

Abstract
Here, we review melanocyte development and how the embryonic melanoblast, although specified to become a melanocyte, is prone to cellular plasticity and is not fully committed to the melanocyte lineage. Even fully differentiated and pigment-producing melanocytes do not always have a stable phenotype. The gradual lineage restriction of neural crest cells toward the melanocyte lineage is determined by both cell-intrinsic and extracellular signals in which differentiation and pathfinding ability reciprocally influence each other. These signals are leveraged by subtle differences in timing and axial positioning. The most extensively studied migration route is the dorsolateral path between the dermomyotome and the prospective epidermis, restricted to melanoblasts. In addition, the embryonic origin of the skin dermis through which neural crest derivatives migrate may also affect the segregation between melanogenic and neurogenic cells in embryos. It is widely accepted that, irrespective of the model organism studied, the immediate precursor of both melanoblast and neurogenic populations is a glial-melanogenic bipotent progenitor. Upon exposure to different conditions, melanoblasts may differentiate into other neural crest-derived lineages such as neuronal cells and vice versa. Key factors that regulate melanoblast migration and patterning will regulate melanocyte homeostasis during different stages of hair cycling in postnatal hair follicles.
Keywords: Neural crest, Cellular plasticity, Migration, Melanocytes, EMT, ZEB proteins
Introduction
Discovered in the nineteenth century by Wilhelm His, the neural crest was described as a “Zwischenstrang”, a band of migratory cells arising between the ectoderm and the neural tube [1, 2]. Its motility is required to complete the characteristic patterning of the peripheral nervous system and many craniofacial structures and is considered as an important innovation during evolution, unique to vertebrates. Neural crest cells migrate throughout the embryo along defined pathways to specific destinations in different tissues and organs where they complete their differentiation. The multipotent neural crest gives rise to numerous cell types including melanocytes, most of the peripheral neurons and glial cells, craniofacial bone and cartilage, adipose tissue, cardiac smooth muscle cells and secretory adrenal cells [2–4]. Neural crest development comprises several key steps: induction, delamination, specification and migration. Melanoblast cells subsequently colonize the skin epidermis and hair follicles where they give rise to distinct and spatially distributed populations including melanocyte stem cells, differentiated hair follicle melanocytes and epidermal melanocytes in close contact with keratinocytes. Throughout the entire spectrum from neural crest to melanoma cells, several molecular players and signaling pathways are involved in the (dys)regulation of the melanocyte lineage.
Induction and delamination of the neural crest
The formation of the neural crest starts upon closure of the neural tube during neurulation. After the neuroectoderm transforms into the neural plate, it invaginates and the neural folds that arose at the neural plate borders fuse to form the neural tube at embryonic day 8 in mice or day 22 in humans (Fig. 1). During or right after the neuroectodermal folds have fused, neural crest is dispersed from the neural folds. In general, the surrounding structures of the mesoderm and ectoderm at the interface of the neural plate and adjacent ectoderm induce the formation and delamination of the neural crest. These signaling molecules, collectively referred to as ‘neural crest inducers’, mainly consist of WNT ligands, FGF, NOTCH and BMP signaling [4, 5]. The inductive signals originate from the surrounding structures such as the neural plate (NOTCH), lateral plate mesoderm (FGF) and ectoderm (WNT). Many of the signaling events converge to the expression of SLUG, a main driver of emigration occurring at E9–E9.5 in mice [6, 7]. Critical cell-intrinsic signaling events specifying the neural crest potential include PAX3, ZIC1/2 and MSX1/2, collectively referred to as ‘neural crest specifiers (Fig. 1) [4]. A critical step for delamination of neural crest cells is an epithelial-to-mesenchymal transition (EMT) orchestrated by various EMT-inducing transcription factors such as SNAIL, SLUG and ZEB proteins. Slug has been recognized as a nuclear factor contributing to the migration of neural crest cells. While Slug alone is dispensable in the mouse for neural crest generation and differentiation, Slug homozygous knockout mice show hypopigmentation alterations on their extremities [8]. In line, Slug gene deletion in humans has been associated with Waardenburg syndrome and piebaldism [9, 10]. EMT-inducing transcription factors directly repress a plethora of epithelial marker genes involved in cellular adhesion, polarity and cytoskeleton (re-) organization. At the same time, various mesenchymal marker genes are directly or indirectly upregulated [11]. Epithelia are closely connected cell layers held together by tight junctions, gap junctions and adherens junctions with apico-basal polarity. The main determinants of epithelial integrity consist of E-cadherin, proteins from the occludin and claudin families, the junctional adhesion molecules (JAMs) and the polarity factors grouped as the Crumbs, the Par, and the Scribble complex [11–13]. Mesenchymal cells have a spindle-shaped cell morphology and only interact at focal adhesion points. Mesenchymal cells are not fixed to intercellular adhesion complexes, allowing them to move as individual cells in response to environmental signals [11]. EMT is responsible for both detachment and migratory properties of neural crest cells and coincides with a cadherin switch in which N-cadherin is substituted for weaker type II cadherins [14]. Only upon SNAIL-associated enhanced activity of matrix metalloproteinases, which allows digestion of the extracellular matrix (ECM), movement of neural crest cells is initiated.
Fig. 1.

Neural crest specification and delamination. Neuroectodermal patterning is a very dynamic developmental program in which Notch signaling and secreted ligands such as BMP, WNT and FGF will specify the posterior neural border and the neural crest arising at the neural folds upon closure of the neural tube. During different stages, different genes are expressed that mark the neural plate border and premigratory and dispersed neural crest cells
Although EMT and delamination are sometimes interchangeably used in neural crest biology, delamination does not necessarily overlap with the completion of EMT.
Migration and specification of the melanocyte lineage
The migration of neural crest cells, in particular trunk neural crest cells that almost exclusively contribute to melanogenic and neurogenic lineages, has been studied extensively in different model organisms including chick, quail, zebrafish and mouse embryos [15–19]. Neural crest development and migration show considerable overlap between these different model organisms, yet differ significantly in timing and spatial pathways of morphogenetic events [19]. Species-specific migration routes and timing differences for onset of neurulation, migration and cell fate specification are prevalent as described below. While the generation of several neural crest derivatives is limited to specific axial levels along the neural tube, melanogenic and neurogenic cells are produced along the entire axis. Nevertheless, neural crest cells isolated from distinct axial positions possess a similar potency to generate several lineages when subjected to appropriate signals [20].
A stereotypic model of melanoblast development and migration is based on chick embryo studies and stipulates that lineage specification is an early event of neural crest development [3, 20]. As such, emerging avian trunk neural crest cells travel either ventrally or dorsolaterally with predominantly neurogenic or melanogenic cell fates, respectively. After delamination along the dorsal side of the neural tube, neural crest cells transiently accumulate in an interstitial ‘migration staging area’ (MSA) from which they disperse in two distinct migration routes [21–26]. A large fraction of immature melanocyte precursor cells, referred to as melanoblasts, migrate along a dorsolateral route away from the neural tube between the somites and ectoderm before invading and colonizing the epidermis. On the other hand, neurogenic cell populations, giving rise to glial cells and peripheral sensory neurons, are restricted to the ventral pathway (Fig. 2). Melanoblasts from late-stage embryos transplanted to the MSA of younger embryos, which are in an embryonic stage before neural crest cells have entered the MSA, are restricted to migrate along the dorsolateral pathway. Similar transplantation assays demonstrate that neurogenic populations are unable to enter the dorsolateral route, regardless of embryonic stage or position along the neural tube from which they were isolated [26, 27]. The dorsolateral versus ventral dichotomy of migration routes is mainly supported by experiments using quail and chick transplantation assays, and more extensive molecular mechanisms on c-Kit and adhesion molecules have been addressed by grafting ES cells in chick embryos [28].
Fig. 2.

Model for mouse melanoblast migration. Melanocytes differentiating from melanoblast cells in the body trunk arise via the dorsolateral migration route between the ectoderm and dermomyotome. Neurogenic populations differentiating from ventrally migrating neural crest cells. In addition, ventrally migrating neural crest cells give rise to numerous other lineages, whereas the dorsolateral pathway is restricted to melanoblasts only (see text). MSA migratory staging area
In contrast, zebrafish melanoblasts exploit both ventral and dorsolateral routes, whereas neurogenic populations are restricted to ventral migration pathways, illustrating species-dependent differences. A study by Ernfors et al. suggested that during mouse embryogenesis, melanoblasts can differentiate from neural crest-derived Schwann cell precursor (SCP) cells, which exploit the ventral migration route [29, 30]. After detaching from the distal ends of nerves innervating the skin dermis, SCP cells can differentiate into melanocytes. The authors show that SCP cells in close proximity to the distal nerve endings within the dermis may serve as a secondary cellular source for differentiated melanocytes, which is also preserved postnatally upon wounding and repair. Earlier studies already suggested the presence of the bipotent progenitor in peripheral nerves, and that surgical injury of nerves causes accumulation of cytokines which direct lineage switching from glia to melanocytes as demonstrated by hyperpigmentation around the cut nerve fragment [29, 31–35]. Moreover, the study suggests that after surgical ablation of the dorsolateral melanoblast population in avian embryos, ventral migration routes can compensate for melanocyte populations [30]. In addition, the authors claim that a large proportion of murine postnatal skin melanocytes are derived from these ventrally migrating SCP cells based on fluorescent fate mapping using the proteolipid protein (Plp) promoter-driven CreERT2 (Plp-CreERT2) line. Unfortunately, the quantitative assessment of the relative contributions of the different migration paths and reservoirs to postnatal melanocyte populations in this study is obscured by a lack of specificity of the adopted Plp-CreERT2 line. Three independent studies challenge the specificity of the Plp-CreERT2 line, demonstrating that also postnatal melanocytes from the dorsolateral pathway can be tagged [36–38]. In another experimental approach by Sommer and co-workers, the glial-specific Dhh-Cre line was adopted, specifically targeting SCP cells, but not melanoblasts from the dorsolateral pathway. In Dhh-Cre mice crossed with a fate-mapping reporter line, no fluorescently labeled postnatal melanocytes could be traced back to the SCP lineage [37].
The discrepancy obtained with the Dhh-Cre and Plp-CreERT2 lines remains unclear. While Dhh-Cre and Plp-CreERT2 might be expressed in distinct neuronal subpopulations, also differences in animal species and anatomical regions that are being analyzed are confounding. Indeed, Nitzan et al. illustrate how neural crest-derived versus Schwann cell progenitor cell-derived melanocytes are two spatially segregated melanocyte populations in avian embryos [39]. The authors demonstrate that anatomical differences in embryonic skin development form the basis of the specificity of these distinct populations. Neural crest-derived melanoblasts that exploit the dorsolateral pathway will migrate through and populate only dermal connective tissue derived from the paraxial mesoderm (somite, dermomyotome), while SCP-derived melanocytes from a ventral migration route are solely navigated through dermis originating from the lateral plate mesoderm. It remains to be shown in other animal species whether melanocyte populations respect a sharp boundary between somite- and lateral plate mesoderm-derived dermis. In favor of this, a similar restricted distribution of mesodermal patterning in ventral a dorsal mouse skin dermis has been documented in other studies [40, 41]. Therefore, it is tempting to speculate that also in mice, somite-derived dermis (positioned at the dorsolateral skin in mice) is populated with melanocytes from the dorsolateral migration route, while lateral plate-derived dermis (contributing to ventral skin) are homed by SCP-derived melanocytes arising via ventral migration routes. To reveal whether such a restricted distribution pattern occurs, which may also be very relevant for limb and cranial melanocytes, there is a strong need for advanced tools, specific reporters, Cre-lines and time-lapse single cell analyses. The studies mentioned above provide a new model for avian melanocyte development via two defined migration routes with restricted distribution patterns depending on the anatomical location and origin of the dermis through which the melanoblasts migrate and reside (Fig. 3). However, the Plp-CreERT2 line cannot be used with confidence to assess the distribution of melanoblasts arising via ventrally migrating routes in mice. As such, further work needs to be done to dissect the lineage relationship between SCP cells and melanocytes in mice, yet a common glial–melanogenic bipotent progenitor is not in doubt. Complementarily, several studies indicate that neural crest-derived melanocytes and Schwann cells are phenotypically unstable in vitro, illustrated by the transition from Schwann cells to melanocytes via an intermediate bipotent stem cell and vice versa [35].
Fig. 3.

New hypothetical model for avian neural crest migration. Melanocytes differentiating from melanoblast cells arising via the dorsolateral migration route migrate through and populate the developing dermis derived from somites (dermomyotome, DM), whereas melanocytes differentiating from ventrally migrating Schwann cell precursors (SCPs) are restricted to skin composed of dermis originating from the lateral plate mesoderm (LPM). The differential mesodermal patterning of the skin is delineated by the ectodermal notch. In addition to SCPs, ventrally migrating neural crest cells give rise to numerous other lineages, whereas the dorsolateral pathway is restricted to melanoblasts only (see text). No notochord (derived from paraxial mesoderm), Nt neural tube, MSA migratory staging area, S sclerotome, DM dermomyotome (somite derived from paraxial mesoderm), LPM lateral plate mesoderm, SCP Schwann cell precursor
The developmental window of start of migration and cell fate specification encompasses only a few hours and has been studied in different organisms. Therefore, different models on how neural crest cells migrate and are specified toward distinct derivatives have emerged. Lineage specification in avian embryos may occur before they exit the neural tube or very shortly after in the MSA, proposing that their fate is predetermined before they enter their distinct migratory pathways. As such, the choice of migration path is a consequence of cell fate. Indeed, some studies suggest that lineage specification already occurs prior to emigration of avian neural crest cells and multipotency is confined to premigratory neural crest cells in an early neuroepithelial stage. In contrast, mice embryonic studies undoubtfully have proven the multipotency of murine neural crest cells instead of a predetermined lineage restriction, with a retained cellular plasticity that allows transitioning between melanogenic and neurogenic populations [20].
Irrespective of the animal species, it is widely accepted that within the vertebrate organism, microenvironmental signals which neural crest cells encounter during their migration are potent drivers of specification [42]. Interestingly, the start of migration and the appearance of differentiation markers show subtle differences in timing for both neurogenic and melanogenic populations. Generally, specification in various lineages is a combination of a predetermined cell-intrinsic cell fates and extracellular signals, of which both are subjected to spatio-temporal influences. Thus, both positional information and timing are determinants for cell fate specification. A temporal switch from ventral to dorsal migration accounts for melanocyte specification during avian embryogenesis. Here, neural progenitors migrate via the ventral route, while melanocyte progenitor cells transiently stall in the previously described migration staging area before migrating on a dorsolateral path.
Moreover, spatio-temporal patterning of the extracellular cues also contributes to fate specification. The surrounding levels of extracellular factors and signaling molecules may be distributed in gradients and fluctuate over time. In avian and zebrafish embryos for example, BMP4 secretion by the notochord promotes neuronal specification, but gradually drops over time, possibly explaining the initial bias toward neurogenic populations preceding melanoblast emergence. Similarly, explants from the quail neural tubes with emigration of neural crest cells will instantly differentiate into neurons, followed by a delayed differentiation wave of melanocytes [5, 26, 43]. In addition, some studies suggest that both melanogenic and neurogenic populations may have subtle differences in spatial emergence along the neuronal tube in some species.
Melanoblast development and colonization of the skin
Recent studies suggest that melanocytes may originate from different neural crest precursor cells from dorsolateral and ventral migration routes. Nevertheless, the dorsolateral pathway has been studied most extensively over the past decades to analyze how and when melanoblasts colonize the epidermis and differentiate into mature melanocytes in mice. Interestingly, melanocytes can be found in various other locations within the body such as the inner ear, heart, adipose tissue and brain and or referred to as nonclassical melanocytes [44].
A summary of the main events and key molecular markers during embryonic melanoblast differentiation is depicted in Fig. 4. During mouse embryonic development, delamination of neural crest cells and entry in the MSA takes place at E9. Migrating cells are observed from E10 onward at the dorsolateral migration path between the ectoderm, which will give rise to the prospective epidermis and the dorsal surface of the somites. The earliest melanoblast markers appear between E10.5 and E11.5 [21, 45]. The first melanocyte-specific gene that is expressed in melanoblasts is the microphthalmia-associated transcription factor (Mitf), considered as the master gene regulator of the melanocyte lineage. Mitf consists of at least five isoforms differing at their N-termini and tissue-specific expression pattern [46]. In this review, we refer specifically to the Mitf-M isoform strongly expressed in melanocytes. Mitf is crucial for many cellular processes including survival, cell cycle progression, proliferation and differentiation of melanocytes and regulates expression of the genes required for melanin pigment synthesis. The first Mitf mutation was discovered after irradiation of mice with X rays. These mutant mice were completely white with small eyes due to defects in melanocytes and retinal pigment epithelial cells. Other defects were associated with failure of secondary bone resorption, the onset of deafness and reduced numbers of mast cells, indicating the pivotal role of the Mitf locus for the development of many cell lineages [47]. Mitf is regulated by many molecular players involved in neural crest and melanoblast development, such as Pax3, Sox10, FoxD3 and CREB [48–52]. Initially, melanoblasts migrate through and proliferate within the dermis. Interestingly, the doubling time of the migrating population that retains within the dermis remains constant between E10 and E15. From E11.5 onward, the first cells that transmigrate from the dermal to epidermal compartment can be observed, which continues up to E15.5 or later. Around E12.5, nearly 50% of all melanoblasts have passed the basement membrane, the physical barrier that separates the dermis from the epidermis. While the melanoblasts within the dermis continue to cycle, the total number is kept constant when a constant flow across the membrane from the dermis to epidermis is maintained [21]. Interestingly, the dermal melanoblasts sustain an asymmetric division pattern given that only one of the two daughter cells is competent to transmigrate, while the other melanoblast remains slow cycling in the dermis. The epidermis is more favorable for melanoblast proliferation compared to the dermis, as the melanoblasts that enter the epidermis start proliferating and differentiating massively. From E15.5 to E16, melanoblasts distribute throughout the epidermis and start clustering into the developing hair follicles. Melanoblasts that reach the epidermis and the hair follicles will be segregated into distinct populations during hair follicle morphogenesis. One population will localize in the lower part of the hair follicle where they differentiate into mature melanocytes and immediately contribute to hair pigmentation during the first hair cycle (Fig. 4b). A second population consists of melanocyte stem cells that reside within a specialized stem cell niche, referred to as the hair follicle bulge, and serve as reservoir to replenish the regenerating adult hair follicles with new melanocytes [53–56]. The hair bulge becomes an anatomically distinct zone during the first postnatal days and is populated by undifferentiated melanocyte stem cells as early as postnatal day 0.5. A third population consists of melanocytes that populate the interfollicular epidermis, responsible for skin pigmentation (Fig. 4b). As mentioned above, neural crest cells have a retained plasticity that allows transitioning between melanogenic and neurogenic populations rather than having a predetermined lineage restriction. The signaling network of neural crest cells is a flexible and adaptable module and not all neural crest derivatives have a stable phenotype. Melanocyte specification is not 100% fixed, and melanoblast and melanocyte cells are able to revert toward a bipotent neurogenic–melanogenic precursor within a landscape of plasticity and uncommitted progenitors [20]. Collectively, functional pigment-producing melanocytes may arise from different precursor cells during embryonic development and postnatal melanocyte homeostasis (Fig. 5). The connection between melanocytes and Schwann cells is also apparent from the neural-like differentiation pattern and structures in some human malignant melanomas, resembling a conventional malignant peripheral nerve sheath tumor (MPNST) [57]. Neurotization of melanocytic lesions has been confirmed and reported in mouse melanoma models as well by different research groups [58–60].
Fig. 4.

Melanoblast development. a Schematic overview of different stages of melanoblast migration and development in mouse embryos. Details are described within the text. MSA migratory staging area. b Melanoblasts that reach the epidermis and the hair follicles will be segregated into distinct populations during hair follicle morphogenesis. Epidermal interfollicular melanocytes are responsible for skin pigmentation, while hair follicle melanocytes synthesize pigment for the hair
Fig. 5.

Sources of differentiated melanocytes. During embryonic development, melanoblast originate from a bipotent melanoblast/glial precursor cell. Melanoblasts that reach developing hair follicles will be segregated into two distinct and physically separated populations during hair follicle morphogenesis. One population will localize in the hair follicle bulb, where they differentiate into mature melanocytes and immediately contribute to hair pigmentation during the first hair cycle. The other population consists of melanocyte stem cells that reside within a specialized stem cell niche, referred to as the hair follicle bulge, and serve as reservoir to replenish the cycling hair follicles with new melanocytes. Schwann cell precursor cells (SCP) may also differentiate into mature melanocytes in vitro or after wounding in vivo. While the relative contribution of SCPs to embryonic and physiological melanocytes in mice remains to be elucidated, in chick embryos SCPs contribute to melanocytes from early embryonic development
Localization, patterning, total number and proliferation of melanoblasts are tightly controlled over time. During skin development, they colonize sequentially the dermis, epidermis and finally the hair follicles. In all of these compartments, they are subjected to distinct developmental cues. The latter implies that the surrounding environmental context will dictate the proliferation rate, survival and differentiation of melanoblast cells. As such, melanoblasts have an unstable phenotype highly susceptible to their location within the skin [61, 62]. Colonization of the epidermis and hair follicles is an active process requiring the activity of KIT/KITL signaling [62, 63]. In contrast, the melanoblast and melanocyte populations present within the dermis are highly dependent on ET3 signaling instead of KIT signaling [21].
Melanocytes within postnatal skin
The adult skin epidermis and dermis constitute two compartments separated by a physical barrier, the basement membrane. The epidermis provides a barrier protecting us against dehydration and all forms of physical, thermal and chemical stress. Next to this spatial segregation, the epidermis and dermis originate from different embryonic structures, exert different functions and can be regarded as distinct communities of cells. The epidermis consisting of keratinocytes is derived from the embryonic ectoderm, whereas melanocytes can be regarded as a product of embryonic EMT by which (neuro)ectodermal cells transition to neural crest cells and subsequently differentiate into melanocytes. As described above, the trunk skin dermis has a mesodermal origin with subtle differences in ventral and dorsal mesodermal origins. The epidermis comprises multiple keratinocyte layers and includes the interfollicular epidermis, sebaceous glands, sweat glands and hair follicles. The epidermis is separated from the underlying collagen-rich dermis via the basement membrane. The dermal papilla is a specialized cluster of fibroblast cells within the dermis that instructs epidermal cells to grow downward during hair follicle development around E14.5 and during postnatal hair follicle regeneration (Fig. 4) [64].
In the hairy part of mouse skin, differentiated melanocytes are confined to the bulb area of the hair follicles where they are responsible for pigmentation of the hair. In contrast, in non-hairy skin such as the tail, ears and paws, the melanocytes are located within the interfollicular epidermis at the epidermal junction, in close contact with the basement membrane [65] (Fig. 8). In humans, melanin-containing melanosomes are transferred from interfollicular melanocytes to keratinocytes to protect the DNA against harmful effects of solar UV radiation. UV radiation reaching the upper layers of the epidermis will release a signaling response in the keratinocytes mediated by p53 activation [66]. Several keratinocyte-secreted factors will in turn promote melanin synthesis and melanosome trafficking. The sun tanning response is mediated by c-KIT and MC1R-signaling converging to MITF. In non-sun-exposed cells, the differentiation and melanin synthesis is reduced by TGF-β signaling via pSmad2/3 inhibition of Pax3 expression (Fig. 6).
Fig. 8.

Overview of melanocyte stem cell quiescence and activation. Schematic overview of autocrine and paracrine signaling factors affecting melanocyte stem cell differentiation as described in the text. UPP upper permanent portion, LPP lower permanent portion, TP transient portion, Diff. differentiated
Fig. 6.

Sun tanning response. Upon sunlight exposure, the upper layers of epidermis become exposed to UV radiation. Several signaling cascades following p53 activation are initiated. These pathways include the secretion of growth factors such as a-MSH, FGF2, KITL and EDN1, which in turn will bind their cognate receptors on melanocytes and activate a differentiation program mediated by PAX3 and MITF. MITF is a main activator of genes involved in pigment synthesis, melanosome generation and trafficking. In the absence of sunlight and UV exposure, TGF-β secreted from the keratinocytes keep the melanocytes in a quiescent state
Melanocyte stem cells during hair follicle cycling
The hair follicle is often compared to a mini-organ which is continuously renewed in periodic cycles of growth and retraction. Therefore, the hair follicle is regarded as a unique model system to study the process of physiological tissue regeneration. The hair follicles undergo numerous hair cycles consisting of different phases of anagen (growth phase), catagen (regression phase) and telogen (resting phase). The lower transient part of the hair follicle is completely regenerated over the hair cycle, whereas the remaining upper permanent portion is maintained throughout life (Fig. 7). The duration of anagen varies depending on the anatomical location of the hairs. In human scalp follicles that can generate long hairs, anagen may last up to 8 years, whereas within the eyebrow, anagen may last as long as 3 months. Signaling cues between the underlying dermal papilla and the bulge region provide the necessary signaling to restart anagen and regulate the length of the telogen resting phase [64]. Hair follicle growth and retraction are also coupled with regenerative cycles of melanocyte differentiation and melanogenesis. At the onset of anagen, actively proliferating and differentiating melanocytes appear in the hair matrix. The entire melanocyte population in the lower portion of the hair follicle is depleted via apoptosis during catagen and melanocytes remain absent in telogen hair follicles until the subsequent anagen phase is re-initiated. Given these regenerative cycles of hair follicle self-renewal and hair growth, the presence of different types of stem cells giving rise to the epithelial and melanocyte lineages had been suggested many years ago. Cotsarelis et al. were the first to identify a label-retaining slow-cycling population residing in the hair follicle bulge [67]. Neonatal mice were injected repeatedly with the nucleotide analog 3H-thymidine, so that all cells during hair follicle morphogenesis incorporate the label in newly synthesized DNA and cells. The label is then chased for several weeks and only slow-cycling cells such as stem cells retain the label in adulthood.
Fig. 7.

Schematic overview of mouse hair cycle. Description in text (DP dermal papilla)
Later, several lineage-tracing experiments have put forward the concept of ‘bulge activation’ which states that bulge epithelial stem cells are induced at the onset of anagen and migrate toward the bulb matrix where they proliferate and differentiate in epithelial lineages that constitute the hair follicle [68–70]. Characterization of the exact epithelial bulge stem cell populations has been under debate for many years [71].
Despite obvious depletion of differentiated melanocytes during hair follicle catagen, pigmentation is restored in the hair cycles that follow. This regeneration of functional pigmented melanocytes suggests that also repopulation of melanocytes stems from a residing melanocyte stem cell pool within the hair follicle. A melanocyte stem cell population, co-housing with epithelial stem cells within the hair follicle bulge, was identified in a study centered on c-Kit [53, 72]. As mentioned above, c-Kit signaling is instrumental for proliferation and differentiation of hair follicle melanocytes during embryogenesis and postnatal differentiation. Melanocytes can be depleted by administration of a neutralizing c-Kit antibody into neonatal mice. Although a prominent loss of pigmentation occurs in the first hair cycle, pigmentation is restored from the second hair cycle onward, indicating the presence of melanocyte stem cells resistant to c-Kit-antibody treatment [53, 72]. Nishimura et al. took advantage of the Dct-LacZ reporter mouse line in which expression of LacZ under the control of the dopachrome tautomerase (Dct) promotor can be used as a tracer for the entire melanocyte lineage [62]. They identified a small subset of slow-cycling LacZ-positive cells that remained localized within the bulge region upon c-Kit antibody treatment of Dct-LacZ neonates [72]. In this landmark paper, melanocyte stem cells were identified in the hair follicle bulge and lower bulge, which are prompted to proliferate and differentiate into mature melanocytes during anagen. These melanocyte stem cells are undifferentiated, slow cycling and are able to regenerate pigmented cells upon engraftment into neonatal albino skins. Many genes involved in melanocyte differentiation that are expressed in melanoblast and/or melanocytes are weakly expressed in melanocyte stem cells. This disparate expression of melanogenic genes is prominent from postnatal day 4 onward as indicated by the loss of Mitf, Tyr, Sox10, Tyrp1 and Ki67 protein expression within presumptive melanocyte stem cells [73]. However, Tyrp2 or Dct expression is maintained and has been used as a bona fide marker for the entire melanocyte lineage including the stem cells [62]. Besides the use of immunohistochemistry and microdissection of the hair follicles, understanding of the melanocyte stem cell pool has been driven by studies involving hair graying caused by inappropriate maintenance of the stem cell pool. Only a few pathways and molecular players have been identified that regulate melanocyte stem cell homeostasis and quiescence as summarized in Fig. 8. When epithelial stem cells regenerate the hair follicle during anagen, activation and differentiation of melanocyte stem cells is required in sync to repopulate the matrix and ensure the generation of pigmented hairs. Increased Wnt-signaling couples the activation of both epithelial and melanocyte stem cells and ensures the coordinated stem cell behavior of these two distinct stem cell populations [74, 75].
At the onset of anagen, both epithelial and melanocyte stem cells activate Wnt-signaling, demonstrated by nuclear β-catenin activity [76]. In contrast, Wnt/β-catenin signaling is suppressed in bulge stem cells during catagen and telogen phase hair follicles. Thus, except for anagen phase, the bulge stem cell niche is devoid of canonical Wnt-signaling to maintain quiescent melanocyte and epithelial stem cells [77, 78]. Ectopic expression of a dominant active form of β-catenin in melanocytes, resulting in forced activation of canonical Wnt-signaling, leads to premature differentiation and ectopic pigmentation within the bulge [74]. This premature differentiation of melanocyte stem cells is at the expense of self-renewal and quiescence leading to gradual loss of this stem cell pool, ultimately causing hair graying after several hair cycles. Interestingly, also conditional loss of β-catenin during postnatal hair cycles inhibits melanocyte proliferation and differentiation during anagen demonstrating that canonical Wnt-signaling is crucial for melanocyte differentiation during hair follicle cycling. The source of Wnt ligands during anagen that regulate the coordinated activation of canonical Wnt-signaling in both compartments can be attributed to the dermal papilla and epithelial stem cells, but not melanocyte stem cells [74, 79] (Fig. 8). In addition, K15-positive epithelial stem cells, but not melanocyte stem cells, show increased expression and secretion of endothelin-1 and -2 that further promote melanocyte stem cell activation. By disrupting the Endn/EndrB signaling using the chemical inhibitor BQ788, Rabbani et al. demonstrated the strong Edn dependency of this Wnt-driven differentiation [74].
Collectively, Wnt-activated epithelial stem cells secrete Wnt and Edn1/Edn2 ligands and facilitate as such the coordinated proliferation and differentiation of melanocyte stem cells. On the other hand, elevated TGF-β signaling maintains quiescent melanocyte stem cells during telogen. Loss of TGF-β receptor signaling in melanocyte stem cells disrupts stem cell maintenance resulting in premature differentiation and graying [56]. Similarly, TGF-β attenuates melanocyte differentiation of interfollicular melanocytes in the absence of sunlight exposure as depicted in Fig. 6. The physiological regression of hair follicles is induced by several mechanisms such as withdrawal of growth factors and differential expression of death receptors [80]. Apoptosis is regulated in a distinct fashion for every hair follicle compartment and lineage. Melanocyte stem cells are believed to be protected against catagen-induced apoptosis due to absence of the Fas receptor and increased expression of Bcl2 [80, 81]. Nishimura et al. proposed that Bcl2 deficiency results in apoptotic clearance of melanocyte stem cells and premature hair graying in Bcl-2 mice [55]. However, Bcl2 is required for development and survival of melanoblasts prior to melanocyte stem cell development, thus the Bcl2-null phenotype does not enable studying correctly the role of Bcl2 in adult melanocyte stem cells [82]. Nonetheless, TGF-β signaling might be a trigger for apoptosis in the setting of Bcl2 deficiency in melanocyte stem cells, as administration of a TGF-β blocking antibody preserves the viability of bulge melanoblasts in Bcl2-null mice [56]. Interestingly, similar to Wnt-deficient epithelial stem cells, also loss of Col17a1 and Nf1B in epithelial stem cells corrupts the niche and leads to a premature differentiation of the co-housing melanocyte stem cells [83]. Nf1b is a transcription factor negatively regulating Edn2 transcription in quiescent epithelial stem cells, which inhibits melanocyte stem cell differentiation in a Kit-dependent fashion [84]. Several studies have highlighted a role for Notch1 and Notch2 signaling as gatekeepers of the melanocyte stem cell pool, also pointing toward the strong cell–cell interactions within the stem cell niche that are required for proper melanocyte homeostasis [85–87].
Interfollicular melanocyte stem cells
Although they contribute to wound healing, ablation of the hair follicle epithelial stem cells does not impact normal interfollicular epidermal homeostasis [88]. The upper epidermal layer of terminally differentiated keratinocytes is continuously shed and replaced by cells from the underlying proliferating basal layer rather than originating from slowly cycling cells. Human skin mostly resembles the non-hairy skin parts of mice with melanocytes residing in between the basal keratinocytes from the epidermis. Experimental and clinical data suggest that epidermal interfollicular melanocytes in mouse and human may originate from both follicular melanocyte stem cells and melanoblasts within the epidermis and dermis (Fig. 9) [61, 89]. The upward migration of melanocytes may be incited by environmental stimuli such as wounding and UV exposure [90]. On the other hand, intradermal cells with high endogenous expression of the neural crest markers NGFRp75 and nestin are able to migrate toward a basal lamina and differentiate into melanocytes in a three-dimensional human skin equivalent model [91]. Similarly, Glover et al. recently identified a melanocyte stem cell population within the mouse tail interfollicular epidermis, able to regenerate functional melanocytes [89]. In vitiligo, a condition where interfollicular melanocytes are depleted from the skin, repigmentation can occur in a perifollicular manner and autologous transplantation of pigmented follicles is performed as therapeutic strategy [92–94]. While the cellular and molecular composition of hair follicle stem cells in the bulge and progenitor cells in the hair germ has been studied in detail in the mouse, the human hair follicle stem cell niche cannot be easily identified anatomically, undermining the detailed analysis and isolation of human hair follicle stem cells [71, 95]. Moreover, as human and mouse skin are manifestly diverse, conclusions drawn from murine experiments do not necessarily coincide with the human hair follicle. Nevertheless, taking advantages of readily visible alterations, mouse melanocyte stem cells offer an ideal model to study stem cell regulation. Besides vitiligo, several other melanocyte-associated disorders such as Waardenburg syndrome, piebaldism, vitiligo and melanoma have been described and linked to specific genetic defects (Fig. 10). Finally, dysregulated stem cell homeostasis likely accounts for melanoma plasticity and intratumoral heterogeneity. Melanoma progression does not rely solely on irreversible clonal changes or lineage-driven remodeling, but can be driven by reversible and functional reprogramming of signaling routes. Nodal embryonic signaling, critical in the maintenance of hESC pluripotency, is elevated in malignant melanoma [96] and contributes to the formation of heterogeneous cellular states like melanoma-derived endothelial-like cells that participate in tumor-associated vascular mimicry [97]. In line with this is the recent discovery that a retinoid X receptor-γ (RXRG)-controlled neural crest stem cell state in melanoma minimal residual disease is an important driver of therapy resistance against RAF/MEK-inhibition [98]. Together, these latter examples indicate that a better understanding of melanocyte biology may constitute the basis of novel therapeutic strategies for different neural crest cell-derived associated pathologies.
Fig. 9.
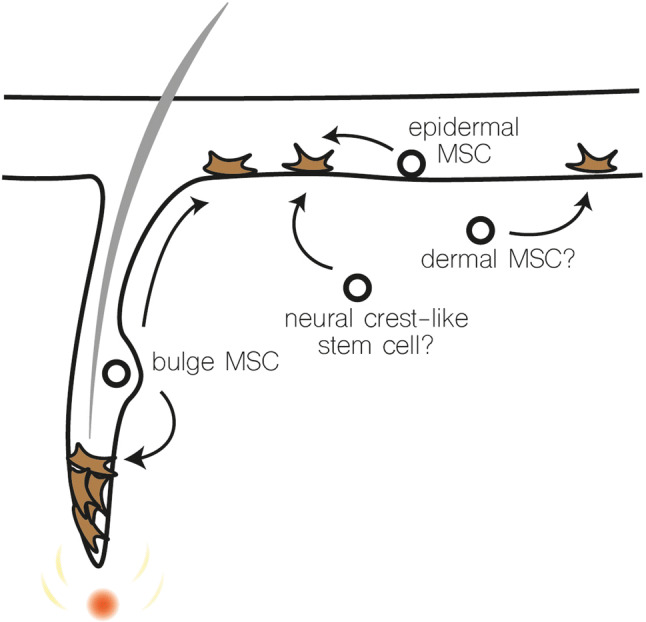
Possible sources of melanocyte stem cells that replenish interfollicular melanocytes. MSC melanocyte stem cells. Interfollicular melanocytes may originate from bulge melanocyte stem cells after upward migration, an epidermal melanocyte stem cell, a dermal melanocyte stem cell or a neural crest-like stem cell
Fig. 10.

Disorders and syndromes associated with melanocyte dysfunction. Piebaldism photograph courtesy of Fleischman [99]
Acknowledgements
G.B.’s laboratory is supported by the Fonds Wetenschappelijk Onderzoek (3G050217W), the Geconcerteerde Onderzoeksacties Ghent University (GOA-01GB1013W), Vlaamse Liga tegen Kanker (365U8914U) and the Stichting tegen Kanker (FAF-F/2016/814).
Abbreviations
- Bcl2
B-cell lymphoma 2
- BMP
Bone morphognetic protein
- BQ778
Selective EDNRB antagonist
- Col17a1
Collagen type XVII alpha 1
- CREB
cAMP-responsive element binding protein
- Dct
Dopachrome tautomerase
- Dhh
Hedgehog
- ECM
Extracellular matrix
- EDN1
Endothelin 1
- EDNRB
Endothelin receptor type B
- ET3
Endothelin 3
- FGF
Fibroblast growth factor
- FOXD3
Forkhead box D3
- hESC
Human embryonic stem cells
- JAMs
Junctional adhesion molecules
- KIT/KITL
c-kit/Kit ligand
- KIT
Kit proto-oncogene
- MC1R
Melanocortin 1 receptor
- Mitf
Microphthalmia-associated transcription factor
- MPNST
Malignant peripheral nerve sheath tumor
- MSA
Migration staging area
- MSX1/2
Msh homeobox 1/2
- Nf1B
Neurofibromin 1b
- NGFRp75
Nerve growth factor receptor
- PAX3
Paired Box 3
- PLP
Proteolipid protein
- SCP
Schwann cell precursor
- SLUG
Snail family transcription factor 2
- Smad
Mother against decapentaplegic homolog
- SNAIL
Snail family transcription factor 1
- SOX10
(Sex determining region Y)-box 10
- TGFβ
Transforming growth factor beta
- Tyr
Tyrosinase
- Tyrp1/2
Tyrosinase-related protein 1/2
- WNT
Wingless-type MMTV integration site family
- ZIC1/2
Zinc finger of the cerebellum 1/2
- ZEB1/2
Zinc finger E-box binding homeobox 1/2
Compliance with ethical standards
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Huang X, Saint-Jeannet JP. Induction of the neural crest and the opportunities of life on the edge. Dev Biol. 2004;275(1):1–11. doi: 10.1016/j.ydbio.2004.07.033. [DOI] [PubMed] [Google Scholar]
- 2.Le Douarin NM, Kalcheim C. The neural crest. Cambridge: Cambridge University Press; 1999. [Google Scholar]
- 3.Knecht AK, Bronner-Fraser M. Induction of the neural crest: a multigene process. Nat Rev Genet. 2002;3(6):453–461. doi: 10.1038/nrg819. [DOI] [PubMed] [Google Scholar]
- 4.Milet C, Monsoro-Burq AH. Neural crest induction at the neural plate border in vertebrates. Dev Biol. 2012;366(1):22–33. doi: 10.1016/j.ydbio.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 5.Duband JL. Diversity in the molecular and cellular strategies of epithelium-to-mesenchyme transitions: insights from the neural crest. Cell Adh Migr. 2010;4(3):458–482. doi: 10.4161/cam.4.3.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Locascio A, et al. Modularity and reshuffling of Snail and Slug expression during vertebrate evolution. Proc Natl Acad Sci USA. 2002;99(26):16841–16846. doi: 10.1073/pnas.262525399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.del Barrio MG, Nieto MA. Overexpression of Snail family members highlights their ability to promote chick neural crest formation. Development. 2002;129(7):1583–1593. doi: 10.1242/dev.129.7.1583. [DOI] [PubMed] [Google Scholar]
- 8.Perez-Losada J, et al. Zinc-finger transcription factor Slug contributes to the function of the stem cell factor c-kit signaling pathway. Blood. 2002;100(4):1274–1286. [PubMed] [Google Scholar]
- 9.Sanchez-Martin M, et al. SLUG (SNAI2) deletions in patients with Waardenburg disease. Hum Mol Genet. 2002;11(25):3231–3236. doi: 10.1093/hmg/11.25.3231. [DOI] [PubMed] [Google Scholar]
- 10.Sanchez-Martin M, et al. Deletion of the SLUG (SNAI2) gene results in human piebaldism. Am J Med Genet A. 2003;122A(2):125–132. doi: 10.1002/ajmg.a.20345. [DOI] [PubMed] [Google Scholar]
- 11.De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- 12.Skrypek N, et al. ZEB2 stably represses RAB25 expression through epigenetic regulation by SIRT1 and DNMTs during epithelial-to-mesenchymal transition. Epigenet Chromatin. 2018;11(1):70. doi: 10.1186/s13072-018-0239-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vandewalle C, Van Roy F, Berx G. The role of the ZEB family of transcription factors in development and disease. Cell Mol Life Sci. 2009;66(5):773–787. doi: 10.1007/s00018-008-8465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Theveneau E, Mayor R. Neural crest delamination and migration: from epithelium-to-mesenchyme transition to collective cell migration. Dev Biol. 2012;366(1):34–54. doi: 10.1016/j.ydbio.2011.12.041. [DOI] [PubMed] [Google Scholar]
- 15.Le Douarin N. A biological cell labeling technique and its use in experimental embryology. Dev Biol. 1973;30(1):217–222. doi: 10.1016/0012-1606(73)90061-4. [DOI] [PubMed] [Google Scholar]
- 16.Creuzet S, et al. Reciprocal relationships between Fgf8 and neural crest cells in facial and forebrain development. Proc Natl Acad Sci USA. 2004;101(14):4843–4847. doi: 10.1073/pnas.0400869101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klymkowsky MW, Rossi CC, Artinger KB. Mechanisms driving neural crest induction and migration in the zebrafish and Xenopus laevis. Cell Adh Migr. 2010;4(4):595–608. doi: 10.4161/cam.4.4.12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sauka-Spengler T, et al. Ancient evolutionary origin of the neural crest gene regulatory network. Dev Cell. 2007;13(3):405–420. doi: 10.1016/j.devcel.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 19.Aybar MJ, Mayor R. Early induction of neural crest cells: lessons learned from frog, fish and chick. Curr Opin Genet Dev. 2002;12(4):452–458. doi: 10.1016/S0959-437X(02)00325-8. [DOI] [PubMed] [Google Scholar]
- 20.Sommer L. Generation of melanocytes from neural crest cells. Pigment Cell Melanoma Res. 2011;24(3):411–421. doi: 10.1111/j.1755-148X.2011.00834.x. [DOI] [PubMed] [Google Scholar]
- 21.Larue L, de Vuyst F, Delmas V. Modeling melanoblast development. Cell Mol Life Sci. 2013;70(6):1067–1079. doi: 10.1007/s00018-012-1112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mort RL, Jackson IJ, Patton EE. The melanocyte lineage in development and disease. Development. 2015;142(7):1387. doi: 10.1242/dev.123729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wehrle-Haller B, Weston JA. Soluble and cell-bound forms of steel factor activity play distinct roles in melanocyte precursor dispersal and survival on the lateral neural crest migration pathway. Development. 1995;121(3):731–742. doi: 10.1242/dev.121.3.731. [DOI] [PubMed] [Google Scholar]
- 24.Simoes-Costa M, Bronner ME. Establishing neural crest identity: a gene regulatory recipe. Development. 2015;142(2):242–257. doi: 10.1242/dev.105445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ernfors P. Cellular origin and developmental mechanisms during the formation of skin melanocytes. Exp Cell Res. 2010;316(8):1397–1407. doi: 10.1016/j.yexcr.2010.02.042. [DOI] [PubMed] [Google Scholar]
- 26.Krispin S, et al. Evidence for a dynamic spatiotemporal fate map and early fate restrictions of premigratory avian neural crest. Development. 2010;137(4):585–595. doi: 10.1242/dev.041509. [DOI] [PubMed] [Google Scholar]
- 27.Harris ML, Erickson CA. Lineage specification in neural crest cell pathfinding. Dev Dyn. 2007;236(1):1–19. doi: 10.1002/dvdy.20919. [DOI] [PubMed] [Google Scholar]
- 28.Beauvais-Jouneau A, et al. A novel model to study the dorsolateral migration of melanoblasts. Mech Dev. 1999;89(1–2):3–14. doi: 10.1016/S0925-4773(99)00191-4. [DOI] [PubMed] [Google Scholar]
- 29.Adameyko I, Lallemend F. Glial versus melanocyte cell fate choice: schwann cell precursors as a cellular origin of melanocytes. Cell Mol Life Sci. 2010;67(18):3037–3055. doi: 10.1007/s00018-010-0390-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adameyko I, et al. Schwann cell precursors from nerve innervation are a cellular origin of melanocytes in skin. Cell. 2009;139(2):366–379. doi: 10.1016/j.cell.2009.07.049. [DOI] [PubMed] [Google Scholar]
- 31.Rizvi TA, et al. A novel cytokine pathway suppresses glial cell melanogenesis after injury to adult nerve. J Neurosci. 2002;22(22):9831–9840. doi: 10.1523/JNEUROSCI.22-22-09831.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nataf V, Le Douarin NM. Induction of melanogenesis by tetradecanoylphorbol-13 acetate and endothelin 3 in embryonic avian peripheral nerve cultures. Pigment Cell Res. 2000;13(3):172–178. doi: 10.1034/j.1600-0749.2000.130309.x. [DOI] [PubMed] [Google Scholar]
- 33.Nichols DH, Weston JA. Melanogenesis in cultures of peripheral nervous tissue. I. The origin and prospective fate of cells giving rise to melanocytes. Dev Biol. 1977;60(1):217–225. doi: 10.1016/0012-1606(77)90120-8. [DOI] [PubMed] [Google Scholar]
- 34.Nichols DH, Kaplan RA, Weston JA. Melanogenesis in cultures of peripheral nervous tissue. II. Environmental factors determining the fate of pigment-forming cells. Dev Biol. 1977;60(1):226–237. doi: 10.1016/0012-1606(77)90121-X. [DOI] [PubMed] [Google Scholar]
- 35.Dupin E, et al. Reversal of developmental restrictions in neural crest lineages: transition from Schwann cells to glial-melanocytic precursors in vitro. Proc Natl Acad Sci USA. 2003;100(9):5229–5233. doi: 10.1073/pnas.0831229100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colombo S, et al. Transcriptomic analysis of mouse embryonic skin cells reveals previously unreported genes expressed in melanoblasts. J Invest Dermatol. 2012;132(1):170–178. doi: 10.1038/jid.2011.252. [DOI] [PubMed] [Google Scholar]
- 37.Hari L, et al. Temporal control of neural crest lineage generation by Wnt/beta-catenin signaling. Development. 2012;139(12):2107–2117. doi: 10.1242/dev.073064. [DOI] [PubMed] [Google Scholar]
- 38.Leone DP, et al. Tamoxifen-inducible glia-specific Cre mice for somatic mutagenesis in oligodendrocytes and Schwann cells. Mol Cell Neurosci. 2003;22(4):430–440. doi: 10.1016/S1044-7431(03)00029-0. [DOI] [PubMed] [Google Scholar]
- 39.Nitzan E, et al. Neural crest and Schwann cell progenitor-derived melanocytes are two spatially segregated populations similarly regulated by Foxd3. Proc Natl Acad Sci USA. 2013;110(31):12709–12714. doi: 10.1073/pnas.1306287110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Candille SI, et al. Dorsoventral patterning of the mouse coat by Tbx15. PLoS Biol. 2004;2(1):E3. doi: 10.1371/journal.pbio.0020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lowe LA, Yamada S, Kuehn MR. HoxB6-Cre transgenic mice express Cre recombinase in extra-embryonic mesoderm, in lateral plate and limb mesoderm and at the midbrain/hindbrain junction. Genesis. 2000;26(2):118–120. doi: 10.1002/(SICI)1526-968X(200002)26:2<118::AID-GENE5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 42.Schartl M, et al. What is a vertebrate pigment cell? Pigment Cell Melanoma Res. 2016;29(1):8–14. doi: 10.1111/pcmr.12409. [DOI] [PubMed] [Google Scholar]
- 43.Kuo BR, Erickson CA. Regional differences in neural crest morphogenesis. Cell Adh Migr. 2010;4(4):567–585. doi: 10.4161/cam.4.4.12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Colombo S, Berlin I, Larue L. Classical and nonclassical melanocytes in vertebrates. In: Boranovsky J, Riley PA, editors. Melanins and melanosomes. Weinheim: Wiley; 2011. p. 407. [Google Scholar]
- 45.Thomas AJ, Erickson CA. FOXD3 regulates the lineage switch between neural crest-derived glial cells and pigment cells by repressing MITF through a non-canonical mechanism. Development. 2009;136(11):1849–1858. doi: 10.1242/dev.031989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shibahara S, et al. Microphthalmia-associated transcription factor (MITF): multiplicity in structure, function, and regulation. J Investig Dermatol Symp Proc. 2001;6(1):99–104. doi: 10.1046/j.0022-202x.2001.00010.x. [DOI] [PubMed] [Google Scholar]
- 47.Moore KJ. Insight into the microphthalmia gene. Trends Genet. 1995;11(11):442–448. doi: 10.1016/S0168-9525(00)89143-X. [DOI] [PubMed] [Google Scholar]
- 48.Watanabe A, et al. Epistatic relationship between Waardenburg syndrome genes MITF and PAX3. Nat Genet. 1998;18(3):283–286. doi: 10.1038/ng0398-283. [DOI] [PubMed] [Google Scholar]
- 49.Verastegui C, et al. Regulation of the microphthalmia-associated transcription factor gene by the Waardenburg syndrome type 4 gene, SOX10. J Biol Chem. 2000;275(40):30757–30760. doi: 10.1074/jbc.C000445200. [DOI] [PubMed] [Google Scholar]
- 50.Kos R, et al. The winged-helix transcription factor FoxD3 is important for establishing the neural crest lineage and repressing melanogenesis in avian embryos. Development. 2001;128(8):1467–1479. doi: 10.1242/dev.128.8.1467. [DOI] [PubMed] [Google Scholar]
- 51.Bertolotto C, et al. Microphthalmia gene product as a signal transducer in cAMP-induced differentiation of melanocytes. J Cell Biol. 1998;142(3):827–835. doi: 10.1083/jcb.142.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawakami A. DE Fisher, The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab Invest. 2017;97:649. doi: 10.1038/labinvest.2017.9. [DOI] [PubMed] [Google Scholar]
- 53.Nishikawa-Torikai S, Osawa M, Nishikawa S. Functional characterization of melanocyte stem cells in hair follicles. J Invest Dermatol. 2011;131(12):2358–2367. doi: 10.1038/jid.2011.195. [DOI] [PubMed] [Google Scholar]
- 54.Nishimura EK. Melanocyte stem cells: a melanocyte reservoir in hair follicles for hair and skin pigmentation. Pigment Cell Melanoma Res. 2011;24(3):401–410. doi: 10.1111/j.1755-148X.2011.00855.x. [DOI] [PubMed] [Google Scholar]
- 55.Nishimura EK, Granter SR, Fisher DE. Mechanisms of hair graying: incomplete melanocyte stem cell maintenance in the niche. Science. 2005;307(5710):720–724. doi: 10.1126/science.1099593. [DOI] [PubMed] [Google Scholar]
- 56.Nishimura EK, et al. Key roles for transforming growth factor beta in melanocyte stem cell maintenance. Cell Stem Cell. 2010;6(2):130–140. doi: 10.1016/j.stem.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.King R, Busam K, Rosai J. Metastatic malignant melanoma resembling malignant peripheral nerve sheath tumor: report of 16 cases. Am J Surg Pathol. 1999;23(12):1499–1505. doi: 10.1097/00000478-199912000-00007. [DOI] [PubMed] [Google Scholar]
- 58.Luo C, et al. Expression of oncogenic BRAFV600E in melanocytes induces Schwannian differentiation in vivo. Pigment Cell Melanoma Res. 2015;28(5):603–606. doi: 10.1111/pcmr.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marsh Durban V, et al. Differential AKT dependency displayed by mouse models of BRAFV600E-initiated melanoma. J Clin Invest. 2013;123(12):5104–5118. doi: 10.1172/JCI69619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Damsky W, et al. mTORC1 activation blocks BrafV600E-induced growth arrest but is insufficient for melanoma formation. Cancer Cell. 2015;27(1):41–56. doi: 10.1016/j.ccell.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aoki H, et al. Two distinct types of mouse melanocyte: differential signaling requirement for the maintenance of non-cutaneous and dermal versus epidermal melanocytes. Development. 2009;136(15):2511–2521. doi: 10.1242/dev.037168. [DOI] [PubMed] [Google Scholar]
- 62.Mackenzie MA, et al. Activation of the receptor tyrosine kinase Kit is required for the proliferation of melanoblasts in the mouse embryo. Dev Biol. 1997;192(1):99–107. doi: 10.1006/dbio.1997.8738. [DOI] [PubMed] [Google Scholar]
- 63.Jordan SA, Jackson IJ. A late wave of melanoblast differentiation and rostrocaudal migration revealed in patch and rump-white embryos. Mech Dev. 2000;92(2):135–143. doi: 10.1016/S0925-4773(99)00332-9. [DOI] [PubMed] [Google Scholar]
- 64.Alonso L, Fuchs E. The hair cycle. J Cell Sci. 2006;119(Pt 3):391–393. doi: 10.1242/jcs.02793. [DOI] [PubMed] [Google Scholar]
- 65.Mayer TC. The migratory pathway of neural crest cells into the skin of mouse embryos. Dev Biol. 1973;34(1):39–46. doi: 10.1016/0012-1606(73)90337-0. [DOI] [PubMed] [Google Scholar]
- 66.Cui R, et al. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell. 2007;128(5):853–864. doi: 10.1016/j.cell.2006.12.045. [DOI] [PubMed] [Google Scholar]
- 67.Cotsarelis G, Sun TT, Lavker RM. Label-retaining cells reside in the bulge area of pilosebaceous unit: implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell. 1990;61(7):1329–1337. doi: 10.1016/0092-8674(90)90696-C. [DOI] [PubMed] [Google Scholar]
- 68.Blanpain C, Fuchs E. Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol. 2009;10(3):207–217. doi: 10.1038/nrm2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blanpain C, Fuchs E. Epidermal stem cells of the skin. Annu Rev Cell Dev Biol. 2006;22:339–373. doi: 10.1146/annurev.cellbio.22.010305.104357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Blanpain C, Horsley V, Fuchs E. Epithelial stem cells: turning over new leaves. Cell. 2007;128(3):445–458. doi: 10.1016/j.cell.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jaks V, et al. Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nat Genet. 2008;40(11):1291–1299. doi: 10.1038/ng.239. [DOI] [PubMed] [Google Scholar]
- 72.Nishimura EK, et al. Dominant role of the niche in melanocyte stem-cell fate determination. Nature. 2002;416(6883):854–860. doi: 10.1038/416854a. [DOI] [PubMed] [Google Scholar]
- 73.Osawa M, et al. Molecular characterization of melanocyte stem cells in their niche. Development. 2005;132(24):5589–5599. doi: 10.1242/dev.02161. [DOI] [PubMed] [Google Scholar]
- 74.Rabbani P, et al. Coordinated activation of Wnt in epithelial and melanocyte stem cells initiates pigmented hair regeneration. Cell. 2011;145(6):941–955. doi: 10.1016/j.cell.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lowry WE, et al. Defining the impact of beta-catenin/Tcf transactivation on epithelial stem cells. Genes Dev. 2005;19(13):1596–1611. doi: 10.1101/gad.1324905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bertolotto C. Melanoma: from melanocyte to genetic alterations and clinical options. Scientifica (Cairo) 2013;2013:635203. doi: 10.1155/2013/635203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Latil M, et al. Cell-type-specific chromatin states differentially prime squamous cell carcinoma tumor-initiating cells for epithelial to mesenchymal transition. Cell Stem Cell. 2017;20(2):191 e5–204 e5. doi: 10.1016/j.stem.2016.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Greco V, et al. A two-step mechanism for stem cell activation during hair regeneration. Cell Stem Cell. 2009;4(2):155–169. doi: 10.1016/j.stem.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhou L, et al. CD133-positive dermal papilla-derived Wnt ligands regulate postnatal hair growth. Biochem J. 2016;473(19):3291–3305. doi: 10.1042/BCJ20160466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Botchkareva NV, Ahluwalia G, Shander D. Apoptosis in the hair follicle. J Invest Dermatol. 2006;126(2):258–264. doi: 10.1038/sj.jid.5700007. [DOI] [PubMed] [Google Scholar]
- 81.Botchkareva NV, Botchkarev VA, Gilchrest BA. Fate of melanocytes during development of the hair follicle pigmentary unit. J Investig Dermatol Symp Proc. 2003;8(1):76–79. doi: 10.1046/j.1523-1747.2003.12176.x. [DOI] [PubMed] [Google Scholar]
- 82.Mak SS, et al. Indispensable role of Bcl2 in the development of the melanocyte stem cell. Dev Biol. 2006;291(1):144–153. doi: 10.1016/j.ydbio.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 83.Tanimura S, et al. Hair follicle stem cells provide a functional niche for melanocyte stem cells. Cell Stem Cell. 2011;8(2):177–187. doi: 10.1016/j.stem.2010.11.029. [DOI] [PubMed] [Google Scholar]
- 84.Chang CY, et al. NFIB is a governor of epithelial-melanocyte stem cell behaviour in a shared niche. Nature. 2013;495(7439):98–102. doi: 10.1038/nature11847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schouwey K, et al. Notch1 and Notch2 receptors influence progressive hair graying in a dose-dependent manner. Dev Dyn. 2007;236(1):282–289. doi: 10.1002/dvdy.21000. [DOI] [PubMed] [Google Scholar]
- 86.Kumano K, et al. Both Notch1 and Notch2 contribute to the regulation of melanocyte homeostasis. Pigment Cell Melanoma Res. 2008;21(1):70–78. doi: 10.1111/j.1755-148X.2007.00423.x. [DOI] [PubMed] [Google Scholar]
- 87.Moriyama M, et al. Notch signaling via Hes1 transcription factor maintains survival of melanoblasts and melanocyte stem cells. J Cell Biol. 2006;173(3):333–339. doi: 10.1083/jcb.200509084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ito M, et al. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat Med. 2005;11(12):1351–1354. doi: 10.1038/nm1328. [DOI] [PubMed] [Google Scholar]
- 89.Glover JD, et al. Maintenance of distinct melanocyte populations in the interfollicular epidermis. Pigment Cell Melanoma Res. 2015;28(4):476–480. doi: 10.1111/pcmr.12375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gilchrest BA. Molecular aspects of tanning. J Invest Dermatol. 2011;131(E1):E14–E17. doi: 10.1038/skinbio.2011.6. [DOI] [PubMed] [Google Scholar]
- 91.Li L, et al. Human dermal stem cells differentiate into functional epidermal melanocytes. J Cell Sci. 2010;123(Pt 6):853–860. doi: 10.1242/jcs.061598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Falabella R. Vitiligo and the melanocyte reservoir. Indian J Dermatol. 2009;54(4):313–318. doi: 10.4103/0019-5154.57604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Falabella R, Barona MI. Update on skin repigmentation therapies in vitiligo. Pigment Cell Melanoma Res. 2009;22(1):42–65. doi: 10.1111/j.1755-148X.2008.00528.x. [DOI] [PubMed] [Google Scholar]
- 94.Rusfianti M, Wirohadidjodjo YW. Dermatosurgical techniques for repigmentation of vitiligo. Int J Dermatol. 2006;45(4):411–417. doi: 10.1111/j.1365-4632.2006.02486.x. [DOI] [PubMed] [Google Scholar]
- 95.Watt FM, Jensen KB. Epidermal stem cell diversity and quiescence. EMBO Mol Med. 2009;1(5):260–267. doi: 10.1002/emmm.200900033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Topczewska JM, et al. Embryonic and tumorigenic pathways converge via Nodal signaling: role in melanoma aggressiveness. Nat Med. 2006;12(8):925–932. doi: 10.1038/nm1448. [DOI] [PubMed] [Google Scholar]
- 97.Hendrix MJ, et al. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer. 2003;3(6):411–421. doi: 10.1038/nrc1092. [DOI] [PubMed] [Google Scholar]
- 98.Rambow F, et al. Toward minimal residual disease-directed therapy in melanoma. Cell. 2018;174(4):843 e19–855 e19. doi: 10.1016/j.cell.2018.06.025. [DOI] [PubMed] [Google Scholar]
- 99.Fleischman RA, et al. Deletion of the c-kit protooncogene in the human developmental defect piebald trait. Proc Natl Acad Sci USA. 1991;88(23):10885–10889. doi: 10.1073/pnas.88.23.10885. [DOI] [PMC free article] [PubMed] [Google Scholar]