMed] [Google Scholar]
- 13.Jun JI, Lau LF. Cellular senescence controls fibrosis in wound healing. Aging (Albany NY) 2010;2(9):627–631. doi: 10.18632/aging.100201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. 2010;12(7):676–685. doi: 10.1038/ncb2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134(4):657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marazita MC, Dugour A, Marquioni-Ramella MD, Figueroa JM, Suburo AM. Oxidative stress-induced premature senescence dysregulates VEGF and CFH expression in retinal pigment epithelial cells: implications for age-related macular degeneration. Redox Biol. 2016;7:78–87. doi: 10.1016/j.redox.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kozlowski MR. RPE cell senescence: a key contributor to age-related macular degeneration. Med Hypotheses. 2012;78(4):505–510. doi: 10.1016/j.mehy.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 18.Kozlowski MR. Senescent retinal pigment epithelial cells are more sensitive to vascular endothelial growth factor: implications for "wet" age-related macular degeneration. J Ocular Pharmacol Ther. 2015;31(2):87–92. doi: 10.1089/jop.2014.0071. [DOI] [PubMed] [Google Scholar]
- 19.Matsunaga H, Handa JT, Aotaki-Keen A, Sherwood SW, West MD, Hjelmeland LM. Beta-galactosidase histochemistry and telomere loss in senescent retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1999;40(1):197–202. [PubMed] [Google Scholar]
- 20.Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37(3):614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 21.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192(4):547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Childs BG, Baker DJ, Kirkland JL, Campisi J, van Deursen JM. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. 2014;15(11):1139–1153. doi: 10.15252/embr.201439245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prieur A, Peeper DS. Cellular senescence in vivo: a barrier to tumorigenesis. Curr Opin Cell Biol. 2008;20(2):150–155. doi: 10.1016/j.ceb.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 24.Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, Sansom OJ, Zender L, Keyes WM. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017;31(2):172–183. doi: 10.1101/gad.290635.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coppé J-P, Desprez P-Y, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campisi J, Robert L. Cell senescence: role in aging and age-related diseases. Interdiscip Topics Gerontol. 2014;39:45–61. doi: 10.1159/000358899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodier F, Munoz DP, Teachenor R, Chu V, Le O, Bhaumik D, Coppe JP, Campeau E, Beausejour CM, Kim SH, Davalos AR, Campisi J. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci. 2011;124(Pt 1):68–81. doi: 10.1242/jcs.071340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, von Zglinicki T. A senescent cell bystander effect: senescence-induced senescence. Aging Cell. 2012;11(2):345–349. doi: 10.1111/j.1474-9726.2012.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat A, Saretzki G, Rudolph KL, Kirkwood TB, von Zglinicki T. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol. 2010;6:347. doi: 10.1038/msb.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Investig. 2018;128(4):1238–1246. doi: 10.1172/jci95148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479(7372):232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509(7501):439–446. doi: 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, Ross OA. Age and age-related diseases: role of inflammation triggers and cytokines. Front Immunol. 2018;9:586–586. doi: 10.3389/fimmu.2018.00586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yin Y, Zhou Z, Liu W, Chang Q, Sun G, Dai Y. Vascular endothelial cells senescence is associated with NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome activation via reactive oxygen species (ROS)/thioredoxin-interacting protein (TXNIP) pathway. Int J Biochem Cell Biol. 2017;84:22–34. doi: 10.1016/j.biocel.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 36.Huy H, Song HY, Kim MJ, Kim WS, Kim DO, Byun JE, Lee J, Park YJ, Kim TD, Yoon SR, Choi EJ, Lee CH, Noh JY, Jung H, Choi I. TXNIP regulates AKT-mediated cellular senescence by direct interaction under glucose-mediated metabolic stress. Aging Cell. 2018;17(6):e12836. doi: 10.1111/acel.12836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11(8):973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rabanal-Ruiz Y, Otten EG, Korolchuk VI. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017;61(6):565–584. doi: 10.1042/EBC20170027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito A, Georgilis A, Montoya A, Wolter K, Dharmalingam G, Faull P, Carroll T, Martinez-Barbera JP, Cutillas P, Reisinger F, Heikenwalder M, Miller RA, Withers D, Zender L, Thomas GJ, Gil J. Erratum: mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. 2015;17(10):1370. doi: 10.1038/ncb3243. [DOI] [PubMed] [Google Scholar]
- 40.Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, Limbad C, Demaria M, Li P, Hubbard GB, Ikeno Y, Javors M, Desprez PY, Benz CC, Kapahi P, Nelson PS, Campisi J. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015;17(8):1049–1061. doi: 10.1038/ncb3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mosieniak G, Sliwinska MA, Alster O, Strzeszewska A, Sunderland P, Piechota M, Was H, Sikora E. Polyploidy formation in doxorubicin-treated cancer cells can favor escape from senescence. Neoplasia (New York, NY) 2015;17(12):882–893. doi: 10.1016/j.neo.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sikora E, Mosieniak G, Sliwinska MA. Morphological and functional characteristic of senescent cancer cells. Curr Drug Targets. 2016;17(4):377–387. doi: 10.2174/1389450116666151019094724. [DOI] [PubMed] [Google Scholar]
- 43.Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavare S, Arakawa S, Shimizu S, Watt FM, Narita M. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23(7):798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roggia MF, Ueta T. Alphavbeta5 Integrin/FAK/PGC-1alpha pathway confers protective effects on retinal pigment epithelium. PLoS ONE. 2015;10(8):e0134870. doi: 10.1371/journal.pone.0134870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, Lazarowski ER, Damian VA, Masliah E, La Spada AR. PGC-1alpha rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012;4(142):142ra197. doi: 10.1126/scitranslmed.3003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kang C, Elledge SJ. How autophagy both activates and inhibits cellular senescence. Autophagy. 2016;12(5):898–899. doi: 10.1080/15548627.2015.1121361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, Elledge SJ. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science (New York, NY) 2015;349(6255):aaa5612. doi: 10.1126/science.aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hussain AA, Lee Y, Marshall J. Understanding the complexity of the matrix metalloproteinase system and its relevance to age-related diseases: age-related macular degeneration and Alzheimer's disease. Prog Retinal Eye Res. 2019 doi: 10.1016/j.preteyeres.2019.100775. [DOI] [PubMed] [Google Scholar]
- 49.Karwatowski WS, Jeffries TE, Duance VC, Albon J, Bailey AJ, Easty DL. Preparation of Bruch's membrane and analysis of the age-related changes in the structural collagens. Br J Ophthalmol. 1995;79(10):944–952. doi: 10.1136/bjo.79.10.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hussain AA, Lee Y, Zhang JJ, Marshall J. Disturbed matrix metalloproteinase activity of Bruch's membrane in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52(7):4459–4466. doi: 10.1167/iovs.10-6678. [DOI] [PubMed] [Google Scholar]
- 51.Guymer RH, Wu Z, Hodgson LAB, Caruso E, Brassington KH, Tindill N, Aung KZ, McGuinness MB, Fletcher EL, Chen FK, Chakravarthy U, Arnold JJ, Heriot WJ, Durkin SR, Lek JJ, Harper CA, Wickremasinghe SS, Sandhu SS, Baglin EK, Sharangan P, Braat S, Luu CD. Subthreshold nanosecond laser intervention in age-related macular degeneration: the LEAD randomized controlled clinical trial. Ophthalmology. 2019;126(6):829–838. doi: 10.1016/j.ophtha.2018.09.015. [DOI] [PubMed] [Google Scholar]
- 52.Lee Y, Hussain AA, Seok JH, Kim SH, Marshall J. Modulating the transport characteristics of Bruch's membrane with steroidal glycosides and its relevance to age-related macular degeneration (AMD) Invest Ophthalmol Vis Sci. 2015;56(13):8403–8418. doi: 10.1167/iovs.15-16936. [DOI] [PubMed] [Google Scholar]
- 53.Cao L, Wang H, Wang F, Xu D, Liu F, Liu C. Abeta-induced senescent retinal pigment epithelial cells create a proinflammatory microenvironment in AMD. Invest Ophthalmol Vis Sci. 2013;54(5):3738–3750. doi: 10.1167/iovs.13-11612. [DOI] [PubMed] [Google Scholar]
- 54.Johnson LV, Leitner WP, Rivest AJ, Staples MK, Radeke MJ, Anderson DH. The Alzheimer's A beta-peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc Natl Acad Sci USA. 2002;99(18):11830–11835. doi: 10.1073/pnas.192203399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cao S, Walker GB, Wang X, Cui JZ, Matsubara JA. Altered cytokine profiles of human retinal pigment epithelium: oxidant injury and replicative senescence. Mol Vis. 2013;19:718–728. [PMC free article] [PubMed] [Google Scholar]
- 56.Chaum E, Winborn CS, Bhattacharya S. Genomic regulation of senescence and innate immunity signaling in the retinal pigment epithelium. Mamm Genome. 2015;26(5–6):210–221. doi: 10.1007/s00335-015-9568-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu D, Wu J, Spee C, Ryan SJ, Hinton DR. BMP4 mediates oxidative stress-induced retinal pigment epithelial cell senescence and is overexpressed in age-related macular degeneration. J Biol Chem. 2009;284(14):9529–9539. doi: 10.1074/jbc.M809393200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Akiguchi I, Pallas M, Budka H, Akiyama H, Ueno M, Han J, Yagi H, Nishikawa T, Chiba Y, Sugiyama H, Takahashi R, Unno K, Higuchi K, Hosokawa M. SAMP8 mice as a neuropathological model of accelerated brain aging and dementia: Toshio Takeda's legacy and future directions. Neuropathology. 2017;37(4):293–305. doi: 10.1111/neup.12373. [DOI] [PubMed] [Google Scholar]
- 59.Feng L, Cao L, Zhang Y, Wang F. Detecting Abeta deposition and RPE cell senescence in the retinas of SAMP8 mice. Discov Med. 2016;21(115):149–158. [PubMed] [Google Scholar]
- 60.Jadeja RN, Powell FL, Jones MA, Fuller J, Joseph E, Thounaojam MC, Bartoli M, Martin PM. Loss of NAMPT in aging retinal pigment epithelium reduces NAD(+) availability and promotes cellular senescence. Aging. 2018;10(6):1306–1323. doi: 10.18632/aging.101469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stefanova NA, Kozhevnikova OS, Vitovtov AO, Maksimova KY, Logvinov SV, Rudnitskaya EA, Korbolina EE, Muraleva NA, Kolosova NG. Senescence-accelerated OXYS rats: a model of age-related cognitive decline with relevance to abnormalities in Alzheimer disease. Cell Cycle (Georgetown, Tex) 2014;13(6):898–909. doi: 10.4161/cc.28255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kozhevnikova OS, Telegina DV, Devyatkin VA, Kolosova NG. Involvement of the autophagic pathway in the progression of AMD-like retinopathy in senescence-accelerated OXYS rats. Biogerontology. 2018;19(3–4):223–235. doi: 10.1007/s10522-018-9751-y. [DOI] [PubMed] [Google Scholar]
- 63.Arend N, Wertheimer C, Laubichler P, Wolf A, Kampik A, Kernt M. Idebenone prevents oxidative stress, cell death and senescence of retinal pigment epithelium cells by stabilizing BAX/Bcl-2 ratio. Ophthalmologica. 2015;234(2):73–82. doi: 10.1159/000381726. [DOI] [PubMed] [Google Scholar]
- 64.Chae SY, Park SY, Park G. Lutein protects human retinal pigment epithelial cells from oxidative stress-induced cellular senescence. Mol Med Rep. 2018;18(6):5182–5190. doi: 10.3892/mmr.2018.9538. [DOI] [PubMed] [Google Scholar]
- 65.Dvashi Z, Green Y, Pollack A. TAK1 inhibition accelerates cellular senescence of retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2014;55(9):5679–5686. doi: 10.1167/iovs.14-14349. [DOI] [PubMed] [Google Scholar]
- 66.Lazzarini R, Nicolai M, Pirani V, Mariotti C, Di Primio R. Effects of senescent secretory phenotype acquisition on human retinal pigment epithelial stem cells. Aging. 2018;10(11):3173–3184. doi: 10.18632/aging.101624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sun Q, Qing W, Qi R, Zou M, Gong L, Liu Y, Li DW. Inhibition of sumoylation alleviates oxidative stress-induced retinal pigment epithelial cell senescence and represses proinflammatory gene expression. Curr Mol Med. 2018;18(9):575–583. doi: 10.2174/1566524019666190107154250. [DOI] [PubMed] [Google Scholar]
- 68.Sun Y, Zheng Y, Wang C, Liu Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018;9(7):753. doi: 10.1038/s41419-018-0794-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hyttinen JMT, Blasiak J, Niittykoski M, Kinnunen K, Kauppinen A, Salminen A, Kaarniranta K. DNA damage response and autophagy in the degeneration of retinal pigment epithelial cells-Implications for age-related macular degeneration (AMD) Ageing Res Rev. 2017;36:64–77. doi: 10.1016/j.arr.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 70.Pinto M, Moraes CT. Mechanisms linking mtDNA damage and aging. Free Radic Biol Med. 2015;85:250–258. doi: 10.1016/j.freeradbiomed.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kaarniranta K, Pawlowska E, Szczepanska J, Jablkowska A, Blasiak J. Role of mitochondrial dna damage in ROS-mediated pathogenesis of age-related macular degeneration (AMD) Int J Mol Sci. 2019 doi: 10.3390/ijms20102374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Karunadharma PP, Nordgaard CL, Olsen TW, Ferrington DA. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2010;51(11):5470–5479. doi: 10.1167/iovs.10-5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ferrington DA, Kapphahn RJ, Leary MM, Atilano SR, Terluk MR, Karunadharma P, Chen GK, Ratnapriya R, Swaroop A, Montezuma SR, Kenney MC. Increased retinal mtDNA damage in the CFH variant associated with age-related macular degeneration. Exp Eye Res. 2016;145:269–277. doi: 10.1016/j.exer.2016.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Laberge RM, Adler D, DeMaria M, Mechtouf N, Teachenor R, Cardin GB, Desprez PY, Campisi J, Rodier F. Mitochondrial DNA damage induces apoptosis in senescent cells. Cell Death Dis. 2013;4:e727. doi: 10.1038/cddis.2013.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rubsam LZ, Davidson BL, Shewach DS. Superior cytotoxicity with ganciclovir compared with acyclovir and 1-beta-D-arabinofuranosylthymine in herpes simplex virus-thymidine kinase-expressing cells: a novel paradigm for cell killing. Cancer Res. 1998;58(17):3873–3882. [PubMed] [Google Scholar]
- 76.Lee HC, Yin PH, Chi CW, Wei YH. Increase in mitochondrial mass in human fibroblasts under oxidative stress and during replicative cell senescence. J Biomed Sci. 2002;9(6 Pt 1):517–526. doi: 10.1007/bf02254978. [DOI] [PubMed] [Google Scholar]
- 77.Williams AB, Schumacher B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb Perspect Med. 2016 doi: 10.1101/cshperspect.a026070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hashimoto Y, Niikura T, Tajima H, Yasukawa T, Sudo H, Ito Y, Kita Y, Kawasumi M, Kouyama K, Doyu M, Sobue G, Koide T, Tsuji S, Lang J, Kurokawa K, Nishimoto I. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer's disease genes and Abeta. Proc Natl Acad Sci USA. 2001;98(11):6336–6341. doi: 10.1073/pnas.101133498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bodzioch M, Lapicka-Bodzioch K, Zapala B, Kamysz W, Kiec-Wilk B, Dembinska-Kiec A. Evidence for potential functionality of nuclearly-encoded humanin isoforms. Genomics. 2009;94(4):247–256. doi: 10.1016/j.ygeno.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 80.Cobb LJ, Lee C, Xiao J, Yen K, Wong RG, Nakamura HK, Mehta HH, Gao Q, Ashur C, Huffman DM, Wan J, Muzumdar R, Barzilai N, Cohen P. Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging. 2016;8(4):796–809. doi: 10.18632/aging.100943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Minasyan L, Sreekumar PG, Hinton DR, Kannan R. Protective mechanisms of the mitochondrial-derived peptide humanin in oxidative and endoplasmic reticulum stress in RPE cells. Oxid Med Cell Longev. 2017;2017:1675230. doi: 10.1155/2017/1675230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nashine S, Cohen P, Chwa M, Lu S, Nesburn AB, Kuppermann BD, Kenney MC. Humanin G (HNG) protects age-related macular degeneration (AMD) transmitochondrial ARPE-19 cybrids from mitochondrial and cellular damage. Cell Death Dis. 2017;8(7):e2951. doi: 10.1038/cddis.2017.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gong Z, Tasset I, Diaz A, Anguiano J, Tas E, Cui L, Kuliawat R, Liu H, Kuhn B, Cuervo AM, Muzumdar R. Humanin is an endogenous activator of chaperone-mediated autophagy. J Cell Biol. 2018;217(2):635–647. doi: 10.1083/jcb.201606095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kaarniranta K, Sinha D, Blasiak J, Kauppinen A, Vereb Z, Salminen A, Boulton ME, Petrovski G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy. 2013;9(7):973–984. doi: 10.4161/auto.24546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sreekumar PG, Ishikawa K, Spee C, Mehta HH, Wan J, Yen K, Cohen P, Kannan R, Hinton DR. The mitochondrial-derived peptide humanin protects RPE cells from oxidative stress, senescence, and mitochondrial dysfunction. Invest Ophthalmol Vis Sci. 2016;57(3):1238–1253. doi: 10.1167/iovs.15-17053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McLeod DS, Grebe R, Bhutto I, Merges C, Baba T, Lutty GA. Relationship between RPE and choriocapillaris in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50(10):4982–4991. doi: 10.1167/iovs.09-3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McLeod DS, Taomoto M, Otsuji T, Green WR, Sunness JS, Lutty GA. Quantifying changes in RPE and choroidal vasculature in eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2002;43(6):1986–1993. [PubMed] [Google Scholar]
- 88.Mullins RF, Johnson MN, Faidley EA, Skeie JM, Huang J. Choriocapillaris vascular dropout related to density of drusen in human eyes with early age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52(3):1606–1612. doi: 10.1167/iovs.10-6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Whitmore SS, Sohn EH, Chirco KR, Drack AV, Stone EM, Tucker BA, Mullins RF. Complement activation and choriocapillaris loss in early AMD: implications for pathophysiology and therapy. Prog Retin Eye Res. 2015;45:1–29. doi: 10.1016/j.preteyeres.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mullins RF, Schoo DP, Sohn EH, Flamme-Wiese MJ, Workamelahu G, Johnston RM, Wang K, Tucker BA, Stone EM. The membrane attack complex in aging human choriocapillaris: relationship to macular degeneration and choroidal thinning. Am J Pathol. 2014;184(11):3142–3153. doi: 10.1016/j.ajpath.2014.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cabrera AP, Bhaskaran A, Xu J, Yang X, Scott HA, Mohideen U, Ghosh K. Senescence increases choroidal endothelial stiffness and susceptibility to complement injury: implications for choriocapillaris loss in AMD. Invest Ophthalmol Vis Sci. 2016;57(14):5910–5918. doi: 10.1167/iovs.16-19727. [DOI] [PubMed] [Google Scholar]
- 92.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (New York, NY) 2013;339(6121):786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science (New York, NY) 2013;339(6121):826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kato K, Omura H, Ishitani R, Nureki O. Cyclic GMP-AMP as an endogenous second messenger in innate immune signaling by cytosolic DNA. Annu Rev Biochem. 2017;86:541–566. doi: 10.1146/annurev-biochem-061516-044813. [DOI] [PubMed] [Google Scholar]
- 95.Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15(12):760–770. doi: 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gluck S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, Ablasser A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19(9):1061–1070. doi: 10.1038/ncb3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wu Y, Wei Q, Yu J. The cGAS/STING pathway: a sensor of senescence-associated DNA damage and trigger of inflammation in early age-related macular degeneration. Clin Interv Aging. 2019;14:1277–1283. doi: 10.2147/cia.S200637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, Heymann M, van der Goot FG, Turcatti G, Behrendt R, Ablasser A. Targeting STING with covalent small-molecule inhibitors. Nature. 2018;559(7713):269–273. doi: 10.1038/s41586-018-0287-8. [DOI] [PubMed] [Google Scholar]
- 99.Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci USA. 2017;114(23):E4612–e4620. doi: 10.1073/pnas.1705499114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nowak JZ. Oxidative stress, polyunsaturated fatty acids-derived oxidation products and bisretinoids as potential inducers of CNS diseases: focus on age-related macular degeneration. Pharmacol Rep. 2013;65(2):288–304. doi: 10.1016/S1734-1140(13)71005-3. [DOI] [PubMed] [Google Scholar]
- 101.Di Guardo G. Lipofuscin, lipofuscin-like pigments and autofluorescence. Eur J Histochem. 2015;59(1):2485. doi: 10.4081/ejh.2015.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mazzitello KI, Arizmendi CM, Family F, Grossniklaus HE. Formation and growth of lipofuscin in the retinal pigment epithelium cells. Phys Rev E Stat Nonlin Soft Matter Phys. 2009;80(5 Pt 1):051908–051908. doi: 10.1103/PhysRevE.80.051908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Boyer NP, Higbee D, Currin MB, Blakeley LR, Chen C, Ablonczy Z, Crouch RK, Koutalos Y. Lipofuscin and N-retinylidene-N-retinylethanolamine (A2E) accumulate in retinal pigment epithelium in absence of light exposure: their origin is 11-cis-retinal. J Biol Chem. 2012;287(26):22276–22286. doi: 10.1074/jbc.M111.329235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hohn A, Jung T, Grimm S, Grune T. Lipofuscin-bound iron is a major intracellular source of oxidants: role in senescent cells. Free Radic Biol Med. 2010;48(8):1100–1108. doi: 10.1016/j.freeradbiomed.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 105.Chowers I, Wong R, Dentchev T, Farkas RH, Iacovelli J, Gunatilaka TL, Medeiros NE, Presley JB, Campochiaro PA, Curcio CA, Dunaief JL, Zack DJ. The iron carrier transferrin is upregulated in retinas from patients with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2006;47(5):2135–2140. doi: 10.1167/iovs.05-1135. [DOI] [PubMed] [Google Scholar]
- 106.Dunaief JL, Richa C, Franks EP, Schultze RL, Aleman TS, Schenck JF, Zimmerman EA, Brooks DG. Macular degeneration in a patient with aceruloplasminemia, a disease associated with retinal iron overload. Ophthalmology. 2005;112(6):1062–1065. doi: 10.1016/j.ophtha.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 107.Hahn P, Milam AH, Dunaief JL. Maculas affected by age-related macular degeneration contain increased chelatable iron in the retinal pigment epithelium and Bruch's membrane. Arch Ophthalmol (Chicago, Ill) 2003;121(8):1099–1105. doi: 10.1001/archopht.121.8.1099. [DOI] [PubMed] [Google Scholar]
- 108.Blasiak J, Szaflik J, Szaflik JP. Implications of altered iron homeostasis for age-related macular degeneration. Front Biosci (Landmark edition) 2011;16:1551–1559. doi: 10.2741/3804. [DOI] [PubMed] [Google Scholar]
- 109.Garcia-Castineiras S. Iron, the retina and the lens: a focused review. Exp Eye Res. 2010;90(6):664–678. doi: 10.1016/j.exer.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ugarte M, Osborne NN, Brown LA, Bishop PN. Iron, zinc, and copper in retinal physiology and disease. Surv Ophthalmol. 2013;58(6):585–609. doi: 10.1016/j.survophthal.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 111.Wong RW, Richa DC, Hahn P, Green WR, Dunaief JL. Iron toxicity as a potential factor in AMD. Retina (Philadelphia, Pa) 2007;27(8):997–1003. doi: 10.1097/IAE.0b013e318074c290. [DOI] [PubMed] [Google Scholar]
- 112.Konig J, Ott C, Hugo M, Jung T, Bulteau AL, Grune T, Hohn A. Mitochondrial contribution to lipofuscin formation. Redox Biol. 2017;11:673–681. doi: 10.1016/j.redox.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hyttinen JMT, Viiri J, Kaarniranta K, Blasiak J. Mitochondrial quality control in AMD: does mitophagy play a pivotal role? Cell Mol Life Sci. 2018;75(16):2991–3008. doi: 10.1007/s00018-018-2843-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Katz ML. Potential role of retinal pigment epithelial lipofuscin accumulation in age-related macular degeneration. Arch Gerontol Geriatr. 2002;34(3):359–370. doi: 10.1016/S0167-4943(02)00012-2. [DOI] [PubMed] [Google Scholar]
- 115.Sparrow JR, Zhou J, Cai B. DNA is a target of the photodynamic effects elicited in A2E-laden RPE by blue-light illumination. Invest Ophthalmol Vis Sci. 2003;44(5):2245–2251. doi: 10.1167/iovs.02-0746. [DOI] [PubMed] [Google Scholar]
- 116.Sun S, Cai B, Li Y, Su W, Zhao X, Gong B, Li Z, Zhang X, Wu Y, Chen C, Tsang SH, Yang J, Li X. HMGB1 and Caveolin-1 related to RPE cell senescence in age-related macular degeneration. Aging. 2019;11(13):4323–4337. doi: 10.18632/aging.102039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liu J, Itagaki Y, Ben-Shabat S, Nakanishi K, Sparrow JR. The biosynthesis of A2E, a fluorophore of aging retina, involves the formation of the precursor, A2-PE, in the photoreceptor outer segment membrane. J Biol Chem. 2000;275(38):29354–29360. doi: 10.1074/jbc.M910191199. [DOI] [PubMed] [Google Scholar]
- 118.Washington I, Saad L. The rate of vitamin A dimerization in lipofuscinogenesis, fundus autofluorescence, retinal senescence and degeneration. In: Bowes Rickman C, LaVail MM, Anderson RE, Grimm C, Hollyfield J, Ash J, editors. Retinal degenerative diseases. Cham: Springer International Publishing; 2016. pp. 347–353. [DOI] [PubMed] [Google Scholar]
- 119.Alaimo A, Linares GG, Bujjamer JM, Gorojod RM, Alcon SP, Martinez JH, Baldessari A, Grecco HE, Kotler ML. Toxicity of blue led light and A2E is associated to mitochondrial dynamics impairment in ARPE-19 cells: implications for age-related macular degeneration. Arch Toxicol. 2019;93(5):1401–1415. doi: 10.1007/s00204-019-02409-6. [DOI] [PubMed] [Google Scholar]
- 120.Cong Y-S, Wright WE, Shay JW. Human telomerase and its regulation. Microbiol Mol Biol Rev. 2002;66(3):407–425. doi: 10.1128/mmbr.66.3.407-425.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wellinger RJ. In the end, what's the problem? Mol Cell. 2014;53(6):855–856. doi: 10.1016/j.molcel.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 122.Heinrichs A. Telomere overhang processing. Nat Struct Mol Biol. 2012;19(7):663–663. doi: 10.1038/nsmb.2342. [DOI] [Google Scholar]
- 123.Zhang C, Liu H-H, Zheng K-W, Hao Y-H, Tan Z. DNA G-quadruplex formation in response to remote downstream transcription activity: long-range sensing and signal transducing in DNA double helix. Nucleic Acids Res. 2013;41(14):7144–7152. doi: 10.1093/nar/gkt443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sparrow JR, Vollmer-Snarr HR, Zhou J, Jang YP, Jockusch S, Itagaki Y, Nakanishi K. A2E-epoxides damage DNA in retinal pigment epithelial cells. Vitamin E and other antioxidants inhibit A2E-epoxide formation. J Biol Chem. 2003;278(20):18207–18213. doi: 10.1074/jbc.M300457200. [DOI] [PubMed] [Google Scholar]
- 125.Wang J, Feng Y, Han P, Wang F, Luo X, Liang J, Sun X, Ye J, Lu Y, Sun X. Photosensitization of A2E triggers telomere dysfunction and accelerates retinal pigment epithelium senescence. Cell Death Dis. 2018;9(2):178. doi: 10.1038/s41419-017-0200-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Feng L, Liao X, Zhang Y, Wang F. Protective effects on age-related macular degeneration by activated autophagy induced by amyloid-beta in retinal pigment epithelial cells. Discov Med. 2019;27(148):153–160. [PubMed] [Google Scholar]
- 127.Masuda N, Tsujinaka H, Hirai H, Yamashita M, Ueda T, Ogata N. Effects of concentration of amyloid beta (Abeta) on viability of cultured retinal pigment epithelial cells. BMC Ophthalmol. 2019;19(1):70. doi: 10.1186/s12886-019-1076-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tsao SW, Gabriel R, Thaker K, Kuppermann BD, Kenney MC. Effects of brimonidine on retinal pigment epithelial cells and muller cells exposed to amyloid-beta 1–42 peptide in vitro. Ophthalmic Surg Lasers Imaging Retina. 2018;49(10):S23–s28. doi: 10.3928/23258160-20180814-04. [DOI] [PubMed] [Google Scholar]
- 129.Ye Z, He SZ, Li ZH. Effect of Abeta protein on inhibiting proliferation and promoting apoptosis of retinal pigment epithelial cells. Int J Ophthalmol. 2018;11(6):929–934. doi: 10.18240/ijo.2018.06.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liu C, Cao L, Yang S, Xu L, Liu P, Wang F, Xu D. Subretinal injection of amyloid-beta peptide accelerates RPE cell senescence and retinal degeneration. Int J Mol Med. 2015;35(1):169–176. doi: 10.3892/ijmm.2014.1993. [DOI] [PubMed] [Google Scholar]
- 131.Mitter SK, Song C, Qi X, Mao H, Rao H, Akin D, Lewin A, Grant M, Dunn W, Jr, Ding J, Bowes Rickman C, Boulton M. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy. 2014;10(11):1989–2005. doi: 10.4161/auto.36184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kaarniranta K, Tokarz P, Koskela A, Paterno J, Blasiak J. Autophagy regulates death of retinal pigment epithelium cells in age-related macular degeneration. Cell Biol Toxicol. 2017;33(2):113–128. doi: 10.1007/s10565-016-9371-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mitter SK, Rao HV, Qi X, Cai J, Sugrue A, Dunn WA, Jr, Grant MB, Boulton ME. Autophagy in the retina: a potential role in age-related macular degeneration. Adv Exp Med Biol. 2012;723:83–90. doi: 10.1007/978-1-4614-0631-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Short S, Fielder E, Miwa S, von Zglinicki T. Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine. 2019;41:683–692. doi: 10.1016/j.ebiom.2019.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, Prata L, Masternak MM, Kritchevsky SB, Musi N, Kirkland JL. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine. 2019;40:554–563. doi: 10.1016/j.ebiom.2018.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]