

Abstract
‘A disintegrin and metalloproteases’ (ADAMs) are a family of transmembrane proteins with diverse functions in multicellular organisms. About half of the ADAMs are active metalloproteases and cleave numerous cell surface proteins, including growth factors, receptors, cytokines and cell adhesion proteins. The other ADAMs have no catalytic activity and function as adhesion proteins or receptors. Some ADAMs are ubiquitously expressed, others are expressed tissue specifically. This review highlights functions of ADAMs in the mammalian nervous system, including their links to diseases. The non-proteolytic ADAM11, ADAM22 and ADAM23 have key functions in neural development, myelination and synaptic transmission and are linked to epilepsy. Among the proteolytic ADAMs, ADAM10 is the best characterized one due to its substrates Notch and amyloid precursor protein, where cleavage is required for nervous system development or linked to Alzheimer’s disease (AD), respectively. Recent work demonstrates that ADAM10 has additional substrates and functions in the nervous system and its substrate selectivity may be regulated by tetraspanins. New roles for other proteolytic ADAMs in the nervous system are also emerging. For example, ADAM8 and ADAM17 are involved in neuroinflammation. ADAM17 additionally regulates neurite outgrowth and myelination and its activity is controlled by iRhoms. ADAM19 and ADAM21 function in regenerative processes upon neuronal injury. Several ADAMs, including ADAM9, ADAM10, ADAM15 and ADAM30, are potential drug targets for AD. Taken together, this review summarizes recent progress concerning substrates and functions of ADAMs in the nervous system and their use as drug targets for neurological and psychiatric diseases.
Keywords: ADAM, Ectodomain shedding, Metalloprotease, Neural development, Neurological disease
Introduction
The nervous system serves as the control center of an organism to detect, interpret, and respond to external or internal changes. These processes involve cell–cell communication as well as intra- and intercellular signaling events, which require proteins expressed on the cell surface to carry out their functions at the front lines. This includes the ADAM (a disintegrin and metalloprotease) protein family, which is a group of type I transmembrane proteins that are present across different species. They are characterized by the presence of a metalloprotease domain for their potential protease activity to cleave membrane proteins at their extracellular juxtamembrane region. This process, which is referred to as “protein ectodomain shedding”, can activate or inactivate the substrate protein, or initiate further cleavage of the substrate protein at or within the transmembrane region by intramembrane proteases, such as γ-secretase, and lead to various functional consequences (reviewed in Ref. [1]). Around 30 ADAM family members are identified in mouse and human (Table 1) with almost half of them being specifically or predominantly expressed in testis or epididymis, indicating an important role of these ADAMs in mammalian reproduction and fertilization (reviewed in Ref. [2]). Other ADAMs are expressed in a wide variety of cells and tissues and are involved in various biological processes, including development, inflammation, and cancer (reviewed in Refs. [3, 4]). Importantly, many of these ADAMs are highly expressed in the nervous system with slightly different spatial or temporal expression profiles, suggesting their important, yet diverse roles in the functions of the nervous system.
Table 1.
ADAMs
| Gene name | Gene synonyms | UniProt accession number a | Metalloprotease activityb | Detected in the mammalian nervous system | |
|---|---|---|---|---|---|
| Mouse | Human | ||||
| Adam1 | Adam1a, Ftna | Q60813 | n/a | + | + [259] |
| Adam2 | Ftnb | Q60718 | Q99965 | − | + [259, 260] |
| Adam3 | Cyrn1 | F8VQ03 | n/a | − | − |
| Adam4 | Q8CGQ2 | n/a | − | − | |
| Adam5 | Tmdc2, ADAM5P, TMDC2 | Q3TTE0 | Q6NVV9 | − | + [261] |
| Adam6 | Q32NZ3 | n/a | − | − | |
| Adam7 | GP83 | O35227 | Q9H2U9 | − | + [262] |
| Adam8 | Ms2, CD156 | Q05910 | P78325 | + | + [73] |
| Adam9 | Kiaa0021, Mdc9, Mltng, Mcmp | Q61072 | Q13443 | + | + [82] |
| Adam10 | Kuz, Madm | O35598 | O14672 | + | + [263] |
| Adam11 | Mdc | Q9R1V4 | O75078 | − | + [15, 17] |
| Adam12 | Mltna | Q61824 | O43184 | + | + [162, 235] |
| Adam13 | Not expressed in mammals | + | − | ||
| Adam14 | Not expressed in mammals | − | − | ||
| Adam15 | Mdc15 | O88839 | Q13444 | + | + [86] |
| Adam16 | Mdc16 | Not expressed in mammals | + | − | |
| Adam17 | CSVP, TACE, CD156b | P78536 | Q9Z0F8 | + | + [102, 169] |
| Adam18 | Adam27, Dtgn3, Tmdc3 | Q9R157 | Q9Y3Q7 | − | − |
| Adam19 | Mltnb,FKSG34 | O35674 | Q9H013 | + | + [235] |
| Adam20 | n/a | O43506 | + | − | |
| Adam21 | Adam31 | Q9JI76 | Q9UKJ8 | + | + [245] |
| Adam22 | Mdc2 | Q9R1V6 | Q9P0K1 | − | + [15] |
| Adam23 | Mdc3 | Q9R1V7 | O75077 | − | + [15] |
| Adam24 | Q9R160 | n/a | + | − | |
| Adam25 | Q9R159 | n/a | + | − | |
| Adam26 | Adam26a | Q9R158 | n/a | + | − |
| Adam27 | Adam18, Dtgn3, Tmdc3 | Q9R157 | Q9Y3Q7 | − | − |
| Adam28 | Mdcl | Q9JLN6 | Q9UKQ2 | + | − |
| Adam29 | Q811Q4 | Q9UKF5 | − | − | |
| Adam30 | n/a | Q9UKF2 | + | + [247] | |
| Adam31 | Adam21 | Q9JI76 | Q9UKJ8 | − | + [245] |
| Adam32 | Q8K410 | Q8TC27 | − | − | |
| Adam33 | Q923W9 | Q9BZ11 | + | + [253] | |
ADAMs that are discussed in this review are indicated in bold
aSwiss-Prot reviewed only
bBased on the presence of metalloprotease active site consensus sequence: HEXGHXXGXXHD
ADAM family proteins share a common multi-domain structure comprising an N-terminal signal peptide, a prodomain, followed by metalloprotease, disintegrin, cysteine-rich, transmembrane, and cytoplasmic domains (Fig. 1). Some family members contain an additional epidermal growth factor (EGF)-like domain between the cysteine-rich and the transmembrane domain. The consensus sequence in the metalloprotease domain that defines an active ADAM protease is HEXGHXXGXXHD, in which histidines bind zinc and glutamic acid helps in the catalytic process [5]. Some members of the ADAM family, such as ADAM11, ADAM22, and ADAM23, do not have the consensus sequence in their metalloprotease domain and are thus inactive ADAMs that are primarily involved in direct cell–cell signaling by binding to other proteins. The cytoplasmic domain of ADAMs, which is relatively short as compared to the extracellular domain, specifies binding sites for various intracellular signal transducing proteins [6].
Fig. 1.

Typical domain structure of ADAM proteins. The region within the catalytic region essential for active ADAMs is highlighted. Note that the EGF domain is not found in all ADAMs, e.g., in ADAM10
The prodomains may help in protein folding upon protein synthesis and have an autoinhibitory effect on the catalytically active members of the ADAM family [7], ensuring that they are not active in the early secretory pathway. Furin and furin-like proprotein convertase 7 (PC7) are considered as two major enzymes that cleave the prodomains of ADAMs during their transit through the secretory pathway, thus regulating their maturation and proteolytic activity. As an exception, ADAM8 is able to release its prodomain by an autocatalytic mechanism [8]. Interestingly, an alternative autocatalytic activity was also reported for ADAM10 and ADAM17. However, this does not involve the prodomain removal and protease activation under physiological conditions, but causes degradation of the mature catalytically active forms during cell lysis in experimental settings [9, 10].
In this review, we focus on those ADAMs that are expressed in the nervous system. We exclude the ones with a predominant expression and function in testis or epididymis, which have been reviewed recently [2]. We will summarize the expression patterns and discuss the known physiological as well as pathological functions of the ADAMs in the nervous system (Fig. 2). Many of these ADAMs are also expressed outside the nervous system, where their function and regulation have been covered by other excellent reviews [11–14]. We will also discuss implications for developing potential therapies that target ADAMs in neurological disorders. We start with the discussion of the non-proteolytic ADAMs ADAM11, 22, and 33. Afterwards, we describe the proteolytic ADAMs.
Fig. 2.

Overview of ADAMs functions in the nervous system. ADAM proteins play diverse roles in the mammalian nervous system. Relevant ADAMs in the regulation of indicated physiological or pathological conditions are listed. Some functions have been well established either through in vivo studies or genetics. Other functions are more suggestive to date. For details, see the corresponding paragraph on each ADAM family member.
The pictures were modified from https://smart.servier.com
Non-proteolytic ADAMs
ADAM11, ADAM22 and ADAM23: a major function in nervous system development and epilepsy
ADAM11, ADAM22 and ADAM23 are catalytically inactive ADAM family members, which lack the consensus sequence HEXGHXXGXXHD in their metalloprotease domain and are predominantly expressed in brain [15]. They act as receptors for leucine-rich glioma-inactivated (LGI) protein family members (summarized in Kegel [16]). All four LGIs (LGI1-4) are neuronal proteins that are secreted, e.g., into the synaptic cleft (Fig. 3). The interaction of LGIs, in particular LGI1 and 4 with inactive ADAMs, which is well understood for ADAM22 and ADAM23, is involved in key processes in the brain, such as myelination and synaptic transmission, and diseases, such as epilepsy. The interaction of ADAMs with LGI2/LGI3 is less understood and remains to be elucidated.
Fig. 3.
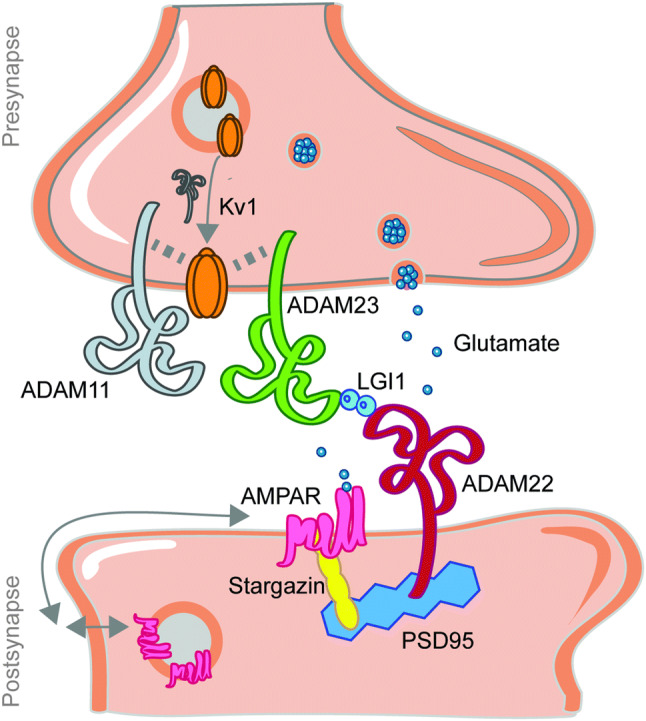
The function of inactive ADAM11, 22 and 23 in the nervous system. Presynaptic ADAM11 and ADAM23 regulate Kv1 channels. ADAM23 binds LGI1, bridging the synaptic cleft to ADAM22. Postsynaptic ADAM22 interacts extracellularly with LGI1 and intracellularly with PSD-95. This interaction is important for synapse maturation and regulates AMPA receptors (AMPAR)
ADAM11—trafficking of potassium channels
ADAM11, also known as MDC (metalloproteinase-like, disintegrin-like, and cysteine-rich protein), is highly expressed in neurons of the peripheral nervous system (PNS) and the central nervous system (CNS) during development and in adult mice. High expression was found in the spinal cord, hippocampus (pyramidal neurons of CA1–CA3 and granular cells of the dentate gyrus) and the cerebellum (granular cells) [17]. In the cerebellum, ADAM11 plays an essential role in synaptic transmission by controlling the trafficking of Kv1-potassium ion channels to the presynaptic side of a special type of synapse, termed pinceau [18]. In this structure, cerebellar basket cells form an unusual type of synapse onto neighboring Purkinje cells, allowing inhibitory synaptic transmission through the neurotransmitter GABA and additional ultrarapid electrical signaling [19]. The Kv1 channel family members Kv1.1 and Kv1.2 cluster in the presynaptic membrane of basket cell axons (Fig. 3), but are absent from the synapse in mice expressing a truncated, non-functional mutant of ADAM11 (ADAM11Δ12−18)[18]. While this deficit did not alter GABA release, it prevented the ultrarapid electrical signaling. Additionally, postsynaptic changes were observed in the mutant mice, such as reduced levels of ADAM22 and postsynaptic density protein-95 (PSD-95). Importantly, ADAM11 was shown to interact with Kv1.1 and Kv1.2 upon co-immunoprecipitation from whole-brain lysates, suggesting that ADAM11 targets, anchors or stabilizes Kv1 channels to the presynaptic membrane in the pinceau structure. Further evidence for the functional interaction between ADAM11 and Kv1 channels comes from the following observation. ADAM11Δ12−18 mutant mice show ataxia and tremor upon stress, such as a cold water swim [18], which is reminiscent of the phenotype of Kv1.1- and Kv1.2-deficient mice [20, 21]. Taken together, ADAM11 is the first protein known to be essential for potassium channel trafficking in cerebellar pinceaux.
In general, ADAM11-deficient mice are viable, fertile and develop normally [22, 23], but show additional phenotypes beyond the stress-induced phenotypes. For example, ADAM11-deficient mice have defects in learning and motor coordination [23], reflecting the high expression of ADAM11 in hippocampus and cerebellum [17]. Moreover, ADAM11-deficient mice revealed abnormalities in pain transmission [22], but the underlying molecular mechanisms remain to be elucidated. In fact, given the wide expression of ADAM11 in the nervous system, it is likely that only a subset of the phenotypes depends on the Kv1 channels.
Proteomic interaction studies identified contactin-associated protein-like 2 (Caspr2, CNTNAP2) [24], LGI family members [25] and integrin α4 [26] as additional ADAM11-binding partners, but the physiological consequences of these interactions await further investigations.
ADAM22: regulator of myelination and epilepsy
ADAM22 (MDC2) is the best studied inactive ADAM and plays a key role in processes such as synapse maturation, synaptic transmission and myelination in the nervous system, where ADAM22 is exclusively expressed [27]. ADAM22 has a wide expression throughout the CNS with the highest levels in cerebellum (granule cells) and hippocampus (CA1 pyramidal neurons), whereas in the spinal cord its expression is restricted to the grey matter [28]. While ADAM22 expression is high in adult mice, it is low during embryogenesis and in new born pups, but increases strongly starting around postnatal day 7 (P7), coinciding with increased synapse maturation and the onset of myelination. In fact, at the same time point the expression levels of other synaptic proteins such as PSD-95 involved in synapse maturation start rising [29]. Concomitant with the time point of this upregulation, ADAM22 knockout mice develop diverse phenotypes, such as ataxia, severe seizures, reduced body weight and uncoordinated movements, and die prematurely between P10 and P25. In addition, PNS nerves are hypomyelinated, whereas CNS myelination appears normal, demonstrating a specific function for ADAM22 in peripheral myelination [28].
Receptor function at the synapse
ADAM22 acts as a receptor for LGI1 [30] and is assumed to be located at the postsynaptic membrane, where it regulates synaptic transmission through formation of a multi-protein complex (Fig. 3). The soluble ligand LGI1 binds to the extracellular domain of its receptor ADAM22, which in turn binds to the scaffolding protein PSD-95 (postsynaptic density protein 95) through its C-terminal PDZ-binding motif [30]. PSD-95 links the complex LGI1/ADAM22/PSD-95 to stargazin, a regulatory subunit of the AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor. Thus, LGI1 binding induces formation of the super-complex and is required for AMPA receptor-dependent excitatory synapse maturation and maintenance [31]. The absence of super-complex formation, for example in ADAM22- or LGI1-deficient mice [31, 32], lowers AMPA receptor function and has detrimental neurological consequences such as epileptic seizures [30]. In fact, the two complex members LGI1 and ADAM22 have been genetically linked to epilepsy in patients. Dozens of mutations were discovered in LGI1 that prevent its secretion or the direct binding to ADAM22 and have been linked to a form of epilepsy called ADLTE/ADPEAF (Autosomal Dominant Lateral Temporal Epilepsy; also referred to as Autosomal Dominant Partial Epilepsy with Auditory Features) [33, 34]; mutations reviewed in detail elsewhere [35, 36]. Two compound heterozygous mutations in ADAM22 (Cys401Tyr and Ser799IlefsTer96) have recently been reported in a patient suffering from encephalopathy with epilepsy. Cys401Tyr is a missense mutation that prevents LGI1 binding to ADAM22, whereas Ser799IlefsTer96 is a frameshift mutation, which prevents maturation and trafficking of ADAM22 to the cell surface where it normally binds LGl1 [37]. Also stargazin, another super-complex member, is linked to epilepsy, when its gene is disrupted in mice [38, 39]. Stargazin is a transmembrane AMPA receptor regulatory protein (TARP), which affects AMPA receptor trafficking, targeting and gating [40]
Besides the genetic links, epilepsy can also be induced by an autoimmune reaction that affects the super-complex containing LGI1 and ADAM22. This specific epileptic disorder is referred to as limbic encephalitis. In this case, autoantibodies against LGI1 have been found in the patient serum [41, 42]. They disturb the receptor–ligand interaction between ADAM22 and LGI1 and, thereby, reduce synaptic AMPA receptor levels [43]. How exactly the reduced AMPA receptor function causes epilepsy remains a matter of debate. One hypothesis refers to the neuronal network activity in brains and states that the impaired AMPA receptor function is particularly relevant in so-called inhibitory interneurons. As a consequence, they would be less able to inhibit excitatory neurons, which in turn can be “hyperactive”, which might be the cause of epilepsy [35]. Also, ADAM22 may regulate the trafficking and stability of Kv1 channel as in the case of ADAM11 [44]. Taken together, the link to human disease establishes an important function for ADAM22 as a LGI1 receptor crucial for AMPA receptor function and a healthy network activity of the brain.
Transsynaptic complexes and crystal structure
Interestingly, the postsynaptic super-complex containing LGI1, ADAM22 and PSD-95 may form an even larger complex bridging the postsynaptic with the presynaptic membrane. Proteomic interaction studies suggested that LGI1/ADAM22/PSD-95 binds to the presynaptic proteins ADAM11, ADAM23 and the Kv1 potassium channels [32] [45] (Fig. 3). Such a synapse-bridging complex is in line with the finding that mice lacking ADAM22, ADAM23, LGI1 or Kv1 channels share highly similar epileptic phenotypes [20, 21, 28, 32, 46–48] and suggest a common underlying cause. Further evidence comes from a recent study, which determined the crystal structure of the ADAM22 ectodomain in complex with LGI1 [49]. LGI1 is known to form dimers [50] and this was also seen in the structure. Importantly, the LGI1 dimer bound one ADAM22 molecule on each side, resulting in a sort of a linear structure consisting of ADAM22–LGI1–LGI1–ADAM22 (Fig. 3). This is consistent with the assumed synapse-bridging function of LGI1 with the exception that two ADAM22 molecules were seen in the crystal structure, whereas one ADAM22 and one ADAM23 molecule are expected to bind the LGI1 dimer at real synapses. Further studies will be needed to validate the synaptic LGI–ADAM-bridge and understand its exact role in synaptic transmission and epilepsy and the extent to which ADAM11 is involved [49]. In any case, the crystal structure of LGI1–ADAM22 is a major step forward towards structure-based anti-epileptic drug design.
The crystal structure of the ectodomain of mature ADAM22 reveals a four-leaf clover shape where each leaf contains either the EGF-like, the cysteine-rich, or the disintegrin and metalloprotease-like domain [49, 51]. While the individual domains have a similar structure as the same domains in the recently published structure of ADAM10, the three-dimensional organization of the domains relative to one another is strikingly different between ADAM22 and ADAM10 [49, 52]. This demonstrates that the structure of individual ADAMs cannot simply be modeled based on the known ADAM structures and that the structure of non-catalytic ADAMs may generally differ from catalytically active ones.
Other ADAM22 interaction partners control myelination and trafficking
LGI4 is another LGI-family member binding ADAM22. Schwann cells (SCs), the myelinating cells in the PNS, secrete LGI4, which binds to neuronal ADAM22. This interaction is essential for peripheral nerve myelination [53]. As a result, mice with a spontaneous LGI4 loss-of-function mutation (claw paw) as well as ADAM22−/− mice show peripheral hypomyelination [53]. Moreover, a patient carrying splicing mutations in LGI4 lacks myelin sheaths on the sciatic nerve [54]. Besides its role in peripheral myelination, the LGI4–ADAM22 complex has also been suggested to contribute to gliogenesis, including glia lineage cell differentiation and proliferation in the PNS [55].
Other interaction partners are 14-3-3 proteins. ADAM22 contains two 14-3-3 binding sites within its cytoplasmic tail and all six 14-3-3 brain-specific proteins bind ADAM22 in a phosphorylation-dependent manner. 14-3-3 protein binding is essential for the efficient cell surface expression of ADAM22 in non-neuronal cells by covering endoplasmic reticulum retention signals present in the ADAM22 cytoplasmic tail [56]. This regulated trafficking is reminiscent of the similar regulation of another ADAM family member, ADAM17. As described in more detail in the section on ADAM17, maturation and activity of this active metalloprotease require binding of ADAM17 to iRhom proteins, which in turn bind 14-3-3 proteins in their cytoplasmic tails. Importantly, iRhoms lacking those sites or the ability to phosphorylate these sites fail to regulate ADAM17 activity [57, 224]. Taken together, 14-3-3 binding proteins and the phosphorylation of 14-3-3 binding sites play an important role in regulating the function of ADAM22 and of the ADAM17/iRhom complex. Thus, it is tempting to speculate that 14-3-3 proteins may also control the trafficking or function of other ADAM family members, such as the active protease ADAM10.
ADAM23: involved in differentiation and synaptic transmission
ADAM23 (MDC3) is predominantly expressed in brain [27], revealing the highest levels in hippocampus (pyramidal cells in CA1 and CA3 but not in dentate gyrus), basal ganglia and cerebellar Purkinje cells [58]. ADAM23, which is assumed to be mainly located at the presynaptic membrane, is involved in synaptic transmission by binding to LGI-family members in a similar manner to ADAM22 on the postsynaptic membrane [49] (Fig. 3). In addition, ADAM23 was genetically linked to epilepsy in mice and dogs [59]. However, the underlying molecular mechanism remains elusive and further studies are needed to understand the role of the synaptic ADAM23 protein in epilepsy and how it is influencing, e.g., ion channels and ADAM22.
Another function of ADAM23 is in neuronal differentiation. Neurons lacking ADAM23 show reduced neurite outgrowth upon LGl1 stimulation in vitro and in vivo. On the other hand, the overexpression of ADAM23 triggers neurite growth and differentiation of human neural progenitor cells [60]. ADAM23−/− mice are smaller compared to wild-type littermates, suffer from ataxia, tremor, severe seizures and die by 2 weeks of age [47, 48].
ADAM23 also interacts directly with the cellular prion protein (PrPc) [61], which upon misfolding is a key pathologic agent in prion diseases. This interaction occurs through the extracellular disintegrin-like domain of ADAM23. ADAM23 uses the same domain also to mediate cell adhesion through integrin binding, namely the integrin-alphavbeta3 (αvβ3) [62, 63] and the α4-integrins α4β7 and α4β1 [26]. Interestingly, ADAM23 is silenced in many cancers by promoter hypermethylation, including head cancer [64] and glioma [65]. It is assumed that the reduced ADAM23 levels lead to increased levels of active αvβ3, enhancing metastatic progression [66].
ADAM23 has also been linked to two infectious diseases, but it remains unclear whether ADAM23 has a causal role. Malaria is caused by Plasmodium parasites and the infection triggers oxidative stress and inflammation, hemorrhage and cerebral brain damage including Purkinje cell damage. During the infection, ADAM23 levels increase significantly and reduce upon antimalarial drug treatment [67, 68]. The other disease is schistosomiasis, which is caused by parasitic flatworms and also causes increased ADAM23 levels in the brain [67].
Taken together, ADAM23 plays a fundamental role in neuronal processes such as neuronal excitability, neuronal differentiation, cell adhesion by integrin binding, differentiation and neurite outgrowth, but is also linked to cancer metastasis and epilepsy.
Although ADAM11, ADAM22 and ADAM23 lack catalytic activity, they are involved in key processes of the nervous system. Further research is needed to elucidate their full repertoire of biological functions. Especially promising seems to be the super-complex, which is formed at synapses between ADAM22/LGI1/ADAM23/PSD-95, and a deeper understanding of where it is formed and how it regulates receptor levels has the potential to advance the understanding of synaptic regulation and maturation.
Proteolytic ADAMs
ADAM8: a role in neuroinflammation
ADAM8 was first identified in macrophages and macrophage-like cell lines by screening a cDNA library comprising genes that were upregulated in response to lipopolysaccharides (LPS) [69]. In addition, its expression was observed in human immune cells [70]. During early development, ADAM8 is expressed in cells derived from the trophoblast and, at later stages, in the developing cartilage, bones, gonads, thymus and CNS [71]. ADAM8 is linked to tumors and inflammation [72]. In the CNS of adult mice, ADAM8 is found at low levels under steady-state conditions. However, in a murine model of neurodegeneration, ADAM8 levels increase in response to tumor necrosis factor α (TNFα), predominantly in the areas of brain and spinal cord where neurodegeneration occurs [73]. Additionally, ADAM8 was proven to dampen TNFα-stimulated signaling by shedding its receptor TNFR1 in a protease-regulated feedback loop [74]. ADAM8 has also been suggested to cleave the prion protein, at least in muscle cells [75], and to be involved in angiogenesis that follows spinal cord injury [76, 77].
ADAM8 is a catalytically active protease. Different from other ADAMs that require furin for cleaving off the pro-domain and get thereby activated, ADAM8 releases its pro-domain by an autocatalytic mechanism [8]. ADAM8 can cleave the myelin basic protein and CD23 in vitro. Similarly, it cleaves peptides comprising the potential cleavage sites of a number of prototypic ADAM substrates, including kit ligand-1 (KL1), TNFα and amyloid precursor protein (APP) [78]. In addition, the neural cell adhesion molecule “close homologue of L1” (CHL1) is shed by ADAM8 when both enzyme and substrate are co-transfected in COS7 cells and conditioned media containing shed CHL1 induce neurite outgrowth, thus implying a role for ADAM8 in this process [79]. In addition to its shedding potential, ADAM8 can also cleave extracellular matrix (ECM) components, including fibronectin [80]. Indeed, after cleavage of the pro-domain, a second autocatalytic event can release a soluble form of ADAM8 catalytic domain, which in turn is capable of ECM degradation [80].
In conclusion, ADAM8 is expressed in different areas of the CNS and in different cell types, including neurons, microglia and astrocytes. Although the physiological relevance of ADAM8-mediated shedding has to be proven in vivo, ADAM8 expression pattern and its ability to process proteins involved in neurodevelopment, myelination and plasticity suggest a central role for this enzyme in the brain. In murine models of neurodegeneration, ADAM8 was found linked to astro/microgliosis and proven to be neuroprotective [74]. This indicates a crucial role for this enzyme in modulating neuroinflammation.
ADAM9 and ADAM15: potential regulators of ADAM10
ADAM9 and ADAM15 are two catalytically active ADAMs with overlapping spatial expression profiles in the adult mouse brain. mRNAs of both ADAMs are highly expressed in hippocampus and the cerebellar Purkinje cell layer [81, 82], suggesting that under physiological conditions ADAM9 and ADAM15 may functionally compensate each other. However, this is not yet clear because triple knockout mice of ADAM9/12/15 do not reveal evident abnormalities [83]. Moreover, expression of ADAM9 and ADAM15 mRNAs can be differentially regulated upon different stimuli that mimic pathological conditions. In kainic acid-induced status epilepticus rat brains, for example, ADAM9 expression is upregulated in dentate gyrus of hippocampus, while ADAM15 expression remained unaltered [84]. In addition, expression of ADAM15 is upregulated in response to severe hypoxia to induce neuronal death [85] and within the first day following peripheral nerve lesions [86].
While ADAM9 and ADAM15 have numerous links to cancer, retinopathy, inflammation and bone formation [87–89], relatively little is known about their functions in the nervous system. Noteworthy, mice with a constitutive knock-out of ADAM9 or ADAM15 are viable and do not have major dysfunctions [81, 82]. ADAM9 has been linked to the beneficial α-secretase cleavage of APP, which occurs within the amyloid β (Aβ) domain of APP and prevents generation of the neurotoxic Aβ peptide, a key molecule in AD [90]. Under constitutive, non-stimulated conditions the α-secretase cleavage of APP in the nervous system is only mediated by ADAM10, but not by ADAM9 [82, 91–93]. Yet, it is well established that α-secretase cleavage can be stimulated above constitutive levels, and this may occur through different metalloproteases, including ADAM9, which cleaves APP two amino acids upstream of the main α-secretase cleavage site [94]. Specifically, overexpression of ADAM9 in COS7 cells stimulated APP α-secretase cleavage. In line with this, promoter polymorphisms with increased ADAM9 transcription were found to be protective against AD [95]. If a pharmacological activation of ADAM9 becomes possible, it would be interesting to test this approach for increasing APP α-cleavage and lowering Aβ levels. In principle, this appears possible, because reactive oxygen species increase ADAM9 expression [96, 97]. Another link of ADAM9 and even ADAM15 to α-secretase comes through the finding that both proteases can shed the constitutive α-secretase ADAM10, thereby inactivating ADAM10 through release of the soluble ADAM10 ectodomain (sADAM10) [98]. This suggests that ADAM10 activity may be regulated by ADAM9 and ADAM15. Supporting this notion, treating SH-SY5Y neuroblastoma cells overexpressing APP with recombinant ADAM9 prodomains to inhibit ADAM9 activity leads to decreased sADAM10 and a concomitant increase in membrane-bound ADAM10, resulting in more APP α-secretase cleavage [99]. Thus, both increasing (through direct cleavage of APP) and blocking ADAM9 (through reduced cleavage of ADAM10) may possibly be an approach to increase the beneficial α-secretase cleavage of APP. Whether both processes together neutralize their effects on APP α-cleavage remains to be tested.
ADAM9 is also linked to the shedding of another protein involved in neurodegeneration, the cellular prion protein (PrPc). While it does not directly cleave PrPc, it was found in different cell types to enhance PrPc shedding in an ADAM10-dependent manner. The exact underlying mechanism is not yet clear, but may involve ADAM9-mediated ADAM10 shedding, because in this case not only the membrane-bound ADAM10, but also the shed sADAM10 is assumed to process PrPc [100]. What remains to be determined, however, is the percentage of shed sADAM10 versus membrane-bound ADAM10 under physiological/pathological conditions and the relative contribution of both forms on substrate shedding. Currently, it is assumed that ADAM10 activity is mostly mediated by the membrane-bound form.
ADAM10: a constitutive protease with major functions in the nervous system
ADAM10 is ubiquitously expressed in all major cell types in brain [101]. As revealed by in situ hybridization, ADAM10 mRNA is highly expressed in the mouse telencephalon, mesencephalon and cerebellum [102]. Among all ADAMs, ADAM10 has been most intensively studied in the brain, mainly because of the physiological and pathological importance of two well-characterized ADAM10 substrates: the Notch receptors for development and the amyloid precursor protein (APP) in AD. In addition, numerous neuronal substrates were identified for ADAM10 [103] as well as phenotypes in ADAM10-deficient mice [104]. Recently, the structure of the mature ADAM10 ectodomain was determined by X-ray crystallography. It revealed that ADAM10 adopts a closed conformation in which the cysteine-rich domain precludes the access of substrates to the active site [52], suggesting that ADAM10 needs to be structurally activated before efficient substrate cleavage. Such activation may also depend on the cytoplasmic and transmembrane domain of ADAM10 [105, 106]. In the following, we will give an overview on the roles of ADAM10 in several key processes during neural development and disease (Fig. 4). The focus will be on recent studies and on substrates where the function is altered by ADAM10 cleavage. Other substrates with potential relevance to the brain have been reviewed recently [107]. Finally, we will discuss trafficking and subcellular localization of ADAM10 in neurons and how this may be regulated by ADAM10 protein-binding partners. Expression regulation of ADAM10 has been covered in recent reviews [108, 109] and will not be discussed here.
Fig. 4.

Key substrates and functions of ADAM10 in the nervous system. a ADAM10 (represented by black scissors) cleaves the Notch receptor at the Notch juxtamembrane region. Subsequently, the C-terminal fragment (CTF) of Notch is further processed by the protease γ-secretase, generating the Notch intracellular domain (NICD), which translocates to the nucleus and activates target gene expression relevant during neural development. b In the CNS (left), ADAM10 cleaves several cell adhesion molecules, e.g., EphrinA5, NCAM, NrCAM, and Nrp1, and thus regulates axon targeting and neurite growth. In the PNS (right), ADAM10 is the major protease to release the soluble DR6 ectodomain (sDR6), which in turn negatively regulates axonal myelination by Schwann cells (SCs). c ADAM10 regulates synapse formation and maintenance by cleaving several key synaptic proteins localized to both pre-synapse and post-synapse, e.g., N-cadherin, neuroligin-1 (NLGN1), and neurexin (NRXN). d ADAM10 cleaves APP within the Aβ sequence, which precludes the formation of pathological Aβ plaques in AD (left). ADAM10 plays a dual role in prion disease (right), as on one hand ADAM10 inhibits the transition of cellular prion protein (PrPc) to pathological prion protein (PrPsc), but on the other hand promotes the spreading of PrPsc
ADAM10 in neurogenesis and neuronal migration
Signaling from the Notch receptors is critical for early embryogenesis, cell fate specification and is also required in adult organisms [110–112]. Ligand binding to Notch triggers ADAM10 cleavage within the Notch extracellular juxtamembrane region. The remaining, membrane-bound fragment is then further cleaved by γ-secretase within the transmembrane domain, releasing the Notch intracellular domain (NICD), which in turn leads to target gene activation in the nucleus (reviewed in Ref. [113]). The role of ADAM10 in ligand-dependent Notch signaling was first shown in Drosophila, where ADAM10 is referred to as Kuzbanian [114, 115]. In mice, constitutive knock-out of ADAM10 causes early lethality at around embryonic day 9.5 (E9.5) due to the impaired Notch signaling [116]. Conditional knock-out of ADAM10 in neural progenitor cells (ADAM10f/f;Nestin-Cre) of mice prevents the very early defects in embryogenesis, but still results in perinatal lethality and precocious neuronal fate specification at the expense of glial cells, confirming the defected Notch signaling in ADAM10-deficient neural progenitor cells [92]. In utero electroporation of Cre recombinase into conditional ADAM10-deficient mouse brains revealed another Notch-dependent phenotype. ADAM10-deficient neurons fail to migrate to the cortical plate, whereas the concomitant overexpression of NICD in ADAM10-deficient neurons rescued the phenotype [117]. Together, these studies indicate that ADAM10 plays a key role in early neural developmental events by initiating Notch signaling.
ADAM10 in axon targeting and neurite growth
Neurons extend axons, often over long distances, to reach their targets in order to form a functional network. This process is controlled by guidance cues (ligands) that interact with their specific cell surface receptors to trigger adhesive, attractive, or repulsive responses of the axon (reviewed in Ref. [118]). ADAM10 appears to be a key regulator in axon targeting as ADAM10 cleaves many of the relevant cell surface proteins in neurons [103]. Additionally, mice with a postnatal deletion of ADAM10 in the nervous system show numerous phenotypes indicative of defective synaptic functions [104].
One example is the ephrin-Eph axis. The membrane-tethered ligand ephrin binds to its Eph receptor, a transmembrane protein, to trigger downstream signaling events. In Drophophila, a dominant-negative form of Kuzbanian inhibits EphrinA2 ectodomain shedding [119], indicating a role of ADAM10 in ephrin-Eph signaling. A further study in mammalian cells suggested that ADAM10 is able to cleave ephrin in trans from Eph-expressing cells [120]. However, the trans-shedding activity of ADAM10 and other ADAM proteases has been little studied so far and evidences from endogenous proteases and substrates are lacking. Another study demonstrated that upon binding of ephrinA5 to EphA3 in cultured cortical neurons, the neuronal cell adhesion molecule (NCAM) is cleaved by ADAM10 [121], which may have a role in neurite branching and neurite outgrowth [122]. Similar roles of ADAM10 were also observed for the semaphorin family of guidance cues. Semaphorin3A (Sema3A) is a secreted ligand, which functionally interacts with neuropilin1 (Nrp1) and the class A plexin receptor complex. ADAM10 cleaves Nrp1 and knock-out of ADAM10 in proprioceptive sensory axons led to increased levels of Nrp1 at the growth cones and, as a consequence, increased responsiveness to Sema3A [123]. Of note, the “neural glial-related cell adhesion molecule” (NrCAM) was identified as a new substrate of ADAM10 in an unbiased proteomic screening [10, 103]. NrCAM knockout and ADAM10 knockout (ADAM10f/f;CamKIIα-Cre) mice share a similar phenotype. Both mutants had axonal targeting deficits within their olfactory bulbs with axons overshooting their mark, suggesting a functional interaction between ADAM10 and its substrate NrCAM to regulate axon targeting [103, 124, 125]. Collectively, ADAM10 cleaves a number of cell adhesion molecules including guidance cues and their receptors, which are important for axon targeting and neurite growth during neuronal development.
ADAM10 in CNS synapse formation and maintenance
In the mammalian CNS, synapses are asymmetric cell–cell adhesion sites between nerve cells, which are designed to mediate the efficient uni-directional transmission of signals from the presynaptic site to the postsynaptic site. The correct formation and maintenance of synapses are required for proper functions of the nervous system and are tightly controlled by cell-surface adhesion molecules, including cadherins and the neurexin–neuroligin complex (reviewed in Ref. [126]). Many of these molecules are cleaved by ADAM10 [103], indicating an important role for ADAM10 in regulating these processes. In line with this conclusion, post-mitotic excitatory neuron-specific ADAM10 knock-out mice (ADAM10f/f;CamKIIα-Cre) were born normally and survived beyond the early developmental stage but displayed altered dendritic spine morphology and multiple defects in synaptic functions [104]. Conversely, moderate overexpression of ADAM10 in a transgenic mouse model led to increased presynaptic boutons [127].
The neuronal cadherin (N-cadherin) was validated as a substrate of ADAM10 using ADAM10-deficient fibroblasts, where decreased N-cadherin C-terminal fragments and increased cell surface expression were observed [128, 129]. The ectodomain of N-cadherin mediates calcium-dependent cell–cell adhesion through homophilic interaction, whereas its cytoplasmic domain interacts with β-catenin, which in turn plays a key role in the transduction of Wnt signaling. Reduced ectodomain shedding of N-cadherin in ADAM10-deficient cells, therefore, influenced cell adhesion processes and led to altered β-catenin-dependent signaling [128]. In neurons, altered localization or inhibition of ADAM10 activity led to an N-cadherin-dependent increase in the size of dendritic spines and a change in the number and current of synaptic AMPA receptors [130].
Neuroligins (NLGN) are a family of potent synaptogenic adhesion proteins located at the postsynapse, which transsynaptically bind to the presynaptic partner, neurexin (NRXN). Proteolytic processing of NLGN1 by ADAM10 is considered as a way to terminate the synaptogenic activity of NLGN1 in an activity-dependent manner through NMDA receptor activation or by binding to the secreted form of NRXN1 [131]. Of note, the secreted form of different NRXN family members and isoforms may also be generated by ADAM10 or ADAM17 cleavage [132, 133]. Therefore, it is tempting to assume that the proteolytic cleavage of NRXN at the presynapse may be functionally linked to the processing of NLGN at the postsynapse, as soluble NRXNs function as negative regulators of NLGNs via induction of full-length NLGs shedding. Interestingly, only NLGN1, which is expressed on glutamatergic neurons, but not NLGN2, expressed on GABAergic neurons, is a substrate of ADAM10 [103, 131]. Another ADAM10 substrate in the NLGN family is NLGN3. However, the relevant function of ADAM10 cleavage on NLGN3 is that the shed NLGN3 ectodomain promotes the growth of high-grade glioma, a common type of brain tumor [134]. Therefore, the proteolytic cleavage of NLGNs by ADAM10 may not only be a way to terminate the functions of full-length proteins [131], but may generate functional shed products. Of note, although ADAM10 was first reported to bind synapse-associated protein 97, a protein that is responsible for trafficking ADAM10 to postsynaptic compartment in excitatory neurons [135], a more recent study reported that in cultured hippocampal neurons, ADAM10 is in close proximity with synaptophysin, a presynaptic vesicle protein [136]. In addition to neurons, oligodendrocyte precursor cells (OPCs), which differentiate into oligodendrocytes to form myelin sheaths on neurons, are also able to receive direct synaptic input from neurons. OPCs express nerve–glia antigen 2 (NG2), whose shedding is triggered by synaptic activity in an ADAM10 activity-dependent manner. The ectodomain of NG2, in turn, can modulate synaptic transmission between neurons [137]. Taken all together, ADAM10 cleaves synaptic cell adhesion molecules located at both presynapse and postsynapse of neurons and NG2 expressed on OPCs, thereby acting as a key regulator in the dynamic processes of synapse formation and maintenance.
ADAM10 in Alzheimer’s and prion diseases
ADAM10 has a tight link to neurodegenerative diseases as it plays a central role in Alzheimer’s and prion diseases through cleavage of APP and the prion protein. ADAM10 acts as the α-secretase of APP in the nervous system [91, 92, 138]. It cleaves APP within the Aβ domain and, thus, precludes the formation of the pathogenic, AD-relevant Aβ peptides, suggesting that activation of ADAM10 may be a way for AD prevention or treatment. Moreover, ADAM10 cleavage of APP generates a soluble APP fragment (sAPPα) that has neuroprotective functions [139], which may be an additional benefit of therapeutic ADAM10 activation. Of note, two mutations in the ADAM10 prodomain were found to be associated with late-onset AD [140]. Overexpression of these mutated forms of human ADAM10 in an AD mouse model (Tg2576) showed enhanced Aβ plaque load and reactive gliosis as compared to overexpression of wildtype human ADAM10, indicating that the mutations attenuated the ADAM10 α-secretase activity toward APP [141]. Mechanistically, these mutations may impair the chaperon function of ADAM10 prodomain for the correct maturation of active ADAM10, although controversial results showed that ADAM10 prodomain may act as a specific inhibitor for ADAM10 activity [141, 142]. Conversely, moderate neuronal overexpression of wildtype ADAM10 in an AD mouse model [human APP (V717I)] showed increased secretion of sAPPα, reduced formation of Aβ peptides and plaques, and alleviated impaired long-term potentiation and cognitive deficits [143], suggesting that therapeutic ADAM10 activation should be beneficial for AD. Of note, as an approved drug for the treatment of psoriasis, acitretin was found to transcriptionally upregulate ADAM10 expression in cell lines [144]. In a phase II clinical trial, acitretin mildly increased sAPPα level in the cerebrospinal fluid (CSF) of AD patients [145]. While it is not yet clear whether the drug improves memory in the patients, this sets the stage for further clinical tests with acitretin-mediated ADAM10 activation in AD [125].
The cellular prion protein (PrPc) is a GPI-anchored protein, which undergoes a conformational change into the pathologic isoform (PrPSc) in prion disease. In ADAM10f/f;Nestin-Cre conditional knockout mice, shedding of PrPc was drastically reduced with a concomitant accumulation of PrPc in the secretory pathway of neurons, indicating that ADAM10 is the physiological sheddase for PrPc [146, 147]. Interestingly, ADAM10 may protect PrPc from conversion into pathologic PrPSc, but on the other hand also accelerate the spread of shed PrPSc. This dual role of ADAM10 in prion disease was evidenced when infecting mice with a conditional loss of ADAM10 in post-mitotic neurons (ADAM10f/f;CamKIIα-Cre) with PrPSc. A shortened time to develop prion disease symptoms was observed, but concomitantly a reduced PrPSc spreading [148]. How ADAM10 regulates these processes is not entirely clear, but may involve active ADAM10 released in membrane vesicles. A number of studies outside the nervous system suggest the presence of proteolytic active ADAM10 in secreted membrane vesicles such as exosomes, where ADAM10 also contributes to substrates cleavage and has functional correlations [149–151]. However, whether ADAM10 is delivered in exosomes in the nervous system is currently not well characterized. Moreover, given that ADAM10 is ubiquitously expressed in almost all the cell types, the functional significance of exosomal ADAM10 remains to be determined.
ADAM10 in PNS axonal growth and myelination
Myelination of axons speeds up the transmission of electrical impulses, and Schwann cell (SC) is the myelinating cell in the PNS, where roles of ADAM10 in axon growth and myelination have been studied both in vitro and in vivo. Inhibition or knockdown of ADAM10 in the co-culture of dorsal root ganglion neurons and SC reduced axonal length without affecting myelination [152–154]. As many ADAM10 substrates are not only cleaved by ADAM10, but also by other proteases [103], it is possible that even in the absence of ADAM10 enough substrate is shed to prevent the detection of a strong myelination phenotype. Contributions of ADAM10 in PNS neurons and SCs in different contexts, i.e., during development and after injury, were further dissected in an in vivo study, where ADAM10 was conditionally knocked out in a cell-type specific manner. SC-specific ADAM10 knockout mice (ADAM10f/f;P0-Cre) did not show a significant difference in axon myelination during development or re-myelination after injury. Motor neuron-specific ADAM10 knockout mice (ADAM10f/f;HB9-Cre) also did not show overt defects in myelination, but showed reduced outgrowth of small-caliber myelinated axons after traumatic injury [155]. Of note, a recent study demonstrated a role of another ADAM10 substrate in PNS myelination during development. Death receptor 6 (DR6) is a member of the TNF receptor (TNFR) superfamily and was found to be shed by ADAM10 to 50% of total DR6 shedding. In contrast to other TNFR superfamily members, where shedding inactivates the receptors [1], the shed DR6 ectodomain from PNS neurons is a new functional molecule and acts as a paracrine molecule to restrain SC proliferation and axon myelination, implicating ADAM10 in PNS myelination by releasing the DR6 ectodomain [156].
Substrate specificity of ADAM10 may depend on interaction with tetraspanins
Recent work suggested that ADAM10 may work in a complex with selected members of the tetraspanin (TSPAN) family (reviewed in Ref. [157]). TSPANs are evolutionarily conserved membrane proteins with four transmembrane domains and two extracellular loops. The larger extracellular loop (LEL) of TSPANs is able to associate dynamically with numerous partner proteins in TSPAN-enriched microdomains. Of note, ADAM10 interacts with the LEL of a subgroup of six TSPANs (TSPAN5, TSPAN10, TSPAN14, TSPAN15, TSPAN17, TSPAN33), which contain eight cysteines in a conserved motif of the LEL (TSPAN-C8). Overexpression of one of the TSPAN-C8s is sufficient to increase levels of the mature, active and prodomain-lacking form of ADAM10 in various cell types. The overexpressed TSPAN-C8 may force ADAM10 to adopt different conformations, or target ADAM10 to different membrane localizations [158–160]. Therefore, TSPAN-C8s may control the substrate specificity of ADAM10, such that ADAM10 only cleaves some of its substrates when in complex with one of the six TSPAN-C8s. This idea suggests that TSPAN-C8s might be interesting drug targets for therapeutic purposes, e.g., for AD, as the α-secretase cleavage of APP may be specifically upregulated without concomitantly affecting all the other ADAM10 substrates. Indeed, while TSPAN15 knockout brains have less mature ADAM10, two ADAM10 substrates (N-cadherin and PrPc) showed reduced shedding but cleavage of Notch and APP was not altered [161]. However, the report on TSPAN15 knockout mice has been so far the only in vivo study working on endogenous TSPAN-C8s to support the hypothesis that different TSPAN-C8s may regulate ADAM10 substrate selectivity. Moreover, it will be required to identify which TSPAN-C8 is responsible for regulating APP cleavage by ADAM10 in neurons.
ADAM12: a link to Alzheimer’s disease and ischemia
ADAM12 is an active protease, but little is known about its substrates. ADAM12 is expressed in a number of tissues, including cartilage, bone, liver and brain [162]. Its expression in tissues characterized by active cell growth and repair indicates roles of ADAM12 in cell signaling, cell adhesion and ECM turnover. Although some functions of ADAM12 have been characterized, including modulation of chondrocyte proliferation [163] and myogenic differentiation [164], its physiological role in the brain still remains elusive. The function does not appear to be essential, because overall, ADAM12 and ADAM9/12/15 triple knockout mice appear normal and reveal no major evident deficiencies [83, 165]. Yet, many specialized neuronal functions may require specific tests to be uncovered. Nevertheless, evidence emerged for a potential role of ADAM12 in the pathology of Alzheimer’s disease (AD). Malinin et al. showed that Aβ-induced neurotoxicity is dependent on the metalloprotease activity of ADAM12 [166]. In agreement with their hypothesis, authors demonstrated that ADAM12 levels are altered in AD brains. Although it is not fully clear how ADAM12 can mechanistically promote neurotoxicity, the authors suggested the enzyme as a valuable drug target for a therapy of AD [166]. ADAM12 may have a second link to AD in that it sheds the low density lipoprotein receptor-related protein 1 (LRP-1) [167], which acts as an Aβ endocytic receptor. However, comprehensive studies to establish whether ADAM12-mediated processing of LRP-1 affects Aβ turnover and consequently plaque deposition are currently missing. Furthermore, ADAM12 is involved in the impairment of neural vascular barriers in response to hypoxia, a pathological condition associated with neurological diseases, including ischemic cerebral attack [168]. Indeed, hypoxia stimulated ADAM12-dependent shedding of claudin-5, a component of the tight junctions, thus destabilizing the vascular endothelium.
Taken together, catalytic activity of ADAM12 is associated with a number of neurological conditions, including AD and ischemia, which render the enzyme a potential drug target for the therapy of these diseases. However, its physiological roles in neurodevelopment and in the adult brain have not been elucidated yet.
ADAM17: a protease with major roles in the nervous system in response to stimuli
ADAM17 was first identified as the enzyme responsible for the proteolytic cleavage of TNFα, a pro-inflammatory cytokine that needs to be proteolytically released from the cell surface to elicit its pro-inflammatory potential [169, 170]. Since then, more than 80 substrates of ADAM17 have been identified, although many have not yet been validated in vivo. The substrates span from signaling molecules, such as cytokines, growth factors and their receptors, to adhesion molecules and endocytic proteins [171]. In the following, several substrates with relevance to the nervous system and their functions will be described in more detail. In agreement with its high number of substrates, ADAM17 plays a crucial role in many biological processes, including development, immune responses, cell proliferation and survival (reviewed in Ref. [171]). Many of these ADAM17 substrates can be constitutively shed by other proteases, including ADAM10 and BACE1. Constitutive shedding occurs in the absence of specific stimuli and may often be a biological mean to maintain turnover of proteins at the cell surface. Conversely, ADAM17 is known to be a regulated protease, which acts in response to precise stimuli [172–174].
ADAM17 is ubiquitously expressed in mammals, including in the CNS [102]. Its expression in human fetal brains is higher than in adults, suggesting that ADAM17 has key functions in brain development [169, 175]. Nevertheless, no evident abnormalities in the developing CNS are associated with ablation of ADAM17 in mouse embryos [176]. Ablation of ADAM17 in mouse is perinatally lethal [176], which makes investigating physiological functions of ADAM17 in the adult brain difficult. The majority of ADAM17 functions in the brain were studied by targeted approaches and indirect evidence. For instance, EGF receptor (EGFR) is expressed in different areas of the brain and linked to a number of biological functions, including development, synaptic plasticity and memory formation. Given the crucial role of ADAM17 in triggering EGFR signaling by shedding its ligands, it is easy to speculate that the enzyme plays an essential role in all these processes [177]. In support of this hypothesis, ADAM17 knockout mice resemble mice that lack the EGFR in that they are born with open eyes and die perinatally, suggesting that ADAM17-mediated shedding of EGFR ligands such as TGFα and heparin-binding EGF-like growth factor (HB-EGF) and subsequent EGFR activation is the major pathway controlled by ADAM17 during development [176, 178–181]. The ADAM17 substrate HB-EGF plays a pivotal role in the synaptic plasticity and memory formation in adult mice, implying that ADAM17 can be involved in these processes [182], although proofs of its direct relevance in vivo are still missing. In contrast, a clear link with ADAM17 was found in HB-EGF-dependent oligodendrocyte development and myelination [183].
ADAM17 in myelination
Myelination is the process of forming a myelin sheath around neuronal axons to allow high-speed nerve conduction. In the CNS, the myelin sheath is a plasma membrane extension of glial cells called oligodendrocytes. Genetic deletion of ADAM17 in oligodendrocyte progenitor cells induces premature cell cycle exit and reduces oligodendrocyte cell survival [183]. In addition, ADAM17 is also required for supporting oligodendrocyte regeneration following demyelination [184]. Thus, ADAM17 ablation leads to deficits in myelination and motor behavior in vivo. Mechanistically, this is due to lack of TGFα and HB-EGF shedding and, consequently, EGFR signaling activation in oligodendrocyte lineage cells [183]. On the other hand, ADAM17 was also shown to be a negative regulator of myelination in the PNS. Neuregulin-1 (NRG1) type III is known to induce myelination. ADAM17 processes NRG1 type III, thus decreasing axonal levels of this functional protein and thereby acting as a negative regulator of myelination in the PNS [185]. This process can be induced by glutamate [186], whereas tissue inhibitor of metalloproteases-3 (TIMP-3), an endogenous inhibitor for ADAM17, reverses this process [187]. Additionally, a different study reported that the soluble ectodomain of NRG1 type III, which is liberated by ADAM17 cleavage, can conversely induce myelination in a paracrine manner by triggering signaling through the ErbB3 receptor [188].
ADAM17 regulates neural plasticity
An additional function for ADAM17 is in neural plasticity, which is the ability of the brain to change its neural connections throughout life. This is linked to the shedding of neuronal pentraxin receptor (NPR) by ADAM17 [189]. NPR is primarily expressed at excitatory synapses, where it associates with AMPA-type glutamate receptors and contributes to synapse formation. In addition, ADAM17 can support axon regeneration by processing neogenin, a receptor for repulsive guidance molecules that inhibits neurite growth [190, 191]. A number of brain proteins have been identified as ADAM17 substrates, including p75 neurotrophin receptor, L1, CADM1 and NCAM [192, 193, 194]. Shedding of these proteins by ADAM17 regulates several processes in the brain, such as neuron survival, neural cell adhesion, synaptic formation and neurite outgrowth. Moreover, ADAM17 is involved in the regulation of brain renin-angiotensin system, as it mediates shedding of angiotensin converting enzyme 2 (ACE2) in neurons, and its aberrant activity leads to neurogenic hypertension [195, 196].
ADAM17 in glioblastoma
ADAM17 plays a key role in the progression of glioblastoma, the most common form of primary malignant brain tumor [197]. This is most likely due to its ability to shed EGF-like growth factors and trigger EGFR signaling. In agreement, inhibitors of this pathway have been proven to decrease glioblastoma migration and invasiveness [198]. TNFα release and the subsequent NF-κB activation are also associated with poor prognosis for glioblastoma. Fibulin-3 was found to trigger the NF-κB pathway in this tumor. Mechanistically, this occurs by Fibulin-3 binding to TIMP-3 and prevents ADAM17 from inhibition by TIMP-3, resulting in the sustained release of TNFα [199].
ADAM17 in Alzheimer’s disease: a double edged sword?
ADAM17 is known to be the “stimulated α-secretase” for its ability to cleave the Alzheimer’s disease (AD)-linked APP in response to different stimuli, including phorbol esters and M1 muscarinic receptor activation, thus playing a potentially crucial role in the pathophysiology of AD [200–203]. Similarly to ADAM10, which acts as the neuronal α-secretase under non-stimulated, constitutive conditions [91, 92, 138], ADAM17-mediated cleavage of APP releases its soluble ectodomain (sAPPα), thereby initiating the neuroprotective non-amyloidogenic pathway of APP processing and potentially reducing generation of the pathogenic Aβ peptide. Although this indicates a protective role for neuronal ADAM17 in the pathogenesis of AD, a study ultimately confirming these findings in vivo is still missing, mainly due to the perinatal lethality of ADAM17 knockout mice. Nevertheless, in support of this hypothesis it was recently reported that a rare variant leading to loss-of-function of ADAM17 is associated with the pathogenesis of AD in human [204], and berberine, an alkaloid compound extracted from plants, has neuroprotective effects on AD patients by stimulating the α-secretase activity [205].
Interestingly, while neuronal ADAM17 may have protective effects on the development of AD by triggering the non-amyloidogenic pathway, microglial ADAM17 may have the opposite effects. In agreement with this hypothesis, the ADAM17 substrate TNFα has emerged to be involved in the pathogenesis of AD, where neuroinflammation is a crucial component of the disease, linking amyloid deposition to neuronal loss. Amyloid induces microglia to release pro-inflammatory cytokines, including TNFα, that induce neuronal death. Ablation of TNFα in a mouse model of AD (PDAPP) lowered cognitive decline without affecting deposition of Aβ plaques [206]. Similarly, ablation of the TNFα target receptor TNFR1 in murine models of AD proved beneficial in preventing learning and memory deficits associated with the disease [207]. Finally, therapies based on TNFα inhibitors have been tested in AD mouse models and AD patients in a phase II clinical trial with encouraging results in slowing down the progression of the disease [208–210]. However, the number of treated patients was low and larger cohorts will be needed to allow more definitive conclusions. Furthermore, ADAM17 can contribute to the stimulated shedding of triggering receptor expressed in myeloid cells (TREM2), which—under constitutive conditions—is cleaved by ADAM10 [211–213]. Trem2 is a genetic risk factor for AD and major regulator of neuroinflammatory component of the disease and a reduction of its shedding may be beneficial [214–216]. Other than TNFα and TREM2, ADAM17 regulates a number of microglial proteins that act in neuroinflammation and progression of the disease, including AXL [217], CSF1R [218, 219] and the Aβ receptor LRP-1 [167, 220], indicating that the role of microglial ADAM17 in the development of AD may go beyond the cleavage of TNFα and TREM2.
iRhoms
ADAM17 plays a critical role in several biological processes. Its activity has to be finely regulated and this occurs at different levels, including post-translational activation of the pro-enzyme by furin convertase and inhibition by endogenous inhibitor TIMP-3 [187, 221–223]. Interestingly, while these regulatory mechanisms are shared by different ADAMs, new exciting insights into the regulation of ADAM17 function have recently emerged, revealing ADAM17-specific regulators that have high relevance in the brain. Two inactive homologues of rhomboid proteases, also known as iRhom1 (RHBDF1) and iRhom2 (RHBDF2), have been identified as crucial upstream regulators of ADAM17. They bind ADAM17 and guide ADAM17 maturation throughout the secretory pathway and are essential for ADAM17 activity. iRhoms mediate the activation of ADAM17 in response to G-protein coupled receptor (GPCR) stimulation [57, 224, 225]. Furthermore, iRhoms can address ADAM17 activity towards specific membrane proteins for their subsequent shedding, thus controlling substrate selectivity of the enzyme [225]. This evidence suggests that iRhoms can be considered not only as regulators of ADAM17 activity, but as a non-proteolytic, regulatory subunit of an ADAM17/iRhom complex. In agreement, mice lacking both iRhoms resemble mice lacking ADAM17, which have open eyes at birth and die perinatally [226]. Interestingly, mice lacking iRhom2 do not display major abnormalities [227, 228]. For iRhom1 two different knockout mouse lines were generated. One of them did not show a phenotype but the other one showed increased lethality after birth [226, 229]. Yet, both mice have a less severe phenotype than ADAM17 knockout mice, suggesting that either iRhom can have compensatory or redundant functions in the absence of the other. Both iRhoms are ubiquitously expressed in all tissues. The brain represents an interesting case, where iRhom2 is mainly expressed in microglia and iRhom1 is mainly expressed in neurons, astrocytes and oligodendrocytes [101, 226]. This separation may imply differential functions for either iRhom in the brain, with iRhom2 playing a role in microglial processes and neuroinflammation, and iRhom1 regulating physiological molecular mechanisms in other brain cells [230].
iRhom2 emerged as an epigenetic risk factor in AD, where altered promoter methylation leads to higher iRhom2 expression in AD patients [231]. Although the role of iRhom2 in AD is not elucidated at this point, it is conceivable that higher expression of iRhom2 leads to increased TNFα release by ADAM17 in microglia and subsequently enhanced neuroinflammation. An iRhom2-targeted therapy would also be preferential to an anti-TNFα therapy. Anti-TNFα molecules prevent the binding of soluble TNFα to TNFR1, but also block the interaction between membrane-tethered TNF and TNFR2, which activates an anti-inflammatory pathway that plays a beneficial role in the disease [232]. On the other hand, inactivation of iRhom2 would only impact the pro-inflammatory function of TNFα, but not its anti-inflammatory properties.
In conclusion, direct evidences of ADAM17 functions in the brain have been difficult to uncover due to the lethality of ADAM17 knockout mice. Many ADAM17 substrates were initially identified outside the CNS, but a number of them are also known to play important roles in the CNS. This suggests that ADAM17 functions in the brain will be broader than the few elucidated so far. ADAM17 is a key actor in the development of AD for its ability to cleave APP and other proteins involved in neuroinflammation, including TNFα and TREM2. The differential expression of ADAM17 regulators iRhom1 and iRhom2 in different brain cells may offer a unique therapeutic opportunity in the context of AD.
ADAM19: a role in PNS regeneration
ADAM19, also known as meltrin-β, is a catalytically active metalloproteinase that cleaves a number of canonical ADAM substrates in vitro, including KL1 and TNFα [233]. Unlike most of the ADAMs, the activity of ADAM19 is not inhibited by TIMPs [233]. Although ADAM17 is the established major physiological TNFα sheddase, the constitutive release of TNFα is increased in cells overexpressing ADAM19, suggesting that the enzyme may contribute to its release in cells or tissues where it is highly expressed [234].
During mouse development, ADAM19 is highly expressed in craniofacial and dorsal root ganglia and ventral horns of the spinal cord, where peripheral neuronal cell lineages differentiate [235]. This expression pattern overlaps with that of neuregulin-1, a member of a group of growth factors that are essential for CNS development [236, 237].
ADAM19 mediates the ectodomain shedding of α-type neuregulins in dorsal root ganglia neurons [238]. Due to the overlapping expression pattern during embryogenesis, it is conceivable that ADAM19 may play a role in the biological processes involving neuregulins, although such hypothesis has to be proven in vivo [238] and a role of ADAM19 in processing neuregulins could not be confirm yet [237]. In addition, ADAM19 plays a key role in nerve regeneration after a crush injury to the sciatic nerves [239]. Although the molecular mechanism is not entirely elucidated, ablation of ADAM19 delayed sciatic functional recovery after injury.
ADAM19 also plays a role in the ephrin-A5–EphA4 signaling pathway through a non-proteolytic pattern and influences formation of the neuromuscular junction in murine embryos [240]. Finally, overexpressed ADAM19 possesses α-secretase activity in A172 cells, i.e., it is able to cleave the AD-linked APP within the Aβ domain. Nevertheless, its contribution to the total APP shedding appears to be minor compared to that of ADAM10, as RNA interference-mediated knock-down of ADAM19 only mildly reduced APP cleavage by about 20% [241].
In conclusion, ADAM19 is a catalytically active ADAM that presents some unique features among members of this family of proteinases. It cleaves prototypic ADAM substrates, including TNFα and APP, but to a lesser extent as compared to other ADAMs. Nevertheless, cleavage of such molecules may have functional consequences in specific cells or conditions in which ADAM19 is expressed at high levels.
ADAM21: a connection to neuroregeneration?
ADAM21 (also called ADAM31) is an ADAM family member with an intact active site for proteolysis, but proteolytic substrates have not yet been identified. ADAM21 was initially thought to be predominantly expressed in testis [242–244], but is also linked to hepatocellular carcinoma [198] and is expressed in other tissues, including brain [245]. In rodent brain, ADAM21 is robustly expressed in neuronal precursor cells of the subventricular zone (SVZ) and in processes of neurons and neuroblasts in the rostral migratory stream [245]. ADAM21 expression is found during development as well as in the adult rodent brain. In the embryo, it is found in developing axons whereas in the adult brain it is highly expressed in the olfactory nerve—a region known for constant neuronal regeneration. It is not yet clear whether the protease function or rather the presumed integrin-binding ability of ADAM21—through its disintegrin domain—is required for these neuronal functions. Yet, the tissue inhibitor of metalloproteases 3 (TIMP-3), which blocks distinct ADAM proteases, shows a partial co-expression with ADAM21 in brain [245], suggesting that the proteolytic function of ADAM21 may indeed be contributing to its function in brain. Together, these results suggest a function for ADAM21 in axon outgrowth and synapse formation, but more studies are needed to better understand the function of ADAM21.
ADAM30: a link to Alzheimer’s disease
ADAM30 is an understudied member of the ADAM family and is proteolytically active. While a first report suggested a testis-specific expression [246], ADAM30 is now known to also be expressed in human brain and specifically in neurons. Interestingly, ADAM30 is not expressed in mouse brains and may not be expressed in mice altogether. ADAM30 has been linked to AD. In postmortem AD brains, the expression level of ADAM30 is reduced up to 50% and negatively correlates with brain levels of Aβ [247], the highly aggregating and neurotoxic peptide in AD [248]. Conversely, overexpression of ADAM30 in mouse forebrain neurons of an AD mouse model lowered Aβ generation. Mechanistically, ADAM30 triggers trafficking of full-length APP to lysosomes, activation of the lysosomal protease cathepsin D and increased APP degradation, thus lowering the amount of APP being available for Aβ production [247]. Taken together, reduced ADAM30 levels may contribute to AD pathogenesis, whereas ADAM30 activation may be a potential new therapeutic approach for AD.
ADAM33: potential roles in cranial neural crest migration
ADAM33 is a catalytically active ADAM. In Xenopus, the gene is also referred to as ADAM13 [249], where its function is required for cranial neural crest migration [250]. In humans, ADAM33 has been mostly studied in the context of asthma [251, 252]. Although it is widely expressed in adult mice with the highest levels detected in the brain, little is known about the ADAM33 functions in mammals, especially in the nervous system [253]. ADAM33 cleaves a number of canonical ADAM substrates in vitro, including APP, KL-1, and TNFα [254, 255]. The catalytic activity of ADAM33, its expression pattern and its similarity to Xenopus ADAM13, whose metalloprotease activity is necessary for cranial neural crest-cell migration [256], suggest a role for this enzyme in the development of mammalian brain.
Conclusions and outlook
A few ADAMs, in particular the proteolytic ADAM10 and the non-proteolytic ADAM22 and 23, have been functionally relatively well characterized in the nervous system. Yet, even for those ADAMs new functions and/or substrates continue to be identified, partly due to methodological advances in proteomics. For many other ADAMs, considerable advances in recent years demonstrated that these ADAMs are not only expressed in the mammalian nervous system, but also have important roles in developmental processes or be linked to pathological conditions, ranging from glioma to neurodegeneration and presumably more in the future. Thus, ADAMs may become potential drug targets for different disorders in the nervous system. Yet, in many cases, such as for ADAM30 and its newly recognized role in AD, the underlying molecular mechanisms need to be elucidated. Moreover, some ADAMs may be particularly relevant under specific conditions, such as during inflammation. For the proteolytic ADAMs, ADAM10 and a few other ones may be mostly shedding membrane proteins under constitutive, non-stimulated conditions, whereas ADAM17 may be particularly relevant as regulated proteases that enhance substrates cleavage upon a given stimulus. It will also be interesting to see whether the proteolytic ADAMs additionally have non-proteolytic functions, similar to what has been described for matrix metalloprotease 12 (MMP-12) [257] and β-site APP cleaving enzyme 1 (BACE1) [258]. Given that proteolytic and non-proteolytic ADAMs have similar domain structures, this possibility is not unlikely. Taken together, while the function of ADAMs in the nervous system remains—with a few exceptions—a relatively uncharted territory, new exciting functions are emerging and we can await new discoveries over the years to come.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (German Research Foundation) within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy) and the research unit FOR2290, by the Centers of Excellence in Neurodegeneration, and the Boehringer Ingelheim Fonds.
Abbreviations
- ADAM
A disintegrin and metalloprotease
- LGI
Leucine-rich glioma-inactivated protein
- PSD-95
Postsynaptic density protein-95
- CNS
Central nervous system
- PNS
Peripheral nervous system
- AMPA
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- EGF
Epidermal growth factor
- SC
Schwann cell
- PrPc
Cellular prion protein
- PrPsc
Pathological prion protein
- ECM
Extracellular matrix
- APP
Amyloid precursor protein
- AD
Alzheimer’s disease
- NICD
Notch intracellular domain
- NCAM
Neuronal cell adhesion molecule
- Nrp1
Neuropilin1
- NrCAM
Neural glial-related cell adhesion molecule
- NLGN
Neuroligin
- NRXN
Neurexin
- OPCs
Oligodendrocyte precursor cells
- NG2
Nerve–glia antigen 2
- Aβ
Amyloid β
- TSPAN
Tetraspanin
- LRP-1
Low density lipoprotein receptor-related protein 1
- HB-EGF
Heparin-binding epidermal growth factor-like growth factor
- NRG1
Neuregulin-1
- TREM2
Triggering receptor expressed in myeloid cells 2
- TNFα
Tumor necrosis factor α
- TIMP
Tissue inhibitor of metalloproteases
- NPR
Neuronal pentraxin receptor
- KL1
Kit ligand-1
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lichtenthaler SF, Lemberg MK, Fluhrer R. Proteolytic ectodomain shedding of membrane proteins in mammals-hardware, concepts, and recent developments. EMBO J. 2018;37(15):e99456. doi: 10.15252/embj.201899456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cho C. Testicular and epididymal ADAMs: expression and function during fertilization. Natre Rev Urol. 2012;9(10):550–560. doi: 10.1038/nrurol.2012.167. [DOI] [PubMed] [Google Scholar]
- 3.Weber S, Saftig P. Ectodomain shedding and ADAMs in development. Development. 2012;139(20):3693–3709. doi: 10.1242/dev.076398. [DOI] [PubMed] [Google Scholar]
- 4.Mullooly M, McGowan PM, Crown J, Duffy MJ. The ADAMs family of proteases as targets for the treatment of cancer. Cancer Biol Ther. 2016;17(8):870–880. doi: 10.1080/15384047.2016.1177684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stocker W, Grams F, Baumann U, Reinemer P, Gomis-Ruth FX, McKay DB, Bode W. The metzincins—topological and sequential relations between the astacins, adamalysins, serralysins, and matrixins (collagenases) define a superfamily of zinc-peptidases. Protein Sci. 1995;4(5):823–840. doi: 10.1002/pro.5560040502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolfsberg TG, White JM. ADAMs in fertilization and development. Dev Biol. 1996;180(2):389–401. doi: 10.1006/dbio.1996.0313. [DOI] [PubMed] [Google Scholar]
- 7.Stone AL, Kroeger M, Sang QX. Structure–function analysis of the ADAM family of disintegrin-like and metalloproteinase-containing proteins (review) J Protein Chem. 1999;18(4):447–465. doi: 10.1023/A:1020692710029. [DOI] [PubMed] [Google Scholar]
- 8.Schlomann U, Wildeboer D, Webster A, Antropova O, Zeuschner D, Knight CG, Docherty AJ, Lambert M, Skelton L, Jockusch H, Bartsch JW. The metalloprotease disintegrin ADAM8. Processing by autocatalysis is required for proteolytic activity and cell adhesion. J Biol Chem. 2002;277(50):48210–48219. doi: 10.1074/jbc.M203355200. [DOI] [PubMed] [Google Scholar]
- 9.Schlondorff J, Becherer JD, Blobel CP. Intracellular maturation and localization of the tumour necrosis factor alpha convertase (TACE) Biochem J. 2000;347(Pt 1):131–138. doi: 10.1042/bj3470131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brummer T, Pigoni M, Rossello A, Wang H, Noy PJ, Tomlinson MG, Blobel CP, Lichtenthaler SF. The metalloprotease ADAM10 (a disintegrin and metalloprotease 10) undergoes rapid, postlysis autocatalytic degradation. FASEB J. 2018;32(7):3560–3573. doi: 10.1096/fj.201700823RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seals DF, Courtneidge SA. The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 2003;17(1):7–30. doi: 10.1101/gad.1039703. [DOI] [PubMed] [Google Scholar]
- 12.Giebeler N, Zigrino P. A disintegrin and metalloprotease (ADAM): historical overview of their functions. Toxins (Basel) 2016;8(4):122. doi: 10.3390/toxins8040122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klein T, Bischoff R. Active metalloproteases of the A Disintegrin and Metalloprotease (ADAM) family: biological function and structure. J Proteome Res. 2011;10(1):17–33. doi: 10.1021/pr100556z. [DOI] [PubMed] [Google Scholar]
- 14.Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med. 2008;29(5):258–289. doi: 10.1016/j.mam.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sagane K, Yamazaki K, Mizui Y, Tanaka I. Cloning and chromosomal mapping of mouse ADAM11, ADAM22 and ADAM23. Gene. 1999;236(1):79–86. doi: 10.1016/S0378-1119(99)00253-X. [DOI] [PubMed] [Google Scholar]
- 16.Kegel L, Aunin E, Meijer D, Bermingham JR. LGI proteins in the nervous system. ASN Neuro. 2013;5(3):167–181. doi: 10.1042/AN20120095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rybnikova E, Karkkainen I, Pelto-Huikko M, Huovila AP. Developmental regulation and neuronal expression of the cellular disintegrin ADAM11 gene in mouse nervous system. Neuroscience. 2002;112(4):921–934. doi: 10.1016/S0306-4522(02)00124-0. [DOI] [PubMed] [Google Scholar]
- 18.Kole MJ, Qian J, Waase MP, Klassen TL, Chen TT, Augustine GJ, Noebels JL. Selective loss of presynaptic potassium channel clusters at the cerebellar basket cell terminal pinceau in Adam11 mutants reveals their role in ephaptic control of Purkinje cell firing. J Neurosci. 2015;35(32):11433–11444. doi: 10.1523/JNEUROSCI.1346-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blot A, Barbour B. Ultra-rapid axon-axon ephaptic inhibition of cerebellar Purkinje cells by the pinceau. Nat Neurosci. 2014;17(2):289–295. doi: 10.1038/nn.3624. [DOI] [PubMed] [Google Scholar]
- 20.Xie G, Harrison J, Clapcote SJ, Huang Y, Zhang JY, Wang LY, Roder JC. A new Kv1.2 channelopathy underlying cerebellar ataxia. J Biol Chem. 2010;285(42):32160–32173. doi: 10.1074/jbc.M110.153676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou L, Zhang CL, Messing A, Chiu SY. Temperature-sensitive neuromuscular transmission in Kv1.1 null mice: role of potassium channels under the myelin sheath in young nerves. J Neurosci. 1998;18(18):7200–7215. doi: 10.1523/JNEUROSCI.18-18-07200.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takahashi E, Sagane K, Nagasu T, Kuromitsu J. Altered nociceptive response in ADAM11-deficient mice. Brain Res. 2006;1097(1):39–42. doi: 10.1016/j.brainres.2006.04.043. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi E, Sagane K, Oki T, Yamazaki K, Nagasu T, Kuromitsu J. Deficits in spatial learning and motor coordination in ADAM11-deficient mice. BMC Neurosci. 2006;7:19. doi: 10.1186/1471-2202-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen N, Koopmans F, Gordon A, Paliukhovich I, Klaassen RV, van der Schors RC, Peles E, Verhage M, Smit AB. Li KW (2015) Interaction proteomics of canonical Caspr2 (CNTNAP2) reveals the presence of two Caspr2 isoforms with overlapping interactomes. Biochim Biophys Acta. 1854;7:827–833. doi: 10.1016/j.bbapap.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 25.Sagane K, Ishihama Y, Sugimoto H. LGI1 and LGI4 bind to ADAM22, ADAM23 and ADAM11. Int J Biol Sci. 2008;4(6):387–396. doi: 10.7150/ijbs.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Hoggard JA, Korleski ED, Long GV, Ree BC, Hensley K, Bond SR, Wolfsberg TG, Chen J, Zeczycki TN, Bridges LC. Multiple non-catalytic ADAMs are novel integrin alpha4 ligands. Mol Cell Biochem. 2018;442(1–2):29–38. doi: 10.1007/s11010-017-3190-y. [DOI] [PubMed] [Google Scholar]
- 27.Sagane K, Ohya Y, Hasegawa Y, Tanaka I. Metalloproteinase-like, disintegrin-like, cysteine-rich proteins MDC2 and MDC3: novel human cellular disintegrins highly expressed in the brain. Biochem J. 1998;334(Pt 1):93–98. doi: 10.1042/bj3340093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sagane K, Hayakawa K, Kai J, Hirohashi T, Takahashi E, Miyamoto N, Ino M, Oki T, Yamazaki K, Nagasu T. Ataxia and peripheral nerve hypomyelination in ADAM22-deficient mice. BMC Neurosci. 2005;6:33. doi: 10.1186/1471-2202-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng S, Gray EE, Chawla G, Porse BT, O’Dell TJ, Black DL. PSD-95 is post-transcriptionally repressed during early neural development by PTBP1 and PTBP2. Nat Neurosci. 2012;15(3):381–388, s381. doi: 10.1038/nn.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science (New York, NY) 2006;313(5794):1792–1795. doi: 10.1126/science.1129947. [DOI] [PubMed] [Google Scholar]
- 31.Lovero KL, Fukata Y, Granger AJ, Fukata M, Nicoll RA. The LGI1-ADAM22 protein complex directs synapse maturation through regulation of PSD-95 function. Proc Natl Acad Sci USA. 2015;112(30):E4129–4137. doi: 10.1073/pnas.1511910112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fukata Y, Lovero KL, Iwanaga T, Watanabe A, Yokoi N, Tabuchi K, Shigemoto R, Nicoll RA, Fukata M. Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc Natl Acad Sci USA. 2010;107(8):3799–3804. doi: 10.1073/pnas.0914537107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dazzo E, Leonardi E, Belluzzi E, Malacrida S, Vitiello L, Greggio E, Tosatto SC, Nobile C. Secretion-positive LGI1 mutations linked to lateral temporal epilepsy impair binding to ADAM22 and ADAM23 receptors. PLoS Genet. 2016;12(10):e1006376. doi: 10.1371/journal.pgen.1006376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yokoi N, Fukata Y, Kase D, Miyazaki T, Jaegle M, Ohkawa T, Takahashi N, Iwanari H, Mochizuki Y, Hamakubo T, Imoto K, Meijer D, Watanabe M, Fukata M. Chemical corrector treatment ameliorates increased seizure susceptibility in a mouse model of familial epilepsy. Nat Med. 2015;21(1):19–26. doi: 10.1038/nm.3759. [DOI] [PubMed] [Google Scholar]
- 35.Fukata Y, Yokoi N, Miyazaki Y, Fukata M. The LGI1-ADAM22 protein complex in synaptic transmission and synaptic disorders. Neurosci Res. 2017;116:39–45. doi: 10.1016/j.neures.2016.09.011. [DOI] [PubMed] [Google Scholar]
- 36.Kegel L, Jaegle M, Driegen S, Aunin E, Leslie K, Fukata Y, Watanabe M, Fukata M, Meijer D. Functional phylogenetic analysis of LGI proteins identifies an interaction motif crucial for myelination. Development. 2014;141(8):1749–1756. doi: 10.1242/dev.107995. [DOI] [PubMed] [Google Scholar]
- 37.Muona M, Fukata Y, Anttonen AK, Laari A, Palotie A, Pihko H, Lonnqvist T, Valanne L, Somer M, Fukata M, Lehesjoki AE. Dysfunctional ADAM22 implicated in progressive encephalopathy with cortical atrophy and epilepsy. Neurol Genet. 2016;2(1):e46. doi: 10.1212/NXG.0000000000000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turetsky D, Garringer E, Patneau DK. Stargazin modulates native AMPA receptor functional properties by two distinct mechanisms. J Neurosci. 2005;25(32):7438–7448. doi: 10.1523/jneurosci.1108-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noebels JL, Qiao X, Bronson RT, Spencer C, Davisson MT. Stargazer: a new neurological mutant on chromosome 15 in the mouse with prolonged cortical seizures. Epilepsy Res. 1990;7(2):129–135. doi: 10.1016/0920-1211(90)90098-G. [DOI] [PubMed] [Google Scholar]
- 40.Nicoll RA, Tomita S, Bredt DS. Auxiliary subunits assist AMPA-type glutamate receptors. Science (New York, NY) 2006;311(5765):1253–1256. doi: 10.1126/science.1123339. [DOI] [PubMed] [Google Scholar]
- 41.Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L, Balice-Gordon R, Cowell JK, Dalmau J. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol. 2010;9(8):776–785. doi: 10.1016/s1474-4422(10)70137-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuliani L, Peles E, Buckley C, Lang B, Vincent A. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain. 2010;133(9):2734–2748. doi: 10.1093/brain/awq213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohkawa T, Fukata Y, Yamasaki M, Miyazaki T, Yokoi N, Takashima H, Watanabe M, Watanabe O, Fukata M. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1–ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci. 2013;33(46):18161–18174. doi: 10.1523/jneurosci.3506-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ogawa Y, Oses-Prieto J, Kim MY, Horresh I, Peles E, Burlingame AL, Trimmer JS, Meijer D, Rasband MN. ADAM22, a Kv1 channel-interacting protein, recruits membrane-associated guanylate kinases to juxtaparanodes of myelinated axons. J Neurosci. 2010;30(3):1038–1048. doi: 10.1523/jneurosci.4661-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schulte U, Thumfart JO, Klocker N, Sailer CA, Bildl W, Biniossek M, Dehn D, Deller T, Eble S, Abbass K, Wangler T, Knaus HG, Fakler B. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron. 2006;49(5):697–706. doi: 10.1016/j.neuron.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 46.Boillot M, Huneau C, Marsan E, Lehongre K, Navarro V, Ishida S, Dufresnois B, Ozkaynak E, Garrigue J, Miles R, Martin B, Leguern E, Anderson MP, Baulac S. Glutamatergic neuron-targeted loss of LGI1 epilepsy gene results in seizures. Brain. 2014;137(Pt 11):2984–2996. doi: 10.1093/brain/awu259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mitchell KJ, Pinson KI, Kelly OG, Brennan J, Zupicich J, Scherz P, Leighton PA, Goodrich LV, Lu X, Avery BJ, Tate P, Dill K, Pangilinan E, Wakenight P, Tessier-Lavigne M, Skarnes WC. Functional analysis of secreted and transmembrane proteins critical to mouse development. Nat Genet. 2001;28(3):241–249. doi: 10.1038/90074. [DOI] [PubMed] [Google Scholar]
- 48.Owuor K, Harel NY, Englot DJ, Hisama F, Blumenfeld H, Strittmatter SM. LGI1-associated epilepsy through altered ADAM23-dependent neuronal morphology. Mol Cell Neurosci. 2009;42(4):448–457. doi: 10.1016/j.mcn.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamagata A, Miyazaki Y, Yokoi N, Shigematsu H, Sato Y, Goto-Ito S, Maeda A, Goto T, Sanbo M, Hirabayashi M, Shirouzu M, Fukata Y, Fukata M, Fukai S. Structural basis of epilepsy-related ligand-receptor complex LGI1-ADAM22. Nat Commun. 2018;9(1):1546. doi: 10.1038/s41467-018-03947-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leonardi E, Andreazza S, Vanin S, Busolin G, Nobile C, Tosatto SC. A computational model of the LGI1 protein suggests a common binding site for ADAM proteins. PLoS One. 2011;6(3):e18142. doi: 10.1371/journal.pone.0018142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu H, Shim AH, He X. Structural characterization of the ectodomain of a disintegrin and metalloproteinase-22 (ADAM22), a neural adhesion receptor instead of metalloproteinase: insights on ADAM function. J Biol Chem. 2009;284(42):29077–29086. doi: 10.1074/jbc.M109.014258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seegar TCM, Killingsworth LB, Saha N, Meyer PA, Patra D, Zimmerman B, Janes PW, Rubinstein E, Nikolov DB, Skiniotis G, Kruse AC, Blacklow SC. Structural basis for regulated proteolysis by the alpha-secretase ADAM10. Cell. 2017;171(7):1638–1648.e1637. doi: 10.1016/j.cell.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ozkaynak E, Abello G, Jaegle M, van Berge L, Hamer D, Kegel L, Driegen S, Sagane K, Bermingham JR, Jr, Meijer D. Adam22 is a major neuronal receptor for Lgi4-mediated Schwann cell signaling. J Neurosci. 2010;30(10):3857–3864. doi: 10.1523/JNEUROSCI.6287-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xue S, Maluenda J, Marguet F, Shboul M, Quevarec L, Bonnard C, Ng AY, Tohari S, Tan TT, Kong MK, Monaghan KG, Cho MT, Siskind CE, Sampson JB, Rocha CT, Alkazaleh F, Gonzales M, Rigonnot L, Whalen S, Gut M, Gut I, Bucourt M, Venkatesh B, Laquerriere A, Reversade B, Melki J. Loss-of-function mutations in LGI4, a secreted ligand involved in Schwann cell myelination, are responsible for arthrogryposis multiplex congenita. Am J Hum Genet. 2017;100(4):659–665. doi: 10.1016/j.ajhg.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nishino J, Saunders TL, Sagane K, Morrison SJ. Lgi4 promotes the proliferation and differentiation of glial lineage cells throughout the developing peripheral nervous system. J Neurosci. 2010;30(45):15228–15240. doi: 10.1523/JNEUROSCI.2286-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Godde NJ, D’Abaco GM, Paradiso L, Novak U. Efficient ADAM22 surface expression is mediated by phosphorylation-dependent interaction with 14-3-3 protein family members. J Cell Sci. 2006;119(Pt 16):3296–3305. doi: 10.1242/jcs.03065. [DOI] [PubMed] [Google Scholar]
- 57.Grieve AG, Xu H, Kunzel U, Bambrough P, Sieber B, Freeman M. Phosphorylation of iRhom2 at the plasma membrane controls mammalian TACE-dependent inflammatory and growth factor signalling. Elife. 2017;6:e23968. doi: 10.7554/eLife.23968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goldsmith AP, Gossage SJ, ffrench-Constant C. ADAM23 is a cell-surface glycoprotein expressed by central nervous system neurons. J Neurosci Res. 2004;78(5):647–658. doi: 10.1002/jnr.20320. [DOI] [PubMed] [Google Scholar]
- 59.Koskinen LL, Seppala EH, Weissl J, Jokinen TS, Viitmaa R, Hanninen RL, Quignon P, Fischer A, Andre C, Lohi H. ADAM23 is a common risk gene for canine idiopathic epilepsy. BMC Genet. 2017;18(1):8. doi: 10.1186/s12863-017-0478-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Markus-Koch A, Schmitt O, Seemann S, Lukas J, Koczan D, Ernst M, Fuellen G, Wree A, Rolfs A, Luo J. ADAM23 promotes neuronal differentiation of human neural progenitor cells. Cell Mol Biol Lett. 2017;22:16. doi: 10.1186/s11658-017-0045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Costa MD, Paludo KS, Klassen G, Lopes MH, Mercadante AF, Martins VR, Camargo AA, Nakao LS, Zanata SM. Characterization of a specific interaction between ADAM23 and cellular prion protein. Neurosci Lett. 2009;461(1):16–20. doi: 10.1016/j.neulet.2009.05.049. [DOI] [PubMed] [Google Scholar]
- 62.Cal S, Freije JM, Lopez JM, Takada Y, Lopez-Otin C. ADAM 23/MDC3, a human disintegrin that promotes cell adhesion via interaction with the alphavbeta3 integrin through an RGD-independent mechanism. Mol Biol Cell. 2000;11(4):1457–1469. doi: 10.1091/mbc.11.4.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Elizondo DM, Andargie TE, Marshall KM, Zariwala AM, Lipscomb MW. Dendritic cell expression of ADAM23 governs T cell proliferation and cytokine production through the alpha(v)beta(3) integrin receptor. J Leukoc Biol. 2016;100(5):855–864. doi: 10.1189/jlb.2HI1115-525R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Calmon MF, Colombo J, Carvalho F, Souza FP, Filho JF, Fukuyama EE, Camargo AA, Caballero OL, Tajara EH, Cordeiro JA, Rahal P. Methylation profile of genes CDKN2A (p14 and p16), DAPK1, CDH1, and ADAM23 in head and neck cancer. Cancer Genet Cytogenet. 2007;173(1):31–37. doi: 10.1016/j.cancergencyto.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 65.Kwon NS, Kim DS, Yun HY. Leucine-rich glioma inactivated 3: integrative analyses support its prognostic role in glioma. OncoTargets Ther. 2017;10:2721–2728. doi: 10.2147/ott.s138912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Verbisck NV, Costa ET, Costa FF, Cavalher FP, Costa MD, Muras A, Paixao VA, Moura R, Granato MF, Ierardi DF, Machado T, Melo F, Ribeiro KB, Cunha IW, Lima VC, Maciel Mdo S, Carvalho AL, Soares FF, Zanata S, Sogayar MC, Chammas R, Camargo AA. ADAM23 negatively modulates alpha(v)beta(3) integrin activation during metastasis. Cancer Res. 2009;69(13):5546–5552. doi: 10.1158/0008-5472.can-08-2976. [DOI] [PubMed] [Google Scholar]
- 67.Dkhil MA, Bauomy AA, Diab MS, Wahab R, Delic D, Al-Quraishy S. Impact of gold nanoparticles on brain of mice infected with Schistosoma mansoni. Parasitol Res. 2015;114(10):3711–3719. doi: 10.1007/s00436-015-4600-2. [DOI] [PubMed] [Google Scholar]
- 68.Mubaraki MA, Hafiz TA, Al-Quraishy S, Dkhil MA. Oxidative stress and genes regulation of cerebral malaria upon Zizyphus spina-christi treatment in a murine model. Microb Pathog. 2017;107:69–74. doi: 10.1016/j.micpath.2017.03.017. [DOI] [PubMed] [Google Scholar]
- 69.Yoshida S, Setoguchi M, Higuchi Y, Akizuki S, Yamamoto S. Molecular cloning of cDNA encoding MS2 antigen, a novel cell surface antigen strongly expressed in murine monocytic lineage. Int Immunol. 1990;2(6):585–591. doi: 10.1093/intimm/2.6.585. [DOI] [PubMed] [Google Scholar]
- 70.Yoshiyama K, Higuchi Y, Kataoka M, Matsuura K, Yamamoto S. CD156 (human ADAM8): expression, primary amino acid sequence, and gene location. Genomics. 1997;41(1):56–62. doi: 10.1006/geno.1997.4607. [DOI] [PubMed] [Google Scholar]
- 71.Kelly K, Hutchinson G, Nebenius-Oosthuizen D, Smith AJ, Bartsch JW, Horiuchi K, Rittger A, Manova K, Docherty AJ, Blobel CP. Metalloprotease–disintegrin ADAM8: expression analysis and targeted deletion in mice. Dev Dyn. 2005;232(1):221–231. doi: 10.1002/dvdy.20221. [DOI] [PubMed] [Google Scholar]
- 72.Koller G, Schlomann U, Golfi P, Ferdous T, Naus S, Bartsch JW. ADAM8/MS2/CD156, an emerging drug target in the treatment of inflammatory and invasive pathologies. Curr Pharm Des. 2009;15(20):2272–2281. doi: 10.2174/138161209788682361. [DOI] [PubMed] [Google Scholar]
- 73.Schlomann U, Rathke-Hartlieb S, Yamamoto S, Jockusch H, Bartsch JW. Tumor necrosis factor alpha induces a metalloprotease–disintegrin, ADAM8 (CD 156): implications for neuron-glia interactions during neurodegeneration. J Neurosci. 2000;20(21):7964–7971. doi: 10.1523/JNEUROSCI.20-21-07964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bartsch JW, Wildeboer D, Koller G, Naus S, Rittger A, Moss ML, Minai Y, Jockusch H. Tumor necrosis factor-alpha (TNF-alpha) regulates shedding of TNF-alpha receptor 1 by the metalloprotease–disintegrin ADAM8: evidence for a protease-regulated feedback loop in neuroprotection. J Neurosci. 2010;30(36):12210–12218. doi: 10.1523/JNEUROSCI.1520-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liang J, Wang W, Sorensen D, Medina S, Ilchenko S, Kiselar J, Surewicz WK, Booth SA, Kong Q. Cellular prion protein regulates its own alpha-cleavage through ADAM8 in skeletal muscle. J Biol Chem. 2012;287(20):16510–16520. doi: 10.1074/jbc.M112.360891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mahoney ET, Benton RL, Maddie MA, Whittemore SR, Hagg T. ADAM8 is selectively up-regulated in endothelial cells and is associated with angiogenesis after spinal cord injury in adult mice. J Comp Neurol. 2009;512(2):243–255. doi: 10.1002/cne.21902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shin HY, Kim H, Kwon MJ, Hwang DH, Lee K, Kim BG. Molecular and cellular changes in the lumbar spinal cord following thoracic injury: regulation by treadmill locomotor training. PLoS One. 2014;9(2):e88215. doi: 10.1371/journal.pone.0088215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Naus S, Reipschläger S, Wildeboer D, Lichtenthaler SF, Mitterreiter S, Guan Z, Moss ML, Bartsch JW (2006) Identification of candidate substrates for ectodomain shedding by the metalloprotease-disintegrin ADAM8. Biol Chem 387(3):337–346 [DOI] [PubMed]
- 79.Naus S, Richter M, Wildeboer D, Moss M, Schachner M, Bartsch JW. Ectodomain shedding of the neural recognition molecule CHL1 by the metalloprotease–disintegrin ADAM8 promotes neurite outgrowth and suppresses neuronal cell death. J Biol Chem. 2004;279(16):16083–16090. doi: 10.1074/jbc.M400560200. [DOI] [PubMed] [Google Scholar]
- 80.Zack MD, Malfait AM, Skepner AP, Yates MP, Griggs DW, Hall T, Hills RL, Alston JT, Nemirovskiy OV, Radabaugh MR, Leone JW, Arner EC, Tortorella MD. ADAM-8 isolated from human osteoarthritic chondrocytes cleaves fibronectin at Ala(271) Arthritis Rheum. 2009;60(9):2704–2713. doi: 10.1002/art.24753. [DOI] [PubMed] [Google Scholar]
- 81.Horiuchi K, Weskamp G, Lum L, Hammes HP, Cai H, Brodie TA, Ludwig T, Chiusaroli R, Baron R, Preissner KT, Manova K, Blobel CP. Potential role for ADAM15 in pathological neovascularization in mice. Mol Cell Biol. 2003;23(16):5614–5624. doi: 10.1128/MCB.23.16.5614-5624.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weskamp G, Cai H, Brodie TA, Higashyama S, Manova K, Ludwig T, Blobel CP. Mice lacking the metalloprotease–disintegrin MDC9 (ADAM9) have no evident major abnormalities during development or adult life. Mol Cell Biol. 2002;22(5):1537–1544. doi: 10.1128/MCB.22.5.1537-1544.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol. 2004;164(5):769–779. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ortiz RM, Karkkainen I, Huovila AP, Honkaniemi J. ADAM9, ADAM10, and ADAM15 mRNA levels in the rat brain after kainic acid-induced status epilepticus. Brain Res Mol Brain Res. 2005;137(1–2):272–275. doi: 10.1016/j.molbrainres.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 85.Rybnikova E, Gluschenko T, Galeeva A, Tulkova E, Nalivaeva NN, Makova NZ, Turner AJ, Samoilov M. Differential expression of ADAM15 and ADAM17 metalloproteases in the rat brain after severe hypobaric hypoxia and hypoxic preconditioning. Neurosci Res. 2012;72(4):364–373. doi: 10.1016/j.neures.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 86.Bosse F, Petzold G, Greiner-Petter R, Pippirs U, Gillen C, Muller HW. Cellular localization of the disintegrin CRII-7/rMDC15 mRNA in rat PNS and CNS and regulated expression in postnatal development and after nerve injury. Glia. 2000;32(3):313–327. doi: 10.1002/1098-1136(200012)32:3<313::AID-GLIA100>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 87.Dreymueller D, Uhlig S, Ludwig A. ADAM-family metalloproteinases in lung inflammation: potential therapeutic targets. Am J Physiol Lung Cell Mol Physiol. 2015;308(4):L325–343. doi: 10.1152/ajplung.00294.2014. [DOI] [PubMed] [Google Scholar]
- 88.Marzia M, Guaiquil V, Horne WC, Blobel CP, Baron R, Chiusaroli R. Lack of ADAM15 in mice is associated with increased osteoblast function and bone mass. Biol Chem. 2011;392(10):877–885. doi: 10.1515/BC.2011.080. [DOI] [PubMed] [Google Scholar]
- 89.Parry DA, Toomes C, Bida L, Danciger M, Towns KV, McKibbin M, Jacobson SG, Logan CV, Ali M, Bond J, Chance R, Swendeman S, Daniele LL, Springell K, Adams M, Johnson CA, Booth AP, Jafri H, Rashid Y, Banin E, Strom TM, Farber DB, Sharon D, Blobel CP, Pugh EN, Jr, Pierce EA, Inglehearn CF. Loss of the metalloprotease ADAM9 leads to cone-rod dystrophy in humans and retinal degeneration in mice. Am J Hum Genet. 2009;84(5):683–691. doi: 10.1016/j.ajhg.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Koike H, Tomioka S, Sorimachi H, Saido TC, Maruyama K, Okuyama A, Fujisawa-Sehara A, Ohno S, Suzuki K, Ishiura S. Membrane-anchored metalloprotease MDC9 has an alpha-secretase activity responsible for processing the amyloid precursor protein. Biochem J. 1999;343(Pt 2):371–375. doi: 10.1042/bj3430371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, Kremmer E, Rossner S, Lichtenthaler SF. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010;29(17):3020–3032. doi: 10.1038/emboj.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jorissen E, Prox J, Bernreuther C, Weber S, Schwanbeck R, Serneels L, Snellinx A, Craessaerts K, Thathiah A, Tesseur I, Bartsch U, Weskamp G, Blobel CP, Glatzel M, De Strooper B, Saftig P. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J Neurosci. 2010;30(14):4833–4844. doi: 10.1523/JNEUROSCI.5221-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Colombo A, Wang H, Kuhn PH, Page R, Kremmer E, Dempsey PJ, Crawford HC, Lichtenthaler SF. Constitutive alpha- and beta-secretase cleavages of the amyloid precursor protein are partially coupled in neurons, but not in frequently used cell lines. Neurobiol Dis. 2013;49:137–147. doi: 10.1016/j.nbd.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Amour A, Knight CG, English WR, Webster A, Slocombe PM, Knauper V, Docherty AJ, Becherer JD, Blobel CP, Murphy G. The enzymatic activity of ADAM8 and ADAM9 is not regulated by TIMPs. FEBS Lett. 2002;524(1–3):154–158. doi: 10.1016/S0014-5793(02)03047-8. [DOI] [PubMed] [Google Scholar]
- 95.Cong L, Jia J. Promoter polymorphisms which regulate ADAM9 transcription are protective against sporadic Alzheimer’s disease. Neurobiol Aging. 2011;32(1):54–62. doi: 10.1016/j.neurobiolaging.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 96.Guaiquil V, Swendeman S, Yoshida T, Chavala S, Campochiaro PA, Blobel CP. ADAM9 is involved in pathological retinal neovascularization. Mol Cell Biol. 2009;29(10):2694–2703. doi: 10.1128/mcb.01460-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sung SY, Kubo H, Shigemura K, Arnold RS, Logani S, Wang R, Konaka H, Nakagawa M, Mousses S, Amin M, Anderson C, Johnstone P, Petros JA, Marshall FF, Zhau HE, Chung LW. Oxidative stress induces ADAM9 protein expression in human prostate cancer cells. Cancer Res. 2006;66(19):9519–9526. doi: 10.1158/0008-5472.Can-05-4375. [DOI] [PubMed] [Google Scholar]
- 98.Tousseyn T, Thathiah A, Jorissen E, Raemaekers T, Konietzko U, Reiss K, Maes E, Snellinx A, Serneels L, Nyabi O, Annaert W, Saftig P, Hartmann D, De Strooper B. ADAM10, the rate-limiting protease of regulated intramembrane proteolysis of Notch and other proteins, is processed by ADAMS-9, ADAMS-15, and the gamma-secretase. J Biol Chem. 2009;284(17):11738–11747. doi: 10.1074/jbc.M805894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Moss ML, Powell G, Miller MA, Edwards L, Qi B, Sang QX, De Strooper B, Tesseur I, Lichtenthaler SF, Taverna M, Zhong JL, Dingwall C, Ferdous T, Schlomann U, Zhou P, Griffith LG, Lauffenburger DA, Petrovich R, Bartsch JW. ADAM9 inhibition increases membrane activity of ADAM10 and controls alpha-secretase processing of amyloid precursor protein. J Biol Chem. 2011;286(47):40443–40451. doi: 10.1074/jbc.M111.280495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cisse MA, Sunyach C, Lefranc-Jullien S, Postina R, Vincent B, Checler F. The disintegrin ADAM9 indirectly contributes to the physiological processing of cellular prion by modulating ADAM10 activity. J Biol Chem. 2005;280(49):40624–40631. doi: 10.1074/jbc.M506069200. [DOI] [PubMed] [Google Scholar]
- 101.Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, Kongi K, Cantuti L, Hanisch UK, Philips MA, Rossner MJ, Mann M, Simons M. Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci. 2015;18(12):1819–1831. doi: 10.1038/nn.4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Karkkainen I, Rybnikova E, Pelto-Huikko M, Huovila AP. Metalloprotease–disintegrin (ADAM) genes are widely and differentially expressed in the adult CNS. Mol Cell Neurosci. 2000;15(6):547–560. doi: 10.1006/mcne.2000.0848. [DOI] [PubMed] [Google Scholar]
- 103.Kuhn PH, Colombo AV, Schusser B, Dreymueller D, Wetzel S, Schepers U, Herber J, Ludwig A, Kremmer E, Montag D, Muller U, Schweizer M, Saftig P, Brase S, Lichtenthaler SF. Systematic substrate identification indicates a central role for the metalloprotease ADAM10 in axon targeting and synapse function. Elife. 2016;5:e12748. doi: 10.7554/eLife.12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Prox J, Bernreuther C, Altmeppen H, Grendel J, Glatzel M, D’Hooge R, Stroobants S, Ahmed T, Balschun D, Willem M, Lammich S, Isbrandt D, Schweizer M, Horre K, De Strooper B, Saftig P. Postnatal disruption of the disintegrin/metalloproteinase ADAM10 in brain causes epileptic seizures, learning deficits, altered spine morphology, and defective synaptic functions. J Neurosci. 2013;33(32):12915–12928, 12928a. doi: 10.1523/JNEUROSCI.5910-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Maretzky T, Evers A, Le Gall S, Alabi RO, Speck N, Reiss K, Blobel CP. The cytoplasmic domain of a disintegrin and metalloproteinase 10 (ADAM10) regulates its constitutive activity but is dispensable for stimulated ADAM10-dependent shedding. J Biol Chem. 2015;290(12):7416–7425. doi: 10.1074/jbc.M114.603753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Deng W, Cho S, Su PC, Berger BW, Li R. Membrane-enabled dimerization of the intrinsically disordered cytoplasmic domain of ADAM10. Proc Natl Acad Sci USA. 2014;111(45):15987–15992. doi: 10.1073/pnas.1409354111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Saftig P, Lichtenthaler SF. The alpha secretase ADAM10: A metalloprotease with multiple functions in the brain. Prog Neurobiol. 2015;135:1–20. doi: 10.1016/j.pneurobio.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 108.Endres K, Fahrenholz F. Regulation of alpha-secretase ADAM10 expression and activity. Exp Brain Res. 2012;217(3–4):343–352. doi: 10.1007/s00221-011-2885-7. [DOI] [PubMed] [Google Scholar]
- 109.Vincent B. Regulation of the alpha-secretase ADAM10 at transcriptional, translational and post-translational levels. Brain Res Bull. 2016;126(Pt 2):154–169. doi: 10.1016/j.brainresbull.2016.03.020. [DOI] [PubMed] [Google Scholar]
- 110.Alabi RO, Farber G, Blobel CP. Intriguing roles for endothelial ADAM10/notch signaling in the development of organ-specific vascular beds. Physiol Rev. 2018;98(4):2025–2061. doi: 10.1152/physrev.00029.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Farber G, Parks MM, Lustgarten Guahmich N, Zhang Y, Monette S, Blanchard SC, Di Lorenzo A, Blobel CP. ADAM10 controls the differentiation of the coronary arterial endothelium. Angiogenesis. 2018 doi: 10.1007/s10456-018-9653-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Farber G, Hurtado R, Loh S, Monette S, Mtui J, Kopan R, Quaggin S, Meyer-Schwesinger C, Herzlinger D, Scott RP, Blobel CP. Glomerular endothelial cell maturation depends on ADAM10, a key regulator of Notch signaling. Angiogenesis. 2018;21(2):335–347. doi: 10.1007/s10456-018-9599-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bray SJ. Notch signalling in context. Nat Rev Mol Cell Biol. 2016;17(11):722–735. doi: 10.1038/nrm.2016.94. [DOI] [PubMed] [Google Scholar]
- 114.Pan D, Rubin GM. Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell. 1997;90(2):271–280. doi: 10.1016/S0092-8674(00)80335-9. [DOI] [PubMed] [Google Scholar]
- 115.Rooke J, Pan D, Xu T, Rubin GM. KUZ, a conserved metalloprotease–disintegrin protein with two roles in Drosophila neurogenesis. Science (New York, NY) 1996;273(5279):1227–1231. doi: 10.1126/science.273.5279.1227. [DOI] [PubMed] [Google Scholar]
- 116.Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, Umans L, Lubke T, Lena Illert A, von Figura K, Saftig P. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet. 2002;11(21):2615–2624. doi: 10.1093/hmg/11.21.2615. [DOI] [PubMed] [Google Scholar]
- 117.Yang Z, Li PF, Chen RC, Wang J, Wang S, Shen Y, Wu X, Fang B, Cheng X, Xiong ZQ. ADAM10-initiated release of notch intracellular domain regulates microtubule stability and radial migration of cortical neurons. Cereb Cortex. 2017;27(2):919–932. doi: 10.1093/cercor/bhx006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chilton JK. Molecular mechanisms of axon guidance. Dev Biol. 2006;292(1):13–24. doi: 10.1016/j.ydbio.2005.12.048. [DOI] [PubMed] [Google Scholar]
- 119.Hattori M, Osterfield M, Flanagan JG. Regulated cleavage of a contact-mediated axon repellent. Science (New York, NY) 2000;289(5483):1360–1365. doi: 10.1126/science.289.5483.1360. [DOI] [PubMed] [Google Scholar]
- 120.Janes PW, Saha N, Barton WA, Kolev MV, Wimmer-Kleikamp SH, Nievergall E, Blobel CP, Himanen JP, Lackmann M, Nikolov DB. Adam meets Eph: an ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell. 2005;123(2):291–304. doi: 10.1016/j.cell.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 121.Brennaman LH, Moss ML, Maness PF. EphrinA/EphA-induced ectodomain shedding of neural cell adhesion molecule regulates growth cone repulsion through ADAM10 metalloprotease. J Neurochem. 2014;128(2):267–279. doi: 10.1111/jnc.12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hinkle CL, Diestel S, Lieberman J, Maness PF. Metalloprotease-induced ectodomain shedding of neural cell adhesion molecule (NCAM) J Neurobiol. 2006;66(12):1378–1395. doi: 10.1002/neu.20257. [DOI] [PubMed] [Google Scholar]
- 123.Romi E, Gokhman I, Wong E, Antonovsky N, Ludwig A, Sagi I, Saftig P, Tessier-Lavigne M, Yaron A. ADAM metalloproteases promote a developmental switch in responsiveness to the axonal repellant Sema3A. Nat Commun. 2014;5:4058. doi: 10.1038/ncomms5058. [DOI] [PubMed] [Google Scholar]
- 124.Heyden A, Angenstein F, Sallaz M, Seidenbecher C, Montag D. Abnormal axonal guidance and brain anatomy in mouse mutants for the cell recognition molecules close homolog of L1 and NgCAM-related cell adhesion molecule. Neuroscience. 2008;155(1):221–233. doi: 10.1016/j.neuroscience.2008.04.080. [DOI] [PubMed] [Google Scholar]
- 125.Brummer T, Mueller SA, Pan-Montojo F, Yoshida F, Fellgiebel A, Tomita T, Endres K, Lichtenthaler SF (2019) NrCAM is a marker for substrate-selective activation of ADAM10 in Alzheimer’s disease. EMBO Mol Med 32(7):e9695 [DOI] [PMC free article] [PubMed]
- 126.Dalva MB, McClelland AC, Kayser MS. Cell adhesion molecules: signalling functions at the synapse. Nat Rev Neurosci. 2007;8(3):206–220. doi: 10.1038/nrn2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bell KF, Zheng L, Fahrenholz F, Cuello AC. ADAM-10 over-expression increases cortical synaptogenesis. Neurobiol Aging. 2008;29(4):554–565. doi: 10.1016/j.neurobiolaging.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 128.Reiss K, Maretzky T, Ludwig A, Tousseyn T, de Strooper B, Hartmann D, Saftig P. ADAM10 cleavage of N-cadherin and regulation of cell-cell adhesion and beta-catenin nuclear signalling. EMBO J. 2005;24(4):742–752. doi: 10.1038/sj.emboj.7600548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Uemura K, Kihara T, Kuzuya A, Okawa K, Nishimoto T, Ninomiya H, Sugimoto H, Kinoshita A, Shimohama S. Characterization of sequential N-cadherin cleavage by ADAM10 and PS1. Neurosci Lett. 2006;402(3):278–283. doi: 10.1016/j.neulet.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 130.Malinverno M, Carta M, Epis R, Marcello E, Verpelli C, Cattabeni F, Sala C, Mulle C, Di Luca M, Gardoni F. Synaptic localization and activity of ADAM10 regulate excitatory synapses through N-cadherin cleavage. J Neurosci. 2010;30(48):16343–16355. doi: 10.1523/JNEUROSCI.1984-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Suzuki K, Hayashi Y, Nakahara S, Kumazaki H, Prox J, Horiuchi K, Zeng M, Tanimura S, Nishiyama Y, Osawa S, Sehara-Fujisawa A, Saftig P, Yokoshima S, Fukuyama T, Matsuki N, Koyama R, Tomita T, Iwatsubo T. Activity-dependent proteolytic cleavage of neuroligin-1. Neuron. 2012;76(2):410–422. doi: 10.1016/j.neuron.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 132.Bot N, Schweizer C, Ben Halima S, Fraering PC. Processing of the synaptic cell adhesion molecule neurexin-3beta by Alzheimer disease alpha- and gamma-secretases. J Biol Chem. 2011;286(4):2762–2773. doi: 10.1074/jbc.M110.142521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Borcel E, Palczynska M, Krzisch M, Dimitrov M, Ulrich G, Toni N, Fraering PC. Shedding of neurexin 3beta ectodomain by ADAM10 releases a soluble fragment that affects the development of newborn neurons. Sci Rep. 2016;6:39310. doi: 10.1038/srep39310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Venkatesh HS, Tam LT, Woo PJ, Lennon J, Nagaraja S, Gillespie SM, Ni J, Duveau DY, Morris PJ, Zhao JJ, Thomas CJ, Monje M. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature. 2017;549(7673):533–537. doi: 10.1038/nature24014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Marcello E, Gardoni F, Mauceri D, Romorini S, Jeromin A, Epis R, Borroni B, Cattabeni F, Sala C, Padovani A, Di Luca M. Synapse-associated protein-97 mediates alpha-secretase ADAM10 trafficking and promotes its activity. J Neurosci. 2007;27(7):1682–1691. doi: 10.1523/JNEUROSCI.3439-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lundgren JL, Ahmed S, Schedin-Weiss S, Gouras GK, Winblad B, Tjernberg LO, Frykman S. ADAM10 and BACE1 are localized to synaptic vesicles. J Neurochem. 2015;135(3):606–615. doi: 10.1111/jnc.13287. [DOI] [PubMed] [Google Scholar]
- 137.Sakry D, Neitz A, Singh J, Frischknecht R, Marongiu D, Biname F, Perera SS, Endres K, Lutz B, Radyushkin K, Trotter J, Mittmann T. Oligodendrocyte precursor cells modulate the neuronal network by activity-dependent ectodomain cleavage of glial NG2. PLoS Biol. 2014;12(11):e1001993. doi: 10.1371/journal.pbio.1001993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA. 1999;96(7):3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, Rydel RE. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron. 1993;10(2):243–254. doi: 10.1016/0896-6273(93)90315-I. [DOI] [PubMed] [Google Scholar]
- 140.Kim M, Suh J, Romano D, Truong MH, Mullin K, Hooli B, Norton D, Tesco G, Elliott K, Wagner SL, Moir RD, Becker KD, Tanzi RE. Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate {alpha}-secretase activity. Hum Mol Genet. 2009;18(20):3987–3996. doi: 10.1093/hmg/ddp323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Suh J, Choi SH, Romano DM, Gannon MA, Lesinski AN, Kim DY, Tanzi RE. ADAM10 missense mutations potentiate beta-amyloid accumulation by impairing prodomain chaperone function. Neuron. 2013;80(2):385–401. doi: 10.1016/j.neuron.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Moss ML, Bomar M, Liu Q, Sage H, Dempsey P, Lenhart PM, Gillispie PA, Stoeck A, Wildeboer D, Bartsch JW, Palmisano R, Zhou P. The ADAM10 prodomain is a specific inhibitor of ADAM10 proteolytic activity and inhibits cellular shedding events. J Biol Chem. 2007;282(49):35712–35721. doi: 10.1074/jbc.M703231200. [DOI] [PubMed] [Google Scholar]
- 143.Postina R, Schroeder A, Dewachter I, Bohl J, Schmitt U, Kojro E, Prinzen C, Endres K, Hiemke C, Blessing M, Flamez P, Dequenne A, Godaux E, van Leuven F, Fahrenholz F. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J Clin Investig. 2004;113(10):1456–1464. doi: 10.1172/JCI20864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tippmann F, Hundt J, Schneider A, Endres K, Fahrenholz F. Up-regulation of the alpha-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J. 2009;23(6):1643–1654. doi: 10.1096/fj.08-121392. [DOI] [PubMed] [Google Scholar]
- 145.Endres K, Fahrenholz F, Lotz J, Hiemke C, Teipel S, Lieb K, Tuscher O, Fellgiebel A. Increased CSF APPs-alpha levels in patients with Alzheimer disease treated with acitretin. Neurology. 2014;83(21):1930–1935. doi: 10.1212/WNL.0000000000001017. [DOI] [PubMed] [Google Scholar]
- 146.Altmeppen HC, Prox J, Puig B, Kluth MA, Bernreuther C, Thurm D, Jorissen E, Petrowitz B, Bartsch U, De Strooper B, Saftig P, Glatzel M. Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo. Mol Neurodegener. 2011;6:36. doi: 10.1186/1750-1326-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Linsenmeier L, Mohammadi B, Wetzel S, Puig B, Jackson WS, Hartmann A, Uchiyama K, Sakaguchi S, Endres K, Tatzelt J, Saftig P, Glatzel M, Altmeppen HC. Structural and mechanistic aspects influencing the ADAM10-mediated shedding of the prion protein. Mol Neurodegener. 2018;13(1):18. doi: 10.1186/s13024-018-0248-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Altmeppen HC, Prox J, Krasemann S, Puig B, Kruszewski K, Dohler F, Bernreuther C, Hoxha A, Linsenmeier L, Sikorska B, Liberski PP, Bartsch U, Saftig P, Glatzel M. The sheddase ADAM10 is a potent modulator of prion disease. Elife. 2015;4:e04260. doi: 10.7554/eLife.04260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Stoeck A, Keller S, Riedle S, Sanderson MP, Runz S, Le Naour F, Gutwein P, Ludwig A, Rubinstein E, Altevogt P. A role for exosomes in the constitutive and stimulus-induced ectodomain cleavage of L1 and CD44. Biochem J. 2006;393(Pt 3):609–618. doi: 10.1042/BJ20051013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Padro CJ, Shawler TM, Gormley MG, Sanders VM. Adrenergic regulation of IgE involves modulation of CD23 and ADAM10 expression on exosomes. J Immunol. 2013;191(11):5383–5397. doi: 10.4049/jimmunol.1301019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhang W, Zhang J, Cheng L, Ni H, You B, Shan Y, Bao L, Wu D, Zhang T, Yue H, Chen J. A disintegrin and metalloprotease 10-containing exosomes derived from nasal polyps promote angiogenesis and vascular permeability. Mol Med Rep. 2018;17(4):5921–5927. doi: 10.3892/mmr.2018.8634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Freese C, Garratt AN, Fahrenholz F, Endres K. The effects of alpha-secretase ADAM10 on the proteolysis of neuregulin-1. FEBS J. 2009;276(6):1568–1580. doi: 10.1111/j.1742-4658.2009.06889.x. [DOI] [PubMed] [Google Scholar]
- 153.Jangouk P, Dehmel T, Meyer Zu Horste G, Ludwig A, Lehmann HC, Kieseier BC. Involvement of ADAM10 in axonal outgrowth and myelination of the peripheral nerve. Glia. 2009;57(16):1765–1774. doi: 10.1002/glia.20889. [DOI] [PubMed] [Google Scholar]
- 154.Luo X, Prior M, He W, Hu X, Tang X, Shen W, Yadav S, Kiryu-Seo S, Miller R, Trapp BD, Yan R. Cleavage of neuregulin-1 by BACE1 or ADAM10 protein produces differential effects on myelination. J Biol Chem. 2011;286(27):23967–23974. doi: 10.1074/jbc.M111.251538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Meyer Zu Horste G, Derksen A, Stassart R, Szepanowski F, Thanos M, Stettner M, Boettcher C, Lehmann HC, Hartung HP, Kieseier BC. Neuronal ADAM10 promotes outgrowth of small-caliber myelinated axons in the peripheral nervous system. J Neuropathol Exp Neurol. 2015;74(11):1077–1085. doi: 10.1097/NEN.0000000000000253. [DOI] [PubMed] [Google Scholar]
- 156.Colombo A, Hsia HE, Wang M, Kuhn PH, Brill MS, Canevazzi P, Feederle R, Taveggia C, Misgeld T, Lichtenthaler SF. Non-cell-autonomous function of DR6 in Schwann cell proliferation. EMBO J. 2018;37(7):e97390. doi: 10.15252/embj.201797390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Matthews AL, Szyroka J, Collier R, Noy PJ, Tomlinson MG. Scissor sisters: regulation of ADAM10 by the TspanC8 tetraspanins. Biochem Soc Trans. 2017;45(3):719–730. doi: 10.1042/BST20160290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Haining EJ, Yang J, Bailey RL, Khan K, Collier R, Tsai S, Watson SP, Frampton J, Garcia P, Tomlinson MG. The TspanC8 subgroup of tetraspanins interacts with A disintegrin and metalloprotease 10 (ADAM10) and regulates its maturation and cell surface expression. J Biol Chem. 2012;287(47):39753–39765. doi: 10.1074/jbc.M112.416503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Noy PJ, Yang J, Reyat JS, Matthews AL, Charlton AE, Furmston J, Rogers DA, Rainger GE, Tomlinson MG. TspanC8 tetraspanins and A Disintegrin and Metalloprotease 10 (ADAM10) interact via their extracellular regions: evidence for distinct binding mechanisms for different TspanC8 proteins. J Biol Chem. 2016;291(7):3145–3157. doi: 10.1074/jbc.M115.703058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jouannet S, Saint-Pol J, Fernandez L, Nguyen V, Charrin S, Boucheix C, Brou C, Milhiet PE, Rubinstein E. TspanC8 tetraspanins differentially regulate the cleavage of ADAM10 substrates, Notch activation and ADAM10 membrane compartmentalization. Cell Mol Life Sci. 2016;73(9):1895–1915. doi: 10.1007/s00018-015-2111-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Seipold L, Altmeppen H, Koudelka T, Tholey A, Kasparek P, Sedlacek R, Schweizer M, Bar J, Mikhaylova M, Glatzel M, Saftig P. In vivo regulation of the A disintegrin and metalloproteinase 10 (ADAM10) by the tetraspanin 15. Cell Mol Life Sci. 2018;75(17):3251–3267. doi: 10.1007/s00018-018-2791-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bernstein HG, Keilhoff G, Bukowska A, Ziegeler A, Funke S, Dobrowolny H, Kanakis D, Bogerts B, Lendeckel U. ADAM (a disintegrin and metalloprotease) 12 is expressed in rat and human brain and localized to oligodendrocytes. J Neurosci Res. 2004;75(3):353–360. doi: 10.1002/jnr.10858. [DOI] [PubMed] [Google Scholar]
- 163.Kveiborg M, Albrechtsen R, Rudkjaer L, Wen G, Damgaard-Pedersen K, Wewer UM. ADAM12-S stimulates bone growth in transgenic mice by modulating chondrocyte proliferation and maturation. J Bone Miner Res. 2006;21(8):1288–1296. doi: 10.1359/jbmr.060502. [DOI] [PubMed] [Google Scholar]
- 164.Cao Y, Zhao Z, Gruszczynska-Biegala J, Zolkiewska A. Role of metalloprotease disintegrin ADAM12 in determination of quiescent reserve cells during myogenic differentiation in vitro. Mol Cell Biol. 2003;23(19):6725–6738. doi: 10.1128/MCB.23.19.6725-6738.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kurisaki T, Masuda A, Sudo K, Sakagami J, Higashiyama S, Matsuda Y, Nagabukuro A, Tsuji A, Nabeshima Y, Asano M, Iwakura Y, Sehara-Fujisawa A. Phenotypic analysis of Meltrin alpha (ADAM12)-deficient mice: involvement of Meltrin alpha in adipogenesis and myogenesis. Mol Cell Biol. 2003;23(1):55–61. doi: 10.1128/MCB.23.1.55-61.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Malinin NL, Wright S, Seubert P, Schenk D, Griswold-Prenner I. Amyloid-beta neurotoxicity is mediated by FISH adapter protein and ADAM12 metalloprotease activity. Proc Natl Acad Sci USA. 2005;102(8):3058–3063. doi: 10.1073/pnas.0408237102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Selvais C, D’Auria L, Tyteca D, Perrot G, Lemoine P, Troeberg L, Dedieu S, Noel A, Nagase H, Henriet P, Courtoy PJ, Marbaix E, Emonard H. Cell cholesterol modulates metalloproteinase-dependent shedding of low-density lipoprotein receptor-related protein-1 (LRP-1) and clearance function. FASEB J. 2011;25(8):2770–2781. doi: 10.1096/fj.10-169508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Cui D, Arima M, Takubo K, Kimura T, Horiuchi K, Minagawa T, Matsuda S, Ikeda E. ADAM12 and ADAM17 are essential molecules for hypoxia-induced impairment of neural vascular barrier function. Sci Rep. 2015;5:12796. doi: 10.1038/srep12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385(6618):729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 170.Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, Hoffman CR, Kost TA, Lambert MH, Leesnitzer MA, McCauley P, McGeehan G, Mitchell J, Moyer M, Pahel G, Rocque W, Overton LK, Schoenen F, Seaton T, Su JL, Becherer JD, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385(6618):733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- 171.Zunke F, Rose-John S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim Biophys Acta. 2017;1864(11 Pt B):2059–2070. doi: 10.1016/j.bbamcr.2017.07.001. [DOI] [PubMed] [Google Scholar]
- 172.Reddy P, Slack JL, Davis R, Cerretti DP, Kozlosky CJ, Blanton RA, Shows D, Peschon JJ, Black RA. Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J Biol Chem. 2000;275(19):14608–14614. doi: 10.1074/jbc.275.19.14608. [DOI] [PubMed] [Google Scholar]
- 173.Le Gall SM, Maretzky T, Issuree PD, Niu XD, Reiss K, Saftig P, Khokha R, Lundell D, Blobel CP. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J Cell Sci. 2010;123(Pt 22):3913–3922. doi: 10.1242/jcs.069997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Doedens JR, Mahimkar RM, Black RA. TACE/ADAM-17 enzymatic activity is increased in response to cellular stimulation. Biochem Biophys Res Commun. 2003;308(2):331–338. doi: 10.1016/S0006-291X(03)01381-0. [DOI] [PubMed] [Google Scholar]
- 175.Li Q, Zhang Z, Li Z, Zhou M, Liu B, Pan L, Ma Z, Zheng Y. ADAM17 is critical for multipolar exit and radial migration of neuronal intermediate progenitor cells in mice cerebral cortex. PLoS One. 2013;8(6):e65703. doi: 10.1371/journal.pone.0065703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ, Black RA. An essential role for ectodomain shedding in mammalian development. Science (New York, NY) 1998;282(5392):1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- 177.Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005;6(1):32–43. doi: 10.1038/nrm1548. [DOI] [PubMed] [Google Scholar]
- 178.Jackson LF, Qiu TH, Sunnarborg SW, Chang A, Zhang C, Patterson C, Lee DC. Defective valvulogenesis in HB-EGF and TACE-null mice is associated with aberrant BMP signaling. EMBO J. 2003;22(11):2704–2716. doi: 10.1093/emboj/cdg264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Mine N, Iwamoto R, Mekada E. HB-EGF promotes epithelial cell migration in eyelid development. Development. 2005;132(19):4317–4326. doi: 10.1242/dev.02030. [DOI] [PubMed] [Google Scholar]
- 180.Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC, et al. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science (New York, NY) 1995;269(5221):230–234. doi: 10.1126/science.7618084. [DOI] [PubMed] [Google Scholar]
- 181.Miettinen PJ, Berger JE, Meneses J, Phung Y, Pedersen RA, Werb Z, Derynck R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature. 1995;376(6538):337–341. doi: 10.1038/376337a0. [DOI] [PubMed] [Google Scholar]
- 182.Oyagi A, Moriguchi S, Nitta A, Murata K, Oida Y, Tsuruma K, Shimazawa M, Fukunaga K, Hara H. Heparin-binding EGF-like growth factor is required for synaptic plasticity and memory formation. Brain Res. 2011;1419:97–104. doi: 10.1016/j.brainres.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 183.Palazuelos J, Crawford HC, Klingener M, Sun B, Karelis J, Raines EW, Aguirre A. TACE/ADAM17 is essential for oligodendrocyte development and CNS myelination. J Neurosci. 2014;34(36):11884–11896. doi: 10.1523/JNEUROSCI.1220-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Palazuelos J, Klingener M, Raines EW, Crawford HC, Aguirre A. Oligodendrocyte regeneration and CNS remyelination require TACE/ADAM17. J Neurosci. 2015;35(35):12241–12247. doi: 10.1523/JNEUROSCI.3937-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.La Marca R, Cerri F, Horiuchi K, Bachi A, Feltri ML, Wrabetz L, Blobel CP, Quattrini A, Salzer JL, Taveggia C. TACE (ADAM17) inhibits Schwann cell myelination. Nat Neurosci. 2011;14(7):857–865. doi: 10.1038/nn.2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Iwakura Y, Wang R, Inamura N, Araki K, Higashiyama S, Takei N, Nawa H. Glutamate-dependent ectodomain shedding of neuregulin-1 type II precursors in rat forebrain neurons. PLoS One. 2017;12(3):e0174780. doi: 10.1371/journal.pone.0174780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kim J, Elias A, Lee T, Maurel P, Kim HA. Tissue inhibitor of metalloproteinase-3 promotes schwann cell myelination. ASN Neuro. 2017;9(6):1759091417745425. doi: 10.1177/1759091417745425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Fleck D, van Bebber F, Colombo A, Galante C, Schwenk BM, Rabe L, Hampel H, Novak B, Kremmer E, Tahirovic S, Edbauer D, Lichtenthaler SF, Schmid B, Willem M, Haass C. Dual cleavage of neuregulin 1 type III by BACE1 and ADAM17 liberates its EGF-like domain and allows paracrine signaling. J Neurosci. 2013;33(18):7856–7869. doi: 10.1523/JNEUROSCI.3372-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cho RW, Park JM, Wolff SB, Xu D, Hopf C, Kim JA, Reddy RC, Petralia RS, Perin MS, Linden DJ, Worley PF. mGluR1/5-dependent long-term depression requires the regulated ectodomain cleavage of neuronal pentraxin NPR by TACE. Neuron. 2008;57(6):858–871. doi: 10.1016/j.neuron.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Okamura Y, Kohmura E, Yamashita T. TACE cleaves neogenin to desensitize cortical neurons to the repulsive guidance molecule. Neurosci Res. 2011;71(1):63–70. doi: 10.1016/j.neures.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 191.van Erp S, van den Heuvel DMA, Fujita Y, Robinson RA, Hellemons A, Adolfs Y, Van Battum EY, Blokhuis AM, Kuijpers M, Demmers JAA, Hedman H, Hoogenraad CC, Siebold C, Yamashita T, Pasterkamp RJ. Lrig2 negatively regulates ectodomain shedding of axon guidance receptors by ADAM proteases. Dev Cell. 2015;35(5):537–552. doi: 10.1016/j.devcel.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 192.Weskamp G, Schlondorff J, Lum L, Becherer JD, Kim TW, Saftig P, Hartmann D, Murphy G, Blobel CP. Evidence for a critical role of the tumor necrosis factor alpha convertase (TACE) in ectodomain shedding of the p75 neurotrophin receptor (p75NTR) J Biol Chem. 2004;279(6):4241–4249. doi: 10.1074/jbc.M307974200. [DOI] [PubMed] [Google Scholar]
- 193.Kalus I, Bormann U, Mzoughi M, Schachner M, Kleene R. Proteolytic cleavage of the neural cell adhesion molecule by ADAM17/TACE is involved in neurite outgrowth. J Neurochem. 2006;98(1):78–88. doi: 10.1111/j.1471-4159.2006.03847.x. [DOI] [PubMed] [Google Scholar]
- 194.Shirakabe K, Omura T, Shibagaki Y, Mihara E, Homma K, Kato Y, Yoshimura A, Murakami Y, Takagi J, Hattori S, Ogawa Y (2017) Mechanistic insights into ectodomain shedding: susceptibility of CADM1 adhesion molecule is determined by alternative splicing and O-glycosylation. Sci Rep 7:46174. 10.1038/srep46174 [DOI] [PMC free article] [PubMed]
- 195.Xia H, Sriramula S, Chhabra KH, Lazartigues E. Brain angiotensin-converting enzyme type 2 shedding contributes to the development of neurogenic hypertension. Circ Res. 2013;113(9):1087–1096. doi: 10.1161/CIRCRESAHA.113.301811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Xu J, Sriramula S, Lazartigues E. Excessive glutamate stimulation impairs ACE2 activity through ADAM17-mediated shedding in cultured cortical neurons. Cell Mol Neurobiol. 2018 doi: 10.1007/s10571-018-0591-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Siney EJ, Holden A, Casselden E, Bulstrode H, Thomas GJ, Willaime-Morawek S. Metalloproteinases ADAM10 and ADAM17 mediate migration and differentiation in glioblastoma sphere-forming cells. Mol Neurobiol. 2017;54(5):3893–3905. doi: 10.1007/s12035-016-0053-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Honda H, Takamura M, Yamagiwa S, Genda T, Horigome R, Kimura N, Setsu T, Tominaga K, Kamimura H, Matsuda Y, Wakai T, Aoyagi Y, Terai S. Overexpression of a disintegrin and metalloproteinase 21 is associated with motility, metastasis, and poor prognosis in hepatocellular carcinoma. Sci Rep. 2017;7(1):15485. doi: 10.1038/s41598-017-15800-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Nandhu MS, Kwiatkowska A, Bhaskaran V, Hayes J, Hu B, Viapiano MS. Tumor-derived fibulin-3 activates pro-invasive NF-kappaB signaling in glioblastoma cells and their microenvironment. Oncogene. 2017;36(34):4875–4886. doi: 10.1038/onc.2017.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Vincent B, Cisse MA, Sunyach C, Guillot-Sestier MV, Checler F. Regulation of betaAPP and PrPc cleavage by alpha-secretase: mechanistic and therapeutic perspectives. Curr Alzheimer Res. 2008;5(2):202–211. doi: 10.2174/156720508783954749. [DOI] [PubMed] [Google Scholar]
- 201.Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H, Fisher A, LaFerla FM. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron. 2006;49(5):671–682. doi: 10.1016/j.neuron.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 202.Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science (New York, NY) 1992;258(5080):304–307. doi: 10.1126/science.1411529. [DOI] [PubMed] [Google Scholar]
- 203.Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998;273(43):27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- 204.Hartl D, May P, Gu W, Mayhaus M, Pichler S, Spaniol C, Glaab E, Bobbili DR, Antony P, Koegelsberger S, Kurz A, Grimmer T, Morgan K, Vardarajan BN, Reitz C, Hardy J, Bras J, Guerreiro R, Balling R, Schneider JG, Riemenschneider M, AESG Consortium A rare loss-of-function variant of ADAM17 is associated with late-onset familial Alzheimer disease. Mol Psychiatry. 2018 doi: 10.1038/s41380-018-0091-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Cai Z, Wang C, He W, Chen Y. Berberine alleviates amyloid-beta pathology in the brain of APP/PS1 transgenic mice via inhibiting beta/gamma-secretases activity and enhancing alpha-secretases. Curr Alzheimer Res. 2018 doi: 10.2174/1567205015666180702105740. [DOI] [PubMed] [Google Scholar]
- 206.Giuliani F, Vernay A, Leuba G, Schenk F. Decreased behavioral impairments in an Alzheimer mice model by interfering with TNF-alpha metabolism. Brain Res Bull. 2009;80(4–5):302–308. doi: 10.1016/j.brainresbull.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 207.Lourenco MV, Clarke JR, Frozza RL, Bomfim TR, Forny-Germano L, Batista AF, Sathler LB, Brito-Moreira J, Amaral OB, Silva CA, Freitas-Correa L, Espirito-Santo S, Campello-Costa P, Houzel JC, Klein WL, Holscher C, Carvalheira JB, Silva AM, Velloso LA, Munoz DP, Ferreira ST, De Felice FG. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab. 2013;18(6):831–843. doi: 10.1016/j.cmet.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 208.Shi JQ, Shen W, Chen J, Wang BR, Zhong LL, Zhu YW, Zhu HQ, Zhang QQ, Zhang YD, Xu J. Anti-TNF-alpha reduces amyloid plaques and tau phosphorylation and induces CD11c-positive dendritic-like cell in the APP/PS1 transgenic mouse brains. Brain Res. 2011;1368:239–247. doi: 10.1016/j.brainres.2010.10.053. [DOI] [PubMed] [Google Scholar]
- 209.Tobinick EL, Gross H. Rapid cognitive improvement in Alzheimer’s disease following perispinal etanercept administration. J Neuroinflammation. 2008;5:2. doi: 10.1186/1742-2094-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Shi JQ, Wang BR, Jiang WW, Chen J, Zhu YW, Zhong LL, Zhang YD, Xu J. Cognitive improvement with intrathecal administration of infliximab in a woman with Alzheimer’s disease. J Am Geriatr Soc. 2011;59(6):1142–1144. doi: 10.1111/j.1532-5415.2011.03445.x. [DOI] [PubMed] [Google Scholar]
- 211.Feuerbach D, Schindler P, Barske C, Joller S, Beng-Louka E, Worringer KA, Kommineni S, Kaykas A, Ho DJ, Ye C, Welzenbach K, Elain G, Klein L, Brzak I, Mir AK, Farady CJ, Aichholz R, Popp S, George N, Neumann U. ADAM17 is the main sheddase for the generation of human triggering receptor expressed in myeloid cells (hTREM2) ectodomain and cleaves TREM2 after Histidine 157. Neurosci Lett. 2017;660:109–114. doi: 10.1016/j.neulet.2017.09.034. [DOI] [PubMed] [Google Scholar]
- 212.Schlepckow K, Kleinberger G, Fukumori A, Feederle R, Lichtenthaler SF, Steiner H, Haass C. An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol Med. 2017;9(10):1356–1365. doi: 10.15252/emmm.201707672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Thornton P, Sevalle J, Deery MJ, Fraser G, Zhou Y, Stahl S, Franssen EH, Dodd RB, Qamar S, Gomez Perez-Nievas B, Nicol LS, Eketjall S, Revell J, Jones C, Billinton A, St George-Hyslop PH, Chessell I, Crowther DC. TREM2 shedding by cleavage at the H157-S158 bond is accelerated for the Alzheimer’s disease-associated H157Y variant. EMBO Mol Med. 2017;9(10):1366–1378. doi: 10.15252/emmm.201707673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Song WM, Joshita S, Zhou Y, Ulland TK, Gilfillan S, Colonna M. Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J Exp Med. 2018;215(3):745–760. doi: 10.1084/jem.20171529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368(2):107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J, Alzheimer Genetic Analysis G. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Weinger JG, Omari KM, Marsden K, Raine CS, Shafit-Zagardo B. Up-regulation of soluble Axl and Mer receptor tyrosine kinases negatively correlates with Gas6 in established multiple sclerosis lesions. Am J Pathol. 2009;175(1):283–293. doi: 10.2353/ajpath.2009.080807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Qing X, Rogers L, Mortha A, Lavin Y, Redecha P, Issuree PD, Maretzky T, Merad M, McIlwain D, Mak TW, Overall CM, Blobel CP, Salmon JE. iRhom2 regulates CSF1R cell surface expression and non-steady state myelopoiesis in mice. Eur J Immunol. 2016;46(12):2737–2748. doi: 10.1002/eji.201646482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Sassi C, Nalls MA, Ridge PG, Gibbs JR, Lupton MK, Troakes C, Lunnon K, Al-Sarraj S, Brown KS, Medway C, Lord J, Turton J, Bras J, ARUK Consortium. Blumenau S, Thielke M, Josties C, Freyer D, Dietrich A, Hammer M, Baier M, Dirnagl U, Morgan K, Powell JF, Kauwe JS, Cruchaga C, Goate AM, Singleton AB, Guerreiro R, Hodges A, Hardy J. Mendelian adult-onset leukodystrophy genes in Alzheimer’s disease: critical influence of CSF1R and NOTCH3. Neurobiol Aging. 2018;66:179 e117–179 e129. doi: 10.1016/j.neurobiolaging.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Liu Q, Zhang J, Tran H, Verbeek MM, Reiss K, Estus S, Bu G (2009) LRP1 shedding in human brain: roles of ADAM10 and ADAM17. Mol Neurodegeneration 4(1):17 [DOI] [PMC free article] [PubMed]
- 221.Wong E, Maretzky T, Peleg Y, Blobel CP, Sagi I. The functional maturation of a disintegrin and metalloproteinase (ADAM) 9, 10, and 17 requires processing at a newly identified proprotein convertase (PC) cleavage site. J Biol Chem. 2015;290(19):12135–12146. doi: 10.1074/jbc.M114.624072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Amour A, Slocombe PM, Webster A, Butler M, Knight CG, Smith BJ, Stephens PE, Shelley C, Hutton M, Knauper V, Docherty AJ, Murphy G. TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 1998;435(1):39–44. doi: 10.1016/S0014-5793(98)01031-X. [DOI] [PubMed] [Google Scholar]
- 223.Capone C, Dabertrand F, Baron-Menguy C, Chalaris A, Ghezali L, Domenga-Denier V, Schmidt S, Huneau C, Rose-John S, Nelson MT, Joutel A. Mechanistic insights into a TIMP3-sensitive pathway constitutively engaged in the regulation of cerebral hemodynamics. Elife. 2016;5:e17536. doi: 10.7554/eLife.17536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Cavadas M, Oikonomidi I, Gaspar CJ, Burbridge E, Badenes M, Felix I, Bolado A, Hu T, Bileck A, Gerner C, Domingos PM, von Kriegsheim A, Adrain C. Phosphorylation of iRhom2 controls stimulated proteolytic shedding by the metalloprotease ADAM17/TACE. Cell Rep. 2017;21(3):745–757. doi: 10.1016/j.celrep.2017.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Maretzky T, McIlwain DR, Issuree PD, Li X, Malapeira J, Amin S, Lang PA, Mak TW, Blobel CP. iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proc Natl Acad Sci USA. 2013;110(28):11433–11438. doi: 10.1073/pnas.1302553110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Li X, Maretzky T, Weskamp G, Monette S, Qing X, Issuree PD, Crawford HC, McIlwain DR, Mak TW, Salmon JE, Blobel CP. iRhoms 1 and 2 are essential upstream regulators of ADAM17-dependent EGFR signaling. Proc Natl Acad Sci USA. 2015;112(19):6080–6085. doi: 10.1073/pnas.1505649112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Adrain C, Zettl M, Christova Y, Taylor N, Freeman M. Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science (New York, NY) 2012;335(6065):225–228. doi: 10.1126/science.1214400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.McIlwain DR, Lang PA, Maretzky T, Hamada K, Ohishi K, Maney SK, Berger T, Murthy A, Duncan G, Xu HC, Lang KS, Haussinger D, Wakeham A, Itie-Youten A, Khokha R, Ohashi PS, Blobel CP, Mak TW. iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science (New York, NY) 2012;335(6065):229–232. doi: 10.1126/science.1214448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Christova Y, Adrain C, Bambrough P, Ibrahim A, Freeman M. Mammalian iRhoms have distinct physiological functions including an essential role in TACE regulation. EMBO Rep. 2013;14(10):884–890. doi: 10.1038/embor.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Lichtenthaler SF, O’Hara BF, Blobel CP. iRhoms in the brain—a new frontier? Cell Cycle. 2015;14(19):3003–3004. doi: 10.1080/15384101.2015.1084187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L, Eaton ML, Keenan BT, Ernst J, McCabe C, Tang A, Raj T, Replogle J, Brodeur W, Gabriel S, Chai HS, Younkin C, Younkin SG, Zou F, Szyf M, Epstein CB, Schneider JA, Bernstein BE, Meissner A, Ertekin-Taner N, Chibnik LB, Kellis M, Mill J, Bennett DA. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014;17(9):1156–1163. doi: 10.1038/nn.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Veroni C, Gabriele L, Canini I, Castiello L, Coccia E, Remoli ME, Columba-Cabezas S, Arico E, Aloisi F, Agresti C. Activation of TNF receptor 2 in microglia promotes induction of anti-inflammatory pathways. Mol Cell Neurosci. 2010;45(3):234–244. doi: 10.1016/j.mcn.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 233.Chesneau V, Becherer JD, Zheng Y, Erdjument-Bromage H, Tempst P, Blobel CP. Catalytic properties of ADAM19. J Biol Chem. 2003;278(25):22331–22340. doi: 10.1074/jbc.M302781200. [DOI] [PubMed] [Google Scholar]
- 234.Zheng Y, Saftig P, Hartmann D, Blobel C. Evaluation of the contribution of different ADAMs to tumor necrosis factor alpha (TNFalpha) shedding and of the function of the TNFalpha ectodomain in ensuring selective stimulated shedding by the TNFalpha convertase (TACE/ADAM17) J Biol Chem. 2004;279(41):42898–42906. doi: 10.1074/jbc.M403193200. [DOI] [PubMed] [Google Scholar]
- 235.Kurisaki T, Masuda A, Osumi N, Nabeshima Y, Fujisawa-Sehara A. Spatially- and temporally-restricted expression of meltrin alpha (ADAM12) and beta (ADAM19) in mouse embryo. Mech Dev. 1998;73(2):211–215. doi: 10.1016/S0925-4773(98)00043-4. [DOI] [PubMed] [Google Scholar]
- 236.Meyer D, Birchmeier C. Distinct isoforms of neuregulin are expressed in mesenchymal and neuronal cells during mouse development. Proc Natl Acad Sci USA. 1994;91(3):1064–1068. doi: 10.1073/pnas.91.3.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Zhou HM, Weskamp G, Chesneau V, Sahin U, Vortkamp A, Horiuchi K, Chiusaroli R, Hahn R, Wilkes D, Fisher P, Baron R, Manova K, Basson CT, Hempstead B, Blobel CP. Essential role for ADAM19 in cardiovascular morphogenesis. Mol Cell Biol. 2004;24(1):96–104. doi: 10.1128/MCB.24.1.96-104.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Shirakabe K, Wakatsuki S, Kurisaki T, Fujisawa-Sehara A. Roles of Meltrin beta /ADAM19 in the processing of neuregulin. J Biol Chem. 2001;276(12):9352–9358. doi: 10.1074/jbc.M007913200. [DOI] [PubMed] [Google Scholar]
- 239.Wakatsuki S, Yumoto N, Komatsu K, Araki T, Sehara-Fujisawa A. Roles of meltrin-beta/ADAM19 in progression of Schwann cell differentiation and myelination during sciatic nerve regeneration. J Biol Chem. 2009;284(5):2957–2966. doi: 10.1074/jbc.M803191200. [DOI] [PubMed] [Google Scholar]
- 240.Yumoto N, Wakatsuki S, Kurisaki T, Hara Y, Osumi N, Frisen J, Sehara-Fujisawa A. Meltrin beta/ADAM19 interacting with EphA4 in developing neural cells participates in formation of the neuromuscular junction. PLoS One. 2008;3(10):e3322. doi: 10.1371/journal.pone.0003322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Tanabe C, Hotoda N, Sasagawa N, Sehara-Fujisawa A, Maruyama K, Ishiura S. ADAM19 is tightly associated with constitutive Alzheimer’s disease APP alpha-secretase in A172 cells. Biochem Biophys Res Commun. 2007;352(1):111–117. doi: 10.1016/j.bbrc.2006.10.181. [DOI] [PubMed] [Google Scholar]
- 242.Hooft van Huijsduijnen R. ADAM 20 and 21; two novel human testis-specific membrane metalloproteases with similarity to fertilin-alpha. Gene. 1998;206(2):273–282. doi: 10.1016/S0378-1119(97)00597-0. [DOI] [PubMed] [Google Scholar]
- 243.Poindexter K, Nelson N, DuBose RF, Black RA, Cerretti DP. The identification of seven metalloproteinase-disintegrin (ADAM) genes from genomic libraries. Gene. 1999;237(1):61–70. doi: 10.1016/S0378-1119(99)00302-9. [DOI] [PubMed] [Google Scholar]
- 244.Yi C, Woo JM, Han C, Oh JS, Park I, Lee B, Jin S, Choi H, Kwon JT, Cho BN, Kim DH, Cho C. Expression analysis of the Adam21 gene in mouse testis. Gene Expr Patterns. 2010;10(2–3):152–158. doi: 10.1016/j.gep.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 245.Yang P, Baker KA, Hagg T. A disintegrin and metalloprotease 21 (ADAM21) is associated with neurogenesis and axonal growth in developing and adult rodent CNS. J Comp Neurol. 2005;490(2):163–179. doi: 10.1002/cne.20659. [DOI] [PubMed] [Google Scholar]
- 246.Cerretti DP, DuBose RF, Black RA, Nelson N. Isolation of two novel metalloproteinase-disintegrin (ADAM) cDNAs that show testis-specific gene expression. Biochem Biophys Res Commun. 1999;263(3):810–815. doi: 10.1006/bbrc.1999.1322. [DOI] [PubMed] [Google Scholar]
- 247.Letronne F, Laumet G, Ayral AM, Chapuis J, Demiautte F, Laga M, Vandenberghe ME, Malmanche N, Leroux F, Eysert F, Sottejeau Y, Chami L, Flaig A, Bauer C, Dourlen P, Lesaffre M, Delay C, Huot L, Dumont J, Werkmeister E, Lafont F, Mendes T, Hansmannel F, Dermaut B, Deprez B, Herard AS, Dhenain M, Souedet N, Pasquier F, Tulasne D, Berr C, Hauw JJ, Lemoine Y, Amouyel P, Mann D, Deprez R, Checler F, Hot D, Delzescaux T, Gevaert K, Lambert JC. ADAM30 downregulates APP-linked defects through cathepsin D activation in Alzheimer’s disease. EBioMedicine. 2016;9:278–292. doi: 10.1016/j.ebiom.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Vassar R, Kuhn PH, Haass C, Kennedy ME, Rajendran L, Wong PC, Lichtenthaler SF. Function, therapeutic potential and cell biology of BACE proteases: current status and future prospects. J Neurochem. 2014;130(1):4–28. doi: 10.1111/jnc.12715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Yoshinaka T, Nishii K, Yamada K, Sawada H, Nishiwaki E, Smith K, Yoshino K, Ishiguro H, Higashiyama S. Identification and characterization of novel mouse and human ADAM33s with potential metalloprotease activity. Gene. 2002;282(1–2):227–236. doi: 10.1016/S0378-1119(01)00818-6. [DOI] [PubMed] [Google Scholar]
- 250.Cousin H, Abbruzzese G, McCusker C, Alfandari D. ADAM13 function is required in the 3 dimensional context of the embryo during cranial neural crest cell migration in Xenopus laevis. Dev Biol. 2012;368(2):335–344. doi: 10.1016/j.ydbio.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Tripathi P, Awasthi S, Gao P. ADAM metallopeptidase domain 33 (ADAM33): a promising target for asthma. Mediat Inflamm. 2014;2014:572025. doi: 10.1155/2014/572025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Van Eerdewegh P, Little RD, Dupuis J, Del Mastro RG, Falls K, Simon J, Torrey D, Pandit S, McKenny J, Braunschweiger K, Walsh A, Liu Z, Hayward B, Folz C, Manning SP, Bawa A, Saracino L, Thackston M, Benchekroun Y, Capparell N, Wang M, Adair R, Feng Y, Dubois J, FitzGerald MG, Huang H, Gibson R, Allen KM, Pedan A, Danzig MR, Umland SP, Egan RW, Cuss FM, Rorke S, Clough JB, Holloway JW, Holgate ST, Keith TP. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature. 2002;418(6896):426–430. doi: 10.1038/nature00878. [DOI] [PubMed] [Google Scholar]
- 253.Gunn TM, Azarani A, Kim PH, Hyman RW, Davis RW, Barsh GS. Identification and preliminary characterization of mouse Adam33. BMC Genet. 2002;3:2. doi: 10.1186/1471-2156-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Zou J, Zhu F, Liu J, Wang W, Zhang R, Garlisi CG, Liu YH, Wang S, Shah H, Wan Y, Umland SP. Catalytic activity of human ADAM33. J Biol Chem. 2004;279(11):9818–9830. doi: 10.1074/jbc.M309696200. [DOI] [PubMed] [Google Scholar]
- 255.Zou J, Zhang R, Zhu F, Liu J, Madison V, Umland SP. ADAM33 enzyme properties and substrate specificity. Biochemistry. 2005;44(11):4247–4256. doi: 10.1021/bi0476230. [DOI] [PubMed] [Google Scholar]
- 256.Alfandari D, Cousin H, Gaultier A, Smith K, White JM, Darribere T, DeSimone DW. Xenopus ADAM 13 is a metalloprotease required for cranial neural crest-cell migration. Curr Biol. 2001;11(12):918–930. doi: 10.1016/S0960-9822(01)00263-9. [DOI] [PubMed] [Google Scholar]
- 257.Marchant DJ, Bellac CL, Moraes TJ, Wadsworth SJ, Dufour A, Butler GS, Bilawchuk LM, Hendry RG, Robertson AG, Cheung CT, Ng J, Ang L, Luo Z, Heilbron K, Norris MJ, Duan W, Bucyk T, Karpov A, Devel L, Georgiadis D, Hegele RG, Luo H, Granville DJ, Dive V, McManus BM, Overall CM. A new transcriptional role for matrix metalloproteinase-12 in antiviral immunity. Nat Med. 2014;20(5):493–502. doi: 10.1038/nm.3508. [DOI] [PubMed] [Google Scholar]
- 258.Huth T, Schmidt-Neuenfeldt K, Rittger A, Saftig P, Reiss K, Alzheimer C. Non-proteolytic effect of beta-site APP-cleaving enzyme 1 (BACE1) on sodium channel function. Neurobiol Dis. 2009;33(2):282–289. doi: 10.1016/j.nbd.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 259.Gerst JL, Raina AK, Pirim I, McShea A, Harris PL, Siedlak SL, Takeda A, Petersen RB, Smith MA. Altered cell-matrix associated ADAM proteins in Alzheimer disease. J Neurosci Res. 2000;59(5):680–684. doi: 10.1002/(SICI)1097-4547(20000301)59:5<680::AID-JNR11>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 260.Murase S, Cho C, White JM, Horwitz AF. ADAM2 promotes migration of neuroblasts in the rostral migratory stream to the olfactory bulb. Eur J Neurosci. 2008;27(7):1585–1595. doi: 10.1111/j.1460-9568.2008.06119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Wolfsberg TG, Straight PD, Gerena RL, Huovila AP, Primakoff P, Myles DG, White JM. ADAM, a widely distributed and developmentally regulated gene family encoding membrane proteins with a disintegrin and metalloprotease domain. Dev Biol. 1995;169(1):378–383. doi: 10.1006/dbio.1995.1152. [DOI] [PubMed] [Google Scholar]
- 262.Cornwall GA, Hsia N. ADAM7, a member of the ADAM (a disintegrin and metalloprotease) gene family is specifically expressed in the mouse anterior pituitary and epididymis. Endocrinology. 1997;138(10):4262–4272. doi: 10.1210/endo.138.10.5468. [DOI] [PubMed] [Google Scholar]
- 263.Marcinkiewicz M, Seidah NG. Coordinated expression of beta-amyloid precursor protein and the putative beta-secretase BACE and alpha-secretase ADAM10 in mouse and human brain. J Neurochem. 2000;75(5):2133–2143. doi: 10.1046/j.1471-4159.2000.0752133.x. [DOI] [PubMed] [Google Scholar]