

Abstract
RIPK4 is a key player in epidermal differentiation and barrier formation. RIPK4 signaling pathways controlling keratinocyte proliferation and differentiation depend on its kinase activity leading to Dvl2, Pkp1 and IRF6 phosphorylation and NF-κB activation. However, the mechanism regulating RIPK4 activity levels remains elusive. We show that cultured keratinocytes display constitutive active phosphorylated RIPK4 while PKC signaling can trigger RIPK4 activation in various non-keratinocyte cell lines, in which RIPK4 is present in a non-phosphorylated state. Interestingly, we identified the SCFβ-TrCP ubiquitin E3 ligase complex responsible for regulating the active RIPK4 protein level. The SCFβ-TrCP complex binds to a conserved phosphodegron motif in the intermediate domain of RIPK4, subsequently leading to K48-linked ubiquitinylation and degradation. The recruitment of β-TrCP is dependent on RIPK4 activation and trans-autophosphorylation. β-TrCP knock-down resulted in RIPK4-dependent formation of actin stress fibers, cell scattering and increased cell motility, suggesting that tight control of RIPK4 activity levels is crucial to maintain cell shape and behavior in keratinocytes.
Electronic supplementary material
The online version of this article (10.1007/s00018-018-2763-6) contains supplementary material, which is available to authorized users.
Keywords: RIPK4, β-TrCP, Keratinocytes, Proteasome, Degradation, PKC
Introduction
Based on the homology of the kinase domain, RIPK4 is classified as a member of the serine/threonine receptor-interacting protein kinases (RIPKs). In general, various signaling pathways from stress sensors, such as death receptors, pathogen pattern receptors and antigen-receptors converge to the RIPKs, which activate NF-κB and MAP kinase signaling, leading to inflammatory responses or cell death [1]. NF-κB and MAP kinase activation also play a key role in epidermal development and homeostasis [2]. The importance of RIPK4 for skin development has been witnessed by the fact that in humans RIPK4 mutations cause the rare autosomal-recessive Bartsocas-Papas syndrome (BPS), typically characterized by popliteal webbing, oligosyndactyly, genital abnormalities, and cleft palate/lip, causing perinatal death [3, 4]. We previously reported that RIPK4-deficient mice exhibit phenotypic features of BPS patients, like cleft palate and skin webbings between the hind limbs [5]. The epithelial malformations observed in RIPK4−/− mice are characterized by aberrant cortical actin organization and E-cadherin mislocalization at the apical site of epithelial cells [5]. Recently, it was shown that keratinocyte-specific deletion of E-cadherin in RIPK4 full KO mice was able to rescue the tail-to-body fusion and fusion of oral epithelia [6]. Furthermore, RIPK4-deficiency leads to impaired epidermal differentiation and barrier formation [5–7]. RIPK4−/− primary keratinocytes are unable to differentiate in vitro indicating a crucial role for RIPK4 in the keratinocyte intrinsic differentiation capacity [5]. RIPK4 can probably act at different levels to promote keratinocyte differentiation. RIPK4 has been shown to phosphorylate and thereby activate the IRF6 transcription factor leading to keratinocyte differentiation [8]. Recently, Lee et al. found that RIPK4 can also promote keratinocyte differentiation by phosphorylating the desmosome protein plakophilin-1 (Pkp1) and that loss of RIPK4 in the epidermis promotes tumorigenesis [9].
RIPK4 was initially identified as an interaction partner of two isozymes of the protein kinase C family, being PKCβ and PKCδ [10, 11]. PKC isozymes are involved in the regulation of differentiation, growth, cell death and motility, and are categorized in three subclasses (conventional, novel and atypical PKC kinases) based on the dependency of Ca2+ and/or diacylglycerol for their activation [12, 13]. In the epidermis PKCs are important regulators of keratinocyte differentiation. The small molecule mimic of diacylglycerol, phorbol 12-myristate 13-acetate (PMA), is a potent activator of conventional and novel PKCs and triggers in vitro keratinocyte differentiation [14]. Recent studies showed that RIPK4 mediates PMA-induced keratinocyte differentiation through activation of the IRF6 transcription factor and mediates PMA-induced NF-κB activation [8, 15, 16].
Targeted protein degradation of misfolded or damaged proteins is essential to maintain cellular proteostasis. Besides this, targeted protein degradation fulfills a crucial regulatory role in numerous signal transduction pathways [17]. The ubiquitin–proteasome system (UPS) is the primary mechanism executing selective protein degradation [18]. The UPS makes use of a two-step process in which substrates are first tagged with K48-linked ubiquitin moieties that are subsequently recognized by the 26S proteasome where the proteolysis of the substrates occurs [19]. The ubiquitin modification is catalyzed by an enzymatic cascade involving a ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3). E3 ligases bind and modify only a subset of substrates, providing specificity to the UPS system. The Skp1-Cul1-F-box protein (SCF) E3 ubiquitin ligase complex is a RING-type ubiquitin ligase composed of three core components; the scaffolding protein Cullin-1, the E2 conjugating enzyme Rbx1 and the Skp1 adaptor protein [20]. Skp1 in turn binds one of the 69 available F-box proteins, which are the substrate specificity subunits of the SCF complex. Beta-transducin repeat-containing protein (β-TrCP), the best characterized F-box protein with over 50 known substrates, recognizes phosphorylated serine residues within degron sequences and was shown to be involved in the regulation of many signaling cascades; e.g. NF-κB and Wnt signaling, or cellular processes such as mitosis [21].
Besides the rare Bartsocas-Papas syndrome caused by the recessive autosomal mutations in RIPK4, deregulation of RIPK4 expression levels and its kinase activity have been linked to several types of cancers and inflammation in adult non-syndromic persons and mice studies [9, 16, 22–26]. Therefore, a better understanding of how RIPK4 levels are regulated is valuable. Here, we identified the SCFβ-TrCP ubiquitin ligase as a bona fide E3 ligase for activated RIPK4. We show that upon PKC activation RIPK4 is hyper-phosphorylated and degraded by the SCFβ-TrCP-mediated proteasomal degradation pathway. Additionally, we found that SCFβ-TrCP controls the levels of constitutively activated RIPK4 in keratinocytes, thereby preventing actin cytoskeletal reorganizations that lead to increased motility in in vitro cultured keratinocytes.
Materials and methods
Cell lines
HEK293T, A431, A549 and HaCaT cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated fetal calf serum and l-glutamine. Cells were cultured at 37 °C in a humidified atmosphere of 5% CO2 and were tested for the absence of mycoplasma contamination. HaCaT cells were subcultured at approximately 80% confluence. The generation of stable doxycycline-inducible RIPK4 expressing HaCaT cell lines was described in De Groote et al. [5].
Reagents and antibodies
The following drugs were used at indicated concentration: cycloheximide (Sigma-Aldrich; 25 µg/ml), MG132 (Calbiochem; 10 µM) and phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich; 0.5 µM), SP600125 (Cayman Chemical; 10 µM), SB202190 (Sigma-Aldrich; 10 µM), D4476 (Sigma-Aldrich; 10 µM), CX-4945 (APExBIO; 10 µM), TPCA (Tocris Bioscience; 10 µM), CHIR99021 (Miltenyi Biotec; 10 µM) dissolved in dimethyl sulfoxide (DMSO) (Sigma-Aldrich). Tet-on inducible cells were induced with doxycycline dissolved in Milli-Q water (Sigma-Aldrich, 1 µg/ml). The following antibodies were used throughout this study: anti-β-TrCP1/2 (Cell Signaling Technology, #4394), anti-RIPK4 (Abnova, clone 2G3), anti-RIPK4 (Cell Signaling Technology, #12636), anti-IKKα (Cell Signaling Technology, #2682), anti-IKKβ (Cell Signaling Technology, #2684), anti-JNK1/2 (Cell Signaling Technology, #9252), anti-β-tubulin (Abcam, #21058), anti-Flag-tag HRP coupled (Sigma-Aldrich, clone M2), anti-VSV-tag (Sigma-Aldrich, clone P5D4) and anti-HA-tag (Covance, clone HA.11). Recombinant Wnt3a was purchased from R&D Systems (5036-WN).
Plasmid constructions and mutagenesis
We obtained pENTR223 vectors containing the open reading frames without stop codon for β-TrCP1, β-TrCP2, cullin-2, cullin-3, cullin-4b, Rbx1, Skp1 from the V8.1 collection provided by the BCCM/GeneCorner plasmid collection [27]. The plasmids encoding RIPK4 mutants were generated from a pENTR3C-hRIPK4 construct using the QuikChange mutagenesis kit (Stratagene). The primer pairs used to generate the RIPK4 mutations can be found in Supplementary Fig. 5. cDNA sequences encoding human PKCα, -βII, γ, -ε and -δ were cloned into pENTR3C using the cloneEZ PCR cloning kit (GenScript). The primer pairs used to clone the PKC isoforms can be found in Supplementary Fig. 5. A PKCβI ORF shuttle clone (IOH43969; in pENTR221) was purchased from ImaGenes. Expression vectors were generated by LR Gateway Cloning (Thermo Fischer Scientific) into modified Gateway compatible vectors pcDNA-FlagMyc (LMBP #04705) and pLenti6-puro-VSV (LMPB # 09165). Expression vectors pcDNA3.1-Flag-CUL1 (LMBP #08315), pcDNA3-HA-hUB-WT (LMMBP #5893), pcDNA3-HA-hUB-K48R (LMBP #5898) and pcDNA3-Flag-hPKCη-Myc (LMBP #09972) were obtained from the BCCM/GeneCorner plasmid collection (http://www.genecorner.ugent.be).
RNA interference
A reverse transfection protocol was used for the transfection of HaCaT and A549 cells. 90,000 cells were seeded in a six-well plate and transfected with siRNA (final concentration: 20 nM) using Polyplus INTERFERin transfection reagent (Westburg B.V.) according to the manufacturer’s protocol. After 48 h, transfected cells were lysed for further analysis. The following ON-TARGETplus SMART pool siRNAs were purchased from Dharmacon (GE Healthcare): non-targeting siRNA (D-001810-10-20), JNK1 (L-003514-00), JNK2 (L-003505-00), IKK1 (L-003473-00) and IKK2 (L-003503-00). RIPK4 targeting siRNA was purchased from Ambion (s28865, Thermo Fisher Scientific). Oligonucleotides were designed for targeting both β-TrCP1 and β-TrCP2 and synthesized by Ambion (Thermo Fisher Scientific); sense sequence 5′-GUGGAAUUUGUGGAACAUC-3′ and antisense sequence 5′-GAUGUUCCACAAAUUCCAC-3′.
RT-qPCR
Total RNA was purified using an RNAeasy mini kit (Qiagen). 1 µg of total RNA was reverse-transcribed using iScript cDNA Synthesis Kit (Biorad). Real-time PCR was performed using SensiFast SYBR No-Rox Kit (GC Biotech), 10 ng of cDNA and 300 nM primers. RIPK4 qPCR primer sequences are as follows: 5′-TGGCTGCCACTGCACTACG-3′ and 5-CGTCCTCCCATCCAGCGTCT-3′. Messenger RNA levels were normalized to the geometric mean of HPRT1 and RPL13a expression using primers described earlier [28]. Real-time PCR was performed using a LightCycler 480 (Roche).
Immunoprecipitation
1.5 × 106 HEK293T cells were transfected transiently with the indicated expression plasmids using a standard calcium phosphate protocol (CaPO4). 24 h after transfection, cells were washed with PBS, harvested by scraping in ice-cold PBS and pelleted by centrifugation. The pelleted cells were lysed in 200 μl NP-40 lysis buffer [1% NP40; 10% glycerol; 150 mM NaCl; 5 mM HEPES pH 7.4; 5 mM EDTA, supplemented with complete protease inhibitor cocktail (Roche) and PhosSTOP (Roche)]. Lysates were cleared from debris by centrifugation and 20 μl of cleared lysate was used for input control. The rest of the cleared lysate was then incubated for overnight at 4 °C on a rotating wheel using anti-FLAG M2 Affinity Gel beads (Sigma, #A2220). Then, beads were washed three times with NP-40 lysis buffer. Finally, the beads were resuspended in 1× Laemmli buffer (50 mM Tris–HCl pH 6.8; 2% SDS; 0.1% bromophenol blue; 4% beta-mercapto-ethanol) and heated for 7 min at 95 °C. Samples were analyzed by standard SDS-PAGE and immunoblotting protocols. In experiments where ubiquitinylation of immunoprecipitated proteins was analyzed by western blotting, cells were lysed in RIPA buffer [20 mM Hepes pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 10 mM N-ethylmaleimide, supplemented with complete protease inhibitor cocktail (Roche) and PhosSTOP (Roche)]. Washing steps after immunoprecipitation were done with high-salt RIPA buffer (500 mM NaCl).
Lambda-phosphatase treatment of immunoprecipitated Flag-tagged RIPK4
Immunoprecipitation of Flag-RIPK4 was performed as described above. After overnight incubation, the anti-FLAG M2 Affinity Gel was divided into three reaction tubes, washed three times with NP40 buffer and two times in lambda-phosphatase reaction buffer (50 mM Tris–HCl, pH 7.5, 100 mM NaCl, 2 mM DTT, 0.1 mM EGTA, 0.01% Brij-35). Two of the IP reactions were then incubated with 100 U of lambda-phosphatase (PO753S, New England Biolabs) with or without PhosSTOP (Roche) for 30 min at room temperature. Following the dephosphorylation reaction, the beads were washed three times with NP40 buffer. HA-tagged β-TrCP2 was separately transfected in HEK293T cells, which were lysed in NP40 buffer. Equal amounts of the lysate were incubated with the washed IP beads from the dephosphorylation reaction at 4 °C for 4 h. IP beads were then washed three times with NP40 buffer and processed as mentioned above.
Phosphatase treatment of whole cell lysates
Cells were treated with the proteasome inhibitor MG132 (10 µM) to stabilize the fraction of phosphorylated RIPK4 and were subsequently lysed in NP40 buffer without phosphatase inhibitors. Lysates were then divided into different reaction tubes and 10× NEB phosphatase buffer was added with or without 400 U lambda-phosphatase (PO753S, New England Biolabs). All reactions were incubated at 37 °C for 5 min and subsequently quenched by adding Laemmli buffer.
Immunofluorescence
HaCaT keratinocytes were seeded and reverse transfected with siRNA on collagen-coated chambered coverslips (iBidi µ-slide 8-well). 48 h after transfection cells were fixed with 3% paraformaldehyde for 15 min at room temperature, permeabilized using 0.2% Triton-X100 (Roche) in PBS for 5 min at room temperature and blocked in 0.5% bovine serum albumin (Sigma-Aldrich). F-actin was stained with Acti-stain 488 phalloidin (Cytoskeleton Inc, # PHDG1-A, 100 nM final concentration), nuclei were stained with Hoechst 33342 (Invitrogen, #H21492) and covered with n-propyl gallate anti-fade. Confocal images were taken with a Leica SP5, making use of a 63×, 1.4 N.A. oil immersion lens.
Live-cell imaging
HaCaT cells were reverse transfected with siRNA and seeded on collagen-coated cell culture plates. See ‘RNA interference’ section in “Materials and methods”. 16 h after transfection the cells were refreshed and imaged every 15 min with an IncuCyte microscope (Essen Bioscience).
TAP-tag purification and mass spectrometry
HEK293T cells were cultured in DMEM supplemented with 10% fetal calf serum, penicillin (100 IU/ml), streptomycin (0.1 mg/ml), l-glutamine (0.03%) and sodium pyruvate (0.4 mM). Cells were plated at 3.7 million cells/14 cm dish, and 24 h after seeding, cells were transfected by CaPO4-precipitation with 10 μg of pCEMM N-TAP(GS)hRIPK4 or 10 μg of empty pCEMM N-TAP(GS) as a negative control. Four 14-cm dishes were transfected per TAP-purification. 24 h after transfection, cells were harvested by scraping in ice-cold PBS. All consecutive steps were performed at 4 °C. Cells were washed three times with PBS, lysed on ice for 15 min in 10 ml TAP-lysis buffer [50 mM Tris HCl pH 7.5, 100 mM NaCl, 5% glycerol, 0.2% NP40, 1.5 mM MgCl2, 1 mM DTT, 50 mM NaF, 1 mM Na3VO4, Complete protease inhibitor cocktail (Roche)]. The supernatant was incubated with 150 μl of IgG-Sepharose beads (Amersham) for 2 h on a rotating platform. The sample was centrifuged and the beads were transferred to a small filter column (Biorad polyprep 10 ml) and washed by gravity flow with 10 ml of TAP-lysis buffer. Consequently, the column was washed with 5 ml of TEV-cleavage buffer (10 mM Tris HCl pH 7.5, 100 mM NaCl, 0.5 mM EDTA, 0.1% NP-40). Next, the beads were incubated for 1 h at 16 °C, while shaking, with 100 U of TEV-protease (Gibco) in 400 μl TEV-cleavage buffer, to elute the bound complexes. The sample was centrifuged, and the collected supernatant represented the TEV-eluate. Next, the beads were washed with another 400 μl of TEV-cleavage buffer from which the supernatant was added to the former TEV-eluate. This 800 μl eluate was consequently incubated for 1 h at 4 °C with 200 μl slurry of streptavidin beads (Ultra-link, Pierce) in TEV-cleavage buffer. The sample was centrifuged and the beads were transferred to small filter columns (Biorad polyprep 10 ml), washed by gravity flow with 10 ml of TEV-cleavage buffer. Finally, complexes were eluted with 1.5 ml TEV-cleavage buffer supplemented with 5 mM biotin (Fluka). This constituted the TAP-eluate that was consequently precipitated with 10% TCA, after which the pellet was washed with cold acetone and resolubilized in Laemmli buffer. Samples were run on gradient gels (NuPAGE 4-12% Bis–Tris gel, Invitrogen) and subsequently blotted to nitrocellulose or silver stained (Silver stain plus, Bio-Rad), or stained with colloidal Coomassie (Coomassie brilliant blue G-250, Invitrogen). Lanes from Coomassie-stained gels from RIPK4 and control purifications (TAP-tag construct without RIPK4 sequence) were cut and tryptic peptides were identified by MS/MS. Briefly, the gel bands were excised and transferred to Biopure® Eppendorf tubes (Eppendorf AG, Hamburg, Germany). After washing with water (HPLC graded, Baker Malinckrodt B.V., Amsterdam, The Netherlands), 50% (v/v) acetonitrile (Baker Malinckrodt B.V.) in water and 100% acetonitrile, they were submerged in 50 mM ammonium bicarbonate at pH 8.0 and digested using trypsin. The digestion proceeded overnight at 37 °C and was stopped by acidification through addition of formic acid.
Following a brief centrifugation step, the peptide mixture was transferred to a new Eppendorf tube, dried and re-dissolved in 20 µl of 0.1% formic acid in 2/98 (v/v) acetonitrile/water (solvent A). 10 μl of this peptide mixture was applied for nano-LC–MS/MS analysis on an Ultimate (Dionex, Amsterdam, The Netherlands) in-line connected to an Esquire HCT mass spectrometer (Bruker, Bremen, Germany). The sample was first trapped on a PepMap™ C18 column, 0.3 mm I.D. (internal diameter) × 5 mm (Dionex, Amsterdam, The Netherlands). After back-flushing from the trapping column, the sample was loaded on a 75-μm I.D. × 150 mm PepMap™ C18 reverse-phase column (Dionex, Amsterdam, The Netherlands). The peptides were eluted with a linear gradient of 3% solvent B [0.1% formic acid in water/acetonitrile (3/7, v/v)] increase per minute at a constant flow rate of 200 nl/min. Using data-dependent acquisition, multiply charged ions with intensities above the threshold of 100,000 were selected for fragmentation. During MS/MS analysis, an MS/MS fragmentation amplitude of 0.7 V and a scan time of 40 ms were used.
The fragmentation spectra were converted to mgf files using the Automation Engine software (version 3.2, Bruker) and were searched using the MASCOT database search engine (http://www.matrixscience.com) against the human IPI database. Peptide mass tolerance and peptide fragment mass tolerance were set at ± 0.5 Da, with the ESI-IT as selected instrument for peptide fragmentation rules. Variable modifications were set to methionine oxidation, pyro-glutamate formation of amino-terminal glutamine, acetylation of the N-terminus and propionamide modification of cysteines.
Wnt reporter assay
HEK293T cells were transfected with TOP/FOP flash luciferase reporter plasmids [29]. Twenty-four hours after transfection the cells were stimulated with 200 ng/ml recombinant Wnt3a. Twenty-four hours later, the cells were lysed in luciferase lysis buffer (25 mM Tris phosphate pH 7.8, 2 mM DTT, 2 mM CDTA, 10% glycerol and 1% Triton-X-100), substrate buffer was added (658 mM luciferin, 378 mM coenzyme A and 742 mM ATP) and luciferase activity was assayed in a GloMax 96 Microplate Luminometer (Promega).
Results
Active RIPK4 is targeted for proteasomal degradation in keratinocytes
RIPK4 is essential for epidermal development and keratinocyte differentiation [5]. When examining the RIPK4 protein levels in the human HaCaT keratinocyte cell line and in the epidermoid carcinoma cell line A431, we noticed that RIPK4 is consistently present as two protein bands, both with low abundance (Fig. 1a). The upper RIPK4 band has been shown to be, at least in part, an auto-phosphorylated form of RIPK4 which we confirmed by performing dephosphorylation experiments (Fig. 1b, compare lanes 1 and 3) [11]. The non-keratinocyte HEK293T and A549 cells only contain non-phosphorylated RIPK4 (Fig. 1a). These data suggest that RIPK4 is continuously activated, and possibly degraded, in in vitro keratinocyte cultures. Therefore, we sought to determine the stability of RIPK4. Treatment of HaCaT and A431 cell lines with the proteasome inhibitor MG132 resulted in substantial accumulation of the phospho-RIPK4 protein band (Fig. 1a), in contrast to other cell lines such as HEK293T and A549 where RIPK4 is present as a stable non-phosphorylated form (Fig. 1a). In the presence of the protein synthesis inhibitor cycloheximide (CHX), RIPK4 levels (both the non-phospho- and phospho-form) drop significantly in HaCaT cells after a 2 h treatment, while its expression was not affected in HEK293T cells, which only express non-active RIPK4 (Fig. 1c). These experiments indicate that the low abundance of activated RIPK4 in keratinocytes is a consequence of proteasome-dependent degradation. Because the presence of phospho-RIPK4 depends on its kinase activity [11], we compared the turnover of ectopically expressed wild-type and kinase-inactive (K51R) RIPK4. Wild-type RIPK4 protein abundance decreased after blocking de novo protein synthesis with CHX, while RIPK4-K51R levels were not affected by this treatment (Fig. 1d). Applying a combination of CHX and the proteasome inhibitor MG132 rescued the degradation of phospho-RIPK4, but did not have a significant effect on non-phospho-RIPK4 levels of the RIPK4-K51R kinase-dead mutant (Fig. 1d). Our data suggest that RIPK4 resides in an active, hyper-phosphorylated state in in vitro cultured keratinocytes, and is continuously targeted for proteasomal degradation.
Fig. 1.
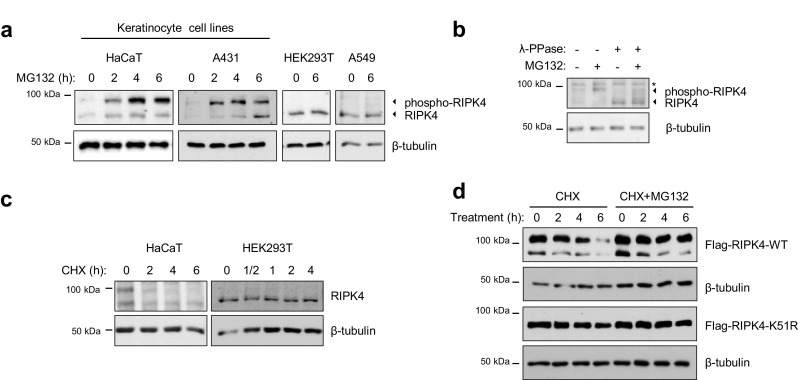
Active RIPK4 is targeted for proteasomal degradation in keratinocytes. a HaCaT, A431, HEK293T and A549 cells were treated with the proteasome inhibitor MG132 (10 µM) for the indicated time points. Whole cell lysates were analyzed by immunoblotting with the indicated antibodies. b HaCaT cells were treated with the proteasome inhibitor MG132 for 4 h. Wholes cell lysates were treated with lambda-phosphatase for 5 min and analyzed by immunoblotting with the indicated antibodies, * aspecific band. c HaCaT and HEK293T cells were treated with cycloheximide (CHX, 25 µg/ml) and lysed at the indicated time points. Whole cell lysates were analyzed by immunoblotting with the indicated antibodies. d HEK293T cells were transfected with expression vectors coding for Flag-tagged RIPK4-WT or -K51R. Twenty-four hours after transfection, cells were treated with CHX (25 µg/ml) with or without MG132 (10 µM) for the indicated time. Whole cell lysates were analyzed by immunoblotting with the indicated antibodies
Kinase active RIPK4 binds to the SCFβ-TrCP ubiquitin ligase complex
To identify proteins involved in the regulation of RIPK4, we performed a tandem affinity purification (TAP) followed by mass spectrometry (MS) analysis of RIPK4-binding partners (Supplementary Fig. 1a, b). We identified two unique peptides corresponding to β-TrCP2 and one peptide shared between β-TrCP1 and 2 (Supplementary Fig. 1c). β-TrCP is an F-BOX protein that adds substrate specificity to the SKP1-CUL1-F-box protein (SCF) E3 ubiquitin ligase complex. β-TrCP1 and β-TrCP2 are paralogs with redundant biochemical characteristics and share identical substrates [21]; therefore, β-TrCP will be used to refer to both proteins unless specified otherwise. The interaction between RIPK4 and β-TrCP1 and β-TrCP2 was confirmed by co-immunoprecipitation of the overexpressed proteins in HEK293T cells (Fig. 2a). To verify that RIPK4 interacts with the conventional SCF complex through β-TrCP, we performed co-immunoprecipitation of various cullin proteins and found that RIPK4 specifically interacts with the cullin-1 scaffold complexes (Fig. 2b). Consequently, we could demonstrate that RIPK4 interacts with the invariable Skp1 adaptor protein and the Rbx1 E2 enzyme of the SCFβ-TrCP complex (Fig. 2c). These results suggest that the SCFβ-TrCP complex is involved in the regulation of RIPK4 stability.
Fig. 2.
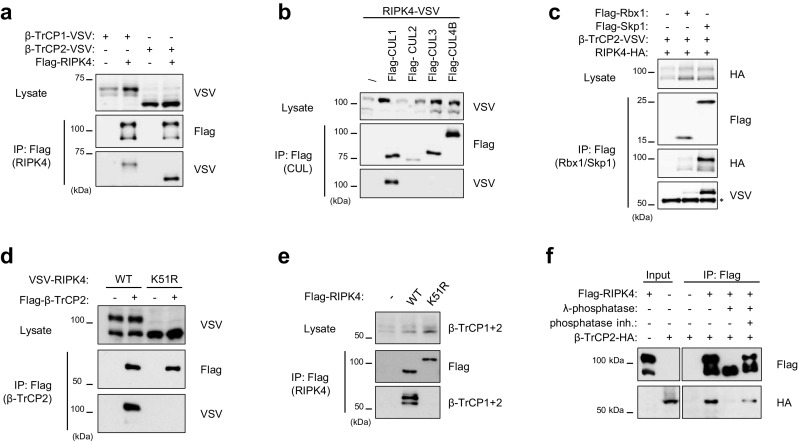
Kinase-active RIPK4 binds to the SCFβ-TrCP ubiquitin ligase complex. a–c HEK293T cells were transfected with expression vectors coding for the indicated tagged proteins. Twenty-four hours after transfection, cells were lysed and immunoprecipitated using anti-Flag beads. Immunoprecipitates (IP) and cell lysates were analyzed by immunoblotting using the indicated antibodies. d HEK293T cells were transfected with VSV-RIPK4-wild-type (WT) or -kinase-dead (K51R) expression vectors in combination with Flag-tagged β-TrCP2. Twenty-four hours after transfection, cells were lysed and immunoprecipitated using anti-Flag beads. IP and cell lysate were subjected to immunoblotting using the indicated antibodies. e HEK293T cells were transfected with Flag-RIPK4-wild-type (WT) or -kinase-dead (K51R) expression vectors. Twenty-four hours after transfection, cells were lysed and immunoprecipitated using anti-Flag beads. IP and cell lysate were subjected to immunoblotting using the indicated antibodies to probe for co-IP of endogenous β-TrCP1/2. f HEK293T cells were transfected with a Flag- RIPK4 expression vector. Twenty-four hours after transfection, cells were lysed and immunoprecipitated using anti-Flag beads. Immunoprecipitated RIPK4 was left untreated or treated with lambda-phosphatase with or without phosphatase inhibitor cocktail. Subsequently, the immunoprecipitates were incubated with total lysate extracts of HA-β-TrCP2-transfected HEK293T cells and further immunoprecipitated with anti-Flag beads. The input and immunoprecipitates were immunoblotted with the indicated antibodies
We observed that immunoprecipitation of the different components of the SCFβ-TrCP complex mainly co-precipitated phospho-RIPK4 (Fig. 2b, c). Because RIPK4 hyper-phosphorylation depends on its kinase activity, we investigated whether RIPK4 kinase activity is required for the interaction with β-TrCP2. Wild-type RIPK4 interacted with ectopically expressed β-TrCP2, while kinase-dead RIPK4-K51R did not (Fig. 2d). In addition, only wild-type RIPK4 was able to pull down endogenous β-TrCP (Fig. 2e). To prove that the RIPK4-β-TrCP2 interaction is RIPK4 phosphorylation-dependent, we performed a lambda-phosphatase treatment of immunoprecipitated RIPK4, which was subsequently incubated with the lysate of HEK239T cells expressing HA-β-TrCP2. Dephosphorylation of RIPK4 prevented the interaction with β-TrCP2 (Fig. 2f). This is in agreement with our previous results showing that hyper-phosphorylated RIPK4 is specifically targeted for proteasomal degradation (Fig. 1d). Taken together, these data indicate that RIPK4 kinase activity is necessary for RIPK4 phosphorylation-dependent recruitment of the SCFβ-TrCP complex.
RIPK4 autophosphorylation recruits β-TrCP to a conserved degron
β-TrCP binds its substrates through its WD40 repeats that recognize a phosphodegron motif in the substrate. The canonical phosphodegron motif has the consensus DSGXXS sequence in which phosphorylation of the two serine residues is required for β-TrCP binding [30]. However, many identified β-TrCP substrates have some degree of variation within the phosphodegron sequence (Fig. 3a) [30]. Upon scanning the RIPK4 protein sequence, we identified three potential phosphodegron motifs, namely 340-DSGVS-344, 378-DSAFSS-383 and 436-DSGAS-440. Alignment of RIPK4 orthologues revealed that the sequence of the DSGVS and DSAFSS motif is highly conserved (Fig. 3b). Because serine phosphorylation within the degron is crucial for recognition by β-TrCP we generated mutants in which we substituted all serine residues within the putative degron motifs by alanine. Subsequently, the RIPK4 phosphodegron mutants were tested for their ability to pull down endogenous β-TrCP from HEK293T cells. The RIPK4 S379A/S382A/S383A mutant lost its ability to bind β-TrCP, while the other putative RIPK4 phosphodegron mutants had similar binding to β-TrCP as wild-type RIPK4 (Fig. 3c). Mutating S379 or S383, serine residues corresponding to the canonical phosphodegron, is sufficient to abrogate the interaction with β-TrCP, while S382 is not involved in β-TrCP recruitment (Fig. 3d). Note that mutating the phosphodegron serine residues does not affect the band shift induced by RIPK4 autophosphorylation (Fig. 3d). These experiments show that the 378-DSAFSS-383 amino acid stretch is a functional phosphodegron crucial for β-TrCP recruitment to active RIPK4.
Fig. 3.
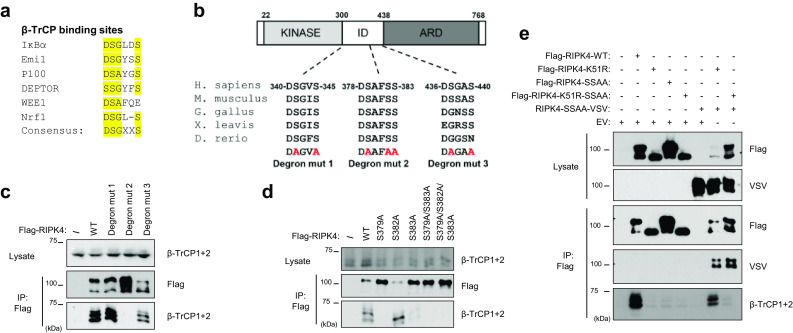
RIPK4 trans-autophosphorylation recruits β-TrCP to a conserved degron. a Phosphodegron alignment of previously described β-TrCP substrates with consensus phosphodegron motif. “X” represents any amino acid, “en dash” represents no amino acid. b Schematic representation of RIPK4 functional domains (KINASE kinase domain, ID intermediate domain, ARD ankyrin repeat domain) with indication of three putative phosphodegron motifs, their alignment with RIPK4 orthologs and their corresponding mutated (mut) forms. c HEK293T cells were transfected with expression vectors for Flag-tagged wild-type (WT) RIPK4 or RIPK4 containing the phosphodegron mutations indicated in b. Twenty-four hours after transfection, cells were lysed and subjected to immunoprecipitation (IP) with anti-Flag beads followed by immunoblotting with the indicated antibodies. d HEK293T cells were transfected with a Flag-tagged RIPK4 expression vector containing single and multiple alanine mutants of the DSAFSS motif. Twenty-four hours after transfection, cells were lysed and subjected to immunoprecipitation with anti-Flag beads followed by immunoblotting with the indicated antibodies. e HEK293T cells were transfected with expression vectors coding for Flag- or VSV-tagged RIPK4 WT or kinase-dead (K51R) versions with a functional or non-functional phosphodegron (SS/AA refers to S379A/S383A), or with empty vector (EV). Twenty-four hours after transfection, cells were lysed and subjected to immunoprecipitation with anti-Flag beads followed by immunoblotting with the indicated antibodies
Our previous results suggested that RIPK4 kinase activity triggers RIPK4 autophosphorylation leading to β-TrCP recruitment. If RIPK4 trans-autophosphorylation would be responsible for hyper-phosphorylation and recruitment of β-TrCP, kinase-active RIPK4 should be able to convert kinase-inactive RIPK4 into a β-TrCP-binding protein upon co-expression. To avoid interference in the co-IP experiments, we used a kinase-active RIPK4 phosphodegron (379A/S383A) mutant that cannot bind β-TrCP instead of wild-type RIPK4. Immunoprecipitation of RIPK4-K51R was indeed able to successfully pull down endogenous β-TrCP when co-expressed with the RIPK4 phosphodegron mutant (Fig. 3e), demonstrating that trans-autophosphorylation confers β-TrCP-binding properties to RIPK4.
β-TrCP ubiquitinylates active RIPK4 and triggers its proteasomal degradation
The SCFβ-TrCP E3 ubiquitin ligase complex catalyzes ubiquitinylation upon binding of its substrates [20]. To assess if RIPK4 is a bona fide substrate of the SCFβ-TrCP ubiquitin ligase complex, we performed ubiquitinylation experiments in HEK293T cells. Cells were treated with the proteasome inhibitor MG132 to enrich the ubiquitinylated species, which otherwise would be targeted for degradation. RIPK4 was significantly ubiquitinylated when overexpressed in the presence of HA-tagged wild-type ubiquitin, while overexpression of RIPK4 in the presence of HA-tagged K48R mutant ubiquitin resulted in very weak ubiquitinylation, suggesting that the bulk of the observed RIPK4 ubiquitinylation was K48-linked (Fig. 4a). Co-expression of β-TrCP increased RIPK4 K48-ubiquitin chains (Fig. 4a). As expected, ectopic β-TrCP2 expression did neither induce ubiquitinylation of kinase-inactive RIPK4 nor of the RIPK4 phosphodegron mutant, in line with the fact that β-TrCP was unable to bind these mutant RIPK4 forms (Fig. 4b). Consequently, the protein levels of the RIPK4 phosphodegron mutant were not affected upon inhibition of de novo protein synthesis (Fig. 4c). Depletion of endogenous β-TrCP1 and β-TrCP2, using siRNA targeting, stabilized the degradation of ectopic RIPK4 when protein translation was inhibited (Fig. 4d). We conclude that RIPK4 recruitment to the SCFβ-TrCP complex catalyzes its ubiquitinylation eventually leading to proteasomal degradation.
Fig. 4.
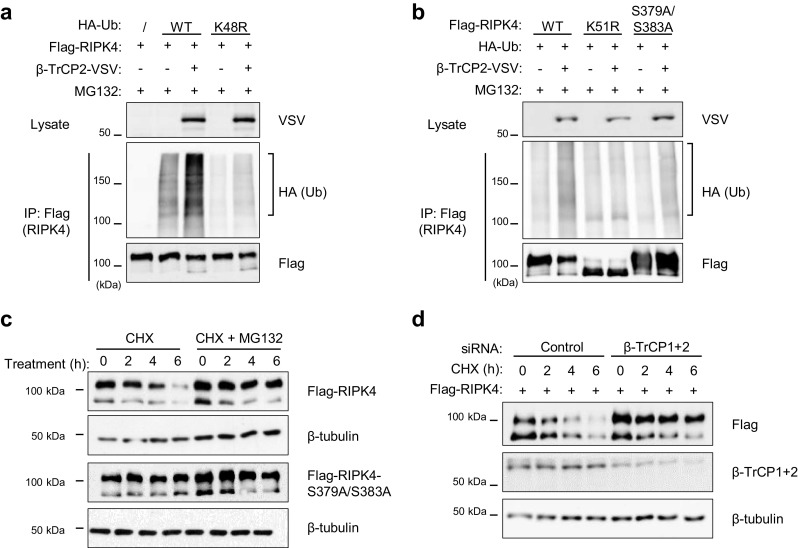
β-TrCP ubiquitinylates active RIPK4 and triggers its proteasomal degradation. a, b HEK293T cells were transfected with expression vectors coding for Flag-tagged RIPK4-wild-type (WT), -kinase-dead (K51R), or -phosphodegron mutant (S379A/S383A), VSV-tagged β-TrCP2 and HA-tagged ubiquitin or ubiquitin with mutated lysine-48 (K48R). Twenty-four hours after transfection, the cells were treated for 6 h with the MG132 (10 µM) to prevent proteasomal degradation and enrich the ubiquitinylated fraction. Then, cell lysates were subjected to immunoprecipitation (IP) with anti-Flag beads followed by immunoblotting with the indicated antibodies. c HEK293T cells were transfected with expression vectors coding for Flag-tagged RIPK4-WT or -S379A/S383A. Twenty-four hours after transfection, cells were treated with CHX (25 µg/ml) with or without MG132 (10 µM) for the indicated time. Whole cell lysates were analyzed by immunoblotting with the indicated antibodies. d HEK293T cells were seeded and transfected with non-targeting (control) or β-TrCP1+2 targeting siRNA (20 nM). Forty-eight hours later the cells were transfected with an expression vector coding for Flag-tagged RIPK4. Next day the cells were treated for the indicated time with CHX (25 µg/ml). Whole cell lysates were analyzed by immunoblotting with the indicated antibodies. Ub ubiquitin
PKC signaling activates RIPK4 and triggers β-TrCP-dependent degradation
Because we observed continuous RIPK4 phosphorylation in keratinocytes in contrast to HEK293T cells, we wanted to identify the signaling pathway that initiates RIPK4 activation. HEK293T and A549 cell lines only express non-phosphorylated RIPK4 (Fig. 1a), therefore we used those cell lines to test which stimuli induce RIPK4 phosphorylation and degradation. Since RIPK4 has been identified as a PKC-interacting protein, we tested if the PKC activator phorbol 12-myristate 13-acetate (PMA), a small molecule mimic of the physiological PKC activator diacylglycerol, triggers RIPK4 phosphorylation and degradation. PMA induced phosphorylation of RIPK4 within minutes of stimulation followed by degradation of phospho-RIPK4 within 2-4 h (Fig. 5a, b). This PMA-induced phospho-RIPK4 degradation could be effectively inhibited by treatment with the proteasome inhibitor MG132 (Fig. 5a, b). The mobility shift observed upon PMA treatment was indeed proven to be assigned to RIPK4 phosphorylation events as lambda-phosphatase treatment converted the upper RIPK4 protein band to the size of non-phosphorylated RIPK4 (Fig. 5c). By knocking down β-TrCP, we could rescue PMA-induced RIPK4 degradation in HEK293T cells (Fig. 5d), suggesting that PKC signaling is activating RIPK4, directly or indirectly, leading to its proteasomal degradation. Because PMA can activate all classical and novel PKC isoforms, we tested which classical and novel PKC isoforms were able to phosphorylate RIPK4, as witnessed by a mobility shift in a western blotting experiment. We found that PKCε, PKCδ and PKCη can induce a mobility shift of RIPK4-K51R upon co-expression in HEK293T cells (Fig. 5e).
Fig. 5.
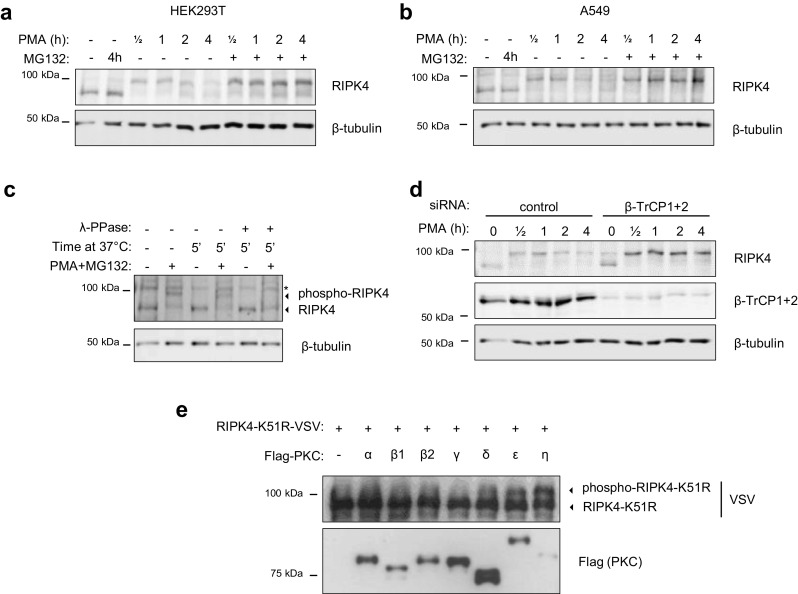
PKC signaling activates RIPK4 and triggers β-TrCP-dependent degradation. HEK293T cells (a) and A549 cells (b) were cultured for 16 h in growth medium containing 1% serum followed by stimulation with 0.5 µM phorbol 12-myristate 13-acetate (PMA) with or without MG132 (10 µM). Whole cell lysates were analyzed by immunoblotting with the indicated antibodies. c A549 cells were treated with a combination of PMA (0.5 µM) and MG132 (10 µM) for 3 h. The cells were lysed and a fraction of the lysate was treated with or without lambda-phosphatase at 37 °C for 5 min and subsequently analyzed by immunoblotting with the indicated antibodies. d HEK293T cells were seeded and transfected with non-targeting (control) or β-TrCP1+2 targeting siRNA (20 nM). Next day the cells were reseeded and cultured for 16 h in 1% serum growth medium followed by stimulation with 0.5 µM PMA for the indicated time. Whole cell lysates were analyzed by immunoblotting with the indicated antibodies. e HEK293T cells were transfected with expression vectors coding for VSV-tagged RIPK4 kinase-dead (K51R) together with Flag-tagged individual PKC isoforms. Twenty-four hours after transfection, cells were lysed and analyzed by immunoblotting with the indicated antibodies. The RIPK4-K51R-VSV proteins were enriched by a 5 h anti-VSV IP before western blotting analysis
Although RIPK4 has also been reported to signal downstream of the Wnt3a receptor LRP6 in HEK293T cells leading to DVL2 phosphorylation and β-catenin stabilization, we could not observe an effect of Wnt3a stimulation on RIPK4 phosphorylation and degradation (Supplementary Fig. 2). Further, we tested for the involvement of kinases previously described to be able to phosphorylate phosphodegron sequences, such as IKKs, Casein Kinases, GSK3β [31–33], or kinases described to prime phosphorylation of these phosphodegron kinases, such as JNK and p38, and found none of them to be involved in PMA-induced RIPK4 degradation (Supplementary Fig. 3) [34, 35].
Inability to degrade RIPK4 in HaCaT keratinocytes results in cell scattering
RIPK4 fulfills a crucial role in the formation of cortical filamentous actin organization at cell–cell contacts in the epidermis of mice [5, 6]. Furthermore, Xenopus embryos depleted from RIPK4 failed to gastrulate, likely due to the absence of cortical actin at the animal cap [5]. In line with this observation, it was shown that ectopic RIPK4-expressing HaCaT cells acquired a scattered and fusiform morphology caused by cytoskeletal rearrangements (Fig. 6a) [5]. Therefore, we hypothesized that accumulation of active endogenous RIPK4 in HaCaT cells could result in similar morphological changes. Depletion of β-TrCP1/2 resulted in a scattered keratinocyte cell morphology and was dependent on RIPK4 as the phenotype could be reverted by additional depletion of RIPK4 (Fig. 6b, c). Knockdown of β-TrCP in HaCaT cells led to accumulation of the phospho-RIPK4 form (Fig. 6d) without affecting the RIPK4 mRNA expression levels (Supplementary Fig. 4), suggesting an effect on protein stabilization. Keratinocytes adopting a scattered morphology upon β-TrCP depletion are characterized by altered F-actin stress fibers that form RIPK4-dependent lamellipodia and filopodia protrusions and distort cortical actin organization (Fig. 6e). Interestingly, time-lapse imaging revealed that β-TrCP knockdown leads to increased RIPK4-dependent keratinocyte motility as single scattered cells detach from the cell colony to migrate to other colonies (Fig. 6f, Corresponding movies are available in Online Resource 1). We conclude that β-TrCP is essential to maintain low levels of activated RIPK4 in keratinocytes to control cytoskeletal organization and motility.
Fig. 6.
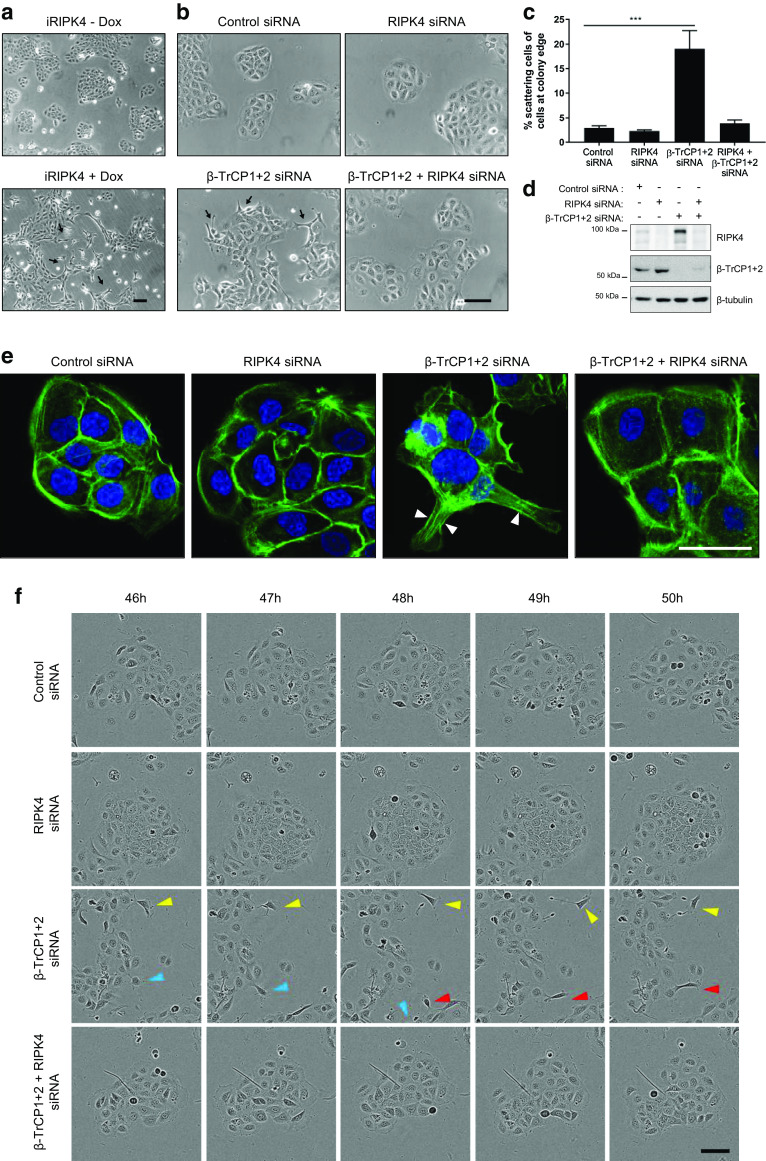
Inability to degrade RIPK4 in HaCaT keratinocytes results in cell scattering. a HaCaT cells transduced with a dox-inducible expression vector for hRIPK4 were grown with or without 1 μg/ml doxycycline (Dox). Forty-eight hours later phase-contrast pictures were taken. Scale bar: 100 µm. b, c HaCaT cells were transfected with non-targeting (control), RIPK4 and/or β-TrCP1+2 siRNA (20 nM). Forty-eight hours later phase-contrast pictures were taken. Scale bar: 100 µm. c Quantification of cellular scattering. Data points represent the average percentage of keratinocytes with elongated protrusions at the edge of cell colonies counted in three fields of three independent experiments (n = 3) and normalized to the number of non-scattered cells at colony edges. A minimum of 300 cells per independent experiment were counted. The error bars represent standard error of the mean. Statistical test: One-way ANOVA, p = 0.0008. d Immunoblotting analysis of cell lysates from b. e HaCaT cells were transfected with non-targeting (control), RIPK4 and/or β-TrCP1+2 siRNA (20 nM). Forty-eight hours later, cells were stained for filamentous actin with Acti-stain 488 phalloidin. Arrowheads point to actin stress fibers forming lamellipodia or filopodia. Scale bar: 50 µm. f HaCaT cells were transfected with non-targeting (control), RIPK4 and/or β-TrCP1+2 siRNA (20 nM). Representative pictures from the same field were taken every hour from 46 h after transfection. Colored arrowheads indicate a single cells over time. Scale bar: 100 µm
Discussion
RIPK4 plays a pivotal role during epidermal development and keratinocyte differentiation [5, 7–9]. Although mutations in RIPK4 cause the developmental Bartsocas-Papas syndrome [3–5, 7] and deregulation of RIPK4 levels has been associated with several cancer types [9, 22, 23, 36], there is little knowledge about the molecular pathways activating and regulating RIPK4 activity. Here, we identified the SCFβ-TrCP ubiquitin ligase complex as a crucial regulatory mechanism that controls the levels of activated RIPK4. This tight control of RIPK4 activity is crucial to maintain cortical actin organization in keratinocytes, which is lost in RIPK4-deficient mice while overexpression of RIPK4 in keratinocytes results in actin stress fiber formation [5].
We previously reported that membrane-bound cortical F-actin is absent in epidermal cells of RIPK4−/− mice [5]. Interestingly, Lee et al. found a significant reduction in phosphopeptides derived from proteins involved in anchoring junctions, desmosomes, adherence junctions, and focal adhesion in differentiated RIPK4-deficient keratinocytes, compared to wild-type cells [9]. These cell–cell interacting complexes link to the actin cytoskeleton [37]. This demonstrates the importance of RIPK4 kinase activity in keratinocyte homeostasis by regulating cell adhesion and actin dynamics. We observed that part of the RIPK4 protein pool is constitutively phosphorylated in in vitro keratinocyte cultures, which is not observed in a number of non-keratinocyte cell lines. This phosphorylated RIPK4 protein band is currently thought to represent the active RIPK4 pool because this phospho-RIPK4 band can only be detected in case of kinase-active RIPK4 forms [11]. This points to the presence of a keratinocyte-intrinsic factor that triggers RIPK4 activation in in vitro monolayer culture conditions.
Further, we found that proteasomal degradation of RIPK4 is mediated by the SCFβ-TrCP E3 ubiquitin ligase complex. RIPK4 contains a DSpAFSSp β-TrCP phosphodegron motif at position 378 that is very similar to the canonical DSpGXXSp motif [30]. Both S379 and S383 were found to be equally important for the recruitment of β-TrCP to RIPK4, whereas mutating the non-canonical S382 did not prevent β-TrCP binding. Although the kinase-dead (K51R) form of RIPK4 and the RIPK4-S379A/S383A mutant did not bind β-TrCP, RIPK4-K51R/RIPK4-S379A/S383A multimers did, suggesting that RIPK4-S379A/S383A could autophosphorylate RIPK4-K51R in trans. Once bound, β-TrCP facilitates K48-linked RIPK4 ubiquitinylation and proteasomal degradation, representing a mechanism for auto-regulation of RIPK4 activity levels in the cell. Higher levels of RIPK4 activity, as observed under conditions of βTrCP1/2 knock-down, resulted in increased cell scattering and motility. Therefore, it may be beneficial for epidermal integrity to keep the active RIPK4 levels under a certain threshold in homeostatic conditions. In accordance with this, we previously observed that HaCaT cells with stable expression of the RIPK4-S376X Bartsocas-Papas mutant, lacking the degron motif, showed higher protein levels and NF-κB activation compared to wild-type RIPK4 and resulted in cell scattering [4, 5]. Because the RIPK4-S376X mutant retains its kinase activity but lacks the C-terminal ankyrin domain, the disease phenotype is probably due to loss of the ankyrin domain. Interestingly, the phosphodegron motif contains a functional caspase cleavage site at Asp 378 [38]. It is thus conceivable that caspase activation during apoptosis generates a stabilized RIPK4 kinase domain that could promote actin-dependent membrane blebbing [39].
A key question remains on the kinase that phosphorylates the phosphodegron motif of RIPK4. Although we provide evidence for the involvement of RIPK4 trans-autophosphorylation in targeting it for proteasomal degradation, the exact sequence of phosphorylation events leading to RIPK4 degradation remains to be determined. We propose two possible models how RIPK4 autophosphorylation could regulate its stability (schematic representation in Fig. 7). A first possibility is that RIPK4 directly autophosphorylates its own phosphodegron and acts as a suicide kinase. Polo-like kinase 4 (PLK4), in which autophosphorylation of the PLK4 phosphodegron is crucial for β-TrCP docking, is a well-studied example of such a type of suicide kinases [40]. This model implies that every condition that triggers RIPK4 activation induces RIPK4 degradation. In a second model, RIPK4 autophosphorylation generates a priming phosphorylation that recruits other degron-phosphorylating kinases. The phosphodegron of RIPK4 is located in a serine-rich sequence stretch in the intermediate domain, of which several serine residues have been reported to be phosphorylated [41, and data not shown]. Hence, it is plausible that some serine residues up- or downstream of the RIPK4 phosphodegron are autophosphorylation sites priming the phosphorylation of the degron by other kinases. The requirement of multiple sequential phosphorylation events could ensure that active RIPK4 is only degraded in certain cellular conditions or provide a time window for RIPK4 to phosphorylate downstream substrates before RIPK4 is degraded. Therefore, we tested various inhibitors of known ‘degron kinases’; however, we did not observe a stabilizing effect on PMA-induced RIPK4 degradation. This argues in favor of the first model in which RIPK4 trans-autophosphorylates its own phosphodegron sequence, but does not exclude the possible involvement of other degron kinases activated upstream or downstream of RIPK4. An example of the latter mechanism can be found in the regulation of NIK (NF-κB-inducing kinase) proteasomal degradation [42].
Fig. 7.

Schematic representation of the proposed RIPK4 degradation mechanisms. In model I, we propose that RIPK4 directly trans-autophosphorylates the phosphodegron motif possibly together with other sites in the flanking serine-rich region. In model II, we propose that RIPK4 autophosphorylation sites, possibly in the serine-rich region flanking the phosphodegron motif, function as priming sites for the recruitment of other kinases which then will phosphorylate the degron
RIPK4 was initially identified as a PKCβ and -δ interacting protein [10, 11]. PKCβ1 was shown to be able to phosphorylate RIPK4; however, whether this phosphorylation resulted in a RIPK4 mobility shift in western blotting was not clear from the presented results [11]. We show that the novel PKC isoforms PKCδ, -ε, and -η can induce a RIPK4 phosphorylation band shift upon overexpression with RIPK4-K51R, suggesting that these PKC family members could be involved in TPA-induced RIPK4 activation. Our data do not exclude the possibility that other PKCs phosphorylate RIPK4 in a way that does not induce a mobility shift in RIPK4. Therefore, more biochemical experiments should be required using quality-controlled purified recombinant PKCs. Recently, Kwa et al. demonstrated that RIPK4 knockdown in normal human primary keratinocytes hampered the expression of differentiation markers upon PKC activation [8]. This led them to conclude that PKCs are upstream of RIPK4 in a pathway responsible for keratinocyte differentiation. Here, we found that PKC activation by PMA results in RIPK4 hyper-phosphorylation and concomitant RIPK4 proteasomal degradation. Because PKCδ can phosphorylate RIPK4 in vitro [11], and RIPK4 hyper-phosphorylation is dependent on its kinase activity, we hypothesize that PKC phosphorylation of RIPK4 could activate RIPK4 eventually leading to autophosphorylation. Therefore, it would be interesting to identify at which site(s) PKCs phosphorylate RIPK4 and how this initiates its kinase activity. Interestingly, transgenic mice overexpressing RIPK4 in the epidermis react hypersensitively to PKC activation by PMA [25]. This is in accordance with our finding that PKCs can activate RIPK4 and that increased RIPK4 levels could trigger uncontrolled inflammatory responses.
Interfering with the degradation of RIPK4 could be a profitable feature for cancer cells. β-TrCP has been implicated in tumor development and progression because of its function in numerous signaling pathways controlling cell growth, survival, death and differentiation [21]. Although β-TrCP has been assigned both oncogenic and tumor-suppressive roles depending on the cellular context, it has mainly been described as an oncogene with increased expression in various cancers including epidermal tumors [43]. This could result in enhanced RIPK4 degradation mimicking RIPK4 depletion, which was recently shown to enhance epidermal tumor formation in mice [9, 44]. On the other hand, RIPK4 has also been suggested to exert oncogenic functions by, for example, promoting cervical squamous cell carcinoma progression [23]. In this case, stabilization of RIPK4 would be a cost-effective strategy of cancer cells to increase the protein levels of RIPK4 and could favor tumor cell motility during tumor invasion. In agreement with this, mutations in the phosphodegron of RIPK4 have been found in kidney cancer (D378V) and malignant melanoma (A380V) [45].
It is becoming increasingly evident that RIPK4 fulfills a crucial role in epidermal homeostasis and pathogenesis besides its developmental role. However, insights in RIPK4 activation and regulation are lagging behind. We identified a molecular mechanism fine-tuning the levels of RIPK4 activation and its role in keratinocyte cytoskeletal organization. Future research into the activation mechanism and downstream signaling pathways of RIPK4 will give valuable information on how RIPK4 functions at the biochemical level and how it controls epidermal differentiation, barrier formation and cancer.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Inability to degrade RIPK4 in HaCaT keratinocytes results in increased cell motility. a–dHaCaT cells were reverse transfected with the following siRNAs (20nm): (a) non-targeting (control), (b)RIPK4, (c) β-TrCP1+2, (d) RIPK4 + β-TrCP1+2 and seeded on collagen-coated tissue culture plates.Imaging was started 24 h after transfection for the following 24 h with one image acquisition every 15 minwith an IncuCyte microscope
Supplementary Fig. 1 Tandem affinity purification of RIPK4 and identification of β-TrCP as binding partner. a-b HEK293T cells were transiently transfected with pCEMM N-TAP(GS)hRIPK4 or empty pCEMM N-TAP(GS) as a negative control. Purified TAP-tagged protein complexes were separated by gel electrophoresis followed by Coomassie staining (a), and subsequently gel fragments were excised for analysis by mass spectrometry (MS) analysis. For each gel fragment a corresponding gel fragment from the negative control purification was analyzed to allow the identification of false positives. Proteins that were identified by at least two peptides and which were absent from the negative control were retained as potential RIPK4 interactors (b). Raw data is available upon request. c Identified peptides in MS/MS analysis from samples in a corresponding to β-TrCP1 and β-TrCP2. Supplementary Fig. 2 Wnt3a stimulation does not induce RIPK4 hyper-phosphorylation. HEK293T cells were cultured for 16 h in growth medium containing 1% serum followed by stimulation with 200 ng/ml recombinant Wnt3a with or without MG132 (10 µM). Whole cell lysates were analyzed by immunoblotting with the indicated antibodies. Simultaneously Wnt3a activity was measured using a Wnt reporter assay. HEK293T cells were transfected with the Wnt reporter one day before stimulation with 200 ng/ml Wnt3a. Lysates were made 24 h hours after stimulation and luciferase activity was measured. Supplementary Fig. 3 Inhibition of potential degron kinases does not affect PMA-induced RIPK4 phosphorylation and degradation. a HEK293T cells were cultured for 16 h in growth medium containing 1% serum followed by 1 h pretreatment with inhibitors for JNK (SP600125), p38 (SB202190), casein kinase I (D4476), casein kinase 2 (CX4945), IKKα and IKKβ (TPCA), GSK3β (CHIR99021) (at 10 µM final concentration) before stimulation with PMA (0.5 µM) with or without MG132 (10 µM). Whole cell lysates were analyzed by immunoblotting with the indicated antibodies. b HEK293T cells were seeded and transfected with non-targeting (control) or with the indicated siRNA (20 nM). Next day the cells refreshed with 1% serum growth medium. 16 h later the cells were stimulated with 0.5 µM PMA with or without MG132 (10 µM) for the indicated time. Whole cell lysates were analyzed by immunoblotting with the indicated antibodies. Supplementary Fig. 4 β-TrCP knockdown does not induce RIPK4 mRNA expression levels. a HaCaT cells were transfected with non-targeting (control) or β-TrCP1+2 siRNA (20 nM). RIPK4, β-TrCP1 and β-TrCP2 mRNA levels were analyzed by qPCR analysis. Error bars represent standard error of the means (n = 2). Supplementary Fig. 5 RIPK4 mutagenesis and PKC isoform cloning primers used in this study. Online Resource 1: Inability to degrade RIPK4 in HaCaT keratinocytes results in increased cell motility. HaCaT cells were reverse transfected with non-targeting (control), RIPK4 and/or β-TrCP1+2 siRNA (20 nM) and seeded on collagen-coated tissue culture plates. Imaging was started 24 h after transfection for the following 24 h with one image acquisition every 15 min with a IncuCyte microscope. (PDF 478 kb)
Acknowledgements
This research has been supported by the Flanders Institute for Biotechnology (VIB); Belgian Grants: Interuniversity Attraction Poles, IAP7/32; Stichting tegen Kanker (2010-162 and FAF-F/2016/868); Flemish Grants: FWO-Vlaanderen (G.0544.11) and a Methusalem Grant (BOF09/01M00709) from the Flemish Government to Peter Vandenabeele; a UGent Grant (GOA-01G01914). G.T. received a Ph.D. fellowship from FWO-Vlaanderen and C.U-R obtained a predoctoral Strategic Research fellowship from IWT-Vlaanderen. We also thank the VIB Bioimaging core facility for excellent assistance.
Compliance with ethical standards
Conflict of interest
The authors state no conflict of interest.
References
- 1.Meylan E, Tschopp J. The RIP kinases: crucial integrators of cellular stress. Trends Biochem Sci. 2005;30(3):151–159. doi: 10.1016/j.tibs.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Lippens S, Hoste E, Vandenabeele P, Agostinis P, Declercq W. Cell death in the skin. Apoptosis Int J Progr Cell Death. 2009;14(4):549–569. doi: 10.1007/s10495-009-0324-z. [DOI] [PubMed] [Google Scholar]
- 3.Kalay E, Sezgin O, Chellappa V, Mutlu M, Morsy H, Kayserili H, Kreiger E, Cansu A, Toraman B, Abdalla EM, Aslan Y, Pillai S, Akarsu NA. Mutations in RIPK4 cause the autosomal-recessive form of popliteal pterygium syndrome. Am J Hum Genet. 2012;90(1):76–85. doi: 10.1016/j.ajhg.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitchell K, O’Sullivan J, Missero C, Blair E, Richardson R, Anderson B, Antonini D, Murray JC, Shanske AL, Schutte BC, Romano RA, Sinha S, Bhaskar SS, Black GC, Dixon J, Dixon MJ. Exome sequence identifies RIPK4 as the Bartsocas-Papas syndrome locus. Am J Hum Genet. 2012;90(1):69–75. doi: 10.1016/j.ajhg.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Groote P, Tran HT, Fransen M, Tanghe G, Urwyler C, De Craene B, Leurs K, Gilbert B, Van Imschoot G, De Rycke R, Guerin CJ, Holland P, Berx G, Vandenabeele P, Lippens S, Vleminckx K, Declercq W. A novel RIPK4–IRF6 connection is required to prevent epithelial fusions characteristic for popliteal pterygium syndromes. Cell Death Differ. 2015;22(6):1012–1024. doi: 10.1038/cdd.2014.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Urwyler-Rosselet C, Tanghe G, Leurs K, Gilbert B, De Rycke R, De Bruyne M, Lippens S, Bartunkova S, De Groote P, Niessen C, Haftek M, Vandenabeele P, Declercq W. Keratinocyte-specific ablation of RIPK4 allows epidermal cornification but impairs skin barrier formation. J Investig Dermatol. 2018 doi: 10.1016/j.jid.2017.12.031. [DOI] [PubMed] [Google Scholar]
- 7.Holland P, Willis C, Kanaly S, Glaccum M, Warren A, Charrier K, Murison J, Derry J, Virca G, Bird T, Peschon J. RIP4 is an ankyrin repeat-containing kinase essential for keratinocyte differentiation. Curr Biol. 2002;12(16):1424–1428. doi: 10.1016/S0960-9822(02)01075-8. [DOI] [PubMed] [Google Scholar]
- 8.Kwa MQ, Huynh J, Aw J, Zhang L, Nguyen T, Reynolds EC, Sweet MJ, Hamilton JA, Scholz GM. Receptor-interacting protein kinase 4 and interferon regulatory factor 6 function as a signaling axis to regulate keratinocyte differentiation. J Biol Chem. 2014;289(45):31077–31087. doi: 10.1074/jbc.M114.589382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee P, Jiang S, Li Y, Yue J, Gou X, Chen SY, Zhao Y, Schober M, Tan M, Wu X. Phosphorylation of Pkp1 by RIPK4 regulates epidermal differentiation and skin tumorigenesis. EMBO J. 2017 doi: 10.15252/embj.201695679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhr C, Rohwer A, Stempka L, Rincke G, Marks F, Gschwendt M. DIK, a novel protein kinase that interacts with protein kinase Cdelta. Cloning, characterization, and gene analysis. J Biol Chem. 2000;275(46):36350–36357. doi: 10.1074/jbc.M004771200. [DOI] [PubMed] [Google Scholar]
- 11.Chen L, Haider K, Ponda M, Cariappa A, Rowitch D, Pillai S. Protein kinase C-associated kinase (PKK), a novel membrane-associated, ankyrin repeat-containing protein kinase. J Biol Chem. 2001;276(24):21737–21744. doi: 10.1074/jbc.M008069200. [DOI] [PubMed] [Google Scholar]
- 12.Rosse C, Linch M, Kermorgant S, Cameron AJ, Boeckeler K, Parker PJ. PKC and the control of localized signal dynamics. Nat Rev Mol Cell Biol. 2010;11(2):103–112. doi: 10.1038/nrm2847. [DOI] [PubMed] [Google Scholar]
- 13.Igumenova TI. Dynamics and membrane interactions of protein kinase C. Biochemistry. 2015;54(32):4953–4968. doi: 10.1021/acs.biochem.5b00565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parkinson EK, Emmerson A. Non-promoting hyperplasiogenic agents do not mimic the effects of phorbol, 12-myristate, 13-acetate on terminal differentiation of normal and transformed human keratinocytes. Carcinogenesis. 1984;5(5):687–690. doi: 10.1093/carcin/5.5.687. [DOI] [PubMed] [Google Scholar]
- 15.Muto A, Ruland J, McAllister-Lucas LM, Lucas PC, Yamaoka S, Chen FF, Lin A, Mak TW, Nunez G, Inohara N. Protein kinase C-associated kinase (PKK) mediates Bcl10-independent NF-kappa B activation induced by phorbol ester. J Biol Chem. 2002;277(35):31871–31876. doi: 10.1074/jbc.M202222200. [DOI] [PubMed] [Google Scholar]
- 16.Kim SW, Oleksyn DW, Rossi RM, Jordan CT, Sanz I, Chen L, Zhao J. Protein kinase C-associated kinase is required for NF-kappaB signaling and survival in diffuse large B-cell lymphoma cells. Blood. 2008;111(3):1644–1653. doi: 10.1182/blood-2007-05-088591. [DOI] [PubMed] [Google Scholar]
- 17.Komander D. The emerging complexity of protein ubiquitination. Biochem Soc Trans. 2009;37(Pt 5):937–953. doi: 10.1042/BST0370937. [DOI] [PubMed] [Google Scholar]
- 18.Cohen-Kaplan V, Livneh I, Avni N, Cohen-Rosenzweig C, Ciechanover A. The ubiquitin–proteasome system and autophagy: coordinated and independent activities. Int J Biochem Cell Biol. 2016;79:403–418. doi: 10.1016/j.biocel.2016.07.019. [DOI] [PubMed] [Google Scholar]
- 19.Kwon YT, Ciechanover A. The ubiquitin code in the ubiquitin–proteasome system and autophagy. Trends Biochem Sci. 2017;42(11):873–886. doi: 10.1016/j.tibs.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 20.Zheng N, Zhou Q, Wang Z, Wei W. Recent advances in SCF ubiquitin ligase complex: clinical implications. Biochem Biophys Acta 1866. 2016;1:12–22. doi: 10.1016/j.bbcan.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol. 2013;14(6):369–381. doi: 10.1038/nrm3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heim D, Cornils K, Schulze K, Fehse B, Lohse AW, Brummendorf TH, Wege H. Retroviral insertional mutagenesis in telomerase-immortalized hepatocytes identifies RIPK4 as novel tumor suppressor in human hepatocarcinogenesis. Oncogene. 2015;34(3):364–372. doi: 10.1038/onc.2013.551. [DOI] [PubMed] [Google Scholar]
- 23.Liu DQ, Li FF, Zhang JB, Zhou TJ, Xue WQ, Zheng XH, Chen YB, Liao XY, Zhang L, Zhang SD, Hu YZ, Jia WH. Increased RIPK4 expression is associated with progression and poor prognosis in cervical squamous cell carcinoma patients. Sci Rep. 2015;5:11955. doi: 10.1038/srep11955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kopparam J, Chiffelle J, Angelino P, Piersigilli A, Zangger N, Delorenzi M, Meylan E. RIP4 inhibits STAT3 signaling to sustain lung adenocarcinoma differentiation. Cell Death Differ. 2017 doi: 10.1038/cdd.2017.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rountree RB, Willis CR, Dinh H, Blumberg H, Bailey K, Dean C, Jr, Peschon JJ, Holland PM. RIP4 regulates epidermal differentiation and cutaneous inflammation. J Investig Dermatol. 2010;130(1):102–112. doi: 10.1038/jid.2009.223. [DOI] [PubMed] [Google Scholar]
- 26.Oleksyn D, Zhao J, Vosoughi A, Zhao JC, Misra R, Pentland AP, Ryan D, Anolik J, Ritchlin C, Looney J, Anandarajah AP, Schwartz G, Calvi LM, Georger M, Mohan C, Sanz I, Chen L. PKK deficiency in B cells prevents lupus development in Sle lupus mice. Immunol Lett. 2017;185:1–11. doi: 10.1016/j.imlet.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, Green TM, Johannessen CM, Silver SJ, Nguyen C, Murray RR, Hieronymus H, Balcha D, Fan C, Lin C, Ghamsari L, Vidal M, Hahn WC, Hill DE, Root DE. A public genome-scale lentiviral expression library of human ORFs. Nat Methods. 2011;8(8):659–661. doi: 10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Staal FJ, van Noort M, Strous GJ, Clevers HC. Wnt signals are transmitted through N-terminally dephosphorylated beta-catenin. EMBO Rep. 2002;3(1):63–68. doi: 10.1093/embo-reports/kvf002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Low TY, Peng M, Magliozzi R, Mohammed S, Guardavaccaro D, Heck AJ. A systems-wide screen identifies substrates of the SCFbetaTrCP ubiquitin ligase. Sci Signal. 2014;7(356):rs8. doi: 10.1126/scisignal.2005882. [DOI] [PubMed] [Google Scholar]
- 31.Wu X, Fukushima H, North BJ, Nagaoka Y, Nagashima K, Deng F, Okabe K, Inuzuka H, Wei W. SCFbeta-TRCP regulates osteoclastogenesis via promoting CYLD ubiquitination. Oncotarget. 2014;5(12):4211–4221. doi: 10.18632/oncotarget.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Magliozzi R, Low TY, Weijts BG, Cheng T, Spanjaard E, Mohammed S, van Veen A, Ovaa H, de Rooij J, Zwartkruis FJ, Bos JL, de Bruin A, Heck AJ, Guardavaccaro D. Control of epithelial cell migration and invasion by the IKKbeta- and CK1alpha-mediated degradation of RAPGEF2. Dev Cell. 2013;27(5):574–585. doi: 10.1016/j.devcel.2013.10.023. [DOI] [PubMed] [Google Scholar]
- 33.Hart M, Concordet JP, Lassot I, Albert I, del los Santos R, Durand H, Perret C, Rubinfeld B, Margottin F, Benarous R, Polakis P. The F-box protein beta-TrCP associates with phosphorylated beta-catenin and regulates its activity in the cell. Curr Biol. 1999;9(4):207–210. doi: 10.1016/S0960-9822(99)80091-8. [DOI] [PubMed] [Google Scholar]
- 34.Zeke A, Misheva M, Remenyi A, Bogoyevitch MA. JNK signaling: regulation and functions based on complex protein–protein partnerships. Microbiol Mol Biol Rev. 2016;80(3):793–835. doi: 10.1128/MMBR.00043-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rojo AI, Medina-Campos ON, Rada P, Zuniga-Toala A, Lopez-Gazcon A, Espada S, Pedraza-Chaverri J, Cuadrado A. Signaling pathways activated by the phytochemical nordihydroguaiaretic acid contribute to a Keap1-independent regulation of Nrf2 stability: role of glycogen synthase kinase-3. Free Radic Biol Med. 2012;52(2):473–487. doi: 10.1016/j.freeradbiomed.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 36.Poligone B, Gilmore ES, Alexander CV, Oleksyn D, Gillespie K, Zhao J, Ibrahim SF, Pentland AP, Brown MD, Chen L. PKK suppresses tumor growth and is decreased in squamous cell carcinoma of the skin. J Investig Dermatol. 2015;135(3):869–876. doi: 10.1038/jid.2014.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simpson CL, Patel DM, Green KJ. Deconstructing the skin: cytoarchitectural determinants of epidermal morphogenesis. Nat Rev Mol Cell Biol. 2011;12(9):565–580. doi: 10.1038/nrm3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meylan E, Martinon F, Thome M, Gschwendt M, Tschopp J. RIP4 (DIK/PKK), a novel member of the RIP kinase family, activates NF-kappa B and is processed during apoptosis. EMBO Rep. 2002;3(12):1201–1208. doi: 10.1093/embo-reports/kvf236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cotter TG, Lennon SV, Glynn JM, Green DR. Microfilament-disrupting agents prevent the formation of apoptotic bodies in tumor cells undergoing apoptosis. Can Res. 1992;52(4):997–1005. [PubMed] [Google Scholar]
- 40.Holland AJ, Lan W, Niessen S, Hoover H, Cleveland DW. Polo-like kinase 4 kinase activity limits centrosome overduplication by autoregulating its own stability. J Cell Biol. 2010;188(2):191–198. doi: 10.1083/jcb.200911102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Razani B, Zarnegar B, Ytterberg AJ, Shiba T, Dempsey PW, Ware CF, Loo JA, Cheng G. Negative feedback in noncanonical NF-kappaB signaling modulates NIK stability through IKKalpha-mediated phosphorylation. Sci Signal. 2010;3(123):ra41. doi: 10.1126/scisignal.2000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bhatia N, Herter JR, Slaga TJ, Fuchs SY, Spiegelman VS. Mouse homologue of HOS (mHOS) is overexpressed in skin tumors and implicated in constitutive activation of NF-kappaB. Oncogene. 2002;21(10):1501–1509. doi: 10.1038/sj.onc.1205311. [DOI] [PubMed] [Google Scholar]
- 44.Chen L, Hayden MS, Gilmore ES, Alexander-Savino C, Oleksyn D, Gillespie K, Zhao J, Poligone B. PKK deletion in basal keratinocytes promotes tumorigenesis after chemical carcinogenesis. Carcinogenesis. 2017 doi: 10.1093/carcin/bgx120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, Stefancsik R, Harsha B, Kok CY, Jia M, Jubb H, Sondka Z, Thompson S, De T, Campbell PJ. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45(D1):D777–D783. doi: 10.1093/nar/gkw1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Inability to degrade RIPK4 in HaCaT keratinocytes results in increased cell motility. a–dHaCaT cells were reverse transfected with the following siRNAs (20nm): (a) non-targeting (control), (b)RIPK4, (c) β-TrCP1+2, (d) RIPK4 + β-TrCP1+2 and seeded on collagen-coated tissue culture plates.Imaging was started 24 h after transfection for the following 24 h with one image acquisition every 15 minwith an IncuCyte microscope
Supplementary Fig. 1 Tandem affinity purification of RIPK4 and identification of β-TrCP as binding partner. a-b HEK293T cells were transiently transfected with pCEMM N-TAP(GS)hRIPK4 or empty pCEMM N-TAP(GS) as a negative control. Purified TAP-tagged protein complexes were separated by gel electrophoresis followed by Coomassie staining (a), and subsequently gel fragments were excised for analysis by mass spectrometry (MS) analysis. For each gel fragment a corresponding gel fragment from the negative control purification was analyzed to allow the identification of false positives. Proteins that were identified by at least two peptides and which were absent from the negative control were retained as potential RIPK4 interactors (b). Raw data is available upon request. c Identified peptides in MS/MS analysis from samples in a corresponding to β-TrCP1 and β-TrCP2. Supplementary Fig. 2 Wnt3a stimulation does not induce RIPK4 hyper-phosphorylation. HEK293T cells were cultured for 16 h in growth medium containing 1% serum followed by stimulation with 200 ng/ml recombinant Wnt3a with or without MG132 (10 µM). Whole cell lysates were analyzed by immunoblotting with the indicated antibodies. Simultaneously Wnt3a activity was measured using a Wnt reporter assay. HEK293T cells were transfected with the Wnt reporter one day before stimulation with 200 ng/ml Wnt3a. Lysates were made 24 h hours after stimulation and luciferase activity was measured. Supplementary Fig. 3 Inhibition of potential degron kinases does not affect PMA-induced RIPK4 phosphorylation and degradation. a HEK293T cells were cultured for 16 h in growth medium containing 1% serum followed by 1 h pretreatment with inhibitors for JNK (SP600125), p38 (SB202190), casein kinase I (D4476), casein kinase 2 (CX4945), IKKα and IKKβ (TPCA), GSK3β (CHIR99021) (at 10 µM final concentration) before stimulation with PMA (0.5 µM) with or without MG132 (10 µM). Whole cell lysates were analyzed by immunoblotting with the indicated antibodies. b HEK293T cells were seeded and transfected with non-targeting (control) or with the indicated siRNA (20 nM). Next day the cells refreshed with 1% serum growth medium. 16 h later the cells were stimulated with 0.5 µM PMA with or without MG132 (10 µM) for the indicated time. Whole cell lysates were analyzed by immunoblotting with the indicated antibodies. Supplementary Fig. 4 β-TrCP knockdown does not induce RIPK4 mRNA expression levels. a HaCaT cells were transfected with non-targeting (control) or β-TrCP1+2 siRNA (20 nM). RIPK4, β-TrCP1 and β-TrCP2 mRNA levels were analyzed by qPCR analysis. Error bars represent standard error of the means (n = 2). Supplementary Fig. 5 RIPK4 mutagenesis and PKC isoform cloning primers used in this study. Online Resource 1: Inability to degrade RIPK4 in HaCaT keratinocytes results in increased cell motility. HaCaT cells were reverse transfected with non-targeting (control), RIPK4 and/or β-TrCP1+2 siRNA (20 nM) and seeded on collagen-coated tissue culture plates. Imaging was started 24 h after transfection for the following 24 h with one image acquisition every 15 min with a IncuCyte microscope. (PDF 478 kb)