


Abstract
In DNA, the deamination of dAMP generates 2′-deoxyinosine 5′-monophosphate (dIMP). Hypoxanthine (HX) residues are mutagenic since they give rise to A·T→G·C transition. They are excised, although with different efficiencies, by an activity of the 3-methyladenine (3-meAde)-DNA glycosylases from Escherichia coli (AlkA protein), human cells (ANPG protein), rat cells (APDG protein) and yeast (MAG protein). Comparison of the kinetic constants for the excision of HX residues by the four enzymes shows that the E.coli and yeast enzymes are quite inefficient, whereas for the ANPG and the APDG proteins they repair the HX residues with an efficiency comparable to that of alkylated bases, which are believed to be the primary substrates of these DNA glycosylases. Since the use of various substrates to monitor the activity of HX-DNA glycosylases has generated conflicting results, the efficacy of the four 3-meAde-DNA glycosylases of different origin was compared using three different substrates. Moreover, using oligonucleotides containing a single dIMP residue, we investigated a putative sequence specificity of the enzymes involving the bases next to the HX residue. We found up to 2–5-fold difference in the rates of HX excision between the various sequences of the oligonucleotides studied. When the dIMP residue was placed opposite to each of the four bases, a preferential recognition of dI:T over dI:dG, dI:dC and dI:dA mismatches was observed for both human (ANPG) and E.coli (AlkA) proteins. At variance, the yeast MAG protein removed more efficiently HX from a dI:dG over dI:dC, dI:T and dI:dA mismatches.
INTRODUCTION
The rules that govern the recognition of a damaged base by the enzyme in charge of its elimination, in vitro and in vivo, would be important for the understanding of the molecular mechanisms involved in DNA repair.
The hydrolytic deamination of dCMP and dAMP residues in DNA yields dUMP and dIMP residues, respectively (1,2). In Escherichia coli, the removal of uracil from DNA is performed by the uracil-DNA glycosylase (3) and by the mismatch-specific G/U-DNA glycosylase (4) whereas the excision of hypoxanthine (HX) is performed by an activity associated with the 3-methyladenine (3-meAde)-DNA glycosylase (5). This latter activity is associated in human cells with the ANPG protein (6,7), in rat cells with the APDG protein (8), in yeast with the MAG protein (9) and in E.coli with the AlkA protein coded for by the alkA gene (10). The mutagenic properties of dIMP residues have been ascertained by site-specific mutagenesis in vivo (11,12). A single dIMP residue, inserted in vitro at a specific locus in a M13mp9 RF molecule, exhibits miscoding properties leading to mutagenesis in E.coli (11). Furthermore, in mammalian cells, a synthetic c-Ha-ras gene containing HX resulted in A·T→G·C transition (12).
The presence of hot spots of mutagenesis in E.coli, as well as in human cells, increases the possibility that the base context next to the lesion could lower the efficacy of the repair of a modified base. A significant variation in the rate of repair, depending upon the surrounding sequence context, has been shown in the case of repair of uracil (13), AP-sites (14), formamidopyrimidine residues (15), O6-methylguanine (16), N-methylated bases (17) and UV-induced photoproducts (18). The slow rate of repair of cyclobutane dimers in the human p53 gene at mutational hotspots in skin cancer is documented, suggesting that hotspots for mutations arise partly as a consequence of heterogeneous repair within specific sequences (19).
The aim of the present study was to investigate the structural requirements for the interaction of the 3-meAde-DNA glycosylases of different origin with dIMP residues, when present in different sequence contexts. The best substrate is a double-stranded oligonucleotide containing a single HX residue. The results show that pure preparations of human, rat, yeast and E.coli 3-meAde-DNA glycosylases have dramatically different HX excision abilities according to the sequence context. Up to 2–5-fold difference in the rates of HX removal between various sequences was measured. Moreover, the bacterial, yeast and mammalian enzymes show definite sequence preference.
MATERIALS AND METHODS
Bacterial strains and plasmids
Escherichia coli BH290 (X::tagA1, alkA1, thy-hsdR) is a derivative of AB1157 (20) and was from laboratory stocks. The pALK10 plasmid (8) is a subclone from pYN1000 (10) containing the E.coli alkA gene. The pBKY143 plasmid containing the yeast mag gene (9), coding for the yeast 3-methyladenine-DNA glycosylase, was a gift from Dr E. Seeberg (University of Oslo, Norway).
Enzymes
The E.coli Fpg protein was purified as described (21). The AlkA protein prepared as described (22) was a gift from Dr B. Tudek (this laboratory). The ANPG40 protein, the truncated 26 kDa, 230 amino acids human enzyme (6) and the APDG protein, the rat enzyme (8) were prepared by G. Chyzak (this laboratory). The MAG protein was purified as described (23).
Materials
Nucleic acids and nucleotides were purchased from Roche (Mannheim, Germany). Radiolabeled reagents were obtained from the following sources: [3H]dimethyl-sulfate (DMS) (3.8 Ci/mmol) from New England Nuclear, [γ-32P]dATP (3000 Ci mmol) and [1′,2′,2,8,-3H]dATP (92 Ci/mmol) from Amersham-Pharmacia. [3H]dITP was prepared by deamination of [3H]dATP with nitrous acid and the products were purified and analyzed as described (24).
Substrates
The preparation of [3H]dIMP double-stranded DNA (160 000 c.p.m./µg) was as described (5) and had a specific activity of 51 c.p.m./fmol of dIMP residue. This substrate will hereafter be referred to as [3H]dIMP-M13mp8 DNA. Poly(dGC-dGC) containing [3H]dIMP residues (80 000 c.p.m./µg) was prepared as described for the preparation of [3H]dIMP-M13mp8 substrate but using poly(dGC-dGC). Its specific activity was 51 c.p.m./fmol of dIMP residue. Alkylated DNA, [3H]DMS-DNA, was prepared as described using [3H]DMS (25,26). The specific activities of the 3H-methylated substrates were 1995 c.p.m./pmol of methylated bases.
Radioactively labeled double-stranded oligonucleotides
Single-stranded oligonucleotides containing or without dIMP residues were synthesized by Dr E. Lescot (this laboratory) or purchased from Genset (Paris, France). The sequence of duplex oligonucleotides that we chose was part of the gpt gene (27). The sequences of the various oligonucleotides used are shown in Table 1.
Table 1. Sequences of duplex oligonucleotides used for measuring the HX-DNA glycosylase activity.

aI, deoxyinosine residue; N, one of the four natural deoxynucleosides T, dA, dG and dC, respectively.
bSequence used by Dianov and Lindahl (32).
Aliquots of 30 pmol of single-stranded oligonucleotide were 5′-end labeled with [γ-32P]ATP then annealed with a non-radioactive complementary oligonucleotide, as described (5). The formation of double-stranded oligonucleotides was monitored by 15% PAGE under non-denaturing conditions (28).
DNA glycosylase assays
HX-DNA glycosylase standard reaction mixture (100 µl) contained 1 pmol of 32P-5′-end labeled oligonucleotide duplex, 50 mM HEPES–KOH pH 7.8, 1 mM EDTA, 5 mM β-mercaptoethanol, 100 µg/ml BSA (Molecular Biology Grade, Roche Mannheim), 20 U E.coli Fpg protein (in order to incise DNA at abasic sites) (29) and limiting amounts of enzyme. In the case of human, rat and yeast 3-meAde-DNA glycosylases, the incubation mixture was supplemented with 100 mM KCl. Incubations were for 30 min at 37°C unless otherwise stated. The reaction was stopped by addition of 12 µl of 3 M NaCl, followed by extraction with an equal volume of phenol–chloroform (1:1). The samples were centrifuged for 3 min, the aqueous phase was collected and the DNA precipitated by addition of 2 µl of 0.25% linear polyacrylamide (30) and 300 µl of ethanol (–20°C). The precipitate was recovered by centrifugation, dried and dissolved in 10 µl of formamide, heated for 3 min at 90°C, loaded onto 20% polyacrylamide gel containing 7 M urea and electrophoresed in Tris–Borate–EDTA buffer. The gels were subjected to autoradiography. For quantification, the gels were exposed to a Storm 840 Phosphor Screen and the amounts of radioactivity in the bands were quantified using the ImageQuaNTTM Software. The data given are from a single experiment, but replicates were consistently within 5%. 3-meAde-DNA glycosylase standard assays were performed in the incubation mixture described above to determine the HX-DNA glycosylase activity, but using as substrate [3H]DMS-DNA and measuring the ethanol-soluble radioactive products (6–8,25).
HPLC chromatography
HPLC chromatography was performed as already described (5) to isolate and subsequently measure [3H]HX after reaction of the enzyme with [3H]dIMP-M13mp8 DNA or [3H]dIMP- poly(dGC–dGC).
Surface plasmon resonance analysis of the interaction between 3-meAde-DNA glycosylase and the immobilized oligonucleotide HX20-34/T
Kinetics of interactions were determined using BIAcore technology (BIAcore AB, Uppsala, Sweden) which allows real-time analysis of unlabeled compounds. Biotinylated oligonucleotide HX20-34/T (5 µg/ml in HBS buffer containing 10 mM HEPES–KOH pH 7.4, 150 mM NaCl, 3.4 mM EDTA, 0.005% BIAcore surfactant P20) was injected at a flow rate of 5 µl/min onto a streptavidin coated sensorchip (SA sensor chip, BIAcore). The amount of immobilized oligonucleotide was found to be ~30–50 pg/mm2 (30–50 RU). Different concentrations of the enzymes (ANPG40 and AlkA) were then injected at a flow rate of 10 µl/min and at the end of the association time (300 s), dissociation occurred by adding running buffer (HBS) for 300 s. The kinetic parameters of the binding reactions were determined using BIAevaluation 2.1 software (BIAcore).
RESULTS
Human ANPG, rat APDG, yeast MAG and E.coli AlkA proteins excise HX residues from duplex oligodeoxyribonucleotide containing dIMP
As shown in Figure 1 the double-stranded oligodeoxyribonucleotide containing a dIMP residue at position 20 migrates as a single species at position 34mer (lane 1). When it is treated either with piperidine (lane 2) or by an excess of Fpg protein (lane 3), treatments that will reveal AP sites, it is not incised, showing that it does not contain the AP site. When this double-stranded oligodeoxyribonucleotide is treated either with the ANPG40 protein (lane 4) or the AlkA protein (lane 10), it is only minimally incised due to the instability of the generated abasic site under the experimental conditions used. However, when the duplex DNA is treated first by the ANPG40 protein (lanes 5–8) or by the AlkA protein (lanes 10–14) then by the FPG (lanes 5 and 11), the Nfo (lanes 7 and 13), the Nth (lanes 8 and 14) proteins or with hot piperidine (lanes 6 and 12), ~70% of the 34mer oligonucleotide are converted to a product migrating at the position of the 19mer. This is due to the excision of HX residue followed by the incision of the oligonucleotide at the AP site according either to a β–δ elimination mechanism (lanes 5, 6, 11 and 12), or a hydrolytic mechanism (lanes 7 and 13), or a β-elimination mechanism (lanes 8 and 14) (31). It should be noted that both proteins have an absolute requirement for a duplex oligonucleotide, since no detectable activity was observed when the ANPG40 (lane 9) or the AlkA proteins (data not shown) act on a single-stranded oligonucleotide containing a dIMP residue. In conclusion, both the ANPG40 and the AlkA proteins act upon DNA containing dIMP residues as a DNA glycosylase leaving an AP site. This also holds true for the rat APDG and the yeast MAG proteins (data not shown).
Figure 1.
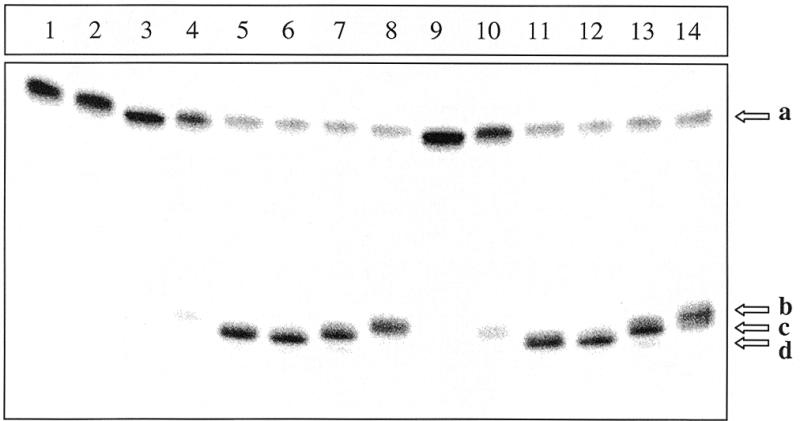
Activity of the ANPG40 and AlkA proteins on duplex deoxyribonucleotides containing dIMP residues. The 32P-5′-end labeled double-stranded oligonucleotide HX20-34/T, after various treatments, was analyzed by PAGE under denaturing conditions. Lane 1, no treatment; lane 2, treated with piperidine (1 M, 30 min, 90°C); lane 3, treated with the Fpg protein (100 ng, 20 min, 37°C); lanes 4–8, HX20-34/T was treated with the ANPG40 protein (0.5 U, 20 min, 37°C); lanes 10–14, treated with the AlkA protein (5 U, 60 min, 37°C). These samples were then further treated (lanes 5 and 11) either with piperidine (1 M, 30 min, 90°C) or with abasic site nicking enzymes (30 min at 37°C). Lanes 6 and 12, with the Fpg protein (100 ng); lanes 7 and 13, with the Nfo protein (500 ng); lanes 8 and 14, with the Nth protein (100 ng). Lane 9, the 32P-5′-end labeled single-stranded oligonucleotide HX20-34/T was treated with the ANPG40 and Fpg proteins as above. Arrow a indicates the 34mer oligonucleotide. Arrow b points to the 19mer oligonucleotide with an α–β unsaturated aldehyde at the 3′-terminus. Arrow c points to the 19mer oligonucleotide with 3′-OH group at the 3′-terminus. Arrow d points to the 19mer oligonucleotide with a phosphate group at the 3′-terminus. For details see Materials and Methods.
Comparison of the various substrates used to monitor the HX-DNA glycosylase activity and kinetic constants of the various 3-meAde-DNA glycosylases for substrates containing dIMP residues
Various substrates have been used, in different laboratories, to evaluate the HX-DNA glycosylase activity, including DNA of M13mp8 or poly(dGC–dGC) containing [3H]dIMP residues and a duplex oligodeoxynucleotide containing a unique dIMP residue at a precise location. These substrates were used in order to compare and to ascertain their relative properties. As shown in Table 2, the duplex oligonucleotide containing a dIMP residue is by far the best substrate for all the three enzymes. This observation leads to the use of this latter substrate to establish the kinetic parameters and the influence of the sequence context upon the excision of HX by the various DNA repair proteins.
Table 2. Comparison of the activity of 3-meAde-DNA glycosylases of various origin on [3H]dIMP M13mp8 DNA, [3H]dIMP poly(dGC–dGC) or duplex oligonucleotide HX20-34/T 34mer.
| Substrate | Hypoxanthine released (fmol/min) by 1 Ua of | ||
|---|---|---|---|
| ANPG40 protein | MAG protein | AlkA protein | |
| [3H]dIMP M13mp8 DNAb | 1.05 | ~0.1 × 10–3 | 5 × 10–3 |
| [3H]dIMP poly(dGC–dGC)b | 0.5 | ~0.1 × 10–3 | 12 × 10–3 |
| Oligonucleotide containing a single dIMP residuec | 311d | 0.4d | 1.2d |
aOne unit of 3-meAde-DNA glycosylase (of any origin) is the amount of protein liberating 1.0 pmol of methylated base as ethanol-soluble products from [3H]DMS-DNA, in 1 min at 37°C.
bAfter incubation of the various substrates containing [3H]dIMP residues (2 nM) with the respective enzyme, the products of the reaction were separated by HPLC and the fractions analyzed for radioactivity. For details see Materials and Methods.
cSubstrate is HX20-34/T and the concentration of dIMP residues is 10 nM.
dData taken from (5).
The results presented in Figure 2A show the time course release of the HX residues from the duplex oligonucleotide containing a dIMP residue treated by the ANPG40 protein. It is measured by the appearance of a new band, migrating at the position of the 19mer, generated by the β-lyase activity of the Fpg protein at the AP site produced by the DNA glycosylase activity. The quantification of the amount of radioactivity migrating at the position of the 19mer shows that it increased linearly during the initial part of the reaction (Fig. 2B). Similar kinetics were obtained with the other proteins and allowed the determination of the initial velocities of the enzymatic reactions used to determine the kinetic constants. The DNA glycosylase kinetics constants of the ANPG40, the MAG and the AlkA proteins, using HX20-34/T oligodeoxynucleotide as substrate, are listed in Table 3. These data show a dramatic difference between the three enzymes, the human one being by far the most efficient. For comparison purposes, it should be recalled that the Km for excision of 3-meAde by the ANPG protein is 8 nM, the turnover number kcat = 11 min–1, the specificity constant kcat/Km = 1.4 nM–1 min–1 and for the excision of the 7-MeGua, Km = 25 nM, kcat = 0.35 min–1 and kcat/Km = 0.014 nM–1 min–1 (7). Therefore the kcat/Km value for the excision of HX present in the HX20-34/T oligodeoxynucleotide shows that the specificity of the reaction is comparable to the release of 7-meGua residues but it is less efficient than for the release of 3-meAde residues.
Figure 2.
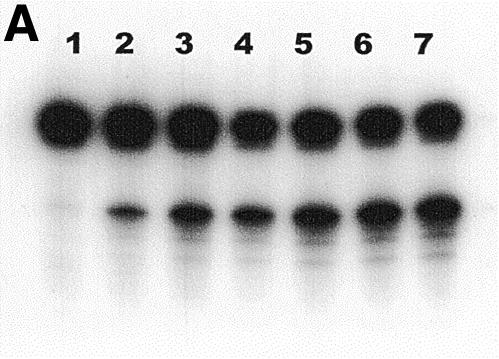

Kinetics of excision, by the ANPG40 protein, of HX from an oligonucleotide duplex containing thymine opposite a deoxyinosine residue. (A) The oligonucleotide HX20-34/T (10 nM) was incubated with the ANPG40 protein (0.2 U) for increasing periods of time in the presence of Fpg protein. The products of the reaction were separated by electrophoresis on a 20% polyacrylamide gel containing 7 M urea and visualized. Lanes 1–7, incubation time 0, 5, 10, 15, 20, 25 and 30 min, respectively. (B) The radioactivity of the products of the reaction shown in (A) was quantified and plotted as a function of time. For details see Materials and Methods.
Table 3. Kinetic constants for the ANPG40, MAG and AlkA proteins for excision of HX from duplex oligonucleotide containing dIMP residue.
| Protein | Km (nM)a | kcat (min–1) | kcat /Km | KD (nM)b |
|---|---|---|---|---|
| ANPG40 | 6 | 21 × 10–2 | 35 × 10–3 | 23.5 |
| MAG | ~1000 | ~15 × 10–3 | ~15 × 10–6 | nd |
| AlkA | 420 | 0.89 × 10–3 | 2 × 10–6 | 232 |
aThe apparent Km of the 3-meAde-DNA glycosylases of different origin were determined by incubating the respective protein with increasing concentrations of HX20-34/T oligonucleotide. The oligonucleotide concentrations range were 5–40 nM for the ANPG40 protein and 100–1000 nM for the MAG and AlkA proteins. The initial velocities measured for excision of HX were treated as described by Lineweaver and Burk.
b30–50 pg/mm2 of biotinylated HX20-34/T was immobilized onto a streptavidin coated sensor chip in a BIAcore. Various concentrations of each enzyme was then injected at a flow rate of 10 µl/min. The kinetic parameters ka and kd were determined using BIAevaluation 2.1 software (BIAcore) and KD calculated as kd/ka. nd, Not determined.
Effect of the sequence context and the nature of the base opposite the dIMP residue on the activity of the mammalian ANPG and APDG proteins
The initial rate of cleavage of various substrates having different sequence context by the mammalian 3-meAde-DNA glycosylases was compared. Moreover, this study was extended by using, for each oligonucleotide, duplexes containing dC, dG or dA positioned opposite the dIMP residue. Since it has been shown that the activity of a purified fraction of calf thymus HX-DNA glycosylase was not affected by the base composition of the oligomer being either A·T or G·C rich (32), this study was focused on a random sequence, by using a stretch of a natural gene (27).
The potential importance of sticky as compared to blunt ends was investigated using HX20-34 which is identical to the core part (31 bases) of HX20-40, but does not contain the 5′ protruding ends and has different 3′ and 5′ flanking bases: in this case the efficiency of the ANPG40 protein is only 1.6-fold higher than that observed when using sticky ends (Table 4A). The sequence context effect was further explored using the oligonucleotides HX20-34/T and HX13-34/T which are identical but contain the dIMP residue in a different context: 5′-GpIpC-3′ and 5′-CpIpC-3′, respectively. The latter substrate is recognized 1.5-fold more efficiently.
Table 4. Initial velocities of cleavage, by various mammalian 3-methyladenine DNA glycosylases, of oligonucleotide duplexes having different structures and containing different bases opposite to the dIMP residue.
|
A. The human ANPG40 protein | ||||
|---|---|---|---|---|
| Oligonucleotides | HX20-40 | HX20-34 | HX13-34 | HX20-DLa |
| Mismatchb | Hypoxanthine released by 1 U of the ANPG40 protein (fmol/min) | |||
| dI:T | 132 | 212 | 316 | 728 |
| dI:dC | 82 | 111 | nd | 82 |
| dI:dG | 85 | 128 | nd | 309 |
| dI:dA | 64 | 68 | nd | 94 |
|
B. The rat APDG protein | ||||
|---|---|---|---|---|
| Oligonucleotides | HX20-40 | HX20-34 | HX13-34 | HX20-DL |
| Mismatchb | Hypoxanthine released by 1 U of the ANPG40 protein (fmol/min) | |||
| dI:T | 85 | 101 | 122 | 141 |
| dI:dC | 43 | 53 | nd | 61 |
| dI:dG | 45 | 48 | nd | 123 |
| dI:dA | 19 | 20 | nd | 53 |
a Sequences from Dianov and Lindahl (32).
bDifferent oligonucleotides containing a single dIMP at a defined position in various sequence context were annealed with the complementary oligonucleotides in order to generate the mismatches dI:T, dI:dC, dI:dG and dI:dA, and were used as substrates for the ANPG40 or APDG proteins. Initial velocities were calculated from the linear part of the curve where the amount of HX released is plotted as a function of time. For details see Materials and Methods. nd, Not determined.
For comparison purpose with the previous investigation of Dianov and Lindahl (32) using purified preparations from calf thymus extracts, their a, c, d and e sequences (32) were included and are in this work sequences HX20-DL/T, HX20-DL/A, HX20-DL/C and HX20-DL/C, respectively.
The potential importance of the sequence context, next to the lesion, was investigated using HX20-DL/T which has sticky ends, the same length, the same location of the dIMP residue in the duplex oligonucleotide as HX20-40/T but has different overall sequence and lesion context (a dG 5′ to the lesion instead of a dC). As shown in Table 4A, the human ANPG40 protein is the most active when using as substrate HX20-DL/T, in this sequence dIMP residue is flanked by two dCs and the duplex has two sticky ends. The results show a dramatic change (5.5-fold) in the efficiency of the repair that could be attributed to the sequence context.
However, the present data do not give a clear explanation concerning the specific features that make HX20-DL the best substrate for the ANPG40 and APDG proteins. It is clear that the two flanking bases are not the main reason for the good or poor removal of HX (compare HX20-DL and HX13-34), but it should be noted that the stability of the duplex containing inosine flanked by pyrimidines is lower than when inosine is flanked by purines (33). The behavior of the rat enzyme (Table 4B) shows a similar profile of activity when compared to the human enzyme. The oligonucleotide HX20-DL/T is the best substrate followed by HX13-34/T, HX20-34/T and HX20-40/T. However, the rat enzyme had a lower activity on all the substrates tested.
The analysis of the efficiency of excision of HX when opposite to each of the four different bases by mammalian 3-meAde-DNA glycosylases reveals that dI:T and dI:dG are the best repaired.
Effect of the sequence context and the nature of the base opposite the dIMP residue on E.coli AlkA protein and Saccharomyces cerevisiae MAG protein activity
In order to compare the properties of 3-meAde-DNA glycosylases from E.coli and yeast to those of the mammalian enzymes, the initial velocity of the reaction was measured when the AlkA and MAG proteins were acting on the substrates described above. The results presented in Table 5 show that the AlkA protein does not exhibit any clear preference for a given substrate, which is at variance with the mammalian enzymes, although in most cases dI:T mismatch was the best repaired. Interestingly, the HX20-DL substrate for all the mismatches shows again in the case of the mismatch dI:dG, peculiar behavior. It is very poorly recognized. Among the substrates tested, the HX20-40 was the best.
Table 5. Initial velocities of cleavage by the E.coli AlkA protein of oligonucleotide duplexes containing different bases opposite the dIMP residue.
| Oligonucleotides | HX20-40 | HX20-34 | HX13-34 | HX20-DL |
|---|---|---|---|---|
| Mismatch | Hypoxanthine released by 1 U of the AlkA protein (fmol/min) | |||
| dI:T | 1.35 | 1.19 | 0.50 | 1.18 |
| dI:dC | 0.92 | 0.67 | nd | 0.48 |
| dI:dG | 0.83 | 0.66 | nd | 0.16 |
| dI:dA | 0.68 | 0.76 | nd | 0.77 |
For details see Table 4.
The results presented in Table 6 show that as already observed for the AlkA protein, the MAG protein has no clear preference for any of the substrates tested. However, regardless of the sequence context, the excision of HX by the Mag protein present in a dI:dG mismatch was the most efficient. Among the substrates tested, the HX13-34 oligonucleotide was the best. The distinctive behavior of the yeast 3-meAde-DNA glycosylase compared to the other enzymes tested should be noted.
Table 6. Initial velocities of cleavage by the yeast MAG protein of oligonucleotide duplexes containing a different base opposite the dIMP residue.
| Oligonucleotides | HX20-40 | HX20-34 | HX13-34 | HX20-DL |
|---|---|---|---|---|
| Mismatch | Hypoxanthine released by 1 U of the MAG protein (fmol/min) | |||
| dI:T | 0.36 | 0.40 | 0.53 | 0.26 |
| dI:dC | 0.34 | 0.42 | nd | 0.26 |
| dI:dG | 0.42 | 0.47 | nd | 0.31 |
| dI:dA | 0.28 | 0.30 | nd | 0.21 |
For details see Table 4.
DISCUSSION
The DNA repair proteins 3-meAde-DNA glycosylases, regardless of their origin, have the ability to release a number of alkylbases (7,8,34), cyclic etheno-adducts (23,35,36), adducts of nitrogen mustards (37) and HX from DNA containing dIMP residues (5). Concerning the excision of HX residues, the human enzyme is by far the most efficient and the yeast MAG and E.coli AlkA proteins perform this reaction very poorly (5).
The purpose of this study was to investigate in detail the factors that influence the efficacy of four 3-meAde-DNA gly-cosylases of different origin for the same substrate. Since the glycosidic bond of akylated bases is quite unstable and does not allow such detailed studies, we chose to study the activity of these 3-meAde-DNA glycosylases on substrates containing HX residues. This study was facilitated by the use of duplex oligonucleotides containing dIMP residues at a precise location and the availability of data concerning the structure of duplex oligonucleotides containing mismatched dIMP residue (38–42). The evidence from crystal structure and NMR studies reveals that (i) the HX base can form stable hydrogen-bonded mismatches with all four normal bases T, G, A and C, in DNA and (ii) a duplex oligonucleotide containing mismatches made of dIMP residues mainly takes a B-DNA helix conformation and does not show large perturbations at the local level (38–42).
The influence of the base opposite the dIMP residue upon the excision of HX residues by the various 3-meAde-DNA glycosylases was first investigated. Studies of the thermal stability of oligonucleotides containing deoxyinosine have shown that the order of the stability is independent of sequence effects, and is: dI:dC > dI:dA > dI:dG > dI:T (33,43). The comparison of the efficiency of HX excision by the mammalian 3-meAde-DNA glycosylases shows that it correlates with the thermal stability of the mismatch: the less stable dI:T and dI:dG are those best repaired. The bacterial enzyme also preferentially repairs dI:dT mismatch, although repair of dI:dC, dI:dG and dI:dA base pairs does not correspond to the thermal stability of the mismatch in the duplex DNA. In the case of the MAG protein, the dI:dG base pair was the best repaired. This unusual repair preference for the yeast 3-meAde-DNA glycosylase makes it different from the 3-meAde-DNA glycosylases of other species. It should be recalled that NMR studies of the dI:dG mismatch in DNA indicate the formation of a dI(syn):dG(anti) base pair in a B-DNA helix (42). As Syn conformation of dI takes place only in the dI:dG base pair, it may be the mechanism that explains the preferential repair of the dI:dG base pair by the MAG protein. Since the recognition of a relatively short oligodeoxyribonucleotide by a DNA gly-cosylase could be modified by the termini of such molecules, the activity of the various enzymes was measured by using the same sequence in the core of the oligonucleotide but having blunt or sticky ends. The results show that oligonucleotides having blunt ends are somehow better recognized.
The comparison of the kinetic constants for the excision of HX by the three different enzymes shows that the efficiency of repair by E.coli and yeast 3-meAde-DNA glycosylases are extremely poor. This is in agreement with the previous in vivo data showing that, in E.coli, the repair of a site-specifically incorporated dIMP residue was not efficient (11). Taken together these results strongly suggest that, in vivo, HX, as a DNA lesion, is not physiologically important for the fast propagating monocellular organisms, perhaps due to the slow rate of adenine deamination in duplex DNA (44). Nevertheless, the difference between the binding constants of E.coli (AlkA) and human (ANPG40) proteins to an oligonucleotide containing a dIMP residue was only one order of magnitude. This data possibly means that the inefficient repair of dIMP residue in DNA by bacterial and yeast proteins could be due to an extremely slow catalysis.
The interaction of the human truncated ANPG protein with oligodeoxyribonucleotides containing either HX or abasic sites has been investigated (45). The region of direct protein–DNA interactions consists only of one turn of helix. In order to form a stable complex between the ANPG protein and HX-DNA, the minimal length of the oligonucleotide should be at least 20 bp (45). However, the crystal structures of the AlkA protein (46,47) and the truncated ANPG protein complexed to a pyrrolidine-containing 13 bp duplex oligonucleotide have been reported (48). AlkA is a globular protein consisting of three equal-sized domains. Its active site is located in a large hydrophobic cleft rich in aromatic residues between domains 2 and 3 (46,47). The ANPG protein consists of a single domain, the core of the protein has a curved antiparallel β sheet with a protruding β hairpin which intercalates into the minor groove of DNA, causing the abasic pyrrolidine nucleotide to flip into the enzyme active site (48). Recognition of damaged bases by both proteins involves π-electron stacking interactions between electron-rich aromatic side chains of the protein and electron-deficient alkylated bases (46,48). Both enzymes utilize activated water molecules for nucleophilic attack. Based on the crystal structures, it was proposed that in the active site of the AlkA protein, the conserved residue Asp238 (46) and in ANPG protein, the conserved amino acid Glu125, deprotonate the water, resulting in a hydroxide nucleophil attacks of the C1′ carbon of the nucleotide (48). According to the general acid and general base catalysis, Asp238 of the AlkA protein and Glu125 of the ANPG protein act as a general base to abstract a proton from bound water. If one assumes that the catalytic mechanism utilized by the ANPG40 and AlkA proteins is similar to that of the uracil-DNA glycosylase for excision of uracil (49), the nucleophilic attack by water may not be sufficient in itself to displace damaged bases like HX or 1,N6-ethenoadenine (ɛA), and some other factor would be required to increase the efficiency of the leaving group of the base. One may speculate that this could be achieved by the protonation of the damaged base and that in the active site of the ANPG protein, the totally conserved His136 could act as a general acid to protonate the HX or ɛA residues. This hypothesis might explain the difference of kinetic constants for the HX repair between bacterial and human enzymes. Possible absence in the active site of the AlkA protein of an amino acid residue which could act as a general acid to protonate HX may explain the slow rate of catalysis observed. Since the kinetic constants for the removal of alkylated bases by the AlkA and ANPG proteins are quite similar (7,50), this could be explained by the fact that alkylated bases have in common a positive charge and a labilized glycosidic bond. Therefore one can tentatively propose that for excision of alkylated bases such as 3meAde and 7meGua, nucleophilic attack by water activated by the general base could be sufficient for catalysis. If it is true, the lower kcat/Km value for the repair of HX and ɛA residues by the AlkA protein could be explained if the active site of the AlkA protein lacks the general acid capable to protonate the base.
The second aim of the present investigation was to evaluate the influence of the sequence context, namely the nucleotide sequence 3′ and 5′ next to the dIMP residue, upon its excision by the various 3-meAde-DNA glycosylases. The results show that the bacterial, yeast and mammalian enzymes investigated have different sequence preferences. The more efficient removal of HX from the HX20-DL/T oligonucleotide was performed by the mammalian proteins, whereas the HX20-40/T was preferred by the E.coli AlkA and the HX20-34/G by the yeast MAG proteins. Since the ANPG40 and APDG proteins show, as described above, a clear preference for less thermodynamically stable mismatches, one can speculate that HX20-DL/T has a lower Tm than the others. Neighboring groups can have a large effect on thermal stability of duplex DNA containing dIMP residues (33), therefore affecting the repair of the lesion. The peculiar properties of the HX20-DL/N oligonucleotides, when substrates for the mammalian and E.coli enzymes, might be explained by the larger differences in Tm values between mismatches, as compared with other oligonucleotides. The thermal stability and NMR studies of the oligonucleotides used can give insight into consensus sequences for good and poor repair of HX from duplex DNA. However, the sequence effect on the repair of HX was studied in vitro on naked DNA, and it is possible that repair in vivo on coated DNA could be different.
The generation of 3-meAde-DNA glycosylase deficient (APDG ko) mice (51,52) will allow the assessment of the role of this enzyme in the repair of HX and the physiological relevance of such damage in vivo. Unexpectedly, these APDG ko mice do not exhibit any particular sensitivity to alkylating agents (53). Therefore, it will be interesting to test if such animals will be sensitive to agents generating HX or ɛA, since these two modified bases are excised in vitro with high efficiency by the APDG protein (5,23).
Acknowledgments
ACKNOWLEDGEMENTS
This work has been supported by grants from European Communities, Association pour la Recherche sur le Cancer and Fondation Franco-Norvégienne pour la Recherche Scientifique et Technique et le Développement Industriel.
REFERENCES
- 1.Lindahl T. and Nyberg,B. (1974) Biochemistry, 13, 3405–3410. [DOI] [PubMed] [Google Scholar]
- 2.Karran P. and Lindahl,T. (1980) Biochemistry, 19, 6005–6011. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl T., Ljungquist,S., Siegert,W., Nyberg,B. and Sperens,B. (1977) J. Biol. Chem., 252, 3286–3294. [PubMed] [Google Scholar]
- 4.Gallinari P. and Jiriçny,J. (1996) Nature, 383, 735–738. [DOI] [PubMed] [Google Scholar]
- 5.Saparbaev M. and Laval,J. (1994) Proc. Natl Acad. Sci. USA, 91, 5873–5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Connor T.R. and Laval,J. (1991) Biochem. Biophys. Res. Commun., 176, 1170–1177. [DOI] [PubMed] [Google Scholar]
- 7.O’Connor T.R. (1994) Nucleic Acids Res., 21, 5561–5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Connor T.R. and Laval,F. (1990) EMBO J., 9, 3337–3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berdal K.G., Bjoras,M., Bjelland,S. and Seeberg,E. (1990) EMBO J., 9, 4563–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakabeppu Y., Miyala,T., Kondo,H., Iwanaga,S. and Sekiguchi,M. (1984) J. Biol. Chem., 259, 13730–13736. [PubMed] [Google Scholar]
- 11.Hill-Perkins M., Jones,M.D. and Karran,P. (1986) Mutat. Res., 162, 153–163. [DOI] [PubMed] [Google Scholar]
- 12.Kamiya H., Miura,H., Kato,H., Nishimura,S., Ohtsuka,E. (1992) Cancer Res., 52, 1836–1839. [PubMed] [Google Scholar]
- 13.Nilsen H., Yazdankhah,S.P., Eftedal,I. and Krokan,H.E. (1995) FEBS Lett., 362, 205–209. [DOI] [PubMed] [Google Scholar]
- 14.Haukanes B.I., Helland,D.E. and Kleppe,K. (1989) Nucleic Acids Res., 17, 1493–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lagravere C., Malfoy,B., Leng,M. and Laval,J. (1984) Nature, 310, 798–800. [DOI] [PubMed] [Google Scholar]
- 16.Georgiadis P., Smith,C.A. and Swann,P.F. (1991) Cancer Res., 51, 5843–5850. [PubMed] [Google Scholar]
- 17.Ye N., Holmquist,G.P. and O’Connor,T.R. (1998) J. Mol. Biol., 284, 269–285. [DOI] [PubMed] [Google Scholar]
- 18.Brash D.E., Seetharam,S., Kraemer,K.H., Seidman,M.M. and Bredberg,A. (1987) Proc. Natl Acad. Sci. USA, 84, 3782–3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tornaletti S. and Pfeifer,G.P. (1994) Science, 263, 1436–1438. [DOI] [PubMed] [Google Scholar]
- 20.Boiteux S., Huisman,O. and Laval,J. (1984) EMBO J., 3, 2569–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boiteux S., O’Connor,T.R., Lederer,F., Gouyette,A. and Laval,J. (1990) J. Biol. Chem., 265, 3916–3922. [PubMed] [Google Scholar]
- 22.Tudek B., Van Zeeland,A.A., Kusmierek,J.T. and Laval,J. (1998) Mutat. Res., 407, 169–176. [DOI] [PubMed] [Google Scholar]
- 23.Saparbaev M., Kleibl,K. and Laval,J. (1995) Nucleic Acids Res., 23, 3750–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karran P. (1981) In Friedberg,E.C. and Hanawalt,P.C. (eds), DNA Repair: A Laboratory Manual of Research Procedures, Vol. 1. Marcel Dekker, New York, pp. 265–273.
- 25.Laval J. (1977) Nature, 269, 829–833. [DOI] [PubMed] [Google Scholar]
- 26.Graves R.J., Felzenswalb,I., Laval,J. and O’Connor,T.R. (1992) J. Biol. Chem., 267, 14429–14435. [PubMed] [Google Scholar]
- 27.Palombo F., Kohfeldt,E., Calcagnile,A., Nehls,P. and Dogliotti,E. (1992) J. Mol. Biol., 223, 587–594. [DOI] [PubMed] [Google Scholar]
- 28.Castaing B., Geiger,A., Seliger,H., Nehls,P., Laval,J., Zelwer,C. and Boiteux,S. (1993) Nucleic Acids Res., 21, 2899–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Connor T.R. and Laval,J. (1989) Proc. Natl Acad. Sci. USA, 86, 5222–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaillard C. and Strauss,F. (1990) Nucleic Acids Res., 18, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bailly V., Verly,W.G., O’Connor,T. and Laval,J. (1989) Biochem. J., 262, 581–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dianov G. and Lindahl,T. (1991) Nucleic Acids Res., 19, 3829–3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin F.H., Castro,M.M., Aboul-ela,F. and Tinoco,I.,Jr (1985) Nucleic Acids Res., 13, 8927–8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakabeppu Y., Kondo,H. and Sekiguchi,M. (1984) J. Biol. Chem., 259, 13723–13729. [PubMed] [Google Scholar]
- 35.Oesch F., Adler,S., Rettelbach,R. and Doerjer,G. (1986) In Singer,B. and Bartsch,H. (eds), The Role of Cyclic and Nucleic Acid Adducts in Carcinogenesis and Mutagenesis, IARC Scientific Publications no. 70. Oxford University Press, New York, NY, pp. 373–379.
- 36.Singer B., Antoccia,A., Basu,A.K., Dosanjh,M.K., Fraenkel-Conrat,H., Gallagher,P.E., Kusmierek,J.T., Qiu,Z.H. and Rydberg,B. (1992) Proc. Natl Acad. Sci. USA, 89, 9386–9390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mattes W.B., Lee,C.S., Laval,J. and O’Connor,T.R. (1996) Carcinogenesis, 17, 643–648. [DOI] [PubMed] [Google Scholar]
- 38.Uesugi S., Oda,Y., Ikehara,M., Kawase,Y. and Ohtsuka,E. (1987) J. Biol. Chem., 262, 6965–6968. [PubMed] [Google Scholar]
- 39.Corfield P.W.R., Hunter,W.N., Brown,T., Robinson,P. and Kennard,O. (1987) Nucleic Acids Res., 15, 7935–7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cruse W.B.T., Aymani,J., Kennard,O., Brown,T., Jack,A.G.C. and Leonard,G.A. (1989) Nucleic Acids Res., 17, 55–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carbonnaux C., Fazakerley,G.V. and Sowers,L. (1990) Nucleic Acids Res., 18, 4075–4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oda Y., Uesugi,S., Ikehara,M., Kawase,Y. and Ohtsuka,E. (1991) Nucleic Acids Res., 19, 5263–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Case-Green S.C. and Southern,E.M. (1994) Nucleic Acids Res., 22, 131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lindahl T. (1993) Nature, 362, 709–714. [DOI] [PubMed] [Google Scholar]
- 45.Miao F., Bouziane,M. and O’Connor,T.R. (1998) Nucleic Acids Res., 26, 4034–4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Labahn J., Scharer,O.D., Long,A., Ezaz-Nikpay,K., Verdine,G.L. and Ellenberger,T.E. (1996) Cell, 86, 321–329. [DOI] [PubMed] [Google Scholar]
- 47.Yamagata Y., Kato,M., Odawara,K., Tokuno,Y., Nakashima,Y., Matsushima,N., Yasumura,K., Tomita,K., Ihara,K., Fujii,Y., Nakabeppu,Y., Sekiguchi,M. and Fujii,S. (1996) Cell, 86, 311–319. [DOI] [PubMed] [Google Scholar]
- 48.Lau A.Y., Schrärer,O.D., Samson,L., Verdine,G.L. and Ellenberger,T. (1998) Cell, 95, 249–258. [DOI] [PubMed] [Google Scholar]
- 49.Savva R., McAuley-Hecht,K., Brown,T. and Pearl,L. (1995) Nature, 373, 487–493. [DOI] [PubMed] [Google Scholar]
- 50.Bjelland S., Birkeland,N.K., Benneche,T., Volden,G. and Seeberg,E. (1994) J. Biol. Chem., 269, 30489–30495. [PubMed] [Google Scholar]
- 51.Hang B., Singer,B., Margison,G.P. and Elder,R.H. (1997) Proc. Natl Acad. Sci. USA, 94, 12869–12784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Engelward B.P., Weeda,G., Wyatt,M.D., Broekhof,J.L., de Wit,J., Donker,I., Allan,J.M., Gold,B., Hoeijmakers,J.H. and Samson,L.D. (1997) Proc. Natl Acad. Sci. USA, 94, 13087–13092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elder R.H., Jansen,J.G., Weeks,R.J., Willington,M.A., Deans,B., Watson,A.J., Mynett,K.J., Bailey,J.A., Cooper,D.P., Rafferty,J.A, Heeran,M.C., Wijnhoven,S.W., van Zeeland,A.A. and Margison,G.P. (1998) Mol. Cell Biol., 18, 5828–5837. [DOI] [PMC free article] [PubMed] [Google Scholar]