

Abstract
Snyder-Robinson syndrome (SRS) is a rare X-linked recessive disorder characterized by a collection of clinical features including mild to severe intellectual disability, hypertonia, marfanoid habitus, facial asymmetry, osteoporosis, developmental delay and seizures. Whole genome sequencing (WGS) identified a mutation in the spermine synthase (SMS) gene (c.746 A>G, p.Tyr249Cys) in a male with kyphosis, seizures, and osteoporosis. His phenotype is unique in that he does not have intellectual disability (ID) but does have a mild learning disability. This case demonstrates a milder presentation of SRS and expands the phenotype beyond the reported literature.
Keywords: Snyder-Robinson syndrome, spermine synthase, spermidine, spermine, osteoporosis, seizures
Introduction
Snyder-Robinson syndrome (SRS), is an X-linked recessive disorder characterized by developmental delay, mild to severe ID, seizures, facial dysmorphism (prominent lower lip, asymmetry), osteoporosis, kyphoscoliosis, hypotonia, speech abnormalities, and an asthenic build (long limbs and fingers) [1]. The syndrome is seen exclusively in males and considered rare, with 26 variants published in the literature or listed in ClinVar (Figure 1). Of significance, all of the published cases have mild to severe intellectual disability [1]. Females are typically unaffected carriers, with one report of mild kyphosis in a carrier mother [2]. Currently, there is no specific treatment for SRS, and management is focused on supportive care and prevention of complications. Speech, physical, and occupational therapy can help with development, while anti-epileptic drugs are used to manage seizures. Osteoporosis usually develops during the first decade, leading to non-traumatic fractures; calcium supplementation has been used in a few individuals to improve bone mineral density [1]. The use of bisphosphonates is controversial with mixed results [3].
Fig. 1.
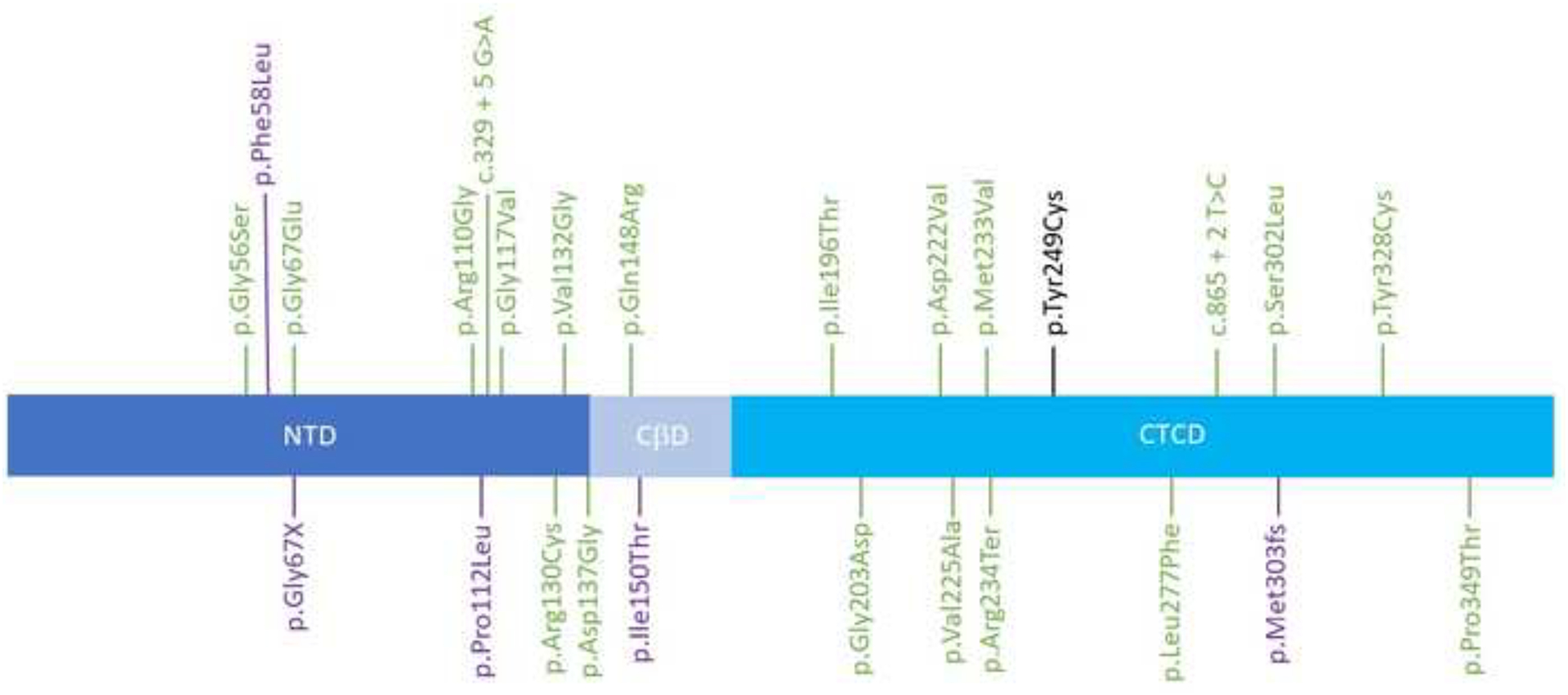
Schematic representation of the SMS gene (transcript NM_004595.5) and the pathogenic and likely pathogenic variants. Green: ClinVar variants. Purple: Variants reported in the literature and not ClinVar. Black: Proband’s variant. NTD: N-Terminal Domain, CßD: Central Beta-strand Domain, CTCD: C-Terminal Catalytic Domain
SRS is caused by hemizygous mutations in the spermine synthase (SMS) gene, resulting in decreased or absent SMS enzyme activity. SMS converts spermidine to spermine, a polyamine essential for normal cell growth and development [1, 4]. Spermine is essential for numerous cellular processes, including proliferation, protein and nucleic acid synthesis, cell adhesion, signaling, and ion channel regulation. Spermine plays a critical role in the central nervous system, including nerve growth and regeneration, response to neuronal injury and stress, regulation of ionic flux and neuronal ion channels, and modulation of the excitatory N-methyl-d-aspartate (NMDA) glutamate receptor [5].
This paper presents a case report describing a male with a novel missense mutation c.746 A>G, p.Tyr249Cys (NM_004595.5) in the SMS gene, causing a milder phenotype. While he presents with many of the symptoms of SRS, including osteopenia, learning disabilities, fractures, seizures, and hypotonia, he lacks ID, one of the most common feature of SRS.
Clinical Summary
The patient is a 19-year-old male who is a fraternal twin. The pedigree is shown in Figure 2. He was born vaginally at 34.5 weeks, with a birth weight of 2.5 kg and a height of 48.3 cm. He received care in the NICU for nine days due to problems regulating his temperature and feeding difficulties. He sat up at 8 months, started crawling at 12 months, and began walking at 20 months. An MRI of the brain and chromosomal microarray at 2 years was normal. His parents noticed that his development was delayed compared to his fraternal twin. He began receiving speech therapy (SLP) and occupational therapy (OT) at the age of 2. His evaluation at 2 years and 6 months showed moderate to severe expressive language delay, oral facial hypotonia, hypernasal speech, significant gross and fine motor delay, hypotonia, truncal weakness, and a wide-based gait. At 2 years and 9 months, he developed a mild strabismus. His weight was 12.8 kg (25th percentile), height was 93.98 cm (50th percentile), and occipital frontal circumference (OFC) was 51.5 cm (90th percentile). At 3 years and 5 months, neurodevelopmental testing was suggestive of a learning disability. An academic assessment at 5 years showed his skills in the average to above average range, yet concerns still existed for an ongoing learning disability. A neuropsychological evaluation at 9 years (3rd grade) assessed his cognitive ability using the Wechsler Intelligence Scale for Children, Fourth Edition (WISC-IV). His Full-Scale IQ was 91 (average is 90–109). His verbal comprehension skills (98), perceptual reasoning skills (94), and processing speed (97) all fell in the normal range. Working memory ability fell into the low average range (88). The Woodcock-Johnson III Tests of Achievement (WJ-III) indicated weaknesses in reading fluency, writing, and math calculation (performing at Grade Equivalents of 2.8, 2.4, and 2.9, respectively). He was diagnosed with anxiety, mild attention deficit disorder (inattentive type), and poor social skills.
Figure 2.
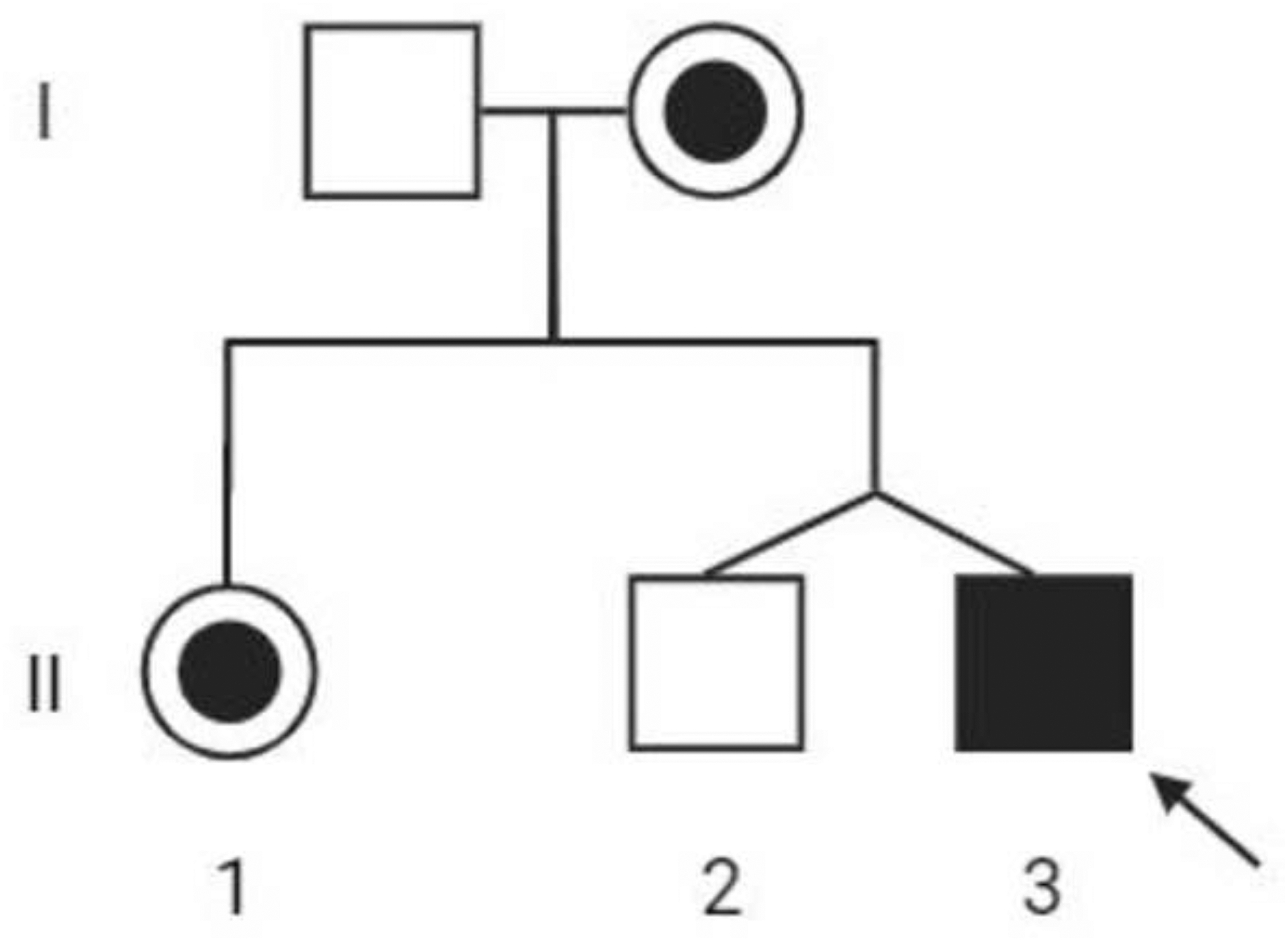
Pedigree of the family. The proband (II3) is indicated by a black square and arrow. The circle with a dot indicates a female carrier. Individuals I1 and II2 do not have the p.Y249C variant. Created with BioRender.com
At 4 years old, he had a bilateral inguinal hernia repair, and at age 13 he had bilateral orchiopexy for cryptorchidism. He suffered a hairline fracture of the right shoulder at age 4; the cause was never determined. His first seizure at age 13 was a partial seizure, evolving into a generalized convulsion. CT and MRI of the brain were normal. EEG did not capture a seizure but was consistent with an underlying encephalopathy. He began taking levetiracetam and remained seizure-free for a few years. Zonisamide was eventually added for breakthrough seizures.
When he was 16 years old, he had a seizure and a fall, fracturing his right proximal femur, which required open reduction and internal fixation. During the postoperative period, a nuclear medicine bone scan showed avascular necrosis of the femoral head, and vertebral compression fractures at T7 and T8. Due to the complications of his femoral fracture, he underwent a total hip arthroplasty.
At 18 years, a DEXA scan of vertebral levels L1–L4 showed a bone mineral density of 0.572 g/cm2 (Z-score −4.4). He was diagnosed with osteoporosis and placed on the bisphosphonate, zoledronic acid.
At 19 years old, his height was 185 cm (88th percentile), and his weight was 61.8 kg (20th percentile). He is tall and slender with mild scoliosis, hyperkyphosis, and mild pectus carinatum. His OFC was 58 cm (98th percentile), with a long face and a slightly prominent jaw. A psychological examination showed his full-scale IQ was 85, with the lowest score for processing speed of 80 and the highest score of 103 for comprehension. In addition, he was diagnosed with moderately impaired reading fluency and mild inattention. He graduated high school and is currently attending college. He is in an independent living program, where he is focusing on developing career and life skills.
His family history is negative for other individuals with a phenotype of SRS (Figure 2). His mother and sister are both heterozygous carriers of the mutation. Of interest, his mother has scoliosis and a history of absence seizures during her childhood, which resolved during puberty. The proband’s fraternal twin brother was negative for the mutation and has a history of learning disabilities.
Methods
Written informed consent for sequencing, data usage, skin biopsy, and publication of clinical details was obtained from participants, legally authorized representatives and/or their guardians if under the age of 18 for all of the family members. The study protocol and consent procedure were approved by the WGC Institutional Review Board (WGC IRB; study number 20120789).
WGS was performed from peripheral blood isolated genomic DNA. The SMS variant and a variant of uncertain significance (VUS) in SEM6B were confirmed by Sanger sequencing in a CLIA lab (GeneDx, Gaithersburg, MD).
Fibroblast cell lines from the proband were established after collecting a 3-mm skin punch biopsy and cultured for 2 weeks in primary fibroblast media (as previously described [6]). Polyamine concentrations in fibroblast lysates were determined in duplicate following derivatization with dansyl chloride and HPLC analyses based on the method of Kabra [7]. The spermidine/spermine ratio was compared to two sex-matched wildtype cell lines and two previously identified SRS lines established in Dr. Tracy Murray Stewart’s lab at Johns Hopkins University (Table 1). Polyamine levels were normalized according to protein concentration, with 1,7-diaminoheptane as the internal standard.
Table 1.
Polyamine concentrations in fibroblast lysates were determined in duplicate following derivatization with dansyl chloride and HPLC analyses.
| Sample ID | Genotype | PUT | SPD | SPM | SPD/SPM |
|---|---|---|---|---|---|
| (nmol/mg Protein) | ratio | ||||
| Control 1 | wt | 0.715 | |||
| 5.82 | 0.701 | ||||
| Control 2 | wt | 0.557 | |||
| 4.84 | 0.488 | ||||
| Proband Sample 1 | p.Y249C | 3.428 | |||
| 3.05 | 3.403 | ||||
| Proband Sample 2 | p.Y249C | 2.963 | |||
| 3.87 | 2.956 | ||||
| Individual 1 with SRS | p.M35R | 4.335 | |||
| 1.86 | 4.312 | ||||
| Individual 2 with SRS | P.L277F | 4.194 | |||
| 2.13 | 4.277 | ||||
The spermidine/spermine ratio was compared to two sex-matched wildtype (wt) cell lines and two previously identified SRS lines. Polyamine levels were normalized according to protein concentration, with 1,7-diaminoheptane as the internal standard. PUT = putrescine, SPD = spermidine, SPM = spermine.
Results
WGS identified the maternally inherited hemizygous variant c.746 A>G, p.Y249C (NM_004595.5) in the SMS gene. The p.Y249C variant was not seen in gnomAD [8], and in silico analysis (CADD of 29.8) supports that this variant has a deleterious effect on protein structure and function. The patient was also heterozygous for a de novo VUS in the SEMA6B gene: c.1367 A>G, p.K456R (NM_032108.3).
The patient’s average spermidine/spermine (SPD/SPM) ratio of 3.2 (3.0–3.4, SD=0.26) was higher than the average control ratio of 0.62 (0.49–0.72, SD=0.11) but was lower than the average ratio of 4.3 (4.2–4.3; SD=0.06) for the two individuals previously identified with SRS (Table 1). These results were consistent with low spermine synthase enzyme activity and consistent with an SRS diagnosis.
It is also worth noting that putrescine was readily detectable in this patient’s cells, averaging 1.08 nmol/mg protein (SD = 0.46). Putrescine is the diamine precursor for spermidine and spermine synthesis and was undetectable in the SRS reference cell lines, consistent with previous reports [9]. Consequently, the total amount of intracellular polyamines in this patient is elevated (15.4 ± 1.5 v. 10.6 ± .8 and 8.7 ± 1.5 in referenced SRS and WT cells, respectively).
Discussion
We present a unique case of SRS in a patient with many of the classic symptoms of the disorder, including osteoporosis, speech problems, kyphoscoliosis, slender habitus, recurrent fractures, and epileptic seizures, but without intellectual disability. His clinical presentation expands the phenotype to include individuals with average cognitive ability. Historically, 100% of SRS patients present with mild to severe intellectual disability, with an IQ that ranges from immeasurable to 77 (with performance and verbal IQs as high as the mid-80s) [1, 10]. Comparatively, our patient’s neurobehavioral and neuropsychological evaluations show a male with average cognitive ability with a learning disability and speech delay. His SMS gene mutation was initially interpreted as a variant of uncertain significance until polyamine analysis confirmed the diagnosis of SRS. His SPD/SPM ratio was higher than a control group but was lower than the two individuals with SRS, thus indicating decreased SMS activity.
Several potential explanations exist for his average cognitive ability and mild learning disability. His SPD/SPM ratio was not as elevated as the two other SRS individuals, implying that while he has reduced SMS enzyme activity, it may not be as severe as previously published patients. Genetic modifiers may have a protective effect. The early intervention of SLP and OT he received when he was 2 years old possibly impacted the severity of his cognitive ability. In addition, his seizures began at age 13 and were relatively well-controlled. Comparatively, the patient described in Peron et al. [2] began having focal motor seizures at age 1 and had an IQ of 41. His earlier onset and frequent seizures may have been a factor in his more severe presentation of intellectual disability [2]. Interestingly, our patient’s mother had absence seizures during childhood that resolved when she grew older. It is uncertain if her seizures are related to her SMS mutation. Similarly, in another case report, the patient’s mother and grandmother presented with kyphosis [2]. The authors noted that this finding could be incidental but could also be evidence that female carriers may present with milder symptoms of SRS.
SRS does not have a clear genotype-phenotype correlation. The first reported case of SRS was a family of 6 symptomatic males and 3 female carriers with the same gene variant (c.329 + 5 G>A), where there was variability in cognitive levels (mild-to-moderate ID) and neurological involvement. IQ levels ranged from 46–77, and only one of the individuals had seizures [10]. Zhang et al. [11] reported another case of borderline to mild ID in a patient with a mutation at p.Tyr328Cys with no seizures, mild kyphosis, and a Full-Scale IQ of 74 (with a verbal IQ of 72 and a performance IQ of 86). He was not evaluated for osteoporosis.
Spermine synthase is a dimer, with each monomer containing a C-terminal domain (CTD) and an N-terminal domain (NTD)[12]. The NTD is critical for binding the two monomers together, and the CTD codes for the protein’s active site. The CTD contains critical residues at D201 and D276, which are important for catalytic activity [12]. The proband’s mutation, located in the CTD and in the N-terminal end of the beta-14 turn, is a replacement of a tyrosine with a cysteine, a less bulky amino acid. This substitution may alter the protein structure, displacing these critical residues. The patient’s mutation is not near the protein’s active site and may not impair the enzyme’s function as severely as other mutations. Four other mutations are located in the same exon 7, but phenotypic information about these individuals is absent or limited. Conducting an analysis of the protein structure would provide greater insight into the mechanisms of the mutation.
The patient also had a heterozygous, de novo mutation in the SEMA6B gene. Pathogenic variants in this gene cause early-onset progressive myoclonic epilepsy and ID [13]. The missense variant is located in exon 13 of the gene, while most pathogenic variants in this gene are nonsense and located in exon 17. It is uncertain if this variant contributes to any of his symptoms.
In conclusion, our proband’s case helps expand the phenotypic spectrum of SRS to include patients without intellectual disability. This report may help diagnose patients with a similar phenotype to our proband and provides evidence that females with mutations in the SMS gene may have symptoms.
Acknowledgments
We would like to thank the family for their support and participation.
Disclosures
This study was supported by donations made to the TGen Foundation.
References
- [1].Schwartz CE, Peron A, & Kutler MJ (2013). Snyder-Robinson Syndrome. In Adam MP (Eds.) et al. , GeneReviews®. University of Washington, Seattle. [Google Scholar]
- [2].Peron A, Spaccini L, Norris J, Bova SM, Selicorni A, Weber G, Wood T, Schwartz CE, & Mastrangelo M (2013). Snyder-Robinson syndrome: a novel nonsense mutation in spermine synthase and expansion of the phenotype. American journal of medical genetics. Part A, 161A(9), 2316–2320. 10.1002/ajmg.a.36116 [DOI] [PubMed] [Google Scholar]
- [3].Manas FNU, and Mols-Kowalczewski B (2022). PMON20 Snyder Robinson Syndrome: A Rare Syndrome. Journal of the Endocrine Society, 6 (Supplement_1), A457–A458. 10.1210/jendso/bvac150.952 [DOI] [Google Scholar]
- [4].Valera Ribera C, Martinez-Ferrer À, Flores Fernández E, Vázquez Gómez I, Orenes Vera A, Valls Pascual E, Ybáñez García D, & Alegre Sancho JJ (2022). Snyder-Robinson syndrome: differential diagnosis of osteogenesis imperfecta. Osteoporosis international: a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA, 33(5), 1177–1180. 10.1007/s00198-021-06228-3 [DOI] [PubMed] [Google Scholar]
- [5].Larcher L, Norris JW, Lejeune E, Buratti J, Mignot C, Garel C, Keren B, Schwartz CE, & Whalen S (2020). The complete loss of function of the SMS gene results in a severe form of Snyder-Robinson syndrome. European journal of medical genetics, 63(4), 103777. 10.1016/j.ejmg.2019.103777 [DOI] [PubMed] [Google Scholar]
- [6].Llaci L, Ramsey K, Belnap N, Claasen AM, Balak CD, Szelinger S, Jepsen WM, Siniard AL, Richholt R, Izat T, Naymik M, De Both M, Piras IS, Craig DW, Huentelman MJ, Narayanan V, Schrauwen I, & Rangasamy S (2019). Compound heterozygous mutations in SNAP29 is associated with Pelizaeus-Merzbacher-like disorder (PMLD). Human genetics, 138(11–12), 1409–1417. 10.1007/s00439-019-02077-7 [DOI] [PubMed] [Google Scholar]
- [7].Kabra PM, Lee HK, Lubich WP, & Marton LJ (1986). Solid-phase extraction and determination of dansyl derivatives of unconjugated and acetylated polyamines by reversed-phase liquid chromatography: improved separation systems for polyamines in cerebrospinal fluid, urine and tissue. Journal of chromatography, 380(1), 19–32. 10.1016/s0378-4347(00)83621-x [DOI] [PubMed] [Google Scholar]
- [8].Chen S, Francioli LC, Goodrich JK, Collins RL, Wang Q, Alföldi J, Watts NA, Vittal C, Gauthier LD, Poterba T, Wilson MW, Tarasova Y, Phu W, Yohannes MT, Koenig Z, Farjoun Y, Banks E, Donnelly S, Gabriel S, Gupta N, Ferriera S, Tolonen C, Novod S, Bergelson L, Roazen D, Ruano-Rubio V, Covarrubias M, Llanwarne C, Petrillo N, Wade G, Jeandet T, Munshi R, Tibbetts K, gnomAD Project Consortium, O’Donnell-Luria A, Solomonson M, Seed C, Martin AR, Talkowski ME, Rehm HL, Daly MJ, Tiao G, Neale BM, MacArthur DG & Karczewski KJ A genome-wide mutational constraint map quantified from variation in 76,156 human genomes. bioRxiv 2022.03.20.485034 (2022). 10.1101/2022.03.20.485034 [DOI] [Google Scholar]
- [9].Murray Stewart T, Khomutov M, Foley JR, Guo X, Holbert CE, Dunston TT, Schwartz CE, Gabrielson K, Khomutov A, & Casero RA Jr (2020). (R,R)-1,12-Dimethylspermine can mitigate abnormal spermidine accumulation in Snyder-Robinson syndrome. The Journal of biological chemistry, 295(10), 3247–3256. 10.1074/jbc.RA119.011572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Arena JF, Schwartz C, Ouzts L, Stevenson R, Miller M, Garza J, Nance M, & Lubs H (1996). X-linked mental retardation with thin habitus, osteoporosis, and kyphoscoliosis: linkage to Xp21.3-p22.12. American journal of medical genetics, 64(1), 50–58. [DOI] [PubMed] [Google Scholar]
- [11].Zhang Z, Norris J, Kalscheuer V, Wood T, Wang L, Schwartz C, Alexov E, & Van Esch H (2013). A Y328C missense mutation in spermine synthase causes a mild form of Snyder-Robinson syndrome. Human molecular genetics, 22(18), 3789–3797. 10.1093/hmg/ddt229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wu H, Min J, Zeng H, McCloskey DE, Ikeguchi Y, Loppnau P, Michael AJ, Pegg AE, & Plotnikov AN (2008). Crystal structure of human spermine synthase: implications of substrate binding and catalytic mechanism. The Journal of biological chemistry, 283(23), 16135–16146. 10.1074/jbc.M710323200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Castellotti B, Canafoglia L, Freri E, Tappatà M, Messina G, Magri S, DiFrancesco JC, Fanella M, Di Bonaventura C, Morano A, Granata T, Gellera C, Franceschetti S, & Michelucci R (2023). Progressive myoclonus epilepsies due to SEMA6B mutations. New variants and appraisal of published phenotypes. Epilepsia open, 8(2), 645–650. 10.1002/epi4.12697 [DOI] [PMC free article] [PubMed] [Google Scholar]