


Abstract
Using site-specific incorporation of the photochemical cross-linking reagent 4-thiouridine, we demonstrate the previously unknown association of two proteins with yeast 3′ splice sites. One of these is an unidentified ~122 kDa protein that cross-links to 3′ splice sites during formation of the pre-spliceosome. The other factor is the DExH-box RNA helicase, Prp22p. With substrates functional in the second step of splicing, only very weak cross-linking of Prp22p to intron sequences at the 3′ splice site is observed. In contrast, substrates blocked at the second step exhibit strong cross-linking of Prp22 to intron sequences at the 3′ splice site, but not to adjacent exon sequences. In vitro reconstitution experiments also show that the association of Prp22p with intron sequences at the 3′ splice site is dependent on Prp16p and does not persist when release of mature mRNA from the spliceosome is blocked. Taken together, these results suggest that the 3′ splice site of yeast introns is contacted much earlier than previously envisioned by a protein of ~120 kDa, and that a transient association of Prp22p with the 3′ splice site occurs between the first and second catalytic steps.
INTRODUCTION
The accurate recognition of both the 5′ splice site and branch site sequences during the first step of nuclear pre-mRNA splicing is accomplished through the use of multiple recognition steps involving specific RNA–RNA and RNA–protein interactions (1,2). The mechanism and factors involved in recognition or selection of the 3′ splice site are not completely defined, however (3). For the second step of nuclear pre-mRNA splicing to occur, the 3′ splice site in the intron-lariat intermediate must be selected and correctly positioned relative to the 3′-OH group of the excised 5′ exon. In yeast, 3′ splice sites are defined by the conserved sequence YAG, and are usually located 10–60 nt downstream from the branch site sequence (4). Generally, the first AG dinucleotide downstream from the pre-mRNA branch site sequence is chosen as the 3′ splice site (5–7). Selection of the 3′ splice site is thus tightly coupled to recognition of the branch site sequence, which occurs during the first catalytic step. Genetic and biochemical studies in yeast support the existence of a complex network of RNA–RNA interactions that may be involved in 3′ splice site recognition (3,8). In addition, six yeast proteins have been identified that have roles in the second catalytic step and may be involved in 3′ splice site selection (3,9–12). Following the first catalytic step, Prp16p appears to function as part of a complex involving Prp17p, Slu7p, Prp18p and Prp22p to facilitate a conformational rearrangement in the spliceosome that is thought to reflect the identification of the 3′ splice site and its positioning within the active site for the second catalytic step. This conformational rearrangement occurs in two stages, an initial ATP-dependent stage involving Prp16p and Prp17p, and a subsequent ATP-independent stage involving Slu7p, Prp18p and Prp22p (3,10,13).
Of the six proteins thus far implicated in events associated with the second catalytic step however, only two, Prp16p and Prp8p appear to have essential roles. PRP17 is non-essential for growth, and splicing extracts depleted of Prp17p exhibit only a partial second step defect (13). In vitro studies have established that Slu7p, Prp18p and Prp22p are required during the second step of splicing with substrates containing 3′ splice sites distal to the branch site. Curiously however, they are not essential for the second step of splicing with substrates containing 3′ splice sites located close (proximal) to the branch site sequence (10,14,15). Thus, although these factors are likely to have important roles in 3′ splice site selection, they are unlikely to be directly involved in binding of the 3′ splice by the active site of the spliceosome.
In yeast splicing extracts, Prp8p, Prp16p and Slu7p have previously been shown to cross-link to the 3′ splice site region. Prp8p is a U5 snRNP associated protein that interacts extensively with the 5′ exon prior to the first step of splicing and with the 3′ splice site region prior to the second step (7,16,17). Mutants in Prp8 that are able to suppress the effects of 5′ and 3′ splice site mutations have also been isolated (18–20). It was proposed that Prp8p functions at the catalytic core of the spliceosome to stabilize interactions between the loop of the U5 snRNA and exon sequences at the 5′ and 3′ splice sites, and to align these sequences during the second catalytic step of splicing. In mammalian splicing extracts, cross-linking of the 3′ splice site to a 100 kDa protein (AG100) in pre-spliceosomes (21) and to either a 75 kDa (AG75) or 70 kDa protein in mature spliceosomes has been reported (21,22). More recent experiments have shown that during pre-spliceosome formation, the 3′ splice site AG in metazoan introns is specifically recognized by U2AF35 (23–25). In yeast however, no homolog of U2AF35 has been reported (26), and possible homologs of the other mammalian proteins that cross-link to the 3′ splice site have not been identified, to our knowledge.
In the present study, we have examined the interactions of proteins in whole cell yeast splicing extracts with the 3′ splice site YAG↓ using site-specific incorporation of the photochemical cross-linking reagent 4-thiouridine (s4U) in derivatives of the yeast actin intron. Although previous cross-linking studies have established the interactions of several proteins with the 3′ splice site region during yeast pre-mRNA splicing (7,16), our experiments demonstrate the association of yeast 3′ splice sites with two previously undetected proteins. One of these proteins associates with the 3′ splice site during formation of the pre-spliceosome, suggesting contacts with the 3′ splice site may be established much earlier than previously envisaged in yeast (27). The other protein is the DExH-box RNA helicase, Prp22p, which plays roles in both the second catalytic step of splicing and in release of mRNA from the spliceosome (10).
MATERIALS AND METHODS
Synthetic oligonucleotides and PCR. Oligodeoxyribonucleotides were obtained from Cybersyn, Inc. (Aston, PA). PCR-generated templates for in vitro transcription were prepared using Vent DNA polymerase (New England Biolabs) according to the manufacturers’ guidelines using the wild-type SP6 actin plasmid (28) as template. A large number of oligodeoxyribonucleotides were used for PCR amplification to create templates for in vitro transcription of RNA fragments, as bridging oligos for in vitro RNA ligation, and for the synthesis of short unmodified or s4U containing RNAs. A complete list of the oligodeoxyribonucleotide sequences used is available upon request. Synthetic RNAs used for in vitro ligations were obtained from TriLink BioTechnologies, Inc. (San Diego, CA) and were gel purified prior to use. The following oligonucleotides were used for RNase H mediated depletion of specific snRNAs in yeast splicing extracts: L19B (U1): 5′-atcttaaggtaagtatcctatagtgagtcgtattag-3′, SRU2 (U2): 5′-cagatactacacttg-3′, U6d1 (U6): 5′-atctctgtattgtttcaaattgaccaa-3′.
Synthesis of RNAs and in vitro RNA ligation
In vitro transcription reactions using PCR derived DNA templates were done using either T7 or SP6 RNA polymerase (New England Biolabs). RNAs were then gel purified prior to further manipulation. The purified 3′ fragment RNAs were dephosphorylated using 0.5 U Bacterial Alkaline Phosphatase (Amersham) per 10 pmol 5′ ends for 1 h at 37°C in 50 mM Tris–Cl pH 8.0, phenol extracted three times, ether extracted twice, and ethanol precipitated. In vitro transcription reactions (300 µl) with synthetic oligodeoxyribonucleotides using T7 RNA polymerase were performed essentially as described in (29) using either 0.2 mM UTP or s4UTP (Amersham). Following ethanol precipitation, the RNA products were treated with 400 U RNase T1 (Life Technologies, Inc.) for 4 h at 37°C in 20 mM Tris–Cl pH 7.5, 1 mM EDTA, and purified on 20% polyacrylamide gels prior to use in in vitro ligation reactions. T4 polynucleotide kinase (New England Biolabs) was used to phosphorylate RNAs with either unlabeled ATP or [γ-32P]ATP (~7000 Ci/mmol, ICN). In vitro RNA ligations were performed essentially as described in (30) using 2.0 µM 5′ RNA fragment, 1.5 µM oligo RNA(s), 0.5 µM 5′ 32P-labeled 3′ RNA fragment, and 0.5 µM bridging oligo for 2 h at 30°C. Following gel purification of the ligated RNAs, concentrations were calculated by assuming the RNAs had the same specific activity as the [γ-32P]ATP used for phosphorylation of the 5′ end-labeled RNA fragment.
Splicing and UV cross-linking reactions
Yeast splicing extracts were prepared as described in (31) from the strain YHM111 (32). Oligonucleotide-directed RNase H depletions in yeast extracts were performed as described in (31) using oligos L19B, SRU2 and U6d1 at final concentrations of 550, 500 and 425 nM, respectively. Mock-depleted, Prp16-depleted and Prp22-depleted extracts were prepared from strain BJ2168 as described (10,11). Recombinant His-Prp16p (0.1 mg/ml in 50 mM Tris–Cl pH 7.5, 2 mM DTT, 1 mM EDTA, 50 mM KCl) was purified from BL21(DE3) by ammonium sulfate precipitation, followed by Ni-NTA affinity chromatography and ion exchange chromatography (CM Sepharose; B.Schwer, unpublished data). The Prp22 proteins were expressed in BL21(DE3) and purified from bacterial lysates essentially as described (10), except that all buffers contained KCl instead of NaCl. The His-tagged Prp22 proteins used for the splicing complementation assays are fractions eluted from the phosphocellulose column with 50 mM Tris–Cl pH 7.5, 2 mM DTT, 1 mM EDTA, 400 mM KCl. Prp22p (0.2 mg/ml) was used directly, whereas Prp22p-K512A (0.12 mg/ml in 50 mM Tris–Cl pH 7.5, 2 mM DTT, 1 mM EDTA, 50 mM KCl) was dialyzed. Splicing reactions (10–26 µl) contained 0.4 nM 32P-labeled pre-mRNA substrate, and were performed as described in (31), except that only 3.0 µl whole cell extract was used per 10 µl reaction. Following incubation at 23°C for the times indicated, reactions were stopped in an ice-water bath prior to cross-linking. For UV cross-linking, 6–24 µl of each splicing reaction was spotted onto a piece of parafilm covering an ice-cold aluminum block. Droplets were then irradiated for 5 min at a distance of ~8 cm with a 365 nm UV lamp (Model B-100AP, UV Products). Subsequently, 0.6 vol of a mixture containing 23.5 U RNase T1/µl, 12.8 mM Tris–Cl pH 7.5, 20 mM EDTA, and 2.7× Complete™ protease inhibitor mix (Roche) was added and reactions were incubated at 37°C for 30 min. An equal volume of 2× SDS protein gel loading buffer was then added and the reactions frozen prior to analysis by SDS–PAGE (33).
Immunoprecipitations
Prior to use, 40 µl of packed Protein A–agarose beads (Roche) were washed three times with 0.8 ml ice-cold IP500 [20 mM Tris–Cl pH 7.5, 500 mM NaCl, 0.12% (w/v) NP-40], and resuspended in 400 µl IP500. Anti-sera (6–8 µl) was then added and the tube containing the mixture rotated for 1.5 h at 4°C. The beads were then washed four times with 0.8 ml ice-cold IP150 [20 mM Tris–Cl pH 7.5, 150 mM NaCl, 0.12% (w/v) NP-40] and stored at 4°C before use. Following cross-linking and RNase T1 digestion, an equal volume of 2× IP150 (24 µl) + 15 µl packed protein A–agarose beads were added to the reaction mixes and the volume adjusted to 400 µl with ice-cold IP150. These mixtures were rotated at 4°C for 1 h. The beads were then washed four times with 0.8 ml ice-cold IP150, resuspended in 15 µl 2× sample loading buffer, boiled, and analyzed by SDS–PAGE. For immunoprecipitations with anti-Prp8 antibodies, the bead suspension and all buffers were modified to include 100 µg/ml heparin and 200 µg/ml BSA to reduce non-specific binding.
RESULTS
Strategy for site-specific RNA–protein cross-linking at the 3′ splice site
In order to examine proteins closely associated with the 3′ splice site in splicing complexes, the photochemical cross-linking reagent s4U was incorporated in a site-specific fashion at three different positions in the 3′ splice site region of yeast actin pre-mRNA derivatives. The nucleotide analog, s4U, is a useful short-range (2–3 Å) cross-linking reagent, which can be specifically photoactivated using long wavelength UV light and efficiently forms cross-links to both proteins and RNA (34). For each of the actin pre-mRNA derivatives used in this study (Fig. 1), single s4U substitutions were achieved through in vitro transcription of a short RNA that contained only a single uridine residue in the presence of s4UTP, followed by in vitro RNA ligation to create full-length splicing substrates (30).
Figure 1.

Sequences of the branch site-3′ splice site region of the yeast actin pre-mRNA derivatives. The sequence of the region encompassing the branch site/3′ splice site region of the wild-type yeast actin pre-mRNA is shown for reference with the branch site adenosine in upper case. For each pre-mRNA, the branch site and 3′ splice site consensus sequences are underlined, the position of 3′ splice site cleavage is indicated by a downward arrow, and the 3′ exon sequence is shown in italics. Spacing between the branch sequence and 3′ splice site is measured from the branch site adenosine to the position of 3′ splice site cleavage. For each pre-mRNA constructed by three- or four-way in vitro RNA ligations, the sequence of the 5′ and 3′ RNA fragments are shown in lower case, the sequence of the unmodified, or s4U containing, oligo RNAs are shown in bold-face upper case and the position of the single 32P label is indicated by an asterisk. The ACT13(s4UCAA↓) and ACT13(s4UACG↓) substrates are identical to the ACT13(s4UCAG↓) substrate depicted here, except for a one or two base substitution in the 3′ splice site. The 3′ splice site mutant ACM13 (ACG↓) substrates are identical to the wild-type ACM13(CAG↓) substrates shown here, except for a two base substitution in the 3′ splice site.
Initially, two different derivatives of the yeast actin pre-mRNA containing a single s4U at position –3 of the actin 3′ splice site (s4U–3A–2G–1↓) were constructed (Fig. 1). These actin pre-mRNA derivatives differ from the wild-type actin pre-mRNA in two important aspects. First, while the distance between the branch site adenosine and 3′ splice site (YAG↓) in the wild-type yeast actin pre-mRNA is 43 bases, the corresponding distance in the actin pre-mRNA derivatives used in our experiments was either 13 or 47 bases; these are referred to as the ACT13(s4UAG↓) and ACT47(s4UAG↓) substrates, respectively. Second, in yeast pre-mRNAs where the spacing between the branch sequence and 3′ splice site is greater than ~20 bases, the 3′ splice site is usually preceded by a uridine-rich sequence (35) which enhances the ability of the 3′ splice site to compete as a splice acceptor in vivo (6). In each of our ligated actin pre-mRNA substrates (Fig. 1), the 3′ splice site sequence is preceded by an adenosine-rich sequence that may affect its use (see below). The adenosine-rich sequence near the 3′ splice site of our cross-linking substrates was necessary in our strategy for the site-specific incorporation of s4U and was also used to avoid the unintentional introduction of any stable secondary structure(s) near the 3′ splice site and/or branch site sequences.
To assess the effects of the changes in branch-site to 3′ splice site distance and the adenosine-rich tract preceding the 3′ splice site on splicing, in vitro splicing assays with the ligated ACT13(s4UAG↓) and ACT47(s4UAG↓) substrates were performed (Fig. 2). In both of these substrates, a unique 32P-label located between the A and G of the 3′ splice site was used, so that only intron containing species are detected. The ACT13(s4UAG↓) substrate efficiently undergoes both steps of splicing as evidenced by the production of intron lariat-3′ exon and intron lariat RNAs (lanes 1–6). In contrast, the ACT47(s4UAG↓) substrate accumulates the intron lariat-3′ exon intermediate and only very small amounts of intron lariat product (lanes 7–12). The accumulation of the lariat intermediate indicates that this substrate efficiently undergoes the first step of splicing, but is largely blocked at the second step. RT–PCR analysis of RNAs produced in splicing reactions with the ACT47(s4UAG↓) substrate confirmed that small amounts of correctly spliced mRNA were produced (data not shown). Comparison to the splicing activities of unmodified versions of these substrates demonstrated that the s4U substitution at the 3′ splice site did not affect the splicing of these substrates (data not shown). The inhibition of the second step with the ACT47(s4UAG↓) substrate must therefore be caused by either the increased distance between the branch sequence and 3′ splice site, or by a combination of this increased distance and the adenosine-rich sequence preceding the 3′ splice site.
Figure 2.
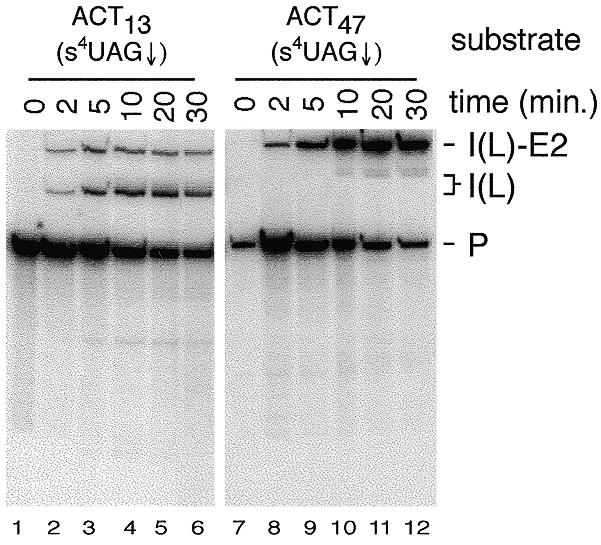
Splicing activity of actin pre-mRNA derivatives containing s4U in the 3′ splice site sequence. The ACT13(s4UAG↓) and ACT47(s4UAG↓) substrates were incubated in splicing extract for the indicated times in the presence of 2 mM ATP. RNA species are labeled as follows: P, pre-mRNA; I(L)-E2, intron lariat-exon2 intermediate; I(L), intron lariat product.
Novel, as well as previously identified proteins, cross-link to the 3′ splice site in yeast splicing extracts
To identify proteins associated with the 3′ splice site, s4U-containing 32P-labeled actin pre-mRNAs were incubated with whole cell yeast splicing extracts under in vitro splicing conditions for various times, irradiated, treated with RNase T1, and analyzed by SDS–PAGE. A time course analysis of the 32P-labeled proteins resulting from cross-linking to the ACT13(s4UAG↓) and ACT47(s4UAG↓) substrates in the presence and absence of ATP is shown in Figure 3A. Cross-linked proteins specifically associated with splicing complexes were initially identified on the basis of their time of appearance relative to splicing products (Fig. 2) and their ATP-dependence. No cross-linked proteins were detected when substrates containing uridine in place of s4U were used (data not shown). With both substrates, three prominent ATP-dependent, cross-linked species with apparent molecular weights of ~122, ~132 and ~300 kDa are observed. Much weaker, ATP-dependent cross-links to an ~260 kDa species are also observed with both substrates. Small amounts of the cross-linked ~122 kDa species can also be seen after prolonged incubation in the absence of exogenous ATP (Fig. 3A, lane 19) and reflect a very low ATP requirement for interaction of this protein with the 3′ splice site region (D.S.McPheeters, unpublished data). Two species of ~180 and 190 kDa also cross-link to these and related cross-linking substrates (Figs 3A, 4B and 5A) in a somewhat ATP-dependent fashion, but cross-linking of these species was highly variable and was not investigated further. Strikingly, with the ACT47(s4UAG↓) substrate, a strong ATP-dependent cross-link to an ~140 kDa species is also observed (Fig. 3A, lanes 8–13). Weak cross-linking to the ~140 kDa species is also detected in reactions using the ACT13(s4UAG↓) substrate (lane 7). For both substrates, analysis of lower molecular weight proteins on higher percentage SDS–polyacrylamide gels revealed no other prominent ATP-dependent cross-linked species (data not shown).
Figure 3.

(A) Analysis of proteins that cross-link to the 3′ splice sites of the ACT13(s4UAG↓) and ACT47(s4UAG↓) substrates. Aliquots of the splicing reactions shown in Figure 2 were subjected to cross-linking and the resulting 32P-labeled proteins analyzed on a 6% SDS–polyacrylamide gel. The positions of molecular weight markers are shown on the right and the approximate apparent molecular weights of the major ATP-dependent cross-linked proteins (see text) indicated on the left. Lane 7 shows a longer exposure of lane 5 to clearly illustrate the weak cross-linking of the ~140 kDa species to the ACT13(s4UAG↓) substrate. (B) Analysis of 3′ splice site cross-linking using splicing extracts depleted of the U1, U2 or U6 snRNAs. Extracts were pretreated with the indicated oligonucleotides, followed by incubation with the indicated substrates for 10 min, cross-linking and analysis on a 6% SDS–polyacrylamide gel. (C) Immunoprecipitation analysis of proteins that cross-link to the 3′ splice site. Samples from either 5 min splicing reactions done with the ACT13(s4UAG↓) substrate or a 20 min splicing reaction done with the ACT47(s4UAG↓) substrate were used for immunoprecipitation following cross-linking. For each substrate, a portion of each reaction was removed for visualization of the total cross-linked proteins (lanes 1 and 5) prior to immunoprecipitation with either anti-Prp16p or Prp22p anti-sera. In lanes 2 and 6 anti-sera was omitted to detect non-specific binding of 32P-labeled, cross-linked proteins to the protein A–agarose alone. A 6% SDS–polyacrylamide gel was used.
Figure 4.
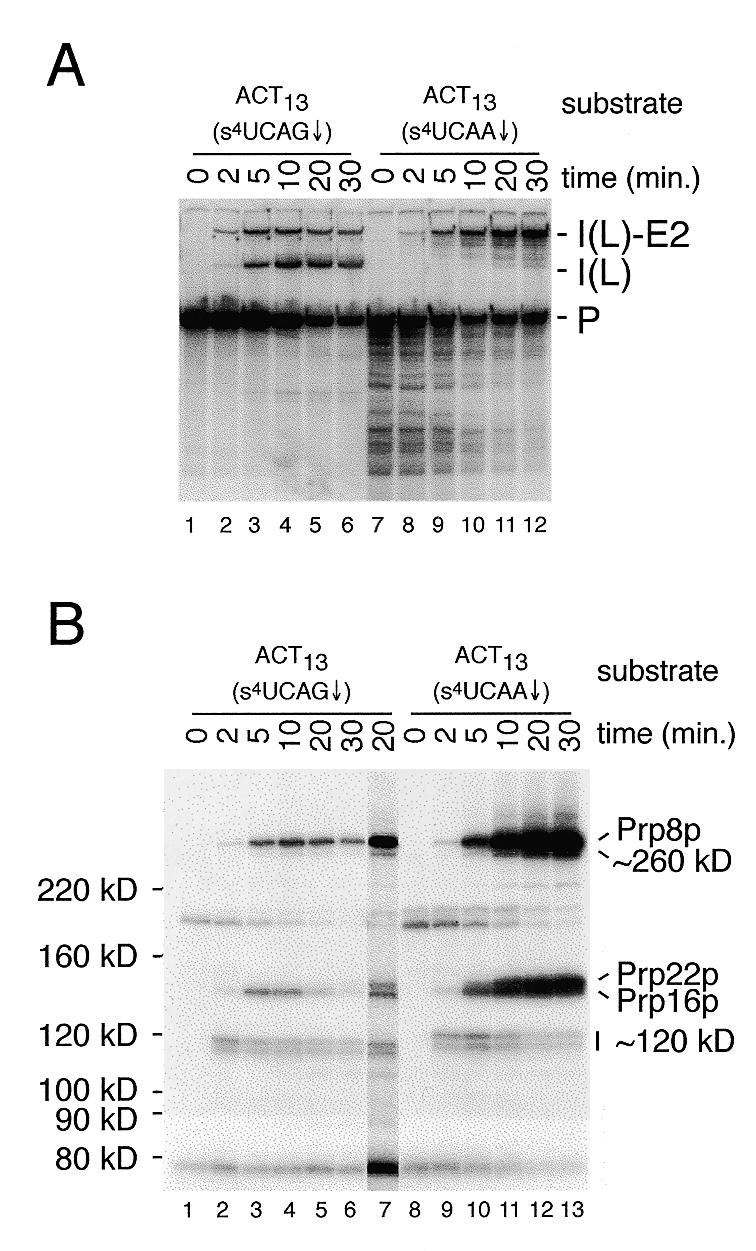
Splicing activity and analysis of proteins that cross-link to a wild-type and a 3′ splice site mutant pre-mRNA substrate. (A) Splicing assays were performed for the times indicated with either the wild-type ACT13(s4UCAG↓) substrate (lanes 1–6) or the 3′ splice site mutant ACT13(s4UCAA↓) substrate (lanes 7–12) in the presence of 2 mM ATP. RNA species are labeled as in Figure 2. (B) Aliquots of each of the splicing reactions shown in (A) were cross-linked and the resulting 32P-labeled proteins analyzed on a 6% SDS–polyacrylamide gel. Lane 7 is from a darker exposure of the 20 min time point with the ACT13(s4UCAG↓) substrate. On the original autoradiograph, clear separation of Prp16p and Prp22p is visible. In the comparison of proteins cross-linked to the ACT13(s4UCAG↓) and ACT13(s4UCAA↓) substrates, note that the relative migration of some of the cross-linked species seen with the 3′ splice site mutant is decreased due to the increased size (18 versus 24 nt) of the 32P-labeled RNase T1 fragment.
Figure 5.
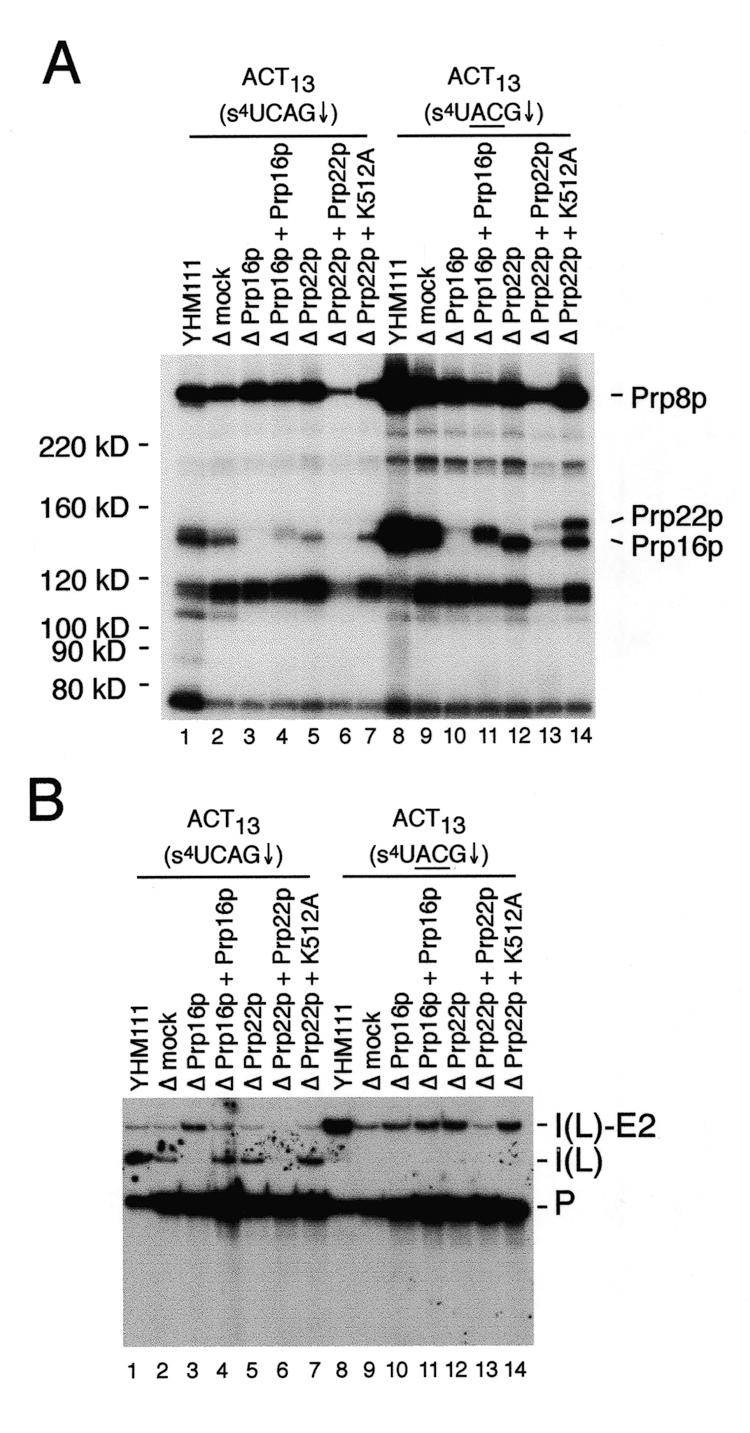
Cross-linking analysis in depleted extracts. (A) Cross-linking assays were performed in the indicated control (YHM111), mock depleted (Δmock), Prp16p (ΔPrp16p) or Prp22p depleted (ΔPrp22p), or reconstituted extracts using the indicated substrates. All samples were analyzed following a 10 min incubation at 23°C. Cross-linked 32P-labeled proteins in control and reconstituted extracts were analyzed on a 6% SDS–polyacrylamide gel. In this experiment, an undiluted, undialyzed sample of recombinant His-tagged Prp22p protein was used that caused an overall inhibition of splicing activity as well as cross-linking. In other experiments (data not shown), normal splicing activity and cross-linking to the HIS-tagged Prp22p at levels comparable to the K512A mutant protein are observed using 5- or 10-fold dilutions of the recombinant protein. On the original autoradiograph, clear separation of the His-tagged Prp16p and Prp22p in the tightly spaced doublet in lane 11 is visible. (B) Splicing assays of the samples used for cross-link analysis. RNA species are labeled as in Figure 2.
Detailed time course analysis revealed that cross-links to the ~122 kDa species appears within the first 30 s, followed by cross-links to the ~132 kDa species at 1 min, and the ~260 and ~300 kDa species at 1.5–2 min with the ACT13(s4UAG↓) substrate, and that cross-links to the ~132 kDa species precede those to the ~140 kDa species with ACT47(s4UAG↓) substrate (data not shown). The very early appearance of cross-links to the ~122 kDa species was not anticipated, since in yeast, the 3′ splice site is not required for the first step of splicing in vitro (27). To further characterize the association of the ~122 kDa species with early splicing complexes, cross-linking in extracts depleted of specific snRNAs by oligonucleotide directed RNase H cleavage was examined (Fig. 3B). No cross-linking of the ~122 kDa species with either substrate is observed in extracts depleted of either the U1 or U2 snRNAs (lanes 2, 3, 6 and 7). In contrast, cross-linking of the ~122 kDa species to both substrates is observed in extracts depleted of the U6 snRNAs (lanes 4 and 8), and demonstrates the ~122 kDa species associates with the 3′ splice site in pre-spliceosomes. The stronger cross-linking of the ~122 kDa species in extracts depleted of the U6 snRNA (lanes 4 and 8), compared to that observed in untreated extracts (lanes 1 and 4), suggests that the association of this factor with the 3′ splice site is transitory, and does not persist in later splicing complexes.
Previous studies have demonstrated cross-linking of Prp8p (279 kDa), Prp16p (121 kDa) and Slu7p (44 kDa) to the region surrounding the 3′ splice site of yeast introns following the first catalytic step (7,16). The apparent molecular weights and timing of the cross-links observed in Figure 3A were consistent with the ~132 and ~300 kDa species representing Prp16p and Prp8p cross-linked to a 19–21 nt. RNase T1 fragment, respectively. As shown in Figure 3C (lane 3), antibodies directed against Prp16p immunoprecipitate the ~132 kDa species cross-linked to the ACT13(s4UAG↓) substrate. Under the conditions used in Figure 3C, the ~300 kDa protein was retained in the absence of added antibody (lanes 2 and 6). Using more stringent conditions (see Materials and Methods), we have found that antibodies directed against Prp8p immunoprecipitate both the ~260 and ~300 kDa species cross-linked to the ACT13(s4UAG↓) substrate (data not shown).
The strong cross-linking of the ~140 kDa species to the ACT47(s4UAG↓) substrate (Fig. 3A, lanes 10–13) is correlated with the strong second step block of this substrate (Fig. 2). Recently, a requirement for the DExH-box helicase Prp22p in the second step of pre-mRNA splicing has been described (10). The apparent molecular weight of the ~140 kDa species is close to that predicted for Prp22p (130 kDa) cross-linked to the labeled 21 nt. RNase T1 fragment (~6 kDa) of the ACT47(s4UAG↓) substrate. Immunoprecipitation of the cross-linked ~140 kDa species from reactions containing the ACT47(s4UAG↓) substrate with anti-Prp22p sera demonstrate that this band is Prp22p (Fig. 3C, lane 8; see Discussion).
The profile of proteins that cross-link to a 3′ splice site mutant changes
The very weak cross-linking of Prp22p to the ACT13(s4UAG↓) substrate suggests that the association of Prp22p with the 3′ splice site may occur transiently prior to the second catalytic step, and not persist following its completion. To examine this possibility, we constructed several 3′ splice site mutant derivatives of the ACT13 substrate to determine if the cross-linking of Prp22p could be enhanced by blocking the second step. To facilitate the construction of mutant substrates, the position of the s4U was shifted upstream by one base to position –4, and the wild-type actin 3′ splice site consensus sequence changed from UAG↓ to CAG↓. Four-way in vitro RNA ligations using a second short downstream RNA were used to create a 3′ splice site mutant (s4UCAA↓) and the corresponding wild-type (s4UCAG↓) versions of the ACT13 substrate (Fig. 1). Comparison of the splicing activity of these substrates (Fig. 4A) demonstrates that while the ACT13(s4UCAG↓) substrate was active in both steps of splicing, the ACT13(s4UCAA↓) substrate was completely blocked for the second step.
Comparison of the pattern of proteins that cross-link to the ACT13(s4UCAG↓) and ACT13(s4UCAA↓) substrates (Fig. 4B), to those with the ACT13(s4UAG↓) substrate (Fig. 3A, lanes 1–6), shows that the shift of the s4U from position –3 to –4 results in an almost identical pattern of cross-linked proteins. The most notable difference is the replacement of the unidentified ~122 kDa species by a doublet with slightly increased mobility. In other experiments, we have determined both bands in this doublet to be identical to the ~122 kDa species (D.S.McPheeters, unpublished data). Mutation of the 3′ splice site in this substrate [ACT13(s4UCAA↓), lanes 9–12], results in the accumulation of cross-links to Prp16p and Prp8p, and a dramatic accumulation of cross-links to the ~140 kDa species (lanes 8–13). Antibodies directed against Prp22p were found to efficiently immunoprecipitate this ~140 kDa cross-linked species (data not shown, see Discussion). Weak cross-linking of this ~140 kDa species to the wild-type ACT13(s4UCAG↓) substrate is also observed (Fig. 4B, lane 7). The large increase in cross-linking of Prp22p is consistent with the weak cross-linking of Prp22p to functional 3′ splice sites (Figs 3A and 4B) resulting from a transient interaction prior to the second catalytic step. With the ACT13(s4UCAA↓) substrate, the cross-linking of the ~122 kDa doublet decreases at later time points in concert with the appearance of cross-links to Prp16p, Prp8p and Prp22p (Fig. 4B, lanes 8–13). A similar trend in cross-linking of Prp16p, Prp8p, Prp22p and the ~122 kDa species is observed with the ACT47(s4UAG↓) substrate (Fig. 3A, lanes 8–13). The reciprocal nature of these cross-links suggests a possible precursor–product relationship, i.e. that the association of Prp16p, Prp8p or Prp22p with the 3′ splice site may normally displace the ~122 kDa species.
The interaction of Prp22p with the 3′ splice site is dependent on Prp16p, but not on ATP-hydrolysis by Prp22p
As noted in the introduction, genetic and biochemical studies have demonstrated that Prp16p, Prp17p, Slu7p, Prp18p and Prp22p function prior to the second catalytic step of splicing. On the basis of differential ATP requirements, Prp16p and Prp17p appear to function in an ATP-dependent step that precedes an ATP-independent step(s) involving Slu7p, Prp18p and Prp22p (10,13,16). To determine if the cross-linking of Prp22p to the 3′ splice site coincides with the biochemical requirement for Prp22p in the ATP-independent step that occurs following ATP hydrolysis by Prp16p, cross-linking was performed in splicing extracts immunodepleted of either Prp16p (Prp16Δ) or Prp22p (Prp22Δ). Using the ACT13(s4UCAG↓) substrate, strong cross-links to the ~122 kDa species, Prp16p and Prp8p are observed in control extracts (Fig. 5A, lanes 1 and 2). Depletion of Prp16p blocks the second step with the wild-type ACT13(s4UCAG↓) substrate (Fig. 5B, lane 3) and abolishes cross-linking to Prp16p (Fig. 5A, lane 3), but does not noticeably affect the cross-linking of other species. Despite the second step block with this substrate in the Prp16Δ extract, no strong cross-linking of Prp22p is observed (Fig. 5A, lane 3). As a control, reconstitution of splicing activity in the Prp16Δ extract by the addition of purified Prp16p results in cross-links to a species with slightly increased molecular weight due to the presence of a His-tag on the purified protein, and allows completion of the second step (Fig. 5A and B, lane 4). This result shows that interaction of Prp22p with the 3′ splice site is not simply a consequence of the second step block, but is dependent on Prp16p. Consistent with this, the cross-linking of Prp16p was not affected in Prp22Δ extracts (Fig. 5A, lane 5; data not shown). In agreement with previous experiments showing that Prp22p is not required for the second step of splicing with substrates having branch site-3′ splice site intervals of <21 nt. (10), depletion of Prp22p does not block the second step of splicing with the wild-type ACT13(s4UCAG↓) substrate (Fig. 5B, lane 5).
Because only weak cross-linking of Prp22p to the 3′ splice sites of pre-mRNAs functional in the second step is observed, the effects of Prp16p and Prp22p depletion were also examined using an ACT13(s4UACG↓) mutant substrate. As expected from the second step block with this substrate (Fig. 5B, lanes 8–14), strong cross-linking of Prp8p, Prp16p and Prp22p is observed in control extracts (Fig. 5A, lanes 8 and 9). Depletion of Prp16p abolishes cross-linking to both Prp16p and Prp22p, but does not appreciably affect the cross-linking to Prp8p (lane 10). This confirms that the association of Prp22p, but not Prp8p, with the 3′ splice site is absolutely dependent on Prp16p. Depletion of Prp22p does not abolish cross-linking of either Prp16p or Prp8p (lane 12), demonstrating the association of Prp16p and Prp8p with the 3′ splice site is independent of Prp22p. Addition of His-tagged Prp16p to the Prp16Δ extract results in the appearance of a tightly spaced doublet in which the upper band corresponds to Prp22p and the lower band corresponds to the His-tagged Prp16p (Fig. 5A, lane 11). In contrast to previous studies (16), no significant increase in cross-linking of Prp8p to the 3′ splice site was observed following addition of purified Prp16p. Addition of purified Prp22p to the Prp22-depleted extract results in cross-links to a species with slightly increased molecular weight due to the presence of a His-tag on the purified protein (Fig. 5A, lane 13). This result confirms the identity of the ~140 kDa species as Prp22p and agrees with previous biochemical studies demonstrating Prp22p functions after Prp16p in the second step (10).
Previous biochemical studies have established that ATP-hydrolysis by Prp22p is not required prior to the second step of splicing (10). To verify that ATP-hydrolysis by Prp22p is not required for the observed cross-linking of Prp22p to the 3′ splice site, we tested the effects of an ATPase defective version of Prp22p (K512A). Addition of purified His-tagged Prp22p-K512A to a Prp22pΔ extract resulted in strong cross-linking of the protein to the ACT13(s4UACG) substrate (Fig. 5A, lane 14), confirming that ATP-hydrolysis by Prp22p is not required for cross-linking.
Following the second catalytic step, ATP-hydrolysis by Prp22p is required for release of mRNA from the spliceosome (10). To determine if the observed interaction of Prp22p with intron sequences at the 3′ splice site may be involved in mRNA release from the spliceosome, we also examined the effects of the K512A mutation on cross-linking to the wild-type ACT13(s4UCAG↓) substrate. Consistent with the proposed transient association of Prp22p with 3′ splice sites during normal splicing, only very weak cross-linking of wild-type Prp22p is observed in control extracts (Fig. 5A, lanes 1 and 2; data not shown). If this interaction between Prp22p and intron sequences at the 3′ splice site is involved in mRNA release following completion of the second catalytic step, addition of the mutant Prp22p-K512A protein to a Prp22Δ extract should result in a dramatic accumulation of cross-links in reactions with the wild-type ACT13(s4UCAG↓) substrate because mRNA release is blocked. However, no accumulation of cross-links to the mutant protein are observed (lane 7), suggesting the association of Prp22p with intron sequences at the 3′ splice does not persist following completion of the second catalytic step.
To partially define the extent of Prp22p’s association with the 3′ splice site prior to the second catalytic step, we examined the cross-linking of Prp22p to the exon sequences adjacent to the 3′ splice site. A derivative of the ACT13 substrate was constructed in which the 3′ exon sequences adjacent to the 3′ splice site were altered to allow placement of a single s4U residue at either position –4 in the intron, or at position +2 in the 3′ exon. The wild-type and 3′ splice site mutant derivatives of these substrates are designated ACM13(CAG↓) and ACM13(ACG↓), respectively (Fig. 1). Cross-linking assays with these substrates containing s4U at either position –4 or +2 at the 3′ splice site are shown in Figure 6. Consistent with our previous results (Figs 3–5), cross-linking of the ~122 kDa species, Prp16p, and Prp8p to the wild-type ACM13(CAG↓) substrate is observed with s4U at position –4 (Fig. 6, lane 2). Mutation of the 3′ splice site in the ACM13 (ACG↓) substrate with s4U at position –4 results in a strong block to the second step of splicing (data not shown) and the appearance of strong cross-links to Prp22p (lane 1). When the position of the s4U in these substrates is shifted to position +2 within the 3′ exon, cross-linking to the ~122 kDa species, Prp16p and Prp8p is observed with the both the wild-type and mutant ACM13 substrates (lanes 3 and 4). In the 3′ splice site mutant substrate with the s4U at position +2 however, no accumulation of cross-links to Prp22p is observed (lane 3). With the wild-type ACM13 substrate, very weak cross-linking to position +2 within the 3′ exon by a species with mobility similar to Prp22p is observed (lane 4), but the identity of this species has not been established. Overall, these results suggest that at prior to the second catalytic step, the interaction of Prp22p with intron sequences at the 3′ splice site does not extend into the adjacent 3′ exon sequences.
Figure 6.

Cross-linking of proteins to the wild-type and mutant ACM13 substrates containing s4U at position –4 within the intron (lanes 1 and 2) or position +2 within the 3′ exon (lanes 3 and 4). Cross-linking assays were performed after incubation of reaction in the presence of 2 mM ATP for 12 min and samples were analyzed on a 7% SDS–polyacrylamide gel.
DISCUSSION
Using site-specific incorporation of a single 32P-label and the photoactivatable cross-linking reagent s4U, we have explored the association of factors with the 3′ splice site region of the yeast actin pre-mRNA during nuclear pre-mRNA splicing. It is unknown at precisely which stage of spliceosome assembly the 3′ splice site of introns is first recognized in yeast, although it is clear that cleavage of the 5′ splice site and formation of the lariat intermediate can occur in the absence of a 3′ splice site (27). In mammalian splicing extracts, a 3′ splice site AG is required prior to the first catalytic step if no long polypyrimidine tract is present (36) and the 3′ splice site AG can be cross-linked to an unidentified 100 kDa protein in pre-spliceosomes (21). Recent experiments have established that in metazoan introns, the 3′ splice site AG is recognized in a sequence-specific manner during pre-spliceosome formation by U2AF35 (23–25). Although no homolog of U2AF35 exists in Saccharomyces cerevisiae (26), the experiments presented here demonstrate that in the yeast pre-spliceosome, the 3′ splice site is contacted in an ATP-dependent manner by an unidentified ~122 kDa protein. Notably, this interaction is independent of the distance between the 3′ splice site and branch site, as it occurs with both the ACT47(s4UAG↓) as well as ACT13(s4UAG↓) substrates (Fig. 3A). This independence suggests the 3′ splice site of yeast introns may be specifically contacted much earlier than previously envisaged (27). Whereas cross-linking of 100 kDa mammalian protein to the 3′ splice site AG in pre-spliceosomes is sensitive 3′ splice site mutations (21), cross-links to the ~122 kDa yeast pre-spliceosome protein are not (Figs 4 and 5), and suggest these factors may be unrelated. Experiments to identify and further characterize this ~122 kDa component of the yeast pre-spliceosome are currently in progress.
Previous cross-linking studies have established that both Prp16p and Prp8p are closely associated with the 3′ splice site following the first catalytic step (7,16). Our experiments have shown that an additional ~140 kDa species is associated with intron sequences at the 3′ splice site following the first catalytic step (Figs 3–6). Our identification of this ~140 kDa band as Prp22p is based upon several lines of evidence. First, the observed size (~140 kDa) is consistent with that predicted for Prp22p (130 kDa) cross-linked to an 18–24 nt RNase T1 fragment. Second, the ~140 kDa species is efficiently immunoprecipitated by antibodies directed against Prp22p. Finally, while depletion of Prp22p from splicing extracts results in loss of cross-linking to the ~140 kDa species, addition of purified Prp22p results in the restoration of cross-linking to a species with slightly decreased mobility consistent with the presence of a His-tag on the recombinant protein.
In vitro studies have defined at least two distinct stages that occur prior to the second catalytic step. The first stage involves Prp17 and Prp16p and is ATP-dependent (13,37), while the second stage is ATP-independent and involves Slu7p, Prp18p and Prp22p (10,11,15). ATP-hydrolysis by Prp16p in the presence of Slu7, Prp18p and Prp22p leads to a conformational change in the spliceosome that results in the protection of the 3′ splice site region from oligonucleotide-mediated RNase H cleavage (10,11,15,37). These protection experiments have utilized an actin intron with a 3′ splice site mutation in order to circumvent the normally rapid conversion of lariat intermediates to mature mRNA (37). Our experiments have shown that Prp22p cross-links to the 3′ splice site (Figs 3–5) in a manner dependent on Prp16p (Fig. 5). The strong cross-linking of Prp22p to non-functional 3′ splice sites suggests that the protection of the 3′ splice site region from oligonucleotide-mediated RNase H cleavage previously observed may be a direct consequence of the interaction of Prp22p with the 3′ splice site region. With our cross-linking substrates (Fig. 1), we have not detected cross-linking of Slu7p to the 3′ splice site region as has been previously reported (D.S.McPheeters, unpublished data; 16). This apparent discrepancy, as well as differences in cross-links to the 3′ splice site between yeast and mammalian systems (7,16,17,21,22), is most likely due to differences in the photochemistry of the cross-linking reagents used. This, and differences in splicing substrates, may also explain our failure to detect the previously observed increase in Prp8 cross-linking following addition of Prp16p to a Prp16p-depleted extract (16). Whatever the reason for these differences, it is clear that use of a variety of cross-linking reagents, substrates, and other approaches will be necessary to fully uncover the many interactions involved in 3′ splice site selection.
It has been proposed that prior to the second catalytic step, Slu7p, Prp18p and Prp22p may act to either form a bridge, or stabilize interactions of factors bound to the branch site and distal 3′ splice site sequences (10,11,15). The dependence of the biochemical requirement in the second step for these factors on the distance between the branch site and 3′ splice site suggests they are involved in an early, as opposed to late, step in 3′ splice site selection. The recent finding that metazoan extracts depleted of the human homolog of Slu7p (hSlu7) utilize incorrect 3′ splice sites is consistent with such multiple steps being involved in 3′ splice site recognition (38). It is unknown if or how any of these factors contribute directly to sequence specific recognition of the consensus YAG↓ sequence at the 3′ splice site. Our data show that Prp22p can be cross-linked to varying extents to a variety of different 3′ splice site sequences, including UAG↓, CAG↓, CAA↓ and ACG↓. The ability of Prp22p to cross-link to several different 3′ splice site sequences suggests that this association itself is largely non-sequence specific and likely occurs in part of a larger complex (see below). Although Prp22p is not required for the second step of splicing with our ACT13(s4UCAG↓) substrate, we have repeatedly observed weak cross-linking of Prp22p to this, and related substrates (Fig. 5). Inhibition of the second step by 3′ splice site mutations in the ACT13 substrates, or alteration of the branch site-3′ splice site spacing and intervening sequence in the ACT47(s4UAG↓) substrate, results in greatly enhanced cross-linking of Prp22p (Figs 3–5). This increase in cross-linking of Prp22p upon inhibition of the second step suggests that the weak cross-linking of Prp22p reflects a normal, but transient association of Prp22p with functional 3′ splice sites prior to the second step. It is also possible that the increased cross-linking of Prp22p to substrates unable to undergo the second step represents a non-specific interaction of Prp22p with sequences downstream of the branch site in substrates lacking functional 3′ splice sites. However, such non-specific interaction of Prp22p seems unlikely given the absence of Prp22p cross-linking to exon sequences in a substrate with a mutant 3′ splice site (Fig. 6).
Current evidence suggests the intron and/or exon sequences flanking the 3′ splice site may be recognized prior to the second step by a complex consisting of the U5 snRNA, Prp8p, Prp16p, Slu7p, Prp18p and Prp22p. With functional 3′ splice sites, cross-linking of Prp22p is weak because this 3′ splice site recognition complex assembles only transiently before the 3′ splice site is bound by the active site of the spliceosome. We suggest the strong cross-linking of Prp22p to the mutant 3′ splice sites of our ACT13 and ACM13 substrates, or the non-mutant 3′ splice site of the ACT47 substrate, represents the ‘dead-end’ assembly of this intermediate complex involved in 3′ splice site recognition. In the ACT13 and ACT47 substrates, the next AG dinucleotides downstream from the normal 3′ splice site are 58 and 92 nt, respectively, from the branch site adenosine. Such spacing is near the upper bound of that normally observed in yeast introns (4), and is much greater than the optimal in vivo spacing of 18–22 nt determined by Luukkonen and Seraphin (5). In the absence of an appropriately spaced YAG, a closely related sequence located within a reasonable distance from the branch site may direct assembly of 3′ splice site recognition complexes. In agreement with this interpretation, correct 3′ splice site choice in yeast is frequently maintained even when mutant 3′ splice site sequences are presented (5,39). In a separate study, we have found very efficient and correct in vivo use of mutant 3′ splice sites that are spaced 12 nt from the branch site adenosine (J.S.Chang and D.S.McPheeters, manuscript in preparation). Mutations in Prp8p (18–20), as well as the U5, U2 and U6 snRNAs (40–42; J.S.Chang and D.S.McPheeters, manuscript in preparation) can activate the use of mutant 3′ splice sites. In total, these data are consistent with a hierarchy of multiple, redundant interactions involved in 3′ splice site selection.
Our experiments have shown that the association of Prp22p with the mutant 3′ splice site of the ACM13(ACG↓) substrate is not likely to extend into the 3′ exon prior to the second catalytic step (Fig. 6). Following the second catalytic step, ATP-hydrolysis by Prp22p is required for release of mature mRNA from the spliceosome even when the branch site to 3′ splice site spacing is short (10). We have found that an ATPase deficient version of Prp22p (K512A) fails to accumulate cross-links to a substrate proficient in the second step (Fig. 5A), suggesting that the association of Prp22p with intron sequences at the 3′ splice site changes following completion of the second step. Following the second step, the RNA helicase activity of Prp22p has been proposed to break contacts made by the U5 snRNA and exon sequences adjacent to the 3′ splice site (10). In light of this proposal, one possible interpretation of our data is that the interaction of Prp22p with the 3′ splice site may shift from intron to exon sequences following the second catalytic step. Further experiments will be necessary in order to test this hypothesis.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Jonatha Gott, Wesley Kroeze, Pat Maroney, Tim Nilsen and Jo Ann Wise for their critical reading of this manuscript and to Jean Beggs and Caroline Russell for their generous gift of Prp8p anti-serum. National Institutes of Health grant GM52310 to D.S.M. supported this work.
REFERENCES
- 1.Staley J.P. and Guthrie,C. (1998) Cell, 92, 315–326. [DOI] [PubMed] [Google Scholar]
- 2.Moore M.J., Query,C.C. and Sharp,P.A. (1993) In Gesteland,R. and Atkins,J. (eds), The RNA World. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 303–358.
- 3.Umen J.G. and Guthrie,C. (1995) RNA, 1, 869–885. [PMC free article] [PubMed] [Google Scholar]
- 4.Spingola M., Grate,L., Haussler,D. and Ares,M.,Jr (1999) RNA, 5, 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luukkonen B.G. and Seraphin,B. (1997) EMBO J., 16, 779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patterson B. and Guthrie,C. (1991) Cell, 64, 181–187. [DOI] [PubMed] [Google Scholar]
- 7.Teigelkamp S., Newman,A.J. and Beggs,J.D. (1995) EMBO J., 14, 2602–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nilsen T.W. (1998) In Simons,R.W. and Grunberg-Manago,M. (eds), RNA Structure and Function. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 279–307.
- 9.Frank D. and Guthrie,C. (1992) Genes Dev., 6, 2112–2124. [DOI] [PubMed] [Google Scholar]
- 10.Schwer B. and Gross,C.H. (1998) EMBO J., 17, 2086–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ansari A. and Schwer,B. (1995) EMBO J., 14, 4001–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Umen J.G. and Guthrie,C. (1995) Genes Dev., 9, 855–868. [DOI] [PubMed] [Google Scholar]
- 13.Jones M.H., Frank,D.N. and Guthrie,C. (1995) Proc. Natl Acad. Sci. USA, 92, 9687–9691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brys A. and Schwer,B. (1996) RNA, 2, 707–717. [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X. and Schwer,B. (1997) Nucleic Acids Res., 25, 2146–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Umen J.G. and Guthrie,C. (1995) RNA, 1, 584–597. [PMC free article] [PubMed] [Google Scholar]
- 17.Reyes J.L., Kois,P., Konforti,B.B. and Konarska,M.M. (1996) RNA, 2, 213–225. [PMC free article] [PubMed] [Google Scholar]
- 18.Umen J.G. and Guthrie,C. (1996) Genetics, 143, 723–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collins C.A. and Guthrie,C. (1999) Genes Dev., 13, 1970–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siatecka M., Reyes,J.L. and Konarska,M.M. (1999) Genes Dev., 13, 1983–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiara M.D., Gozani,O., Bennett,M., Champion-Arnaud,P., Palandjian,L. and Reed,R. (1996) Mol. Cell. Biol., 16, 3317–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu S. and Green,M.R. (1997) EMBO J., 16, 4421–4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu S., Romfo,C.M., Nilsen,T.W. and Green,M.R. (1999) Nature, 402, 832–835. [DOI] [PubMed] [Google Scholar]
- 24.Zorio D.A. and Blumenthal,T. (1999) Nature, 402, 835–838. [DOI] [PubMed] [Google Scholar]
- 25.Merendino L., Guth,S., Bilbao,D., Martinez,C. and Valcarcel,J. (1999) Nature, 402, 838–841. [DOI] [PubMed] [Google Scholar]
- 26.Abovich N. and Rosbash,M. (1997) Cell, 89, 403–412. [DOI] [PubMed] [Google Scholar]
- 27.Rymond B.C. and Rosbash,M. (1985) Nature, 317, 735–737. [DOI] [PubMed] [Google Scholar]
- 28.Lin R.J., Newman,A.J., Cheng,S.C. and Abelson,J. (1985) J. Biol. Chem., 260, 14780–14792. [PubMed] [Google Scholar]
- 29.Lowary P., Sampson,J., Milligan,J., Groebe,D. and Uhlenbeck,O.C. (1986) In Knippenberg,P.H. and Hilbers,C.W. (eds), Structure and Dynamics of RNA. Plenum Press, New York, NY, pp. 69–76.
- 30.Moore M.J. and Sharp,P.A. (1992) Science, 256, 992–997. [DOI] [PubMed] [Google Scholar]
- 31.McPheeters D.S., Fabrizio,P. and Abelson,J. (1989) Genes Dev., 3, 2124–2136. [DOI] [PubMed] [Google Scholar]
- 32.Madhani H.D. and Guthrie,C. (1992) Cell, 71, 803–817. [DOI] [PubMed] [Google Scholar]
- 33.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 34.Favre A., Saintome,C., Fourrey,J.L., Clivio,P. and Laugaa,P. (1998) J. Photochem. Photobiol., 42, 109–124. [DOI] [PubMed] [Google Scholar]
- 35.Parker R. and Patterson,B. (1987) The Molecular Biology of RNA: New Perspectives. Academic Press, pp. 133–149.
- 36.Reed R. (1989) Genes Dev., 3, 2113–2123. [DOI] [PubMed] [Google Scholar]
- 37.Schwer B. and Guthrie,C. (1992) EMBO J., 11, 5033–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chua K. and Reed,R. (1999) Nature, 402, 207–210. [DOI] [PubMed] [Google Scholar]
- 39.Ruis B.L., Kivens,W.J. and Siliciano,P.G. (1994) Nucleic Acids Res., 22, 5190–5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Newman A.J. and Norman,C. (1992) Cell, 68, 743–754. [DOI] [PubMed] [Google Scholar]
- 41.Madhani H. and Guthrie,C. (1994) Genes Dev., 8, 1071–1086. [DOI] [PubMed] [Google Scholar]
- 42.Lesser C.F. and Guthrie,C. (1993) Science, 262, 1982–1988. [DOI] [PubMed] [Google Scholar]