

Abstract
Cancer heterogeneity arises during tumor progression as a consequence of genetic insults, environmental cues, and reversible changes in the epigenetic state, favoring tumor cell plasticity. The role of enhancer reprogramming is emerging as a relevant field in cancer biology as it supports adaptation of cancer cells to those environmental changes encountered during tumor progression and metastasis seeding. In this review, we describe the cancer-related alterations that drive oncogenic enhancer activity, leading to dysregulated transcriptional programs. We discuss the molecular mechanisms of both cis- and trans-factors in overriding the regulatory circuits that maintain cell-type specificity and imposing an alternative, de-regulated enhancer activity in cancer cells. We further comment on the increasing evidence which implicates stress response and aging-signaling pathways in the enhancer landscape reprogramming during tumorigenesis. Finally, we focus on the potential therapeutic implications of these enhancer-mediated subverted transcriptional programs, putting particular emphasis on the lack of information regarding tumor progression and the metastatic outgrowth, which still remain the major cause of mortality related to cancer.
Keywords: Enhancer, Cis-regulatory elements, Epigenetic, Signaling pathways, Transcription factors, Reprogramming, DNA damage, Cancer, Tumor progression, Metastasis
Introduction
Cancer is both a genetic and an epigenetic disease, whose outcome is influenced by the tumor microenvironment. These determinants represent the major driving forces of tumorigenesis causing the functional heterogeneity observed in most of the cancer types. Tumor heterogeneity increases the fitness of cancer cells, and currently, it represents a challenge for precise diagnosis and targeted therapy. Different sources of heterogeneity include cell-of-origin, driver oncogenes, genomic alterations, epigenetic changes, cell plasticity, and microenvironment [1]. Recent findings highlighted that among others, epigenomic reprogramming plays a central role in cancer progression and metastasis formation, participating in supporting malignant heterogeneity [2]. Oncogenic transformation frequently involves de novo acquisition of developmental programs, alteration of cell specification, and the aberrant activation of stem-cell-like transcriptional programs. In cancer cells, many factors contribute to override regulatory circuits that, by integrating extrinsic and intrinsic signals, coordinate cell-type-specific transcriptional programs.
Cancer genomics demonstrated that recurrent genetic mutations occur on genes coding for epigenetic regulators [3–5]. In addition to the dis-regulation of these trans-epigenetic factors, genome-wide sequencing of the non-coding genome revealed frequent alterations of cis-regulatory elements such as enhancers and insulators [6]. Different genetic mechanisms can lead to perturbations of enhancer activity, including chromosomal translocations, focal amplifications, and small insertion/deletions or alteration of chromatin topology [7–9]. Finally, many driver mutations impinge on signal transduction genes leading to the improper modulation of signaling cascades and responsiveness to intrinsic and extrinsic cues. Considering that modulation of enhancers’ activity plays a major role in cell identity maintenance, and in controlling cell adaptation to environmental changes, it is conceivable that genetic insults affecting the epigenetic machinery and cis-regulatory sequences may alter enhancer activity, subverting cell fate determination [10]. Specifically, recent works indicate that reprogramming of enhancer function could represent a hallmark of cancer, as it contributes to the de-regulated expression of epi-driver genes, conferring cell growth advantages. In this respect, we define as oncogenic enhancer reprogramming those cancer-related alterations that, independently of their origin, cause aberrant oncogenic activity, leading to de-regulated transcriptional programs, which foster tumor progression and metastasis dissemination (Fig. 1). As a consequence, understanding the molecular basis driving enhancer reprogramming in tumorigenesis could represent a new route towards cancer therapy.
Fig. 1.

Aberrant activity of transcriptional enhancers favors cancer cell plasticity. a Transcriptional enhancers dictate cell- and tissue-specific transcriptional programs, integrating both lineage-determining and signal-dependent transcription factors. Their activity is constrained by physical limits, such as insulators. b Alteration of the physiological activity of transcriptional enhancers may favor cancer cell plasticity and tumor disruption. Aberrant functionality of both cis- and trans-acting factors converge to impose subverted transcriptional landscapes, which might be favorable for promoting the reprogramming of differentiated cells and induce oncogenic features. Black crosses indicate loss of promoter, enhancer or trans-factors activity; red crosses indicate point mutations in cis-regulatory elements. Curved arrows indicate functional interaction of enhancer regions to cognate promoters. LDTF lineage-determining transcription factors, SDTF signal-dependent transcription factors, RNAPII RNA polymerase II
In this review, we will focus on the role of enhancer reprogramming in tumorigenesis, both during the first steps of tumor initiation and in late cancer progression. We will provide an overview of the most recently identified molecular mechanisms and pathways involved in activation of oncogenic enhancers and, finally, we will comment on potential therapeutic perspectives based on oncogenic enhancer reprogramming.
Features and functions of transcriptional enhancers
Spatio-temporal control of gene expression patterns is the key to metazoan development and phenotypic evolution, which is ensured by precise regulatory elements. Among these, cis-regulatory transcriptional enhancers are defined as short non-coding DNA elements (~ 100–2000 bp) capable to boost transcription of related promoters over long genomic distances (up to few megabases), independently of their location and orientation [11, 12]. Over, million putative enhancers have been annotated in the human genome, generating complex modular and combinatorial regulatory networks, in which not only several enhancers influence the expression of a single target gene, but also a specific enhancer may mediate the synchronous transcriptional bursting of multiple promoters, in response to developmental and environmental cues [13–15].
Enhancers share common genomic and epigenetic features. Their distinctive attribute is the presence of arrays of both lineage-determining and signal-dependent transcription factor (LDTFs and SDTFs, respectively) binding sites, functioning as integrators of internal and external signals [16]. Importantly, clusters of enhancers, known as super-enhancers (SEs), function together to regulate cell-type specific genes [17, 18]. To be activated, enhancers need to be cooperatively bound by multiple TFs, either as a multi-protein complex, the so-called ‘enhanceosome’, or as independent units, possibly exploiting the ability of ‘pioneer’ TFs to bind nucleosomal DNA [19–21]. Cooperative TFs’ binding favors nucleosome eviction and thus enhancers correlate with regions of chromatin accessibility, DNA nucleases hypersensitivity, and reduced nucleosome density, as assessed by DNase I-, FAIRE-, and ATAC-sequencing [22, 23]. On the other side, incorporation of hyper-dynamic histone variants at enhancers, such as H2A.Z and H3.3, may render the chromatin less stable and facilitate initial TF access [24–26]. Nucleosomes in enhancer regions are invariantly decorated with mono-methylation of lysine 4 on histone 3 (H3K4me1). H3K4me1 is suggested to prime enhancer regions prior to transcriptional activation. Nonetheless, its functional role is still debated, as it persists even after the end of transcription. Upon enhancer activation, acetylation of lysine 27 is deposited on histone 3 (H3K27ac). Accordingly, binding of p300/CBP, the main histone acetyltransferases which mediate H3K27ac are highly enriched on active enhancers and used for their genome-wide identification. Tri-methylation of lysine 4 on histone 3 (H3K4me3) is a predominant mark of CpG-rich active promoters, yet it is also present at lower level on active enhancers [27, 28]. Consequently, the relative enrichment of H3K4me1 over H3K4me3 is the main epigenetic feature to distinguish enhancers from active promoters [16, 29, 30]. Importantly, tipping of this equilibrium towards abnormal enrichment of H3K4me3 on enhancers might favor tumorigenesis [31].
In their active form, enhancers physically interact with promoter regions through chromatin looping. Although there is broad evidence of interactions from ‘chromosome conformation capture’ technique (3C) and its high-throughput derivatives (4C, 5C, Hi-C, and HiChIP), underlying molecular mechanisms of enhancer–promoter looping are largely unclear. However, cohesin has been demonstrated to stabilize these associations together with Mediator complex and the CTCF insulator protein [32–34]. In addition, physical constraints, such as insulators and distinct topological-associated domains (TADs), limit the activity of transcriptional enhancers [11, 35]. Active enhancers trigger transcription at cognate core promoters thanks to the ability of TFs to recruit co-activators, which lack site-specific DNA-binding competency, but act as histone modifiers (e.g., acetyltransferases, such as p300/CBP and TIP60, and methyltransferases, such as MLL3/4), chromatin remodelers (e.g., BRG1 and CHD7), and long-range interaction facilitators (e.g., Mediator). TFs and co-activator complexes ultimately influence the recruitment and activity of RNA polymerase II (RNAPII) at core promoters, by favoring both the formation of the pre-initiation complex and pause release into productive elongation [36]. Finally, enhancer activity correlates with pervasive transcription from enhancer regions, which leads to the production of non-coding transcripts known as enhancer–RNAs (eRNAs) [37, 38]. Even though increasing evidence are pointing towards functional roles for eRNAs, whether these transcripts are mere by-products of transcriptional activation or serve as crucial regulators of enhancer activity is still debated [38].
Given their prominent role in controlling spatio-temporal restricted patterns of gene expression both during development and in response to intrinsic and extrinsic cues, faulty enhancer function is one of the major drivers in multiple subtypes of tumors [39, 40].
Cis-acting aberrations and oncogenic enhancer activation
Genetic alterations at enhancers are associated with tumors
Genome-wide association studies (GWAS) aim to identify the molecular link between genetic variants and complex diseases, by comparing genetic profiles of large cohorts of human samples characterized by the presence or the absence of a given trait [41, 42]. So far, GWAS have shown that most genetic variants that predispose to cancer preferentially map to the non-coding cis-regulatory elements [43]. Among these, alterations of enhancers play a prominent role in cancer biology, in a tissue- and disease-specific manner. There are different genetic alterations targeting enhancers, which can affect normal gene expression and contribute to cancer development (Fig. 2). They include single-nucleotide polymorphisms (SNPs), small insertions or deletions (INDELs), and larger structural variants such as focal amplifications, large deletions, inversions, and translocation of existing enhancers. In general, germline variants mainly alter the affinity of TF-binding sites. They rarely have a strong effect on gene expression and on the fitness of the organism, even though they can promote oncogenesis. For this reason, cancer development often requires the acquisition of additional somatic mutations such as structural variants, including large genomic rearrangements, causing stronger effects on gene expression deregulation [6, 44].
Fig. 2.
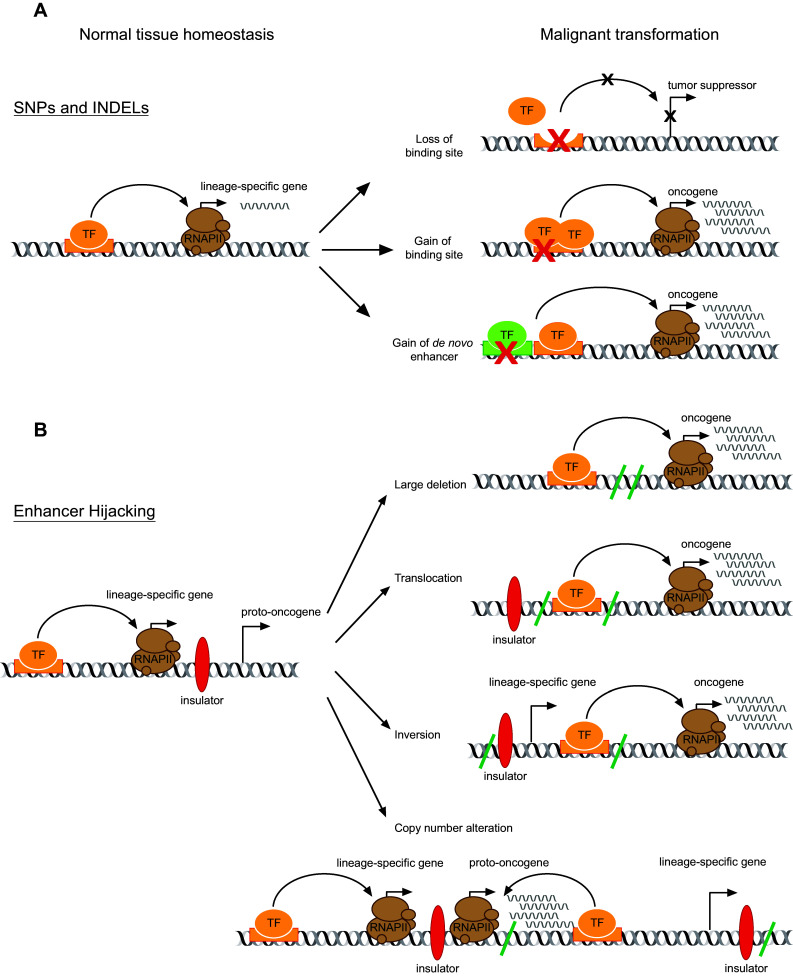
Cis-acting alterations driving oncogenic enhancer activity. a Overview of the possible effects of single-nucleotide polymorphisms and small insertions or deletions on enhancers’ activity. Point mutations may either remove or introduce binding sites at enhancer regions. The following alteration of transcription factor binding leads to enhancer reprogramming, which can cause down-regulation of tumor suppressors or up-regulation of oncogenes (upper and middle panels). Single-nucleotide polymorphisms may also introduce new binding sites in non-enhancer regions, causing nucleation of de novo oncogenic enhancers (lower panel). b Large genomic rearrangements are responsible for enhancers hijacking. Large deletions, translocations, inversions, and copy number alterations can place active enhancers adjacent to proto-oncogenes or remove them from nearby tumor suppression, thus promoting a pro-oncogenic transcriptional program. Black crosses indicate loss of promoter or enhancer activity; red crosses indicate point mutations in cis-regulatory elements. Curved arrows indicate functional interaction of enhancer regions to cognate promoters. Green back slashes indicate the regions affected by large structural variants. SNPs single-nucleotide polymorphisms, INDELs small insertions or deletions, TF transcription factors, RNAPII RNA polymerase II
SNPs that occur in the body of existing enhancers can disrupt TF-binding sites, directly inactivating enhancers and leading to transcriptional down-regulation of the original target gene, therefore, favoring tumor onset [45, 46] (Fig. 2a). Vice versa, gain of extra TF-binding sites can occur and induce over-expression of target proto-oncogenes [45] (Fig. 2a). SNPs and INDELs at non-coding sequences can also generate de novo binding sites for TFs, leading to the formation of aberrant transcriptional enhancer complexes that drive high levels of target genes expression [47–49] (Fig. 2a). This can be exemplified by a work performed in primary patient T-cell acute lymphoblastic leukemias (T-ALLs), which shows that acquisition of heterozygous INDELs, upstream of the transcription start site (TSS) of the TAL1 oncogene, introduces a de novo-binding motif for the MYB TF [50]. MYB binding creates an aberrant SE which drives monoallelic over-expression of TAL1. Accordingly, a work by Navarro et al. shows that insertions at the 5′ of TAL1 in TAL + T-ALLs cause a selective switch from the H3K27me3 repressive histone mark to H3K27ac marks, which trigger reactivation of TAL1 [51].
Mutations that alter insulator sequences of proto-oncogene-containing CTCF–CTCF loops appear to have a pivotal role in the deregulation of gene expression observed in some cancers. Specifically, the CTCF- and cohesin-binding motifs are the most altered TF-binding sites in cancer cells [9, 52]. Furthermore, mutation of chromatin interaction factors–DNA-binding sites localized in promoters can strongly affect enhancer–promoter looping, therefore, subverting normal gene expression. Among the motifs reported to harbor the highest average number of cancer mutations, the DNA-binding site for the zinc finger protein ZNF143 is included [53]. ZNF143 is a chromatin interaction factor which has been proved to directly bind promoters, therefore, contributing to the connection with distal regulatory elements bound by CTCF [54]. SNPs located within ZNF143 DNA recognition sequence can impair its binding and consequently a correct enhancer–promoter interaction frequency, thus affecting the expression of target gene.
Collectively, these studies indicate that various types of mutations targeting non-coding cis-regulatory elements can deregulate enhancer activity to affect normal gene expression and promote tumor development.
Long-range chromosomal structural alterations such as large deletions, translocations, and inversions can cause a phenomenon named ‘enhancer hijacking’, consisting in the repositioning of an enhancer in a new genomic context, where it can activate a proto-oncogene [55–57] (Fig. 2b). For example, in myeloid malignancies, both translocation and inversion events occurring in the long arm of chromosome 3 (3q) can cause the reallocation of a GATA2 enhancer element to the ectopic 3q26.2/EVI1 site, with consequent aberrant activation of the EVI1 gene, involved the in regulation of the stem-cell compartment [58]. This simultaneously causes reduction of GATA2 expression, showing that large chromosomal rearrangements involving enhancers can cause concomitant deregulation of multiple genes.
Recently, two additional mechanisms based on short-range chromosomal structural alterations have been reported to cause enhancer hijacking. Specifically, copy number alterations of enhancers can contribute to tumorigenesis, by increasing proto-oncogenes expression, thereby promoting oncogenic signaling [8, 59, 60]. Moreover, deletion of CTCF-bound insulators between an enhancer and a proto-oncogene can increase the expression of the latter [9]. The merging of structural domains can induce regulatory resetting and alter gene expression. In a recent work, Weischenfeldt et al. explained that colon cancer-related IGF2 amplification and over-expression is caused by tandem duplication of a region comprising the proto-oncogene and a distant enhancer, which results in their placement one next to the other, without intervening insulators [57]. Overall, these studies show that maintenance of chromatin architecture is crucial for the correct enhancer-mediated regulatory function and that disruption of structural domains can alter regulatory interactions and lead to tumorigenesis.
Transposable elements providing enhancer function in cancer
Apart from cis-regulatory enhancers, another class of non-coding elements, the repetitive transposable elements (TEs), may be altered, thus sustaining oncogenesis. TEs represent almost half of the human genome and functionally affect the transcriptome by either providing regulatory sequences or transposing and altering the pre-existent regulatory landscape. Indeed, although mutations and truncations have rendered most TEs unable to transpose, they represent a major source of both promoter and enhancer sequences, which may be co-opted during evolution, to rewire or install alternative regulatory networks [11, 61]. Accordingly, 6–30% of human and mouse TSS initiate within TEs and the majority of primate-specific regulatory sequences are estimated to derive from them [11, 61]. The central role of TEs in regulating cell-specific transcriptional programs has been largely documented in pluripotent stem cells. In this context, TEs are enriched for the binding of pluripotent TFs, serving as alternative promotes and enhancers [62, 63].
Importantly, given their role in gene expression regulation, TEs have been linked to malignant transformation, which may be achieved by DNA hypo-methylation, TE insertion, or point mutations, leading to formation or loss of TF-binding sites [64, 65]. Regarding a possible role of TEs in shaping the oncogenic enhancer landscape, an Alu insertion upstream of an enhancer region of the tumor suppressor CBL has been found associated with its down-regulation, suggesting its functional role in breast cancer tumorigenesis [66]. In K562 leukemia cells, a hyper-methylated LTR TE is bound with low affinity by NF-Y and GATA1/2 TFs and can function as transcriptional enhancer [67]. Finally, in Ewing sarcoma, the oncogenic fusion protein EWS–FLI binds to repetitive GGAA microsatellites, thus functioning as promoter- and enhancer-like structures, which modulate gene expression [68]. We just started to address the putative role of TEs as providers for regulatory sequences involved in cancer initiation and progression. Further research in this direction could help in understanding whether TEs alterations represent a mechanism shared among different tumor types.
Signaling pathways deregulation leads to oncogenic enhancer activation
Enhancer sequences are enriched in clustered binding sites for multiple TFs, including SDTFs that respond to environmental cues, causing dynamic changes in gene expression [16, 69]. Alteration of signaling pathways is a common feature of cancer and de-regulated SDTFs can deeply change the transcriptional program, causing an altered activity of their target enhancers (Fig. 3a). As a consequence, oncogenic enhancers are often enriched in binding sites specific for those SDTFs which lay downstream to the specific signaling pathways upon which cancer cells rely for cell growth advantage. This can be exemplified by colorectal cancer, in which cell transformation depends on hyper-activation of the WNT pathway and the consequent formation of constitutive complexes between β-catenin and the intestinal TCF family member TCF4 [70]. In a recent work, Hnisz et al. show that colorectal cancer cells acquire oncogenic SEs enriched in binding sites for TCF4 and particularly responsive to perturbation of the pathway [71]. Similarly, in mature small B-cell lymphomas characterized by gain-of-function NOTCH mutations, expression of genes that contribute to tumorigenesis is under the control of NOTCH-bound distal enhancers [72]. Among these, the MYC oncogene is responsible of unrestrained cell proliferation and its up-regulation occurs through lineage restricted enhancers.
Fig. 3.
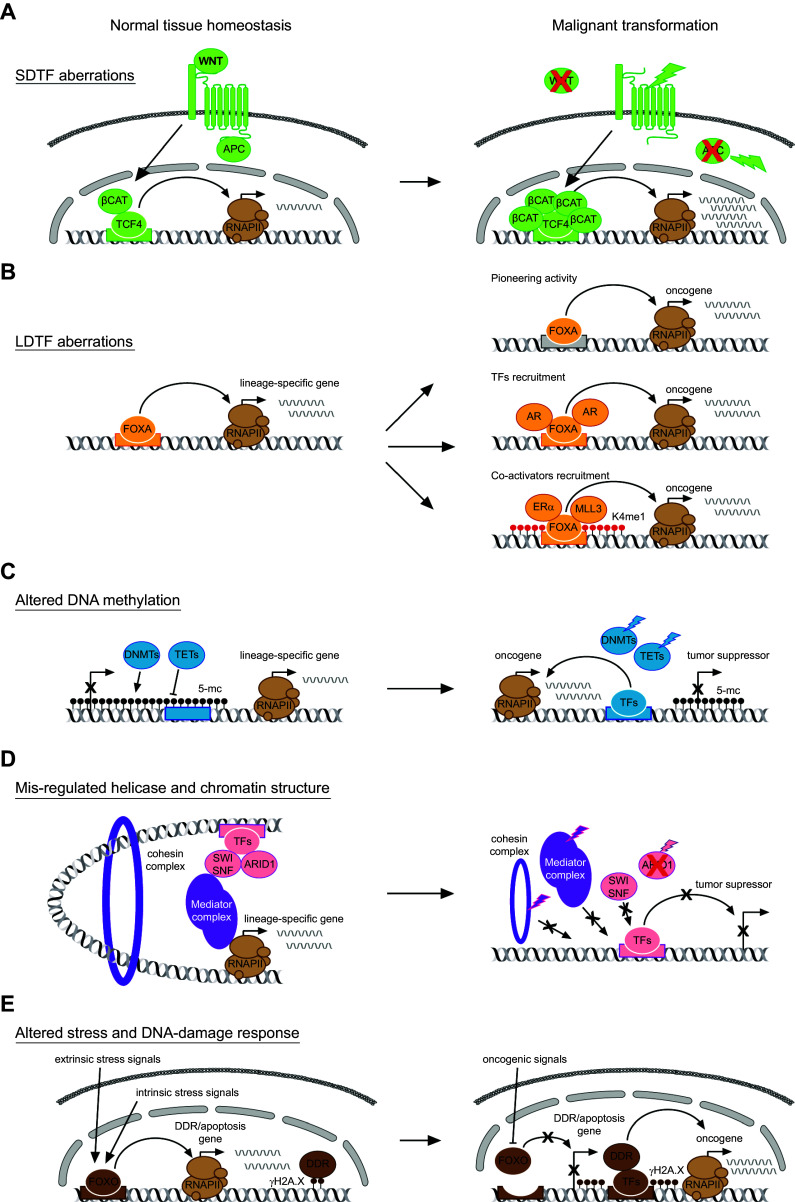
Trans-acting alterations driving oncogenic enhancer activity. a In the intestinal colonic epithelium, WNT pathway activation is responsible for maintaining cell identity. Mutations of multiple factors in the pathway lead to hyper-activation of β-catenin on TCF4-bound enhancer, thus leading to malignant transformation in colorectal cancer cells. b FOXA represents a lineage-determining transcription factor in different tissues. Given its pioneer ability, its mis-regulated DNA binding may favor transcription of oncogenes from oncogenic enhancers (upper panel). Alternatively, its altered ability to recruit other transcription factors or co-factors may promote oncogenic enhancers nucleation (middle and lower panels). c Aberrant methylation landscapes achieved by mutations of DNMTs or TETs represent a major cause of enhancer reprogramming in tumorigenesis. d Three-dimensional structure of the chromatin is favored by helicases and complexes such as mediator and cohesins. Mutations affecting subunits of these multi-protein complexes affects the recruitment of these structural components and the chromatin structure, impairing transcription of tumor suppressors. e In physiological condition, extrinsic and intrinsic stress signals (oxidative, replicative, metabolic, proliferative, and toxic) are sensed and counteracted by specific transcriptional programs. Upon tumorigenesis, new oncogenic signal may affect the activity of transcription factors, such as FOXOs, to activate the DNA-damage response, thus leading on one side to accumulation of DNA damages and on the other side to activation of novel oncogenic enhancers. Black crosses indicate loss of promoter or enhancer activity; red crosses indicate loss of trans-factors activity. Curved arrows indicate functional interaction of enhancer regions to cognate promoters. Colored lightning bolt indicate mutations affecting trans-factors. LDTF lineage-determining transcription factors, SDTF signal-dependent transcription factors, RNAPII RNA polymerase II, TF transcription factors, DDR DNA-damage response
Activation of oncogenic pathways can contribute to malignant transformation by causing a global remodeling of the enhancer landscape and this happens through distinct mechanisms. For example, uncontrolled RTK activation, driven by chronic RAS–ERK signaling, causes dynamic changes in H3K27ac at active enhancers, leading to inappropriate gene activation and promoting tumorigenesis [73]. In this context, HRASG12V-driven transformation requires GATA4 and STAT3 for enhancer marking and PRKGB kinase is a key downstream target responsible for mediation of aberrant gene expression. In addition, oncogenic signaling can modulate enhancer function by directly engaging the transcriptional machinery. This can be exemplified by liver cancer cells, in which deregulation of the Hippo pathway activity results in YAP/TAZ binding to a specific subset of enhancers and SEs, which turn to be potently activated [74]. From these loci, YAP/TAZ modulate transcriptional elongation of growth-promoting genes by regulating promoter–proximal RNAPII pause release, through direct recruitment of the Mediator complex and CDK9 elongating kinase.
Another intriguing aspect of the impact of multiple signaling pathways on enhancer regulation is the crosstalk occurring between different signals, causing an alteration of a pre-established enhancer landscape. In breast cancer, the interplay between TNFα and estrogen pathways, which represent a pro-inflammatory and a mitogenic signaling, respectively, results in a large transcriptional crosstalk [75]. Specifically, TNFα signaling dramatically alters estrogen-regulated transcriptional response by redistributing NF-kB and FOXA1 binding to new genomic loci. In this context, they favor formation of new ERα binding sites, which act as active enhancers. Another study in breast cancer cells shows how YAP/TAZ transcriptional response is mediated by both interaction with TEAD factors, which normally allow their binding to DNA, and chromatin co-occupancy with AP-1 transcription factors [76]. Together, they form a transcription factor complex which binds to distal enhancers carrying both TEAD and AP-1 binding motifs and activates target genes in a joint way. In this context, AP-1 factors are instrumental for YAP/TAZ-induced oncogenic growth.
Deregulation of lineage-determining transcription factors and oncogenic enhancer activation
Cell identity is established and maintained by LDTFs (also referred to as ‘master TFs’), which bind to SEs to regulate cell-type-specific genes [17, 18]. Many LDTFs posses pioneering DNA-binding ability, thus engaging silent chromatin and favoring the recruitment of other TFs and co-factors during cell fate determination [21, 77]. Upon transformation, the transcriptional program dictated by LDTFs in the cell-of-origin is partly conserved in many cancer types, suggesting that tumorigenesis relies on the cooperation between LDTFs and oncogenic factors. Accordingly, aberrant activity of LDTFs is a major driver of multiple cancers and has been demonstrated to modulate binding of oncogenic TFs and activation of tumor-specific enhancers [6, 78].
Among LDTFs, receptors of estrogen and androgen (ERα and AR) are required for differentiation and development of breast ductal and normal prostate epithelia, respectively. At the same time, many breast and prostate cancers rely on the same TFs, whose cistromes are largely reprogrammed during tumorigenesis, thanks to the cooperative binding with other pioneer and LDTFs, such as FOXA1, HOXB13, and progesterone receptor (PR) [79–81]. More recently, an aberrant gastrointestinal-lineage transcriptome has been identified in AR-independent/castration-resistant prostate cancers, which depends on the ability of HNF4G, a pioneer LDTF, to generate and maintain specific enhancers [82]. Seemingly, a wound-induced transcriptome persists in squamous cell carcinoma, in which new oncogenic enhancers are activated by the interplay of stress and LDTFs [83]. In medulloblastoma, specific LDTFs binding at SEs help discriminating cellular origins [84]. These data indicate that the hijack and mis-regulation of LDTF-mediated enhancer activation is a common feature of multiple cancers and may help to explain inter-tumor heterogeneity and treatment resistance.
Tumors may exploit LDTFs to activate oncogenic enhancers in multiple ways including transcriptional modulation, through genetic mutation and amplification or alteration of their DNA-binding sites in cis-regulatory elements. FOX family members exemplify this concept, being involved in multiple cancers and through disparate mechanisms [77, 85] (Fig. 3b). The proto-typical pioneer TFs FOXA1/2 do not only influence the binding of steroid hormones, but are also required to recruit Mediator, cohesin, and MLL3 methyltransferase co-factors to enhancer regions in breast, lung, and liver cancer cells, implying their conserved role in shaping the oncogenic enhancer landscape in multiple tumors [85–87]. In glioblastoma, instead, a mutation in the EGFR-signaling pathway leads to transcriptional activation of FOXG1 and SOX9, which in turn collaborates to induce enhancer reprogramming to sustain tumorigenesis [88]. In the alveolar rhabdomyosarcoma, chromosomal translocation generates the chimeric fusion TF PAX3–FOXO1, which establishes a myogenic and oncogenic SE landscape [89]. In the gastrointestinal stromal tumor, FOXF1 itself transcriptionally activates ETV1 as a pioneer TF and consequently cooperates with this LDTF to dictate a lineage-specific transcriptome through enhancer binding, which is required for tumor growth [90]. Similar mechanisms, in which gene fusion leads to modulation of enhancer and SE landscapes by co-option of LDTFs, have been recently described also in leukemia and prostate cancer [91, 92]. Alternatively, LDTFs may affect oncogenic transcription patterns through eRNA production. In castration-resistant prostate cancers, AR-responsive enhancers produce eRNAs, which in turn favor RNAPII elongation both in cis and in trans. Importantly, their genetic knockdown affects cancer cell growth and chemical inhibition of AR binding reveals specific eRNAs, possibly involved in resistance to therapeutic treatment [93, 94]. During tumorigenesis, clonal selection favors those clones endowed with proliferative advantage. Altogether, these recent evidence illustrate the range of mechanisms involving aberrant LDTF activity, which oncogenic clonal selection may exploit.
Even though cancers may benefit from the utilization of LDTFs, it is also demonstrated that decommissioning of lineage-specific enhancer leads to cell reprogramming and aberrant SE activation drives oncogenic expression in cancer cells [17, 95]. Accordingly, we recently reported a new function for the known pluripotent and oncogenic transcription factor MYC [96, 97]. Specifically, MYC mediates the decommissioning of the luminal transcriptional program in mammary epithelial cells. It down-regulates the expression of GATA3 and ESR1 LDTFs, thus impairing their capacity to bind to lineage-specific enhancers. This leads to a cellular reprogramming towards a progenitor-like phenotype, which favors the consequent activation of distinct oncogenic enhancers during breast cancer tumorigenesis [98]. Similarly, in colorectal tumor, FOXA transcriptional down-regulation correlates with its diminished binding onto epithelial-specific enhancer, ultimately supporting epithelial–mesenchymal transition (EMT) [99].
Finally, LDTF-mediated reprogramming of enhancer landscape has been recently linked to invasiveness and metastatic disruption, indicating its role not only during initial steps of tumorigenesis, but also in tumor progression. FOXA1, TEAD, ETS, and NFIB LDTFs, all support enhancer reprogramming, which confers invasive and metastatic traits to pancreatic cancer, melanoma, squamous cell carcinoma, and small cell lung cancer cells, respectively [100–103].
We just started to investigate the great diversity of LDTF modulation affecting the activation of oncogenic/metastatic-specific enhancers. The increasing availability of genome-wide transcriptional and epigenetic profiling in cancer cells is pushing towards the development of bioinformatics tools to systematically clarify the molecular events leading to enhancer reprogramming during cancer initiation and progression and to identify the causal LDTFs, which may represent new therapeutic options [104, 105].
Role of trans-acting chromatin regulatory factors in oncogenic enhancer activation
Trans-acting chromatin-modifying proteins (CMPs) are ultimate effectors of cis-acting regulatory elements. As a consequence, their mutation and mis-expression/function have been widely associated with both solid and hematological malignancies, representing them as major mediators of cancer disruption and progression (Fig. 3). Nonetheless, they typically exert their tumorigenic function in combination with mutations of other well-defined oncogenes or tumor suppressor genes. While their alteration is normally context- and cancer-type specific, in some cases, the same chromatin player is widely dysregulated in a range of tumors, suggesting a broad role for oncogenesis (e.g., the MLL3/4 complex of the COMPASS family and the SWI/SNF chromatin remodeling complex). The complicated role of CMPs in cancer is also underlined by the fact that the same factor may undergo both loss-of-function and gain-of-function mutations in different cancer types. Indeed, multiple models may explain their driving role in tumorigenesis and include: (1) promoter/enhancer de-activation of tumor suppressor genes; (2) inappropriate promoter/enhancer activation of oncogenes; and (3) aberrant modulation of promoter/enhancer activity of disease-relevant genes, other than cancer genes (e.g., LDTFs). Excellent recent reviews on these topics are referenced [39, 106]. Here, we will mainly focus on the most recent findings regarding the role of CMP malfunction on the activation of oncogenic enhancers.
DNA methylation landscapes
DNA methylation is mediated by DNA methyltransferases (DNMTs), mainly at CpG islands (CGIs), and removed either through passive demethylation during cell division or actively through the action of 10–11 translocation protein (TETs). In physiological condition, CGIs throughout the genome are methylated, keeping these regions silenced, while the promoters of active genes are hypo-methylated. In many cancers, CGIs at promoters of tumor suppressors are frequently hyper-methylated, mediating their transcriptional silencing and giving rise to the so-called ‘CpG island methylator phenotype’ (CIMP). On the contrary, intergenic regions are often globally hypo-methylated [107] (Fig. 3c).
Recent studies indicate that, in virtually all tumor types, enhancers and SEs are among the most affected genomic region in terms of differential DNA methylation between normal and cancer cells (both concerning hyper- and hypo-methylation). Importantly, different DNA methylation patterns at enhancers can be used to stratify cancer types and subtypes. Moreover, hypo-methylation followed by oncogenic TFs’ binding and methylation plasticity at enhancers predict metastatic progression and patient mortality, respectively [108–111].
While aberrant enhancer methylation is a general characteristic in cancer, the CMPs which dictate these alterations are variable. In colorectal cancer, a predominant role for DNMT1 over DNMT3A/B in mediating enhancer methylation has been observed, and putative active distal enhancers are characterized by a dramatic hypo-methylation level [112]. In both acute myeloid (AML) and in T-ALL leukemias, instead, mis-regulation of DNMT3A drives enhancer hypo-methylation, which favors the pathogenesis in combination with additional hits [113, 114]. In other cases, dysregulated DNA methylation alters the functionality of oncogenic enhancers. In IDH mutant gliomas, the activity of TETs is impaired causing CIMP, hyper-methylation of CTCF and cohesin-binding sites and finally resulting in loss of insulator function and mistargeting of oncogenic enhancers [115].
Chromatin-modifying proteins affecting histone methylation and acetylation
The H3K4me1 histone mark, which characterizes transcriptional enhancers, is mainly introduced by MLL3/4 methyltransferases, being part of the large multi-protein COMPASS complex. The H3K27 demethylase UTX is also a subunit of COMPASS and favors enhancer activation by removing H3K27me3, the repressive histone modification introduced by polycomb group proteins (PcG), thus facilitating the deposition of H3K27ac by p300/CBP [116]. Given their prominent role in shaping transcriptional patterns, deregulation of all the above mentioned CMPs is largely frequent in many cancer types. Increasing evidence indicates that MLL3/4 acts as tumor suppressors and is mainly targeted by mis-sense and non-sense mutations; nonetheless, it has also been reported their role as oncogenic factors [116, 117].
Despite their central role in determining enhancer features and their frequent rate of mutation in cancers, we still have scant evidence supporting the role of MLL3/4 and UTX in regulating oncogenic enhancers. In ER-positive breast cancer cells, MLL3 is recruited at enhancer regions by the pioneer LDTF FOXA1 and is required for H3K4me1 deposition and ERα activity on ER-responsive enhancers [87]. In AML, MLL1 activity is mis-regulated by the fusion of its amino-terminal part with different partner genes. In particular, MLL–AF9 and MLL–AF4 are two frequent oncofusion proteins, which have been recently demonstrated to bind distinct active distal regulatory elements, both promoting a transcriptional program similar to the one dictated by the LDTF RUNX1 [118].
Chromatin remodeling complexes and histone variant dynamics
ATP-dependent chromatin remodeling complexes (CRCs) are responsible for maintaining and altering chromatin structure, by moving, ejecting, or restructuring the nucleosome. Four distinct families of CRCs are present in eukaryotes, SWI/SNF, ISWI, CHD, and INO80, which differ for their functional activity, protein domains, and subunits. Importantly, SWI/SNF is frequently implicated in malignant transformation, with more than 20% of human cancers carrying mutations in components of this CRC, and can act both as tumor suppressor and oncogene [119, 120].
Independent studies strongly implicated SWI/SNF in the reprogramming of the enhancer landscape upon tumorigenesis (Fig. 3d). In a mouse model of colon cancer, ARID1A, a subunit of SWI/SNF, acts as tumor suppressor and its loss drives invasive colon adenocarcinoma. This is due to the activity of ARID1A in mediating the recruitment of the SWI/SNF complex at distal enhancers: upon ARID1A loss, the enhancer landscape is reshaped, leading to both de-activation of physiologically active enhancers and activation of new oncogenic enhancers, altering the transcriptional program of the colonic epithelium [121]. Similarly, ARID1A cooperates with ARID1B in dictating enhancer accessibility in colorectal and ovarian cancer cell lines, indicating its conserved role in shaping the enhancer landscape upon tumorigenesis. [122]. Another member of ARID family, ARID5B, is up-regulated in T-ALL cells through activation of an oncogenic super-enhancer and participates to shape the tumor transcriptional program [123]. Likewise, in pediatric rhabdoid tumors, loss of SMARCB1 is the unique genetic alteration which drives the malignancy and is associated with aberrant SWI/SNF targeting at enhancers, with the residual activity of the complex being involved in activating oncogenic SEs and blocking the differentiation. Accordingly, rescue of SMARCB1 in rhabdoid tumor cells causes widespread enhancer activation [124, 125]. Finally, INO80 complex governs the oncogenic transcription from enhancers and SEs in combination with LDTFs in both melanoma and non-small-cell lung cancer, acting as oncogene, and its silencing affects tumorigenesis and tumor maintenance [126, 127].
Apart from CRCs’ activity, the structure of the nucleosome is also determined by the types of histones, of which it is comprised. The canonical histone octamer consists of two copies each of the core histones H2A, H2B, H3, and H4, but these can be replaced in the nucleosome by alternative histone variants. Importantly, these histone variant dynamics are regulated by either CRCs or chaperones. Only in the last years, it has become clear that aberrant histone dynamics, achieved by either histone gene mutations or transcriptional deregulation, could drive oncogenesis [128, 129]. Recent findings identified H2A.Z variant to be an important regulator of enhancer activity in cancer cells. In breast cancer cell lines, H2A.Z is enriched at ERα-responsive enhancers, where its INO80-mediated eviction leads to transcriptional activation. Importantly, H2A.Z depletion impairs RNAPII recruitment and eRNAs’ production, functionally linking H2A.Z occupancy and enhancer activation [130, 131]. Likewise, in prostate cancer cells, acetylated H2A.Z-containing nucleosomes are positioned at oncogenic and AR-responsive enhancers, leading to the formation of nucleosome-depleted regions and eRNA transcripts [132]. We just started to decipher the implications of histone variants and their interplay with CRCs/chaperones on oncogenic enhancer regulation. Nevertheless, given their prominent role in ESC maintenance and in cell reprogramming towards pluripotency [128], it is tempting to speculate that dis-regulation of histone variant dynamics might represent a conserved mechanism to promote malignant transcriptional programs from aberrant enhancer landscapes.
Structural components of the enhancer–promoter loop
As introduced above, the activity of enhancers depends on their ability to physically interact with the transcriptional machinery at promoters, through DNA looping. Different molecules play a major role in bridging TFs and co-activators at enhancers with the pre-initiation complex (PIC) at promoters, among which Mediator, cohesin, and CTCF are the best characterized [32–34]. Given their fundamental role in modulating gene expression patterns, all these structural factors are associated with cancer disruption [133–135]; nonetheless, very few data are available which directly link them to oncogenic enhancer modulation (Fig. 3d).
In multiple myeloma cells, Mediator and BRD4 are highly enriched at oncogenic SEs and BRD4 inhibition leads to loss of Mediator binding and transcriptional shutting-off of these SEs [136]. Similar findings were recapitulated in glioblastoma multiforme and non-small-cell lung cancer cells, suggesting a global role of Mediator in defining the SE landscape of multiple tumor cells [136]. Conversely, in AML, Mediator-associated kinases seem to repress oncogenic SEs, leading to silencing of tumor suppressors. Accordingly, their inhibition disproportionally induces those tumor suppressor genes and suppresses AML progression and cell proliferation [137]. More recently, genome-wide analysis indicates that LDTFs (FOXA1 and ERα) recruit Mediator and cohesin to initially form and activate enhancer regions in breast, liver, and lung cancer cell lines and that they are required for proliferation of these cells [86, 138].
Opposing effects on oncogenic enhancer modulation have also been reported for CTCF. In prostate cancer cell lines, genome-wide studies on the three-dimensional chromatin structure reveal that upon transformation TADs, boundaries are reshaped, forming more numerous and smaller domains. Importantly, the anchor points of these cancer-specific differential interactions are enriched for CTCF binding and enhancer regulatory elements, indicating a functional role of CTCF at oncogenic enhancer regions [139]. On the other side, in breast cancer cells, CTCF negatively regulates ER-responsive enhancers, by binding at the level of eRNAs and interacting with the nuclear matrix. Importantly, CTFC depletion favors transcription of enhancer–cognate genes and breast cancer cell proliferation [140].
Finally, a map of differentially active enhancers between colonic crypt epithelium and colorectal cancer samples has recently identified tumor-specific enhancers, which are enriched for known colorectal cancer risk loci and AP1 and cohesin binding. Impairment of these enhancers affects oncogene expression and tumor growth [141]. Nonetheless, the role of cohesin in oncogenic enhancer activation remains unclear: in colorectal carcinoma cell lines, cohesin loss causes elimination of all chromatin loops, but does not affect global gene expression profile and histone marks. However, it leads to condensations of SEs and down-regulation of nearby genes, suggesting a functional role for cohesin in defining the SEs landscape of colorectal cancer cells [142].
Taking into account their central role for tumorigenesis and modulation of oncogenic enhancers, major efforts must be dedicated in the future to elucidate the mechanistic functions of both cancer-type-specific and globally mis-regulated CMPs. This will hugely help in developing novel potential therapeutic compounds, which will flank and complement the already available inhibitors, currently tested in clinical trials [143, 144].
Oncogenic signals driving enhancer reprogramming
Cancer is a chronic and heterogeneous disease in which aging represents the most recurrent risk factor for solid tumors. Many epidemiological studies showed that the incidence of the most recurrent cancers increases with age [145]. Although we do not have a full comprehension of the aging-associated factors that are causative of tumorigenesis, many processes are commonly altered in these settings. Of importance, age-associated perturbations such as DNA damage, epigenetic alterations, and cell senescence are interconnected, influencing each other [146]. In this respect, alterations of the TFs activity, epigenetic state, and the increment of DNA damage converge on cis-regulatory elements and contribute to changes in gene expression, thus representing common mechanisms involved both in aging and cancer [147].
For example, members of the FOXO TFs are involved both in aging and tumorigenesis, as their activation promotes longevity, while suppressing tumor formation in the oncogenic setting (Fig. 3e). From a molecular point of view, FOXOs can act as pioneer TFs by binding to nucleosome-associated enhancer regions, driving chromatin remodeling and opening, thus facilitating gene activation [148–151]. Of importance, the FOXOs’ transcriptional activity is strongly dependent on both extrinsic and intrinsic signals as they respond to changes in nutrients availability, cellular stresses (oxidative stress and DNA damage), and growth factors. Upon stress-induced activation, FOXOs coordinate transcriptional response, which increases resistance to both cellular and environmental stresses. On the basis of these regulatory mechanisms, together with in vivo functional analyses and histopathology data, FOXOs are considered tumor suppressor genes that activate stress responses by driving enhancer reprogramming (Fig. 3e).
Increase of DNA damage is a hallmark of aged cells, which augment the risk of accumulating DNA lesions that favor the onset of tumorigenesis. Different sources of DNA damage can elicit the accumulation of DNA lesions in pre-neoplastic state, triggering the activation of different DNA-damage repair pathways [152]. Among these lesions, DNA double-strand breaks (DSBs) are of particular importance for cancer progression as their accumulation on fragile sites compromises genomic integrity. DSBs can arise from both exogenous and endogenous sources, including exposure to ionizing radiation and replicative stress. These cues trigger DNA-damage response (DDR) which ensure efficient repair of damage sites by coordinating the DNA repair machinery with chromatin modifiers, thus re-establishing both genetic and epigenetic integrity [153]. Considering that DDR represents a barrier against cancer is not surprising that many genes involved in these signal cascade are mutated in many tumors, which progressively accumulate DNA lesions and genomic alterations. Oncogene-induced DNA damage occurs preferentially at fragile sites, which resides in early replicating regions associated with transcriptionally active genes and cis-regulatory elements [152, 154]. Of importance, DDR takes place within the chromatin context, which represents a barrier for efficient detection and repair of DSBs. To overcome this, the DNA repair machinery induces widespread chromatin rearrangements at DSB sites. Specifically, the access to damaged DNA sites is ensured by a coordinated series of chromatin events which, starting from ATM/ATR-mediated phosphorylation of H2AX, leads to nucleosome eviction, histone modifications, and chromatin remodeling. This cascade determines chromatin decompaction which extends far away from the DSB sites, covering large chromatin regions (up to 100 Kb) [155, 156]. Although many evidence demonstrate that a signal cascade determines chromatin changes at DSB sites, the mechanisms governing the restoration of the epigenetic integrity are poorly defined. More importantly, it is currently unknown whether the epigenome is properly restored after DNA damage or instead the induced-chromatin state could persist on these loci, establishing an epigenetic memory. In this context, DDR could shape the chromatin state, determining a window of opportunity for modulating the epigenetic state that could influence other relevant biological processes such as transcription.
We propose that DNA damage could represent an oncogene-associated cue that participates in enhancer reprogramming. It is plausible that the chromatin rearrangement in response to DSBs could support relaxation of chromatin surrounding enhancers, which otherwise would be embedded in close, poorly accessible chromatin domains. This DDR-induced-chromatin decompaction could favor the association of oncogenic TFs, whose binding affinity is otherwise blocked by nucleosome occupancy. This temporary chromatin opening and the consequent binding of TFs could represent the initiating event for recruiting other chromatin modifiers which in turn will initiate enhancer activation. Further experiments delineating whether sustained DDR could facilitate enhancer reprogramming would help defining a broader pro-oncogenic function of DNA repair.
Beside this proposed mechanisms for de novo enhancers nucleation, recent findings show that the DNA repair machinery supports the activation of oncogenic enhancers. Localized DNA breaks have been functionally associated with transcriptional activation mediated by nuclear hormone receptors’ binding. In response to hormonal stimulus, TFs’ binding elicits local DNA damage, as shown for AR-mediated recruitment of activation-induced cytosine deaminase (AID), which causes cytosine deamination [157], or for ERα binding, that triggers LSD1-mediated DNA oxidation [158]. These reactions lead to damaged DNA bases, whose removal triggers the formation of transient nicks that function as entry points for Topoisomerase 2 (TOP2β) [159] or LINE-1 repeat-encoded ORF2 endonucleases [157]. The resulting DNA cleavage relieves topological stress and facilitates the recruitment of the DNA repair machinery, which supports transcriptional activation [159–161]. For example, it was shown that in prostate cancer cells, AR activation leads to recruitment of TOP1 to enhancers, which were pre-bound by the pioneer TF NKX3.1 [161]. Shortly after TOP1 binding, the components of the DDR pathway such as MRN complex (MRE11, RAD50, and NBS1) and ATR are loaded on the enhancers, followed by the recruitment of DNA repair enzymes belonging to the BER pathway [161]. More importantly, in this work, it has been established that perturbation of the DDR impairs enhancer activation and consequently expression of the regulated genes. Since activation of enhancers is coupled with transcription of eRNAs in cis, the accumulation of local topological stress could affect enhancer activity and the recruitment of DNA repair machinery could resolve it. Alternatively, transcriptional-induced DNA damage could favor the establishment of a permissive chromatin state and stimulate chromatin looping, thus favoring enhancer–promoter communication [162]. Of importance, using advanced approach to map DNA-damage site genome wide, it has been showed that DSBs are enriched at boundaries of enhancer/promoter chromatin loops [154]. Together, these findings support the notion that recruitment of the DNA repair machinery could facilitate enhancer-mediated transcription activation by resolving topological stresses. Recent findings showed that SE activation in thymic stromal cells correlates with widespread binding of TOP1 and the accumulation of γH2AX [163].
These results raise the possibility that the high-density binding of multiple TFs on clustered enhancers and their activation in the oncogenic setting could be supported by the combinatorial action of the DNA repair machinery. In the near future, further investigation should highlight the prevalent function of the crosstalk between TFs, chromatin players, and DDR in coordinating enhancer reprogramming in cancer cells.
Reprogramming of enhancers in the context of cancer progression and metastasis
Despite the substantial progress in understanding the genetic and epigenetic changes driving tumor initiation, the mechanisms driving tumor progression and metastasis formation are largely unknown. Considering that metastasis is the main cause of death among cancer patients, defining those traits that distinguish metastatic capability represents an unmet medical need. Metastasis consists in a series of steps in which cancer cells disseminate from primary tumors to the bloodstream and then seed distant organs, where they can give rise to overt overgrowth, forming macro-metastasis [164]. This long route towards metastasis exposes cancer cells to different hostile microenvironments and their capacity to adapt to these extreme conditions represents a specific feature of pro-metastatic cells. Albeit of its importance, we currently have a limited knowledge of those genetic and/or epigenetic determinants that support the acquisition of metastatic hallmarks. Extensive genome sequencing efforts did not permit to identify driver genes or genetic alterations specifying tumor dissemination and metastasis burden [2, 10, 165, 166]. Considering the continuous and dynamic pressure of the microenvironment on disseminated tumor cells, it is reasonable considering that epigenetic alterations could play a central role in metastasis formation. The invasion–metastasis cascade implies that some features of metastatic cells are acquired and selected early during tumor formation, while others are induced during the colonization phase [164].
The initial step of metastasization involves the acquisition of increased mobility, invasiveness, and degradation of the extracellular matrix, which is supported by the epithelial–mesenchymal transition (EMT). This reversible cellular change is activated by specific TFs (SNAIL, SLUF, TWIST, and ZEB1), which drive the activation of specific transcriptional programs. Of note, activation of a disseminating phenotype may occur in the early phases of tumor formation, being detectable in pre-neoplastic stage. For example, using animal model recapitulating HER2-driven breast cancer, it has been shown that early disseminated tumor cells are supported by the activation of self-reinforcing signaling pathways [167, 168]. In this context, the balance between HER2-induced high proliferative state and the WNT-associated disseminating phenotype determines the pro-metastatic potential of the disseminated tumor cells (DTCs). It would be of extreme importance determining the consequence of these different signals, which govern early dissemination, by defining the downstream transcriptional and epigenetic programs controlling these reversible phenotypes.
In the colonization phase, a combination of cell-autonomous and extrinsic factors governs the capacity of DTCs to seed into distal organs. Both clinical data and animal models highlighted that DTCs persist in indolent state of dormancy, in distal organs. This cellular condition, which is characterized by the non-proliferative states of DTCs, is the result of the unfavorable environments in which immune surveillance represents a major defense against metastasis [164]. To survive in this setting, dormant DTCs activate autocrine signals or corrupt the surrounding stroma to ensure sufficient activation of pro-survival signaling, such as PI3K/AKT pathway. At the same time, DTCs are able to colonize or to set up cellular niches, which determine the activation and reinforcement of stem-cell signaling, such as WNT and NOTCH pathways. The reactivation of these developmental pathways supports cell plasticity, inducing stem-cell-like features. Overall, a balance between DTCs and the local microenvironments determines a certain level of cell plasticity, which permits the maintenance of this indolent state. The causative drivers of overt metastasis growth are poorly defined, although it is plausible to predict that an unbalance of the host microenvironment, combined with the further acquisition of pro-metastatic features, could permit DTCs to exit dormancy, activating a proliferation program. Indeed, recent single-cell RNA-seq data demonstrated that low burden metastasis expresses a specific program associated with EMT, survival, and stemness, which is then reverted in high burden metastasis, that activate an MYC-driven proliferative signature [169]. Of importance, the phenotypic changes associated with the activation of alternative transcriptional programs are not associated with genetic alterations, implying that non-genetic (epigenetic) changes determine these features [169]. These results have relevant implications relative to the contribution of epigenetic modulation, which could support the amplification of pro-metastatic signals or repress metastasis suppressor genes through DNA methylation [170].
In the attempt of defining the epigenetic contribution to DTCs’ colonization and metastasis growth, recent findings suggest that global epigenetic reprogramming may play a central role during this process. Two studies on pancreatic ductal adenocarcinoma (PDAC) evaluated the impact of genetic and epigenetic heterogeneity within the same individual on the formation and growth of distal metastasis [2, 165]. While the authors could not identify known pro-metastatic driver genes [165], they showed that epigenomic reprogramming during tumor progression favors adaptation to metabolic changes occurring at distal metastatic sites [2]. A comparative analysis between primary tumors and local and distal metastasis showed a global reduction of heterochromatic (H3K9me2/3, H4K20me3, and DNA methylation) markers correlated with tumor colonization at distal sites. Of importance, genome-wide mapping showed that specific chromatin reprogramming occurs at large domains (LOCKs), establishing permissive conditions for the local activation of specific promoters. Thus, the epigenomic reprogramming, which was already primed within the primary tumor cells, specifies altered gene expression programs that have been further selected during tumor progression and metastasis growth [2]. Albeit metastatic-associated epigenomic changes include also increment of euchromatin histone marks, the contribution of enhancer reprogramming to the adaptation of metastatic cells to the environmental changes have not been investigated.
Using a genetically engineered mouse model that recapitulates the main stages of PDAC tumor progression, Vakoc and his team evaluated whether enhancer landscape reprogramming could contribute to metastasis formation [101]. By profiling genome-wide enrichment of H3K27Ac in organoids derived from normal ducts, primary tumors, and metastatic lesions, the authors defined a subset of enhancers, which are specifically activated in metastatic cells. Of importance, while both H3K27Ac and H3K4me1 levels specifically increase in metastasis-derived organoids, the same cis-regulatory elements are already primed in primary tumor cells, as detected by the pattern of chromatin accessibility. This finding is surprising as the authors demonstrated that the over-activation of the pioneer transcription factor FOXA1 drives enhancer activation, inducing metastasis properties. However, FOXA1 over-expression is not sufficient to drive enhancer reprogramming, as it requires the synergic action of GATA5 to boost their activation. The metastasis-associated enhancer reprogramming activates an embryonic foregut endoderm transcriptional program, suggesting that de-activation of developmental-associated signals may sustain metastasis formation. These findings suggested that cancer-driven deregulation of TFs and possibly their interplay with microenvironment-activated signals could alter the epigenetic landscape, favoring the activation of alternative transcriptional program in metastatic cells. Similar conclusions were also reached in an independent study in which small cell lung cancer (SCLC) metastasis are associated with widespread increase in chromatin accessibility [100]. By combining genetic model systems of tumor progression with genome-wide analyses of chromatin accessibility, the authors showed that SCLC tumors gain metastatic potential by over-expressing the transcription factor NFIB. Specifically, it has been demonstrated that the metastatic-associated chromatin opening is driven by the binding of NFIB, leading to the activation of a neural transcriptional program. In other words, this neuroendocrine cancer requires the overall rewiring of its lineage-specific signature to drive the NFIB-mediated metastatic ability. Although the augmented chromatin accessibility has been associated with activation of distal cis-regulatory elements, further studies should support the conclusion that NFIB induces enhancer reprogramming. In addition, gaining molecular insights on the mechanisms through which these TFs alter the pattern of enhancer activity and their specificity in driving pro-metastatic states would provide a mean for targeting and possibly eliminating metastatic diseases.
Future perspectives
It has been recognized that beside the prominent role of genetic alterations and environmental cues, epigenetic changes play a central role in tumor evolution and progression. In particular, oncogenic-associated perturbation of the epigenetic machinery causes both molecular and cellular heterogeneity within tumors, increasing the fitness and the plasticity of cancer cells. In this review, we focused on those oncogenic signals that directly or indirectly impinge on enhancer activity. We specifically highlighted recent findings which show that enhancer reprogramming could represent a hallmark of cancer progression and metastasis dissemination. By analyzing the different factors that participate in oncogenic enhancer deregulation, we underlined the importance of considering the combinatorial contribution of genetic, epigenetic, and environmental perturbations. Specifically, we proposed that enhancer reprogramming results from cancer-related perturbations, which could be time-, context-, and cell-type specific. This notion implies that the same oncogenic insults may lead to different outcomes in terms of oncogenic enhancer regulation, leading to different de-regulated transcriptional programs, supporting malignant heterogeneity. Understanding the principle and investigating the molecular insights that govern enhancer reprogramming in tumorigenesis represent a front line in the field of cancer biology, opening the opportunity to define better therapeutic strategy. Despite its relevance, this field is still on its infancy and further research is required to answer to many open questions. In this review, we propose the notion that many cancer-associated processes, which are not directly linked with transcriptional deregulation in cancer, could indeed participate in triggering and sustaining enhancer reprogramming. To define the mechanisms governing the interplay between genetic and non-genetic insults, it would be mandatory to adopt a multi-disciplinary approach that should include molecular, cellular, and functional biology. For example, despite the large amount of genome-wide data showing a correlation between genetic and epigenetic alterations affecting enhancer in cancer cells, there are still scant evidence demonstrating the functional relevance of oncogenic enhancers in driving tumor progression and metastasis growth. One possible route to dissect the contribution of enhancers’ deregulation in tumorigenesis is represented by CRISPR-based epigenome editing [171]. This upcoming approaches, which have been recently adopted to identify functional cis-regulatory elements and their interconnections to modulate gene expression [172–174], could represent a new frontiers as therapeutic strategy to target cancer cells relying on oncogenic enhancer activation. On the same line, despite the therapeutic potential of epigenetic drugs, the current limited knowledge of the mechanisms governing the dynamic, and reversible changes affecting oncogenic enhancers limit their usage to primary tumors otherwise nonresponsive to the standard chemotherapy agents. We postulated that, on the basis of the involvement of different oncogenic signals in enhancer deregulation, new therapeutic regimes should focus on the combinatorial action of epi-drugs with cancer-specific treatments, to elicit cancer cell vulnerability. In addition, these combinatorial treatments may also help preventing the occurrence of drug resistance, which limits the therapeutic benefits of newly designed therapies. Accordingly, recent studies report that the use of CDK7 or BRD4 inhibitors strongly affects the oncogenic enhancer remodeling and hinders the emergence of drug-resistant cancer cell populations [175–177]. These works highlighted that the onset of resistance to therapeutic agents represents an adaptation of cancer cells, which is often driven by epigenetic reprogramming. In this context, defining the common oncogenic signals that support drug-induced cell plasticity and the involvement of enhancer deregulation could represent a new avenue to define alternative therapeutic options.
Acknowledgements
We wish to express our appreciation to Center of Integrative Biology (CIBIO) of the University of Trento for granting a postdoctoral fellowship to Luca Fagnocchi. The authors also wish to acknowledge the Fondazione AIRC for granting a postdoctoral fellowship to Vittoria Poli (AIRC-M ID 21158). Work in Zippo group was supported by grants from the Italian Ministry of Health (GR-2011-02351172), and CARIPLO foundation (2014-0915).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interests.
Contributor Information
Luca Fagnocchi, Email: luca.fagnocchi@unitn.it.
Alessio Zippo, Email: alessio.zippo@unitn.it.
References
- 1.Koren S, Bentires-Alj M. Breast tumor heterogeneity: source of fitness. Hurdle for therapy. Mol Cell. 2015;60:537–546. doi: 10.1016/j.molcel.2015.10.031. [DOI] [PubMed] [Google Scholar]
- 2.McDonald OG, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet. 2017;49:367–376. doi: 10.1038/ng.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fujimoto A, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760–764. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 4.Kandoth C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stephens PJ, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sur I, Taipale J. The role of enhancers in cancer. Nat Rev Cancer. 2016;16:483–493. doi: 10.1038/nrc.2016.62. [DOI] [PubMed] [Google Scholar]
- 7.Northcott PA, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature. 2014;511:428–434. doi: 10.1038/nature13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang X, et al. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat Genet. 2016;48:176–182. doi: 10.1038/ng.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hnisz D, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016;351:1454–1458. doi: 10.1126/science.aad9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vogelstein B, et al. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Long HK, Prescott SL, Wysocka J. Ever-changing landscapes: transcriptional enhancers in development and evolution. Cell. 2016;167:1170–1187. doi: 10.1016/j.cell.2016.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spitz F. Gene regulation at a distance: from remote enhancers to 3D regulatory ensembles. Semin Cell Dev Biol. 2016;57:57–67. doi: 10.1016/j.semcdb.2016.06.017. [DOI] [PubMed] [Google Scholar]
- 13.Andersson R, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Consortium, E.P An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukaya T, Lim B, Levine M. Enhancer control of transcriptional bursting. Cell. 2016;166:358–368. doi: 10.1016/j.cell.2016.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013;49:825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hnisz D, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whyte WA, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Panne D. The enhanceosome. Curr Opin Struct Biol. 2008;18:236–242. doi: 10.1016/j.sbi.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13:613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- 21.Zaret KS, Mango SE. Pioneer transcription factors, chromatin dynamics, and cell fate control. Curr Opin Genet Dev. 2016;37:76–81. doi: 10.1016/j.gde.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Risca VI, Greenleaf WJ. Unraveling the 3D genome: genomics tools for multiscale exploration. Trends Gene TIG. 2015;31:357–372. doi: 10.1016/j.tig.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet. 2014;15:272–286. doi: 10.1038/nrg3682. [DOI] [PubMed] [Google Scholar]
- 24.Chen P, Wang Y, Li G. Dynamics of histone variant H3.3 and its coregulation with H2A.Z at enhancers and promoters. Nucleus. 2014;5:21–27. doi: 10.4161/nucl.28067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin C, Felsenfeld G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007;21:1519–1529. doi: 10.1101/gad.1547707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin C, et al. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat Genet. 2009;41:941–945. doi: 10.1038/ng.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 28.Pekowska A, et al. H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J. 2011;30:4198–4210. doi: 10.1038/emboj.2011.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia-Gonzalez E, Escamilla-Del-Arenal M, Arzate-Mejia R, Recillas-Targa F. Chromatin remodeling effects on enhancer activity. Cell Mol Life Sci CMLS. 2016;73:2897–2910. doi: 10.1007/s00018-016-2184-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim TK, Shiekhattar R. Architectural and functional commonalities between enhancers and promoters. Cell. 2015;162:948–959. doi: 10.1016/j.cell.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen X, et al. ATAC-see reveals the accessible genome by transposase-mediated imaging and sequencing. Nat Methods. 2016;13:1013–1020. doi: 10.1038/nmeth.4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo Y, et al. CTCF/cohesin-mediated DNA looping is required for protocadherin alpha promoter choice. Proc Natl Acad Sci USA. 2012;109:21081–21086. doi: 10.1073/pnas.1219280110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kagey MH, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu Z, Wei G, Chepelev I, Zhao K, Felsenfeld G. Mapping of INS promoter interactions reveals its role in long-range regulation of SYT8 transcription. Nat Struct Mol Biol. 2011;18:372–378. doi: 10.1038/nsmb.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang J, Corces VG. Insulators, long-range interactions, and genome function. Curr Opin Genet Dev. 2012;22:86–92. doi: 10.1016/j.gde.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reiter F, Wienerroither S, Stark A. Combinatorial function of transcription factors and cofactors. Curr Opin Genet Dev. 2017;43:73–81. doi: 10.1016/j.gde.2016.12.007. [DOI] [PubMed] [Google Scholar]
- 37.Kim TK, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016;17:207–223. doi: 10.1038/nrg.2016.4. [DOI] [PubMed] [Google Scholar]
- 39.Herz HM. Enhancer deregulation in cancer and other diseases. BioEssays News Rev Mol Cell Dev Biol. 2016;38:1003–1015. doi: 10.1002/bies.201600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo Z, Lin C. Enhancer, epigenetics, and human disease. Curr Opin Genet Dev. 2016;36:27–33. doi: 10.1016/j.gde.2016.03.012. [DOI] [PubMed] [Google Scholar]
- 41.Maurano MT, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012;22:1748–1759. doi: 10.1101/gr.136127.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang X, Bailey SD, Lupien M. Laying a solid foundation for Manhattan—’setting the functional basis for the post-GWAS era’. Trends Gene TIG. 2014;30:140–149. doi: 10.1016/j.tig.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khurana E, et al. Role of non-coding sequence variants in cancer. Nat Rev Genet. 2016;17:93–108. doi: 10.1038/nrg.2015.17. [DOI] [PubMed] [Google Scholar]
- 45.Oldridge DA, et al. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature. 2015;528:418–421. doi: 10.1038/nature15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kandaswamy R, et al. Genetic predisposition to chronic lymphocytic leukemia is mediated by a BMF super-enhancer polymorphism. Cell Rep. 2016;16:2061–2067. doi: 10.1016/j.celrep.2016.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Z, et al. APOBEC signature mutation generates an oncogenic enhancer that drives LMO1 expression in T-ALL. Leukemia. 2017;31:2057–2064. doi: 10.1038/leu.2017.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu S, et al. Whole-genome noncoding sequence analysis in T-cell acute lymphoblastic leukemia identifies oncogene enhancer mutations. Blood. 2017;129:3264–3268. doi: 10.1182/blood-2017-03-771162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abraham BJ, et al. Small genomic insertions form enhancers that misregulate oncogenes. Nat Commun. 2017;8:14385. doi: 10.1038/ncomms14385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mansour MR, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373–1377. doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Navarro JM, et al. Site- and allele-specific polycomb dysregulation in T-cell leukaemia. Nat Commun. 2015;6:6094. doi: 10.1038/ncomms7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Katainen R, et al. CTCF/cohesin-binding sites are frequently mutated in cancer. Nat Genet. 2015;47:818–821. doi: 10.1038/ng.3335. [DOI] [PubMed] [Google Scholar]
- 53.Ji X, et al. 3D chromosome regulatory landscape of human pluripotent cells. Cell Stem Cell. 2016;18:262–275. doi: 10.1016/j.stem.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bailey SD, et al. ZNF143 provides sequence specificity to secure chromatin interactions at gene promoters. Nat Commun. 2015;2:6186. doi: 10.1038/ncomms7186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bandopadhayay P, et al. MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet. 2016;48:273–282. doi: 10.1038/ng.3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Drier Y, et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat Genet. 2016;48:265–272. doi: 10.1038/ng.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weischenfeldt J, et al. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat Genet. 2017;49:65–74. doi: 10.1038/ng.3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Groschel S, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157:369–381. doi: 10.1016/j.cell.2014.02.019. [DOI] [PubMed] [Google Scholar]
- 59.Zhang X et al (2017) Somatic super-enhancer duplications and hotspot mutations lead to oncogenic activation of the KLF5 transcription factor. Cancer Discov [DOI] [PMC free article] [PubMed]
- 60.Glodzik D, et al. A somatic-mutational process recurrently duplicates germline susceptibility loci and tissue-specific super-enhancers in breast cancers. Nat Genet. 2017;49:341–348. doi: 10.1038/ng.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Faulkner GJ, et al. The regulated retrotransposon transcriptome of mammalian cells. Nat Genet. 2009;41:563–571. doi: 10.1038/ng.368. [DOI] [PubMed] [Google Scholar]
- 62.Gerdes P, Richardson SR, Mager DL, Faulkner GJ. Transposable elements in the mammalian embryo: pioneers surviving through stealth and service. Genome Biol. 2016;17:100. doi: 10.1186/s13059-016-0965-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kunarso G, et al. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet. 2010;42:631–634. doi: 10.1038/ng.600. [DOI] [PubMed] [Google Scholar]
- 64.Burns KH. Transposable elements in cancer. Nat Rev Cancer. 2017;17:415–424. doi: 10.1038/nrc.2017.35. [DOI] [PubMed] [Google Scholar]
- 65.de Souza FS, Franchini LF, Rubinstein M. Exaptation of transposable elements into novel cis-regulatory elements: is the evidence always strong? Mol Biol Evol. 2013;30:1239–1251. doi: 10.1093/molbev/mst045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Clayton EA, et al. Patterns of transposable element expression and insertion in cancer. Front Mol Biosci. 2016;3:76. doi: 10.3389/fmolb.2016.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hu T, et al. Hypermethylated LTR retrotransposon exhibits enhancer activity. Epigenetics. 2017;12:226–237. doi: 10.1080/15592294.2017.1289300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnson KM, Taslim C, Saund RS, Lessnick SL. Identification of two types of GGAA-microsatellites and their roles in EWS/FLI binding and gene regulation in Ewing sarcoma. PLoS ONE. 2017;12:e0186275. doi: 10.1371/journal.pone.0186275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fagnocchi L, Mazzoleni S, Zippo A. Integration of signaling pathways with the epigenetic machinery in the maintenance of stem cells. Stem Cells Int. 2016;2016:8652748. doi: 10.1155/2016/8652748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 71.Hnisz D, et al. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol Cell. 2015;58:362–370. doi: 10.1016/j.molcel.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ryan RJH, et al. A B cell regulome links notch to downstream oncogenic pathways in small B cell lymphomas. Cell Rep. 2017;21:784–797. doi: 10.1016/j.celrep.2017.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nabet B, et al. Deregulation of the Ras-Erk signaling axis modulates the enhancer landscape. Cell Rep. 2015;12:1300–1313. doi: 10.1016/j.celrep.2015.06.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Galli GG, et al. YAP drives growth by controlling transcriptional pause release from dynamic enhancers. Mol Cell. 2015;60:328–337. doi: 10.1016/j.molcel.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Franco HL, Nagari A, Kraus WL. TNFalpha signaling exposes latent estrogen receptor binding sites to alter the breast cancer cell transcriptome. Mol Cell. 2015;58:21–34. doi: 10.1016/j.molcel.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zanconato F, et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol. 2015;17:1218–1227. doi: 10.1038/ncb3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Iwafuchi-Doi M, Zaret KS. Cell fate control by pioneer transcription factors. Development. 2016;143:1833–1837. doi: 10.1242/dev.133900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bradner JE, Hnisz D, Young RA. Transcriptional addiction in cancer. Cell. 2017;168:629–643. doi: 10.1016/j.cell.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bhagwat AS, Vakoc CR. Targeting transcription factors in cancer. Trends Cancer. 2015;1:53–65. doi: 10.1016/j.trecan.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pomerantz MM, et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat Genet. 2015;47:1346–1351. doi: 10.1038/ng.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Singhal H, et al. Genomic agonism and phenotypic antagonism between estrogen and progesterone receptors in breast cancer. Sci Adv. 2016;2:e1501924. doi: 10.1126/sciadv.1501924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shukla S, et al. Aberrant activation of a gastrointestinal transcriptional circuit in prostate cancer mediates castration resistance. Cancer Cell. 2017;32(6):792–806. doi: 10.1016/j.ccell.2017.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ge Y, et al. Stem cell lineage infidelity drives wound repair and cancer. Cell. 2017;169:636–650. doi: 10.1016/j.cell.2017.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boulay G, et al. OTX2 activity at distal regulatory elements shapes the chromatin landscape of group 3 medulloblastoma. Cancer Discov. 2017;7:288–301. doi: 10.1158/2159-8290.CD-16-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Golson ML, Kaestner KH. Fox transcription factors: from development to disease. Development. 2016;143:4558–4570. doi: 10.1242/dev.112672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fournier M, et al. FOXA and master transcription factors recruit Mediator and Cohesin to the core transcriptional regulatory circuitry of cancer cells. Sci Rep. 2016;6:34962. doi: 10.1038/srep34962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jozwik KM, Chernukhin I, Serandour AA, Nagarajan S, Carroll JS. FOXA1 directs H3K4 monomethylation at enhancers via recruitment of the methyltransferase MLL3. Cell Rep. 2016;17:2715–2723. doi: 10.1016/j.celrep.2016.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu F, et al. EGFR mutation promotes glioblastoma through epigenome and transcription factor network remodeling. Mol Cell. 2015;60:307–318. doi: 10.1016/j.molcel.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gryder BE, et al. PAX3-FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov. 2017;7:884–899. doi: 10.1158/2159-8290.CD-16-1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ran L, et al. FOXF1 defines the core-regulatory circuitry in gastrointestinal stromal tumor (GIST) Cancer Discov. 2017;8(2):234–251. doi: 10.1158/2159-8290.CD-17-0468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kron KJ, et al. TMPRSS2-ERG fusion co-opts master transcription factors and activates NOTCH signaling in primary prostate cancer. Nat Genet. 2017;49:1336–1345. doi: 10.1038/ng.3930. [DOI] [PubMed] [Google Scholar]
- 92.Thirant C, et al. ETO2-GLIS2 hijacks transcriptional complexes to drive cellular identity and self-renewal in pediatric acute megakaryoblastic leukemia. Cancer Cell. 2017;31:452–465. doi: 10.1016/j.ccell.2017.02.006. [DOI] [PubMed] [Google Scholar]
- 93.Zhao J, et al. Alterations of androgen receptor-regulated enhancer RNAs (eRNAs) contribute to enzalutamide resistance in castration-resistant prostate cancer. Oncotarget. 2016;7:38551–38565. doi: 10.18632/oncotarget.9535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhao Y, et al. Activation of P-TEFb by androgen receptor-regulated enhancer RNAs in castration-resistant prostate cancer. Cell Rep. 2016;15:599–610. doi: 10.1016/j.celrep.2016.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Whyte WA, et al. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature. 2012;482:221–225. doi: 10.1038/nature10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fagnocchi L, et al. A Myc-driven self-reinforcing regulatory network maintains mouse embryonic stem cell identity. Nat Commun. 2016;7:11903. doi: 10.1038/ncomms11903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fagnocchi L et al (2017) Multiple roles of MYC in integrating regulatory networks of pluripotent stem cells. Front Cell Dev Biol 5 [DOI] [PMC free article] [PubMed]
- 98.Poli V, et al. MYC-driven epigenetic reprogramming favors the onset of tumorigenesis by inducing a stem cell-like state. Nat Commun. 2018;9:1024. doi: 10.1038/s41467-018-03264-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jagle S, et al. SNAIL1-mediated downregulation of FOXA proteins facilitates the inactivation of transcriptional enhancer elements at key epithelial genes in colorectal cancer cells. PLoS Genet. 2017;13:e1007109. doi: 10.1371/journal.pgen.1007109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Denny SK, et al. Nfib promotes metastasis through a widespread increase in chromatin accessibility. Cell. 2016;166:328–342. doi: 10.1016/j.cell.2016.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Roe JS, et al. Enhancer reprogramming promotes pancreatic cancer metastasis. Cell. 2017;170:875–888. doi: 10.1016/j.cell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Verfaillie A, et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat Commun. 2015;6:6683. doi: 10.1038/ncomms7683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yang H et al (2015) ETS family transcriptional regulators drive chromatin dynamics and malignancy in squamous cell carcinomas. eLife 4 [DOI] [PMC free article] [PubMed]
- 104.Rhie SK, et al. Identification of activated enhancers and linked transcription factors in breast, prostate, and kidney tumors by tracing enhancer networks using epigenetic traits. Epigenet Chromatin. 2016;9:50. doi: 10.1186/s13072-016-0102-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yao L, Shen H, Laird PW, Farnham PJ, Berman BP. Inferring regulatory element landscapes and transcription factor networks from cancer methylomes. Genome Biol. 2015;16:105. doi: 10.1186/s13059-015-0668-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Morgan MA, Shilatifard A. Chromatin signatures of cancer. Genes Dev. 2015;29:238–249. doi: 10.1101/gad.255182.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sato T, Issa JJ, Kropf P. DNA hypomethylating drugs in cancer therapy. Cold Spring Harb Perspect Med. 2017;7:1–14. doi: 10.1101/cshperspect.a026948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bell RE, et al. Enhancer methylation dynamics contribute to cancer plasticity and patient mortality. Genome Res. 2016;26:601–611. doi: 10.1101/gr.197194.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fleischer T, et al. DNA methylation at enhancers identifies distinct breast cancer lineages. Nat Commun. 2017;8:1379. doi: 10.1038/s41467-017-00510-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Heyn H, et al. Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol. 2016;17:11. doi: 10.1186/s13059-016-0879-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vidal E, et al. A DNA methylation map of human cancer at single base-pair resolution. Oncogene. 2017;36:5648–5657. doi: 10.1038/onc.2017.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cai Y, et al. Critical threshold levels of DNA methyltransferase 1 are required to maintain DNA methylation across the genome in human cancer cells. Genome Res. 2017;27:533–544. doi: 10.1101/gr.208108.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lu R, et al. Epigenetic perturbations by Arg882-mutated DNMT3A potentiate aberrant stem cell gene-expression program and acute leukemia development. Cancer Cell. 2016;30:92–107. doi: 10.1016/j.ccell.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yang L, et al. DNMT3A loss drives enhancer hypomethylation in FLT3-ITD-associated leukemias. Cancer Cell. 2016;29:922–934. doi: 10.1016/j.ccell.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Flavahan WA, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529:110–114. doi: 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sze CC, Shilatifard A. MLL3/MLL4/COMPASS family on epigenetic regulation of enhancer function and cancer. Cold Spring Harb Perspect Med. 2014;6:1–15. doi: 10.1101/cshperspect.a026427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Herz HM, Hu D, Shilatifard A. Enhancer malfunction in cancer. Mol Cell. 2014;53:859–866. doi: 10.1016/j.molcel.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Prange KHM, et al. MLL-AF9 and MLL-AF4 oncofusion proteins bind a distinct enhancer repertoire and target the RUNX1 program in 11q23 acute myeloid leukemia. Oncogene. 2017;36:3346–3356. doi: 10.1038/onc.2016.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 120.St Pierre R, Kadoch C. Mammalian SWI/SNF complexes in cancer: emerging therapeutic opportunities. Curr Opin Genet Dev. 2017;42:56–67. doi: 10.1016/j.gde.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mathur R, et al. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat Genet. 2017;49:296–302. doi: 10.1038/ng.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kelso TWR et al (2017) Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. eLife 6 [DOI] [PMC free article] [PubMed]
- 123.Leong WZ, et al. ARID5B as a critical downstream target of the TAL1 complex that activates the oncogenic transcriptional program and promotes T-cell leukemogenesis. Genes Dev. 2017;31:2343–2360. doi: 10.1101/gad.302646.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nakayama RT, et al. SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat Genet. 2017;49:1613–1623. doi: 10.1038/ng.3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang X, et al. SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat Genet. 2017;49:289–295. doi: 10.1038/ng.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang S, et al. INO80 is required for oncogenic transcription and tumor growth in non-small cell lung cancer. Oncogene. 2017;36:1430–1439. doi: 10.1038/onc.2016.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhou B, et al. INO80 governs superenhancer-mediated oncogenic transcription and tumor growth in melanoma. Genes Dev. 2016;30:1440–1453. doi: 10.1101/gad.277178.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Buschbeck M, Hake SB. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat Rev Mol Cell Biol. 2017;18:299–314. doi: 10.1038/nrm.2016.166. [DOI] [PubMed] [Google Scholar]
- 129.Zink LM, Hake SB. Histone variants: nuclear function and disease. Curr Opin Genet Dev. 2016;37:82–89. doi: 10.1016/j.gde.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 130.Brunelle M, et al. The histone variant H2A.Z is an important regulator of enhancer activity. Nucleic Acids Res. 2015;43:9742–9756. doi: 10.1093/nar/gkv825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Segala G, Bennesch MA, Pandey DP, Hulo N, Picard D. Monoubiquitination of histone H2B blocks eviction of histone variant H2A.Z from inducible enhancers. Mol Cell. 2016;64:334–346. doi: 10.1016/j.molcel.2016.08.034. [DOI] [PubMed] [Google Scholar]
- 132.Valdes-Mora F, et al. Acetylated histone variant H2A.Z is involved in the activation of neo-enhancers in prostate cancer. Nat Commun. 2017;8:1346. doi: 10.1038/s41467-017-01393-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Losada A. Cohesin in cancer: chromosome segregation and beyond. Nat Rev Cancer. 2014;14:389–393. doi: 10.1038/nrc3743. [DOI] [PubMed] [Google Scholar]
- 134.Marshall AD, Bailey CG, Rasko JE. CTCF and BORIS in genome regulation and cancer. Curr Opin Genet Dev. 2014;24:8–15. doi: 10.1016/j.gde.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 135.Soutourina J. Transcription regulation by the Mediator complex. Nat Rev Mol Cell Biol. 2017;19:262–274. doi: 10.1038/nrm.2017.115. [DOI] [PubMed] [Google Scholar]
- 136.Loven J, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pelish HE, et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature. 2015;526:273–276. doi: 10.1038/nature14904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Murakami S, Nagari A, Kraus WL. Dynamic assembly and activation of estrogen receptor alpha enhancers through coregulator switching. Genes Dev. 2017;31:1535–1548. doi: 10.1101/gad.302182.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Taberlay PC, et al. Three-dimensional disorganization of the cancer genome occurs coincident with long-range genetic and epigenetic alterations. Genome Res. 2016;26:719–731. doi: 10.1101/gr.201517.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fiorito E, et al. CTCF modulates Estrogen Receptor function through specific chromatin and nuclear matrix interactions. Nucleic Acids Res. 2016;44:10588–10602. doi: 10.1093/nar/gkw785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cohen AJ, et al. Hotspots of aberrant enhancer activity punctuate the colorectal cancer epigenome. Nat Commun. 2017;8:14400. doi: 10.1038/ncomms14400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rao SSP, et al. Cohesin loss eliminates all loop domains. Cell. 2017;171:305–320. doi: 10.1016/j.cell.2017.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ribich S, Harvey D, Copeland RA. Drug discovery and chemical biology of cancer epigenetics. Cell Chem Biol. 2017;24:1120–1147. doi: 10.1016/j.chembiol.2017.08.020. [DOI] [PubMed] [Google Scholar]
- 144.Shortt J, Ott CJ, Johnstone RW, Bradner JE. A chemical probe toolbox for dissecting the cancer epigenome. Nat Rev Cancer. 2017;17:160–183. doi: 10.1038/nrc.2016.148. [DOI] [PubMed] [Google Scholar]
- 145.de Magalhaes JP. How ageing processes influence cancer. Nat Rev Cancer. 2013;13:357–365. doi: 10.1038/nrc3497. [DOI] [PubMed] [Google Scholar]
- 146.Zhang R, Chen HZ, Liu DP. The four layers of aging. Cell Syst. 2015;1:180–186. doi: 10.1016/j.cels.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 147.Booth LN, Brunet A. The aging epigenome. Mol Cell. 2016;62:728–744. doi: 10.1016/j.molcel.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Webb AE, et al. FOXO3 shares common targets with ASCL1 genome-wide and inhibits ASCL1-dependent neurogenesis. Cell Rep. 2013;4:477–491. doi: 10.1016/j.celrep.2013.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013;14:83–97. doi: 10.1038/nrm3507. [DOI] [PubMed] [Google Scholar]
- 151.Eijkelenboom A, Mokry M, Smits LM, Nieuwenhuis EE, Burgering BM. FOXO3 selectively amplifies enhancer activity to establish target gene regulation. Cell Rep. 2013;5:1664–1678. doi: 10.1016/j.celrep.2013.11.031. [DOI] [PubMed] [Google Scholar]
- 152.Tubbs A, Nussenzweig A. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017;168:644–656. doi: 10.1016/j.cell.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dabin J, Fortuny A, Polo SE. Epigenome maintenance in response to DNA damage. Mol Cell. 2016;62:712–727. doi: 10.1016/j.molcel.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Canela A, et al. Genome organization drives chromosome fragility. Cell. 2017;170:507–521. doi: 10.1016/j.cell.2017.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Price BD, D’Andrea AD. Chromatin remodeling at DNA double-strand breaks. Cell. 2013;152:1344–1354. doi: 10.1016/j.cell.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hauer MH, et al. Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nat Struct Mol Biol. 2017;24:99–107. doi: 10.1038/nsmb.3347. [DOI] [PubMed] [Google Scholar]
- 157.Lin C, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139:1069–1083. doi: 10.1016/j.cell.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Perillo B, et al. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science. 2008;319:202–206. doi: 10.1126/science.1147674. [DOI] [PubMed] [Google Scholar]
- 159.Ju BG, et al. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 2006;312:1798–1802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- 160.Puc J, et al. Ligand-dependent enhancer activation regulated by topoisomerase-I activity. Cell. 2015;160:367–380. doi: 10.1016/j.cell.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Periyasamy M, et al. APOBEC3B-mediated cytidine deamination is required for estrogen receptor action in breast cancer. Cell Rep. 2015;13:108–121. doi: 10.1016/j.celrep.2015.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Le May N, Fradin D, Iltis I, Bougneres P, Egly JM. XPG and XPF endonucleases trigger chromatin looping and DNA demethylation for accurate expression of activated genes. Mol Cell. 2012;47:622–632. doi: 10.1016/j.molcel.2012.05.050. [DOI] [PubMed] [Google Scholar]
- 163.Bansal K, Yoshida H, Benoist C, Mathis D. The transcriptional regulator Aire binds to and activates super-enhancers. Nat Immunol. 2017;18:263–273. doi: 10.1038/ni.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Massague J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529:298–306. doi: 10.1038/nature17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Makohon-Moore AP, et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat Genet. 2017;49(3):358–366. doi: 10.1038/ng.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Yates LR, et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell. 2017;32:169–184. doi: 10.1016/j.ccell.2017.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Harper KL et al (2016) Mechanism of early dissemination and metastasis in Her2(+) mammary cancer. Nature [DOI] [PMC free article] [PubMed]
- 168.Hosseini H et al (2016) Early dissemination seeds metastasis in breast cancer. Nature [DOI] [PMC free article] [PubMed]
- 169.Lawson DA, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. 2015;526:131–135. doi: 10.1038/nature15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ozturk S, et al. SDPR functions as a metastasis suppressor in breast cancer by promoting apoptosis. Proc Natl Acad Sci USA. 2016;113:638–643. doi: 10.1073/pnas.1514663113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Pulecio J, Verma N, Mejia-Ramirez E, Huangfu D, Raya A. CRISPR/Cas9-based engineering of the epigenome. Cell Stem Cell. 2017;21:431–447. doi: 10.1016/j.stem.2017.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Simeonov DR, et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature. 2017;549:111–115. doi: 10.1038/nature23875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Mumbach MR, et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat Genet. 2017;49:1602–1612. doi: 10.1038/ng.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Fulco CP, et al. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science. 2016;354:769–773. doi: 10.1126/science.aag2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kwiatkowski N, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616–620. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rusan M, et al. Suppression of adaptive responses to targeted cancer therapy by transcriptional repression. Cancer Discov. 2018;8:59–73. doi: 10.1158/2159-8290.CD-17-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zawistowski JS, et al. Enhancer remodeling during adaptive bypass to MEK inhibition is attenuated by pharmacologic targeting of the P-TEFb complex. Cancer Discov. 2017;7:302–321. doi: 10.1158/2159-8290.CD-16-0653. [DOI] [PMC free article] [PubMed] [Google Scholar]