

Abstract
Autophagy is an evolutionarily conserved, multi-step lysosomal degradation process for the clearance of damaged or superfluous proteins and organelles. Accumulating studies have recently revealed that autophagy is closely related to a variety of types of cancer; however, elucidation of its Janus role of either tumor-suppressive or tumor-promoting still remains to be discovered. In this review, we focus on summarizing the context-dependent role of autophagy and its complicated molecular mechanisms in different types of cancer. Moreover, we discuss a series of small-molecule compounds targeting autophagy-related proteins or the autophagic process for potential cancer therapy. Taken together, these findings would shed new light on exploiting the intricate mechanisms of autophagy and relevant small-molecule compounds as potential anti-cancer drugs to improve targeted cancer therapy.
Keywords: Autophagy, Tumor-suppressive, Tumor-promoting, Small-molecule compound, Targeted cancer therapy
Introduction
Autophagy, a highly evolutionarily conserved process, is responsible for degradation and recycling of intracellular components by lysosome system. Under physiological conditions, autophagy is maintained at basal levels which contributes to the successive degradation of superabundant, abnormal, damaged or risk factors [1]. Three major types of autophagy have been characterized: macroautophagy, microautophagy, and chaperone-mediated autophagy. Among them, macroautophagy depends on specialized double-membraned vesicles known as autophagosomes to progressively package autophagic cargo and then deliver them to the lysosomes by membrane fusion. Microautophagy relies on the direct uptake of cytoplasmic material through lysosomal membrane invaginate. And chaperone-mediated autophagy involves the lysosomal-associated membrane protein 2 (LAMP2)-dependent translocation of autophagic substrates bound to cytosolic chaperones of the heat shock protein family across the lysosomal membrane [2]. Although different kinds of autophagy are all closely related to cancer, macroautophagy is the best-characterized form of autophagy and is more closely tied to cancer progression. Interestingly, many signaling pathways related to tumor transformation and progression can dramatically regulate autophagy initiation; thereby making their relationship more fascinating. In this review, autophagy refers to macroautophagy, unless otherwise specified (Fig. 1).
Fig. 1.

Some oncogenic and tumor suppressive signaling pathways related to tumor progression and autophagy initiate. a Oncogenic and tumor suppressive signaling pathways play important roles in the development of the tumor. Of which MAPK, PI3K-AKT and Notch signaling promotes the malignant process while p53 signaling inhibits it. NF-κB signaling responds to inflammation and ROS to inhibit tumor progression. b Oncogenic and tumor suppressive signaling pathways are closely related to autophagy initiate. MAPK signaling can activate autophagy through AMPK activation and promotion of autophagy-related gene translation. PI3K-AKT signaling inhibits autophagy through mTOR activation and p53 inhibition. Notch signaling inhibits autophagy via p53 and PTEN inhibition. NF-κB signaling inhibits ROS aggregation thus to inhibit autophagy
The process of classical autophagy mainly consists of five successive subtle steps, including (I) induction, (II) vesicle nucleation, (III) vesicle elongation and completion, (IV) docking and fusion, and finally, (V) degradation and recycle [3]. Autophagy could be directly rhythmically regulated by various autophagy-modulating genes and proteins (Fig. 2a). In the initiation step of autophagy, the widely accepted sensor is the mechanistic target of rapamycin complex I (mTORCI) and many autophagy inducers trigger autophagy by initiating signal transduction cascades to tactfully inhibit mTORCI [4]. Except for mTORCI, ULK complex members Unc51-like protein kinase 1(ULK1), mATG13, FIP200, and ATG101 are also crucial for the initiation of autophagic responses. AMPK, the sensor of ATP/AMP, is of great importance in this step by directly regulating mTORCI and ULK1 [5]. At the stage of vesicle nucleation, the most important complex is the Vps34 complex and Beclin1. The process of autophagy is mainly dependent on two ubiquitin-like conjugation systems to process the linkage of ATG5 to ATG12 and ATG16L1, and phosphatidylethanolamine to proteins of the microtubule-associated protein 1 light chain 3 (LC3). ATG7 is vital for the formation of ATG5–ATG12–ATG16L complex and the maturation of LC3II [6]. In the last step of autophagy, Lysosome-associated membrane protein 1 and 2 play a crucial role for the regulation of lysosomal motility [7].
Fig. 2.

Autophagy process and the key roles in cancer. a The form of autophagy consists of several successive steps, including (1) induction, (2) vesicle nucleation, (3) vesicle elongation and completion, (4) docking and fusion, and (5) degradation and recycling. Each step can be positively or negatively regulated by key autophagy-related proteins. b On one hand, autophagy helps cancer cell proliferation and maintains carcinogenesis (red) in different stages of cancer. On the other hand, it suppresses its malignant transformation and promotes cancer cell death (blue). In general, autophagy plays a double-edged sword to control the cancer cell fate
Baseline autophagy is the basis for maintaining the health of organisms [1]. Autophagy, which always sustains an adaptive response to stress, is stimulated by a lot of factors. Autophagy maintains organisms on an energetic homeostasis at the starvation state and plays a fatal role when encountering diverse stress conditions, such as oxidative damage, damaged organelles aggregation, dangerous stimulator aggregation, microbial infection [8], etc. Defective autophagy is always accompanied by multiple diseases, such as immunodeficiency disease, geriatric disease, senescence, and cancer [9]. Compared to normal cells, cancer cells commonly display dysregulation of autophagy. It has been demonstrated that BECN1, the pivotal autophagy gene, is deficient in ovarian, breast and prostate cancer cells [10]. Autophagy deficiency commonly results in malignant transformation and poor prognosis of cancer. Heterozygous disruption of BECN1 also increases the risk of malignant transformation and rapidly progress to premalignant lesions. Mice lacking Ambra1 show a higher genetic susceptibility to cancer than wild-type ones [11]. Somatic mutations in ATG genes are frequently observed in malignant cancers [12] and the deficiency of Atg5 or Atg7 also increases the risk of the malignant transformation [13]. Additionally, autophagy suppresses the carcinogenesis through several strategies [14] (Fig. 2b). It is thought that autophagy prevents cancer development, but once cancer is established, autophagy always promotes cancer cells survival, especially in those malignant types. Moreover, autophagy often promotes cancer progression and resistance to treatment, which makes the cancer treatment more difficult [14, 15]. In this review, we focus on providing an exquisite insight into the context-dependent role of autophagy including cancer-suppressive or tumor-promoting roles and elucidating related signaling pathways. Meanwhile, we illustrate a number of small-molecule compounds directly targeting autophagy executors. Together, these inspiring findings may shed light on targeting autophagy with small molecular compounds to improve cancer therapy.
Tumor-promoting role of autophagy in cancer
Autophagy is crucial to support organismal fitness, which also applies to cancer cells. Indeed, autophagy does promote tumor progression or protect tumor cells from stress especially in established cancer. In many aggressive cancers, autophagy often involved in chemotherapy resistance [16]. Herein, we mainly focus on the role of autophagy in stress resistance, necrosis and apoptosis resistance.
Resistance to stress
Energy and nutrients stress
Energy and nutrient stress is the most common but considerable threat to cancer cells. Because of their uncontrolled proliferation, cancer cells require more nutrient and energy than normal cells. Under starvation, autophagy acts as the first protector to avoid energy shortage. Autophagy is activated under energy and nutrient stress by several mechanisms (Fig. 3a). AMPK, the salient energy sensor to maintain energy homeostasis under nutrient starvation, can stimulate autophagy through mTOR in a TSC1/2 dependent pathway [17]. Another key autophagy regulator is mTOR. Induction of autophagy can be easily triggered by mTOR inhibition. Atg1/ULK1 is a central component in autophagy and the autophagy regulator ULK complex is formed by ULK1, ATG13, FIP200, and ATG101. [18]. mTORC1 and ULK1 can be regulated by AMPK through direct phosphorylation. After the induction of autophagy by AMPK, many other key autophagy factors are involved in the latter process. Among them, the Vps34 complex is the most critical one and AMPK can directly regulate Vps34 complex through phosphorylation to initiate autophagy [19]. AMPK and mTOR can activate or regulate autophagy to protect cells from the stress of energy and nutrition under physiological and pathological conditions. During the occurrence of a tumor, higher levels of autophagy may help the malignant growth of tumor cells, so that the inhibition of autophagy can be an effective strategy for the therapy at this stage.
Fig. 3.

Autophagy helps cancer cell response to stress. a Under energy and nutrients stress, autophagy can be activated by several mechanisms. b When cancer cells are exposed to hypoxia, autophagy can be stimulated via HIF1a-regulated pathways. c Cells can remove abnormal mitochondria to keep mitochondria maintenance via mitophagy mechanism, and mitophagy mainly dependent on the PINK1-Parkin pathway
Hypoxia stress
Hypoxia in the tumor microenvironment is the most popular phenomenon during cancer progression and it has been considered as a poor prognosis marker for years. Recent reports show that hypoxia-inducible factors are closely related to cancer invasion and progression in metastatic breast cancer [20]. Tumor cells regulate the hypoxia-inducible factor family of transcription factors (HIFs) to adapt to hypoxia stress. HIFs are always over-expressed in multiple cancers and are associated with tumor resistance and poor prognosis [21]. The HIFs consist of three isoforms, HIF-1, -2 and -3. HIF-1 is ubiquitously expressed, while HIF-2 and -3 are only selectively expressed in most mammalian cells. HIF-1 is a heterodimer formed by HIF-1α and -1β. Under the hypoxia condition, the accumulation and nucleus translocation of HIF-1α contribute to the activation of several transcription factors participating in different biological processes [22].
Autophagy could help tumor cells adapt to hypoxia. It has been demonstrated that autophagy could promote angiogenesis of bone marrow-derived mesenchymal stem cells under hypoxia [23]. And autophagy facilitates the invasion of salivary adenoid cystic carcinoma under the condition of hypoxia [24]. Autophagy increases hypoxia-induced IL6 to promote malignant glioma progression [25]. And the chemotherapy sensitivity in hepatocellular carcinoma cells could be reduced by autophagy induced by hypoxia [26]. Hypoxia induces autophagy mainly via the activation of HIF-1α (Fig. 3b). When HIF-1α is activated under hypoxia, it will induce autophagy via directly up-regulating the expression of BNIP3, which will successively lead to the disruption of Beclin1/Bcl-2 complex, resulting in the releasing of Beclin1 to stimulate autophagy. And hypoxia-sustained tumor cells can maintain their vitality by the degradation of p62 in autophagy [27]. Hypoxia also induces autophagy through microRNAs. For instance, miR-155 can target members of mTOR signaling to promote autophagy in several human cancer cells [28]. And miR-301a/b targets N-myc downstream regulated gene 2 (NDRG2) to increasing cell autophagy which contributes to the survival of prostate cancer cells under hypoxia [29]. Hypoxia induces miR210 up-regulation to enhance autophagy and reduces radio-sensitivity in colon cancer cells [30]. It is worth noting that hypoxia can induce or enhance autophagy via several other pathways. ERK1/2, mTOR, unfolded protein response (UPR) and p38/JNK-dependent pathways are also involved in Hypoxia-induced autophagy [31–34]. The highly activated HIF-1α pathway can help tumor cells resist hypoxia due to the rapid proliferation.
Mitochondria damage
Cells could remove abnormal mitochondria to keep mitochondria in maintenance through mitophagy (Fig. 3c). And the selective degradation of damaged mitochondria via autophagy was first reported as mitophagy in the year of 2007 [35]. The elimination of damaged mitochondria starts with the overexpression of BNIP3L. BNIP3L directly interacts with LC3 at the mitochondrial membranes and causes the dissipation of mitochondrial membrane potential [36]. What is more, it has been confirmed that BNIP3 competitively disrupts the formation of BCL-2/Beclin-1complex to induce mitophagy. PINK1 and Parkin are two famous Parkinson’s disease-related genes and also key mitophagy regulators. PINK1 cooperates with Parkin to sustain mitochondrial in maintenance. PINK1 could directly target the mitochondria with its N-terminus. When mitochondria are damaged, the mitochondrial membrane potential will be decreased, resulting in the accumulation and the activation of PINK1. Activated PINK1 could respond to the decrease in mitochondrial membrane potential by recruiting Parkin from the cytosol to the outer mitochondria membrane [37]. Additionally, PINK1 could recruit the autophagy receptors (such as p62, NBR1, NDP52, Tax1BP1) to induce mitophagy [38]. Newly report identified PHB2 as a crucial mitophagy receptor in Parkin-induced mitophagy. PHB2 binds to LC3 through an LC3-interaction region domain upon mitochondrial depolarization and proteasome-dependent outer membrane rupture [39]. Key autophagy factors such as ULK1 [40], Beclin1 [41], ATG5–ATG12–ATG16L [42], VDAC1 [43] are involved in regulating this process. Beclin-1 could activate Parkin and PINK1 to maintain the level of mitophagy and control the process of autophagy at the same time. LC3 and ATG5–ATG12–ATG16L locate the mitochondrial membrane, then form the structural components of the double-membraned cisterns after conjugation. Other crucial autophagy factors, such as p62 and ATG7, play irreplaceable roles in eliminating the ubiquitinated damaged mitochondria by mitophagy [44].
The function of mitophagy is closely related to tumor stage [45]. Mitophagy could be suppressed to a certain extent during cancer progression, resulting in a decrease of removal of the damaged mitochondria which increased the aggregation of tumor-promoting ROS or other tumorigenic mitochondrial signals. But what is noteworthy is that mitophagy could conduce to stress adaptation and survival of established tumors. The key mitophagy modulator BNIP3 could be upregulated to impair anti-angiogenic therapy in xenograft glioma models [46]. Additionally, oncogenic K-Ras could trigger the up-regulation of mitophagy to eliminate dysfunctional mitochondria, contributing to the rapid proliferation of tumors [47].
In established cancer, tumor cells can often respond to stress through autophagy regulation in conditions of energy and nutrients, hypoxia stress and mitochondria damage. Therefore, inhibition of autophagy in established cancer might a promising strategy that prevents malignant progression of tumor cells. Thus, small molecule inhibitors targeted to autophagy will have a good application prospects.
Resistance to necrosis
The necrosis of tumor cells has been a huge barrier to cancer progression. Necrosis has always been considered as the passive and unregulated form of cell death [48]. During necrosis, the increasing levels of ROS and intracellular calcium will eventually lead to cell death [49]. Recent years, scientists have recognized that necrosis could be regulated by certain factors. For instance, RIP1-dependent regulated necrosis exhibits RIP1 activation and can be suppressed by RIP1 inhibitors [50]. Necroptosis is a programmed necrotic cell death. During necroptosis, one signaling is dependent on RIP activation [51]. PARP-1, a nuclear poly (ADP-ribose) polymerase involved in DNA repair, has been reported as a regulator in TRAIL-induced necroptosis [52].
Necrosis could crosstalk with autophagy through multiple cell metabolism and death pathways such as MAPK, AKT, TGFβ and NF-κB pathways [53, 54] (Fig. 4a) and tumor cells can avoid necrosis by inducing autophagy. Lim et al. discovered that the activation of DR4/JNK pathway-mediated autophagy made tumor cells acquire TRAIL resistance to escape from TRAIL-mediated cell death in HepG2 cells [55]. In human lung cancer cells, AGM130 induced slight autophagy to resist necrosis [56]. Autophagy induced by the ring-DIMs and DIM has a cell protective function to resist necrosis in prostate cancer cells [57]. Thus, autophagy could be a protector for cancer cells to escape from necrosis.
Fig. 4.

Autophagy helps cancer cell resistance to necrosis and apoptosis. a Necrosis could crosstalk with autophagy through multiple cell metabolism patterns and death pathways. When damage factors stimulate cancer cells, necrosis can be initiated mainly through RIP-dependent pathways. To escape from necrosis, cancer cells initiate autophagy via MAPK, AKT, TGF-β and NF-κB pathways. b Two classical apoptosis pathways. Death receptor signaling mainly relies on the activation of death receptors including Fas, TNFα, and TRAF2 by their respective ligands. And the induction of mitochondrial control of apoptosis mainly relies on Bcl-2 family proteins. Blue: anti-apoptosis Bcl-2 family proteins; yellow: pro-apoptosis Bcl-2 family proteins. c Apoptosis could directly crosstalk with autophagy through several key proteins. d The strategies of autophagy positively or negatively regulate apoptosis
Resistance to apoptosis
Apoptosis, also known as type I programmed cell death, is the most widely studied form of cell death. Caspases, a family of cysteine proteases, are the central executor of apoptosis. Death receptor signaling and mitochondrial control of apoptosis are the two classical mechanisms of apoptosis (Fig. 4b). Death receptor signaling mainly relies on the activation of death receptors and their respective ligands. The induction of mitochondrial control of apoptosis mainly relies on Bcl-2 family proteins [58]. Since it plays a crucial role in cell death, apoptosis is a big threat to the survival of cancer cells and targeting apoptosis for developing the cancer therapy has been concerned and carried out for years.
Apoptosis could directly crosstalk with autophagy through several key proteins (Fig. 4c) and autophagy could regulate apoptosis by means of some strategies (Fig. 4d). In many cases, autophagy indeed can help cancer cell escape from apoptosis or at least decrease the degree of apoptosis. Wei et al. discovered that autophagy could play a positive role in promoting the resistance to apoptosis which induced by photodynamic therapy in colorectal cancer stem-like cells [59]. In 2011, it was reported that autophagy was involved in protecting breast cancer cells from apoptosis induced by epirubicin and promoting epirubicin-resistance [60]. Meanwhile, ovarian cancer cells were more susceptible to cisplatin-induced apoptosis when autophagy was down-regulated [61]. Autophagy inhibition could enhance apoptosis in different cancer cells, including breast cancer and lung cancer cells.
Autophagy resists to apoptosis through several pathways. Autophagy could induce degradation of apoptotic components including activation of caspase-8 11 [62] and contribute to the degradation of damaged mitochondria to prevent defective mitochondria-mediated apoptosis. The elimination of ROS by autophagy could block the activation of apoptosis-related factors such as AIF (apoptosis-inducing factor) [63]. Key autophagy regulator Beclin1 could inhibit tBid translocation to the mitochondrial membrane, resulting in apoptosis reverse [64]. In addition, apoptosis could be inhibited by autophagy-related releasing of HMGB1 [65]. Thus, autophagy has the function of supporting tumor cell to survive by apoptosis antagonist. Collectively, autophagy plays a significant role in maintaining tumor cell surviving by stress resistance, necrosis inhibition, and apoptosis antagonist.
Tumor-suppressive role of autophagy in cancer
Autophagy can be tumor-suppressive role by preventing cancer initiation and progression. Several recent studies have demonstrated that autophagy can inhibit malignant transformation in a variety of models by different mechanisms such as maintaining genomic stability, as well as reducing harmful mutations and carcinogenic damage [14]. Autophagy can also inhibit tumor metastasis through a multitude of mechanisms, which we discussed in our previous review in 2016 [66]. Here, we mainly focus on cytotoxic and cytostatic autophagy, anti-inflammation and its synergistic effect in immunotherapy.
Cytotoxic and cytostatic autophagy in cancer
Cytotoxic and cytostatic autophagy are closely associated with grow inhibition and cell death which could increase sensitivity to cancer therapy. Cytotoxic autophagy is the form of autophagy which promotes cell death when induced, and the cell death may be associated with subsequent apoptosis or reduced sensitivity to therapy when blocked. Cytostatic autophagy is the form of autophagy which can mediate growth inhibition, survival reducing or association with senescence [67]. Of all the cytotoxic and cytostatic autophagy, we mainly focus on autophagic cell death and autophagy-dependent cell death in cancer.
Autophagic cell death in cancer
Autophagic cell death or type II cell death is independent of apoptosis or necrosis, which is mediated by autophagy and also can be blocked by autophagy inhibition [68]. Autophagic cell death can be initiated through several factors and pathways (Fig. 5), of which AKT-mTOR pathway, Vps34 complex and p53 are widely studied.
Fig. 5.

Key signaling pathways in autophagic cell death. Several growth factors signaling pathways are involved in cancer progression and have close relationships with autophagic cell death. Key growth factor signaling such as EGFR, Akt, MAPK/ERK signaling can negatively regulate autophagic cell death by inhibiting key autophagy factors such as Beclin-1 and AMPK. BNIP3, Bax, Bcl-2 can also participate in autophagic cell death modulation by disturbing Beclin1–Bcl-2 complex. Tumor suppressor p53 can facilitate autophagic cell death directly through beclin1 activation
AKT-mTOR pathway
Autophagic cell death is always trigged through AKT-mTOR inhibition. In MCF7 cells, PI3K-AKT-mTOR-dependent autophagic cell death is involved in enhancing breast cancer cells sensitivity to fulvestrant and tamoxifen [69]. Autophagic cell death induced by carnosic acid in HepG2 cells resulted from Akt/mTOR inhibition [70]. Autophagic cell death can improve the sensitivity of apoptosis-resistant cancer cells. mTOR dependent autophagic cell death contributes to cell death induced by liensinine and dauricine in multiple apoptosis-resistant cells [71].
Vps34 complex
The Vps 34 complex is also the key regulator of autophagic cell death. Up-regulation of Beclin-1 expression is significant in the JNK- and XAF1-mediated autophagic cell death [72, 73]. Oncogenic Ras-induced up-regulation of autophagy regulator Beclin-1 could promote autophagic cell death which threatens the survival of cells [74]. And sorafenib was reported to induce autophagic cell death through Beclin1 activation in hepatocellular carcinoma cells [75].
p53
p53 is one of the most famous tumor suppressors and it has an outstanding role in promoting autophagic cell death. It has been reported that radiation induces autophagic cell death through the activation of p53-DRAM in breast cancer cells [76]. c-Met inhibitor SU11274 induces autophagic cell death in human lung cancer A549 cells via the p53-ERK-Beclin1 signaling [77]. Autophagic cell death could be induced by p53/AMPK up-regulation after Fangchinoline treatment in human hepatocellular carcinoma cells [78].
In many cases, autophagic cell death is widely involved in cancer therapy, and autophagic cell death modulated by small target molecules has been a promising strategy for decreasing the side effects of chemotherapy.
Autophagy-dependent cell death in cancer
Autophagy-dependent cell death is the type of cell death when it is proven that autophagy is a pre-requisite for the occurrence of cell death, but it is not proven that autophagy mechanistically mediates the switch to cell death [79]. Here, we mainly discuss the autophagy-dependent apoptosis. Autophagy and apoptosis occur in the same cell, and under most circumstances, autophagy precedes apoptosis. In this context, autophagy is sensitive to cellular stress, especially if the level of stress is not lethal to initiate apoptosis. When autophagy can resist the stress, it would inhibit apoptosis to prevent cell death. When autophagy cannot prevent cell death, autophagy may activate apoptosis. It has been confirmed that key autophagy proteins are involved in the induction of apoptosis. Over-expression of autophagy-related genes such as Atg3, Atg4, Atg12, and Atg8 could activate apoptosis through specific signaling pathways [80]. Calpain-mediated Atg5 cleavage generates an ATG5 fragment which could be transported to the mitochondrial membrane to initiate the releasing of Cytochrome C, which leads to the loss of MMP and ultimately mitochondrial apoptosis [81]. ATG12 could bind to anti-apoptosis protein Bcl-2 to promote mitochondrial apoptosis [82]. And c-src could stimulate apoptosis via the activation of caspase-9 [83]. Furthermore, Beclin1 could be regulated by ser/thr kinase, such as DAPK, JNK, and AKT to regulate apoptosis [84].
Except for its suppressive role of autophagy on apoptosis we mentioned above, autophagy does promote apoptosis in many cases. Autophagy could effectively enhance apoptosis in human breast cancer cells after oridonin treatment [85]. LC3 silencing could abolish activation of apoptosis in A549 cells after cisplatin treatment [86]. In TPC-1 cells, the block of autophagy by ATG7 siRNA could desensitize the cells to apoptosis induced by TRAIL [87]. Since the significant role of apoptosis in cancer therapy, modulating autophagy-dependent apoptosis could be another promising adjuvant therapy for cancer.
Anti-inflammation
Inflammation is an important host response to homeostasis imbalance. It plays vital roles in host defense, tissue remodeling, metabolism regulation and cancer development [88]. Inflammatory conditions promote cancer, on the one hand, by promoting oxidative stress and cancer-causing mutations. And on the other hand, inflammation aggregate in tumor microenvironment promotes tumor progression. Inflammation contributes to the maintenance of the cell viability and promotion of angiogenesis, metastasis, insensitive to immune responses and so on [89]. Recently, tumor-associated inflammation has become a potential prognostic tool in some type of cancers.
During inflammation in cancer, autophagy could resist inflammation to control the tumor process (Fig. 6a). First, autophagy could rapidly remove dead cells to prevent unwanted inflammation [90]. It was reported that autophagy-deficient Atg5−/− embryos were susceptible to plentiful inflammation and unable to remove apoptotic cells [91]. Second, autophagy could also remove the damaged mitochondria, which leads to the decrease in the release of inflammation activators such as ROS and mitochondrial DNA thereby resist inflammation. Additionally, autophagy could eliminate the aggregation of inflammasome structures to inhibit pro-inflammatory responses [92]. The most important effect of autophagy on inflammation is to inhibit the inflammasome activation and IL-1β release. Inhibition of autophagy shows higher IL-1β production and the promotion of autophagy presents lower IL-1β production [93]. Lacking ATG16L1 presents higher IL-1β and IL-18 levels in Mice [94]. Autophagy inhibits IL-1β and IL18 production through decreasing ROS release, first. Then autophagy hampers the cleavage of pro- IL-1β and pro-IL18. Finally, autophagy thoroughly removes pro- IL-1β proteins. Autophagy could regulate the activation of caspase1 through regulating NLRP3 inflammasome [95]. Autophagy could inhibit necrosis to prevent the release of inflammatory molecules, such as ATP/UTP, uric acid, HMGB1 and several damage-associated factors [96]. Given the close relationship between autophagy and inflammation, a lot of therapies targeting autophagy-modulating are on its way and some certain achievement have been made, of which immunotherapy is now an emerging and impressive one.
Fig. 6.

Autophagy, inflammation and immunogenic cell death in cancer. a Autophagy resists inflammation to decrease cancer progression. Autophagy inhibits inflammation mainly in four ways. (1) Resistance to necrosis which decreases the production of inflammation factors. (2) Promotion of the degradation of dead cells to remove unwanted inflammation. (3) Promotion of the degradation of damaged mitochondria. Damaged mitochondria produce a large amount of mtDNA and ROS which could induce inflammation through IL-1β and IL-18 activation. (4) Promotion of the degradation of inflammasome and IL-1β. b Autophagy plays a positive role in immunogenic cell death. Several chemotherapeutic agents could induce the autophagy-dependent release of tumor antigens by tumor cells and will lead to the maturation and the activation of antigen-presenting cells. Autophagy in antigen-presenting cells can promote antigen presentation by both MHC class II and I molecules thus initiating immunogenic cell death. Autophagy can also promote the survival of activated T cells. The inhibitory receptors CTLA-4 and PD-1 will limit the activation of CD8+ T cells. Immune checkpoints inhibitors inhibit the inhibitory receptors thus promoting effective CTL-mediated tumor eradication
Autophagy in immunotherapy
Autophagy can stimulate tumor antigen cross-presentation [97], which provides another potential mechanism of autophagy in immunotherapy. Nowadays, immunotherapy has become more and more promising in the cancer treatment, and several cancer immunotherapies have been developed, including vaccines, chimeric antigen receptor (CAR)-expressing T cells, bispecific antibodies, and immune checkpoint inhibitors [92, 98]. For instance, inhibitors of programmed death 1 (PD-1) show good therapeutic activity for a variety of cancers [99]. However, cancer cells could escape from immune destruction by various ways, which results in tumor progression.
Recent studies suggest that autophagy as an important regulator of cellular immune response is closely related to the modulation of immunotherapy (Fig. 6b). Some studies show that the stimulation of autophagy could enhance cancer immunotherapy. Autophagy could promote antigen-specific T cell responses by potentiating the processing and presentation of tumor antigens [100], which is a vital requisite for immunogenic cell death (ICD) and autophagy enhancers may increase the efficacy of cancer immunotherapy [101]. It was reported that the knockout of autophagy genes (ATG5, ATG7, and BECN1) resulted in a significant decrease of chemotherapy-induced immunosurveillance owing to the inhibition of releasing ATP in several human and murine cancer cell lines, which could be reversed by addition of ecto-ATPase inhibitors [102]. Several Oncolytic viruses (OVs) have been utilized in immunotherapy for several cancers. During the process, autophagy stimulates immune responses by promoting antigen presentation [103]. Conversely, autophagy is also regarded as a pro-survival mechanism in some cases. Autophagy was activated after targeting CD47 by SIRPαD1-Fc, which resulted in immunotherapy drug resistance by inhibiting the Akt/mTOR signaling pathway in non-small cell lung cancer [104]. It also impairs cancer immunotherapy by inhibiting iNKT cell activation which plays a key role in cancer immunotherapy [105]. PD-L1/PD1 engagement could induce autophagy in nearby T cells, resulting in decrease effect of immunotherapy and tumor resistance [106].
Cancer immunotherapy has shown great promise for several cancers, and most studies demonstrated autophagy did synergistic in immunotherapy. But we should notice the role of autophagy in cancer immunotherapy remains controversial and the mechanism remains to be investigated. When we use autophagic modulators to improve immunotherapy, a lot of factors should be considered, such as tumor type, staging and immunotherapy agents. Nevertheless, we believe that targeting autophagy is an increasingly attractive strategy for immunotherapeutic.
As mentioned above, there are intractable problems using autophagy regulators to improve cancer therapy: should we try to enhance or inhibit autophagy? When to enhance and when to inhibit it? And how to judge the correct situation? Although these problems are difficult and frustrating, we believe that the proper regulation of autophagy can bring new hope for cancer treatment, and small-molecule compounds targeting autophagy are on their way.
Targeting autophagy by small-molecule compounds in cancer therapy
The step-by-step autophagy pathway provides potentially druggable targets to regulate autophagy. Current efforts in the clinic of autophagy-modulating are mainly focused on inhibitors of mTOR and inhibiting the lysosome using chloroquine (CQ) or the related hydroxychloroquine (HCQ). Other autophagy regulators such as ULK1, ATG4B and VPS34, have been reported to be new druggable targets as small-molecular compounds targeting them showed potential anti-tumor activity. Here, we mainly focus on these small-molecule compounds that can directly target autophagy-related proteins or autophagy process in cancer cells (Fig. 7; Table 1).
Fig. 7.

Small-molecule compounds directly target autophagy-related proteins or autophagic process in cancer. Several autophagy-targeted small molecular compounds have been discovered in cancer therapy. mTOR inhibitors and ULK1 activator can promote autophagy induction, the ATG4 activator can promote vesicle elongation and completion to up-regulate autophagy. ULK1 inhibitors inhibit autophagy induction, the Vps34 inhibitors hinder vesicle nucleation, the ATG4 inhibitors inhibit vesicle elongation and completion, and lysosome-targeted inhibitors inhibit the normal function of lysosomes to inhibit autophagy. The compounds in green frame represent autophagy activators and in red represent autophagy inhibitors
Table 1.
Small-molecule compounds targeting autophagy-related proteins or autophagic process in cancer therapy
| Compound | Target | Structure | Cell type | Reference |
|---|---|---|---|---|
| Rapamycin | mTORC1 |
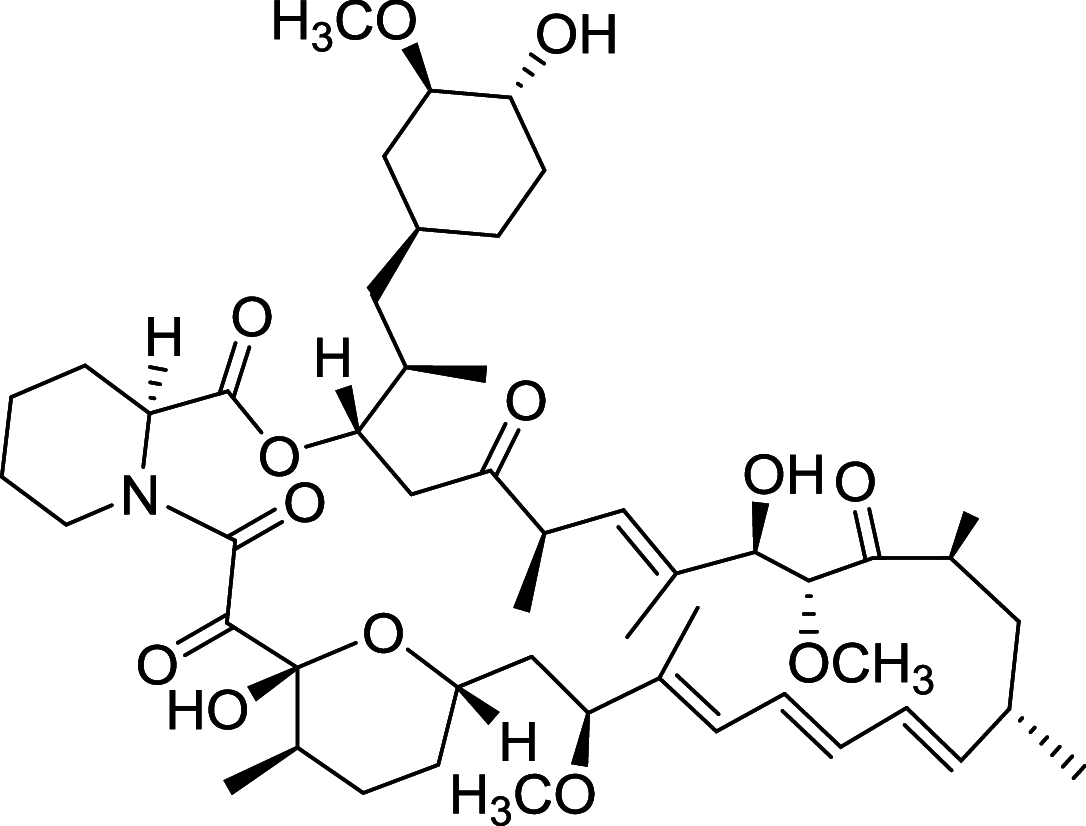
|
HEI193,08031-9,ESC-FC1801 | [108, 153] |
| Temsirolimus | mTORC1 |
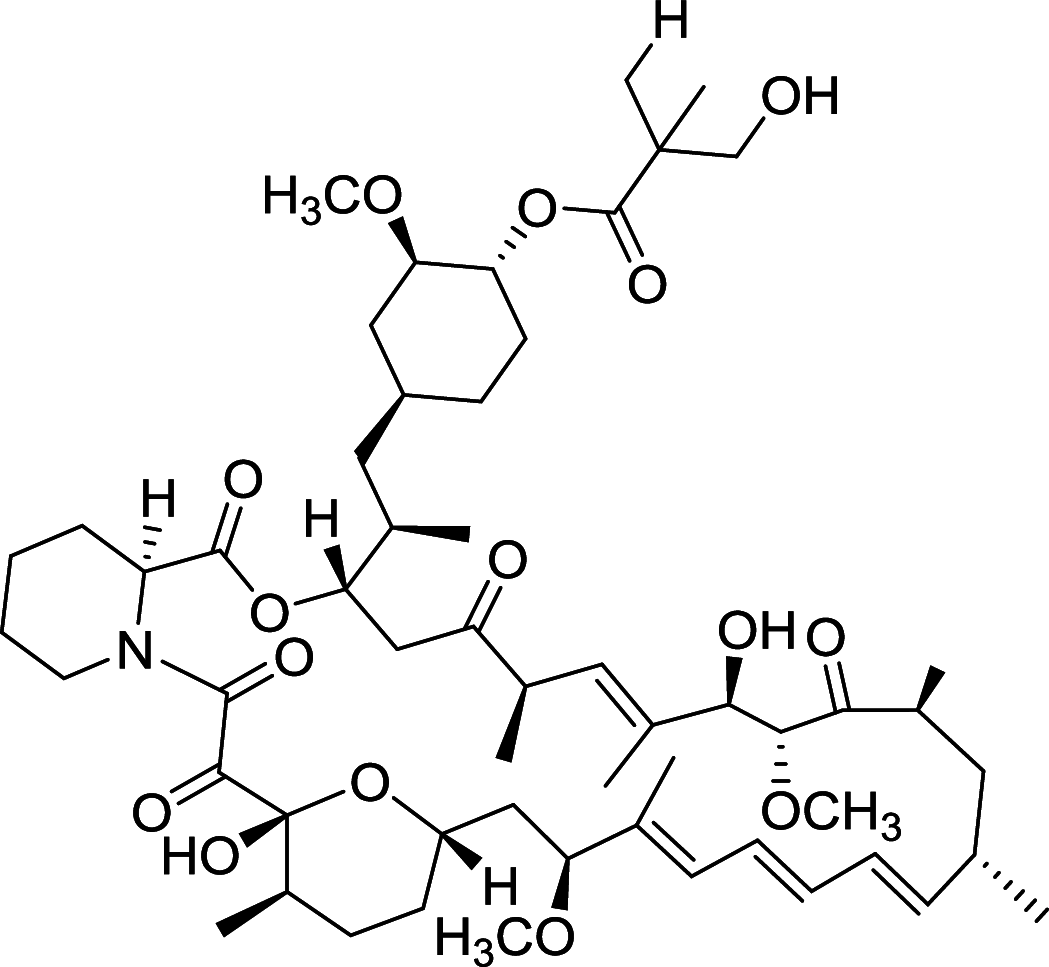
|
MDA-MB-468, MDA-MB-435, MDA-MB-231, MCF-7, T-47D, SKBR-3, BT-474 | [108, 109, 154] |
| Everolimus | mTORC1 |
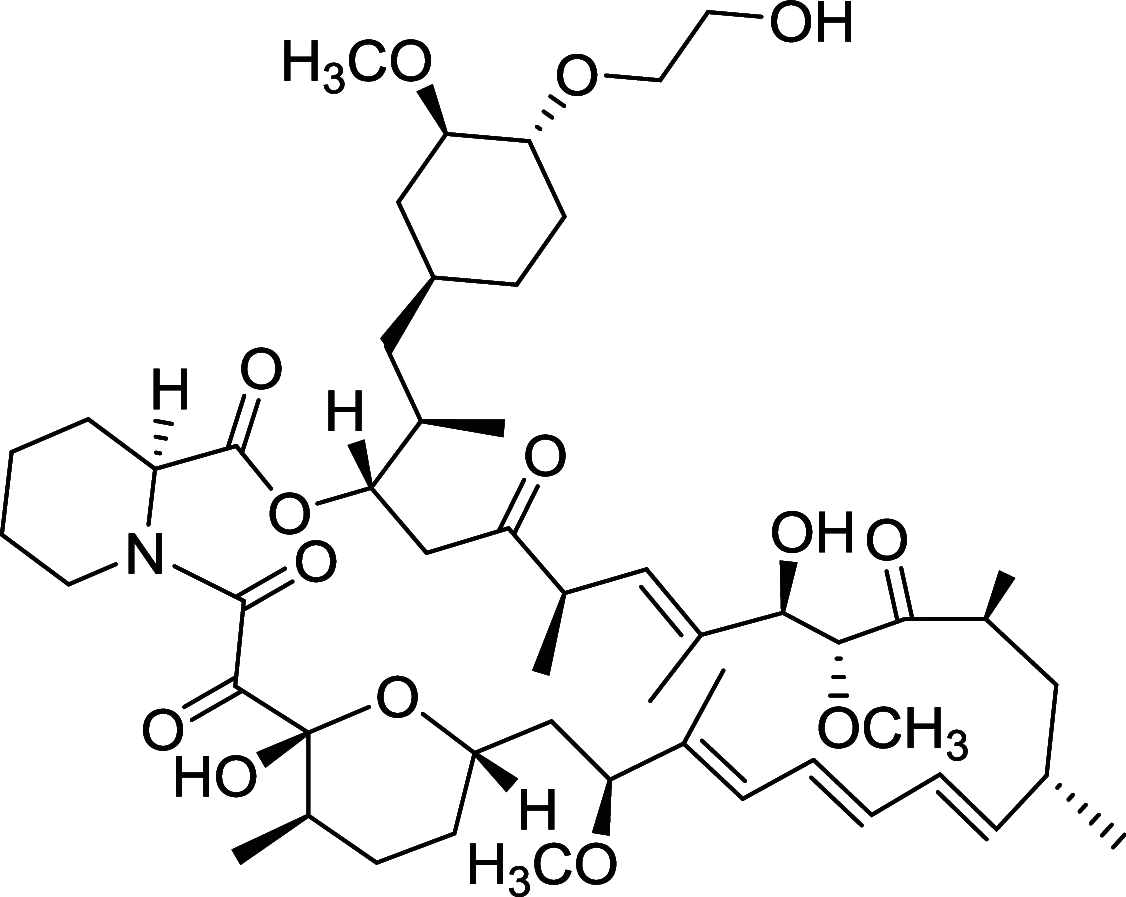
|
HEY, SKOV3, OVCAR5, IGROV1, OV433 | [108, 155] |
| Ridaforolimus | mTORC1 |
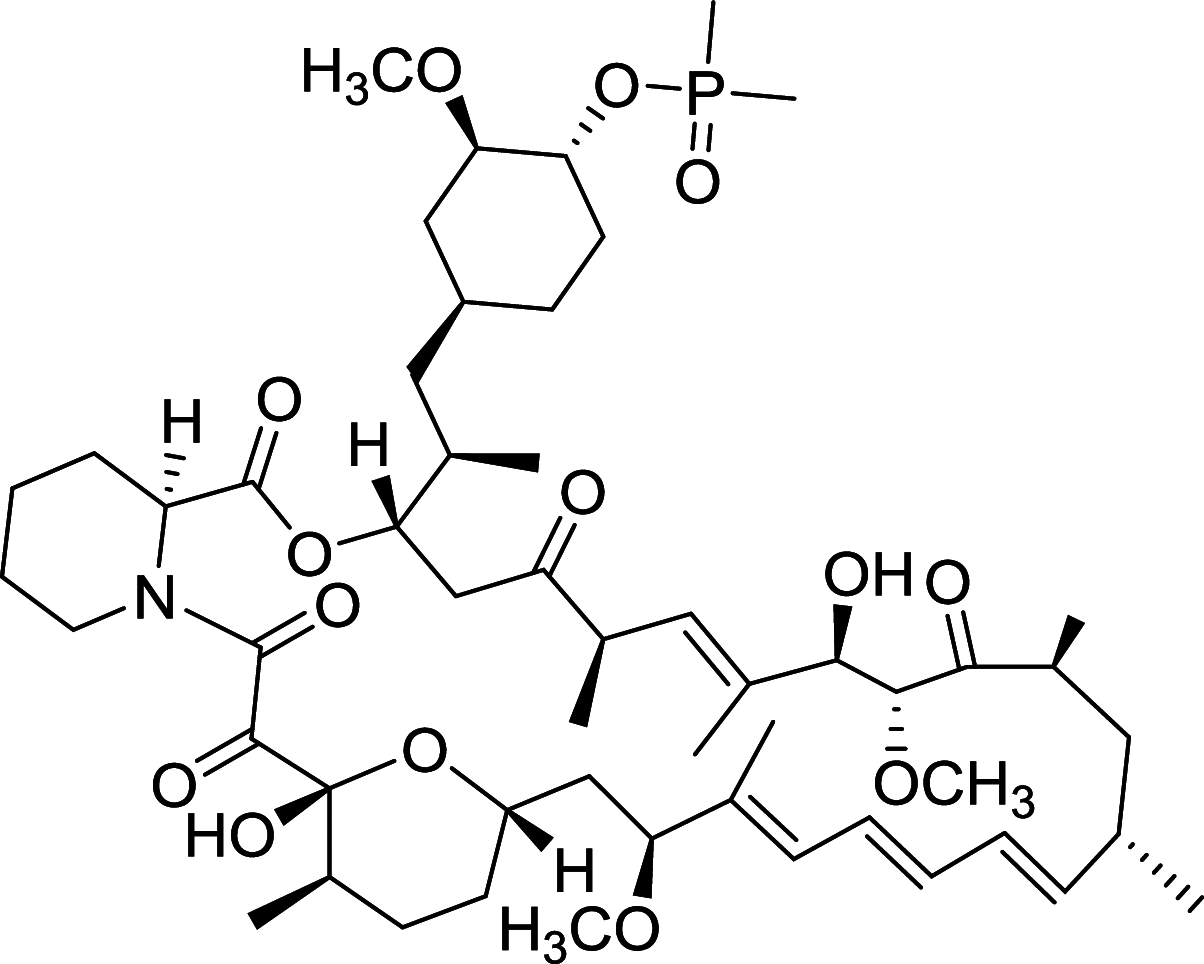
|
MCF-7 | [156] |
| PI103 | mTOR/PI3K |
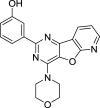
|
PC-3, DU145, LNCaP | [108, 157] |
| PI540 | mTOR/PI3K |
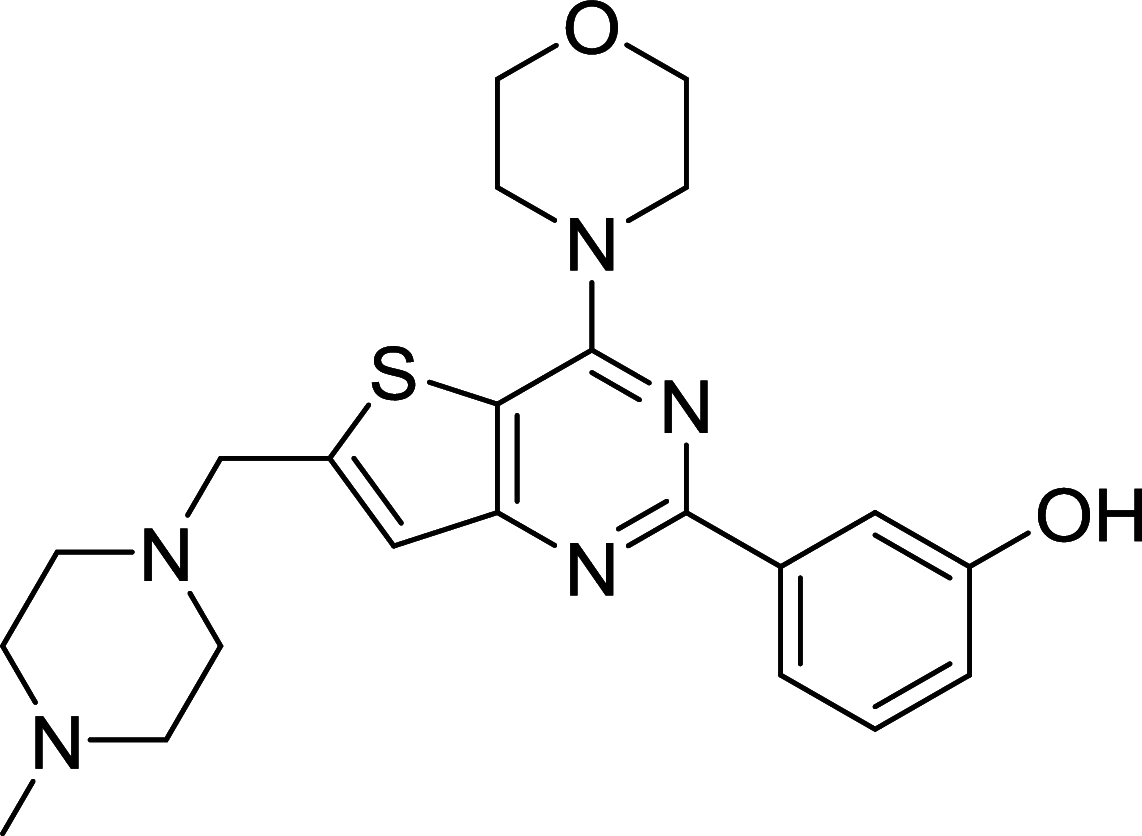
|
– | [112, 157] |
| PI620 | mTOR/PI3K |
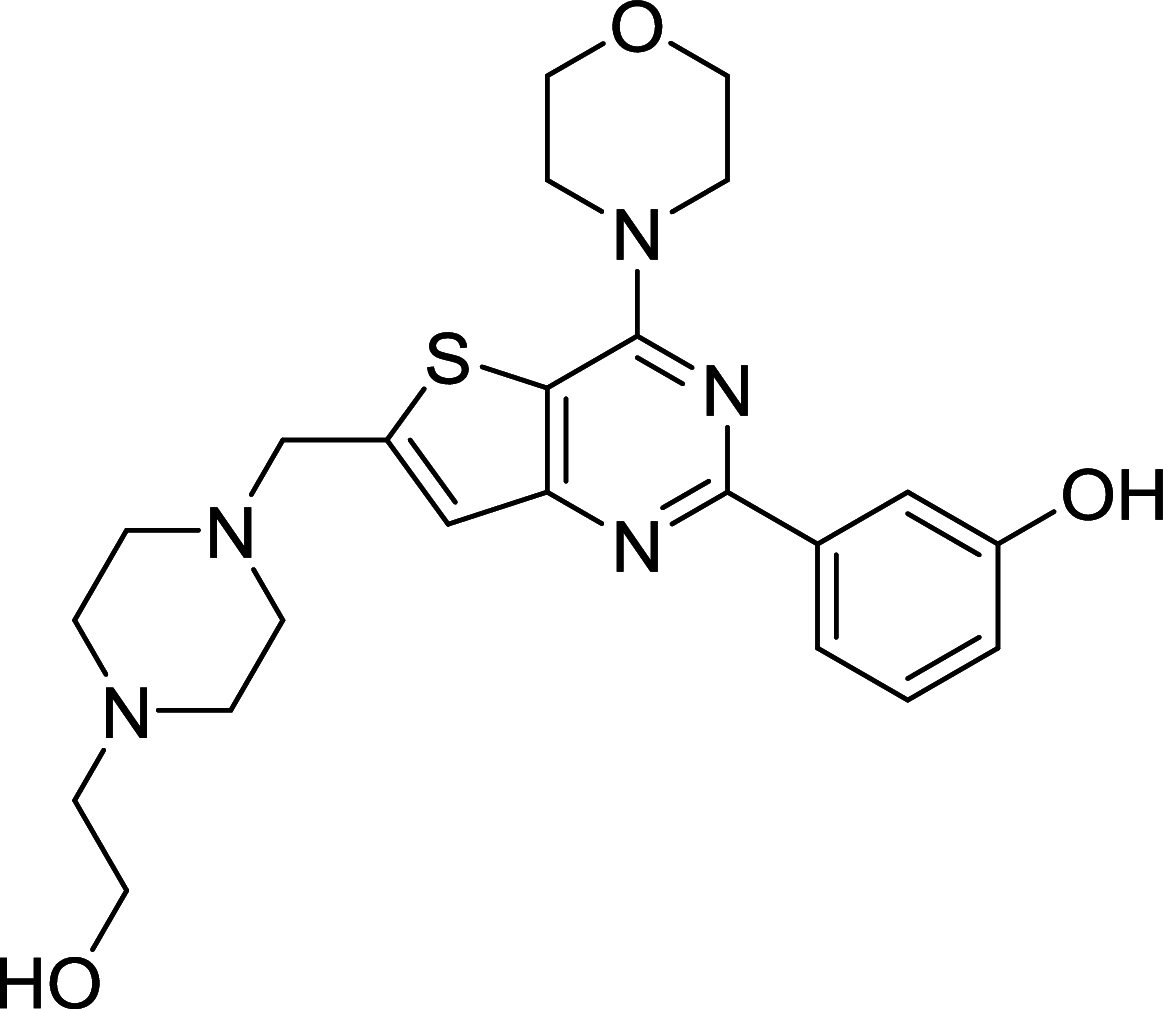
|
– | [112, 157] |
| NVPBEZ235 | mTOR/PI3K |
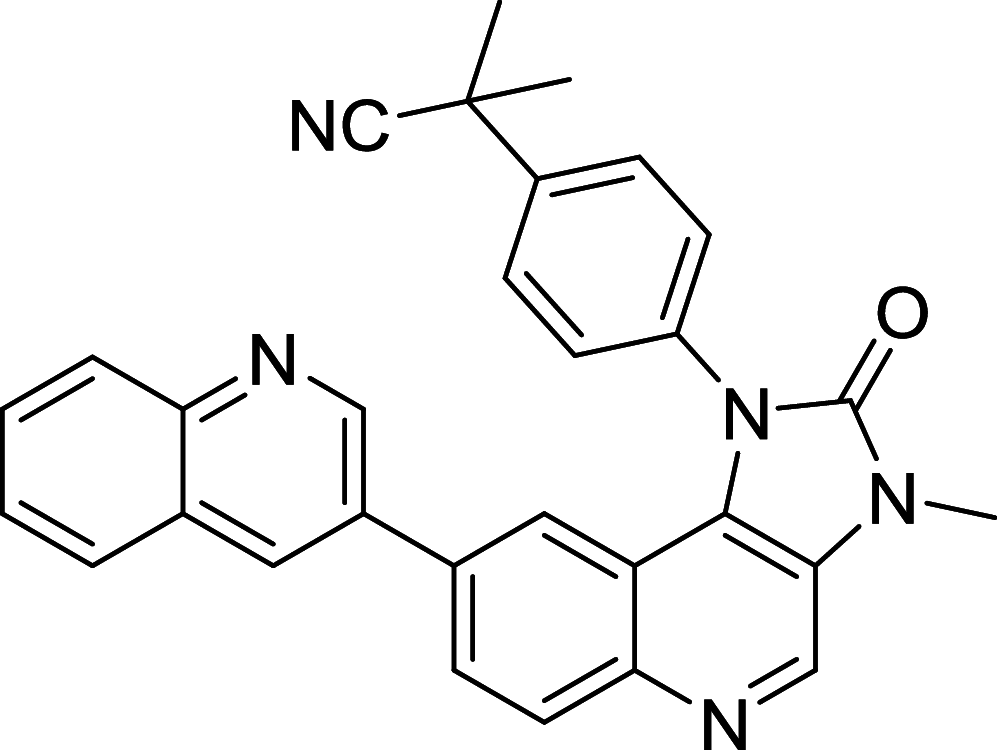
|
NCI-N87, SNU216, MCF-7, BT47, | [158, 159] |
| GSK2126458 | mTOR/PI3K |
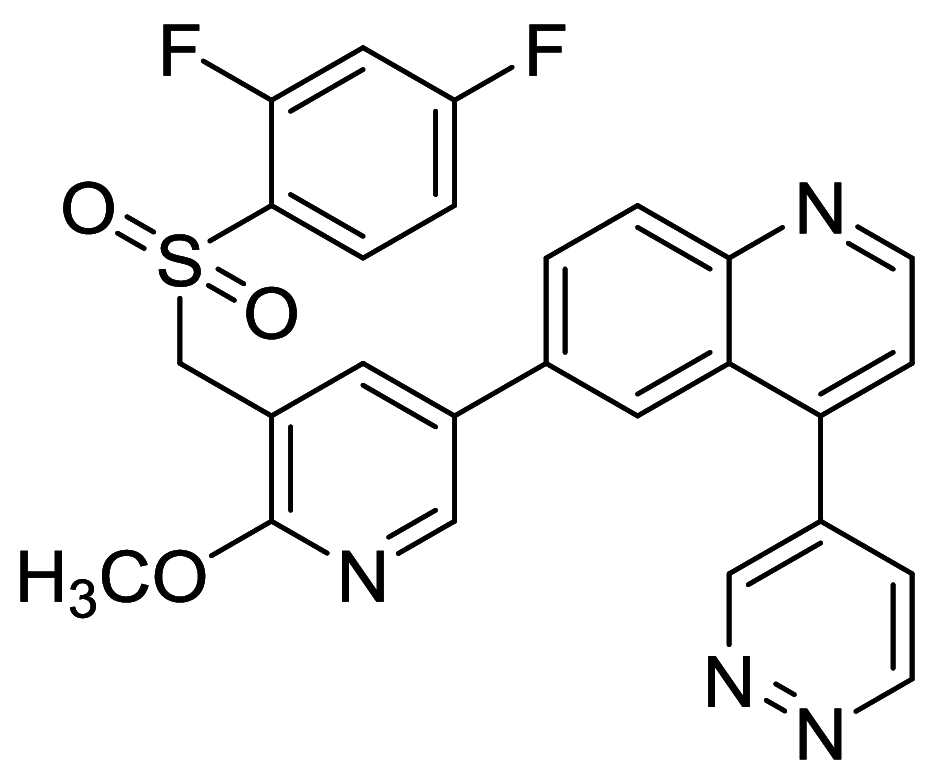
|
CNE-1, CNE-2, 5-8F, 6-10B | [108, 160] |
| BGT226 | mTOR/PI3K |
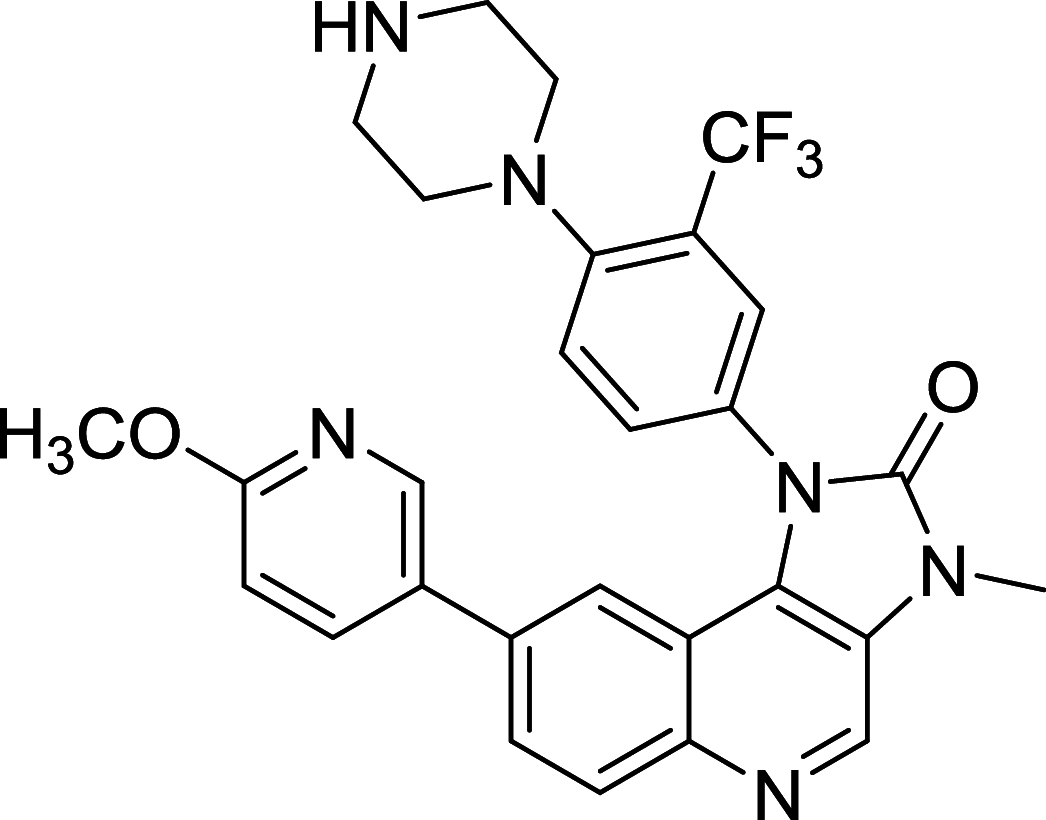
|
Hep3B, HepG2, SNU449, SNU475 | [108, 161] |
| XL765 | mTOR/PI3K |
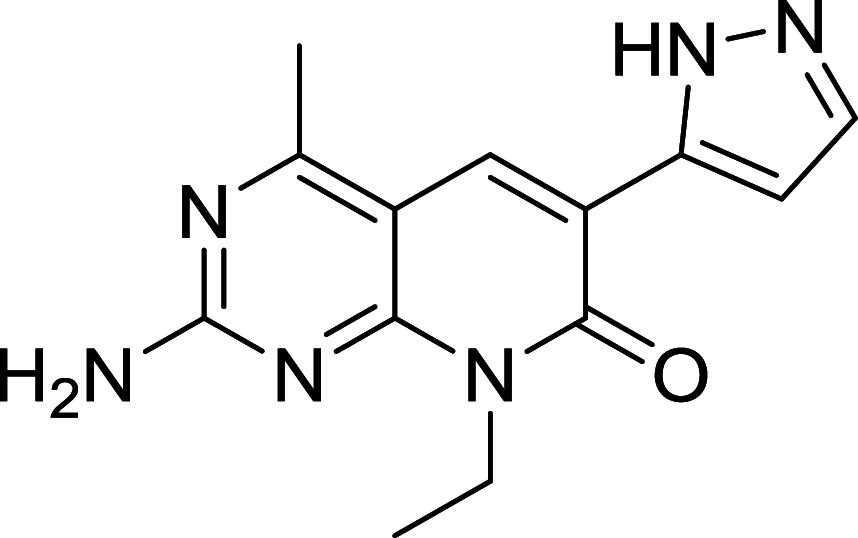
|
CLL | [108, 162] |
| GDC0980 | mTOR/PI3K |
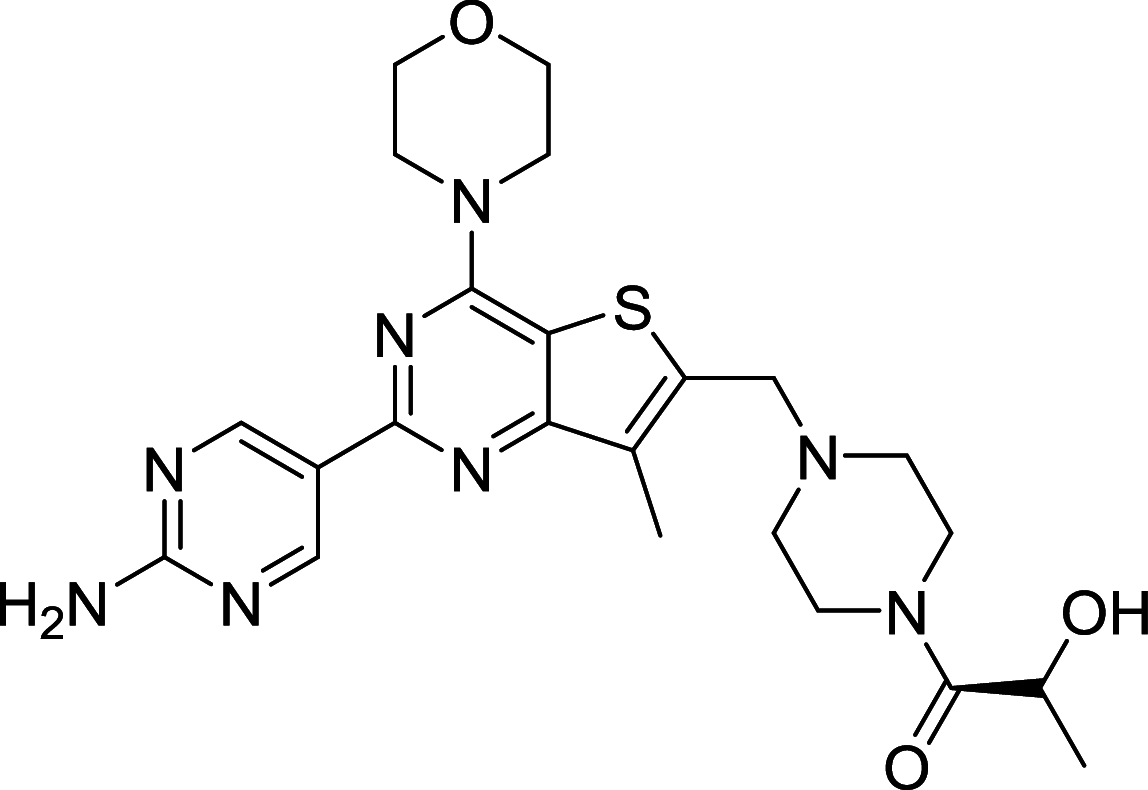
|
LNCaP, Vcap, 22Rv1 | [108, 163] |
| SF1126 | mTOR/PI3K |
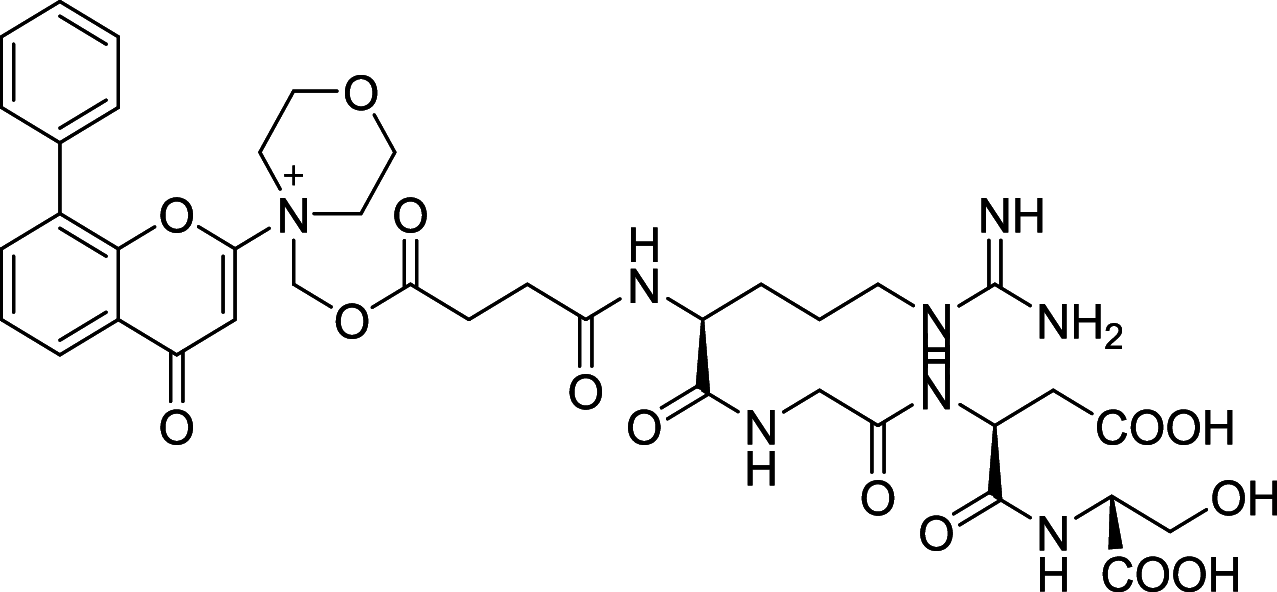
|
Hep3B, HepG2, SK-Hep1, Huh7 | [108, 164] |
| PP242 | mTOR |
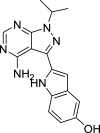
|
OVCAR-3 | [114, 165] |
| AZD8055 | mTOR |
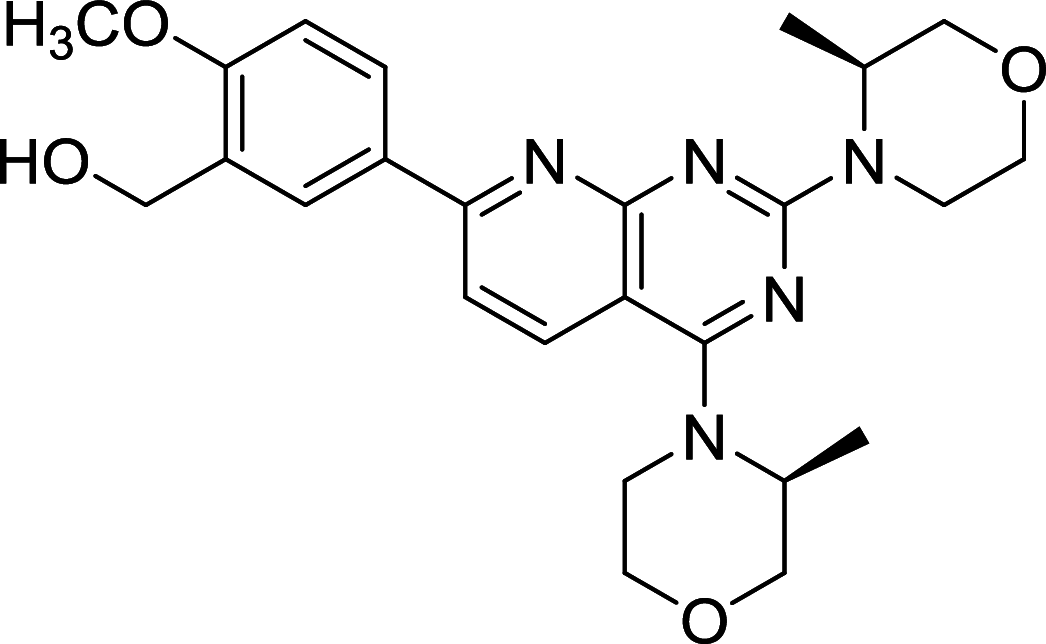
|
L3.6pl, MV4-11 | [117, 166, 167] |
| AZD2014 | mTOR |
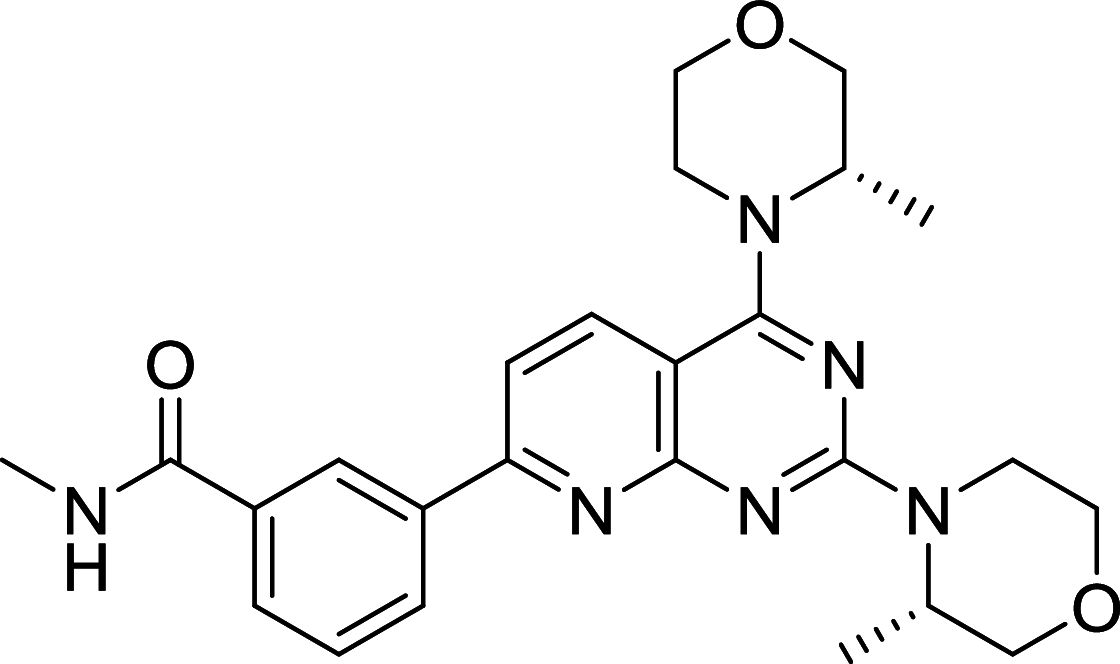
|
MCF7, SCC4, SCC25, HCCLM3, Huh-7, SMMC-7721, HepG2, HL-7702 | [117, 168–170] |
| OSI027 | mTOR |
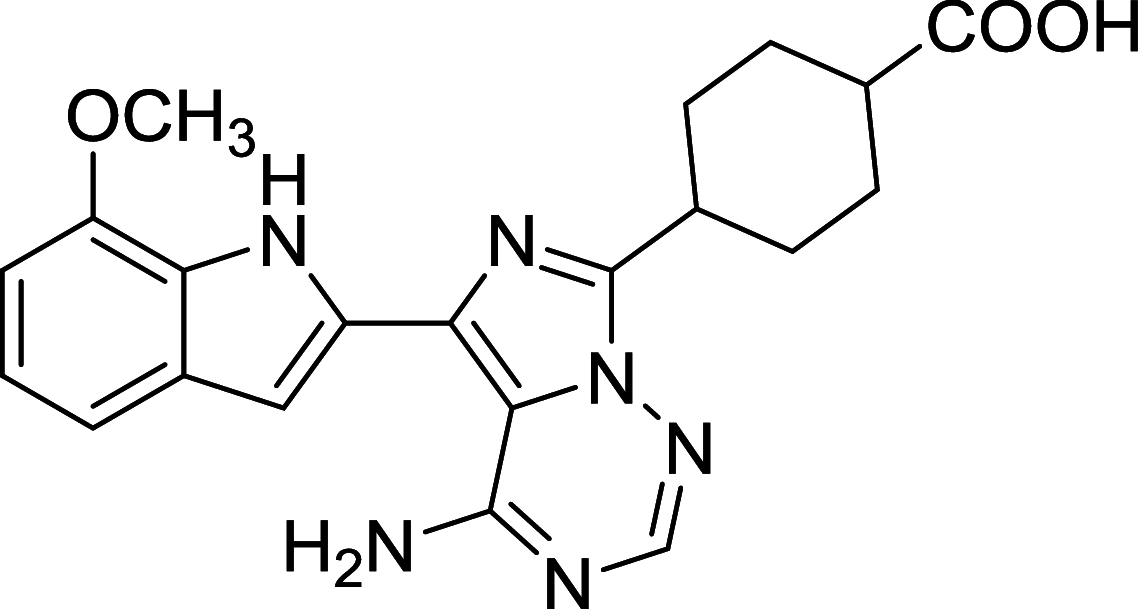
|
Panc-1, BxPC-3, CFPAC-1 | [118, 171] |
| INK128 | mTOR |
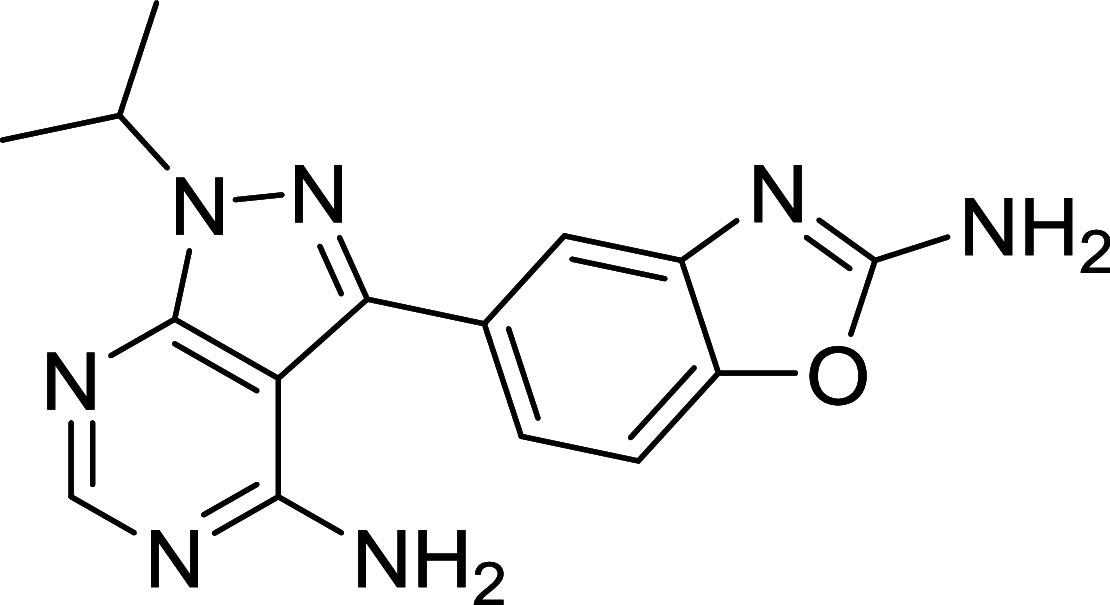
|
CHLA-255, SK-N-AS, SH-SY5Y, IMR32, LA–N-6, CHLA-255, Miapaca-2, Panc1, PSN1, MRC9, RAW264.7,MCF7, SUP-B15,MCC-2, MCC-3, MCC-5 | [115, 116, 172–177] |
| Palomid 529 | mTOR |
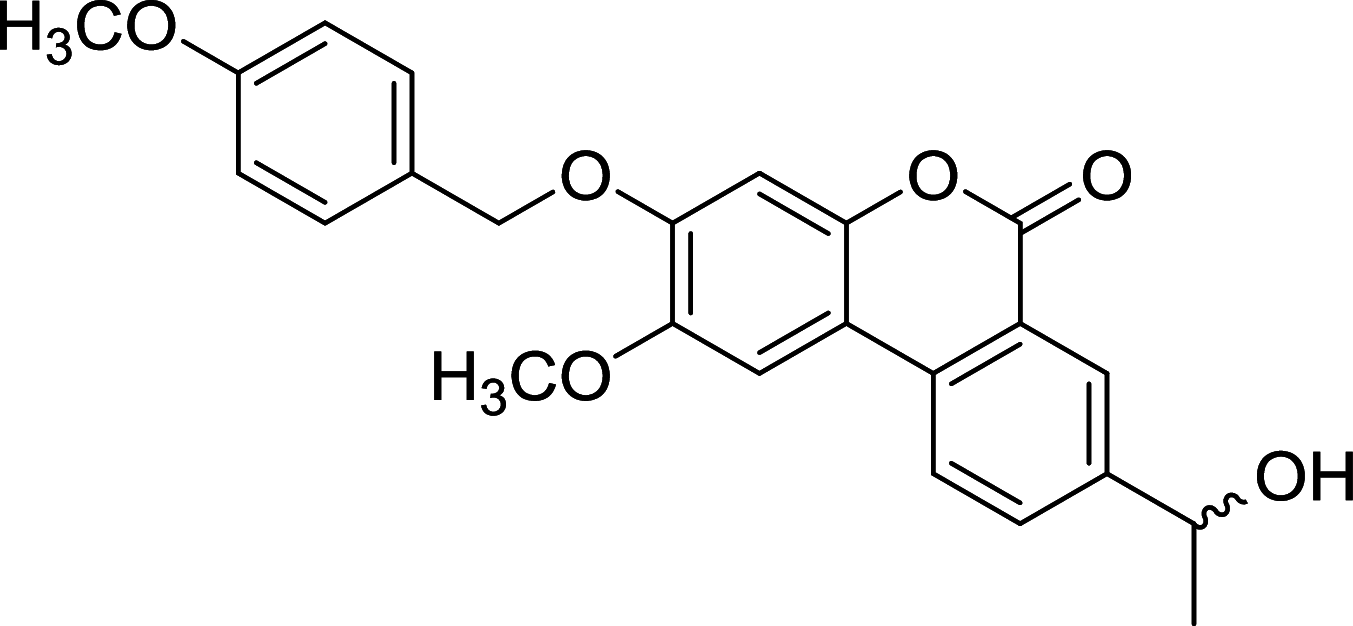
|
CWR22R-2152, CWR22R-2272, CWR22R-2274, LnCaP-104S, LnCaP-104R1, C4-2B, DU145, PC3, VCaP, DuCaP | [119, 126] |
| LYN-1604 | ULK1 |
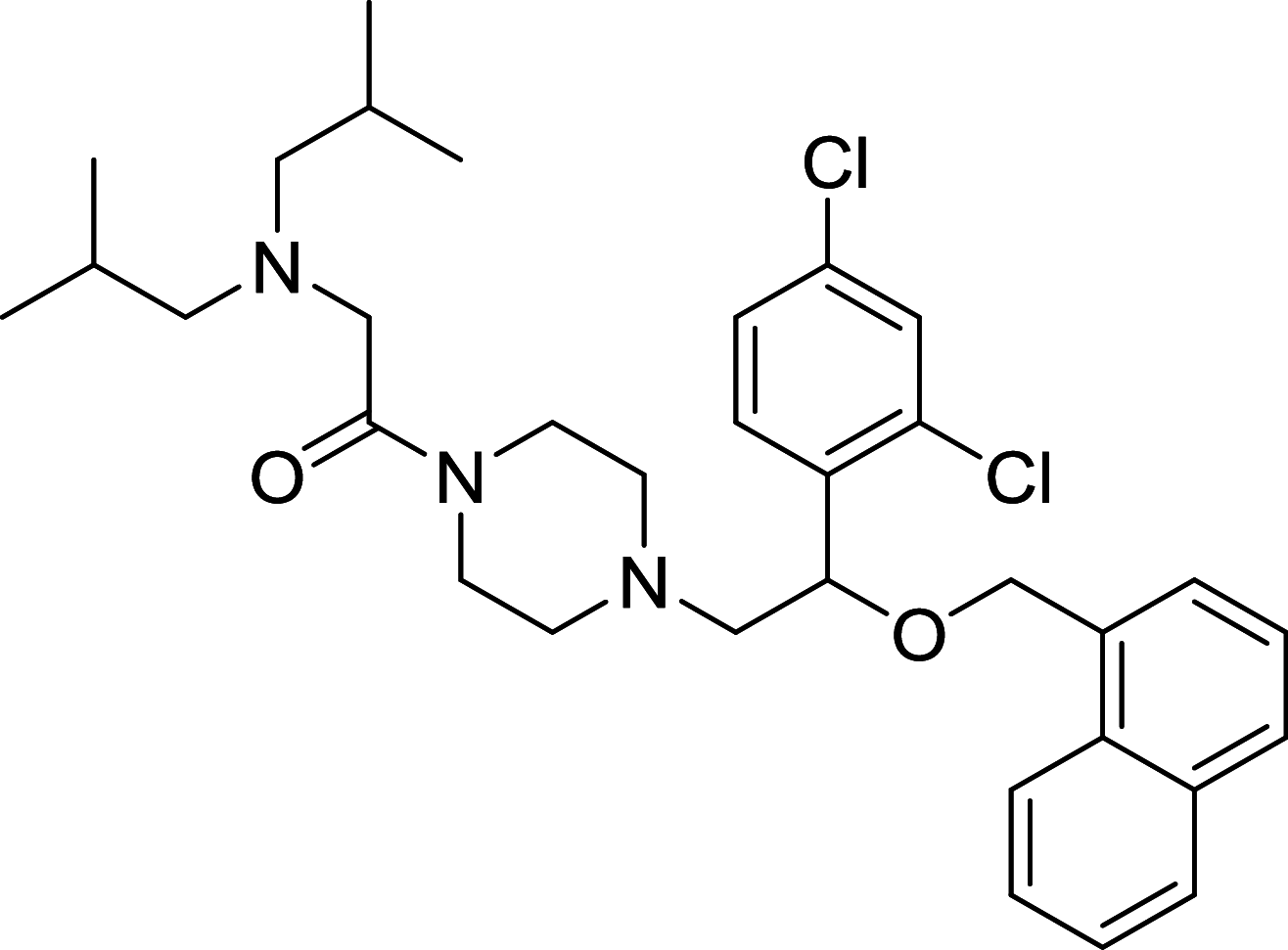
|
MDA-MB-231 | [123, 124] |
| SBI-0206965 | ULK1 |
|
A549 | [125] |
| Compound 6 | ULK1 |
|
– | [126] |
| MRT67307 | ULK1 |
|
– | [127] |
| MRT68921 | ULK1 |
|
– | [127] |
| SAR405 | Vps34 |
|
HeLa, H1299 | [130] |
| PI3KD/V-IN-01 | Vps34 |
|
AML, CLL | [134] |
| VPS34-IN1 | Vps34 |
|
U2OS | [131] |
| PIK-III | Vps34 |
|
H4, HeLa, PSN-1, Panc10.05, RKO | [132] |
| Flubendazole | Atg4 |
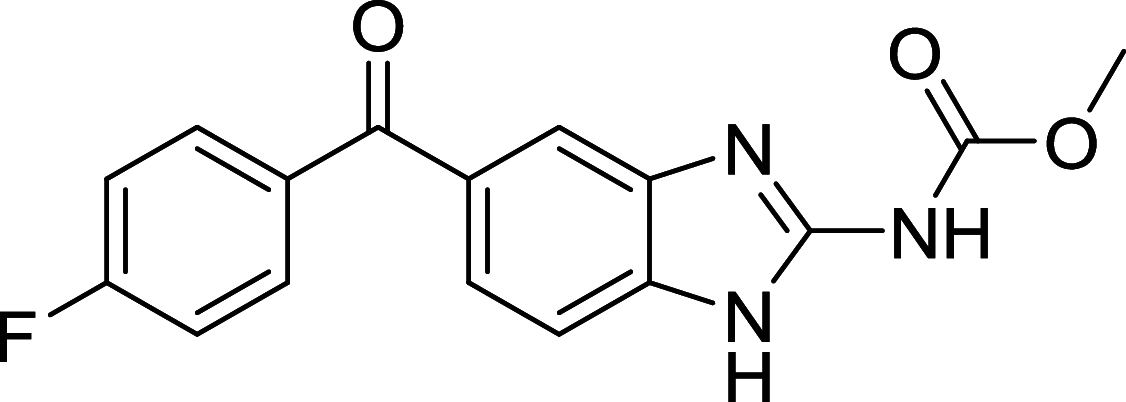
|
MDA-MB-231 | [135, 136] |
| NSC185058 | Atg4 |
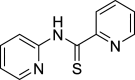
|
293T, HuH7, Saos-2 | [137] |
| Chloroquine | Lysosome |
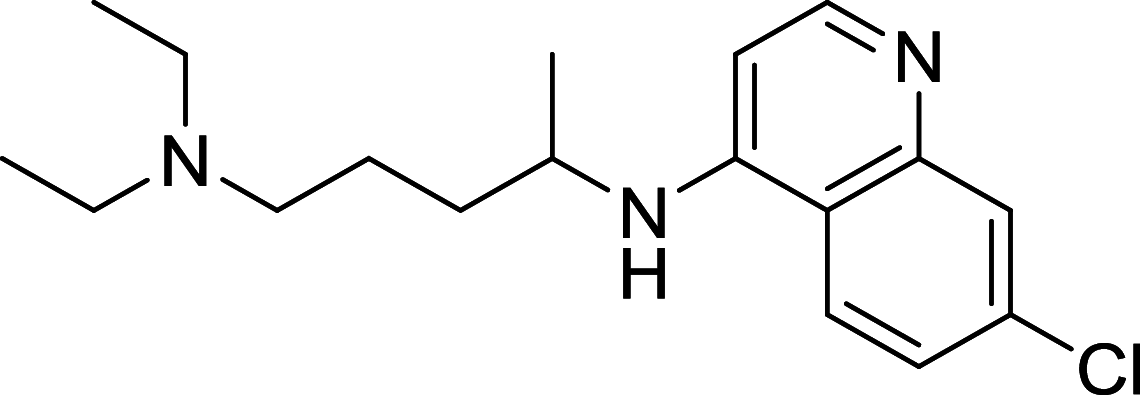
|
Hs578t, MDA-MB-231, SUM159, SW1116, HCT116, HT-29, SW480, NCM460, RT4, T24, PC3, SV-Huc-1 | [140–142] |
| Hydroxychloroquine | Lysosome |
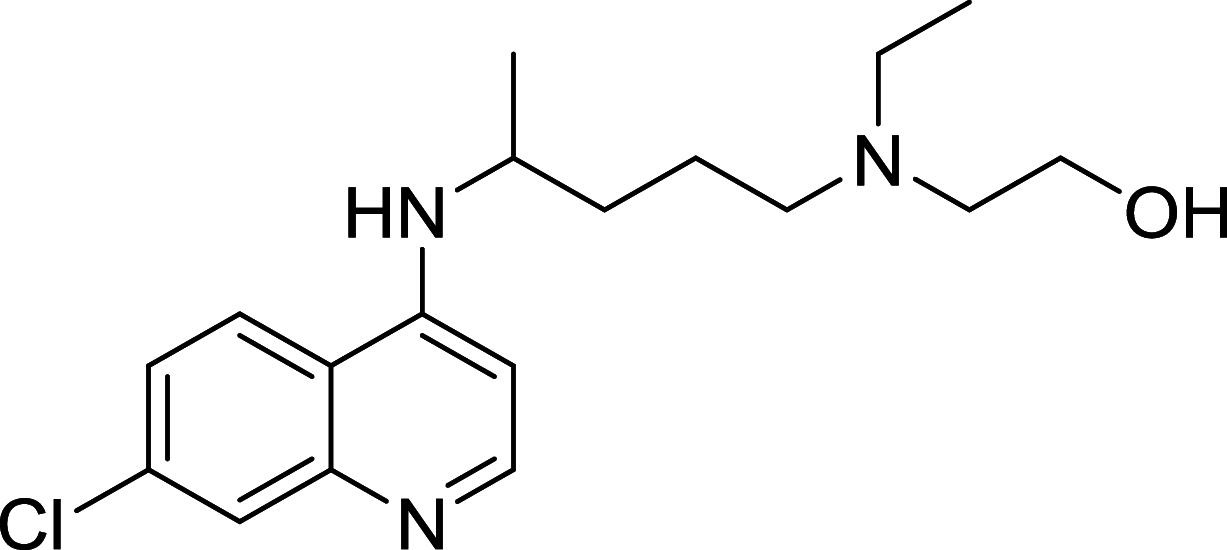
|
MCF-7, HDFs, RT4, 5637, T24, PC3, SV-Huc-1 | [142, 143] |
| Au(I)-loaded NPs | Lysosome |
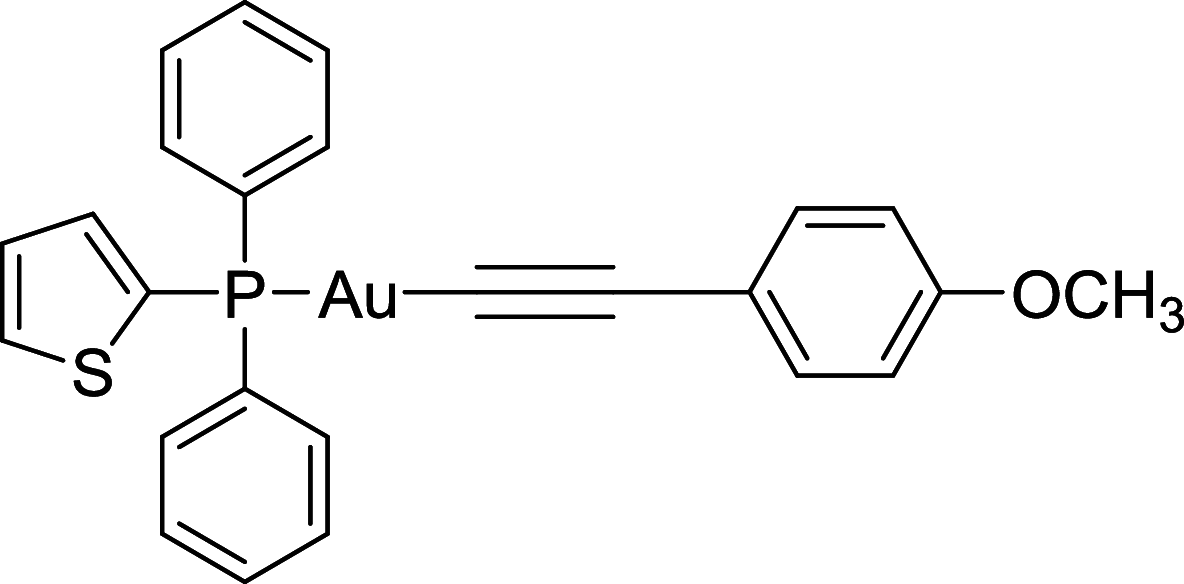
|
MCF-7 | [144] |
| VATG-027 | Lysosome |
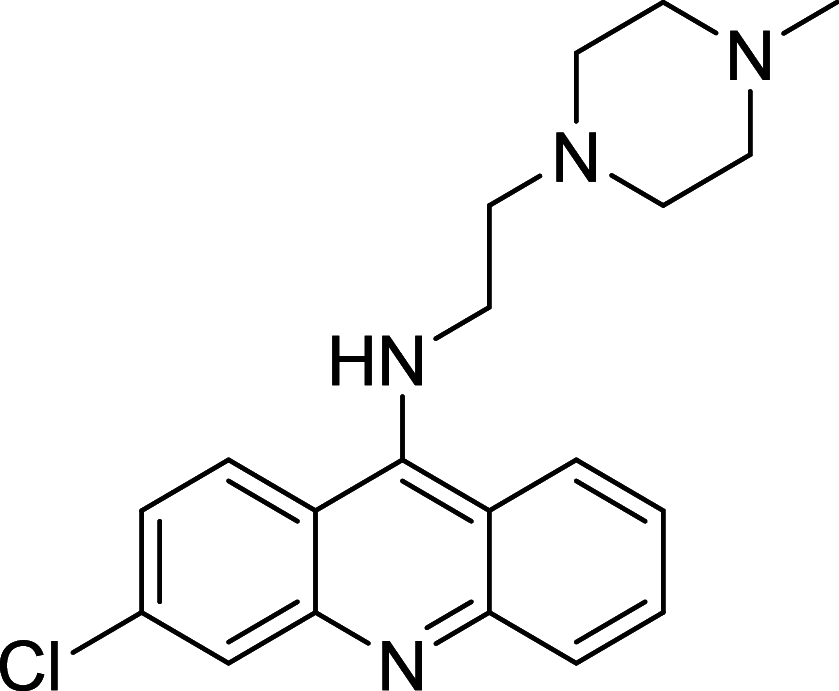
|
U2OS | [145] |
| VATG-032 | Lysosome |
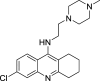
|
U2OS | [145] |
| Lys05 | Lysosome |
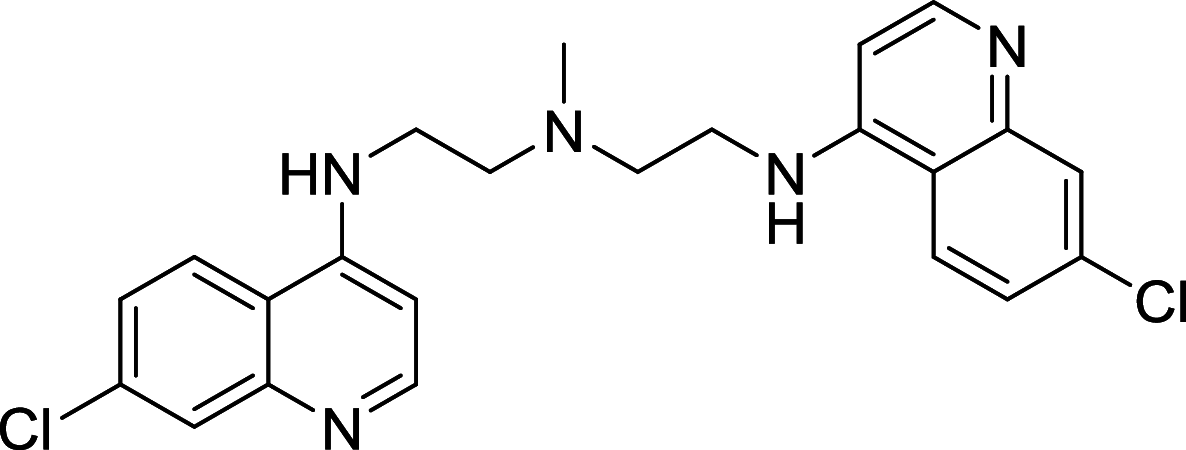
|
HT29 | [146] |
| Matrine | Lysosome |
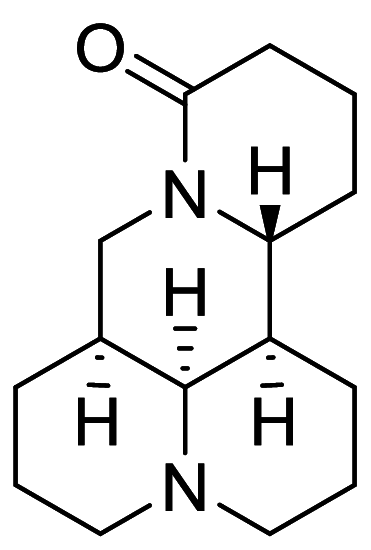
|
SGC7901 | [147] |
Small-molecule compounds targeting mTORC1 in cancer
The best-characterized regulator of autophagy is mTORC1 and it can be activated or inhibited by different strategies [4, 107]. Many mTOR inhibitors have been discovered and tested in clinical trials for years [108].
Rapamycin analogues
Rapamycin is the most widely studied inhibitor of mTOR but with unfavorable pharmacokinetic properties. To improve its practicality, some rapamycin analogues have been designed and discovered, of which Temsirolimus (CCI-779) and Everolimus (RAD001), are two typical compounds [108]. Temsirolimus shows an amazing anti-tumor effect across a wide variety of tumor in preclinical models, particularly those with defective PTEN. Notably, Temsirolimus, as a mTOR inhibitor has received Food and Drug Administration (FDA) approval for Advanced Renal-Cell Carcinoma as first-line therapy since 2007 [109]. Everolimus already has an established role in the United States in oncology. Everolimus now is under phase II trial and Temsirolimus is under phase I trial of non-small-cell lung cancer (NSCLC), respectively. Of note, Everolimus and Temsirolimus are under phase I trial for some advanced solid tumors and metastatic solid tumors, respectively [108].
Dual mTOR/PI3K inhibitors
The dual mTOR/PI3K inhibitors have been developed for years and most of them are in early phases or Phase I/II of clinical trials [110, 111]. For instance, PI103, the first new generation of dual mTOR/PI3K inhibitor, has an outstanding performance in mTOR inhibiting but disappointing for its poor in vivo pharmacokinetic properties [108]. To improve its physicochemical attributes, the second generation of mTOR inhibitors PI540 and PI620 have been designed and developed [112]. Structure-based designed mTOR inhibitor NVPBEZ235 showed limited anticancer activity but performs well in combination with established cancer drugs for cancer therapy [113] Other dual mTOR/PI3K inhibitors such as BGT226, XL765, GDC0980, SF1126 are also been explored and have encouraging performance in cancer treatments [108].
Pan-mTOR inhibitors
PP242 is the first reported comprehensive inhibitor of both mTORC1 and mTORC2. It is effective in both suppressing tumor growth and combination with other anti-tumor drugs [114]. INK128, a derivative of PP242, is currently in Phase I trials in advanced solid tumors as well as multiple myeloma and its combination usage is now in Phase trials [115, 116]. AZD8055 and AZD2014 are now in trials on advanced solid tumors [117]. OSI027 shows promising activity against leukemia [118]. Palomid 529 performs well as a cell proliferation inhibitor as well as in combination with other anti-tumor drugs [119].
mTOR inhibitors are effective in treating tumors harboring alterations in the mTOR pathway, no matter alone or in combination [109, 120]. As time went by, some tumors get acquired resistance to mTOR inhibitors [121], although the mechanisms of resistance remain undefined, mTOR mutation might bear the main responsibility. Thus, except for developing new-generation mTOR inhibitor to overcome mTOR resistance mutations [122], the combination with immunotherapy or other targeted therapy, such as ERK1/2 inhibitors and EGFR inhibitors, might be more feasible.
Small-molecule compounds targeting ULK1 in cancer
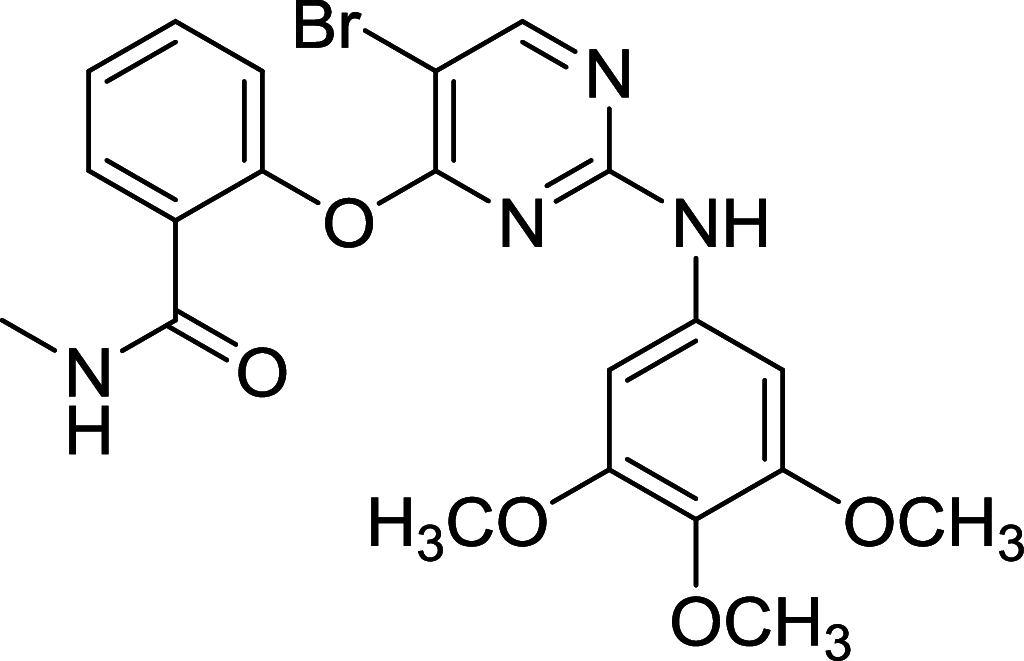
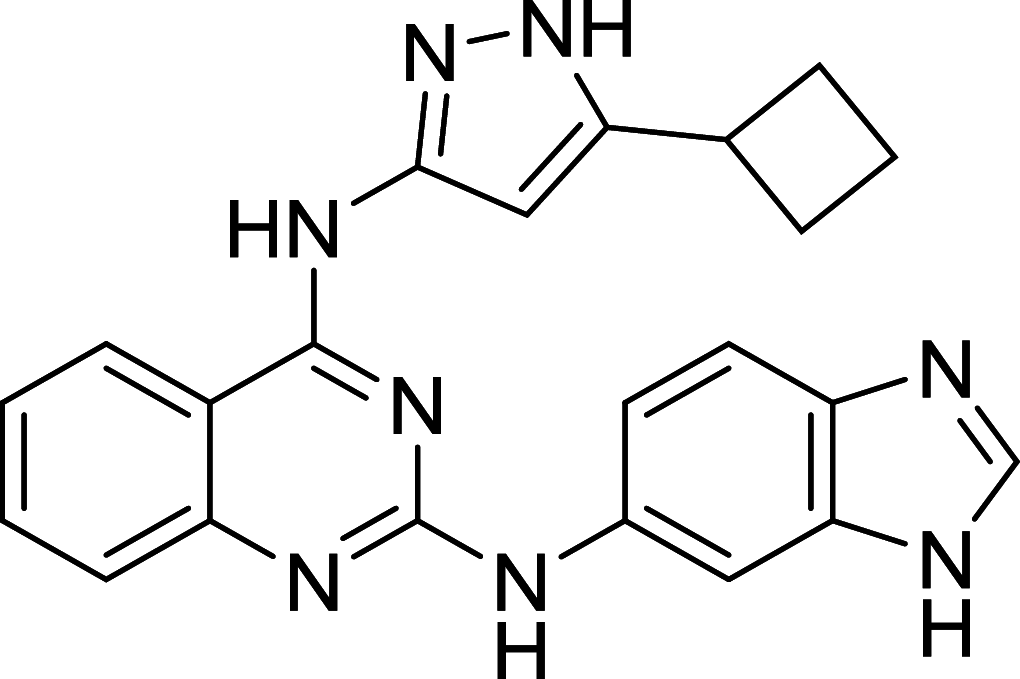
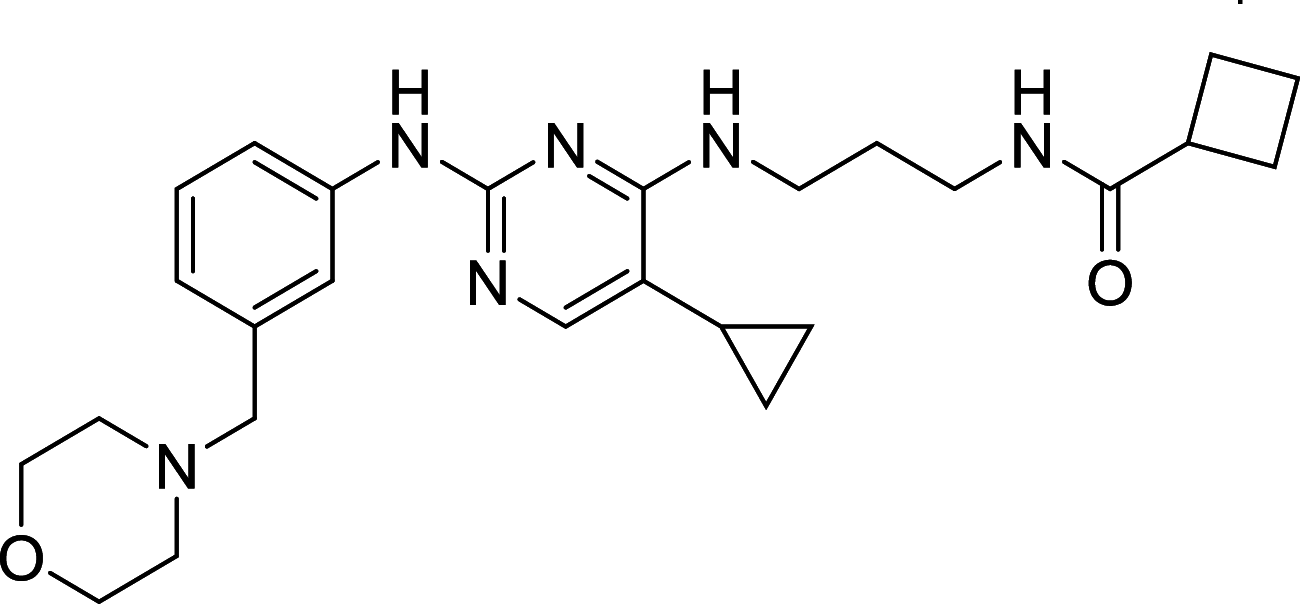
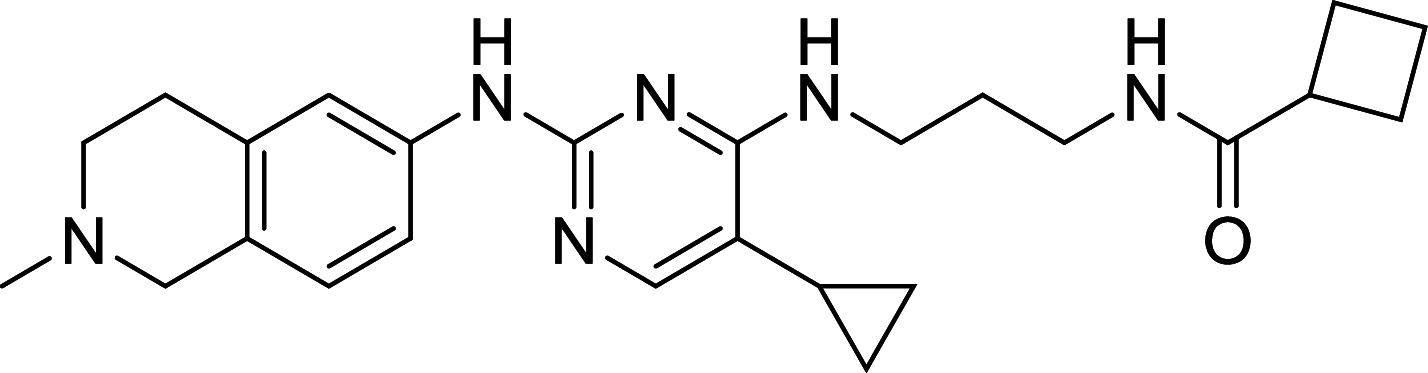
ULK1, the mammalian homolog of ATG1, has been well known as the autophagic initiator that may decide the subsequent cell fate. Recently, accumulating evidence has revealed that down-regulation of ULK1 is often found in most breast cancer tissues, suggesting that ULK1 may be a novel anti-TNBC target [123]. And a ULK1 activator LYN-1604 has been reported to induce triple-negative breast cancer cell death through ULK1 activation [124]. SBI-0206965, a highly selective ULK1 inhibitor, could suppress ULK1-mediated phosphorylation to inhibit autophagy and decrease cell survival [125]. Compound 6, a chemically synthesized small molecular compound, has been identified to induce conformational changes within the ULK1 kinase domain to inhibit ULK1 activity but lack of selectivity for cellular use [126]. MRT67307 and MRT68921 show the autophagy-inhibiting capacity through ULK1 inhibition and they could disrupt autophagosome maturation in MEFs [127]. Although ULK1 is critical in autophagy initiation, there is no approved ULK1 targeted therapy in cancer up to now. Its frustrating, but it also has huge potential for development. No mater autophagy activation or inhibition, ULK1 is an irreplaceable shiny target.
Small-molecule compounds targeting VPS34 in cancer
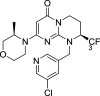
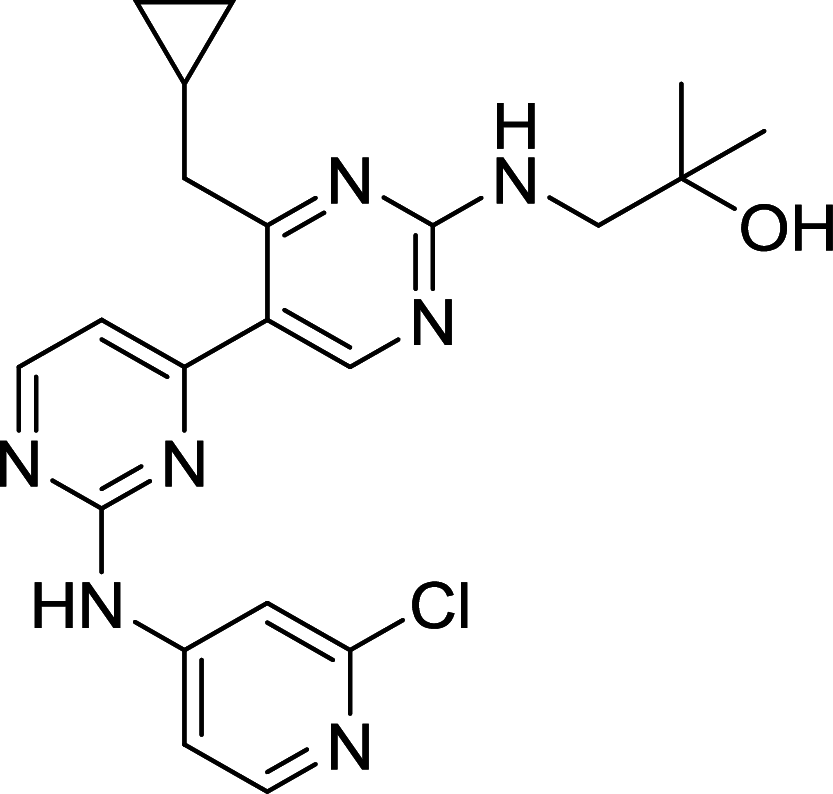
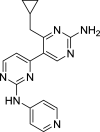
The class III phosphatidylinositol-3 kinase, Vps34, which could convert the phosphatidylinositol (PI) membrane lipid to PI3P, thereby controlling PI3P-mediated intracellular vesicular trafficking and initiate autophagy by forming different complexes. Core components of the Vps34 complexes include Vps34, Vps15, Beclin1, Atg14L/Barkor and UVRAG [19]. Full body deletion of Vps34 is embryonically lethal [128] and deletion of Vps34 inhibits autophagosome formation in different tissue types [129]. In 2010, the structure solution of Vps34 shelled new light on the development of selective Vps34 inhibitors. SAR405, a structure-based designed selective ATP-competitive inhibitor of Vps34, prevents autophagy in HeLa and H1299 cells [130]. VPS34-IN1 can inhibit the phosphorylation of PtdIns in U2OS tumor cells [131]. PIK-III inhibits the catalytic function of VPS34 thus to inhibit autophagy in different tumor cells [132].
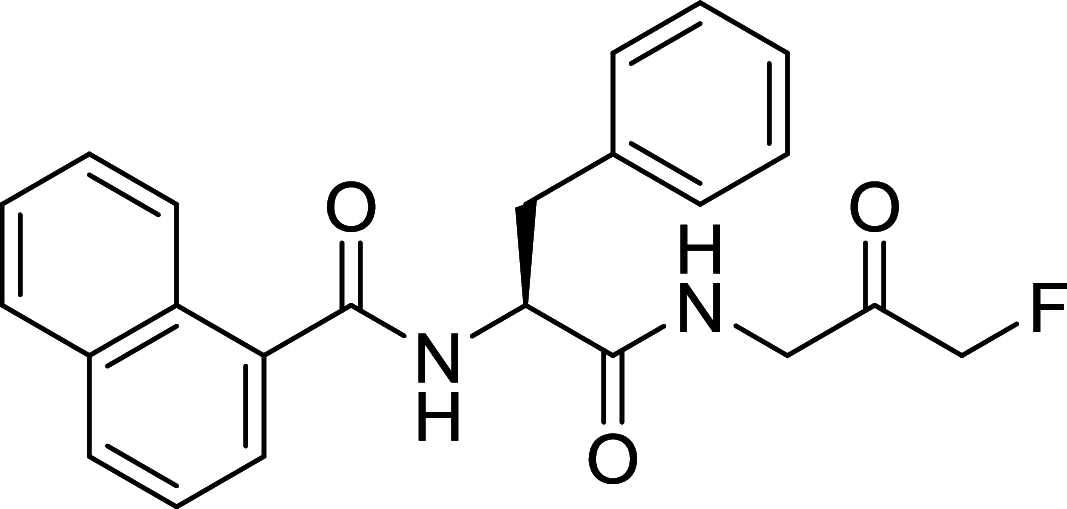
Since the significant role of the Vps34 complex in autophagy and tumorigenesis [133], more small-molecule compounds targeting Vps34 are on their way to cancer therapy. Nowadays, some research has demonstrated that Vps34 inhibitor synergized with mTOR inhibition in tumor cells [134]. For instance, combining Vps34 inhibitor SAR405 with mTOR inhibitor everolimus may have a significant synergy on the reduction of cell proliferation using renal tumor cells. Thus, targeting Vps34 would be a good potential strategy for future cancer therapy.
Small-molecule compounds targeting ATG4 in cancer
ATG4B, a key autophagy protein, cleaves Atg8 to regulate the bind or releases of Atg8-PE into the membrane to control autophagy. It has been reported that abnormal expression levels of some human Atg4 proteins occur in several types of cancer cells, which may be closely related to tumor progression, tumor suppression and cancer therapy resistance [135]. Flubendazole, a potential Atg4B agonist, could induce autophagic cell death and ROS release in breast cancer cells [136]. Except for ATG4B activator, ATG4B inhibitors have been considered to promote the inhibition of autophagy. NSC185058 inhibits autophagy in several different tumor cells by inhibiting ATG4B [137]. To date, a series of highly potent FMK-based covalent ATG4B inhibitors have been discovered with the lack of biological activity data [138]. As the key supervisor of LC3 conjugation system, ATG4B controls the progress of autophagy. The development of ATG4B-targeted small molecular compounds is still in its infancy, but we believe targeting ATG4B would be also a promising strategy.
Small-molecule compounds affecting the lysosome or autophagosome
The formation of autophagosome and autolysosome is two crucial processes during autophagy. Strategies to interfere and prevent these autophagic processes have been proposed to negatively affect tumor growth. Chloroquine (CQ), developed as an antimalarial drug, was discovered that could suppress autophagy through inhibiting lysosomal protease and blocking the fusion of autophagosomes–lysosome [139, 140]. It was reported that CQ could be used to treat colorectal cancer, breast cancer, bladder cancer and so on [140–142]. Hydroxychloroquine (HCQ), the analog of CQ, was also approved to enter the clinical trial phase of many types cancers, such as estrogen receptor positive breast cancer, prostate cancer, non-small cell lung cancer and so on [143]. Au(I)-loaded NPs, a compound that combines pH-sensitive polymeric nanoparticles with gold(I) compound Au(I), can block autophagy to induce cell death [144]. VATG-027 and VATG-032 function through lysosomal deacidification mechanisms and ultimately disrupt autophagosome turnover in U2OS cells [145]. Lys05 inhibits autophagy by deacidifying the Lysosome in HT29 cells [146]. Matrine blocks trafficking and the proteolytic activation of lysosomal proteases to inhibit autophagy in SGC7901 cells [147].
The function of lysosomes is essential to a perfect autophagy process. As the final link to affect autophagy, the inhibition of autophagy by their functional defects is significant, thus small-molecule compounds affecting the lysosome or autophagosome as autophagy inhibitors are very talented, and clinical trials of CQ or HCQ as autophagy inhibitors have demonstrated the safety of targeting autophagy for cancer therapy.
Conclusions
Alterations in multiple signaling pathways reflect a powerful means of adjusting the development, maintenance and overall adaption of cancer cells. Accumulating studies have revealed that nearly all major oncogenic signaling pathways are found to be deregulated and most of which are closely associated with the defective autophagy. Tumor cells often present unusual levels of autophagy that contributes to the survival mechanisms in tumor cells. In cancer cells, content dependent autophagy acts both tumor suppressive and tumor-promoting roles, while how to choose the appropriate regulation to treat a tumor is still a scientific problem. Moreover, autophagy has also been implicated in resistance to multiple standard chemotherapeutic agents [148]. It has also been involved in the survival of dormant tumor cells and may be crucial for their recurrence. Thus, the autophagy-targeted therapy seems to be indispensable and more promising.
With the rapid development of research on autophagy, the mechanism of autophagy controlling cancer cells fate has been gradually unveiled. Modulation of autophagy has been accepted as novel therapeutic approaches for cancer therapy. Since the role of autophagy helps tumor cells respond to different stress conditions, including energy stress, hypoxia and cellular damage, development of autophagy inhibitors is an attractive strategy for cancer therapy. Except for the classical autophagy inhibitors for cancer treatment, CQ and HCQ, several new kinds of autophagy inhibitors, such as ULK1 inhibitors (SBI-0206965, MRT68921), as well as an ATG4B inhibitor (NSC185058) and Vps34 inhibitor (SAR405), have exhibited the promising potential for cancer therapy. Considering the tumor-suppressive role of autophagy, several activators have been applied to improve cancer therapy, of which mTOR inhibitors is the most famous kind. What is more, some classic cancer targets, such as BRD4 and ERK1/2, are closely associated with autophagy. Recently, a small-molecule inhibitor targeting BRD4 could induce AMPK modulated autophagy-associated cell death in breast cancer [149]. It suggests that autophagy-related protein could be the candidate of dual-target cancer therapy. Moreover, some autophagy-modulating compound database or webserver [150, 151], may help us to discover more potential small-molecule drugs targeting autophagy.
The content-dependent role of autophagy in cancer cell fate has provided an insight into the development of novel strategies for cancer therapy; however, we should take care of identifying the conditions which autophagy inhibition will be beneficial or harmful. For example, it has been reported in many cancer cell lines with activated RAS are highly dependent on autophagy for survival [152], in this type of tumor, autophagy inhibition will be beneficial, and autophagy inhibitors combine with ERK1/2 inhibitors will be effective. While in many cancer cell lines lacking the expression of Beclin1, autophagy activation seems advantageous, and autophagy activators combine with chemotherapy drugs might be beneficial.
A new hope of utilizing autophagy for targeted cancer therapy may lie in discovering candidate small-molecule compounds that modulate tumor-promoting or tumor-suppressive autophagic pathways and even the entire autophagic signaling network (the autophagic multiple-target strategy), rather than an individual (single target). On the basis of this viewpoint, further elucidation of the intricate mechanisms of autophagy will be regarded as a promising strategy for the discovery of more and more new small-molecule drugs targeting the autophagic signaling network in future cancer therapy.
Author contributions
All authors read and approved the final manuscript.
Funding
We are grateful to Prof. Canhua Huang (Sichuan University) for his good suggestions on this manuscript. This work was supported by grants from National Key R&D Program of China (Grant No. 2017YFC0909301 and Grant No. 2017YFC0909302) and National Natural Science Foundation of China (Grant No. 81673455, Grant No. 81602130, Grant No. 81473091 and Grant No. 81673290).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no competing interest.
Footnotes
Jin Zhang, Guan Wang, and Yuxin Zhou contributed equally to this work.
Contributor Information
Yi Chen, Email: toddychan@163.com.
Bo Liu, Email: liubo2400@163.com.
References
- 1.Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy–inflammation–cell death axis in organismal aging. Science. 2011;333:1109–1112. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galluzzi L, Bravo-San Pedro JM, Levine B, et al. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2017;16(7):487–511. doi: 10.1038/nrd.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ke B, Tian M, Li J, et al. Targeting programmed cell death using small-molecule compounds to improve potential cancer therapy. Med Res Rev. 2016;36(6):983–1035. doi: 10.1002/med.21398. [DOI] [PubMed] [Google Scholar]
- 4.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 7.Huynh KK, Eskelinen EL, Scott CC, et al. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 2007;26:313–324. doi: 10.1038/sj.emboj.7601511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heath RJ, Xavier RJ. Autophagy, immunity and human disease. Curr Opin Gastroenterol. 2009;25:512–520. doi: 10.1097/MOG.0b013e32833104f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Investig. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cianfanelli V, Fuoco C, Lorente M, et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-MYC dephosphorylation and degradation. Nat Cell Biol. 2014;17:20–30. doi: 10.1038/ncb3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang MR, Kim MS, Oh JE, et al. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J Pathol. 2009;217:702–706. doi: 10.1002/path.2509. [DOI] [PubMed] [Google Scholar]
- 13.Yang A, Rajeshkumar NV, Wang X, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014;4:905–913. doi: 10.1158/2159-8290.CD-14-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galluzzi L, Pietrocola F, Bravo-San Pedro JM, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34:856–880. doi: 10.15252/embj.201490784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30:1913–1930. doi: 10.1101/gad.287524.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang SF, Wang XY, Fu ZQ, et al. TXNDC17 promotes paclitaxel resistance via inducing autophagy in ovarian cancer. Autophagy. 2015;11:225–238. doi: 10.1080/15548627.2014.998931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardie DG. AMP-activated protein kinase as a drug target. Annu Rev Pharmacol Toxicol. 2007;47:185–210. doi: 10.1146/annurev.pharmtox.47.120505.105304. [DOI] [PubMed] [Google Scholar]
- 18.Chan EY, Tooze SA. Evolution of Atg1 function and regulation. Autophagy. 2009;5:758–765. doi: 10.4161/auto.8709. [DOI] [PubMed] [Google Scholar]
- 19.Kim J, Kim YC, Fang C, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152:290–303. doi: 10.1016/j.cell.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brooks DL, Schwab LP, Krutilina R, et al. ITGA6 is directly regulated by hypoxia-inducible factors and enriches for cancer stem cell activity and invasion in metastatic breast cancer models. Mol Cancer. 2016;15:26. doi: 10.1186/s12943-016-0510-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29:625–634. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1) Mol Pharmacol. 2005;33:423–425. doi: 10.1124/mol.106.027029. [DOI] [PubMed] [Google Scholar]
- 23.Liu J, Hao H, Huang H, et al. Hypoxia regulates the therapeutic potential of mesenchymal stem cells through enhanced autophagy. Int J Low Extremity Wounds. 2015;14:63–72. doi: 10.1177/1534734615573660. [DOI] [PubMed] [Google Scholar]
- 24.Wu H, Huang S, Chen Z, et al. Hypoxia-induced autophagy contributes to the invasion of salivary adenoid cystic carcinoma through the HIF-1α/BNIP3 signaling pathway. Mol Med Rep. 2015;12:6467–6474. doi: 10.3892/mmr.2015.4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xue H, Yuan G, Guo X, et al. A novel tumor-promoting mechanism of IL6 and the therapeutic efficacy of tocilizumab: hypoxia-induced IL6 is a potent autophagy initiator in glioblastoma via the p-STAT3–MIR155-3p–CREBRF pathway. Autophagy. 2016;12:1129–1152. doi: 10.1080/15548627.2016.1178446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng WX, Xiong EM, Ge L, et al. Egr-1 promotes hypoxia-induced autophagy to enhance chemo-resistance of hepatocellular carcinoma cells. Exp Cell Res. 2016;340:62–70. doi: 10.1016/j.yexcr.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 27.Jaakkola PM, Pursiheimo JP. p62 degradation by autophagy: another way for cancer cells to survive under hypoxia. Autophagy. 2009;5:410–412. doi: 10.4161/auto.5.3.7823. [DOI] [PubMed] [Google Scholar]
- 28.Wan G, Xie W, Liu Z, et al. Hypoxia-induced MIR155 is a potent autophagy inducer by targeting multiple players in the MTOR pathway. Autophagy. 2013;10:70–79. doi: 10.4161/auto.26534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo YJ, Liu JX, Guan YW. Hypoxia induced upregulation of miR-301a/b contributes to increased cell autophagy and viability of prostate cancer cells by targeting NDRG2. Eur Rev Med Pharmacol Sci. 2016;20:101–108. [PubMed] [Google Scholar]
- 30.Sun Y, Xing X, Liu Q, et al. Hypoxia-induced autophagy reduces radiosensitivity by the HIF-1α/miR-210/Bcl-2 pathway in colon cancer cells. Int J Oncol. 2015;46:750–756. doi: 10.3892/ijo.2014.2745. [DOI] [PubMed] [Google Scholar]
- 31.Wu J, Niu J, Li X, et al. Hypoxia induces autophagy of bone marrow-derived mesenchymal stem cells via activation of ERK1/2. Cell Physiol Biochem. 2014;33:1467–1474. doi: 10.1159/000358711. [DOI] [PubMed] [Google Scholar]
- 32.Fang Y, Tan J, Zhang Q. Signaling pathways and mechanisms of hypoxia-induced autophagy in the animal cells. Cell Biol Int. 2015;39:891–898. doi: 10.1002/cbin.10463. [DOI] [PubMed] [Google Scholar]
- 33.Blagosklonny MV. Hypoxia, MTOR and autophagy: converging on senescence or quiescence. Autophagy. 2013;9:260–262. doi: 10.4161/auto.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Notte A, Rebucci M, Fransolet M, et al. Taxol-induced unfolded protein response activation in breast cancer cells exposed to hypoxia: ATF4 activation regulates autophagy and inhibits apoptosis. Int J Biochem Cell Biol. 2015;62:1–14. doi: 10.1016/j.biocel.2015.02.010. [DOI] [PubMed] [Google Scholar]
- 35.Kim I, Rodriguezenriquez S, Lemasters JJ. Minireview: selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sandoval H, Thiagarajan P, Dasgupta SK, et al. Essential role for Nix in autophagic maturation of red cells. Nature. 2008;454:232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawajiri S, Saiki S, Sato S, et al. PINK1 is recruited to mitochondria with parkin and associates with LC3 in mitophagy. FEBS Lett. 2008;584:1073–1079. doi: 10.1016/j.febslet.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 38.Pickrell AM, Youle RJ. The roles of PINK1, Parkin and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei Y, Chiang WC, Sumpter R, Jr, et al. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017;168:224–238. doi: 10.1016/j.cell.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Michiorri S, Gelmetti V, Giarda E, et al. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ. 2010;17:962–974. doi: 10.1038/cdd.2009.200. [DOI] [PubMed] [Google Scholar]
- 42.Kaufmann A, Beier V, Franquelim HG, et al. Molecular mechanism of autophagic membrane-scaffold assembly and disassembly. Cell. 2014;156:469–481. doi: 10.1016/j.cell.2013.12.022. [DOI] [PubMed] [Google Scholar]
- 43.Geisler S, Holmström KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 44.Zhong Z, Umemura A, Sanchez-Lopez E, et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164:896–910. doi: 10.1016/j.cell.2015.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vyas S, Zaganjor E, Haigis MC. Mitochondria and cancer. Cell. 2016;166:555–566. doi: 10.1016/j.cell.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu YL, DeLay M, Jahangiri A, et al. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012;72:1773–1783. doi: 10.1158/0008-5472.CAN-11-3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo JY, Karsli-Uzunbas G, Mathew R, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27:1447–1461. doi: 10.1101/gad.219642.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Proskuryakov SY, Gabai VL. Mechanisms of tumor cell necrosis. Curr Pharm Des. 2010;16:56–68. doi: 10.2174/138161210789941793. [DOI] [PubMed] [Google Scholar]
- 49.Baek MW, Cho HS, Kim SH, et al. Ascorbic acid induces necrosis in human laryngeal squamous cell carcinoma via ROS, PKC, and calcium signaling. J Cell Physiol. 2016;232:417–425. doi: 10.1002/jcp.25438. [DOI] [PubMed] [Google Scholar]
- 50.He S, Wang L, Miao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 51.Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 52.Jouan-Lanhouet S, Arshad MI, Piquet-Pellorce C, et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 2012;19:2003–2014. doi: 10.1038/cdd.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Galluzzi L, Vitale I, Abrams JM, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Konstantakou EG, Voutsinas GE, Velentzas AD, et al. 3-BrPA eliminates human bladder cancer cells with highly oncogenic signatures via engagement of specific death programs and perturbation of multiple signaling and metabolic determinants. Mol Cancer. 2015;14:1–26. doi: 10.1186/s12943-015-0399-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lim SC, Jeon HJ, Kee KH, et al. Involvement of DR4/JNK pathway-mediated autophagy in acquired TRAIL resistance in HepG2 cells. Int J Oncol. 2016;49:1983–1990. doi: 10.3892/ijo.2016.3699. [DOI] [PubMed] [Google Scholar]
- 56.Ahn MY, Kim TH, Kwon SM, et al. 5-Nitro-5′-hydroxy-indirubin-3′-oxime (AGM130), an indirubin-3′-oxime derivative, inhibits tumor growth by inducing apoptosis against non-small cell lung cancer in vitro and in vivo. Eur J Pharm Sci. 2015;79:122–131. doi: 10.1016/j.ejps.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 57.Goldberg AA, Draz H, Montes-Grajales D, et al. 3,3′-Diindolylmethane (DIM) and its ring-substituted halogenated analogs (ring-DIMs) induce differential mechanisms of survival and death in androgen-dependent and-independent prostate cancer cells. Genes Cancer. 2016;7:59. doi: 10.18632/genesandcancer.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 59.Wei MF, Chen MW, Chen KC, et al. Autophagy promotes resistance to photodynamic therapy-induced apoptosis selectively in colorectal cancer stem-like cells. Autophagy. 2014;10:1179–1192. doi: 10.4161/auto.28679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun WL, Chen J, Wang YP, et al. Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development. Autophagy. 2011;7:1035–1044. doi: 10.4161/auto.7.9.16521. [DOI] [PubMed] [Google Scholar]
- 61.He H, Yu JJ, Xu Q. Downregulation of ATG14 by EGR1-MIR152 sensitizes ovarian cancer cells to cisplatin-induced apoptosis by inhibiting cyto-protective autophagy. Autophagy. 2015;11:373–384. doi: 10.1080/15548627.2015.1009781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hou W, Han J, Lu C, et al. Autophagic degradation of active caspase-8: a crosstalk mechanism between autophagy and apoptosis. Autophagy. 2010;6:891–900. doi: 10.4161/auto.6.7.13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kaminskyy VO, Piskunova T, Zborovskaya IB, et al. Suppression of basal autophagy reduces lung cancer cell proliferation and enhances caspase-dependent and -independent apoptosis by stimulating ROS formation. Autophagy. 2012;8:1032–1044. doi: 10.4161/auto.20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kang R, Zeh HJ, Lotze MT, et al. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhan Z, Li Q, Wu P. Autophagy-mediated HMGB1 release antagonizes apoptosis of gastric cancer cells induced by vincristine via transcriptional regulation of Mcl-1. Autophagy. 2012;8:109–121. doi: 10.4161/auto.8.1.18319. [DOI] [PubMed] [Google Scholar]
- 66.Yao Dahong, Wang Peiqi, Zhang Jin, et al. Deconvoluting the relationships between autophagy and metastasis for potential cancer therapy. Apoptosis. 2016;21:683–698. doi: 10.1007/s10495-016-1237-2. [DOI] [PubMed] [Google Scholar]
- 67.Gewirtz DA. The four faces of autophagy: implications for cancer therapy. Cancer Res. 2014;274(3):647–651. doi: 10.1158/0008-5472.CAN-13-2966. [DOI] [PubMed] [Google Scholar]
- 68.Galluzzi L, Bravo-San Pedro JM, Vitale I, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015;22:58–73. doi: 10.1038/cdd.2014.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu X, Li R, Shi W, et al. Silencing of MicroRNA-21 confers the sensitivity to tamoxifen and fulvestrant by enhancing autophagic cell death through inhibition of the PI3K-AKT-mTOR pathway in breast cancer cells. Biomed Pharmacother. 2016;77:37–44. doi: 10.1016/j.biopha.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 70.Gao Q, Liu H, Yao Y, et al. Carnosic acid induces autophagic cell death through inhibition of the Akt/mTOR pathway in human hepatoma cells. J Appl Toxicol. 2014;35:485–492. doi: 10.1002/jat.3049. [DOI] [PubMed] [Google Scholar]
- 71.Law BY, Chan WK, Xu SW, et al. Natural small-molecule enhancers of autophagy induce autophagic cell death in apoptosis-defective cells. Sci Rep. 2014;4:5510. doi: 10.1038/srep05510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Park KJ, Lee SH, Lee CH, et al. Upregulation of Beclin-1 expression and phosphorylation of Bcl-2 and p53 are involved in the JNK-mediated autophagic cell death. Biochem Biophys Res Commun. 2009;382:726–729. doi: 10.1016/j.bbrc.2009.03.095. [DOI] [PubMed] [Google Scholar]
- 73.Sun PH, Zhu LM, Qiao MM, et al. The XAF1 tumor suppressor induces autophagic cell death via upregulation of Beclin-1 and inhibition of Akt pathway. Cancer Lett. 2011;310:170–180. doi: 10.1016/j.canlet.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 74.Ornelas A, McCullough CR, Lu Z, et al. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell. 2011;42:23–35. doi: 10.1016/j.molcel.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 75.Tai WT, Shiau CW, Chen HL, et al. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis. 2013;4:e485. doi: 10.1038/cddis.2013.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cui L, Song Z, Liang B, et al. Radiation induces autophagic cell death via the p53/DRAM signaling pathway in breast cancer cells. Oncol Rep. 2016;35:1639–3647. doi: 10.3892/or.2016.4752. [DOI] [PubMed] [Google Scholar]
- 77.Liu Y, Yang Y, Ye YC, et al. Activation of ERK-p53 and ERK-mediated phosphorylation of Bcl-2 are involved in autophagic cell death induced by the c-Met inhibitor SU11274 in human lung cancer A549 cells. J Pharmacol Sci. 2012;118:423–432. doi: 10.1254/jphs.11181FP. [DOI] [PubMed] [Google Scholar]
- 78.Wang N, Pan W, Zhu M, et al. Fangchinoline induces autophagic cell death via p53/sestrin2/AMPK signalling in human hepatocellular carcinoma cells. Br J Pharmacol. 2011;164:731–742. doi: 10.1111/j.1476-5381.2011.01349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rdedition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yan C, Yang JM. Autophagy and apoptosis: rivals or mates? Chin J Cancer. 2013;32:103–105. doi: 10.5732/cjc.013.10022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yousefi S, Perozzo R, Schmid I, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–1132. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 82.Rubinstein AD, Eisenstein M, Ber Y, et al. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol Cell. 2011;44:698–709. doi: 10.1016/j.molcel.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 83.Sandilands E, Serrels B, McEwan DG, et al. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat Cell Biol. 2011;14:51–60. doi: 10.1038/ncb2386. [DOI] [PubMed] [Google Scholar]
- 84.Mariño G, Niso-Santano M, Baehrecke EH, et al. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li Y, Wang Y, Wang S, et al. Oridonin phosphate-induced autophagy effectively enhances cell apoptosis of human breast cancer cells. Med Oncol. 2015;32:365. doi: 10.1007/s12032-014-0365-1. [DOI] [PubMed] [Google Scholar]
- 86.Hsin IL, Ou CC, Wu MF, et al. GMI, an immunomodulatory protein from Ganoderma microsporum, potentiates cisplatin-induced apoptosis via autophagy in lung cancer cells. Mol Pharm. 2015;12:1534–1543. doi: 10.1021/mp500840z. [DOI] [PubMed] [Google Scholar]
- 87.Jin SM, Jang HW, Sohn SY, et al. Role of autophagy in the resistance to tumour necrosis factor-related apoptosis-inducing ligand-induced apoptosis in papillary and anaplastic thyroid cancer cells. Endocrine. 2014;45:256–262. doi: 10.1007/s12020-013-9997-8. [DOI] [PubMed] [Google Scholar]
- 88.Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. 2009;9:692–703. doi: 10.1038/nri2634. [DOI] [PubMed] [Google Scholar]
- 89.Shigdar S, Li Y, Bhattacharya S, et al. Inflammation and cancer stem cells. Cancer Lett. 2014;345:271–278. doi: 10.1016/j.canlet.2013.07.031. [DOI] [PubMed] [Google Scholar]
- 90.Maderna P, Godson C. Phagocytosis of apoptotic cells and the resolution of inflammation. Biochim Biophys Acta. 2003;1639:141–151. doi: 10.1016/j.bbadis.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 91.Qu X, Zou Z, Sun Q, et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–946. doi: 10.1016/j.cell.2006.12.044. [DOI] [PubMed] [Google Scholar]
- 92.Netea-Maier RT, Plantinga TS, van de Veerdonk FL, et al. Modulation of inflammation by autophagy: consequences for human disease. Autophagy. 2016;12:245–260. doi: 10.1080/15548627.2015.1071759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Harris J, Hartman M, Roche C, et al. Autophagy controls IL-1{beta} secretion by targeting pro-IL-1{beta} for degradation. J Biol Chem. 2011;286:9587–9597. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 95.Zhou R, Yazdi AS, Menu P, et al. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 96.Krysko O, Løve Aaes T, Bachert C, et al. Many faces of DAMPs in cancer therapy. Cell Death Dis. 2013;4:e631. doi: 10.1038/cddis.2013.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ma Y, Galluzzi L, Zitvogel L, et al. Autophagy and cellular immune responses. Immunity. 2013;39:211–227. doi: 10.1016/j.immuni.2013.07.017. [DOI] [PubMed] [Google Scholar]
- 99.Guzik K, Zak KM, Grudnik P, et al. Small-molecule inhibitors of the Programmed Cell Death-1/Programmed Death-ligand 1 (PD-1/PD-L1) interaction via transiently-induced protein states and dimerization of PD-L1. J Med Chem. 2017;60:5857–5867. doi: 10.1021/acs.jmedchem.7b00293. [DOI] [PubMed] [Google Scholar]
- 100.Bezu L, Gomes-de-Silva LC, Dewitte H, et al. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol. 2015;6:187. doi: 10.3389/fimmu.2015.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Martins I, Wang Y, Michaud M, et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 2014;21:79–91. doi: 10.1038/cdd.2013.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Martins I, Michaud M, Sukkurwala AQ, et al. Premortem autophagy determines the immunogenicity of chemotherapy-induced cancer cell death. Autophagy. 2012;8:413–415. doi: 10.4161/auto.19009. [DOI] [PubMed] [Google Scholar]
- 103.Hu L, Jiang K, Ding C, et al. Targeting autophagy for oncolytic immunotherapy. Biomedicines. 2017;5:5. doi: 10.3390/biomedicines5010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang X, Fan J, Wang S, et al. Targeting CD47 and autophagy elicited enhanced antitumor effects in non-small cell lung cancer. Cancer Immunol Res. 2017;5:363–375. doi: 10.1158/2326-6066.CIR-16-0398. [DOI] [PubMed] [Google Scholar]
- 105.Keller CW, Loi M, Ewert S, et al. The autophagy machinery restrains iNKT cell activation through CD1D1 internalization. Autophagy. 2017;15:1–12. doi: 10.1080/15548627.2017.1297907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Robainas M, Otano R, Bueno S, et al. Understanding the role of PD-L1/PD1 pathway blockade and autophagy in cancer therapy. Onco Targets Ther. 2017;10:1803–1807. doi: 10.2147/OTT.S132508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Benjamin D, Colombi M, Moroni C, et al. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011;10:868–880. doi: 10.1038/nrd3531. [DOI] [PubMed] [Google Scholar]
- 109.Fazio N, Dettori M, Lorizzo K. Temsirolimus for advanced renal-cell carcinoma. N Engl J Med. 2007;357(10):1050. doi: 10.1056/NEJMc071868. [DOI] [PubMed] [Google Scholar]
- 110.Neshat MS, Mellinghoff IK, Tran C, et al. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci USA. 2001;98:10314–10319. doi: 10.1073/pnas.171076798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Beck JT, Ismail A, Tolomeo C. Targeting the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway: an emerging treatment strategy for squamous cell lung carcinoma. Cancer Treat Rev. 2014;40:980–989. doi: 10.1016/j.ctrv.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 112.Khan DMA, Afzal H. Targeting the mTOR kinase domain: the second generation of mTOR inhibitors. Drug Discov Today. 2011;16:325–331. doi: 10.1016/j.drudis.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lu X, Horner JW, Paul E, et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature. 2017;543(7647):728–732. doi: 10.1038/nature21676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hsieh AC, Costa M, Zollo O, et al. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell. 2010;17:249–261. doi: 10.1016/j.ccr.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Janes MR, Limon JJ, So L, et al. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med. 2010;16:205–213. doi: 10.1038/nm.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hsieh AC, Ruggero D. Targeting eukaryotic translation initiation factor 4E (eIF4E) in cancer. Clin Cancer Res. 2010;16:4914–4920. doi: 10.1158/1078-0432.CCR-10-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Marshall G, Howard Z, Dry J, et al. Benefits of mTOR kinase targeting in oncology: pre-clinical evidence with AZD8055. Biochem Soc Trans. 2011;39:456–459. doi: 10.1042/BST0390456. [DOI] [PubMed] [Google Scholar]
- 118.Carayol N, Vakana E, Sassano A, et al. Critical roles for mTORC2- and rapamycin-insensitive mTORC1-complexes in growth and survival of BCR-ABL-expressing leukemic cells. Proc Natl Acad Sci USA. 2010;107:12469–12474. doi: 10.1073/pnas.1005114107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gravina GL, Marampon F, Petini F, et al. The TORC1/TORC2 inhibitor, Palomid 529, reduces tumor growth and sensitizes to docetaxel and cisplatin in aggressive and hormone-refractory prostate cancer cells. Endocr Relat Cancer. 2011;18:385–400. doi: 10.1530/ERC-11-0045. [DOI] [PubMed] [Google Scholar]
- 120.Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366(6):520–529. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wagle N, Grabiner BC, Van Allen EM, et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N Engl J Med. 2014;371(15):1426–1433. doi: 10.1056/NEJMoa1403352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rodrik-Outmezguine VS, Okaniwa M, Yao Z, et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature. 2016;534(7606):272–276. doi: 10.1038/nature17963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ouyang L, Zhang L, Fu L, et al. A small-molecule activator induces ULK1-modulating autophagy-associated cell death in triple negative breast cancer. Autophagy. 2017;13(4):777–778. doi: 10.1080/15548627.2017.1283470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang L, Fu L, Zhang S, et al. Discovery of a small molecule targeting ULK1-modulated cell death of triple negative breast cancer in vitro and in vivo. Chem Sci. 2017;8:2687–2701. doi: 10.1039/C6SC05368H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Egan DF, Chun MG, Vamos M, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. 2015;59:285–297. doi: 10.1016/j.molcel.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lazarus MB, Novotny CJ, Shokat KM. Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. ACS Chem Biol. 2014;10:257–261. doi: 10.1021/cb500835z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Petherick KJ, Conway OJ, Mpamhanga C, et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem. 2015;290:11376–11383. doi: 10.1074/jbc.C114.627778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhou X, Takatoh J, Wang F. The mammalian class 3 PI3K (PIK3C3) is required for early embryogenesis and cell proliferation. PLoS One. 2011;6:e16358. doi: 10.1371/journal.pone.0016358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Parekh VV, Wu L, Boyd KL, et al. Impaired autophagy, defective T cell homeostasis, and a wasting syndrome in mice with a T cell-specific deletion of Vps34. J Immunol. 2013;190:5086–5101. doi: 10.4049/jimmunol.1202071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pasquier B. SAR405, a PIK3C3/Vps34 inhibitor that prevents autophagy and synergizes with MTOR inhibition in tumor cells. Autophagy. 2015;11:725–726. doi: 10.1080/15548627.2015.1033601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bilanges B, Vanhaesebroeck B. Cinderella finds her shoe: the first Vps34 inhibitor uncovers a new PI3K-AGC protein kinase connection. Biochem J. 2014;464:e7–e10. doi: 10.1042/BJ20141218. [DOI] [PubMed] [Google Scholar]
- 132.Dowdle WE, Nyfeler B, Nagel J, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. 2014;16:1069–1079. doi: 10.1038/ncb3053. [DOI] [PubMed] [Google Scholar]
- 133.Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 134.Pasquier B. SAR405, a PIK3C3/Vps34 inhibitor that prevents autophagy and synergizes with mTOR inhibition in tumor cells. Autophagy. 2015;11(4):725–726. doi: 10.1080/15548627.2015.1033601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang L, Li J, Ouyang L, et al. Unraveling the roles of Atg4 proteases from autophagy modulation to targeted cancer therapy. Cancer Lett. 2016;373(1):19–26. doi: 10.1016/j.canlet.2016.01.022. [DOI] [PubMed] [Google Scholar]
- 136.Zhang L, Guo M, Li J, et al. Systems biology-based discovery of a potential Atg4B agonist (Flubendazole) that induces autophagy in breast cancer. Mol BioSyst. 2015;11:2860–2866. doi: 10.1039/C5MB00466G. [DOI] [PubMed] [Google Scholar]
- 137.Akin D, Wang SK, Habibzadegah-Tari P, et al. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy. 2014;10:2021–2035. doi: 10.4161/auto.32229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Qiu Z, Kuhn B, Aebi J, et al. Discovery of fluoromethylketone-based peptidomimetics as covalent ATG4B (autophagin-1) inhibitors. ACS Med Chem Lett. 2016;7:802–806. doi: 10.1021/acsmedchemlett.6b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ch’nq JH, Lee YQ, Gun SY, et al. Validation of a chloroquine-induced cell death mechanism for clinical use against malaria. Cell Death Dis. 2014;5:e1305. doi: 10.1038/cddis.2014.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen PJ, Luo XY, Nie PP, et al. CQ synergistically sensitizes human colorectal cancer cells to SN-38/CPT-11 through lysosomal and mitochondrial apoptotic pathway via p53-ROS cross-talk. Free Radic Biol Med. 2017;104:280–297. doi: 10.1016/j.freeradbiomed.2017.01.033. [DOI] [PubMed] [Google Scholar]
- 141.Liang DH, Choi DS, Ensor JE, et al. The autophagy inhibitor chloroquine targets cancer stem cells in triple negative breast cancer by inducing mitochondrial damage and impairing DNA break repair. Cancer Lett. 2016;376:249–258. doi: 10.1016/j.canlet.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lin YC, Lin JF, Wen SI, et al. Chloroquine and hydroxychloroquine inhibit bladder cancer cell growth by targeting basal autophagy and enhancing apoptosis. Kaohsiung J Med Sci. 2017;33:215–223. doi: 10.1016/j.kjms.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shi TT, Yu XX, Yan LJ, et al. Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother Pharmacol. 2017;79:287–294. doi: 10.1007/s00280-016-3197-1. [DOI] [PubMed] [Google Scholar]
- 144.Lin YX, Gao YJ, Wang Y, et al. pH-sensitive polymeric nanoparticles with gold(I) compound payloads synergistically induce cancer cell death through modulation of autophagy. Mol Pharm. 2015;12:2869–2878. doi: 10.1021/acs.molpharmaceut.5b00060. [DOI] [PubMed] [Google Scholar]
- 145.Goodall ML, Wang T, Martin KR, et al. Development of potent autophagy inhibitors that sensitize oncogenic BRAF V600E mutant melanoma tumor cells to vemurafenib. Autophagy. 2014;10:1120–1136. doi: 10.4161/auto.28594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.McAfee Q, Zhang Z, Samanta A, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci USA. 2012;109:8253–8258. doi: 10.1073/pnas.1118193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang Z, Zhang J, Wang Y, et al. Matrine, a novel autophagy inhibitor, blocks trafficking and the proteolytic activation of lysosomal proteases. Carcinogenesis. 2012;34:128–138. doi: 10.1093/carcin/bgs295. [DOI] [PubMed] [Google Scholar]
- 148.Wang J, Wu GS. Role of autophagy in cisplatin resistance in ovarian cancer cells. J Biol Chem. 2014;289:7163–17173. doi: 10.1074/jbc.M114.558288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ouyang L, Zhang L, Liu J, et al. Discovery of a small-molecule bromodomain-containing protein 4 (BRD4) inhibitor that induces AMP-activated protein kinase-modulated autophagy-associated cell death in breast cancer. J Med Chem. 2017;60(24):9990–10012. doi: 10.1021/acs.jmedchem.7b00275. [DOI] [PubMed] [Google Scholar]
- 150.Deng Y, Zhu L, Cai H, et al (2017) Autophagic compound database: a resource connecting autophagy-modulating compounds, their potential targets and relevant diseases. Cell Prolif (Epub ahead of print) [DOI] [PMC free article] [PubMed]
- 151.Xie T, Zhang L, Zhang S, et al. ACTP: a webserver for predicting potential targets and relevant pathways of autophagy-modulating compounds. Oncotarget. 2016;7(9):10015–10022. doi: 10.18632/oncotarget.7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lock R, Roy S, Kenific CM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2010;22:165–178. doi: 10.1091/mbc.e10-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Giovannini M, Bonne NX, Vitte J, et al. mTORC1 inhibition delays growth of neurofibromatosis type 2 schwannoma. Neuro Oncol. 2014;16:493–504. doi: 10.1093/neuonc/not242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yu K, Toral-Barza L, Discafani C, et al. mTOR, a novel target in breast cancer: the effect of CCI-779, an mTOR inhibitor, in preclinical models of breast cancer. Endocr Relat Cancer. 2001;8:249–258. doi: 10.1677/erc.0.0080249. [DOI] [PubMed] [Google Scholar]
- 155.Guo H, Zhong Y, Jackson AL, et al. Everolimus exhibits anti-tumorigenic activity in obesity-induced ovarian cancer. Oncotarget. 2016;7:20338–20356. doi: 10.18632/oncotarget.7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Becker MA, Hou X, Tienchaianada P, et al. Ridaforolimus (MK-8669) synergizes with Dalotuzumab (MK-0646) in hormone-sensitive breast cancer. BMC Cancer. 2016;16:814. doi: 10.1186/s12885-016-2847-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chang L, Graham PH, Hao J, et al. PI3K|[sol]|Akt|[sol]|mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. 2014;5:e1437. doi: 10.1038/cddis.2014.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhu Y, Tian T, Zou J, et al. Dual PI3K/mTOR inhibitor BEZ235 exerts extensive antitumor activity in HER2-positive gastric cancer. BMC Cancer. 2015;15:1–10. doi: 10.1186/1471-2407-15-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chen X, Zhao M, Hao M, et al. Dual inhibition of PI3K and mTOR mitigates compensatory AKT activation and improves tamoxifen response in breast cancer. Mol Cancer Res. 2013;11:1269–1278. doi: 10.1158/1541-7786.MCR-13-0212. [DOI] [PubMed] [Google Scholar]
- 160.Liu T, Sun Q, Li Q, et al. Dual PI3K/mTOR inhibitors, GSK2126458 and PKI-587, suppress tumor progression and increase radiosensitivity in nasopharyngeal carcinoma. Mol Cancer Ther. 2014;14:429–439. doi: 10.1158/1535-7163.MCT-14-0548. [DOI] [PubMed] [Google Scholar]
- 161.Simioni C, Cani A, Martelli AM, et al. The novel dual PI3K/mTOR inhibitor NVP-BGT226 displays cytotoxic activity in both normoxic and hypoxic hepatocarcinoma cells. Oncotarget. 2015;6:17147–17160. doi: 10.18632/oncotarget.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Thijssen R, Ter Burg J, van Bochove GG, et al. The pan phosphoinositide 3-kinase/mammalian target of rapamycin inhibitor SAR245409 (voxtalisib/XL765) blocks survival, adhesion and proliferation of primary chronic lymphocytic leukemia cells. Leukemia. 2015;30:337–345. doi: 10.1038/leu.2015.241. [DOI] [PubMed] [Google Scholar]
- 163.Qi W, Morales C, Cooke LS, et al. Reciprocal feedback inhibition of the androgen receptor and PI3K as a novel therapy for castrate-sensitive and -resistant prostate cancer. Oncotarget. 2015;6:41976–41987. doi: 10.18632/oncotarget.5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Singh AR, Joshi S, Burgoyne AM, et al. Single agent and synergistic activity of the “first in class” dual PI3K/BRD4 inhibitor SF1126 with Sorafenib in hepatocellular carcinoma. Mol Cancer Ther. 2016;15:2553–2562. doi: 10.1158/1535-7163.MCT-15-0976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Musa F, Alard A, David-West G, et al. Dual mTORC1/2 inhibition as a novel strategy for the re-sensitization and treatment of platinum-resistant ovarian cancer. Mol Cancer Ther. 2016;15:1555–1567. doi: 10.1158/1535-7163.MCT-15-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zeng JY, Sharma S, Zhou YQ, et al. Synergistic activities of MET/RON inhibitor BMS-777607 and mTOR inhibitor AZD8055 to polyploid cells derived from pancreatic cancer and cancer stem cells. Mol Cancer Ther. 2014;13(37):756–765. doi: 10.1158/1535-7163.MCT-13-0242. [DOI] [PubMed] [Google Scholar]
- 167.Willems L, Chapuis N, Puissant A, et al. The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia. 2011;26:1195–1202. doi: 10.1038/leu.2011.339. [DOI] [PubMed] [Google Scholar]
- 168.Guichard SM, Curwen J, Bihani T, et al. AZD2014, an inhibitor of mTORC1 and mTORC2, is highly effective in ER + breast cancer when administered using intermittent or continuous schedules. Mol Cancer Ther. 2015;14:37–44. doi: 10.1158/1535-7163.MCT-15-0365. [DOI] [PubMed] [Google Scholar]
- 169.Yu CC, Huang HB, Hung SK, et al. AZD2014 radiosensitizes oral squamous cell carcinoma by inhibiting AKT/mTOR axis and inducing G1/G2/M cell cycle arrest. PLoS One. 2016;11:e0151942. doi: 10.1371/journal.pone.0151942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Liao H, Huang Y, Guo B, et al. Dramatic antitumor effects of the dual mTORC1 and mTORC2 inhibitor AZD2014 in hepatocellular carcinoma. Am J Cancer Res. 2015;5:125–139. [PMC free article] [PubMed] [Google Scholar]
- 171.Zhi X, Chen W, Xue F, et al. OSI-027 inhibits pancreatic ductal adenocarcinoma cell proliferation and enhances the therapeutic effect of gemcitabine both in vitro and in vivo. Oncotarget. 2015;6:26230–26241. doi: 10.18632/oncotarget.4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zhang H, Dou J, Yu Y, et al. mTOR ATP-competitive inhibitor INK128 inhibits neuroblastoma growth via blocking mTORC signaling. Apoptosis. 2015;20:50–62. doi: 10.1007/s10495-014-1066-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hayman TJ, Wahba A, Rath BH, et al. The ATP-competitive mTOR inhibitor INK128 enhances in vitro and in vivo radiosensitivity of pancreatic carcinoma cells. Clin Cancer Res. 2014;20:110–119. doi: 10.1158/1078-0432.CCR-13-2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Pan H, Xu LH, Ouyang DY, et al. The second-generation mTOR kinase inhibitor INK128 exhibits anti-inflammatory activity in lipopolysaccharide-activated RAW 264.7 cells. Inflammation. 2014;37:756–765. doi: 10.1007/s10753-013-9794-9. [DOI] [PubMed] [Google Scholar]
- 175.Liu ZG, Tang J, Chen Z, et al. The novel mTORC1/2 dual inhibitor INK128 enhances radiosensitivity of breast cancer cell line MCF-7. Int J Oncol. 2016;49:1039–1045. doi: 10.3892/ijo.2016.3604. [DOI] [PubMed] [Google Scholar]
- 176.Janes MR, Vu C, Mallya S, et al. Efficacy of the investigational mTOR kinase inhibitor MLN0128/INK128 in models of B-cell acute lymphoblastic leukemia. Leukemia. 2013;27:586–594. doi: 10.1038/leu.2012.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kannan A, Lin Z, Shao Q, et al. Dual mTOR inhibitor MLN0128 suppresses Merkel cell carcinoma (MCC) xenograft tumor growth. Oncotarget. 2015;7:6576–6592. doi: 10.18632/oncotarget.5878. [DOI] [PMC free article] [PubMed] [Google Scholar]