

Abstract
Bcl-2 proteins have emerged as critical regulators of intracellular Ca2+ dynamics by directly targeting and inhibiting the IP3 receptor (IP3R), a major intracellular Ca2+-release channel. Here, we demonstrate that such inhibition occurs under conditions of basal, but not high IP3R activity, since overexpressed and purified Bcl-2 (or its BH4 domain) can inhibit IP3R function provoked by low concentration of agonist or IP3, while fails to attenuate against high concentration of agonist or IP3. Surprisingly, Bcl-2 remained capable of inhibiting IP3R1 channels lacking the residues encompassing the previously identified Bcl-2-binding site (a.a. 1380–1408) located in the ARM2 domain, part of the modulatory region. Using a plethora of computational, biochemical and biophysical methods, we demonstrate that Bcl-2 and more particularly its BH4 domain bind to the ligand-binding domain (LBD) of IP3R1. In line with this finding, the interaction between the LBD and Bcl-2 (or its BH4 domain) was sensitive to IP3 and adenophostin A, ligands of the IP3R. Vice versa, the BH4 domain of Bcl-2 counteracted the binding of IP3 to the LBD. Collectively, our work reveals a novel mechanism by which Bcl-2 influences IP3R activity at the level of the LBD. This allows for exquisite modulation of Bcl-2’s inhibitory properties on IP3Rs that is tunable to the level of IP3 signaling in cells.
Electronic supplementary material
The online version of this article (10.1007/s00018-019-03091-8) contains supplementary material, which is available to authorized users.
Keywords: Inositol 1,4,5-trisphosphate receptor; Calcium channels; Protein binding; Ligand–receptor interaction; Ligand-binding domain; Inhibition; Mechanism of interaction
Introduction
B-cell lymphoma-2 (Bcl-2) protein is the founding member of the Bcl-2 family of proteins and a well-known inhibitor of apoptosis [1–4]. Bcl-2 harbors all four Bcl-2-homology (BH1-4) domains, which are evolutionary conserved α-helical motifs and a shared feature in the family [5, 6]. Bcl-2 executes its pro-survival functions by a complex interaction network, employing the BH domains in various protein–protein interactions. The canonical mechanism of apoptosis inhibition by Bcl-2 involves the neutralization of the pro-apoptotic members of the family, exemplified by the executioners Bax and Bak or the BH3-only proteins such as Bim. This process occurs by sequestration of the BH3 domain of the pro-apoptotic members in the hydrophobic cleft of Bcl-2, which is formed by its BH3–BH1–BH2 domains [4, 7, 8]. In addition, prevention of Bax activation by Bcl-2 involves an interaction between the N-terminus of Bax and the BH4 domain of Bcl-2 [9]. Another mechanism to block apoptosis that is exploited by Bcl-2 is the modulation of intracellular Ca2+ signals [10–13]. Bcl-2 was shown to target the main Ca2+-release channel in cells, the inositol 1,4,5-trisphosphate (IP3) receptor (IP3R) and to suppress its activity. By decreasing the amount of Ca2+ released from the endoplasmic reticulum (ER), Bcl-2 prevents Ca2+-dependent apoptosis, triggered by various stimuli [14, 15]. The current model of IP3R inhibition by Bcl-2 involves multi-region binding interactions. To date, two distinct Bcl-2-binding regions on IP3R were identified. Bcl-2 targets the modulatory region, particularly the Fragment 3 (a.a. 923–1581, according to mouse IP3R1) [16–18], which results from the in vitro chymotrypsinization or trypsinization of the receptor [19, 20] and which predominantly consists of the ARM2 domain [21]. Bcl-2 also binds to the C-terminal region, particularly to a fragment containing the cytosolic tail and the sixth transmembrane domain (TMD) of IP3R (C-terminus, a.a. 2512–2749, according to mouse IP3R1) [17, 22]. The binding of Bcl-2 to the Fragment 3 of IP3R occurs via its BH4 domain, which appeared to target a particular stretch of 20 residues (a.a. 1389–1408, according to the mouse and rat IP3R1). This interaction is considered to be responsible for the inhibition of IP3Rs by Bcl-2 as various techniques have demonstrated that the isolated BH4 domain of Bcl-2 was sufficient to bind and inhibit the IP3R [18, 23, 24]. A more recent study revealed that the C-terminal TMD of Bcl-2 is required for efficient in-cellulo inhibition of IP3R activity and prevention of subsequent Ca2+-mediated cell death [25]. The latter finding was an important step in clarifying the mechanism of highly effective regulation of Ca2+-dependent apoptosis by Bcl-2 in intact cells. Nevertheless, the full complexity of the interaction between Bcl-2 and IP3R is still not completely understood and requires further investigation. In particular, according to the published cryo-electron microscopic structure of full-length IP3R1 [21, 26], the binding site in the Fragment 3 and the C-terminus are located at relatively large distance apart, making it difficult to explain how one Bcl-2 molecule could occupy both sites at the same time.
Bcl-2 was also reported to distinguish between patterns of distinct cellular Ca2+ signals, inhibiting pro-apoptotic, high-amplitude Ca2+ elevations, but not pro-survival Ca2+ oscillations [27]. However, the mechanism of this process remains unresolved. Interestingly, it was previously demonstrated that Bcl-2 suppresses intracellular Ca2+ signals, triggered by submaximal, but not by supramaximal concentrations of agonist [15]. This suggests that anti-apoptotic Bcl-2 may lose its IP3R-inhibitory effect in conditions of strong IP3R activation, though the underlying mechanisms responsible for this effect remain unknown. Here, we demonstrate that high concentrations of IP3 or of agonists alleviate the inhibitory effect of the BH4 domain of Bcl-2, as well as of the recombinant purified or overexpressed Bcl-2, on IP3R activity in various experimental settings. Furthermore, the deletion of the stretch targeted by Bcl-2 (a.a. 1389–1408) in full-length IP3R1 failed to prevent the Bcl-2-mediated inhibition of IP3R1. These data suggest the presence of an additional region in IP3R as a target of Bcl-2, whose interaction can be modulated by IP3 levels. Using a computational modelling approach, we found that (1) Bcl-2’s BH4 domain could form a complex with the ligand-binding domain (LBD) and (2) the conformational changes in the LBD induced by IP3 binding hindered this interaction. This was supported by different experimental approaches, demonstrating that the LBD (a.a. 1–604), and more particular the IP3-binding core (IBC, a.a. 226–604) of IP3R1 could bind full-length Bcl-2 or its BH4 domain, produced as synthetic peptide. Furthermore, IP3 and adenophostin A (AdA), two IP3R ligands, interfered with the binding of Bcl-2 and its BH4 domain to the LBD. In line, the BH4 domain of Bcl-2 attenuated the binding of IP3 to the LBD. Thus, the present study represents an important contribution towards revealing the molecular mechanism of interaction between the anti-apoptotic protein Bcl-2 and IP3R.
Materials and methods
Peptides
The following peptides, obtained from LifeTein (Hillsborough, NJ, USA) with purity ≥ 85% were used: BH4-Bcl-2: RTGYDNREIVMKYIHYKLSQRGYEW and the control scrambled peptide BH4-Bcl-2-CTR: WYEKQRSLHGIMYYVIEDRNTKGYR. N-terminally biotinylated peptides (biotin-BH4-Bcl-2 and biotin-BH4-Bcl-2-CTR) were used for SPR.
Plasmids, constructs and protein purification
The pCMV construct containing the genetically encoded Ca2+ sensor CEPIA targeted to the ER lumen (G-CEPIA1-ER) was a gift from Dr. Masamitsu Iino (Addgene plasmid #58215) [28]. pCMV24-3xFLAG-Myc constructs for expression of 3xFLAG–Bcl-2 were obtained as previously described [29]. pcDNA3 construct for expression of rat IP3R1 lacking the binding site for BH4 of Bcl-2 in Fragment 3 (IP3R1Δ1389–1408) was developed using Q5 mutagenesis protocol with the following primers pair: Forward: GCTGTGTGCACAGAGGGCAAGGTCACTCATGAAGACTGTATC Reverse: GATACAGTCTTCATGAGTGACCTTGCCCTCTGTGCACACAGC.
BL21(DE3) Escherichia coli bacteria were transformed with pGEX-6p2 constructs containing cDNAs of parental GST, GST-LBD (a.a. 1–604), GST-SD (a.a. 1–225), GST-IBC (a.a. 226–604), GST-Fragment 3 (a.a. 923–1581) or GST-C-terminus (a.a. 2512–2749), which were obtained as previously described [17, 30]. The expressed parental GST or GST-fusion proteins were purified as previously described [17, 30].
BL21(DE3) E. coli bacteria were transformed with pET47-Bcl-2Δ23 plasmid, which was kindly provided by Dr. Varda Shoshan-Barmatz (Ben-Gurion University of the Negev, Israel). After growing overnight at 37 °C, bacteria were diluted to an A600 of 0.2 and incubated at 40 °C for 2 h (heat shock). Protein expression was induced by isopropyl-d-1-thiogalactopyranoside (100 μM) at 20 °C for 2 h. Bacteria were harvested by centrifugation at 5000g for 5 min and lysed in a buffer containing 150 mM NaCl, 10 mM Tris pH 7.4, 20% glycerol, 30 mM imidazole. Samples were sonicated and centrifuged at 35,000 rpm for 40 min. Supernatants were collected and incubated with a nickel-nitrilotriacetic acid resin (Ni–NTA agarose) for 1 h at 4 °C. The recombinant N-terminal His-tagged and C-terminal truncated Bcl-2 (6xHis-Bcl-2) were eluted with lysis buffer containing 500 mM imidazole.
The purified GST- and His-tagged proteins were dialyzed against standard phosphate-buffered saline (PBS) without Ca2+ and Mg2+ (Invitrogen, Merelbeke, Belgium) using Slide-A-Lyzer cassettes with a cutoff of 10 kDa (Thermo Fisher Scientific, Pittsburg, PA, USA). The concentration of the purified and dialyzed proteins was determined using the Bradford assay (Sigma-Aldrich, Munich, Germany). Purity and quality were assessed after SDS–PAGE via total protein staining using the Imperial Protein Stain (Thermo Fisher Scientific, Pittsburg, PA, USA) following the manufacturer’s recommendations.
Cell culture and transfections
All media and supplements used in this paper were purchased from Life Technologies (Ghent, Belgium) unless stated otherwise.
COS-1 cells used for GST-pull downs and single-cell measurements were cultured at 37 °C, 10% CO2 in Dulbecco’s Modified Eagle’s medium (DMEM), containing 10% fetal calf serum (Sigma-Aldrich), 100 IU/ml penicillin, 100 µg/ml streptomycin, 2.5 µg/ml fungizone and 2 mM glutamax. 24 h after seeding COS-1 cells were transiently transfected with empty pCMV24-3xFLAG-Myc (3xFLAGempty) or with 3xFLAG-Bcl-2 plasmids, containing mCherry and the p2a sequence (3xFLAG-p2a-mCherry and 3xFLAG-Bcl-2-p2a-mCherry) [29]. X-tremeGene HP DNA (Roche, Basel, Switzerland) was used as a transfection reagent according to the manufacturer’s instructions.
COS-7 cells used for the competitive fluorescent ligand assay were obtained from the RIKEN Cell Bank (Tokyo, Japan) and were cultured in DMEM with 1000 µg/ml glucose (Sigma-Aldrich), and supplemented with 10% fetal calf serum (Gibco BRL, Rockville, MD, USA), 100 IU/ml penicillin (Gibco BRL) and 100 µg/ml streptomycin (Gibco BRL). Cells were transfected with 5 µg/ml of plasmid using LipofectAMINE 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
MEF cells were cultured at 37 °C in a 10% CO2 incubator in DMEM/Ham’s F12 medium supplemented with 10% fetal calf serum, 3.8 mM l-glutamine, 85 IU/ml penicillin and 85 μg/ml streptomycin.
DT40 cells lacking all three IP3R isoforms (DT40-3KO) with ectopically expressing IP3R1 [31] were used for the nuclear patch-clamp experiments. The cells were cultured at 39 °C in a 5% CO2 incubator in RPMi medium supplemented with 10% fetal calf serum, 1% chicken serum, 2.05 mM l-glutamine, 100 IU/ml penicillin and 100 μg/ml streptomycin.
The previously described HEK cells lacking all three IP3R isoforms (HEK-3KO) [32], were used for single-cells Ca2+ measurements. HEK-3KO were co-transfected with pcDNA3-IP3R or pcDNA3-IP3RΔ1389–1408 and 3xFLAG-p2a-mCherry or 3xFLAG-Bcl-2-p2A-mCherry.
GST-pull down assays
48 h after transfection COS-1 cells overexpressing 3xFLAG-Bcl-2 were harvested and lysed in a buffer containing 25 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1.5 mM MgCl2, 0.5 mM DTT, 1% Triton X-100 and protease inhibitor cocktail tablets (Roche). After 30 min of incubation at 4 °C the clear lysates were collected via centrifugation for 2 min at 10,000 rpm at 4 °C. Parental GST or GST-fused fragments of IP3R1 (0.5 µM) were incubated together with 100 µg lysate in the lysing buffer (final volume 500 µl) at 4 °C. After 1 h, the GST-proteins used as bait, were immobilized on glutathione-Sepharose 4B beads (GE Healthcare, Diegem, Belgium) for 1.5 h at 4 °C. To study the effect of the AdA, 3 µM AdA (Merck Chemicals, Belgium) or the vehicle control (MQ water) was added during the incubation. The beads were washed five times with the Triton X-100 buffer. The GST-complexes were eluted in 40 µl 2 × LDS (Life Technologies) supplemented with 1:200 β-mercaptoethanol by boiling for 5 min at 95 °C. Samples (10 µl) were analyzed via SDS-PAGE, using mouse monoclonal HRP-conjugated anti-FLAG M2 (1:1000; Sigma-Aldrich) and the quantification was performed as previously described.
SPR experiments
SPR analysis was performed using a Biacore T200 (GE Healthcare, Diegem, Belgium). Immobilization of biotin-BH4-Bcl-2 and biotin-BH4-Bcl-2-CTR to the streptavidin-coated sensor chip (BR-1005-31; GE Healthcare) and SPR measurements were performed as described previously [18]. NaOH (50 mM) with 0.0026% SDS was used as a regeneration buffer. The IP3 and AdA were added at the indicated concentration to the analytes.
Single-cell cytosolic Ca2+ imaging
Fura-2-AM [Ca2+] measurements in COS-1 and HEK-3KO cells were performed as previously described [25]. BAPTA (3 mM) was added for 1 min prior to stimulation with ATP or CCH to chelate all free extracellular Ca2+. Cytosolic Ca2+ rises in response to 0.5 μM and 100 μM ATP or 10 μM CCH were measured in mCherry-positive (excitation 546 nm, emission 610 nm) and Fura-2-loaded cells. Intracellular cytoplasmic Ca2+ concentrations were calculated as previously described [18].
Single-cell ER Ca2+ imaging
The G-CEPIA1-ER construct was introduced into COS-1 cells as described above. A Zeiss Axio Observer Z1 Inverted Microscope equipped with a 20 × air objective and a high-speed digital camera (Axiocam Hsm, Zeiss, Jena, Germany) were used for these measurements. Changes in fluorescence were monitored in the GFP channel (480/520 excitation/emission). To chelate extracellular Ca2+, 3 mM BAPTA (Alfa Aesar, Ward Hill, MA, USA) was added. One min later 0.5 or 100 μM ATP was added. All traces were normalized (F/F0) where F0 is the baseline fluorescence of each trace.
Unidirectional 45Ca2+-flux assay
The unidirectional 45Ca2+-flux experiments were performed in permeabilized MEFs as previously described [18]. IICR was triggered during the unidirectional 45Ca2+-efflux phase by the addition of the indicated [IP3] for 2 min. BH4-Bcl-2 peptide was added 2 min before IP3 till 2 min after IP3. IICR was plotted as fractional loss, representing the amount of actively accumulated Ca2+ leaving the stores during the sampling period [33].
Electrophysiology
Isolated nuclei from DT40-3KO cells stably transfected with rabbit IP3R1 were prepared by homogenization as previously described [31]. A 3 μl aliquot of nuclear suspension was placed in 3 ml of bath solution which contained 140 mM KCl, 10 mM Hepes, 500 μM BAPTA and 246 nM free Ca2+, pH 7.1. Nuclei were allowed to adhere to a plastic culture dish for 10 min prior to patching. Single IP3R1 channel potassium currents were measured in the on-nucleus patch-clamp configuration using pCLAMP 9 and an Axopatch 200B amplifier (Molecular Devices, Sunnydale, CA, USA). Pipette solution contained 140 mM KCl, 10 mM Hepes, 1–10 μM IP3, 5 mM ATP, and 200 nM free Ca2+ as well as BH4-Bcl-2 peptide or 6xHis-Bcl-2 where noted. Traces were consecutive 3 s sweeps recorded at − 100 mV, sampled at 20 kHz and filtered at 5 kHz. A minimum of 15 s of recordings were considered for data analyses. Pipette resistances were typically 20 MΩ and seal resistances were > 5 GΩ. Single-channel openings were detected by half-threshold crossing criteria using the event detection protocol in Clampfit 9. We assumed that the number of channels in any particular nuclear patch is represented by the maximum number of discrete stacked events observed during the experiment. Only patches with one apparent channel were considered for analyses. Po was calculated using Clampfit 9 and Origin 6 software (Origin Lab, Northampton, MA, USA). Error bars are SEM.
In silico docking study
The ZDock server version ZD3.0.2 [34] was used to perform a targeted docking experiment with PDB-entry 3UJ4 as template for IP3R1 in its apo-form [35], and PDB-entry 4LXD as template for Bcl-2 [36]. Seven possible complexes were obtained. Analysis of these complexes reveals that only two complexes did not result in clashes between the LBD of IP3R and Bcl-2. FiberDock [37] was then used to refine these complexes. The Fiberdock output statistics are given in Supplementary Material. Next, we superimposed the resulting complex with the IP3-bound structure of IP3R (PDB-entry 3T8S [38]). PyMol (The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC) was used for visual inspection, superposition and generation of the figures.
Competitive fluorescent ligand assay
COS-7 cells were grown in experimental chambers consisting of 7-mm plastic cylinders and fibronectin-coated cover slips. Permeabilization was performed by exposing cells to intracellular-like medium (ICM: 125 mM KCl, 19 mM NaCl, 10 mM Hepes–KOH, pH 7.3, 1 mM EGTA, and 330 µM CaCl2) containing 200 µg/ml (w/v) saponin (ICN, Cleveland, OH, USA) for ~ 90 s. Permeabilized cells were washed with ICM and then exposed to ICM containing various concentrations of IP3 or other reagents. Fluorescence images were captured using a dual-wavelength ratio imaging system consisting of an EM-CCD camera (C9135-special; Hamamatsu photonics, Shizuoka, Japan) and W-View (Hamamatsu photonics) optics coupled to a Nikon TE2000 inverted fluorescence microscope equipped with a Nikon S Fluor 60 oil immersion objective (NA 1.25). Fluorescence was monitored with excitation at 425 nm and dual-emission at 480 nm and 535 nm. Data were analyzed with AQUACOSMOS 2.6 software (Hamamatsu photonics). All the experiments were performed at room temperature.
Statistical analysis
Two-tailed unpaired Student’s t tests were used to compare two conditions. When comparing three or more conditions a repeated-measure ANOVA with Bonferroni post test was performed. *Indicates significantly different results with p < 0.05.
Results
Bcl-2 fails to inhibit IP3R-mediated Ca2+ release triggered by high concentrations of agonist in intact cells
Various studies have demonstrated that Bcl-2 overexpression dampens agonist-induced Ca2+ release in multiple intact cell models [15, 18, 39]. Nevertheless, most of those experiments were performed using submaximal concentrations of agonist. Here, we compared the impact of Bcl-2 overexpression on IP3R-mediated Ca2+ signaling triggered by a range of ATP concentrations. Using the ratiometric fluorescent Ca2+ dye Fura-2, we monitored the cytosolic [Ca2+] rises in COS-1 cells transfected either with an empty vector or with 3xFLAG-Bcl-2. As a selection marker we used mCherry encoded in the same plasmid, but separated by the p2A sequence to eventually yield two separate expressed proteins, 3xFLAG(-Bcl-2) and mCherry [29]. Thus, all cells that express the mCherry protein also express 3xFLAG-Bcl-2. The free extracellular [Ca2+] was reduced to < 10 nM by the addition of BAPTA (3 mM) ensuring that the ATP-induced [Ca2+] rise is only due to Ca2+ release from intracellular stores. As previously shown, 3xFLAG-Bcl-2 decreased the cytosolic [Ca2+] rises in response to a submaximal ATP concentration (0.5 μM) (Fig. 1a). In contrast, when a high concentration (100 μM) of the agonist was applied, 3xFLAG-Bcl-2 failed to inhibit the Ca2+ release (Fig. 1b, Fig. S1), in good agreement with a previous report [15]. In addition to the decreased capacity of Bcl-2 to inhibit agonist-induced [Ca2+] rises at high [ATP] (Fig. 1c), we observed a decrease in the number of responding cells in that condition (Fig. 1d). From our raw data, it is clear that the increase in Fura-2 ratio provoked by the addition of 100 µM ATP is much lower than the increase in Fura-2 ratio provoked by ionomycin, which provokes a maximal intracellular [Ca2+] rise (Fig. S1). This excludes that the lack of Bcl-2-mediated inhibition of Ca2+ release triggered by 100 μM ATP is due to a saturation of the Fura-2 signal.
Fig. 1.

Cytosolic [Ca2+] rises triggered by submaximal, but not supramaximal concentration of the agonist, are suppressed by Bcl-2. a, b Cytosolic Ca2+ release in response to 0.5 (a) or 100 (b) μM ATP was measured in the mCherry-positive Fura-2-loaded COS-1 cells overexpressing 3xFLAG-empty vector or 3xFLAG-Bcl-2. The free extracellular Ca2+ was chelated by addition of 3 mM BAPTA. The obtained Fura-2 fluorescence signals (F340/F380) were calibrated and representative traces are plotted as [Ca2+]. c Quantitative analysis of the amplitude of the ATP-induced Ca2+ signals from at least three independent experiments, which were performed on different days after independent transfections (total number of cells for each [ATP] > 90) is plotted as mean ± SEM. d Quantitative analysis of the number of cells responding to the agonist. e, f Decrease in the ER Ca2+ content in response to 0.5 (e) or 100 (f) μM ATP was monitored using G-CEPIA1-ER in mCherry-positive COS-1 cells, overexpressing 3xFLAG-empty vector or 3xFLAG-Bcl-2. Extracellular Ca2+ was chelated by the addition of 3 mM BAPTA. Typical normalized (F/F0) traces of the obtained fluorescence signals are presented. g Quantitative analysis of the ER Ca2+ decrease in response to ATP. For each trace, the ATP-induced Ca2+ release was determined by subtracting the fluorescence after ATP addition (during plateau phase) from the fluorescence just before ATP addition after normalization. Values depict average ± SEM from at least three independent experiments, which were performed on different days after independent transfections (total number of cells for each [ATP] > 90). h Quantitative analysis of the number of cells responding to the agonist expressed as a % of the total number of measured cells. *Stands for p < 0.05
The overall increase in the cytosolic [Ca2+] triggered by ATP is influenced by the net Ca2+ release from the ER into the cytosol and its extrusion from the cytosol to other compartments such as the mitochondria or to the extracellular medium. Furthermore, in living cells, Bcl-2 is well known to modulate other Ca2+-transport systems beside IP3Rs [11], including the Ca2+ flux into the mitochondria [40]. Therefore, to strengthen the contention that the decrease in ATP-induced [Ca2+] rises in the cytosol caused by Bcl-2 overexpression is due to an inhibition of Ca2+ flux from the ER and not to altered Ca2+ extrusion from the cytosol, we directly monitored the Ca2+ dynamics in the ER using the genetically encoded Ca2+ sensor G-CEPIA1-ER. We challenged COS-1 cells expressing G-CEPIA1-ER together with either 3xFLAG-vector or 3xFLAG-Bcl-2 with 0.5 or 100 μM ATP and determined the decrease in the ER Ca2+ content. Compared to the control cells, Bcl-2-overexpressing cells displayed a lower decrease in ER Ca2+ levels when evoked by 0.5 μM ATP, but not by 100 μM ATP (Fig. 1e–g). Similar results were obtained by analyzing the % of responding cells (Fig. 1h), which indicated that in living cells Bcl-2 inhibits Ca2+ flux from the ER triggered by low, but not by high [ATP]. Nevertheless, the cytosolic [Ca2+] rise measured by Fura-2 appeared more strongly suppressed by Bcl-2 overexpression than the ER [Ca2+] drop measured by G-CEPIA1-ER. This may be due to differences in the dynamic range at which the measurements were performed or may be the result of the impact of Bcl-2 on non-ER Ca2+-transport systems.
The IP3R-inhibitory properties of purified Bcl-2 or of its BH4 domain decline with increasing [IP3] or [agonist]
Next, we aimed to measure the direct effect of Bcl-2 on the activity of IP3R1 channels in response to different concentrations of IP3. We conducted IP3R1 single-channel recordings using the nuclear-membrane patch-clamp technique on isolated nuclei obtained from DT40-3KO cells ectopically expressing IP3R1. When 1 μM IP3 was used to trigger IP3R opening, application of 1 μM purified Bcl-2 (6xHis-Bcl-2) decreased the open probability (Po) of IP3R1 by four fold, while when 10 μM IP3 was applied 6xHis-Bcl-2 did not affect the channel activity (Fig. 2a–c). This observation also strongly advocates a direct effect of Bcl-2 on the IP3R in our results obtained with intact cells (Fig. 1).
Fig. 2.

Recombinant Bcl-2 and BH4-Bcl-2 fail to inhibit IP3R activity induced by high concentrations of IP3 in single-channel measurements. a, b Representative IP3R1 single-channel recordings from DT40-3KO cells ectopically expressing IP3R1. The channel opening was evoked by 1 (a) or 10 (b) μM IP3 at 200 nM Ca2+ and 5 mM ATP, in the presence of PBS (a, b left) or in the presence of 1 μM 6xHis-Bcl-2 purified protein, which was dialysed against PBS (a, b right). c Histogram depicting the Po ± SEM for the IP3R1 under the described conditions. The total number of recordings for each condition is indicated within every bar. d, e Representative IP3R1 single-channel recordings from DT40-3KO cells ectopically expressing IP3R1. The channel opening was evoked by 1 (d) or 5 (e) μM IP3 at 200 nM Ca2+ and 5 mM ATP, in DMSO control condition (d, e left) or in presence of 50 μM BH4-Bcl-2 peptide (d, e right). f Histogram depicting the Po ± SEM for the IP3R1 under the described conditions. The total number of recordings for each condition is indicated within every bar. *Stands for p < 0.05. The single-channel recordings presented on panel d and the corresponding analyzed data on panel f for 1 μM IP3 with and without 50 μM BH4-Bcl-2 were obtained from exactly the same data set, as previously published in [39]. All other results represent newly acquired data
The BH4 domain of Bcl-2 has been documented to be sufficient to inhibit IP3R-mediated Ca2+ release in various experimental settings, including Ca2+ imaging in intact cells, unidirectional 45Ca2+ fluxes in permeabilized cells and single-channel recordings using nuclear patch [18, 23, 39]. In all these conditions, submaximal concentrations of IP3 or agonist were applied to activate IP3R channels. Here, we assessed the ability of Bcl-2’s BH4 domain, produced as a synthetic peptide (BH4-Bcl-2), to inhibit IP3R channels that were activated by varying concentrations of IP3. The obtained results were in line with our data with recombinant Bcl-2. While as previously shown, 50 μM BH4-Bcl-2 strongly decreased the Po triggered by 1 μM IP3 [39], the peptide BH4-Bcl-2 failed to decrease the Po of IP3R1 triggered by 5 μM IP3 (Fig. 2d–f).
Finally, we monitored the effect of the BH4-Bcl-2 on IP3-induced Ca2+ release (IICR) by performing unidirectional 45Ca2+ flux assays in saponin-permeabilized MEFs (Fig. 3). These cells have low endogenous levels of Bcl-2 [16] and as such, they are a good system to study the activity of exogenous Bcl-2-protein functions. The non-mitochondrial Ca2+ stores were loaded with 45Ca2+ and the unidirectional Ca2+ flux was measured in the presence of EGTA (1 mM) and thapsigargin (4 µM). We quantified 45Ca2+ release triggered by sub- and supramaximal concentrations of IP3 in presence or absence of different concentrations of the BH4-Bcl-2 peptide. BH4-Bcl-2 was added from 2 min before until 2 min after IP3 application. The results are plotted as the fractional loss (%/2 min) over time. Consistent with previous reports, in these experiments BH4-Bcl-2 suppressed IICR triggered by a submaximal concentration of IP3 (2 µM) with an IC50 of about 20 μM (Fig. 3a). Surprisingly, the peptide appeared to be a less potent inhibitor of IICR when a higher concentration of IP3 (4 µM) was applied (Fig. 3b). At IP3 concentrations of 10 µM (Fig. 3c) and 50 µM (Fig. 3d), BH4-Bcl-2 failed to inhibit IICR (Fig. 3e).
Fig. 3.

The BH4-Bcl-2 peptide fails to inhibit IICR triggered by high IP3 concentrations. a–d Typical experiment of unidirectional 45Ca2+ fluxes in permeabilized MEFs. Ca2+ release was induced by 2 (a), 4 (b), 10 (c) or 50 (d) μM IP3 (the time of addition is indicated with a black bar) in control condition or in presence of different concentrations of BH4-Bcl-2 peptide (the time of addition is indicated with a grey bar). The results are plotted as fractional loss after 2 min of incubation with IP3 minus the fractional loss before the addition of IP3 (%/2 min) as a function of time. e Concentration–response curve of the IICR as quantified from four independent experiments, performed on independently grown cell cultures. The values of IICR measured as fractional loss were calculated as percentage of the IICR in control condition (vehicle), which was set as 100%. *Stands for p < 0.05
Taken together, our data clearly show that Ca2+ release evoked by high concentrations of IP3 is not a subject to inhibition by Bcl-2 or by its BH4 domain.
Bcl-2 remains capable of inhibiting IP3R1 lacking a.a. 1389–1408
This amino acid stretch (a.a. 1308-1408), previously identified as responsible for the interaction of IP3R1 with Bcl-2, is highly conserved among different IP3R isoforms and species and even other intracellular Ca2+-release channels, such as ryanodine receptors [18, 41, 42]. Therefore, we anticipated that deleting this region in the full-length IP3R1 channel would completely abrogate the ability of Bcl-2 to inhibit IP3R1-mediated Ca2+ release. To test this hypothesis we transfected HEK-3KO cells in which all three IP3R isoforms have been deleted by CRISPR/Cas9 [32] with wild-type IP3R1 or IP3R1Δ1389–1408 and 3xFLAG-Bcl-2-p2A-mCherry or empty plasmid (Fig. 4a). The expression levels of IP3R1Δ1389–1408 appeared lower than those of the wild-type IP3R1. A ratio IP3R1:mCherry 3:1 was used, ensuring that all mCherry expressing cells contain IP3R1 or IP3R1Δ1389–1408. We compared the effect of Bcl-2 overexpression on wild-type IP3R1 versus IP3R1Δ1389–1408-mediated Ca2+ release triggered by 10 μM carbachol (CCH) by performing similar Fura-2-based measurements of [Ca2+] as described above. As expected, 3xFLAG-Bcl-2 decreased the number of responding cells expressing wild-type IP3R1 as well as the magnitude of cytosolic [Ca2+] rises in response to 10 μM CCH (Fig. 4b). Importantly, cells expressing IP3R1Δ1389–1408 were also responsive to CCH stimulation, although less cells responded and the response exhibited a lower amplitude compared to the cells expressing the wild-type IP3R1 (Fig. 4c). This could be due to the lower activity of the IP3R1Δ1389–1408 and/or its lower expression relative to the wild-type IP3R1 (Fig. S2). In any case, these data indicate that the IP3R1Δ1389–1408 is functional and thus can be used to assess whether or not its Ca2+-flux properties can be inhibited by Bcl-2. Importantly, despite the fact that the IP3R1Δ1389–1408 lacks the Bcl-2-binding site in the modulatory region, Ca2+ release mediated by this mutated channel could still be reduced by 3xFLAG-Bcl-2 overexpression (Fig. 4d, e). These findings suggest that Bcl-2 and its BH4 domain might target additional region(s) outside of the a.a. stretch 1389–1408 in IP3R1.
Fig. 4.

Bcl-2 inhibits IP3R lacking the Bcl-2-binding site (a.a. 1389–1408) in the modulatory region. a, b The cytosolic Ca2+ release in response to 10 μM CCH was measured in the mCherry-positive Fura-2 cells, co-transfected with 3xFLAG-empty vector or 3xFLAG-Bcl-2 and IP3R (a) or IP3RΔ1389–1408 (b). The free extracellular Ca2+ was chelated by addition of 3 mM BAPTA. The obtained Fura-2 fluorescence signals (F340/F380) were calibrated and representative traces are plotted as [Ca2+]. c Quantitative analysis of the amplitude of the CCH-induced Ca2+ signals from at least three independent experiments performed on independently transfected cell batches (total number of cells > 100 cells) is plotted as mean ± SEM. d Quantitative analysis of the number of cells responding to the agonist. *Stands for p < 0.05
In silico docking of 3D structures reveals a possible complex formation between the BH4 domain of Bcl-2 with the apo-, but not with the IP3-bound form of the LBD of IP3R1
Based on the functional data, suggesting that (1) IP3 levels can modulate the extent of the action of Bcl-2 on the IP3R and (2) Bcl-2, via its BH4 domain could interact with IP3R outside of the previously described binding site in the modulatory region, we hypothesized that this additional target might be the LBD. We first applied a modeling approach to assess complex formation between the BH4 domain of Bcl-2 and the LBD of IP3R1 and performed a targeted docking experiment with ZDock [34]. Using the PDB-entries of Bcl-2 (4LXD, [36]) and of the apo-form of the LBD of IP3R1 (3UJ4, [35]) as templates, we obtained seven possible complexes. Two of them were very alike, and in both models, Bcl-2 did not clash with the LBD of IP3R. We used FiberDock [37] to determine the most probable complex (the global energy of each complex is given in Supplementary Material). This low-energy model is presented in Fig. 5a. Next, we compared the conformational changes in the LBD after IP3 binding (PDB-entry 3T8S, [38]) with the apo-form of the LBD of IP3R1. IP3 binds in a cleft that is formed by the two domains within the LBD, the suppressor domain (SD) and the IBC. This cleft closes and becomes narrower when IP3 is bound (Fig. 5b). Superposition of the docked complex between Bcl-2 and the apo-form of IP3R1 and the IP3-bound form of IP3R1 revealed that Bcl-2 might not be able to bind to the LBD in the presence of IP3. This might be due to spatial constraints, which result from conformational changes in IP3R1 upon IP3 binding; especially, the cleft between the two domains of the LBD is rendered too narrow for Bcl-2 to bind (Fig. 5c).
Fig. 5.

Molecular docking of Bcl-2 and its BH4 domain on the LBD of human IP3R1. The color scheme: blue—LBD of apo IP3R (PDB-entry 3UJ4, [35]); green—LBD of IP3-bound IP3R (PDB-entry 3T8S, [38]); orange—Bcl-2 (PDB-entry 4LXD, [36]); bright orange—BH4 of Bcl-2. The positively charged K508 residue, which was previously shown to be critical for IP3 binding [45], is indicated. a Proposed model of the interaction between Bcl-2 and the LBD of the apo-form of IP3R1. The BH4 domain of Bcl-2 is colored in bright orange and its residues are represented as sticks. b Superposition of the LBD of IP3R in its apo- and IP3-bound form. IP3 binds the cleft between the two subdomains of the LBD. This cleft is narrower when IP3 is bound. c Binding of Bcl-2 to the IP3-bound form of the LBD of IP3R1 based on the proposed model of the interaction between Bcl-2 and the apo-form of IP3R1. The narrower IP3 binding cleft results in clashes between IP3R and Bcl-2
Bcl-2 targets the IBC of IP3R1 via its BH4 domain
We examined whether Bcl-2 is able to directly bind to LBD and/or its subdomains, IBC and SD. Using purified GST-fused IP3R1 fragments as bait, we performed GST-pull down experiments with lysates from COS-1 cells overexpressing 3xFLAG-Bcl-2. The GST-Fragment 3 and GST-C-terminus served as positive controls for Bcl-2 binding, while the parental GST formed the negative control. 3xFLAG-Bcl-2 bound with similar efficiency to the GST-Fragment 3, GST-C-terminus, GST-LBD and GST-BC, while a less prominent binding to the GST-SD was observed (Fig. 6a, b).
Fig. 6.

The LBD of IP3R is a novel target of full-length Bcl-2 and its BH4 domain. a Representative GST-pull down experiments for assessing the binding of 3xFLAG-Bcl-2 from COS-1 cell lysate to GST-fused IP3R1 fragments corresponding to the Fragment 3, C-terminus, LBD, IBC and SD. The samples were analyzed via Western blot and stained with anti-FLAG antibody. Total COS-1 lysates (0.2 μg) were used as input. b The immunoreactive bands from three independent experiments, using each time independently transfected cells and freshly prepared lysates, were quantified and normalized to the binding of 3xFLAG-Bcl-2 to GST-Fragment 3, which was set as 1. The data are plotted as mean ± SEM. *Stands for p < 0.05. c–e Representative sensorgrams of the surface plasmon resonance experiments expressed in resonance units (RU) as a function of time. The biotin-BH4-Bcl-2 peptide and the scrambled peptide (biotin-BH4-Bcl-2-CTR) were immobilized on different channels of a streptavidin-coated sensor chip. The channels on the chip were exposed to the indicated concentrations of purified GST-LBD (c), GST-IBC (d) and GST-SD (e). Sensorgrams are obtained after background correction for binding to the respective scrambled versions of the biotinylated BH4-domain peptide. f Quantitative analysis of the binding properties of biotin-BH4-Bcl-2 to the different GST-domains of IP3R1. Values were obtained from three independent experiments (i.e., three independent immobilizations of the peptides) and are plotted as mean ± SEM
Second, we aimed to assess whether the BH4 domain of Bcl-2 directly binds to the LBD of IP3R1 using the surface plasmon resonance (SPR) technique. We used a biotinylated peptide that covers the BH4 domain of Bcl-2 (biotin-BH4-Bcl-2), which was immobilized on a streptavidin chip and we monitored its binding to purified GST-LBD (Fig. 6c), GST-IBC (Fig. 6d) and GST-SD (Fig. 6e) of IP3R1. As control analytes, we used parental GST (negative control) and GST-Fragment 3 (positive control), which was described as the main Bcl-2-binding region on IP3R1. The signals were corrected by subtracting the background binding to a control scrambled peptide, immobilized on another channel on the same chip. The generated response curves displayed a concentration-dependent increase in resonance units (RU) for the positive control GST-Fragment 3, GST-LBD and GST-BC, indicating a specific binding of biotin-BH4-Bcl-2 to these fragments (Fig. 6f). With GST-SD the increase in RU was significantly less, while parental GST did not show any substantial binding to the biotin-BH4-Bcl-2.
IP3 and AdA disturb the interaction between BH4-Bcl-2 or full-length Bcl-2 and the LBD of IP3R1
The results obtained above indicated that overexpressed Bcl-2 directly targets the LBD of IP3R1. Furthermore, we narrowed down the interacting region to the IBC, containing the determinants of IP3 binding. To test whether ligands binding to the LBD can disturb the interaction between Bcl-2 and LBD, we performed GST-pull down experiments in the presence of AdA, a more stable and potent IP3R agonist [43]. Our rationale for utilizing AdA over IP3 in those experiments was that the presence of phosphatases and kinases in the cell lysate, which can metabolize IP3 might interfere with IP3 stability [44]. Our results revealed that the binding of full-length Bcl-2 to GST-LBD was severely impaired in the presence of AdA, while the binding to GST-Fragment 3 was not affected (Fig. 7a). Next, we assessed whether IP3 and Bcl-2 bind to the same residues within the IBC. We compared the ability of 3xFLAG-Bcl-2 to bind to GST-LBD and GST-LBD K508Q, a mutant version previously shown to display severely reduced IP3 binding capacity [45]. This analysis did not reveal any significant differences in the binding affinity (Fig. 7b), which suggests that although Bcl-2 targets the IBC of IP3R1, it does not interact with the same amino acids as IP3.
Fig. 7.

Bcl-2 and its BH4 domain compete with IP3 and AdA for LBD, but they do not occupy the exact same site. a Representative GST-pull down experiments for assessing the effect of AdA (5 μM) on the binding of 3xFLAG-Bcl-2 from COS-1 cell lysate to GST-fused IP3R1 fragments corresponding to the Fragment 3 and LBD. The samples were analyzed via Western blot and stained with anti-FLAG antibody. 0.2 μg of total COS-1 lysates was used as input. The immunoreactive bands from three independent experiments, utilizing each time independently transfected cells and freshly prepared lysates, were quantified and normalized to the binding of 3xFLAG-Bcl-2 to GST-Fragment 3, which was set as 1. The data are plotted as mean ± SEM. b Representative GST-pull down experiments for comparison of the binding of 3xFLAG-Bcl-2 from COS-1 cell lysate to GST-LBD and GST-LBD K508Q. The samples were analyzed via Western blot and stained with anti-FLAG antibody. Total COS-1 lysates (1 μg) were used as input. The immunoreactive bands from three independent experiments, utilizing each time independently transfected cells and freshly prepared lysates, were quantified and normalized to the binding of 3xFLAG-Bcl-2 to GST-LBD, which was set as 1. The data are plotted as mean ± SEM. *Stands for p < 0.05. c, d Representative sensorgrams of the surface plasmon resonance experiments expressed in RU as a function of time. The biotin-BH4-Bcl-2 peptide and the scrambled peptide were immobilized on different channels of a streptavidin-coated sensor chip. The channels on the chip were exposed to 1.1 µM purified GST-LBD (c) or 0 and 1.1 μM GST-Fragment 3 d of IP3R1 in presence of IP3 (20 µM) or AdA (5 µM). e The channels on the chip were exposed to different concentrations of GST-LBD or mutated GST-LBD K508Q, which fails to bind IP3
To test whether IP3R ligands could interfere with the interaction between the BH4 domain and LBD, we conducted SPR experiments in the presence of IP3 and AdA. Here, we used purified GST-fragments and peptides, thereby allowing the use of IP3. The sensorgrams, shown in Fig. 7c, reveal that the addition of IP3 or AdA results in a decreased signal. The compounds did not bind directly to the biotin-BH4-Bcl-2 and did not significantly affect the binding of GST-Fragment 3 to the peptide (Fig. 7d). Based on these data, we concluded that IP3 and AdA disturb selectively the interaction between BH4-Bcl-2 and the LBD of IP3R1. To assess whether IP3 and Bcl-2 bind to the same residues within the IBC, we compared the ability of biotin-BH4-Bcl-2 to bind to GST-LBD and GST-LBD K508Q. This analysis did not reveal any significant differences in the binding affinity (Fig. 7e), confirming that like the full-length Bcl-2, the BH4-Bcl-2 targets the IBC of IP3R1, but not the same residues as IP3.
The BH4-Bcl-2 disrupts the interaction between LBD and a fluorescently labeled IP3R ligand
We showed that IP3, as well as AdA, could interfere with BH4-Bcl-2 binding to the LBD of IP3R1. To address the opposite question, namely whether BH4-Bcl-2 might affect the interaction of IP3 with the LBD, we performed FRET-based measurements using a competitive fluorescent ligand assay for IP3 [46]. In this method, binding of a fluorescent low-affinity ligand (FLL) and a fluorescent IP3-binding protein (CFP-coupled LBD) generates FRET signals. Ligands of the IP3R, such as IP3, reduce this FRET signal due to the decrease in the binding of FLL to CFP-coupled LBD, and increase the fluorescence emission ratio of CFP/FLL (480 nm/535 nm) (Fig. 8a). We showed that application of BH4-Bcl-2, but not of a scrambled version, caused an increase in the CFP/FLL signal, indicating that BH4-Bcl-2 decreased the binding of FLL to the LBD (Fig. 8a). IP3 (10 μM) was applied at the end of the experiment to determine the maximal response. BH4-Bcl-2 increased the fluorescence ratio in a concentration-dependent manner, and the IC50 value of BH4-Bcl-2 in the presence of 100 nM FLL could be estimated to be approximatively about 20 μM (Fig. 8b).
Fig. 8.

BH4-Bcl-2 reduces the binding of FLL to LBD. a Typical FRET-based measurements to assess the effect of the BH4-Bcl-2 or the scrambled peptide (BH4-Bcl-2-CTR; scr) on the interaction between FLL and a fluorescent IP3-binding protein (CFP-coupled LBD). The decrease in binding is shown by an increase in the emission ratio of CFP/FLL (480 nm/535 nm). IP3 (10 μM) was added at the end of the experiment to obtain the maximal increase in CFP/FLL (maximal decrease of binding). b Quantification of the increase in the FRET signal due to binding of 3, 10 or 30 μM BH4-Bcl-2 expressed as percentage of the maximal increase obtained after addition of 10 μM IP3. The data from three or four independent experiments are plotted as mean ± SEM. *Stands for p < 0.05
Discussion
The main findings of this study are that (1) IP3R1 inhibition by the anti-apoptotic Bcl-2 protein is modulated by the ligand, IP3, and thus by the level of IP3R activity; (2) Bcl-2, via its BH4 domain, has a novel target, represented by the LBD of IP3R1, a domain distinct from the modulatory region; (3) Bcl-2 and IP3 display a mutual antagonistic impact on their ability to bind the LBD of the IP3R1.
Previous works suggested that IP3Rs inhibition by Bcl-2 is not a static phenomenon, but can be dynamically modulated, perhaps partially by the level of IP3 signaling in response to agonist stimulation [15, 27]. Yet, the molecular mechanisms that could account for the modulatory nature of Bcl-2-mediated IP3R inhibition remained unresolved. After confirming in the present study that the level of IP3 (or of agonist), indeed modulates the inhibitory effect of Bcl-2 and its BH4 domain on IP3R activity, we demonstrated that Bcl-2 is able to target IP3R1 outside of its modulatory region. This unexpected finding sparked the identification of the LBD of IP3R1 as a novel target of Bcl-2 and its BH4 domain.
The original approach for examining the IP3R regions targeted by Bcl-2 was based on GST-pull down experiments [16]. This method makes use of GST-tagged proteins, representing the fragments obtained after limited proteolysis of the full-length IP3R1 channel. However, this approach might miss certain functional regions, since limited proteolysis does not necessarily coincide with functional boundaries. For instance, the LBD covers a.a. 1–604, while limited proteolysis yields the Fragment 1 corresponding to a.a. 1–345 and the Fragment 2 corresponding to a.a. 346–922. As a consequence, an interaction, which would require the intact LBD, might be missed using such an experimental paradigm. Instead, here, we made use of the functional domain (LBD) and its subdomains (IBC and SD) and revealed that Bcl-2 via its BH4 domain targets the same region of the LBD as IP3, namely the IBC. Nevertheless, our SPR and GST-pull down experiments, which show that Bcl-2 and its BH4 domain target the wild-type and the “loss of function” mutant LBD with similar affinity, indicate that Bcl-2 and IP3 have different underlying binding determinants. We strongly believe that future identification of the IP3R residues involved in the Bcl-2 binding is of a great importance. This might prompt the development of novel tools to disturb the Bcl-2/IP3R interaction, which has been shown to play crucial role in the survival of cancer cells [47, 48].
The fact that Bcl-2 and its ligand IP3 target the same region but not necessarily the same residues, suggests that the Bcl-2/IP3R complex might be allosterically modulated by IP3 levels. This is supported by (1) computational modelling showing that Bcl-2 binding to the LBD is sterically hindered by the conformational changes induced by IP3 binding and (2) experimental data demonstrating that IP3 and AdA interfere with the Bcl-2 (BH4 domain)/LBD interaction. Vice versa, it seems that the BH4 domain of Bcl-2 decreased the ligand binding to the LBD, in line with the previously reported ability of full-length Bcl-2 to decrease the affinity of IP3 for binding to the full-length IP3R [14]. Thus, Bcl-2 might serve as an efficient inhibitor of IP3R channels, not only by targeting the modulatory region, thereby interfering with the conformational coupling of IP3 binding to channel opening, but also by directly targeting the LBD and antagonizing the interaction with IP3.
Previous work revealed that IP3 binding to all subunits in a tetrameric IP3R channel is required for proper channel activation and IP3R opening [32]. Thus, it is possible that the binding of the BH4 domain of Bcl-2 to just one of the monomers could be sufficient to inhibit IP3R channel opening. This may account for the very potent inhibition of IP3Rs by Bcl-2 or its BH4 domain. Indeed, the BH4 domain of Bcl-2 is able to completely inhibit IP3R-mediated Ca2+ release. As a consequence, higher concentrations of IP3 would not only increase the probability that all four IP3R subunits are occupied and thus trigger Ca2+ flux through the IP3R, but also decrease the probability that Bcl-2 (or its BH4 domain) accesses the LBD and thus elicits IP3R inhibition.
Our findings do not necessarily contradict the previously reported observations (replicated in this study) on Bcl-2 interaction with the modulatory region of IP3R. In fact, the inhibition of IP3R1Δ1389–1408, lacking the Bcl-2-binding site in the modulatory region, appeared less prominent than the inhibition of the wild-type IP3R1 by Bcl-2. This supports the concept that IP3R modulation by Bcl-2 occurs via both Bcl-2-binding sites, one located in the LBD and the other one located between residues 1389–1408.
An inherent limitation of the current study is the link between the binding of Bcl-2 to the LBD in the full-length IP3R and IP3R inhibition by Bcl-2. The unequivocal proof of such link will require the identification of the amino acid stretch or specific residues within the LBD responsible for Bcl-2 binding, their deletion in the full-length IP3R1 and IP3R1Δ1389–1408 and assessment of Bcl-2's impact on them. Of course, an inherent prerequisite for such analysis is that deleting the Bcl-2-binding site in the LBD in full-length IP3R1 does not render the channel irresponsive to IP3. However, we would like to stress that the complete IP3R sequence was screened in previous studies for potential Bcl-2-binding sites. No other regions than the Fragment 3 and the C-terminus were identified. Fragment 3 is targeted by Bcl-2’s BH4 domain [18], whereas the C-terminus serves to recruit Bcl-2, via its TMD, in the proximity of IP3Rs [25]. Thus, it is very unlikely that other regions besides the LBD could account for the inhibition of IP3R1Δ1389–1408 by Bcl-2.
From the IP3R structure [21, 26, 49] (Fig. 9), it seems that both Bcl-2-binding regions are surface-accessible. Interestingly, in this 3D structure, a.a. 1389–1408, representing the central Bcl-2-binding site, from one subunit appeared to be located in very close proximity of the LBD of the neighboring monomer. Therefore, the 3D structure supports the possibility of having two Bcl-2-binding sites in IP3R1 in close proximity to each other. However, further work is needed to understand whether Bcl-2 can occupy both sites in neighboring monomers, whether binding of Bcl-2 to one site prevents its binding to the neighboring site or whether Bcl-2 can dynamically switch between both binding sites. Moreover, the loss of IP3R inhibition by Bcl-2 in conditions of high [IP3] suggests that in these conditions Bcl-2 binding to the central site may not be sufficient for IP3R inhibition by Bcl-2. In any case, the Bcl-2 inhibitory effects on IP3Rs present at low [IP3] but not at high [IP3] are not solely due to the interaction with the Fragment 3, since (1) Bcl-2 inhibits the channel lacking the binding site in Fragment 3 and (2) the binding of Bcl-2 or its BH4 domain to Fragment 3 was not affected by the presence of IP3R agonists. However, further work is needed to understand IP3R/Bcl-2-complex formation at the molecular level.
Fig. 9.
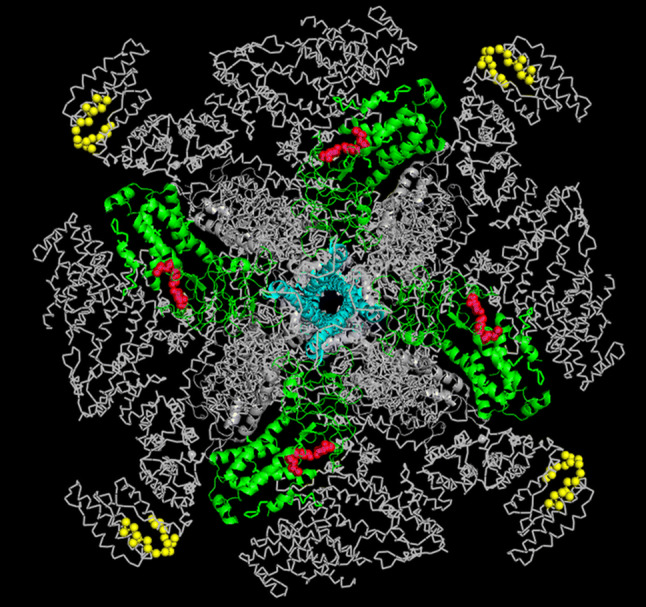
Overview of the Bcl-2-binding sites on IP3R1 structure. Structure of IP3R1 as viewed from the cytosol. The C-terminal 6th TMD of each monomer, which is important for Bcl-2 binding to the C-terminus, are indicated in cyan; the previously identified Bcl-2-binding site (a.a. 1389–1408) in ARM2 is indicated in yellow. The IBC is indicated in green with the crucial IP3-binding amino acids in red. Please note that the Bcl-2-binding site present in the ARM2 domain of one subunit is located in close proximity to the LBD of the neighboring subunit, representing the novel target of Bcl-2 described in present study
It should be noted that the LBD has been previously identified as a binding site for other Bcl-2-family members. Indeed, Bcl-2-like protein (Bcl-2L10) [50] and its zebrafish orthologue Nrz [51], have been shown to decrease IP3 binding to the IP3R, by targeting the LBD. Disruption of this interaction appeared as a potential strategy to target breast cancer [52]. Therefore, our study is revealing yet another human anti-apoptotic Bcl-2 family protein that can target the LBD of IP3Rs, interfering with IP3 binding and suppressing IP3R activity.
To conclude, IP3 and Bcl-2 compete for the LBD, thereby antagonizing each other’s effects on IP3R function. On the one hand, Bcl-2 fails to inhibit IP3R activity provoked by high [IP3] or [agonist] in in vitro or in cellulo systems, respectively. Hence, this shows that IP3R inhibition by Bcl-2 is modulated by the level of agonist stimulation. On the other hand, Bcl-2 or its BH4 domain counteract IP3 binding to the IP3R or LBD. This provides a novel molecular mechanism by which Bcl-2 proteins can inhibit IP3R function, at least in conditions of moderate IP3 signaling or agonist stimulation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank Anja Florizoone and Marina Crabbé for the excellent technical help. The authors are very grateful to Dr. Colin W. Taylor and Dr. Vera Konieczny (Department of Pharmacology, University of Cambridge, England, UK) and to Dr. Llewelyn Roderick (Department of Cardiovascular Sciences, KU Leuven, Belgium) for their assistance in preliminary work. The authors thank all members of the Lab. Molecular and Cellular Signaling in Leuven and Clark W. Distelhorst (Case Western Reserve University, Cleveland OH) for fruitful discussions. The work was supported by Grants from the Research Foundation-Flanders (FWO Grants 6.057.12, G.0819.13, G.0C91.14, G.0A34.16, G.0901.18), by the Research Council of the KU Leuven (OT Grant 14/101) and by the Interuniversity Attraction Poles Program (Belgian Science Policy; IAP-P7/13). GB, JBP and DIY are partners of the FWO Scientific Research Network (CaSign W0.019.17N). HI and EV are recipients of post-doctoral fellowships of the FWO. HI was supported by a mobility Grant from the FWO for a stay in the team of Dr. Yule (Rochester University, NY).
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Tsujimoto Y, et al. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228(4706):1440–1443. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- 2.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335(6189):440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 3.Cuende E, et al. Programmed cell death by bcl-2-dependent and independent mechanisms in B lymphoma cells. EMBO J. 1993;12(4):1555–1560. doi: 10.1002/j.1460-2075.1993.tb05799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Czabotar PE, et al. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15(1):49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 5.Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci. 2009;122(Pt 4):437–441. doi: 10.1242/jcs.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2(9):647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 7.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13(15):1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 8.Antonsson B, et al. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997;277(5324):370–372. doi: 10.1126/science.277.5324.370. [DOI] [PubMed] [Google Scholar]
- 9.Barclay LA, et al. Inhibition of Pro-apoptotic BAX by a noncanonical interaction mechanism. Mol Cell. 2015;57(5):873–886. doi: 10.1016/j.molcel.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baffy G, et al. Apoptosis induced by withdrawal of interleukin-3 (IL-3) from an IL-3-dependent hematopoietic cell line is associated with repartitioning of intracellular calcium and is blocked by enforced Bcl-2 oncoprotein production. J Biol Chem. 1993;268(9):6511–6519. [PubMed] [Google Scholar]
- 11.Vervliet T, Parys JB, Bultynck G. Bcl-2 proteins and calcium signaling: complexity beneath the surface. Oncogene. 2016;35(39):5079–5092. doi: 10.1038/onc.2016.31. [DOI] [PubMed] [Google Scholar]
- 12.Pinton P, Rizzuto R. Bcl-2 and Ca2+ homeostasis in the endoplasmic reticulum. Cell Death Differ. 2006;13(8):1409–1418. doi: 10.1038/sj.cdd.4401960. [DOI] [PubMed] [Google Scholar]
- 13.Rong Y, Distelhorst CW. Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol. 2008;70:73–91. doi: 10.1146/annurev.physiol.70.021507.105852. [DOI] [PubMed] [Google Scholar]
- 14.Chen R, et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol. 2004;166(2):193–203. doi: 10.1083/jcb.200309146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanson CJ, et al. Bcl-2 suppresses Ca2+ release through inositol 1,4,5-trisphosphate receptors and inhibits Ca2+ uptake by mitochondria without affecting ER calcium store content. Cell Calcium. 2008;44(3):324–338. doi: 10.1016/j.ceca.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 16.Rong YP, et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Mol Cell. 2008;31(2):255–265. doi: 10.1016/j.molcel.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Monaco G, et al. Profiling of the Bcl-2/Bcl-XL-binding sites on type 1 IP3 receptor. Biochem Biophys Res Commun. 2012;428(1):31–35. doi: 10.1016/j.bbrc.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 18.Monaco G, et al. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 2012;19(2):295–309. doi: 10.1038/cdd.2011.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshikawa F, et al. Trypsinized cerebellar inositol 1,4,5-trisphosphate receptor. Structural and functional coupling of cleaved ligand binding and channel domains. J Biol Chem. 1999;274(1):316–327. doi: 10.1074/jbc.274.1.316. [DOI] [PubMed] [Google Scholar]
- 20.Maes K, et al. Mapping of the ATP-binding sites on inositol 1,4,5-trisphosphate receptor type 1 and type 3 homotetramers by controlled proteolysis and photoaffinity labeling. J Biol Chem. 2001;276(5):3492–3497. doi: 10.1074/jbc.M006082200. [DOI] [PubMed] [Google Scholar]
- 21.Fan G, et al. Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature. 2015;527(7578):336–341. doi: 10.1038/nature15249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eckenrode EF, et al. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J Biol Chem. 2010;285(18):13678–13684. doi: 10.1074/jbc.M109.096040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rong YP, et al. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci USA. 2009;106(34):14397–14402. doi: 10.1073/pnas.0907555106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rong YP, et al. Targeting Bcl-2 based on the interaction of its BH4 domain with the inositol 1,4,5-trisphosphate receptor. Biochim Biophys Acta. 2009;1793(6):971–978. doi: 10.1016/j.bbamcr.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivanova H, et al. The trans-membrane domain of Bcl-2α, but not its hydrophobic cleft, is a critical determinant for efficient IP3 receptor inhibition. Oncotarget. 2016;7:55704–55720. doi: 10.18632/oncotarget.11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan G, et al. Cryo-EM reveals ligand induced allostery underlying InsP3R channel gating. Cell Res. 2018;28(12):1158–1170. doi: 10.1038/s41422-018-0108-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong F, et al. Bcl-2 differentially regulates Ca2+ signals according to the strength of T cell receptor activation. J Cell Biol. 2006;172(1):127–137. doi: 10.1083/jcb.200506189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki J, et al. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat Commun. 2014;5:4153. doi: 10.1038/ncomms5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vervliet T, et al. Regulation of the ryanodine receptor by anti-apoptotic Bcl-2 is independent of its BH3-domain-binding properties. Biochem Biophys Res Commun. 2015;463(3):174–179. doi: 10.1016/j.bbrc.2015.04.131. [DOI] [PubMed] [Google Scholar]
- 30.Bultynck G, et al. Thimerosal stimulates Ca2+ flux through inositol 1,4,5-trisphosphate receptor type 1, but not type 3, via modulation of an isoform-specific Ca2+-dependent intramolecular interaction. Biochem J. 2004;381(Pt 1):87–96. doi: 10.1042/BJ20040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wagner LE, Yule DI. Differential regulation of the InsP3 receptor type-1 and -2 single channel properties by InsP3, Ca2+ and ATP. J Physiol. 2012;590(Pt 14):3245–3259. doi: 10.1113/jphysiol.2012.228320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alzayady KJ, et al. Defining the stoichiometry of inositol 1,4,5-trisphosphate binding required to initiate Ca2+ release. Sci Signal. 2016;9(422):ra35. doi: 10.1126/scisignal.aad6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Missiaen L, et al. Measurement of intracellular Ca2+ release in intact and permeabilized cells using 45Ca2+ Cold Spring Harb Protoc. 2014;2014(3):263–270. doi: 10.1101/pdb.top066126. [DOI] [PubMed] [Google Scholar]
- 34.Pierce BG, et al. ZDOCK server: interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics. 2014;30(12):1771–1773. doi: 10.1093/bioinformatics/btu097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seo MD, et al. Structural and functional conservation of key domains in InsP3 and ryanodine receptors. Nature. 2012;483(7387):108–112. doi: 10.1038/nature10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Souers AJ, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19(2):202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 37.Mashiach E, Nussinov R, Wolfson HJ. FiberDock: a web server for flexible induced-fit backbone refinement in molecular docking. Nucleic Acids Res. 2010;38(suppl_2):W457–W461. doi: 10.1093/nar/gkq373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin CC, Baek K, Lu Z. Apo and InsP3-bound crystal structures of the ligand-binding domain of an InsP3 receptor. Nat Struct Mol Biol. 2011;18(10):1172–1174. doi: 10.1038/nsmb.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monaco G, et al. Alpha-helical destabilization of the Bcl-2-BH4-domain peptide abolishes its ability to inhibit the IP3 receptor. PLoS One. 2013;8(8):e73386. doi: 10.1371/journal.pone.0073386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fouque A, et al. The apoptotic members CD95, Bcl-Xl, and Bcl-2 cooperate to promote cell migration by inducing Ca2+ flux from the endoplasmic reticulum to mitochondria. Cell Death Differ. 2016;23(10):1702–1716. doi: 10.1038/cdd.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ivanova H, et al. The BH4 domain of Bcl-2 orthologues from different classes of vertebrates can act as an evolutionary conserved inhibitor of IP3 receptor channels. Cell Calcium. 2017;62:41–46. doi: 10.1016/j.ceca.2017.01.010. [DOI] [PubMed] [Google Scholar]
- 42.Vervliet T, et al. Bcl-2 binds to and inhibits ryanodine receptors. J Cell Sci. 2014;127(Pt 12):2782–2792. doi: 10.1242/jcs.150011. [DOI] [PubMed] [Google Scholar]
- 43.Takahashi S, Kinoshita T, Takahashi M. Adenophostins A and B: potent agonists of inositol-1,4,5-trisphosphate receptor produced by Penicillium brevicompactum structure elucidation. J Antibiot (Tokyo) 1994;47(1):95–100. doi: 10.7164/antibiotics.47.95. [DOI] [PubMed] [Google Scholar]
- 44.Sims CE, Allbritton NL. Metabolism of inositol 1,4,5-trisphosphate and inositol 1,3,4,5-tetrakisphosphate by the oocytes of Xenopus laevis. J Biol Chem. 1998;273(7):4052–4058. doi: 10.1074/jbc.273.7.4052. [DOI] [PubMed] [Google Scholar]
- 45.Yoshikawa F, et al. Mutational analysis of the ligand binding site of the inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1996;271(30):18277–18284. doi: 10.1074/jbc.271.30.18277. [DOI] [PubMed] [Google Scholar]
- 46.Oura T, et al. Highly sensitive measurement of inositol 1,4,5-trisphosphate by using a new fluorescent ligand and ligand binding domain combination. ChemBioChem. 2016;17(16):1509–1512. doi: 10.1002/cbic.201600096. [DOI] [PubMed] [Google Scholar]
- 47.Greenberg EF, et al. Synergistic killing of human small cell lung cancer cells by the Bcl-2-inositol 1,4,5-trisphosphate receptor disruptor BIRD-2 and the BH3-mimetic ABT-263. Cell Death Dis. 2015;6:e2034. doi: 10.1038/cddis.2015.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Greenberg EF, Lavik AR, Distelhorst CW. Bcl-2 regulation of the inositol 1,4,5-trisphosphate receptor and calcium signaling in normal and malignant lymphocytes: potential new target for cancer treatment. Biochim Biophys Acta. 2014;1843(10):2205–2210. doi: 10.1016/j.bbamcr.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamada K, et al. IP3-mediated gating mechanism of the IP3 receptor revealed by mutagenesis and X-ray crystallography. Proc Natl Acad Sci USA. 2017;114(18):4661–4666. doi: 10.1073/pnas.1701420114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bonneau B, et al. IRBIT controls apoptosis by interacting with the Bcl-2 homolog, Bcl2l10, and by promoting ER-mitochondria contact. Elife. 2016;5:e19896. doi: 10.7554/eLife.19896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bonneau B, et al. The Bcl-2 homolog Nrz inhibits binding of IP3 to its receptor to control calcium signaling during zebrafish epiboly. Sci Signal. 2014;7(312):ra14. doi: 10.1126/scisignal.2004480. [DOI] [PubMed] [Google Scholar]
- 52.Nougarede A, et al. Breast cancer targeting through inhibition of the endoplasmic reticulum-based apoptosis regulator Nrh/BCL2L10. Cancer Res. 2018;78(6):1404–1417. doi: 10.1158/0008-5472.CAN-17-0846. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.