

Abstract
Genetic and functional studies suggest diverse pathways being affected in the neurodegenerative disease amyotrophic lateral sclerosis (ALS), while knowledge about converging disease mechanisms is rare. We detected a downregulation of microRNA-1825 in CNS and extra-CNS system organs of both sporadic (sALS) and familial ALS (fALS) patients. Combined transcriptomic and proteomic analysis revealed that reduced levels of microRNA-1825 caused a translational upregulation of tubulin-folding cofactor b (TBCB). Moreover, we found that excess TBCB led to depolymerization and degradation of tubulin alpha-4A (TUBA4A), which is encoded by a known ALS gene. Importantly, the increase in TBCB and reduction of TUBA4A protein was confirmed in brain cortex tissue of fALS and sALS patients, and led to motor axon defects in an in vivo model. Our discovery of a microRNA-1825/TBCB/TUBA4A pathway reveals a putative pathogenic cascade in both fALS and sALS extending the relevance of TUBA4A to a large proportion of ALS cases.
Electronic supplementary material
The online version of this article (10.1007/s00018-018-2873-1) contains supplementary material, which is available to authorized users.
Keywords: Amyotrophic lateral sclerosis, Frontotemporal dementia, MicroRNA, TBCE, Microtubules, Zebrafish
Introduction
More than 90% of the patients affected by the neurodegenerative disorder amyotrophic lateral sclerosis (ALS) have no family history of the disease and are considered sporadic (sALS) cases [1]. The remaining approximately 5–10% report a positive family history for ALS or the genetically and neuropathologically linked disease frontotemporal dementia (FTD) [1, 2], and are, therefore, classified as familial (fALS). Despite the diverse functions of the more than two dozen known ALS genes, their physiological functions point to few overarching cell biological topics relevant for ALS pathogenesis [3]. Amongst these, one major topic is the alteration in cytoskeletal dynamics as evidenced by ALS-causing mutations in PFN1 (coding for profilin 1) [4], DCTN1 (dynactin subunit p150) [5], or TUBA4A (tubulin alpha-4A chain) [6]. However, in most patients a mutation cannot be assured. Furthermore, genetic mouse models have not been predictive for the clinical success of pre-clinically effective therapies so far. One reason for this failure in translation might be that the exact relationship between monogenically caused ALS and ALS of sporadic patients without genetic mutations remains largely unclear. Molecular research on ALS pathogenesis mostly focuses on downstream effects of known disease mutations. Hence, knowledge about disease pathways relevant for both genetic and sporadic ALS is largely lacking, but would be important for novel treatment strategies that are effective for the major proportion of ALS patients.
Much of the ALS research interest is centered on TDP-43/TARDBP because cytoplasmic aggregates of hyperphosphorylated TDP-43 are found in post-mortem specimens of most ALS patients [7] and mutations in TARDBP can cause fALS in rare instances [8]. The finding that TDP-43 is involved in the biogenesis of microRNAs (miRNAs) links miRNA dysmetabolism to the pathogenesis of ALS [9, 10]. MiRNAs are small (18–25 nucleotides) non-coding RNAs regulating gene expression post-transcriptionally. A single miRNA is thought to influence the expression of several 100 genes [11, 12]. By hybridization with the 3′ untranslated region (3′ UTR), miRNAs induce either degradation, deadenylation or translational repression of the target mRNAs [13]. Recently, we detected altered miRNAs in serum of fALS and sALS patients using miRNA microarray analyses [14, 15]. According to our previous work, miR-1825 was the only miRNA significantly downregulated in serum of both sALS and fALS and a trend towards reduction of miR-1825 (uncorrected p value = 0.0586) was observed in serum of pre-manifest ALS mutation carriers [14, 15]. This suggested that reduced serum/plasma levels of miR-1825 could be a general feature of most ALS patients and may be part of a common pathogenic cascade.
In this study, we describe a miR-1825/TBCB/TUBA4A pathway that is dysregulated in both sporadic and familial ALS. Our data suggest disturbances of the microtubule cytoskeleton caused by increased levels of TBCB as a common disease mechanism in both fALS and sALS. We, therefore, uncover a miR-1825-dependent pathway that connects genetic and epigenetic aspects of the disease and may be relevant for the mechanistic understanding of a large proportion of ALS cases.
Materials and methods
Ethics statement and patient cohorts
Analyses of post-mortem samples were performed in accordance with the Declaration of Helsinki (WMA, 1964) and study protocols were approved by the local medical ethical review boards. Informed consent was obtained from all individual participants included in the study. Genotyping of SOD1 and C9ORF72 of clinically definite (El-Escorial criteria) ALS patients was performed by Sanger sequencing or repeat-primed PCR and Southern blotting, respectively [16, 17]. ALS cases were considered sporadic when no familial background of the disease was known and mutations in SOD1 or C9ORF72 were not detected. Characteristics of ALS patients and controls are shown in Supplementary Tables 1–4. Procedures involving animals were conducted according to the guidelines of the German animal welfare act and approved by the regional board (Tübingen; Germany).
RNA purification, reverse transcription and quantitative RT-PCR
For miRNA analyses, RNA was isolated using the miRNeasy Mini kit (Qiagen) according to the manufacturers’ instructions. RNA integrity numbers (RIN) were determined using the Agilent 2100 Bioanalyzer (Agilent Technologies). Reverse transcription and quantitative PCRs were performed using the miScript PCR System (Qiagen). MiRNA-specific oligonucleotides were purchased from Qiagen (miScript Primer Assays, Supplementary Table 5). For mRNA analyses, RNA was isolated with the RNeasy Plus Mini kit (Qiagen) and reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad). For qPCRs, we used the iQ SYBR Green Supermix (Bio-Rad). All qPCRs were run on a CFX96 real-time system (Bio-Rad). The method was used to calculate the fold change of RNA or miRNA level compared to control samples. MiRNA levels were normalized to U6 small nuclear RNA while mRNA levels were normalized to U6 or TATA-box-binding protein (TBP), unless otherwise stated. Oligonucleotides used for qRT-PCR are listed in Supplementary Table 5. The order of processed samples was randomized.
In situ hybridization (ISH)
The 5′- and 3′-digoxigenin-labelled miRCURY LNA Detection probe specific for hsa-miR-1825 (#611352-360) and scrambled control (#699004-360) was purchased from Exiqon (Vedbaek, Denmark). The ISH was performed according to the manufacturer’s protocol with following minor modifications: prior to hybridization, samples were blocked with prehybridization buffer (50% formamide, 5 × SSC, 1 × Denhardt’s solution, 500 µg/ml yeast tRNA) for 30 min at hybridization temperature (54 °C). Then, 40 nM probes in hybridization buffer (50% formamide, 5 × SSC, 1 × Denhardt’s solution) were incubated with the samples at 54 °C for 1 h. For probe detection, anti-digoxigenin antibody labelled with horseradish peroxidase (HRP; 5 U/ml, #11207733910, Sigma-Aldrich) and DAB-solution (Dako) were used. Slides were counterstained using Mayer’s hemalum (#109249, Merck Millipore). Bright-field images were recorded using BZ-9000E microscope (Bioreva, Keyence). Sections were processed in a random order.
Plasmids and RNA oligonucleotides
Synthetic mimic-1825 (syn-hsa-miR-1825 miScript miRNA Mimic, #MSY0006765) and antimiR-1825 (anti-hsa-miR-1825 miScript miRNA Inhibitor, #MIN0006765) and their corresponding negative controls (AllStars Negative control siRNA, #1027280; miScript Inhibitor Negative control, #1027271) were purchased from Qiagen (Hilden, Germany). Biotinylated mimic-1825 (hsa-miR-1825 miRCURY LNA microRNA Mimic, #479997-671) and biotinylated mimic-39-3p (cel-miR-39-3p miRCURY LNA microRNA Mimic, #479997-671) were acquired from Exiqon (Vedbaek, Denmark). 6-FAM labelled mimic-control RNA (HMC002, #8021324906-000020; HMC002_as, ##8021324906-000030) was purchased from Sigma-Aldrich (St. Louis, USA).
Plasmids encoding for TBCB-myc were generated by cloning TBCB-myc (transcript variant 1, GeneBank-ID 50428924) into BamHI–EcoRI sites of pcDNA3 (Invitrogen, Carlsbad, USA). The 3′ UTR fragments of CASP3 (transcript variant alpha, GeneBank-ID 73622121, binding site 1: bp 1961–2180, binding site 2: bp 2315–2506) and TBCB (transcript variant 1, GeneBank-ID 50428924, bp 1311–1475) as well as the CDS of TBCB (bp 576–1310) comprising the predicted binding sites for miR-1825 were cloned into SacI–XhoI sites of pmiR-GLO dual-luciferase miRNA target expression vector (Promega). Predicted binding sites of miR-1825 were mutated using the Stratagene Quick-Change Site-directed mutagenesis kit (Agilent Technologies) according to manufacturer’s instructions. Oligonucleotides used for the mutagenesis are listed in Supplementary Table 6.
Cell culture and transfection
Human embryonic kidney (HEK) 293 cells (DSMZ, ACC-305) were cultivated in DMEM containing 10% FBS at 37 °C and 5% CO2. Cells were negatively tested for mycoplasma. 24 h after plating cells were transfected with DNA-plasmids using calcium phosphate precipitation. MiRNA mimic or antimiR transfections were carried out using HiPerfect transfection reagent (Qiagen). Co-transfection of plasmids and RNA-oligonucleotides was performed with Lipofectamine2000 (Thermo Fisher Scientific) as recommended by the manufacturer.
Preparation and transfection of mouse primary cortical neurons
Primary cortical neurons of E15 mouse embryos (C57BL/6J) were prepared as previously described [18]. Using Lipofectamine2000 (Thermo Fisher Scientific), primary neurons (DIV2 or DIV5) were transfected according to the manufacturer’s protocol.
SWATH–mass spectrometry
48 h after transfection of HEK293 cells with 5 nM mimic-1825 or mimic-ctrl., cells were lysed with lysis buffer (25 mM Tris, 6 M guanidine-HCL, pH 8) and subjected to tryptic digestion using the GASP-protocol [19]. Quantitative label-free proteomic analysis was accomplished by nano-HPLC–SWATH–mass spectrometry as published previously [20] except that a TOP40 method was used for the library runs and 60 SWATH-windows of variable size were used for SWATH–MS-runs. In total, 1523 proteins were detected in all samples. For statistical analysis, we focused on downregulated proteins with a fold change < 0.75. A two-tailed Student’s t test was used to identify differentially expressed proteins between the groups and p values were corrected for multiple testing using FDR correction. FDR values < 0.05 were considered statistically significant.
Western blotting
SDS-PAGE and western blotting were performed using the NuPAGE system (Invitrogen). Primary antibodies were rabbit-anti-Caspase3 (#9662, Cell Signaling Technology, 1:1000), rabbit-anti-TBCB (#PA03169A0Rb, Cusabio Life science, 1:1000), rabbit-anti-TUBA4A (#P68366, Abgent, 1:8000), rabbit-anti-TUBA4A (#ab8374, Abcam, 1:500), mouse-anti-myc tag (9B11, #2276, Cell Signaling Technology, 1:2000), rabbit-anti-POLR2H (RPAB3) (#AP20815a, Abgent, 1:1000), rabbit-anti-β-tubulin (D2N5G, #15115, Cell Signaling Technology, 1:1000), rabbit-anti-TUBA1A/TUBA1B/TUBA1C/TUBA8/TUBA4A (#PA-193238, Cusabio Life science, 1:1000), rabbit-anti-TUBA1A (#ab200216, Abcam, 1:1000), rabbit-anti-tuba8l2 (#orb53155, biorbyt, 1:2000), rabbit-anti-GAPDH (#10494-1-AP, proteintech, 1:5000) and rabbit-anti-β-actin (13E5, #4970S, Cell Signaling Technology, 1:2000). Goat-anti-rabbit and goat-anti-mouse HRP-labelled secondary antibodies were purchased from Life Technologies (#G21234, #G21040, respectively, 1:1000). Densitometric analyses were performed using ImageJ (version 1.48) as described previously [21].
RNA microarray analyses
24 h post-transfection with 5 nM mimic-1825 or mimic-ctrl., HEK293 cells were washed with PBS and total RNA was extracted using the RNeasy Plus Mini kit (Qiagen) as recommended by the manufacturer. Microarray analyses were performed on Affymetrix GeneChip Human Gene 2.0 ST Arrays (Affymetrix, Santa Clara, CA, USA). The chips were scanned with an Affymetrix GeneChip Scanner 3000 and subsequent images analyzed using Affymetrix Expression Console Software in combination with BRB-ArrayTools (http://linus.nci.nih.gov/BRB-ArrayTools.html). Raw feature data were normalized using RMA normalization [22]. We identified differentially expressed genes among the two classes using a two-sample t test. Resulting p values were corrected for multiple testing using FDR correction. Genes were considered significant if their FDR was less than 0.05 and displayed a fold change between the two groups of at least 1.5-fold as published previously [23, 24]. Hierarchical cluster analysis (average linkage) was performed using the Genesis software package [25]. Transcriptome data have been deposited in Gene expression omnibus (GEO) database (GSE101046).
Biotinylated miRNA pulldown assay
HEK293 cells in 10-cm dishes were transfected with 20 nM biotinylated mimic-1825 or mimic-39-3p. 24 h post-transfection, cells were washed with PBS, lysed in 500 µl lysis buffer (10 mM Tris–HCl, 300 mM NaCl, 10 mM KCl, 1.5 mM MgCl2, pH 7.5, 2.5 mM DTT, 0.5% NP-40) supplemented with 60 U/ml RiboLock RNase Inhibitor (Thermo Fisher Scientific) and centrifuged for 2 min at 10,000×g and 4 °C. The supernatant was incubated for 2 h at 4 °C with streptavidin agarose beads (#20357, Thermo Fisher Scientific) and pre-blocked in 1 µg/µl BSA and 1 µg/µl yeast tRNA (3 h, 4 °C) under constant rotation. Afterwards, the beads were washed five times in lysis buffer and the precipitated RNA was isolated directly from the beads using the RNeasy Plus Mini kit (Qiagen). Oligonucleotides used for RT-PCR are listed in Supplementary Table 7.
Dual-luciferase reporter assay
HEK293 cells cultured in 96-well plates were co-transfected with 100 ng of the respective pmiRGlow dual-luciferase plasmid and 30 nM mimic-1825, 40 nM antimiR-1825, or the respective negative control RNA. 48 h post-transfection, Renilla and firefly luciferase activity were measured using the Dual-Glow Luciferase Assay System (Promega) according to the manufacturer’s protocol. The Renilla luciferase activity served as an internal transfection control and was used to normalize the activity of the firefly luciferase.
Toxicity assays and cell stress
48 or 72 h after transfection of HEK293 cells in a 96-well plate with mimic-1825 or antimiR-1825 or the respective controls, toxicity was determined by measuring the activity of CASP3 and CASP7 or the cytoplasmic lactate dehydrogenase (LDH) release using the Apo-ONE homogeneous caspase-3/7 assay (Promega) or the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega), respectively. Additionally, 48 h after transfection with mimic-1825 or mimic-control, HEK293 cells were washed with PBS, fixed using Roti Histofix 4% (Roth) and mounted using Fluoromount G with DAPI (Southern biotech). Images were taken with an Axio Observer.A1 microscope (Zeiss) and AxioVision-software (Zeiss; version 4.8). DAPI-positive cells were automatically counted using ImageJ software (version 1.48).
Cellular stress was induced by addition of 400 mM sorbitol or 400 µM sodium arsenite to the culture media for 3 h.
Co-immunoprecipitation
48 h post-transfection, HEK293 cells in 10-cm dishes were washed with PBS and resuspended in 500 µl Triton-X lysis buffer (50 mM Tris-HCl, 100 mM NaCl, 5 mM EDTA, pH 7.5, 0.3% Triton-X-100). Lysates were incubated on ice for 30 min, followed by centrifugation for 5 min at 10,000×g and 4 °C. Supernatants were adjusted to equal protein concentrations and 1 ml of each condition was incubated for 2 h at 4 °C under constant rotation with 20 µl mouse-anti-c-myc-agarose beads (#20168, Thermo Fisher Scientific) equilibrated in Triton-X lysis buffer. Subsequently, the beads were washed five times with 1 ml Triton-X lysis buffer and boiled in protein sample buffer before separation of precipitated proteins by SDS-PAGE and detection by western blotting. The order of processed samples was randomized.
Immunocytochemistry
Immunocytochemistry was performed as previously described [18]. Primary antibodies were rabbit-anti-TUBA4A (#AP13535b, Abgent, 1:100), rabbit-anti-β-tubulin (D2N5G, #15115, Cell Signaling Technology, 1:200), rabbit-anti-TUBA1A/TUBA1B/TUBA1C/TUBA8/TUBA4A (#PA-193238, Cusabio Life science, 1:100), rabbit-anti-TUBA1A (#ab200216, abcam, 1:100) and mouse-anti-myc (9B11, #2276, Cell Signaling Technology, 1:600). Secondary antibodies were donkey-anti-rabbit-Alexa488 and goat-anti-mouse-Alexa647 (#A21206, #A21235, respectively; both Life Technologies, 1:750). For STED microscopy imaging, goat-anti-rabbit-Atto594 and goat-anti-mouse-Atto647N were used (#77671, #50185, respectively, both Sigma-Aldrich, 1:750). Phalloidin-Atto594 (#51927, Sigma-Aldrich) was used as previously described [26]. Cells were mounted using Fluoromount G with DAPI (Southern biotech).
Confocal microscopy
Confocal imaging was performed at room temperature using a laser scanning microscope (Leica TCS SP8) with HC PL APO CS2 20×/0.75 dry and HC PL APO CS2 63×/1.20 water objectives. Images were taken using LAS X microscope software (version 1.1.0.12420). Apart from equal contrast and brightness correction for samples and controls, no additional picture modifications were applied. ImageJ software (version 1.48) was used for measurements of fluorescent intensity and the length and branching of motor neuron axons. The analysis of primary neurites of primary cortical neurons was performed by a blinded investigator.
Stimulated emission depletion (STED) microscopy and analysis
Super-resolution images were obtained using a custom built dual color STED microscope described recently [27]. In brief, the system utilizes a SC-450-PP-HE super-continuum laser source (Fianium) for excitation and depletion beams, a piezo stage for fast sample scanning (733.2DD, Physik Instrumente), a high NA objective for illumination and collection of fluorescence light (HCX PL APO 100x/1.40-0.70 oil CS, Leica) and an avalanche photodiode (SPCMAQRH-13/14-FC, Perkin-Elmer) for fluorescence detection. The microscope operation and image acquisition were controlled by a custom-written LabVIEW program (National Instruments). Pixel dwell times were 400 µs, and 1.6 mW of STED laser power and 0.9 µW of excitation laser power were used. The images were post-processed in ImageJ (version 1.51J8). All the represented images were adjusted for contrast, brightness and processed by Gaussian blurring with a standard deviation fitting to the estimated localization precision (~ 35 nm). The investigator analyzing the TUBA4A contour lengths was blinded.
Biochemical fractionation of polymerized and soluble tubulins
48 h after transfection of HEK293 cells in 10-cm dishes, biochemical fractionation of soluble and polymerized tubulins was performed as previously described [28]. Samples were prepared in a random order.
Microinjection of mRNA into zebrafish embryos and whole mount immunohistochemistry
Adult zebrafishes (Danio rerio) were bred and maintained as previously described [29]. Capped sense mRNA of human TBCB-myc was in vitro transcribed from TBCB-myc pcDNA3 plasmid using the mMESSAGE mMACHINE T7 Transcription Kit (#AM1344, Thermo Fisher Scientific) and purified using the RNeasy Plus Mini kit (Qiagen). MRNA (800 ng/µl in 200 mM KCl) was injected into fertilized wt zebrafish oocytes in the one-cell stage using a FemtoJet microinjector (Eppendorf AG). Embryos injected with 200 mM KCl served as injection control. For motor neuron staining, embryos were fixed at 48 hpf with Dent’s fixative (4:1 MeOH/DMSO) and bleached using 10% (v/v) H2O2 in Dent’s fixative (each over night at room temperature). After rehydration with MeOH/PBT (PBS + 0.1% Tween-20), blocking was performed using 10% FBS in PBT for 90 min at room temperature. Mouse-anti-SV2 antibody (#AB2315387, Developmental Studies Hybridoma Bank, 1:100) and goat-anti-mouse-DyLight488 (#35502, Thermo Fisher Scientific, 1:500) were incubated for 2 days each at 4 °C. The order of processed zebrafish embryos was randomized.
Statistical analysis
Statistical analyses were performed and the corresponding graphs were drawn using the GraphPadPrism software (version 5.06 or version 6.01). Values are presented as mean ± SEM unless otherwise stated. D’Agostino’s K-squared test was used to assess Gaussian distribution. Either two-tailed unpaired Student’s t tests or Mann–Whitney U tests were used for calculation of p values. Statistical significance level was set at p ≤ 0.05. Correlation analysis was performed using the Pearson’s r.
Results
MiR-1825 is systemically downregulated in ALS patients
Downregulation of miR-1825 in serum or plasma of sALS patients has been independently reported by two groups [14, 30]. To assess the systemic expression of miR-1825 also in CNS as well as further extra-CNS tissue, we measured the relative levels of miR-1825 in post-mortem tissue samples of ALS patients by qRT-PCR. As expected for post-mortem samples, RNA integrity numbers (RIN) were low but did not significantly differ between ALS patients and controls for most experimental groups. This indicates that the results of the qRT-PCR analyses of post-mortem tissues were largely unbiased by different RNA qualities of ALS and control samples (Supplementary Table 8). An exception was cerebral cortex samples of fALS patients, which displayed decreased RNA degradation compared to respective controls. Notably, although RNA quality of fALS cortex samples was higher than in the controls (Supplementary Table 8), downregulation of miR-1825 was still evident. Moreover, as shown in Fig. 1a, b, miR-1825 was significantly downregulated also in most other tissues from both sALS and fALS patients examined, including muscle, brainstem and cerebral cortex.
Fig. 1.

Deregulation of miR-1825 and pre-mir-1825 in ALS post-mortem tissues. a–c Relative levels of miR-1825 (a, b) and transcript variant 1 of the protein-O-fucosyltransferase 1 (POFUT1) (c), the host gene of pri-mir-1825, were determined in post-mortem tissue of fALS and sALS patients as well as healthy controls by qRT-PCR and normalized to U6 snRNA. d Relative concentration of pre-mir-1825 in post-mortem tissue of fALS patients and healthy controls as determined by qRT-PCR. Ct values were normalized to miR-15a which was invariable in samples reverse transcribed in pre-mir-specific buffer. Clinical characteristics of ALS patients and controls used for qRT-PCR analyses are shown in Supplementary Tables 1–4 (green dots in a–d indicate individual data points; box plots in a–d represent the 25 and 75% quantiles, horizontal lines indicate the median and whiskers show minimum and maximum values; *p ≤ 0.05, **p < 0.01 in a two-tailed Mann–Whitney-U test for data with non-Gaussian distribution or in a two-tailed Student’s t test for data with Gaussian distribution; ns not significant, bs brainstem, ctx cortex, sc spinal cord, liv liver, lyn lymph node, ki kidney, sm skeletal muscle). e Representative in situ hybridization (ISH) images of miR-1825-specific and scrambled probes in cortex of a fALS patient carrying a C9orf72 repeat expansion and a healthy control individual. Nuclei were counterstained using hematoxylin. 2 controls and three ALS patients were analyzed showing similar results (bars indicate 40 µm)
These data thus indicate a downregulation of miR-1825 in tissues affected in ALS. Additionally, increased levels of pre-mir-1825 and unchanged levels of POFUT1 mRNA, the host gene of miR-1825, in brainstem and spinal cord of fALS patients suggest that a malfunction of pre-mir-1825 nuclear export and/or processing causes the observed downregulation of mature miR-1825 in ALS (Fig. 1c, d). To assess the localization of miR-1825 in the CNS, in situ hybridization experiments of human cerebral cortex and spinal cord tissue were performed and demonstrated robust and high expression of miR-1825 in cortical neurons and motor neurons. Although there seemed to be a higher intensity in controls compared to sALS and fALS patient tissue, a caveat has to be expressed as the DAB-based in situ hybridization experiments are technically not suitable for quantitative evaluation (Fig. 1e, Supplementary Fig. 1).
MiR-1825 influences cell survival
As miR-1825 is downregulated in ALS, we tested whether miR-1825 has an impact on cellular survival. Therefore, HEK293 cells were transfected with synthetic RNA mimicking miR-1825 (mimic-1825) or a negative control RNA (mimic-ctrl.). Efficient transfection was confirmed by qRT-PCR and by transfection of fluorescently labeled mimic-ctrl. (Supplementary Fig. 2). As shown in Fig. 2a, transfection of mimic-1825 in HEK293 cells increased cellular survival under basal conditions as measured by the release of lactate dehydrogenase (LDH) into the culture media. Additionally, activity of CASP3/7 was reduced and the number of surviving cells was increased upon mimic-1825 transfection (Fig. 2b, c). Moreover, HEK293 cells transfected with mimic-1825 showed a slightly decreased CASP3/7 activity upon treatment with sorbitol that confers oxidative and osmotic stress. In contrast, no difference could be detected compared to mimic-control cells when sodium arsenite, an oxidative and metabolic stressor, was applied (Fig. 2d). However, both sorbitol and arsenite treatment led to increased CASP3/7 activity when endogenous miR-1825 was blocked by antimiR-1825 (Fig. 2d). Hence, overexpression of miR-1825 reduced cell death under basal culture conditions, and blockade of miR-1825, functionally corresponding to the downregulation observed in ALS patients, increased cellular vulnerability.
Fig. 2.
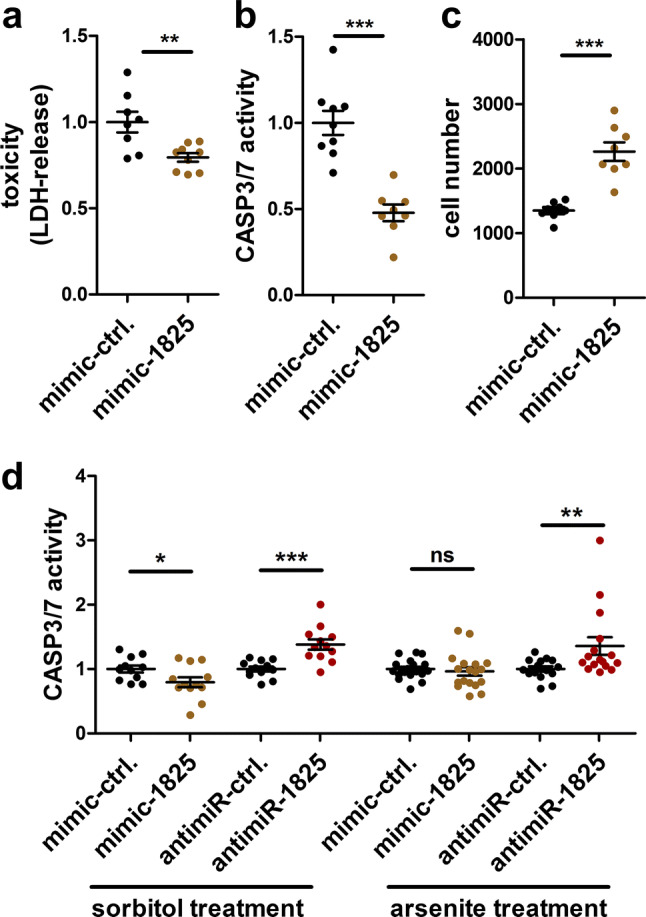
MiR-1825 modulates cell survival. a–c Lactate dehydrogenase (LDH) release (a), CASP3/7 activity (b) and cell number (c) were determined in HEK293 cells 72 h (a) or 48 h (b, c) after transfection of mimic-1825 or the respective control RNA (mimic-ctrl.) (n = 3 biological replicates with 2–3 technical replicates each). d CASP3/7 activity of HEK293 cells 48 h after transfection of mimic-1825, antimiR-1825 or respective control RNA and treatment with 400 mM sorbitol (n = 4 biological replicates with 2–3 technical replicates each) or 400 µM sodium arsenite (n = 6 biological replicates with 2–3 technical replicates each; bars indicate mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 in a two-tailed Student’s t test, ns not significant)
Identification of TBCB and CASP3 as direct targets of miR-1825
To identify the yet unknown mRNA targets of miR-1825, we used a combined proteomic and transcriptomic approach (Fig. 3a). 48 h post-transfection, the cells were first subjected to mass spectrometry using the SWATH methodology, which detected in total 1523 proteins (Supplementary Table 9). Transfection of mimic-1825 caused a significant reduction of eight proteins compared to mimic-ctrl. (fold change < 0.75; FDR < 0.05; Supplementary Table 10). The most strongly significantly downregulated proteins were tubulin-folding cofactor b (TBCB; downregulation 0.56-fold), the DNA-directed RNA polymerases I, II, and III subunit RPABC3 (RPAB3; 0.64-fold) and caspase-3 (CASP3; 0.65-fold). The downregulation of TBCB and CASP3 as observed by mass spectrometry could be confirmed in mimic-1825 transfected HEK293 cells by western blotting, while levels of RPAB3 did not change. Conversely, transfection of an antimiR-1825, which blocks endogenous miR-1825, led to an increase of both, TBCB and CASP3, but not RPAB3 protein (Fig. 3b, c). We thus validated the regulation of TBCB and CASP3 by miR-1825, while RPAB3 turned out to be a false positive hit of the mass spectrometric analysis. Notably, miRWalk2.0 [31] predicts potential binding sites of miR-1825 within TBCB and CASP3 mRNAs, but not in the mRNA of RPAB3.
Fig. 3.

Identification of miR-1825 targets by proteomic and transcriptomic profiling. a Experimental design for miR-1825 target identification. b Representative western blots of HEK293 cells used for densitometric analysis shown in c. c Densitometric quantification of western blots of HEK293 cells 48 h post-transfection with mimic-1825, antimiR-1825 or the corresponding control RNAs. Levels of TBCB (n = 5), CASP3 (n = 5) and RPAB3 (n = 7) were normalized to β-actin. d Hierarchical cluster analysis (average linkage) of all RNAs detected by microarray analysis of HEK293 cells transfected with mimic-1825 or the respective control RNA (mimic-ctrl.; n = 6 biological replicates per condition). e QRT-PCR validation of mRNA levels of proteins found to be downregulated (CASP3, TBCB, RPAB3), unaltered (DDX3, XPO7, PFN1) or upregulated (EDC3, DDX24, CPSF7) upon mimic-1825 transfection by mass spectrometry (n = 6 biological replicates). QRT-PCR measurements were carried out 24 h post-transfection and Ct values were normalized to TBP (bars in c and e indicate mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001 in a two-tailed Student’s t test; ns not significant)
To analyze possible effects of miR-1825 at the transcriptome level, RNA of HEK293 cells transfected for 24 h with mimic-1825 or mimic-ctrl. was investigated with Affymetrix GeneChip™ Human Gene 2.0 ST Arrays. None of the mRNAs detected by the microarray was significantly changed (FDR ≤ 0.05; Supplementary Table 11). Similarly, as shown in Fig. 3d, a hierarchical cluster analysis (average linkage) did not separate between the transcriptomes of HEK293 cells transfected with mimic-1825 or mimic-ctrl., respectively. We validated this result by qRT-PCR quantification of selected mRNAs coding for proteins found to be upregulated, downregulated or unchanged in the above mass spectrometry experiment (Supplementary Table 9). The levels of none of the mRNAs examined, including the TBCB and CASP3 mRNAs, changed upon mimic-1825 transfection (Fig. 3e). This indicates that miR-1825 regulates expression of its target genes by translational repression and not by mRNA degradation, as previously described for several other miRNAs [32–34].
To further confirm TBCB and CASP3 as miR-1825 targets, a biotinylated synthetic miR-1825 or biotinylated C. elegans control miRNA not present in humans (cel-mir-39-3p) was transfected in HEK293 cells, and precipitated from respective cell lysates using streptavidin beads 48 h post-transfection. RT-PCR showed an enrichment of TBCB and CASP3 mRNAs in the pulldown from cells transfected with miR-1825 compared to cel-mir-39-3p (Fig. 4a). This suggests a specific association of TBCB and CASP3 mRNAs with miR-1825 in cells. Additionally, we employed a luciferase reporter assay to confirm TBCB and CASP3 mRNAs as direct targets of miR-1825 and to identify the binding sites. According to miRWalk2.0 [31], CASP3 mRNA contains two predicted binding sites of miR-1825 in its 3′ untranslated region (UTR) while TBCB mRNA comprises one predicted binding site in the coding sequence (CDS). The putative miR-1825 binding sites as well as the complete 3′ UTR of TBCB were cloned in a reporter plasmid downstream of a firefly luciferase, which was co-transfected together with mimic-1825 or mimic-ctrl. in HEK293 cells. Binding site 1 of the CASP3 3′ UTR as well as the binding site in the CDS of TBCB significantly reduced the firefly luciferase signals (Fig. 4b). In contrast, binding site 2 of CASP3 and the complete 3′ UTR of TBCB did not alter the luciferase signal (Fig. 4b). Additionally, mutation of the two confirmed binding sites in CASP3 and TBCB mRNAs abolished the reduction of firefly luciferase by mimic-1825 (Fig. 4c, d). Conversely, inhibiting endogenous miR-1825 by antimiR-1825 increased the respective luciferase signals (Fig. 4e). We could thus validate TBCB and CASP3 mRNAs as direct targets of miR-1825, and confirmed the predicted miR-1825 binding sites in the CASP3 3′ UTR and the TBCB CDS. However, the effects of miR-1825 or antimiR-1825 on the translation of transfected firefly luciferase mRNA fused to CASP3 and TBCB miR-1825 binding sites are less pronounced than the effects on the endogenous mRNAs of CASP3 and TBCB. This suggests additional regulatory regions or structural elements in the endogenous mRNAs of CASP3 and TBCB mRNAs necessary for optimal function of miR-1825.
Fig. 4.

TBCB and CASP3 mRNA are direct targets of miR-1825. a RT-PCR of TBCB, CASP3 and GAPDH mRNA from HEK293 cells transfected with equal amounts of biotinylated miR-1825 or cel-mir-39-3p (input) and in respective streptavidin-pulldown samples. Four independent experiments showed similar results. b, d Relative luciferase activity in HEK293 cells co-transfected with the indicated luciferase reporter plasmids and with miR-1825 or the respective control RNA (mimic-ctrl.). Luciferase activities were normalized to cells transfected with the control vector (pmirGl.). c Sequence of wild-type or mutated miR-1825-binding sites (related to b, d, e) of the CASP3 3′ UTR or the coding sequence (CDS) of TBCB mRNA. e Relative luciferase activity detected in HEK293 cells co-transfected with antimiR-1825 or antimiR-ctrl. as well as with the indicated luciferase reporter plasmids (bars in b, d and e represent mean ± SEM; n = 4 biological replicates with 2–3 technical replicates each; *p < 0.05, **p < 0.01 in a two-tailed Student’s t test; ns not significant, bs binding site, CDS coding sequence, mut mutated, pmirGl. pmirGLO dual-luciferase miRNA target expression vector)
Increased TBCB levels cause depolymerization and reduced TUBA4A levels in HEK293 cells and cortical primary neurons
We showed that TBCB is increased upon decrease of miR-1825. TBCB promotes the folding of alpha-tubulins and is involved in the association/dissociation of alpha-/beta-tubulin heterodimers [35, 36]. As mutations in the alpha-tubulin TUBA4A have been shown to cause ALS [6], we hypothesized that increased TBCB protein expression might affect TUBA4A and/or tubulin dynamics in general. Immunoprecipitation of myc-tagged TBCB from lysates of HEK293 cells quantitatively precipitated TUBA4A demonstrating a close association of both proteins (Fig. 5a). Excess TBCB furthermore decreased the level of TUBA4A as determined by western blotting and quantification of fluorescence intensity in immunocytochemical staining of HEK293 cells as well as primary cortical mouse neurons (Fig. 5b–d). In contrast, the overall levels of alpha-tubulins and beta-tubulins, as determined with pan alpha- or beta-tubulin antibodies, respectively, remained unchanged (Supplementary Fig. 3). Similar to a previous study [6], HEK293 cells showed a less filamentous TUBA4A staining than primary neurons (Fig. 5c–e). Importantly, TBCB-myc overexpression in HEK293 cells (3.4-fold compared to endogenous TBCB levels, Fig. 5b) was comparable to the upregulation of TBCB in ALS post-mortem brain cortex tissue as shown below (fALS: 3.3-fold, sALS: 2.4-fold, Fig. 8b, c).
Fig. 5.

TBCB/TUBA4A interaction and downregulation of TUBA4A protein upon overexpression of TBCB or blocking of endogenous miR-1825. a Anti-myc immunoprecipitation from lysates of HEK293 cells transfected with TBCB-myc or the empty plasmid (pcDNA3) followed by western blotting. Three independent experiments showed similar results. b Densitometric TUBA4A western blot analysis of TBCB-myc and control plasmid (pcDNA3) transfected HEK293 cells (n = 12). Data were normalized to β-actin. A representative western blot is shown in the left panel. c Confocal microscopy images of HEK293 cells expressing TBCB-myc stained for TBCB-myc and TUBA4A using myc- and TUBA4A-specific antibodies, respectively. Mean intensity of TUBA4A immunofluorescence was quantified in TBCB-myc expressing (+) and non-expressing (−) cells on the same slide (n = 14 quantified images from two biological replicates). d Immunostaining of myc and TUBA4A in primary mouse neurons transfected with TBCB-myc and analysis of TUBA4A immunofluorescence intensity in TBCB-myc positive (+) and negative (−) neurons on the same slide (n = 13 quantified images from two biological replicates). e Fluorescence intensity measurement and representative confocal microscopy images of HEK293 cells transfected with antimiR-1825 or the respective control (n = 10 quantified images from two biological replicates) stained for TUBA4A. f Upper panel: representative western blot of HEK293 cells used for quantification shown in the lower panel. Lower panel: densitometric western blot measurement of TUBA4A level relative to β-actin in HEK293 cells transfected with mimic-1825, antimiR-1825 or the respective control RNAs (n = 5; bars in b–f show mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001 in a two-tailed Student’s t test; ns not significant, IP immunoprecipitation; scale bars in c–e represent 20 µm)
Fig. 8.

Increase of TBCB and decrease of TUBA4A protein in post-mortem cortex tissue of fALS and sALS patients. a Exemplary western blots of fALS patient and healthy control cortex tissue used for quantification in b. b, c Densitometric western blot analysis of TBCB, TUBA4A and CASP3 protein levels relative to β-actin in post-mortem cortex tissue from four fALS patients and matched healthy controls (b), as well as of 17 sALS patients and 16 matched healthy controls (c). c Western blots used for quantification shown in c are presented in Supplementary Fig. 6. d Correlation of TBCB and TUBA4A protein levels in post-mortem cortex tissue of 17 sALS patients. Pearson’s correlation coefficient r and p value are shown. Characteristics of ALS patients and controls used for western blot analyses are shown in Supplementary Tables 1 and 2 (bars in b and c represent mean ± SEM; *p < 0.05, **p < 0.01 in a two-tailed Student’s t test)
Furthermore, transfection of antimiR-1825 that causes a reduction of endogenous miR-1825 (see Supplementary Fig. 2b) also decreased TUBA4A levels in HEK293 cells (Fig. 5e, f). Confocal microscopy furthermore showed a less intense perinuclear and more diffuse staining of TUBA4A in cells overexpressing TBCB, while the subcellular distribution of (total) alpha- and beta-tubulins remained unchanged (Fig. 5c, Supplementary Fig. 3a, c). Stimulated emission depletion (STED) microscopy suggested that increased TBCB expression led to shorter TUBA4A skein-like structures in HEK293 cells (Fig. 6a–c). Biochemical fractionation of polymerized and depolymerized tubulins indeed revealed an increase of depolymerized TUBA4A upon overexpression of TBCB while the polymerization of total alpha- and beta-tubulins was not altered (Fig. 6d).
Fig. 6.

Pathologically increased TBCB induces depolymerization of TUBA4A. a Representative STED microscopy image of TBCB-myc transfected HEK293 cells labelled for TUBA4A (green) and TBCB-myc (red) (scale bar = 2 µm). b Magnified sections of TUBA4A architecture in TBCB-myc positive (+) or negative (−) cells (scale bar = 1 µm). c Length measurements of continuous TUBA4A skein-like structures in TBCB-myc expressing (+) and non-expressing (−) HEK293 cells (n = 23–25 quantified cells with three measurements each from seven independent experiments). d Representative western blot (left panel) and densitometric western blot analysis (right panel) of equal volumes of fractionated monomeric (m) and polymerized (p) TUBA4A, total alpha-tubulin and total beta-tubulin in TBCB-myc or control vector (pcDNA3) transfected cells (n = 7–8). Polymeric fractions were standardized to the respective monomeric fractions (bars in c and d represent mean ± SEM. **p < 0.01, ***p < 0.001 in a two-tailed Student’s t test)
To further address the possibility that TBCB, besides TUBA4A, influences also other alpha-tubulin isoforms, we re-analyzed the mass spectrometry results of our discovery experiment (Supplementary Table 9). Three alpha-tubulins other than TUBA4A, namely TUBA1A, TUBA1B and TUBA1C, were detected in HEK293 cells. Only the level of TUBA1A was slightly increased upon transfection of mimic-1825 in the mass spectrometry experiment (fold change = 1.34; uncorrected p = 0.0035; Supplementary Fig. 4a), while validation experiments could not confirm an effect of excess TBCB on the protein level or the polymerization of TUBA1A (Supplementary Fig. 4b, c). Therefore, we assume that the increase of TUBA1A may be a secondary effect of mimic-1825 transfection not related to the TBCB-induced effects on TUBA4A. Taken together, our data suggest that excess TBCB sequesters specifically TUBA4A from the microtubules and leads to its degradation.
Excess TBCB disturbs neuronal process morphology in vitro and in vivo
To test the hypothesis that increased TBCB has functional consequences in neurons, we overexpressed TBCB in cultured neurons and zebrafish. An increased number of neuronal processes were evident in primary cortical mouse neurons at 7 days in vitro (DIV7) after transfection with TBCB-myc at DIV2 (Fig. 7a, b), while later transfection of TBCB-myc at DIV5 did not result in significant neurite outgrowth alterations (Fig. 7b). In parallel, human TBCB was overexpressed in zebrafish embryos by injecting the respective mRNA at the one-cell stage. Zebrafish overexpressing human TBCB displayed no gross developmental phenotype (Fig. 7c). However, excess TBCB resulted in reduced levels of the zebrafish TUBA4A homolog, Tuba8l2 (Fig. 7d). Additionally, TBCB overexpressing zebrafish embryos showed motor neuron defects at 48 h post-injection including reduced motor neuron axon length and increased motor neuron branching compared to control injected embryos, reminiscent to the increased number of primary neurites in cultured mouse neurons transfected at DIV2 (Fig. 7e, f). Our data thus confirm the TBCB-dependent regulation of TUBA4A protein levels in vivo and suggest functional consequences of increased TBCB levels for the neuronal cytoskeleton in vitro and in vivo.
Fig. 7.

Excess TBCB causes neurite growth defects in primary cortical mouse neurons and a zebrafish model. a Representative confocal images of mouse primary cortical neurons transfected with TBCB-myc at 2 days in vitro (DIV) and stained for actin and TBCB-myc using Phalloidin and myc antibody, respectively (scale bar = 20 µm). b Quantification of primary neurites of TBCB-myc expressing (+) and non-expressing (−) primary cortical mouse neurons at DIV7. Neurons were transfected at DIV2 or DIV5. The number of neurons that were quantified is shown in green. c Representative images of zebrafish embryos injected with TBCB-myc mRNA or injection solution (control) at 48 h post-injection (scale bar = 500 µm). d Left panel: representative western blot used for quantification shown in right panel. Right panel: densitometric western blot analysis of zebrafish embryos at 48 h after injection with TBCB-myc mRNA or control injected (n = 4 biological replicates). Tuba8l2 levels were normalized to Gapdh. Additional western blots used for quantification in d are shown in Supplementary Fig. 5. e Confocal average intensity z-projection images of motor neurons stained with SV2 antibody in TBCB-myc mRNA injected and control injected zebrafish embryos at 48 h post-injection. Motor axon length and number of axonal branches were quantified in f as indicated in e (scale bar = 50 µm; numbers of zebrafish embryos used for quantification are indicated in green; bars in b, d and f represent mean ± SEM; *p < 0.05, **p < 0.01 in a two-tailed Student’s t test; ns not significant, transf. transfection, DIV days in vitro)
TBCB is increased and TUBA4A is decreased in the cortex of both fALS and sALS patients
We next aimed to corroborate the validity of our findings in ALS patients. To that end, we quantified the levels of TBCB, TUBA4A and CASP3 protein in post-mortem cortical tissue lysates from fALS (n = 4) and sALS (n = 17) patients as well as from matched control individuals by quantitative western blotting. FALS patients samples were derived from patients with a mutation in C9ORF72 (n = 3) or SOD1 (n = 1). Importantly, the brain cortex samples were derived from the same ALS patients and controls that have been already used to determine the levels of miR-1825 (Fig. 1a, b). We detected alterations with regard to the cytoskeleton-related proteins in agreement with our in vitro and in vivo findings above: a significant upregulation of TBCB protein was observed in the cortex of both fALS and sALS patients (fALS: p = 0.0163; sALS: p = 0.0138; Fig. 8b, c), concomitant with a reduction in TUBA4A levels (fALS: p = 0.0031; sALS: p = 0.0318; Fig. 8b, c). In addition, TBCB and TUBA4A protein levels were inversely correlated in post-mortem cortex tissue of sALS patients (r = − 0.726, p = 0.0295; Fig. 8d). CASP3 showed a trend towards higher levels in both fALS and sALS which, however, did not reach statistical significance (fALS: p = 0.170; sALS: p = 0.206; Fig. 8b, c). Taken together, dysregulation of the three components of the microRNA-1825/TBCB/TUBA4A axis could be confirmed in both sALS and fALS patient brains.
Discussion
In this work, we use in vitro, cell culture and in vivo experiments followed by result validation in human biomaterial to outline a novel ALS-associated molecular cascade (see synopsis in Table 1). We describe how an epigenetic change leads to regulation of the known ALS disease gene and microtubule component TUBA4A via TBCB in both sporadic and familial ALS patients. TBCB is one of five conserved tubulin-folding cofactors (TBCA to TBCE) regulating assembly and disassembly of alpha–beta-tubulin heterodimers [35, 36]. TBCB forms a physiological dimer with TBCE [37] that is associated with neurodegenerative diseases. A mutation in TBCE leads to a loss of protein expression and causes progressive motor neuropathy in mice [38, 39]. Moreover, hypomorphic TBCE mutations cause a mixed neurodevelopmental phenotype that includes a distal spinal muscular atrophy [40]. Therefore, both reduced TBCE function or expression and increased TBCB levels, as described in this study, are linked to neurodegeneration. This is in agreement with the observation that TBCB and TBCE function is directly dependent and mutually balanced by their cognate-binding partner [36]. While mutations in TBCE cause a juvenile neurodegenerative disease, upregulation of TBCB by miR-1825 is associated with adult-onset ALS. A trend towards downregulation of miR-1825 was already evident in pre-clinical ALS mutation carriers [15] and it remains to be determined if and how long downregulation of miR-1825 precedes clinical symptoms. Overall, TBCB and TBCE that regulate the composition of microtubules seem to form the core of a neurodegenerative disease pathway leading to either ALS or hereditary neuropathy when genetically or epigenetically altered (see overview in Fig. 9).
Table 1.
Summary of results obtained from different model organisms as well as validation in ALS patient-derived post-mortem CNS tissue
| Alteration | Effect | Model organism | Evident in | ||
|---|---|---|---|---|---|
| HEK293 | Primary neurons | Zebrafish embryos | Human post-mortem tissue | ||
| miR-1825 ↓ | TBCB ↑ | ✓ | n.a. | n.a. | ✓ |
| CASP3 ↑ | ✓ | – | |||
| miR-1825 ↓ | Cell toxicity ↑ | ✓ | n.a. | n.a. | n.a. |
| TBCB ↑ | TUBA4A ↓ | ✓ | ✓ | ✓ | ✓ |
| miR-1825 ↓ | TUBA4A ↓ | ✓ | n.a. | n.a. | ✓ |
| TBCB ↑ | Neuronal process defects | n.a. | ✓ | ✓ | n.a. |
n.a. not applicable (a miRNA homolog to miR-1825 does not exist in mice or zebrafish)
Fig. 9.

Model of TBCB and TBCE as core of a microtubule-disrupting pathway in neurodegenerative diseases. TBCB and TBCE form a heterodimer and regulate homeostasis of microtubules. A reduction of TBCE protein caused by genetic mutations or increased expression of TBCB, at least partially a consequence of miR-1825 downregulation, ultimately lead to neurodegeneration
The initial observation of reduced blood miR-1825 levels in ALS patients [14, 15, 30] could be extended to ALS patient brain tissue and also extra-CNS organs. Results of miRNA expression studies from patient material are often poorly reproducible and to a large extent dependent on the technical platforms and protocols used [41]. Nevertheless, the ALS-associated downregulation of miR-1825 turned out to be robust. It is consistently observed in data obtained by miRNA screening and qRT-PCR validation experiments from at least three laboratories using different methodologies, patient cohorts and sample types [14, 15, 30, 42]. Regarding that miRNAs may either be actively secreted or passively released by cells [43], our data indicate similar intra- and extracellular alterations of miR-1825. Therefore, extracellular downregulation of miR-1825 may be rather a reflection of intracellular downregulation than indicative for miRNA secretion defects in ALS. Concomitant with the downregulation of miR-1825, we found an increase of pre-mir-1825, most likely reflecting defective nuclear export and/or processing in ALS. This is in line with a previous study demonstrating increased pre-miRNAs levels due to DICER1 defects in ALS as a consequence of mutations in several different genes [44]. Nevertheless, it remains to be determined whether defects in DICER1 function are responsible for the downregulation specifically of miR-1825. Alternatively, mutations in major ALS genes such as C9orf72 [45] or SOD1 [46] as well as the cytoplasmic aggregation of TDP-43 [47] have been shown to impair nucleocytoplasmic transport putatively leading to the accumulation of pre-mirs in the nucleus and consequently lower levels of mature miRNAs in the cytoplasm. However, detailed analyses of the biogenesis of miR-1825 are needed to identify mechanisms resulting in the ALS-related downregulation of specifically miR-1825. Nevertheless, this miR-1825 dysregulation represents another example of systemic pathomolecular changes observed in this neurodegenerative disease [14, 15, 48].
We also show that miR-1825 downregulates the expression of CASP3, besides TBCB, at the translational level. Both target genes could principally be involved in the increased cellular susceptibility and neurodegeneration associated with experimental miR-1825 blockade or miR-1825 downregulation in patients, respectively. CASP3 represents one of the most extensively characterized cell death executors. Elevated levels of proteolytically activated CASP3 have been found in different mutant SOD1 mouse strains [49]. However, evidence for a role of activated CASP3 in ALS patients is scarce and controversially discussed. Activated CASP3 positive neurons were not found to be more frequent in ALS patient post-mortem material compared to controls [50, 51] and enzymatic CASP3 activity was either unchanged, elevated or reduced in ALS patient tissue [52]. This casts doubt on the role of CASP3 as a primary upstream player in ALS pathogenesis. Moreover, CASP3 does not display a concentration-dependent auto-activation [53]. Consequently, alterations of cytoskeletal architecture rather than the increase in full-length CASP3 protein, observed upon blockade of miR-1825, are probably responsible for increased CASP3 activity, which could be a secondary event. Overall, current evidence does not solidly support CASP3 as the main link between reduced miR-1825 and motor neuron degeneration. In contrast, the top hit in our screen for genes upregulated by reduced miR-1825, TBCB, is a protein/protein-binding partner of the known ALS gene product TUBA4A [6, 54]. TBCB sequesters TUBA4A (this work) but also the ALS-associated dynactin subunit p150 from microtubules [55–57]. We showed that TBCB upregulation led to depolymerization and downregulation specifically of TUBA4A without affecting other tubulins. In line with our results, excess TBCB was found to cause microtubule destruction in HeLa cells using a TUBA4A-specific antibody [37]. Functional effects of increased TBCB levels on TUBA4A depolymerization are thus similar to those of ALS-causing genetic mutation of TUBA4A [6]. It is important to emphasize that in our HEK293 cell model overexpression of TBCB-myc (3.4-fold compared to endogenous TBCB levels) as well as increase of endogenous TBCB upon antimiR-1825 transfection (1.5-fold) are not exceeding but are comparable to the upregulation of TBCB in ALS post-mortem brain cortex tissue (fALS: 3.3-fold, sALS: 2.2-fold).
We furthermore showed that TBCB upregulation resulted in axonal pathology in vivo. In accordance with our results, TBCB overexpression leads to growth cone retraction in mouse/rat neuronal cell line while knockdown of TBCB caused longer axons [58]. Importantly, dysregulation of all of the three main components of the miR-1825/TBCB/TUBA4A cascade was successfully validated in ALS patient brain tissue.
The discovery of TBCB as a major target gene of miR-1825 fits generally well with the role of the cytoskeleton in ALS pathogenesis, as evidenced by experimental and human genetic data [4, 5, 59, 60]. Moreover, we demonstrate that TUBA4A can be involved in ALS not only by one of the rare genetic mutations but also by reduced expression of an upstream miRNA. Our findings underline the relevance specifically of TUBA4A for ALS pathogenesis independent of the various genetic factors involved in ALS.
After confirmation in additional, independent cohorts of fALS and sALS patients, the miR-1825/TBCB/TUBA4A axis might turn out to be of broad therapeutic relevance. The ALS field witnessed a vast therapeutic development failure and poor predictive value of the transgenic mutant SOD1 model with regard to success in clinical trials in the past 25 years. This could reflect that monogenic forms of the disease are not representative for the majority of usually sporadic ALS cases. We extend the role of TUBA4A beyond rare familial ALS cases and outline a pathological regulatory pathway from miR-1825 dysregulation to changes in microtubule composition that is shared by genetic and the majority of sporadic forms of ALS. Besides cytoplasmic aggregation of phosphorylated TDP-43, downregulation of miR-1825 and the resulting downstream effects described here belong to the rare known common pathomolecular denominators of sALS and fALS.
We thus discovered a regulatory axis composed of several potential therapeutic target molecules, which could turn out to be relevant for a large proportion of ALS cases and adds important mechanistic information about ALS pathogenesis. Dysregulation of the three main components of this axis was confirmed in post-mortem ALS patient tissue. The core of this molecular cascade involving TBCB/TBCE and TUBA4A is now genetically and epigenetically linked to at least two neurodegenerative diseases, ALS and a neurodevelopmental disorder that also includes spinal muscular atrophy. Finally, our results could be the basis for the development of innovative disease models that are representative also for sporadic ALS in addition to genetic forms of the disease.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We are indebted to the patients and families participating in this study. We are also grateful to Elena Jasovski, Ramona Bück, Nadine Todt and Aline Sage for their excellent technical assistance. We also thank Lüder-Hinrich Meyer (Ulm University) for providing the dual-luciferase miRNA reporter plasmid and Jasmin Breymayer and Angelika Rück of the core facility confocal and multi-photon microscopy in Ulm for their help and technical advice. This work was supported in whole or in parts by grants from the German Federal Ministry of Education and Research (STRENGTH consortium/BMBF, 01GI0704; German network for ALS research (MND-NET)), the Charcot Foundation for ALS Research (ACL, JHW), and the DFG-funded Swabian ALS Registry.
Abbreviations
- ALS
Amyotrophic lateral sclerosis
- C9orf72
Chromosome 9 open reading frame 72
- CASP3
Caspase-3
- CNS
Central nervous system
- DIV
Days in vitro
- fALS
Familial ALS
- miRNA
MicroRNA
- miR-1825
MicroRNA-1825
- POFUT1
Protein O-fucosyltransferase
- pre-miRNA
Precursor microRNA
- pri-miRNA
Primary microRNA
- sALS
Sporadic ALS
- TBCB
Tubulin-folding cofactor b
- TBCE
Tubulin-folding cofactor e
- TDP-43
Transactive response DNA-binding protein 43
- TUBA4A
Tubulin alpha-4A
Author contributions
AMH, SJB, JR, DD, AF and KH performed experiments and analyzed data. AMH, AF and JHW designed and supervised the study and interpreted the results. PMA, SP and DRT provided post-mortem tissue samples. JM, MO, ACL, DB, SJ and KMD helped conducting the study and provided critical input for scientific interpretations. AMH, AF and JHW wrote the manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Axel Freischmidt and Jochen H. Weishaupt: Shared authorship.
References
- 1.Rosenbohm A, Peter RS, Erhardt S, et al. Epidemiology of amyotrophic lateral sclerosis in Southern Germany. J Neurol. 2017;264(4):749–757. doi: 10.1007/s00415-017-8413-3. [DOI] [PubMed] [Google Scholar]
- 2.Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nature reviews. Neurology. 2011;7(11):603–615. doi: 10.1038/nrneurol.2011.150. [DOI] [PubMed] [Google Scholar]
- 3.Weishaupt JH, Hyman T, Dikic I. Common molecular pathways in amyotrophic lateral sclerosis and frontotemporal dementia. Trends Mol Med. 2016;22(9):769–783. doi: 10.1016/j.molmed.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 4.Wu C-H, Fallini C, Ticozzi N, et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. 2012;488(7412):499–503. doi: 10.1038/nature11280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puls I, Jonnakuty C, LaMonte BH, et al. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33(4):455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- 6.Smith BN, Ticozzi N, Fallini C, et al. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron. 2014;84(2):324–331. doi: 10.1016/j.neuron.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (New York, NY) 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 8.Kabashi E, Valdmanis PN, Dion P, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40(5):572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 9.Freischmidt A, Muller K, Ludolph AC, et al. Systemic dysregulation of TDP-43 binding microRNAs in amyotrophic lateral sclerosis. Acta Neuropathol Commun. 2013;1:42. doi: 10.1186/2051-5960-1-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawahara Y, Mieda-Sato A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc Natl Acad Sci USA. 2012;109(9):3347–3352. doi: 10.1073/pnas.1112427109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis BP, I-h Shih, Jones-Rhoades MW, et al. Prediction of mammalian microRNA targets. Cell. 2003;115(7):787–798. doi: 10.1016/S0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 12.Lim LP, Lau NC, Garrett-Engele P, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433(7027):769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 13.Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 14.Freischmidt Axel, Müller Kathrin, Zondler Lisa, Weydt Patrick, Mayer Benjamin, von Arnim Christine A.F., Hübers Annemarie, Dorst Johannes, Otto Markus, Holzmann Karlheinz, Ludolph Albert C., Danzer Karin M., Weishaupt Jochen H. Serum microRNAs in sporadic amyotrophic lateral sclerosis. Neurobiology of Aging. 2015;36(9):2660.e15-2660.e20. doi: 10.1016/j.neurobiolaging.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 15.Freischmidt A, Muller K, Zondler L, et al. Serum microRNAs in patients with genetic amyotrophic lateral sclerosis and pre-manifest mutation carriers. Brain J Neurol. 2014;137(Pt 11):2938–2950. doi: 10.1093/brain/awu249. [DOI] [PubMed] [Google Scholar]
- 16.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Es MA, Dahlberg C, Birve A, et al. Large-scale SOD1 mutation screening provides evidence for genetic heterogeneity in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2010;81(5):562–566. doi: 10.1136/jnnp.2009.181453. [DOI] [PubMed] [Google Scholar]
- 18.Feiler MS, Strobel B, Freischmidt A, et al. TDP-43 is intercellularly transmitted across axon terminals. J Cell Biol. 2015;211(4):897–911. doi: 10.1083/jcb.201504057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fischer R, Kessler BM. Gel-aided sample preparation (GASP)—a simplified method for gel-assisted proteomic sample generation from protein extracts and intact cells. Proteomics. 2015;15(7):1224–1229. doi: 10.1002/pmic.201400436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwarzfischer P, Reinders J, Dettmer K, et al. Comprehensive metaboproteomics of Burkitt’s and diffuse large B-cell lymphoma cell lines and primary tumor tissues reveals distinct differences in pyruvate content and metabolism. J Proteome Res. 2017;16(3):1105–1120. doi: 10.1021/acs.jproteome.6b00164. [DOI] [PubMed] [Google Scholar]
- 21.Helferich AM, Ruf WP, Grozdanov V, et al. α-synuclein interacts with SOD1 and promotes its oligomerization. Mol Neurodegener. 2015;10:66. doi: 10.1186/s13024-015-0062-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics (Oxford, England) 2003;4(2):249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 23.Neuss S, Holzmann K, Speit G. Gene expression changes in primary human nasal epithelial cells exposed to formaldehyde in vitro. Toxicol Lett. 2010;198(2):289–295. doi: 10.1016/j.toxlet.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 24.Bayer H, Lang K, Buck E, et al. ALS-causing mutations differentially affect PGC-1α expression and function in the brain vs. peripheral tissues. Neurobiol Dis. 2017;97((Pt A)):36–45. doi: 10.1016/j.nbd.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 25.Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics (Oxford, England) 2002;18(1):207–208. doi: 10.1093/bioinformatics/18.1.207. [DOI] [PubMed] [Google Scholar]
- 26.Freischmidt A, Schöpflin M, Feiler MS, et al. Profilin 1 with the amyotrophic lateral sclerosis associated mutation T109M displays unaltered actin binding and does not affect the actin cytoskeleton. BMC Neurosci. 2015;16:77. doi: 10.1186/s12868-015-0214-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Osseforth C, Moffitt JR, Schermelleh L, et al. Simultaneous dual-color 3D STED microscopy. Opt Express. 2014;22(6):7028–7039. doi: 10.1364/OE.22.007028. [DOI] [PubMed] [Google Scholar]
- 28.Gundersen GG, Khawaja S, Bulinski JC. Postpolymerization detyrosination of alpha-tubulin: a mechanism for subcellular differentiation of microtubules. J Cell Biol. 1987;105(1):251–264. doi: 10.1083/jcb.105.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Just S, Meder B, Berger IM, et al. The myosin-interacting protein SMYD1 is essential for sarcomere organization. J Cell Sci. 2011;124(Pt 18):3127–3136. doi: 10.1242/jcs.084772. [DOI] [PubMed] [Google Scholar]
- 30.Takahashi I, Hama Y, Matsushima M, et al. Identification of plasma microRNAs as a biomarker of sporadic amyotrophic lateral sclerosis. Mol Brain. 2015;8(1):67. doi: 10.1186/s13041-015-0161-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dweep H, Gretz N. miRWalk2.0: a comprehensive atlas of microRNA–target interactions. Nat Methods. 2015;12(8):697. doi: 10.1038/nmeth.3485. [DOI] [PubMed] [Google Scholar]
- 32.Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75(5):855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- 33.Pillai RS, Bhattacharyya SN, Artus CG, et al. Inhibition of translational initiation by let-7 microRNA in human cells. Science (New York, NY) 2005;309(5740):1573–1576. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
- 34.Brennecke J, Hipfner DR, Stark A, et al. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003;113(1):25–36. doi: 10.1016/S0092-8674(03)00231-9. [DOI] [PubMed] [Google Scholar]
- 35.Lewis SA, Tian G, Cowan NJ. The alpha- and beta-tubulin folding pathways. Trends Cell Biol. 1997;7(12):479–484. doi: 10.1016/S0962-8924(97)01168-9. [DOI] [PubMed] [Google Scholar]
- 36.Serna M, Carranza G, Martin-Benito J, et al. The structure of the complex between alpha-tubulin, TBCE and TBCB reveals a tubulin dimer dissociation mechanism. J Cell Sci. 2015;128(9):1824–1834. doi: 10.1242/jcs.167387. [DOI] [PubMed] [Google Scholar]
- 37.Kortazar D, Fanarraga ML, Carranza G, et al. Role of cofactors B (TBCB) and E (TBCE) in tubulin heterodimer dissociation. Exp Cell Res. 2007;313(3):425–436. doi: 10.1016/j.yexcr.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 38.Schäfer MK, Bellouze S, Jacquier A, et al. Sensory neuropathy in progressive motor neuronopathy (pmn) mice is associated with defects in microtubule polymerization and axonal transport. Brain Pathol (Zurich, Switz) 2017;27(4):459–471. doi: 10.1111/bpa.12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin N, Jaubert J, Gounon P, et al. A missense mutation in Tbce causes progressive motor neuronopathy in mice. Nat Genet. 2002;32(3):443–447. doi: 10.1038/ng1016. [DOI] [PubMed] [Google Scholar]
- 40.Sferra A, Baillat G, Rizza T, et al. TBCE mutations cause early-onset progressive encephalopathy with distal spinal muscular atrophy. Am J Hum Genet. 2016;99(4):974–983. doi: 10.1016/j.ajhg.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ono S, Lam S, Nagahara M, et al. Circulating microRNA biomarkers as liquid biopsy for cancer patients: pros and cons of current assays. J Clin Med. 2015;4(10):1890–1907. doi: 10.3390/jcm4101890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Butovsky O, Jedrychowski MP, Cialic R, et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann Neurol. 2015;77(1):75–99. doi: 10.1002/ana.24304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen X, Liang H, Zhang J, et al. Secreted microRNAs: a new form of intercellular communication. Trends Cell Biol. 2012;22(3):125–132. doi: 10.1016/j.tcb.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 44.Emde A, Eitan C, Liou L-L, et al. Dysregulated miRNA biogenesis downstream of cellular stress and ALS-causing mutations: a new mechanism for ALS. EMBO J. 2015;34(21):2633–2651. doi: 10.15252/embj.201490493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang K, Donnelly CJ, Haeusler AR, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525(7567):56–61. doi: 10.1038/nature14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang J, Ito H, Wate R, et al. Altered distributions of nucleocytoplasmic transport-related proteins in the spinal cord of a mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. 2006;112(6):673–680. doi: 10.1007/s00401-006-0130-4. [DOI] [PubMed] [Google Scholar]
- 47.Chou C-C, Zhang Y, Umoh ME, et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci. 2018;21(2):228–239. doi: 10.1038/s41593-017-0047-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kolde G, Bachus R, Ludolph AC. Skin involvement in amyotrophic lateral sclerosis. Lancet (Lond, Engl) 1996;347(9010):1226–1227. doi: 10.1016/S0140-6736(96)90737-0. [DOI] [PubMed] [Google Scholar]
- 49.Pasinelli P, Houseweart MK, Brown RH, Jr, et al. Caspase-1 and -3 are sequentially activated in motor neuron death in Cu, Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2000;97(25):13901–13906. doi: 10.1073/pnas.240305897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Embacher N, Kaufmann WA, Beer R, et al. Apoptosis signals in sporadic amyotrophic lateral sclerosis: an immunocytochemical study. Acta Neuropathol. 2001;102(5):426–434. doi: 10.1007/s004010100438. [DOI] [PubMed] [Google Scholar]
- 51.Yamazaki M, Esumi E, Nakano I. Is motoneuronal cell death in amyotrophic lateral sclerosis apoptosis? Neuropathol Off J Jpn Soc Neuropathol. 2005;25(4):381–387. doi: 10.1111/j.1440-1789.2005.00648.x. [DOI] [PubMed] [Google Scholar]
- 52.Martin LJ. Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol. 1999;58(5):459–471. doi: 10.1097/00005072-199905000-00005. [DOI] [PubMed] [Google Scholar]
- 53.Colussi PA, Harvey NL, Shearwin-Whyatt LM, et al. Conversion of procaspase-3 to an autoactivating caspase by fusion to the caspase-2 prodomain. J Biol Chem. 1998;273(41):26566–26570. doi: 10.1074/jbc.273.41.26566. [DOI] [PubMed] [Google Scholar]
- 54.Huttlin EL, Bruckner RJ, Paulo JA, et al. Architecture of the human interactome defines protein communities and disease networks. Nature. 2017;545(7655):505–509. doi: 10.1038/nature22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuh GF, Stockmann M, Meyer-Ohlendorf M, et al. Tubulin-binding cofactor B is a direct interaction partner of the dynactin subunit p150(Glued) Cell Tissue Res. 2012;350(1):13–26. doi: 10.1007/s00441-012-1463-z. [DOI] [PubMed] [Google Scholar]
- 56.Stockmann M, Meyer-Ohlendorf M, Achberger K, et al. The dynactin p150 subunit: cell biology studies of sequence changes found in ALS/MND and Parkinsonian syndromes. J Neural Transm (Vienna, Austria: 1996) 2013;120(5):785–798. doi: 10.1007/s00702-012-0910-z. [DOI] [PubMed] [Google Scholar]
- 57.Munch C, Sedlmeier R, Meyer T, et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology. 2004;63(4):724–726. doi: 10.1212/01.WNL.0000134608.83927.B1. [DOI] [PubMed] [Google Scholar]
- 58.Lopez-Fanarraga M, Carranza G, Bellido J, et al. Tubulin cofactor B plays a role in the neuronal growth cone. J Neurochem. 2007;100(6):1680–1687. doi: 10.1111/j.1471-4159.2006.04328.x. [DOI] [PubMed] [Google Scholar]
- 59.Figlewicz DA, Krizus A, Martinoli MG, et al. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum Mol Genet. 1994;3(10):1757–1761. doi: 10.1093/hmg/3.10.1757. [DOI] [PubMed] [Google Scholar]
- 60.Al-Chalabi A, Andersen PM, Nilsson P, et al. Deletions of the heavy neurofilament subunit tail in amyotrophic lateral sclerosis. Hum Mol Genet. 1999;8(2):157–164. doi: 10.1093/hmg/8.2.157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.