

Abstract
MicroRNAs are small endogenous RNAs that pair and bind to sites on mRNAs to direct post-transcriptional repression. However, there is a possibility that microRNAs directly influence protein structure and activity, and this influence can be termed post-translational riboregulation. This conceptual review explores the literature on neurodegenerative disorders. Research on the association between neurodegeneration and RNA-repeat toxicity provides data that support a protein–RNA recognition code. For example, this code explains why hnRNP H and SFPQ proteins, which are involved in amyotrophic lateral sclerosis, are sequestered by the (GGGGCC)n repeat sequence. Similarly, it explains why MNBL proteins and (CTG)n repeats in RNA, which are involved in myotonic dystrophy, are sequestered into RNA foci. Using this code, proteins involved in diseases can be identified. A simple protein BLAST search of the human genome for amino acid repeats that correspond to the nucleotide repeats reveals new proteins among already known proteins that are involved in diseases. For example, the (CAG)n repeat sequence, when transcribed into possible peptide sequences, leads to the identification of PTCD3, Rem2, MESP2, SYPL2, WDR33, COL23A1, and others. After confirming this approach on RNA repeats, in the next step, the code was used in the opposite manner. Proteins that are involved in diseases were compared with microRNAs involved in those diseases. For example, a reasonable correspondence of microRNA 9 and 107 with amyloid-β-peptide (Aβ42) was identified. In the last step, a miRBase search for micro-nucleotides, obtained by transcription of a prion amino acid sequence, revealed new microRNAs and microRNAs that have previously been identified as involved in prion diseases. This concept provides a useful key for designing RNA or peptide probes.
Keywords: Molecular recognition, Non-coding RNA, Huntington’s disease, Alzheimer’s disease, Parkinson’s disease, Prion diseases
Background
Non-coding RNA and its role
The genome, a complete set of genes, has been said to be responsible for the storage of information on how to develop, organize and control all molecular, cellular and multicellular life. The genome, the complete list of nucleotides, is organized into polynucleotides with specific sequences; however, defining exactly what section of the sequence comprises a gene is difficult. The concept “one gene makes one protein” [1] clearly had to be shifted to the concept of “a union of genomic sequences encoding a coherent set of potentially overlapping functional products” [2]. According to the ENCODE project [3], the vast majority (80.4%) of human DNA participates in at least one biochemical RNA or chromatin-associated RNA in at least one cell type. Unsurprisingly, much of the genome is important for regulation, and only a minimal portion (less than 2%) is important for protein-coding [3]. Epigenetics is widely accepted to cause changes in gene expression that do not result from alterations in nucleotide sequences [4]. Multiple epigenetic mechanisms orchestrate neurofunction through coordinated responses to extracellular cues, which determine the spatial and temporal expression of key regulators that control neuron function fate or even neurogenesis (differentiation and proliferation of neural stem cells) [5]. Several epigenetic mechanisms have been extensively investigated: DNA methylation and demethylation, histone methylation and demethylation, histone acetylation and deacetylation, chromatin-remodelling, and regulation mediated by non-coding RNAs [5]. Non-coding RNAs (ncRNAs) are probably critical components for epigenetic control, representing a main part of the transcriptome [3] and a main part of the chromatin network [6, 7]. A length of more or less than 200 nt divides ncRNAs into two main groups—small (sncRNAs) and long (lncRNAs). ncRNAs are particularly abundant in the CNS; an estimated 40% of the genes for lncRNAs are specifically expressed in brain tissue, and sncRNAs have also been reported to be enriched in the CNS [8].
MicroRNAs (miRNAs) are sncRNAs of ~ 22 nt in length that mediate gene silencing by guiding Argonaute (AGO) proteins to target sites in mRNAs [9]. AGOs constitute a large family of proteins that use single-stranded small nucleic acids as guides to complementary sequences in RNA or DNA targeted for silencing [10]. Prokaryotic AGOs participate in host defence by DNA interference, whereas eukaryotic AGOs control a wide range of processes by RNA interference [10]. More than 60% of human protein-coding genes harbour predicted miRNA target sites [11]. miRBase lists 1917 precursor miRNAs and 2654 mature miRNAs in Homo sapiens [12]. miRNAs not only are responsible for gene silencing but also can fine-tune protein expression [13], thereby buffering “noise” at the level of gene expression [14]. The miRNA pool is controlled by various mechanisms, for example, by direct modification of miRNA sequences (mature miRNA variants that differ in length, sequence or both), by editing the sequence of miRNA precursors, by non-templated nucleotide addition, by sequestration, by miRNA transport modulation or by modulation of miRNA turnover [9]. Interestingly, viruses often modulate host miRNAs for their replication [9]. Many of the miRNAs are loaded into extracellular vesicles for cell–cell communication, and miRNA sorting into extracellular vesicles can be regulated by chromosome location, regulation of miRNA biosynthesis, AGO activity, and sequence-specific sorting by RNA-binding proteins (RBPs) [15].
The human genome contains thousands of lncRNAs, but specific biological functions and biochemical mechanisms have been discovered for only about a dozen. lncRNAs maintain features common to protein-coding genes such as promoter regions, intron–exon boundaries and alternative splicing; however, in contrast, lncRNAs are mainly nuclear localized, less polyadenylated and far more tissue-specific than protein-coding genes [16]. It has been reported that approximately 40% of lncRNAs are directly associated with diverse chromatin-modifying complexes; the extensive number of associations between lncRNAs and chromatin-modifying enzymes suggests, in general, that lncRNAs directly guide chromatin-remodelling proteins to target loci and function as scaffolds for the recruitment of transcription factors to activate or repress gene expression [7, 16]. Evolutionarily, lncRNAs appear later than sncRNAs; approximately one-third seem to have arisen within the primate lineage, a large fraction of tissue-specific lncRNAs are expressed in the brain, and lncRNAs are generally less conserved than protein-coding genes [16]. Protein-coding genes are more conserved, and species from worm to man house similar numbers of protein-coding genes, which indicates that many aspects of complex organisms arise from regulation from non-protein-coding regions. lncRNAs are generally expressed at lower levels than protein-coding genes, and their expression positively correlates with the expression of antisense coding genes [16]. In contrast to the readily identifiable open reading frames (ORFs) of protein-coding genes, lncRNA sequence features or functional elements cannot presently be used to identify novel lncRNAs [17]. In addition, their lower conservation during evolution makes identifying their orthologues or paralogues by sequence similarity challenging. Consequently, lncRNA annotation relies almost entirely on physical transcriptomic evidence [17]. Specific lncRNAs activated by DNA damage (NORADs) stand out among lncRNAs. These lncRNAs are more highly conserved than other lncRNAs, are abundantly expressed in many cell types, are upregulated upon DNA damage and induce chromosomal instability and aneuploidy when deleted [18].
Protein–RNA interactome
Interactions between tRNAs and mRNAs to translate genetic code represent basic examples of RNA–RNA interactions [19]. However, interactions between miRNAs and pre-mRNAs [20] and between miRNAs and lncRNAs [21] stand out among the RNA–RNA interactions to enable the basic regulatory functions of RNA.
The main part of the RNA–RNA interactome is mediated by RNA-binding proteins (RBPs), such as AGOs mentioned above. RBPs are typically thought of as proteins that bind RNA through one or multiple globular RNA-binding domains and change the fate or function of the bound RNAs [22]. RBPs participate in the formation of ribonucleoprotein complexes (RNPs) that are principally involved in gene expression and regulation. Pre-mRNAs, containing exons and introns, are subjected to processing by a range of RNPs that include uridine-rich small nuclear RNPs that make up the spliceosome, the large RNP that removes non-coding introns [23]. Protein biosynthesis is mediated by another large RNP, the ribosome, which directs mRNA translation through the combined actions of its small and large RNP subunits. The ribosome further cooperates with the signal recognition particle RNP to translocate some newly synthesized proteins into the endoplasmic reticulum [23]. Indeed, spliceosomes and ribosomes, large RNP machines, reveal that protein–RNA interactions do not require canonical globular RNA-binding domains [22]. Many RBPs or regions in them are found to be intrinsically disordered, and this property helps them to recognize and bind RNA-partner sequences [24]. RBDmap, which was recently developed to determine the RNA-binding sites of native RBPs, identified 1174 binding sites within 529 HeLa cell RBPs; nearly half of the RNA-binding sites mapped to intrinsically disordered regions, uncovering unstructured domains as prevalent partners in protein–RNA interactions [25]. This finding implies that disordered short sequences of a protein could help to bind RNA and adopt stable structures upon binding. In light of this observation, RNA-binding peptides or micropeptides (RBMs) should be included in the RNA interactome; interestingly, functional small peptides have been identified in transcripts previously annotated as lncRNAs [26]. Conventional RBDs change the fate or function of bound RNAs, but there is ample room for considering the possibility that the known biological function of a protein can be altered through riboregulation [22].
Protein–RNA recognition code
A protein–RNA recognition code was proposed after the 3D5A crystal structure, which revealed release factor 1 (RF1) interacting with tRNA [27], or earlier, after crystal structures of aminoacyl transfer RNA synthetases interacting with tRNA [28]. The code is depicted in Fig. 1. The second position in the codon is used for the CCA sequence readout, proposed as the one-letter code [27] or primary code [28]. The first position in the codon is used as “more specifically recognizing addition” and is proposed as the two-letter code [27] or secondary code [28].
Fig. 1.
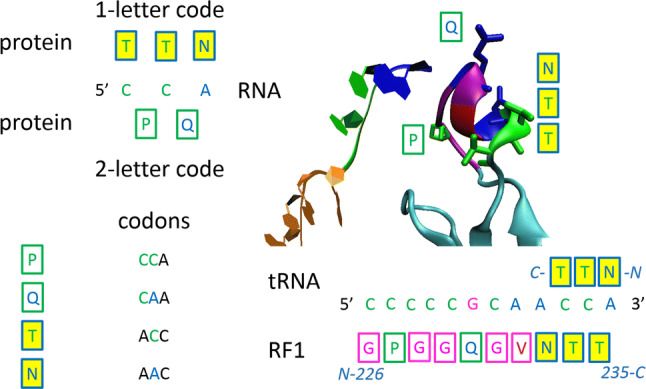
Protein–RNA recognition code. CCA trinucleotide is recognised as two dinucleotides—two-letter code (P-CC, Q-CA), or each nucleotide is recognised separately—one-letter code (T-C, T-C, N-A). One-letter code—second nucleotide in codons; two-letter code—first two nucleotides in codons. 3D5A crystal structure which contain release factor 1 (RF1) interacting with tRNA, as example [27]
The protein–RNA recognition code is still controversial at present; however, as will be shown later, this code can be used to explain or predict the involvement of miRNAs in neurodegeneration. The third position in the final codon code, the three-letter code (triplet), was perhaps also derived from some sort of ancient reactions or protein–RNA interactions [28], especially in the case of amino acids with two codons, for example cysteine, or methionine with the single codon, but in this case, only very low-specific interactions could influence the establishment of the third position.
Neurodegeneration
The protein–RNA interactome is the most abundant cellular phenomenon that regulates cell function and fate and is associated with a number of human diseases, such as cancer [29] and rheumatic diseases [30], but it is especially associated with neurodegeneration [31]. In neurons, basic biological processes that drive neurodegeneration are abnormally altered expression of some disease-driving RNAs and proteins, dysfunction of specific cellular organelles such as mitochondria or lysosomes, and neuroinflammation and altered responses of glia in the brain [32].
As mentioned above, ncRNAs fulfil the regulation of gene expression and are particularly abundant in the CNS. Unsurprisingly, “dysfunction of the regulation software” leads to molecular processes contributing to neurodegeneration. Six basic mechanisms exist by which ncRNAs may affect pathology in neurodegenerative disorders [8]: (1) epigenetic regulation; (2) RNAi (post-transcriptional repression of gene expression by miRNAs or small-interfering RNAs is collectively termed RNAi); (3) alternative splicing (shifting the splicing profiles of transcripts); (4) mRNA stability change; (5) translational regulation (ncRNAs have a direct impact on the translation of mRNA transcripts); and (6) molecular decoys (ncRNAs can act as molecular traps that titrate away RNAs or RBPs).
Toxic, improperly processed RNA usually contains repeat sequences, consisting of 3–6 nucleotides; the expanded RNA adopts unusual secondary structures that affect its function or sequesters RBPs and RNAs specialized to repeated sequence motifs [33]. Non-transported RNA accumulates in the nucleus, it makes nuclear inclusions, or RNA with nuclear function is improperly transported to the cytoplasmic compartment and form cytosolic inclusions, and insoluble RNA or RNP aggregates are observed as RNA foci [33]. Theoretically, toxic RNA can be purposely driven to RNA inclusion bodies to act as a sink for toxic RNA to provide neuroprotection [33]. In 2011, Laura Ranum’s group observed protein synthesis that was initiated not at the canonical ATG start codon but instead directly on the expanded repeats [34]. This repeat-associated non-ATG translation (RAN translation) results in the formation-accumulation of toxic homopolymeric proteins, which should be considered in pathogenic models [34].
Amyotrophic lateral sclerosis (ALS)
ALS, also known as motor neuron disease, is characterized by the degeneration of both upper and lower motor neurons, which leads to muscle weakness and eventual paralysis [35]. Within populations of European descent, up to 20% of individuals with ALS have a family history of either ALS or FTD (frontotemporal dementia, a behavioural variant), and of these, four genes account for up to 70% of all cases of familial ALS, namely, C9orf72 (encoding guanine nucleotide exchange), TARDBP (encoding TAR DNA-binding protein 43, TDP43), SOD1 (encoding superoxide dismutase) and FUS (encoding FUS-RBP) [35].
The (GGGGCC)n hexanucleotide repeat expansion in intron 1 of C9orf72 is a major cause of ALS and FTD [35]. Various interactomes containing C9orf72 RNA and RBPs have been proposed, and a comparative analysis has been performed on RBPs that bind to C9orf72 RNA in five studies [36]. hnRNP H (H component of the heterogeneous nuclear RNP) shows the most overlap across studies, followed by splicing factor proline and glutamine-rich (SFPQ), interleukin enhancer binding factor 2 and myelin basic protein [33]. Figure 2a depicts the reasons why hnRNP H and SFPQ are sequestered by the (GGGGCC)n repeat sequence: the hnRNP H is rich in GGA, and SFPQ is rich in GGP amino acid sequences, and according to the protein–RNA recognition code, GGA and GGP can be transcribed exactly to GGGGC and GGGGCC nucleotide sequences. Both proteins are rich in G and R amino acids for the G nucleotide readout. The C nucleotide readout is based on A and P amino acids, and S is used for both variants, the C and G readout (Fig. 2a).
Fig. 2.

Proteins sequestered into RNA foci (a–c); and the recognition of polyadenylation signal by polyadenylation factor subunit 2 (d WDR33). a H component of the heterogeneous nuclear RNP (hnRNP H), recognising toxic (GGGGCC)n repeat; b muscleblind-like protein 1 (MNBL1), recognising toxic (CTG)n repeat; c the heat shock transcription factor 1 (HSF1), recognising toxic (CAG)n repeat; and d WDR33 recognising polyadenylation signal
A BLAST search of the GGPGGPGGPGGPGGPGGP amino acid sequence shows that various isoforms of Ewing Sarcoma Protein are the first hits. Ewing Sarcoma Protein (EWS, EWSR1 gene) is an RBP that is a well-known player in cancer biology due to its role in the specific translocations occurring in sarcomas [37]. EWS is also known to be involved in ALS, and postmortem analysis of sporadic ALS cases revealed cytoplasmic mislocalization of EWS and identified disease-specific variants that also affected EWS localization in motor neurons [38]. Various isoforms of regulator of nonsense transcripts 1 (UPF1) represent the next hits in the BLAST search. UPF1 is also known to be involved in ALS, showing significant protection of mammalian neurons from TDP43- and FUS-related toxicity [39]. UPF1 is an RNA helicase and master regulator of nonsense-mediated mRNA decay; it contains N-terminal PGGPGGPGGGGAGGPGGA (no other GGP), which perfectly recognizes the (GGGGCC)n repeat sequence and perhaps initiates decay. Another hit, PNUTS, a protein phosphatase 1 (PP1) nuclear targeting subunit (or serine/threonine-protein phosphatase 1 regulatory subunit 10), has PGGP and similar motifs in the C-terminal region. PNUTS is a protein that targets PP1 to the nucleus and inhibits its activity, and its C terminus contains three distinct domains involved in RNA binding [40]. The C-part of the protein seems to well recognize the (GGGGCC)n repeat sequence. However, this protein has not yet been studied in relation to ALS.
Autosomal-dominant myotonic dystrophy (DM)
Myotonic dystrophy (DM) is the most common adult muscular dystrophy and is characterized by autosomal-dominant progressive myopathy, myotonia and multiorgan involvement [41]. To date, two distinct forms caused by similar mutations have been identified. Myotonic dystrophy type 1 (DM1, Steinert’s disease) is caused by a (CTG)n expansion in DMPK (myotonic dystrophy protein kinase gene), while myotonic dystrophy type 2 (DM2) is caused by a (CCTG)n expansion in ZNF9/CNBP (nucleic acid-binding protein—CNBP gene; previously known as zinc finger 9 gene—ZNF9 gene) [41].
Three human RBPs that regulate alternative splicing, muscle blind-like proteins 1–3 (MNBL1–3), are co-localized with RNA foci in DM1 and DM2 [31]. MNBL knockout in mouse models, resulting in loss of MNBL protein function, can mimic the disease, while the overexpression of MNBL proteins can rescue repeat RNA-induced toxicity in both Drosophila and mouse models of DM1, strongly supporting a role for these proteins in DM [31]. Other human RBPs have been identified in connection with DM, including CUG-binding protein 1 (CUGBP1), which can interact with CTG repeats [42]. However, this member of the CELF family of proteins, which plays a role in RNA splicing processing, is not sequestered into RNA foci, inconsistent with the hypothesis that some RBPs may be sequestered and inhibited by repeat-containing RNA [31]. Overexpression of CUGBP1 in mice results in splicing changes that are also observed in patients; increases in CUGBP1 expression can contribute to disease [43].
DM1 and DM2 are now largely believed to be due to defects in alternative splicing that arise as a result of the sequestration of MNBL proteins into RNA foci and a disruption in the expression of other RBPs, including CUGBP1 [31]. Figure 2b depicts why MNBL proteins are sequestered into RNA foci; the proteins are rich in cysteines and LA duplet amino acid sequences. The TGC nucleotide triplet represents exactly one form of a two-cysteine codon, and the LA duplet can be exactly transcribed to the CTGC nucleotide sequence, according to the two-letter recognition code. Interestingly, proteins could also be identified by one LGPV sequence that can be transcribed exactly to the TGCT nucleotide sequence by the one-letter recognition code. Using these sequences for BLAST search, various proteins can be identified, for example, sphingomyelin phosphodiesterase (SMPD1 gene) has similar distributions of cysteines: LA-AL and LP-PL duplets, LGP-PGL triplets, and one LGPV quartet and one (LA)5 repeat. In humans, deficient SMPD1 enzymatic activity leads to the type A and B forms of Niemann-Pick disease. Type A is a rapidly progressing neurodegenerative disorder that generally leads to death by 3 years of age. Patients with type B have little or no neurologic involvement and may survive into late adolescence [44]. No data exist in the literature describing the coexistence of both DM and Niemann-Pick disease. Another protein that shows a similar distribution of cysteine and the mentioned amino acid duplets and triplets identified by BLAST is the DIRC2 protein (disrupted in renal cancer 2). This gene encodes a membrane-bound protein from the major facilitator superfamily of transporters. Disruption of this gene by translocation has been associated with haploinsufficiency and renal cell carcinomas [45]. There is one report describing the coexistence of both DM and renal cancer [46].
Huntington’s disease (HD)
Huntington’s disease is a progressive, fatal, neurodegenerative disorder caused by an expanded (CAG)n repeat in the huntingtin gene, which encodes an abnormally long (n > 36) polyglutamine repeat in the huntingtin protein [47]. Huntington’s disease has served as a model for the study of other more common neurodegenerative disorders, such as Alzheimer’s disease and Parkinson’s disease. As in some other neurodegenerative disorders, neurodegeneration in affected individuals begins many years before the onset of diagnosable signs and symptoms of Huntington’s disease, and it is accompanied by subtle cognitive, motor, and psychiatric changes [47].
Pure (CAG)n repeats have been shown to be responsible for the toxicity; placing a pure expanded (CAG)n repeat in the 3′-untranslated region of a reporter construct leads to neuronal toxicity and behavioural defects in C. elegans, Drosophila, and mice [31]. Moreover, these pure CAG repeats accumulate into RNA foci in these models [31]. Additionally, the levels of the heat shock transcription factor 1 (HSF1) protein were recently shown to be lower in HD models, in differentiated human inducible pluripotent stem cells and in HD patient striatum and cortex, with a concomitant defect in target gene expression [48]. Expression of the cellular protein folding and pro-survival machinery by HSF1 ameliorates the biochemical and neurobiological defects caused by protein misfolding [48].
Figure 2c depicts the relatedness between the HSF1 amino acid sequence and the (CAG)n repeat sequence. DTD and QGQ are interesting triplets, according to the two-letter code. DTD is specific for the 3′–5′ readout, frame (G)AC(G)A, and QGQ is more specific for the 5′–3′ readout, frame (C)AG(C)A. BLAST search for (DTD)n showed pentatricopeptide repeat domain 3 (PTCD3) as the first hit. PTCD3 (MRP-S39) associates with the mitochondrial small ribosomal subunit and regulates translation [49]. Lowering PTCD3 in cells decreased mitochondrial protein synthesis, the activity of complexes III and IV, and mitochondrial respiration [49]. Interestingly, in HD, one putative pathological mechanism reported to play a prominent role in the pathogenesis of this neurological disorder is mitochondrial dysfunction [50]. PTCD3 has an TSDSDTDSSSDSDSDTSEGK –C-end that is well designed for (CAG)n repeat recognition, according to the code. In human neuronal cells, the expression of the (CAG)n-expanded exon 1 of the huntingtin gene caused an increase in (CAG)n repeat-derived sncRNAs, which were found to be cleaved by Dicer, the enzyme that generates mature miRNAs from pre-miRNAs [51]. These (CAG)n repeat-derived miRNAs could potentially dysregulate the functions of many proteins. As mentioned above, there is ample room for considering the possibility that the known biological function of a protein can be altered through riboregulation [22], and the protein–RNA recognition code strongly supports this possibility. The Rem2 GTPase is the second hit in the BLAST search for (DTD)n. Rem2 controls the proliferation and apoptosis of neurons during embryo development [52] and is known to be involved in neuronal development, such as in the control of neuron synapse formation. The expression of Rem2 is enriched in the central extended amygdala, and Rem2 is an important part of a homeostatic mechanism that controls synapse number [53]. REM2 has N-end-MHTDLDTDMDMDTE that is well designed, according to the code, for the recognition of (CAG)n repeat-derived miRNAs.
For the (QGQ)n sequence, the BLAST search identified mesoderm posterior protein 2 (MESP2), a downstream gene in the Notch signalling pathway [54], as the first hit. Mutations in MESP2 were found in spondylocostal dysostosis [55]. Spondylocostal dysostosis is a heterogeneous group of disorders with severe axial skeletal malformation radiologically characterized by multiple vertebral segmentation defects [55]. There is no direct link between MESP2 and HD, but activation of Notch signalling has been demonstrated to result in increased microtubule stability and changes in axonal morphology and branching [56]. In contrast, Notch inhibition leads to an increase in cytoskeleton plasticity with intense neurite remodelling [56]. When microtubules are too destabilized, axonal trafficking is impaired, and synaptic contacts collapse [57]. Indeed, aberrant mobility and trafficking of mitochondria are observed in an early event in HD pathophysiology [58]. In MESP2, the amino- and carboxy-terminal domains are separated by a (GQ)n repeat region, which is also found in MESP1 (n = 2) but expanded in MESP2 (n = 13). Mouse MESP1 and MESP2 do not contain GQ repeats. According to the protein–RNA recognition code, (GQ)13 is an ideal sequence recognized and impaired by toxic (CAG)n repeats (Fig. 2c). Synaptophysin-like 2 (SYPL2) is the second protein identified in the BLAST search of (GQ)n. Downregulation of this gene has been reported in Alzheimer’s disease and multiple sclerosis, and SYPL2 appears to be upregulated in HD [59]. SYPL2 has a (GQ)n-like (CAG)n-compatible sequence at the carboxy-terminus (HGQGQGQDQDQDQDQGQGPSQESAAEQGAVEKQ-C-end), which can be influenced according to the code. According to the two-letter code, DQD can also be transcribed to a GACAGA hexanucleotide, which is another frame in (CAG)n repeats. Synaptophysin (SYP) has an GQ and QG-rich C end and the strongest (CAG)n-readout by QGP, GPQ, PQG, GQP triplets (one-letter code). SYP is able to read (CAG)n in various frames, QGP(AGC), GPQ(GCA), PQG(CAG), YGP(AGC) in the 5′–3′ readout and GQP(GAC), SGY(CGA) in the 3′–5′ readout (see frames in Fig. 2c). The SYP-C-end will be recognized by (CAG)n repeat-derived sncRNAs or (CAG)n repeat-derived miRNAs. In early papers, the decreased contents of SYP, a glycoprotein component of synaptic vesicle membranes, were shown to be closely correlated with synapse loss; therefore, SYP has been frequently used as a sensitive marker in neurological cases and disease models [60].
Using (QGP)n in a BLAST search, polyadenylation factor subunit 2 (WDR33) was found as the first hit. 3′-polyadenylation is a key step in eukaryotic mRNA biogenesis. In mammalian cells, this process is dependent on the recognition of the AAUAAA hexanucleotide in the pre-mRNA, as a polyadenylation signal (PAS), by the cleavage and polyadenylation specificity factor (CPSF) complex. A core CPSF complex comprising CPSF160, WDR33, CPSF30 and Fip1 is sufficient for PAS recognition [61]. The N-terminal KRMRK motif in WDR33 was identified as a critical determinant of specific AAUAAA motif recognition [61]. This year, a 3.1-Å-resolution cryo-EM structure of a core CPSF module bound to the PAS was presented, revealing that the recognition is more complicated [62]. Nevertheless, WDR33 is a critical component for polyadenylation signal recognition, and mRNA 3′-untranslated region isoform changes are a feature of molecular pathology in the HD brain [63]. Isoform shifts in the HD motor cortex are not limited to huntingtin; 11% of alternatively polyadenylated genes change the abundance of their 3′-untranslated region isoforms [63]. Figure 2d depicts how disordered sequences, KRMRK and NKVK, plus the ETILQ-ordered sequence recognize the AAUAAA hexanucleotide. Unfortunately, the most important N-terminal QQQAMQQ sequence, according to the code, is probably disordered and not present in the structure. As shown in Fig. 2d, C-QQM-A-QQQ-N is responsible for the 5′AAUAAA3′ readout and is composed of two triplets spaced by alanine. WDR33 was the first hit in the (QGP)n BLAST search, and the other proteins listed were collagens. (QGP)n provides various frames for the readout; for example, the CAG nucleotide triplet is well recognized by the PQG amino acid triplet. Collagens are rich in combinations of various frames, PQG (18 in COL11A1), QGP (19 in COL11A1), GPQ (18 in COL11A1), GQP (2 in COL11A1), and PGQ (5 in COL11A1). According to the one-letter code and two-letter code, collagens are even rich in the strongest amino acids for the recognition of (CAG)n repeat frames, PKG (eight in COL11A1; codons CCC, AAA, GGG), KGP (five in COL11A1), GPK (eight in COL11A1), GKP (six in COL11A1), and PGK (five in COL11A1), which explains why (CAG)n repeats are so toxic. If direct riboregulation exists, then (CAG)n repeat-derived miRNAs will interfere with miRNAs designed for targeted proteins. Theoretically, there is ample room for considering the possibility that the known biological function of a protein can be directly modulated by miRNAs.
Compared to other repeats, (CAG)n repeats are more toxic, and they provide much more reading frames than (GGGGCC)n repeat sequences in ALS or (CTG)n repeat sequences in DM. For example, a BLAST search for (PKG)n identified COL23A1 (collagen XXIII, alpha-1 chain). Collagen XXIII is a component of adherens junctions, focal adhesions, and synaptic adhesions and is expressed by excitatory neurons in the accessory olfactory bulb and by cells in the retina [64]. In HD, the most prominent early pathological changes are observed in the striatum (dorsal) [65], and the ventral striatum is well connected with the olfactory bulb and olfactory sensory input into the striatum [66]. Collagen XXIII is upregulated in metastatic prostate cancer and is important for anchorage-independent growth and cell seeding to the lung. Collagen XXIII can be used as a tissue and fluid biomarker for non-small cell lung cancer and prostate cancer [67]. Toxic (CAG)n repeat-derived miRNAs may affect collagen function, such as the function of collagen XXIII. This hypothesis is supported by the observation that cancer is rarely reported on the death certificates of patients with HD, and cancer has a significantly lower incidence among patients with HD [68]. This fact has been even recently used to slow tumour growth by (CAG)n repeats in a preclinical mouse model of ovarian cancer with no signs of toxicity to the mice [69]. A lack of collagen VI has also been shown to promote neurodegeneration by impairing autophagy and inducing apoptosis during ageing [70]. COL6A1 was seventh in the (QGP)n BLAST search, and sequestration of collagen VI by (CAG)n is toxic. Therefore, collagens are likely the largest protein group affected by (CAG)n repeat degradation products. Human collagens comprise a family of 28 or more proteins, each of which is characterized by the presence of a triple-helical structure, formed as three separate protein strands that are sometimes different gene products and wind around one another to form a right-handed superhelix [71]. This defining conformation is facilitated by a repetitive (GXX′)n structure, where n may be as large as 350, and the amino acids X and X′ are quite often proline and polar amino acid.
Figure 2c shows the QAS amino acid sequence in contact with the (CAG)n repeat; the (CAG)n repeat sequence is strongly recognized using the two-letter code, and the amino acids have CA(G), GC(A), and AG(C) codons. A BLAST search for the (QAS)n repeat identified the NMP4 (nuclear matrix transcription factor 4), also named ZNF384 (zinc finger protein 384), protein. Interestingly, this protein regulates collagen I expression in osteoblasts [72]; therefore, if riboregulation of proteins exists, then the (CAG)n repeat sequence will influence collagen I and its regulator. Generally, NPM4 is known to function as a regulator of promoters of extracellular matrix genes and suppress bone anabolism [73]. Fusion genes involving NPM4 have been identified in B cell precursor acute lymphoblastic leukaemia [74]. NPM4 has a (QA)10(QAS)2Q16 sequence at the C-end designed for (CAG)n repeat; (AQ)n will also recognize (CAG)n (Fig. 2c). Interestingly, NPM4 has (CAG)14 repeats in its gene sequence. There is a hypothesis that (CAG)n repeats could be part of a mechanism used during evolution to maintain genome integrity and, in the context of multicellular organisms, to prevent cancer formation by producing toxic siRNAs [69]. The clearest connection may be the (CAG)n repeats in the androgen receptor gene and prostate cancer. While longer repeats (n > 20) confer a protective effect among prostate cancer patients, shorter CAG repeats (n < 18) have been shown to result in a twofold increase in cancer risk, a more aggressive disease, and a high risk of distant metastases [69]. Indeed, both the androgen receptor and the huntingtin genes contain some of the longest CAG repeats in the human genome; however, those are not found in the mouse orthologues at the same positions in the ORF [69]. In summary, there is a very close balance between anticancer protection and neurological disorders such as HD or spinocerebellar ataxia (the prevalent types of SCA are connected with (CAG)n repeats—SCA1, 2, 3, 6, 7, 8, 12, and 17 [75]).
Finally, the data from HD literature suggest that riboregulation of proteins exists; proteins involved in HD can be found by BLAST search by amino acid sequence repeats transcribed from (CAG)n repeats. An examination of miRNAs shows that miRNAs that repress prostate cancer have compatible nucleotide sequences to the amino acid sequences of their targets and the CAG triplet. For example, MIR34a was reported as an inhibitor of prostate cancer stem cells and metastasis, directly repressing the CD44 adhesion molecule [76]. Figure 3a shows amino acid sequences that can be recognized in CD44 by the one-letter transcription of MIR34a. The presence of the recognition sequences indicates that MIR34a could directly influence the CD44 protein and regulate its conformation and activity. The second part of Fig. 3a shows that MIR34a could directly influence collagens; MIR34a has only one CAG nucleotide triplet, but collagens have many amino acid sequences that recognize the GCAG nucleotide quartet.
Fig. 3.

Illustrated protein sequences that are recognised by a MIR34a, b MIR9, and c MIR107; and d cartoon of the Aβ42 fibril core, structure (5KK3)
Alzheimer’s disease (AD)
AD is a progressive neurodegenerative disorder that is the most common cause of dementia and imposes immense suffering on patients and their families [77]. AD was initially defined as a clinical-pathologic entity that was definitively diagnosed at autopsy and as possible or probable AD in life; consequently, the term AD is often used to describe two very different entities: prototypical clinical syndromes without neuropathologic verification and AD neuropathologic changes [78]. Currently, the definition of AD in living people is shifted from a syndromal to a biological construct. AD is defined by its underlying pathologic processes that can be documented by biomarkers in vivo or postmortem [78]. Biomarkers are divided into three groups: (1) amyloid deposition, (2) pathologic tau, and (3) neurodegeneration [78]:
In the brain, extracellular deposition of amyloid-β (Aβ) is formed by Aβ-peptides that arise from the sequential cleavage of membrane-spanning amyloid precursor protein (APP) by the beta-secretase APP cleaving enzyme 1 (BACE1) and the γ-secretase complex containing the presenilin (PSEN) proteins in the catalytic domain [79].
Intracellular neurofibrillary tangles, another hallmark of AD, are composed of tau. Tau is a microtubule-associated protein working as scaffolding proteins that are enriched in axons. In pathological conditions, increasing tau hyperphosphorylation renders the protein prone to aggregation, reduces its affinity for microtubules, and thereby influences neuronal plasticity, thus causing neurodegeneration [77].
Aβ deposited in the brain is believed to originate from the brain tissue itself; however, blood-derived Aβ was recently shown to be able to enter the brain, form the Aβ-related pathologies and induce functional deficits of neurons [80]. In vitro, picomolar concentrations of Aβ42-oligomers induced Ca2+ influx into lipid vesicles; therefore, aggregations of the Aβ-peptide itself could be the cause of toxicity [81]. Simulations show that the Aβ42 β-sheet tetramer triggers experimentally observed permeabilization and can further assemble into a pore structure [82]. Aβ-peptide is commonly thought to be intrinsically unstructured, meaning that in solution, it does not acquire a unique tertiary fold but rather forms a set of structures. As such, Aβ-peptide cannot be crystallized, and most structural knowledge on Aβ-peptide comes from NMR and molecular dynamics. However, Aβ-peptide can have significant secondary and tertiary structures [83]. Aβ40 and Aβ42 also seem to feature highly different conformational states, with the C-terminus of Aβ42 being more structured than that of the Aβ40 fragment [84]. The formation of a β-hairpin in the sequence IIGLMVGGVVIA may be responsible for the higher propensity of Aβ42 to form amyloids [84].
Here, we show that the MVGGVVIA sequence is strongly recognized by MIR9 miRNA through the one-letter code. As mentioned above, sncRNA regulators are highly enriched in the brain, where they play key roles in neuronal development, plasticity and disease. In neurodegenerative disorders such as AD, brain miRNA profiles are altered; thus, miRNA dysfunction could be both a cause and a consequence of the disease [85]. Aβ is a powerful regulator of miRNA levels, and the usual downregulation of mature miRNAs is extremely rapid [85]. MIR9 is highly expressed in the hippocampus, the region of the brain associated with memory and learning. MIR9 reduction in AD has been shown in various human AD brain samples, mouse models and neuronal cell culture models, and MIR9 is generally regarded as being neuroprotective [79]. Overexpression of MIR9 was sufficient to restore Aβ42− induced dendritic spine loss and rescued Aβ42 induced tau phosphorylation at Ser 262 mediated by the CAMKK2–AMPK pathway [86]. Figure 3b depicts how MIR9 recognizes Aβ-peptide. The MVGGVVIA C-end is strongly recognized by the one-letter code, and the next DVGSNKGAIIGL sequence shows features of recognition by the two-letter code. The VFFA core of Aβ42 is also recognized by the one-letter code, and the DVGSNKGAIIGL N-terminal sequence also shows features of recognition by the two-letter code. In summary, MIR9 recognizes Aβ42, especially from the C-terminus.
MIR107 is probably the most important miRNA in AD; its expression has a negative correlation with neurofibrillary tangle formation, and its downregulation is a major contributor to AD progression. MIR107 targets BACE1 and the metalloproteinase ADAM10, which is also involved in APP processing, and has also been shown to target cofilin, an actin-binding protein that dissembles actin filaments in dendritic spine heads and is therefore important in memory and learning [79]. Figure 3c shows how MIR107 recognizes Aβ42. The DAEFRHDSGY N-terminus is recognized by the one-letter code, but MIR107 also well recognizes the KGAIIGL sequence at the C-terminus. Aggregation of Aβ42 forms cross-β amyloid fibrils, and the atomic resolution structure (5KK3) of a monomorphic form of Aβ42 shows that the fibril core consists of a dimer of Aβ42 molecules, each containing four β-strands in an S-shaped amyloid fold [87]. The cartoon in Fig. 3d depicts the fibril core and shows regions where MIR9 and MIR107 will interact. MIR107 recognizes more accessible region and free disordered N-terminal region; therefore, MIR107 could be sequestered on the fibril surface. MIR9 is likely more involved in interactions with the monomer during the process of fibril formation, and several intermediate forms, such as nuclei, oligomers, and protofibrils of various sizes, forms, and structures, may exist [87].
Parkinson’s disease (PD)
PD is a progressive neurological disorder characterized by a large number of motor and non-motor features [88]. Loss of substantia nigra neurons and the presence of Lewy body inclusions (insoluble protein aggregates composed mainly but not exclusively of α-synuclein) in some of the remaining neurons are the hallmark pathology seen in the final stages of PD; however, attempts to correlate Lewy body pathology to either cell death or severity of clinical symptoms have not been successful [89]. The clinical symptoms of PD suggest that a failure of synapses (a structure that permits a neuron to pass an electrical or chemical signal to another neuron), not loss of neurons, is the pathophysiological mechanism of the disease [89]. Ninety percent or even more of α-synuclein aggregates are not localized in Lewy bodies, but at the presynapse in the form of aggregates much smaller than Lewy bodies [89]. α-Synuclein is a highly conserved protein encoded by the SNCA gene and is involved in clustering synaptic vesicles at the presynaptic terminals; therefore, primary aggregation in the presynapse is logical.
α-Synuclein has long been defined as a ‘natively unfolded’ monomer of approximately 14 kDa that is believed to acquire an α-helical secondary structure only upon binding to lipid vesicles. Native α-synuclein has been shown to have an α-helical tetramer structure without lipid addition, and this structure has a much greater lipid-binding capacity than the recombinant α-synuclein studied previously [90]. In light of this observation, disruptions in chaperons (chaperons can prevent aggregation by protein folding-facilitation or mediated-degradation, or they can prone aggregation), such as heat shock proteins, are considered to play a key role in α-synuclein aggregation in PD [91]. miRNAs could regulate chaperone pathways [91], or miRNAs could be small molecules that directly stabilize (or destabilize) the physiological tetramer structure. As mentioned above, there is ample room for considering the possibility that the known biological function of a protein can be altered through riboregulation [22].
MIR7 and MIR153 have been identified to regulate α-synuclein levels post-transcriptionally (mRNA sequestration and translational repression) [92]. Their overexpression significantly reduces endogenous α-synuclein levels, whereas their inhibition enhances translation of a luciferase construct bearing the α-synuclein 3-untranslated region in primary neurons; these miRNAs bind specifically to the 3′-untranslated region of α-synuclein and downregulate its mRNA and protein levels [92]. MIR7 and MIR153 can be predicted to regulate α-synuclein aggregation, but whether this is performed only on the post-transcriptional/translational level or if they can also directly control the α-synuclein structure remains an open question. On the post-transcriptional/translational level, the results indicate that MIR7 and MIR153 are co-expressed with α-synuclein in neurons to regulate its levels through a transcription feed-forward loop that fine-tunes rather than blocks α-synuclein mRNA expression and translation [92].
Figure 4a, b depict the relatedness between the α-synuclein amino acid sequence and the sequence of these miRNAs. According to the protein–RNA recognition code, important parts of the α-synuclein sequence can be directly transcribed to MIR7 and MIR153 sequences by the one-letter code. This observation indicates a strong interaction, which suggests that these miRNAs also post-translationally regulate α-synuclein and influence α-synuclein structure. Additionally, overexpression of MIR7 was recently observed to facilitate the degradation of α-synuclein aggregates by promoting autophagy with an unknown mechanism [93]. MIR221 is a serum miRNA that shows a positive correlation with part III of the united Parkinson’s disease rating scale, leading to its proposal as a potential biomarker for PD [94]. MIR221 has also been found to regulate cell viability and apoptosis by targeting PTEN (phosphatase and tensin homologue) [95]; however, Fig. 4c shows that according to the one-letter code, MIR221 could recognize and riboregulate α-synuclein. A number of other miRNAs are predicted to regulate α-synuclein aggregation; many of them will most likely recognize the α-synuclein amino acid sequence according to the protein–RNA recognition code.
Fig. 4.

Illustrated protein sequences that are recognised by a MIR7, b MIR153-1, and c MIR221-3p
Prion diseases (PDs)
Prion diseases, a group of disorders caused by abnormally shaped proteins called prions, occur in sporadic (Jakob–Creutzfeldt disease), genetic (genetic Jakob–Creutzfeldt disease, Gerstmann–Straussler–Scheinker syndrome, and fatal familial insomnia), and acquired (kuru, variant Jakob–Creutzfeldt disease, and iatrogenic Jakob–Creutzfeldt disease) forms [96]. Unfortunately, to date, all human PDs clinical trials have failed to show survival benefit. However, very rare polymorphisms in the prion protein gene have been identified that appear to protect against PDs [96, 97]. The prion disease model states that the pathologic disease-causing misfolded form of the prion protein, PrPSc (in which “Sc” stands for scrapie, the prion disease of sheep and goats) acts as a template, such that when it comes into contact with a prion protein, PrPC (in which “C” stands for the normal, cellular form of the protein), PrPC is transformed into PrPSc, resulting in two PrPSc [96]. The PrPC population, with a primarily α-helical structure, is changed to an abnormal population of PrPSc, which stands for proteinaceous infectious particles (prions), and has a primarily β-pleated sheet structure [96].
Figure 5a shows the crystal structure of wild-type PrPC (5YJ5) and pathogenic mutations in full-length human PrPC [98]. The structure of the huPrPC is similar to that of the E219K mutant [99] and consists of a disordered N-terminal tail and a well-structured C-terminal segment containing two short antiparallel β-strands and three α-helices. Interestingly, the most frequently pathogenically mutated region is between the end of the second and beginning of the third α-helices (yellow in Fig. 5a). The spacer of the second and third α-helices (red in Fig. 5a) is seemingly important for the conformational transition of PrPSc to β-sheets. Transcribing the amino acid sequence of this spacer to the nucleotide sequence using the one-letter code can lead to the identification of miRNAs in the miRBase (http://www.mirbase.org). Table 1 presents examples of miRNAs found in the miRBase. MIR361 represents the best match with the spacer found in the database. The FirePlex Discovery Engine (https://www.fireflybio.com), which assembles a list of the most important miRNAs associated with the prion keyword, did not identify this miRNA. MIR146a is first in the list and seems to be the most important in PDs. However, one study reported an association between MIR361-5p and PDs [100]. In this study, the authors investigated miRNA expression changes in synaptoneurosomes prepared from the forebrains and hippocampi of mice at two time points during PDs. At the preclinical stage (105 days post-infection), MIR146a showed an 1.1-fold increase in the hippocampus and an 0.2-fold decrease in the forebrain, and MIR361 showed an 1.7-fold increase in both the hippocampus and forebrain [100]. At the terminal stage of PDs (167 days post-infection), MIR146a showed an 6.95-fold increase in the forebrain, and MIR361 showed an 2.8-fold decrease in the forebrain [100]. According to the protein–RNA recognition code, MIR361 recognizes the frequently mutated region more strongly (Fig. 5b) than it recognizes the MIR146a (Fig. 5c), and MIR361 even well recognizes the octapeptide repeat region (Fig. 5b). At the end-stage of PDs, MIR361 appears to be sequestered by the amyloid fibrils.
Fig. 5.

Prion diseases. a Pathogenic mutations (red, [98]) shown in the sequence and crystal structure of human wild-type PrPC (5YJ5), short important amino acid sequences can be transcribed into nucleotide sequences and searched via the online miRBase. b, c Illustrated protein sequences that are recognised by MIR361 and MIR146a respectively
Table 1.
Examples of miRNAs found in miRBase
| Searched sequence | Name | MIR sequence | Prion reference |
|---|---|---|---|
| cagaauc | hsa-miR-361-5p | 1 UUAUCAGAAUCUCCAGGGGUAC 22 | [100] |
| hsa-miR-541-3p | 1 UGGUGGGCACAGAAUCUGGACU 22 | – | |
| cagaaauc | hsa-miR-9902 | 1 CCCAGAAAUCUGGUAUGCCAGC 22 | – |
| hsa-miR-342-3p | 1 UCUCACACAGAAAUCGCACCCGU 23 | [101, 102] | |
| cagaauuc | hsa-miR-2115-3p | 1 CAUCAGAAUUCAUGGAGGCUAG 22 | – |
| hsa-miR-1324 | 1 CCAGACAGAAUUCUAUGCACUUUC 24 | – | |
| caggaauc | hsa-miR-145-5p | 1 GUCCAGUUUUCCCAGGAAUCCCU 23 | [100] |
| hsa-miR-1289 | 1 UGGAGUCCAGGAAUCUGCAUUUU 23 | – | |
| hsa-miR-6735-5p | 1 CAGGGCAGAGGGCACAGGAAUCUGA 25 | – | |
| gaaucac | hsa-miR-876-5p | 1 UGGAUUUCUUUGUGAAUCACCA 22 | – |
Mature miRNAs, SSEARCH, E value cutoff (10,000), human
Conclusion
ncRNAs are likely critical components for epigenetic control. An estimated 40% of the genes for lncRNAs are specifically expressed in brain tissue, and miRNAs have also been reported to be enriched in the CNS. More than 60% of human protein-coding genes harbour predicted miRNA target sites [11]. Many of the miRNAs are loaded into extracellular vesicles for cell–cell communication; interestingly, viruses often modulate host miRNAs for their replication. Large RNP machines reveal that protein–RNA interactions do not require canonical globular RNA-binding domains; many regions in RBPs are found to be intrinsically disordered, and this property helps them to recognize and bind RNA-partner sequences, adopting stable structures upon binding. Conventional RBDs change the fate or function of the bound RNAs, but there is ample room for considering the opposite possibility that the known biological function of a protein can be altered through riboregulation, for example, by miRNAs. miRNAs, or sncRNAs that arise as RNA degradation products, can directly influence the activity and structure of proteins. These events can be programmed or coded by nucleotide-amino acid sequences. The protein–RNA recognition code [27], which is still controversial, plays an important role in this case [103]. In the interactome, interruption of protein–RNA recognition harmony leads to molecular processes contributing to neurodegeneration. ALS, also known as motor neuron disease, shows how toxic (GGGGCC)n hexanucleotide repeat expansion improperly sequesters hnRNP H and SFPQ. These proteins are rich in G and GGP and GGA triplets that support the protein–RNA recognition code hypothesis. DM1 is caused by a (CTG)n expansion in the DMPK gene, and repeat RNA-induced toxicity sequesters MNBL1–3 proteins into RNA foci. The proteins are rich in cysteines, L, A and LA amino acid duplets that can be exactly transcribed to the CTGC nucleotide sequence. HD, a neurodegenerative disorder caused by an expanded (CAG)n repeat in the huntingtin gene, teaches us how (CAG)n repeats can be toxic to neurons on one hand but have anticancer properties on the other. The system creates a balance between anticancer properties (n > 20) and neurodegeneration (n > 36). The protein–RNA recognition code explains why (CAG)n repeats can be so toxic. According to the code, many proteins have sequences at the N- or C-terminus that recognize various (CAG)n repeat reading frames. Dysregulation of these proteins can lead to cell death. Theoretically, there is ample room for considering the possibility that the known biological function of a protein can be directly modulated by miRNAs. (CAG)n repeat-derived sncRNAs, which were found to be cleaved by the enzyme that generates mature miRNAs from pre-miRNAs (Dicer), would compete with miRNAs designed for the riboregulation. For example, MIR34a, which directly represses the CD44 adhesion molecule, has one GCAG motif (also MIR107 from AD has one). A reduction in MIR9 in AD has been shown in various human AD brain samples, mouse models and neuronal cell culture models, and MIR9 is generally regarded as being neuroprotective. According to the protein–RNA recognition code, MIR9 strongly recognizes Aβ42, especially the MVGGVVIA C-end sequence, which may be responsible for the higher propensity of Aβ42 to form amyloids. MIR107 also well recognizes Aβ42, especially the disordered N-terminal region. In PD, MIR7 and MIR153 can be predicted to regulate α-synuclein aggregation. According to the protein–RNA recognition code, important parts of the α-synuclein sequence can be directly transcribed to MIR7 and MIR153 sequences. Similarly, in PDs, a group of disorders caused by prions, transcribing amino acid sequences to the nucleotide sequence may lead to the identification of miRNAs potentially involved in the disease. It provides useful key to design miRNA probes.
In my opinion, future research should focus on “contactless” recognition. There is a possibility that protein–RNA recognition can be performed without contact and binding. In that case, co-crystallization or crosslinking and precipitation will not occur. Based on this concept, bioinformatics research could provide evidence and explanations for processes like this in the near future, and all needed datasets for this task likely already exist in public databases. Paradoxically, neurodegenerative disorders and cancer could provide the datasets to comprehend the molecular, cellular and multicellular life in all his details.
Acknowledgements
This work was supported by “Vedecká grantová agentúra MŠVVaŠ SR a SAV” (VEGA 2/0058/17) and the Research and Development Operational Programme (ITMS 26220120054).
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Beadle GW, Tatum EL. Genetic control of biochemical reactions in Neurospora. Genetics. 1941;27:499–506. doi: 10.1073/pnas.27.11.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerstein MB, Bruce C, Rozowsky JS, Zheng D, Du J, Korbel JO, Emanuelsson O, Zhang ZD, Weissman S, Snyder M. What is a gene, post-ENCODE? History and updated definition. Genome Res. 2007;17:669–681. doi: 10.1101/gr.6339607. [DOI] [PubMed] [Google Scholar]
- 3.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, Khatun J, Lajoie BR, Landt SG, Lee B-, Pauli F, Rosenbloom KR, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Felsenfeld G. A brief history of epigenetics. Cold Spring Harbor Perspect Biol. 2014;6:a018200. doi: 10.1101/cshperspect.a018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yao B, Christian KM, He C, Jin P, Ming G-, Song H. Epigenetic mechanisms in neurogenesis. Nat Rev Neurosci. 2016;17:537–549. doi: 10.1038/nrn.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maison C, Bailly D, Peters AHFM, Quivy J-, Roche D, Taddei A, Lachner M, Jenuwein T, Almouzni G. Higher-order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat Genet. 2002;30:329–334. doi: 10.1038/ng843. [DOI] [PubMed] [Google Scholar]
- 7.Chen J, Xue Y. Emerging roles of non-coding RNAs in epigenetic regulation. Sci China Life Sci. 2016;59:227–235. doi: 10.1007/s11427-016-5010-0. [DOI] [PubMed] [Google Scholar]
- 8.Salta E, De Strooper B. Noncoding RNAs in neurodegeneration. Nat Rev Neurosci. 2017;18:627–640. doi: 10.1038/nrn.2017.90. [DOI] [PubMed] [Google Scholar]
- 9.Gebert LFR, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol. 2019;20:21–37. doi: 10.1038/s41580-018-0045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swarts DC, Makarova K, Wang Y, Nakanishi K, Ketting RF, Koonin EV, Patel DJ, Van Der Oost J. The evolutionary journey of argonaute proteins. Nat Struct Mol Biol. 2014;21:743–753. doi: 10.1038/nsmb.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedman RC, Farh KK-, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kozomara A, Griffiths-Jones S. MiRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsang J, Zhu J, van Oudenaarden A. MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol Cell. 2007;26:753–767. doi: 10.1016/j.molcel.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ebert MS, Sharp PA. Roles for microRNAs in conferring robustness to biological processes. Cell. 2012;149:505–524. doi: 10.1016/j.cell.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villarroya-Beltri C, Baixauli F, Gutiérrez-Vázquez C, Sánchez-Madrid F, Mittelbrunn M. Sorting it out: regulation of exosome loading. Semin Cancer Biol. 2014;28:3–13. doi: 10.1016/j.semcancer.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, Ruan Y, Lassmann T, Carninci P, Brown JB, Lipovich L, Gonzalez JM, Thomas M, Davis CA, Shiekhattar R, Gingeras TR, Hubbard TJ, Notredame C, Harrow J, Guigó R. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uszczynska-Ratajczak B, Lagarde J, Frankish A, Guigó R, Johnson R. Towards a complete map of the human long non-coding RNA transcriptome. Nat Rev Gen. 2018;19:535–548. doi: 10.1038/s41576-018-0017-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Munschauer M, Nguyen CT, Sirokman K, Hartigan CR, Hogstrom L, Engreitz JM, Ulirsch JC, Fulco CP, Subramanian V, Chen J, Schenone M, Guttman M, Carr SA, Lander ES. The NORAD lncRNA assembles a topoisomerase complex critical for genome stability. Nature. 2018;561:132–136. doi: 10.1038/s41586-018-0453-z. [DOI] [PubMed] [Google Scholar]
- 19.Holley RW, Apgar J, Everett GA, Madison JT, Marquisee M, Merrill SH, Penswick JR, Zamir A. Structure of a ribonucleic acid. Science. 1965;147:1462–1465. doi: 10.1126/science.147.3664.1462. [DOI] [PubMed] [Google Scholar]
- 20.Engreitz JM, Sirokman K, McDonel P, Shishkin AA, Surka C, Russell P, Grossman SR, Chow AY, Guttman M, Lander ES. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent pre-mRNAs and chromatin sites. Cell. 2014;159:188–199. doi: 10.1016/j.cell.2014.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barry G. Integrating the roles of long and small non-coding RNA in brain function and disease. Mol Psychiatry. 2014;19:410–416. doi: 10.1038/mp.2013.196. [DOI] [PubMed] [Google Scholar]
- 22.Hentze MW, Castello A, Schwarzl T, Preiss T. A brave new world of RNA-binding proteins. Nat Rev Mol Cell Biol. 2018;19:327–341. doi: 10.1038/nrm.2017.130. [DOI] [PubMed] [Google Scholar]
- 23.Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 24.Basu S, Bahadur RP. A structural perspective of RNA recognition by intrinsically disordered proteins. Cell Mol Life Sci. 2016;73:4075–4084. doi: 10.1007/s00018-016-2283-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Castello A, Fischer B, Frese CK, Horos R, Alleaume A-, Foehr S, Curk T, Krijgsveld J, Hentze MW. Comprehensive identification of RNA-binding domains in human cells. Mol Cell. 2016;63:696–710. doi: 10.1016/j.molcel.2016.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mackowiak SD, Zauber H, Bielow C, Thiel D, Kutz K, Calviello L, Mastrobuoni G, Rajewsky N, Kempa S, Selbach M, Obermayer B. Extensive identification and analysis of conserved small ORFs in animals. Genome Biol. 2015;16:179. doi: 10.1186/s13059-015-0742-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nahalka J. Protein-RNA recognition: cracking the code. J Theor Biol. 2014;343:9–15. doi: 10.1016/j.jtbi.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 28.Nahalka J. Quantification of peptide bond types in human proteome indicates how DNA codons were assembled at prebiotic conditions. J Proteom Bioinform. 2011;4:153–159. doi: 10.4172/jpb.1000184. [DOI] [Google Scholar]
- 29.Li Y, McGrail DJ, Xu J, Li J, Liu NN, Sun M, Lin R, Pancsa R, Zhang J, Lee JS, Wang H, Mills GB, Li X, Yi S, Sahni N. MERIT: systematic analysis and characterization of mutational effect on RNA interactome topology. Hepatology. 2018 doi: 10.1002/hep.30242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang Y, Zhou T, Yu X, Xue Z, Shen N. The role of long non-coding RNAs in rheumatic diseases. Nat Rev Rheumatol. 2017;13:657–669. doi: 10.1038/nrrheum.2017.162. [DOI] [PubMed] [Google Scholar]
- 31.Todd TW, Petrucelli L. Neurodegenerative diseases and RNA-mediated toxicity. In: Wolfe MS, editor. The molecular and cellular basis of neurodegenerative diseases: underlying mechanisms. 1. New York: Academic Press; 2018. pp. 441–475. [Google Scholar]
- 32.Katsnelson A, De Strooper B, Zoghbi HY. Neurodegeneration: from cellular concepts to clinical applications. Sci Transl Med. 2016;8:364. doi: 10.1126/scitranslmed.aal2074. [DOI] [PubMed] [Google Scholar]
- 33.Zhang N, Ashizawa T. RNA toxicity and foci formation in microsatellite expansion diseases. Curr Opin Genet Dev. 2017;44:17–29. doi: 10.1016/j.gde.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MAC, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LPW. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci USA. 2011;108:260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, Shaw PJ, Simmons Z, Van Den Berg LH. Amyotrophic lateral sclerosis. Nat Rev Disease Prim. 2017;3:17071. doi: 10.1038/nrdp.2017.71. [DOI] [PubMed] [Google Scholar]
- 36.Haeusler AR, Donnelly CJ, Rothstein JD. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci. 2016;17:383–395. doi: 10.1038/nrn.2016.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paronetto MP. Ewing sarcoma protein: a key player in human cancer. Int J Cell Biol. 2013;2013:642853. doi: 10.1155/2013/642853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Couthouis J, Hart MP, Erion R, King OD, Diaz Z, Nakaya T, Ibrahim F, Kim H-, Mojsilovic-petrovic J, Panossian S, Kim CE, Frackelton EC, Solski JA, Williams KL, Clay-falcone D, Elman L, McCluskey L, Greene R, Hakonarson H, Kalb RG, Lee VMY, Trojanowski JQ, Nicholson GA, Blair IP, Bonini NM, Van Deerlin VM, Mourelatos Z, Shorter J, Gitler AD. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21:2899–2911. doi: 10.1093/hmg/dds116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barmada SJ, Ju S, Arjun A, Batarse A, Archbold HC, Peisach D, Li X, Zhang Y, Tank EMH, Qiu H, Huang EJ, Ringe D, Petsko GA, Finkbeiner S. Amelioration of toxicity in neuronal models of amyotrophic lateral sclerosis by hUPF1. Proc Natl Acad Sci USA. 2015;112:7821–7826. doi: 10.1073/pnas.1509744112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim Y-, Watanabe T, Allen PB, Kim Y-, Lee S-, Greengard P, Nairn AC, Kwon Y- PNUTS, a protein phosphatase 1 (PP1) NUclear targeting subunit: characterization of its PP1 and RNA-binding domains and regulation by phosphorylation. J Biol Chem. 2003;278:13819–13828. doi: 10.1074/jbc.M209621200. [DOI] [PubMed] [Google Scholar]
- 41.Meola G, Cardani R. Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim Biophys Acta Mol Basis Dis. 2015;1852:594–606. doi: 10.1016/j.bbadis.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 42.Timchenko LT, Miller JW, Timchenko NA, Devore DR, Datar KV, Lin L, Roberts R, Thomas Caskey C, Swanson MS. Identification of a (CUG)(n) triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res. 1996;24:4407–4414. doi: 10.1093/nar/24.22.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ho TH, Bundman D, Armstrong DL, Cooper TA. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum Mol Genet. 2005;14:1539–1547. doi: 10.1093/hmg/ddi162. [DOI] [PubMed] [Google Scholar]
- 44.Wan Q, Schuchman EH. A novel polymorphism in the human acid sphingomyelinase gene due to size variation of the signal peptide region. Biochim Biophys Acta Mol Basis Dis. 1995;1270:207–210. doi: 10.1016/0925-4439(95)00050-E. [DOI] [PubMed] [Google Scholar]
- 45.Bodmer D, Eleveld M, Kater-Baats E, Janssen I, Janssen B, Weterman M, Schoenmakers E, Nickerson M, Linehan M, Zbar B, Van Kessel AG. Disruption of a novel MFS transporter gene, DIRC2, by a familial renal cell carcinoma-associated t(2;3)(q35;q21) Hum Mol Genet. 2002;11:641–649. doi: 10.1093/hmg/11.6.641. [DOI] [PubMed] [Google Scholar]
- 46.Addeo A, Bini R, Viora T, Bonaccorsi L, Leli R. Von hippel-lindau and myotonic dystrophy of steinert along with pancreatic neuroendocrine tumor and renal clear cell carcinomal neoplasm: case report and review of the literature. Int J Surg Case Rep. 2013;4:648–650. doi: 10.1016/j.ijscr.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 48.Gomez-Pastor R, Burchfiel ET, Neef DW, Jaeger AM, Cabiscol E, McKinstry SU, Doss A, Aballay A, Lo DC, Akimov SS, Ross CA, Eroglu C, Thiele DJ. Abnormal degradation of the neuronal stress-protective transcription factor HSF1 in huntington’s disease. Nat Commun. 2017;8:14405. doi: 10.1038/ncomms14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davies SMK, Rackham O, Shearwood A-J, Hamilton KL, Narsai R, Whelan J, Filipovska A. Pentatricopeptide repeat domain protein 3 associates with the mitochondrial small ribosomal subunit and regulates translation. FEBS Lett. 2009;583:1853–1858. doi: 10.1016/j.febslet.2009.04.048. [DOI] [PubMed] [Google Scholar]
- 50.Quintanilla RA, Johnson GVW. Role of mitochondrial dysfunction in the pathogenesis of huntington’s disease. Brain Res Bull. 2009;80:242–247. doi: 10.1016/j.brainresbull.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krol J, Fiszer A, Mykowska A, Sobczak K, de Mezer M, Krzyzosiak WJ. Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol Cell. 2007;25:575–586. doi: 10.1016/j.molcel.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 52.Edel MJ, Boué S, Menchon C, Sánchez-Danés A, Belmonte JCI. Rem2 GTPase controls proliferation and apoptosis of neurons during embryo development. Cell Cycle. 2010;9:3414–3422. doi: 10.4161/cc.9.17.12719. [DOI] [PubMed] [Google Scholar]
- 53.Paradis S, Harrar DB, Lin Y, Koon AC, Hauser JL, Griffith EC, Zhu L, Brass LF, Chen C, Greenberg ME. An RNAi-based approach identifies molecules required for glutamatergic and GABAergic synapse development. Neuron. 2007;53:217–232. doi: 10.1016/j.neuron.2006.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Andersson ER, Lendahl U. Therapeutic modulation of notch signalling-are we there yet? Nat Rev Drug Discov. 2014;13:357–378. doi: 10.1038/nrd4252. [DOI] [PubMed] [Google Scholar]
- 55.Whittock NV, Sparrow DB, Wouters MA, Sillence D, Ellard S, Dunwoodie SL, Turnpenny PD. Mutated/MESP2 causes spondylocostal dysostosis in humans. Am J Hum Genet. 2004;74:1249–1254. doi: 10.1086/421053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bonini SA, Ferrari Toninelli G, Montinaro M, Memo M. Notch signalling in adult neurons: a potential target for microtubule stabilization. Ther Adv Neurol Disord. 2013;6:375–385. doi: 10.1177/1756285613490051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trushina E, Heldebrant MP, Perez-Terzic CM, Bortolon R, Kovtun IV, Badger JD, II, Terzic A, Estévez A, Windebank AJ, Dyer RB, Yao J, McMurray CT. Microtubule destabilization and nuclear entry are sequential steps leading to toxicity in huntington’s disease. Proc Natl Acad Sci USA. 2003;100:12171–12176. doi: 10.1073/pnas.2034961100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang DTW, Rintoul GL, Pandipati S, Reynolds IJ. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol Dis. 2006;22:388–400. doi: 10.1016/j.nbd.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 59.Chen S, Lu FF, Seeman P, Liu F. Quantitative proteomic analysis of human substantia nigra in alzheimer’s disease, huntington’s disease and multiple sclerosis. Neurochem Res. 2012;37:2805–2813. doi: 10.1007/s11064-012-0874-2. [DOI] [PubMed] [Google Scholar]
- 60.Goto S, Hirano A. Synaptophysin expression in the striatum in huntington’s disease. Acta Neuropathol. 1990;80:88–91. doi: 10.1007/BF00294227. [DOI] [PubMed] [Google Scholar]
- 61.Clerici M, Faini M, Aebersold R, Jinek M. Structural insights into the assembly and polya signal recognition mechanism of the human CPSF complex. ELife. 2017;6:e33111. doi: 10.7554/eLife.33111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clerici M, Faini M, Muckenfuss LM, Aebersold R, Jinek M. Structural basis of AAUAAA polyadenylation signal recognition by the human CPSF complex. Nat Struct Mol Biol. 2018;25:135–138. doi: 10.1038/s41594-017-0020-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Romo L, Ashar-Patel A, Pfister E, Aronin N. Alterations in mRNA 3′ UTR isoform abundance accompany gene expression changes in human huntington’s disease brains. Cell Rep. 2017;20:3057–3070. doi: 10.1016/j.celrep.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Monavarfeshani A, Knill CN, Sabbagh U, Su J, Fox MA. Region- and cell-specific expression of transmembrane collagens in mouse brain. Front Integr Neurosci. 2017;11:20. doi: 10.3389/fnint.2017.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walker FO. Huntington’s disease. Lancet. 2007;369:218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 66.Xiong A, Wesson DW. Illustrated review of the ventral striatum’s olfactory tubercle. Chem Senses. 2016;41:549–555. doi: 10.1093/chemse/bjw069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spivey KA, Chung I, Banyard J, Adini I, Feldman HA, Zetter BR. A role for collagen XXIII in cancer cell adhesion, anchorage-independence and metastasis. Oncogene. 2012;31:2362–2372. doi: 10.1038/onc.2011.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sørensen SA, Fenger K, Olsen JH. Significantly lower incidence of cancer among patients with huntington disease: an apoptotic effect of an expanded polyglutamine tract? Cancer. 1999;86:1342–1346. doi: 10.1002/(SICI)1097-0142(19991001)86:7<1342::AID-CNCR33>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 69.Murmann AE, Gao QQ, Putzbach WE, Patel M, Bartom ET, Law CY, Bridgeman B, Chen S, McMahon KM, Thaxton CS, Peter ME. Small interfering RNAs based on huntingtin trinucleotide repeats are highly toxic to cancer cells. EMBO Rep. 2018;19:e45336. doi: 10.15252/embr.201745336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cescon M, Chen P, Castagnaro S, Gregorio I, Bonaldo P. Lack of collagen VI promotes neurodegeneration by impairing autophagy and inducing apoptosis during aging. Aging. 2016;8:1083–1101. doi: 10.18632/aging.100924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Farndale RW. Collagen-induced platelet activation. Blood Cells Mol Dis. 2006;36:162–165. doi: 10.1016/j.bcmd.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 72.Thunyakitpisal P, Alvarez M, Tokunaga K, Onyia JE, Hock J, Ohashi N, Feister H, Rhodes SJ, Bidwell JP. Cloning and functional analysis of a family of nuclear matrix transcription factors (NP/NMP4) that regulate type I collagen expression in osteoblasts. J Bone Miner Res. 2001;16:10–23. doi: 10.1359/jbmr.2001.16.1.10. [DOI] [PubMed] [Google Scholar]
- 73.Childress P, Stayrook KR, Alvarez MB, Wang Z, Shao Y, Hernandez-Buquer S, Mack JK, Grese ZR, He Y, Horan D, Pavalko FM, Warden SJ, Robling AG, Yang F-, Allen MR, Krishnan V, Liu Y, Bidwell JP. Genome-wide mapping and interrogation of the Nmp4 antianabolic bone axis. Mol Endocrinol. 2015;29:1269–1285. doi: 10.1210/me.2014-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hirabayashi S, Ohki K, Nakabayashi K, Ichikawa H, Momozawa Y, Okamura K, Yaguchi A, Terada K, Saito Y, Yoshimi A, Ogata-Kawata H, Sakamoto H, Kato M, Fujimura J, Hino M, Kinoshita A, Kakuda H, Kurosawa H, Kato K, Kajiwara R, Moriwaki K, Morimoto T, Nakamura K, Noguchi Y, Osumi T, Sakashita K, Takita J, Yuza Y, Matsuda K, Yoshida T, Matsumoto K, Hata K, Kubo M, Matsubara Y, Fukushima T, Koh K, Manabe A, Ohara A, Kiyokawa N. ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica. 2017;102:118–129. doi: 10.3324/haematol.2016.151035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sun Y-, Lu C, Wu Z- Spinocerebellar ataxia: relationship between phenotype and genotype—a review. Clin Genet. 2016;90:305–314. doi: 10.1111/cge.12808. [DOI] [PubMed] [Google Scholar]
- 76.Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, Patrawala L, Yan H, Jeter C, Honorio S, Wiggins JF, Bader AG, Fagin R, Brown D, Tang DG. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17:211–216. doi: 10.1038/nm.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Du X, Wang X, Geng M. Alzheimer’s disease hypothesis and related therapies. Transl Neurodegener. 2018;7:2. doi: 10.1186/s40035-018-0107-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R, Elliott C, Masliah E, Ryan L, Silverberg N. NIA-AA research framework: toward a biological definition of alzheimer’s disease. Alzheimer’s Dement. 2018;14:535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rajgor D. Macro roles for microRNAs in neurodegenerative diseases. Non-coding RNA Res. 2018;3:154–159. doi: 10.1016/j.ncrna.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bu XL, Xiang Y, Jin WS, Wang J, Shen LL, Huang ZL, Zhang K, Liu YH, Zeng F, Liu JH, Sun HL, Zhuang ZQ, Chen SH, Yao XQ, Giunta B, Shan YC, Tan J, Chen XW, Dong ZF, Zhou HD, Zhou XF, Song W, Wang YJ. Blood-derived amyloid-β protein induces Alzheimer’s disease pathologies. Mol Psychiatry. 2017;2017:204. doi: 10.1038/mp.2017.204. [DOI] [PubMed] [Google Scholar]
- 81.Flagmeier P, De S, Wirthensohn DC, Lee SF, Vincke C, Muyldermans S, Knowles TPJ, Gandhi S, Dobson CM, Klenerman D. Ultrasensitive measurement of Ca2+ influx into lipid vesicles induced by protein aggregates. Angew Chem Int Ed. 2017;56:7750–7754. doi: 10.1002/anie.201700966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nasica-Labouze J, Nguyen PH, Sterpone F, Berthoumieu O, Buchete N-, Coté S, De Simone A, Doig AJ, Faller P, Garcia A, Laio A, Li MS, Melchionna S, Mousseau N, Mu Y, Paravastu A, Pasquali S, Rosenman DJ, Strodel B, Tarus B, Viles JH, Zhang T, Wang C, Derreumaux P. Amyloid β protein and alzheimer’s disease: when computer simulations complement experimental studies. Chem Rev. 2015;115:3518–3563. doi: 10.1021/cr500638n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vivekanandan S, Brender JR, Lee SY, Ramamoorthy A. A partially folded structure of amyloid-beta(1-40) in an aqueous environment. Biochem Biophys Res Commun. 2011;411:312–316. doi: 10.1016/j.bbrc.2011.06.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sgourakis NG, Yan Y, McCallum SA, Wang C, Garcia AE. The alzheimer’s peptides Aβ40 and 42 adopt distinct conformations in water: a combined MD/NMR study. J Mol Biol. 2007;368:1448–1457. doi: 10.1016/j.jmb.2007.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schonrock N, Ke YD, Humphreys D, Staufenbiel M, Ittner LM, Preiss T, Götz J. Neuronal micro RNA deregulation in response to alzheimer’s disease amyloid-β. PLoS One. 2010;5:e11070. doi: 10.1371/journal.pone.0011070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chang F, Zhang L-, Xu WU-, Jing P, Zhan P- microRNA-9 attenuates amyloidβ-induced synaptotoxicity by targeting calcium/calmodulin-dependent protein kinase kinase 2. Mol Med Rep. 2014;9:1917–1922. doi: 10.3892/mmr.2014.2013. [DOI] [PubMed] [Google Scholar]
- 87.Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, Donovan KJ, Michael B, Wall J, Linse S, Griffin RG. Atomic resolution structure of monomorphic Aβ42Amyloid fibrils. J Am Chem Soc. 2016;138:9663–9674. doi: 10.1021/jacs.6b05129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jankovic J. Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79:368–376. doi: 10.1136/jnnp.2007.131045. [DOI] [PubMed] [Google Scholar]
- 89.Schulz-Schaeffer WJ. The synaptic pathology of α-synuclein aggregation in dementia with lewy bodies, parkinson’s disease and parkinson’s disease dementia. Acta Neuropathol. 2010;120:131–143. doi: 10.1007/s00401-010-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bartels T, Choi JG, Selkoe DJ. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–111. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Alvarez-Erviti L, Seow Y, Schapira AHV, Rodriguez-Oroz MC, Obeso JA, Cooper JM. Influence of microRNA deregulation on chaperone-mediated autophagy and α-synuclein pathology in parkinson’s disease. Cell Death Dis. 2013;4:e545. doi: 10.1038/cddis.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Doxakis E. Post-transcriptional regulation of α-synuclein expression by mir-7 and mir-153. J Biol Chem. 2010;285:12726–12734. doi: 10.1074/jbc.M109.086827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Choi DC, Yoo M, Kabaria S, Junn E. MicroRNA-7 facilitates the degradation of alpha-synuclein and its aggregates by promoting autophagy. Neurosci Lett. 2018;678:118–123. doi: 10.1016/j.neulet.2018.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ma W, Li Y, Wang C, Xu F, Wang M, Liu Y. Serum miR-221 serves as a biomarker for parkinson’s disease. Cell Biochem Funct. 2016;34:511–515. doi: 10.1002/cbf.3224. [DOI] [PubMed] [Google Scholar]
- 95.Li L, Xu J, Wu M, Hu JM. Protective role of microRNA-221 in parkinson’s disease. Bratislava Med J. 2018;119:22–27. doi: 10.4149/BLL_2018_005. [DOI] [PubMed] [Google Scholar]
- 96.Geschwind MD. Prion diseases. Continuum. Lifelong Learn Neurol. 2015;21:1612–1638. doi: 10.1212/CON.0000000000000251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Asante EA, Smidak M, Grimshaw A, Houghton R, Tomlinson A, Jeelani A, Jakubcova T, Hamdan S, Richard-Londt A, Linehan JM, Brandner S, Alpers M, Whitfield J, Mead S, Wadsworth JDF, Collinge J. A naturally occurring variant of the human prion protein completely prevents prion disease. Nature. 2015;522:478–481. doi: 10.1038/nature14510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lloyd SE, Mead S, Collinge J. Genetics of prion diseases. Curr Opin Genet Dev. 2013;23:345–351. doi: 10.1016/j.gde.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Biljan I, Giachin G, Ilc G, Zhukov I, Plavec J, Legname G. Structural basis for the protective effect of the human prion protein carrying the dominant-negative E219K polymorphism. Biochem J. 2012;446:243–251. doi: 10.1042/BJ20111940. [DOI] [PubMed] [Google Scholar]
- 100.Boese AS, Saba R, Campbell K, Majer A, Medina S, Burton L, Booth TF, Chong P, Westmacott G, Dutta SM, Saba JA, Booth SA. MicroRNA abundance is altered in synaptoneurosomes during prion disease. Mol Cell Neurosci. 2016;71:13–24. doi: 10.1016/j.mcn.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 101.Montag J, Hitt R, Opitz L, Schulz-Schaeffer WJ, Hunsmann G, Motzkus D. Upregulation of miRNA hsa-miR-342-3p in experimental and idiopathic prion disease. Mol Neurodegener. 2009;4:36. doi: 10.1186/1750-1326-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bellingham SA, Coleman BM, Hill AF. Small RNA deep sequencing reveals a distinct miRNA signature released in exosomes from prion-infected neuronal cells. Nucleic Acids Res. 2012;40:10937–10949. doi: 10.1093/nar/gks832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nahalka J, Hrabarova E, Talafova K. Protein-RNA and protein-glycan recognitions in light of amino acid codes. Biochim Biophys Acta Gen Subj. 2015;1850:1942–1952. doi: 10.1016/j.bbagen.2015.06.013. [DOI] [PubMed] [Google Scholar]