

Abstract
Our molecular understanding of the cystic fibrosis transmembrane conductance regulator (CFTR)—the chloride channel that is mutated in cystic fibrosis—has been greatly enhanced by a number of recent atomic-level structures of the protein in different conformations. One surprising aspect of these structures was the finding that the eighth of CFTR’s 12 membrane-spanning segments (TM8) appeared close to the channel pore. Although functional evidence supports a role for other TMs in forming the pore, such a role for TM8 has not previously been reported. Here, we use patch-clamp recording to investigate the functional role of TM8. Using substituted cysteine accessibility mutagenesis, we find that three amino acid side-chains in TM8 (Y913, Y914, and Y917) are exposed to the extracellular, but not the intracellular, solution. Cysteine cross-linking experiments suggest that Y914 and Y917 are in close proximity to L102 (TM1) and F337 (TM6), respectively, suggesting that TM8 contributes to the narrow selectivity filter region of the pore. Different amino acid substitutions suggest that Y914, and to a lesser extent Y917, play important roles in controlling anion flux through the open channel. Furthermore, substitutions that reduce side-chain volume at Y917 severely affect channel gating, resulting in a channel with an extremely unstable open state. Our results suggest that pore-lining TM8 is among the most important TMs controlling the permeation phenotype of the CFTR channel, and also that movement of TM8 may be critically involved in channel gating.
Keywords: Cystic fibrosis transmembrane conductance regulator, Chloride channel, Substituted cysteine accessibility mutagenesis, Selectivity filter, Channel gating, Channel structure
Introduction
Cystic fibrosis (CF) is caused by genetic mutations that disrupt the function of the cystic fibrosis transmembrane conductance regulator (CFTR), an epithelial cell Cl− channel [1, 2]. CFTR activity is controlled by protein kinase A (PKA)-dependent phosphorylation, and once phosphorylated, channel gating is controlled by cytoplasmic ATP. Detailed biophysical and structure–function analyses have indicated that ATP-dependent dimerization of CFTR’s two cytoplasmic nucleotide-binding domains (NBDs) leads to opening of the Cl− channel pore, and that the pore closes again following ATP hydrolysis and full or partial separation of the NBDs (reviewed in Refs. [2–4]). Consistent with this model, recent atomic-level electron cryo-microscopy (cryo-EM) structures of dephosphorylated, ATP-free zebrafish, and human CFTR show widely separated NBDs and closed, inward-facing membrane-spanning domains (MSDs) [5, 6] (Fig. 1a). In contrast, phosphorylated, ATP-bound CFTR has dimerized NBDs and more closely associated MSDs [7, 8] (Fig. 1a). However, even in these phosphorylated, ATP-bound structures, the putative Cl− permeation pathway appears closed at its extracellular end, suggesting that some further, perhaps local, structural rearrangement is required to attain the fully open, conductive state [7, 8].
Fig. 1.
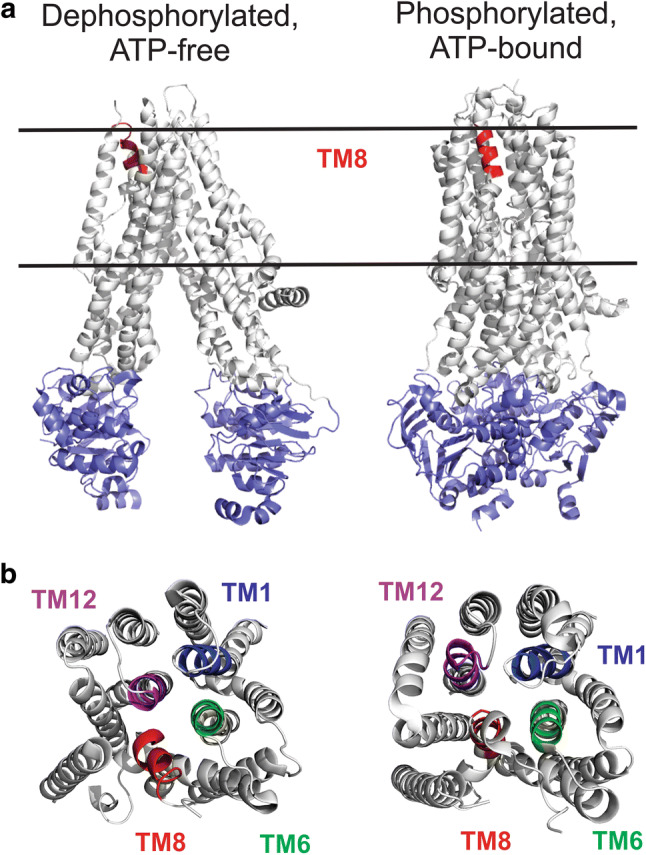
Location of TM8 within the atomic structure of CFTR. a Cryo-EM structures of human CFTR in a dephosphorylated, ATP-free state (left) [6], and in a phosphorylated, ATP-bound, NBD-dimerized state (right) [8]. In both cases, the NBDs are shown in blue. The location of that part of TM8 mutated in the present SCAM study is indicated in red. The approximate location of the membrane is indicated by horizontal black lines. The cytoplasmic R domain, which is mostly unstructured, is not shown in these images. b Views of these same structures from the extracellular side of the membrane, showing the location of the mutated region of TM8 (red) together with that of aligned regions of other TMs (TM1, blue; TM6, green; TM12, purple) as indicated
Amongst other important insights provided by these recent cryo-EM structures are some confirmatory—as well as some more surprising—evidences regarding the possible structure of the Cl− permeation pathway. Functional and homology modelling work had previously suggested that the narrowest region of the channel pore, located close to its extracellular end, is lined by transmembrane (TM) helices 1, 6, and 12 (reviewed in Refs. [2, 9, 10]). This outer part of the pore has also been suggested to be the location of the selectivity filter that allows discrimination between different anions [11–14], as well as the gate that opens and closes to control Cl− permeation through the pore [7, 8, 15–17]. While these ideas, and the contributions of TMs 1, 6, and 12, are supported by currently available cryo-EM structures [6, 8, 18], these structures also make the more unexpected prediction that TM8 may also contribute to this outer part of the pore [5, 7, 18] (Fig. 1). Furthermore, differences in the position of TM8 in different structures were used to suggest that this region may undergo a significant conformational change during ATP-dependent structural rearrangements of the pore, hinting that TM8 may play an important, previously unrecognized role in gating the pore [7, 18, 19]. However, in spite of its potential importance in Cl− permeation and channel gating, to date, TM8 has received scant attention from functional studies.
In the present study, we have addressed this lack of direct experimental information on TM8 using a number of functional approaches. Using substituted cysteine accessibility mutagenesis (SCAM), we show that three amino acid side-chains in TM8 are exposed to the extracellular solution. Disulfide and Cd2+ bridge cross-linking experiments provide some suggestion that these exposed side-chains may be located close to functionally important pore-forming side-chains in TMs 1 and 6. Different amino acid substitutions at these pore-lining TM8 positions illuminate important functional roles in Cl− permeation and channel gating.
Materials and methods
Experiments were carried out on Chinese hamster ovary (CHO) and baby hamster kidney (BHK) cells transiently transfected with human CFTR, prepared using procedures similar to those described previously [20]. CHO cells were used for whole-cell patch-clamp experiments, and BHK cells for excised, inside-out membrane patches. Some experiments (Figs. 2, 3, 4) used a “cys-less” CFTR variant in which all endogenous cysteines have been removed by mutagenesis [21] and includes a mutation in the first NBD (V510A) to increase protein expression in the cell membrane [22]. Additional mutations were introduced using the QuikChange site-directed mutagenesis system (Agilent Technologies, Santa Clara, CA, USA) and verified by DNA sequencing (Macrogen Inc., Seoul, Republic of Korea). For the initial SCAM screening of TM8, cysteines were substituted individually for ten consecutive residues, Y913–V922.
Fig. 2.

Modification of cysteine-substituted TM8 mutants by extracellular MTS reagents. a Examples of the time course of whole-cell currents carried by Y914C, Y917C and Y919C-CFTR (cys-less background). As described in “Materials and methods”, current amplitude was monitored using voltage ramps (see b) and is plotted at membrane potentials of − 50 mV (open circle) and + 50 mV (closed circle). Currents were activated by extracellular application of cAMP stimulatory cocktail (see “Materials and methods”) as indicated by the gray bar marked cAMP. After activation, cells were treated with negatively charged MTSES− (1 mM; red bar for Y914C and Y919C) or positively charged MTSET+ (1 mM; blue bar for Y917C). At the end of the experiment, current was confirmed as CFTR-mediated using GlyH-101 (20 μM; black bar marked GlyH). b Example current (I)–voltage (V) relationships for these cells, recorded during voltage ramps before (control; black lines) and after (red lines for MTSES−, blue line for MTSET+) exposure to MTS reagent. Note the inward rectification of the control I–V curves for Y914C and Y917C. c Mean fraction of control current remaining following exposure to MTSES− (red bars) or MTSET+ (blue bars) for these and other TM8 cysteine-mutant channels. The effects of MTS reagents were not appreciably voltage-dependent, and the effects of these reagents were averaged across the voltage range used. Asterisks indicate a significant difference from control, pre-MTS current levels (p < 0.05). Mean of data from 3–4 cells
Fig. 3.

Single-channel currents carried by cysteine-substituted TM8 mutants in inside-out membrane patches. a Example single-channel currents carried by the named channel variants at a membrane potential of − 50 mV. The closed state is indicated by the line to the far left. Note that only very brief, incompletely resolved openings were observed for Y917C, a finding further illustrated by the expanded part of this trace shown in b (expanded from the part of the trace marked “b” in a). c Mean single-channel current amplitude measured at − 50 mV for these and other TM8 cysteine-mutant channels. Control current amplitude (black bars) at this voltage was significantly different from wild-type only in Y914C and V922C (p < 10−8), as indicated by asterisks. Current amplitude following prolonged (> 2 min) exposure to intracellular MTSES− (200 μM) is indicated by the gray bars. For no channel, variant studied did this exposure to MTSES− result in a significant change in current amplitude (p > 0.05). Note that current amplitude was not quantified for Y917C due to the poor resolution of individual channel openings. Mean of data from 4 to 7 patches
Fig. 4.

Cross-linking of cysteine side-chains introduced into different TMs. a Example whole-cell currents carried by L102C/Y914C (left) and F337C/Y914C (right), under similar experimental conditions as in Fig. 2. As shown in Fig. 2a, currents were activated by exposure to cAMP cocktail (gray bars). Following activation, cells were exposed to CuPhe (10 μM Cu2+: 40 μM phenanthroline; red bars). At the end of the experiment, current was confirmed as CFTR-mediated using GlyH-101 (20 μM; black bars marked GlyH). Example I–V curves for these cells are also shown, before (control) and after exposure to CuPhe. b Example I–V curves for the named channel variants, illustrating the effects of exposure to Cd2+ (100 μM). Note the inward rectification of the control I–V curves for Y914C and Y917C-bearing channels. c Mean fraction of control current remaining following exposure to CuPhe. Asterisk indicates a significant difference from control (p < 0.05). d Mean fraction of control current remaining following exposure to Cd2+. For single-cysteine mutants, asterisks indicate a significant difference from control (p < 0.05). For double mutants, daggers indicate a significant difference from both of the corresponding single mutants (p < 0.002). Mean of data from 3 to 4 cells in c, d
Whole-cell patch-clamp recordings (Figs. 2, 4) were made as described previously [14, 23]. Briefly, the bath (extracellular) solution contained (mM): 145 NaCl, 15 Na glutamate, 4.5 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 5 glucose, pH 7.4, and the pipette (intracellular) solution (mM): 139 CsCl, 2 MgCl2, 5 EGTA, 10 HEPES, 5 glucose, 4 Na2ATP, 1 MgATP, 0.1 GTP, pH 7.2. Current was monitored during depolarizing voltage ramps (from − 50 to + 50 mV) applied every 10 s. Following attainment of the whole-cell configuration and recording of stable baseline currents, CFTR channels were activated by extracellular application of a cyclic AMP stimulatory “cocktail” containing forskolin (10 μM), 3-isobutyl-1-methylxanthine (100 μM), and 8-(4-chlorophenylthio) cyclic AMP (100 μM). As described in previous studies, following stable activation of CFTR currents, cells were treated with one of two cysteine-reactive methanethiosulfonate (MTS) reagents, the negatively charged [2-sulfonatoethyl] MTS (MTSES−) or the positively charged [2-(trimethylammonium)ethyl] MTS (MTSET+) (1 mM) [24, 25], the oxidizing agent copper(II)-o-phenanthroline (CuPhe) (10 μM Cu2+:40 μM phenanthroline) [14, 23], or CdCl2 (100 μM) [23] applied directly to the bath. At the end of the experiment, remaining currents were confirmed as being carried by CFTR by their sensitivity to the specific CFTR inhibitor GlyH-101 (20 μM). For all experiments using MTS reagents, CuPhe, or Cd2+, cells were pre-treated with dithiothreitol (DTT; 5 mM) for 5 min immediately prior to the experiment, to ensure that cysteine side-chains were in a reduced state.
Inside-out membrane patches excised from BHK cells were used to record both macroscopic and single-channel currents. Following patch excision and recording of background currents, CFTR channels were activated by exposure to PKA catalytic subunit (20 nM) plus MgATP (1 mM) in the intracellular solution. Both intracellular (bath) and extracellular (pipette) solutions were based on one containing (mM): 150 NaCl, 2 MgCl2, 10 N-tris[hydroxymethyl]methyl-2-aminoethanesulfonate, pH 7.4. For single-channel recordings (Figs. 3, 5), NaCl in the pipette solution was replaced with Na gluconate to generate an outwardly directed Cl− concentration gradient. MTSES− was applied directly to the cytoplasmic face of excised patches at a concentration of 200 μM, lower than that used for whole-cell recording (1 mM) due to the open-channel blocking effects of intracellular MTSES− [22]. For experiments measuring the relative permeability of SCN− (PSCN/PCl) (Fig. 6), intracellular NaCl was replaced by NaSCN. Measurement of single-channel and macroscopic current amplitudes, and construction of leak-subtracted macroscopic current–voltage relationships were carried out as described previously [20]. Membrane voltages were corrected for liquid junction potentials calculated using PCLAMP software (Molecular Devices, Sunnyvale, CA, USA). For PSCN/PCl measurement, the macroscopic current reversal potential (VREV) was estimated by fitting a polynomial function to the leak-subtracted current–voltage relationship, and then used to calculate PSCN/PCl according to the following equation:
where [SCN−]in= 150 mM, [Cl−]in = 4 mM, and [Cl−]out= 154 mM, and R, T, and F have their usual thermodynamic meanings.
Fig. 5.

Single-channel currents recorded from Y914 and Y917 mutant channels in inside-out membrane patches. a Example single-channel currents carried by the named channel variants at a membrane potential of − 50 mV. The closed state is indicated by the line to the far left. Note that only very brief, incompletely resolved openings were observed for Y917A, as well as for Y917S and Y917V (data not shown). Mean single-channel current (i)–voltage (V) relationships for Y914 (b) and Y917-substituted channels (c) as indicated. d Mean single-channel conductance measured from the slope of the i –V relationship from individual patches. Asterisks indicate a significant difference from wild-type (p < 10−6). Mean of data from 5 to 9 patches
Fig. 6.

Macroscopic current rectification and anion selectivity in Y914 and Y917 mutant channels. a Example macroscopic current (I)–voltage (V) relationships recorded from inside-out patches under symmetrical high (154 mM) [Cl−] conditions, for the CFTR variants named. b Example I–V relationships (normalized to current at the most negative voltage) for these same CFTR variants, recorded (from different patches) with Cl−-containing or SCN−-containing intracellular (bath) solutions as indicated. Note that the current reversal potential (VREV) is ~ 0 mV with Cl−-containing solutions but considerably more positive with SCN−-containing solutions, indicating a high relative SCN− permeability. c Mean rectification ratio (current amplitude at − 80 mV/current amplitude at + 80 mV) measured from individual I–V relationships recorded under symmetrical high [Cl−] conditions such as those shown in a. Asterisks indicate a significant difference from wild-type (p < 0.02). d Mean relative SCN− permeability (PSCN/PCl) quantified from VREV measured from individual I–V relationships such as those shown in b. Asterisks indicate a significant difference from wild-type (p < 0.02). Mean of data from 4 to 7 patches in c, d
Experiments were carried out at room temperature, 21–24 °C. Values are presented as mean ± SEM. For graphical presentation of mean values, error bars represent SEM, and where no error bars are visible, SEM is smaller than the size of the symbol. Tests of significance were carried out using Student’s two-tailed t test, with p < 0.05 being considered statistically significant. All chemicals were from Sigma-Aldrich (Oakville, ON, Canada) except for GlyH-101 (EMD Chemicals, Gibbstown, NJ, USA), MTSES− and MTSET+ (Toronto Research Chemicals, Toronto, ON, Canada), and PKA (Promega, Madison, WI, USA).
Results
External and internal access to TM8
We investigated the accessibility of the outer part of TM8 to the extracellular solution using SCAM. Figure 2a, b shows examples of the effects of exposure to extracellular MTS reagents (MTSES−; MTSET+) on cAMP-stimulated whole-cell currents carried by TM8-mutant channels Y914C, Y917C, and Y919C. Similar experiments indicated that, while currents carried by Y913C, Y914C, and Y917C were significantly reduced in amplitude following exposure to either MTSES− or MTSET+, other TM8 mutants (V915C, F916C, I918C, Y919C, V920C, G921C, and V922C) were unaffected by exposure to either MTSES− or MTSET+ (Fig. 2c). These effects of MTSES− and MTSET+ could be at least partly reversed by exposure to DTT (5 mM; data not shown). Whereas the effects of 1 mM MTSES− and MTSET+ were almost immediate for Y913C and Y914C, full modification of Y917C by both of these reagents always required several minutes of exposure (Fig. 2a, and data not shown). While not investigated in further detail, these apparent differences in the rate of MTS reagent modification may indicate that Y917 is relatively poorly accessible to large extracellular reagents compared to Y913 and Y914. Overall, these MTS modification results suggest that only residues located in the most extracellular part of TM8 (Fig. 1) are exposed to the extracellular solution.
To investigate accessibility of these same sites to intracellular MTSES, we used single-channel recording from inside-out membrane patches, allowing MTSES to be applied directly to the cytoplasmic face of the patch. Control experiments suggested that cysteine mutagenesis of TM8 affected both Cl− permeation and gating of cys-less CFTR channels (Fig. 3). Thus, both Y914C and V922C had significantly smaller single-channel current amplitudes at − 50 mV compared to cys-less (Fig. 3), although Cl− current amplitude in other mutants was apparently unaffected (Fig. 3). However, we were unable to quantify the single-channel current amplitude in Y917C due to apparent instability of the channel open state in this mutant (Fig. 3a, b) that meant that the full-open state could not be properly resolved under our recording conditions. Because we consistently observed only very flickery, poorly resolved openings for this mutant (n = 7 patches), we did not attempt to quantify this apparent drastic change in channel gating.
Previously, we have used a reduction in single-channel current amplitude following exposure to negatively charged MTSES− as evidence for accessibility to the intracellular solution [26, 27]. However, addition of 200 μM MTSES− to the bath (intracellular) solution did not significantly affect single-channel current amplitude of any of nine TM8 cysteine-mutant tested (Fig. 3c), suggesting that this part of TM8 is not exposed to the intracellular solution.
Cysteine cross-linking between TM8 and TMs 1 and 6
Structural data suggest that TM8 might lie close to other TMs that contribute to the narrow region of the CFTR pore in certain configurations [5, 7] (see “Introduction”) (Fig. 1). Because Y914C and Y917C were modified by extracellular MTS reagents (Fig. 2) and showed altered single-channel current properties (Fig. 3), we sought to investigate the proximity of cysteine side-chains substituted at these two positions with side-chains known to contribute to formation of the narrow region of the pore and to the function of the anion selectivity filter, namely L102 in TM1 [14] and F337 in TM6 [11]. As in our previous work [23], we used two different functional approaches to test for the proximity of two cysteine residues introduced into different TMs, namely oxidant-induced disulfide bond formation, and Cd2+ metal bridge formation. As shown in Fig. 4a, c, exposure to extracellular oxidant CuPhe resulted in a slow, modest decrease in whole-cell current amplitude carried by activated L102C/Y914C mutant channels. This inhibition by CuPhe could be at least partly reversed by exposure to DTT (5 mM; data not shown). In contrast, current carried by other double-cysteine-mutant channels (L102C/Y917C, F337C/Y914C, and F337C/Y917C) appeared insensitive to CuPhe under these conditions (Fig. 4a, c). Application of a relatively low concentration of extracellular Cd2+ (100 μM) inhibited Y914C current without significantly affecting current amplitude for other single-cysteine mutants (L102C, F337C, and Y917C) (Fig. 4b, d), suggesting a relatively high Cd2+ binding affinity for a cysteine introduced at position Y914. This concentration of Cd2+ more strongly inhibited F337C/Y917C currents (Fig. 4b, d), suggesting some degree of Cd2+ co-ordination between the two cysteine side-chains introduced in these positions. Other double-cysteine-mutant channels showed Cd2+ inhibition that was similar to, or even weaker than, the corresponding single-cysteine mutants (Fig. 4d) and, thus, failed to provide firm evidence for Cd2+ co-ordination by these cysteine pairs. All inhibitory effects of Cd2+ observed were rapidly and fully reversible on exposure to DTT (5 mM; data not shown), consistent with an effect on exposed cysteine side-chains.
Effect of other mutations at pore-lining sites in TM8
Our results with cys-less CFTR (Figs. 2, 3, 4) suggest that TM8 residues Y914 and Y917 line the outer pore and potentially contribute to channel function in distinct ways. To investigate these possibilities further, we introduced a number of different substitutions at these sites in a wild-type CFTR background. The effect of different substitutions on single-channel current properties of the channel is illustrated in Fig. 5. For Y914, all substitutions introduced (Y914A, Y914F, and Y914S) significantly decreased single-channel conductance (Fig. 5a, b, d), consistent with results with cys-less Y914C (Fig. 3). At Y917C, different substitutions produced an apparently side-chain-size-dependent effect on channel gating. Thus, substituting the native tyrosine with a smaller amino acid [as in Y917A (Fig. 5a) and Y917S and Y917V (data not shown)] resulted in channels that exhibited only very brief, poorly resolved channel openings in inside-out membrane patches. These findings are very similar to those with cys-less Y917C (Fig. 3a, b), another small amino acid substitution, and as described above for Y917C could not be characterized in more detail. In contrast, the largest amino acids substituted (Y917F; Y917L) had no significant effect on channel conductance (Fig. 5a, c, d). Only Y917Q significantly—albeit only modestly compared to Y914 substitutions—caused a measureable reduction in single-channel current amplitude (Fig. 5a, c, d).
Because of potential TM8 contribution to the narrow selectivity filter region (see above), we also investigated the effects of these amino acid substitutions on anion selectivity. Anion selectivity in CFTR is predominantly characterized by a high relative permeability of strongly lyotropic anions such as SCN− and Au(CN)−2 [28, 29], and disruption of anion selectivity is associated with a decrease in the relative permeability of these lyotropic anions [11, 14, 30]. We, therefore, used SCN− relative permeability (PSCN/PCl) as a simple measure of anion selectivity [31, 32]. PSCN/PCl was quantified from the macroscopic current reversal potential (VREV) measured with SCN−-containing bath solutions, as described in “Materials and methods”. During control experiments with Cl−-containing bath solutions, we noticed that several substitutions at both Y914 and Y917 resulted in very strong inward rectification of the current–voltage relationship, in contrast to the near-linear relationship associated with wild-type CFTR (Fig. 6a). This rectification of the current–voltage relationship was quantified as a “rectification ratio”, the ratio of current amplitude measured at − 80 mV divided by that measured at + 80 mV (Fig. 6c). Note that some inward rectification was also observed for Y917C-containing channels under whole-cell recording conditions (Figs. 2, 4). Measurement of VREV indicated that all Y914-substituted channels investigated showed significantly increased PSCN/PCl (p < 0.02) (Fig. 6b, d). PSCN/PCl also appeared very slightly increased in some Y917-substituted channels (Fig. 6b, d), although this apparent change was not statistically significant (p > 0.05).
Discussion
Longstanding functional models of the open CFTR channel pore describe a narrow, uncharged section in the mid-to-outer membrane-spanning part of the protein, connected to the extracellular and intracellular solutions by wider vestibules that are decorated by functionally important positively charged amino acid side-chains (reviewed in Refs. [2, 4, 10]). The central narrow region is thought to be the location of both the anion selectivity filter [11–14] and the channel gate [15–17]. SCAM studies suggest that TMs 1 [33–35], 6 [36–39], 11 [32], and 12 [39–41] contribute structurally to the narrow region of the pore. Structure–function analyses suggest that the functional properties of the narrow region are dominated by side-chains from TM6 [11, 12, 31], with a more supporting role of TM1 [14, 31]. The present work provides functional evidence that TM8 also contributes to the narrow region of the pore, and, furthermore, illustrates how this TM may influence pore function.
Cysteine side-chains introduced into TM1 [33, 35], TM5 [42], TM6 [36, 39], TM11 [32], and TM12 [32, 39] are accessible to extracellular, channel-impermeant MTS reagents, suggesting that each of these TMs contributes to an externally accessible part of the protein, possibly the channel pore. Similarly, cysteines substituted for Y913, Y914, and Y917 in TM8 are accessible to both negatively charged MTSES− and positively charged MTSET+ in the extracellular solution (Fig. 2), consistent with a pore-lining position of part of TM8, at least in some channel conformation(s). However, cysteines introduced at sites more deeply into the membrane (I918–V922) were apparently not accessible to external MTS reagents (Fig. 2), suggesting that Y917 represents the limit of MTS reagent penetration into the pore from the outside, or that parts of TM8 more intracellular than this point are buried from the pore. In contrast, we found no evidence that cysteines introduced into TM8 could be modified by intracellular MTSES− (Fig. 3). Since intracellular MTS reagents can penetrate quite deeply into the open channel to modify cysteines in TMs 1 [34, 35], 5 [42], 6 [37, 38], 11 [25], and 12 [40, 41], this may suggest that the studied segment of TM8 does not contribute to the formation of the wide inner vestibule of the pore. This is consistent with the cryo-EM structure of phosphorylated, ATP-bound human CFTR, where TM8 does not appear to line the inner vestibule of the pore [8]. However, we cannot rule out that residues closer to the intracellular end of TM8 than we have investigated might line this inner vestibule. The location of pore-lining residues Y913, Y914, and Y917—together with nearby key narrow pore-lining residues from other TMs, L102 (TM1) and F337 (TM6)—in both the phosphorylated, ATP-bound and dephosphorylated, ATP-free cryo-EM structures of Fig. 1, are shown in Fig. 7a, b. It is clear from the ATP-bound structure that these three TM8 side-chains are clustered close to the central axis of the channel, close to the putative Cl− permeation pathway and close to L102 and F337 (Fig. 7a, c). In this structure, the side-chains of Y913 and Y914 project towards the central axis of the pore, whereas Y917 appears oriented towards TM12 (Fig. 7c). However, since this is thought to represent a non-conductive (closed) form of the channel [8], it is difficult to correlate these structures with any potential functional role of these residues in anion permeation through the open channel. Interestingly, each of these TM8 side-chains appears further from the central pore in the ATP-free structure (Fig. 7b), consistent with the outer part of TM8 including these residues moving substantially during ATP-dependent channel gating.
Fig. 7.
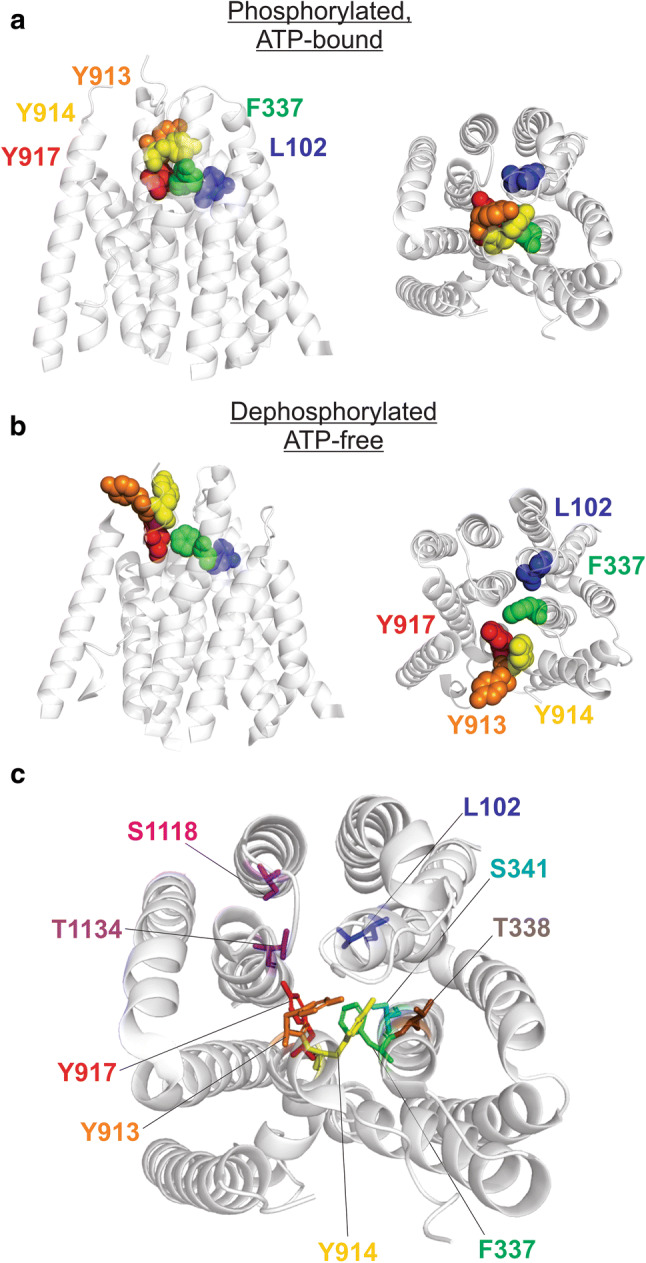
Location of pore-lining TM8 side-chains in the CFTR structure. a, b Relative locations of TM8 residues Y913 (orange), Y914 (yellow), and Y917 (red) compared with L102 (TM1; blue) and F337 (TM6; green). These side-chains only are shown as space-filling models within the overall structures of the membrane-spanning parts of human CFTR in the phosphorylated, ATP-bound state (a) and the dephosphorylated, ATP-free state (b) (see also Fig. 1). c A more detailed view of side-chains from different TMs proposed to play a role in the function of the narrow pore region, viz., L102 (TM1), F337, T338, S341 (TM6), Y913, Y914, Y917 (TM8—present study), S1118 (TM11), and T1134 (TM12). These side-chains only are shown as sticks within a cross section of the pore in the phosphorylated, ATP-bound state (as in a)
Our results also suggest that TM8 residues influence the anion permeation properties of the pore. In a cys-less background, Y914C and V922C were associated with decreased single-channel current amplitudes, although cysteine substitution at most sites in TM8 did not significantly alter Cl− current amplitude (Fig. 3). Since V922 does not appear to be pore-lining (based on SCAM experiments), we suggest that the small decrease in conductance observed in V922C is unlikely to reflect altered anion: side-chain interactions. Instead, mutation of V922 may alter the structure of the open-channel pore, for example, due to a change in the packing of different TMs surrounding the pore, or a change in the interaction between different TMs. For example, different mutations at non-pore-lining C343, at a similar position in mid-TM6, have amino acid side-chain size-dependent effects on Cl− conductance, that may reflect changes in the precise orientation of TM6 in the open state [43]. Other substitutions for Y914 (in a wild-type background) also significantly reduced Cl− conductance, not only Y914A and Y914S but even the relatively conservative Y914F mutation (Fig. 5). Mutation of other residues lining the narrow pore region is also associated with severely reduced conductance, for example Q98, P99, and L102 in TM1 [14, 31] and F337, T338, and S341 in TM6 [11, 20, 44, 45]. Mutation of Y917 appeared to have a much smaller impact on Cl− conductance (Figs. 3, 5), although this effect was somewhat obscured by the apparent strong effect of mutations at this position on channel gating (see below). Nevertheless, a strong impact of mutations at both Y914 and Y917 on Cl− permeation was evidenced by the extreme inward rectification of the macroscopic current–voltage relationship associated with several mutations at both of these sites (in particular, those that introduced relatively small alanine, serine, or valine side-chains). Similar strong inward rectification has previously been observed following charge-neutral amino acid substitutions within the narrow region, most notably for T338N (TM6) [44] and P99A (TM1) [31]. While the origin of this current rectification is not clear, the implication that currents carried by Cl− influx are disrupted far more than those by Cl− efflux suggests a significant alteration in the interaction between permeating Cl− ions and the pore. Thus, on balance, it would appear that both Y914 and Y917 play important roles in Cl− permeation, and that mutations at either of these sites—especially those that reduce side-chain volume—strongly disrupt permeation. However, the effects of mutations on anion selectivity were relatively modest. Using SCN− permeability as a simple marker of the anion selectivity process [31, 32], none of the mutations investigated disrupted normal lyotropic anion selectivity (which would be associated with a decrease in PSCN/PCl [11]). This is, perhaps, not surprising; to date, only mutations at F337 (TM6) [11] and (to a lesser extent) L102 (TM1) [14] have been shown to disrupt lyotropic anion selectivity. Mutation-associated increases in PSCN/PCl have been more commonly observed, for example with mutations in TM1 (A96V) [31], TM6 (T338A, T338N, S341A) [12, 31, 44, 46] and TM11 (S1118C) [32]. The positions of some of these important pore-lining side-chains in the phosphorylated, ATP-bound structure of CFTR are shown in Fig. 7c. Even though this structure appears unlikely to fully reflect an open, conductive channel, it can be seen that residues from TMs 1, 6, 8, and 12 appear to surround a central pore. This structure, therefore, appears consistent with the ability of these side-chains from TM8 being capable of influencing anion permeation through the pore.
Perhaps, more unusual was the clear effect of several substitutions at Y917 on the gating of the channel. Although difficult to study in detail using single-channel recording, several substitutions [Y917C (Fig. 3), Y917A (Fig. 5), Y917S and Y917V (data not shown)] had very similar effects, resulting in a channel that exhibited only very brief sojourns to the open state that were never clearly resolved at the recording bandwidth used. While our inability to analyze quantitatively these results means that we cannot rule out other gating defects, at the first glance, this would appear to suggest an extremely unstable channel open state following the substitution of these relatively small amino acid side-chains for the bulky tyrosine that is found at this position in wild-type CFTR. Consistent with this idea that side-chain size at position 917 influences the stability of the open state, substitution of larger side-chains (in Y917F, Y917L, and Y917Q) did allow the proper resolution of channel openings (Fig. 5).
In fact, based purely on the structural data, it has already been speculated that TM8 may play a role in gating of the pore [7, 8, 18, 19]. In human CFTR (Figs. 1, 7), comparison of different structures suggests that phosphorylation and NBD dimerization results in a rotation of approximately 55° of the outer portion of TM8 that contains Y914 and Y917 [8]. As can be seen by comparison of Fig. 7a, b, this movement of the outer part of TM8 brings it closer to TM1, TM6, and the central pore in the ATP-bound conformation. Furthermore, molecular dynamics simulations based on the structure of zebrafish CFTR suggested that an interaction between the outer segments of TM6 and TM8 might control local gate function [19]. However, even in the ATP-bound conformation (Fig. 7b), an apparent constriction of the pore close to the level of Y917 (TM8), F337 (TM6), and L102 (TM1) is too narrow to allow Cl− ions to pass (Fig. 7a, c). Since this is the only part of the pore expected to prevent Cl− passage in this structure, it is tempting to speculate that this region forms the gate that controls pore opening and closing. Functional evidence from other TMs also suggests that the narrow region of the pore is the physical location of the channel gate [15–17], with a particular emphasis having been placed on the potential role of F337 [15, 17]. While mutations of F337 and L102 have previously been associated with altered channel gating, these reported effects appear quite different from those we have observed with Y917 mutants. Mutations that reduce side-chain volume at position 337 cause a “gain-of-function”, increasing overall open probability and also promoting ATP-independent channel opening [34], consistent, perhaps, with the bulky phenylalanine side-chain contributing to a physical barrier in the closed pore. The L102C mutation caused the open channel to “flicker” frequently to a closed state [35], indicating, perhaps, a more modest instability of the open state than that we observe for Y917 mutants. Modification of L102C with positively charged MTSET+ also resulted in an apparent gain-of-function, causing the channel to become almost permanently locked in the open state [35]. Thus, even if we make the assumption that the narrow region illustrated in Fig. 7c does form the gate, then different TMs might contribute to gate function in different ways.
How, then, might we explain the apparent instability of the open state when the side-chain volume at position 917 is reduced? Without additional structural information (in particular, of a fully open-channel pore), we can only speculate. One possibility suggested by the structure shown in Fig. 7c is that an interaction between Y917 and nearby TM12 is required to stabilize the open state. In this scenario, mutations that reduce side-chain volume at position 917 might then disrupt this interaction with TM12, allowing TM8 to move into a position which interrupts channel openings (perhaps, a position more similar to that observed in the ATP-free conformation of Fig. 7b). If this speculation is correct, then molecular manipulations that stabilize the interaction between the outer parts of TM8 and TM12 might be able to stabilize the open state and, potentially, lead to the potentiation of channel activity.
We also sought to corroborate the apparent proximity of side-chains from different TMs using disulfide cross-linking (Fig. 4). Oxidant-induced disulfide bond formation suggested proximity between L102C and Y914C (Fig. 4a, c), while Cd2+-bridge formation suggested proximity between F337C and Y917C (Fig. 4b, d). These two approaches suggest S–S distances of approximately 2 Å and 5 Å, respectively [47], at least in certain protein conformations achieved at any point in the gating cycle. However, we not find any evidence to support other cross-linkages (i.e., between L102C and Y917C, or between F337C and Y914C) (Fig. 4). Furthermore, the two cross-linking approaches used did not appear to corroborate one another either for L102C/Y914C or for F337C/Y917C (Fig. 4). It should be stressed that a negative result in these experiments does not necessarily exclude cross-link formation; it may be that cross-links can form without significantly altering the overall function of the protein. The two different cross-linking approaches used (disulfide formation and Cd2+ bridge formation) may require different geometric arrangements of the two cysteine side-chains involved, such that corroborative agreement between the two approaches is not necessarily automatic. Furthermore, the assumed altered gating that we would predict for some of these double-cysteine mutants (especially those containing the Y917C mutation) might make cross-link formation difficult, especially if cross-linking occurs only in the open state. Nevertheless, these experiments do provide some support for physical proximity of the outer part of TM8 with both TM1 and TM6, at least at some point in the gating cycle. Previously we used disulfide cross-linking to demonstrate physical proximity between L102 and F337 [14].
The relative locations of these amino acids, in both the phosphorylated, ATP-bound structure and the dephosphorylated, ATP-free structure observed using cryo-EM, are compared in Fig. 7a, b. All four cysteine pairs tested are predicted to be closer together in the ATP-bound structure compared to the ATP-free structure, consistent with movement of the outer end of TM8 towards the central axis of the protein following ATP-dependent NBD dimerization. The closest-approaching pair is F337 and Y917, which are only around 3.3 Å apart in the ATP-bound structure (closest atomic distances for native side-chains). This is consistent with the apparent capability to form a Cd2+ bridge between this cysteine pair. In contrast, L102 and Y914 are farthest apart in both structures, approximately 10 Å in the ATP-bound structure and more than 16Å in the ATP-free structure, seemingly at odds with the apparent ability of CuPhe to form disulfide bonds between cysteines substituted at these two positions. Possible reasons for this apparent discrepancy are that additional conformations may exist (even transiently) that can be “trapped” by rare disulfide bond formation events, and that considerable flexibility may exist in this part of the protein that occasionally brings together parts of the protein that appear far apart in average, static structures such as those shown in Fig. 7.
Overall our results demonstrate that the outer part of TM8, and specifically Y914 and Y917, make a physical contribution to the narrow region of the CFTR pore (Figs. 2, 4) where they significantly impact both anion permeation (Figs. 3, 5, 6) and gating (Figs. 3, 5). It has long been established that amino acid residues in TM6 play a dominant role in controlling the permeation properties of the CFTR pore, in particular its narrow selectivity filter region (reviewed in Ref. [10]). TM1 plays a supporting role, while residues in TMs 11 and 12 make only very minor functional contributions in this part of the pore [10]. While we have not presented a comprehensive, like-for-like comparative study, on balance, we would suggest that, along with TM1, TM8 plays an intermediate role in supporting the permeation phenotype controlled primarily by anion interactions with TM6 (Fig. 7c). However, this role appears to be restricted to a relatively small part close to the extracellular end of TM8.
Acknowledgements
We would like to thank Christina Irving for technical assistance. This work was supported by Cystic Fibrosis Canada.
Abbreviations
- BHK
Baby hamster kidney
- CF
Cystic fibrosis
- CFTR
CF transmembrane conductance regulator
- CHO
Chinese hamster ovary
- Cryo-EM
Electron cryo-microscopy
- CuPhe
Copper(II)-o-phenanthroline
- DTT
Dithiothreitol
- MSD
Membrane-spanning domain
- MTS
Methanethiosulfonate
- MTSES
[2-Sulfonatoethyl] MTS
- MTSET
[2-(Trimethylammonium)ethyl] MTS
- NBD
Nucleotide-binding domain
- PKA
Protein kinase A
- SCAM
Substituted cysteine accessibility mutagenesis
- TM
Transmembrane helix
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Wang Y, Wrennall JA, Cai Z, Li H, Sheppard DN. Understanding how cystic fibrosis mutations disrupt CFTR function: from single molecules to animal models. Int J Biochem Cell Biol. 2014;52:47–57. doi: 10.1016/j.biocel.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Csanády L, Vergani P, Gadsby DC. Structure, gating, and regulation of the CFTR anion channel. Physiol Rev. 2019;99:707–738. doi: 10.1152/physrev.00007.2018. [DOI] [PubMed] [Google Scholar]
- 3.Moran O. The gating of the CFTR channel. Cell Mol Life Sci. 2017;74:85–92. doi: 10.1007/s00018-016-2390-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hwang T-C, Yeh J-T, Zhang J, Yu Y-C, Yeh H-I, Destefano S. Structural mechanisms of CFTR function and dysfunction. J Gen Physiol. 2018;150:539–570. doi: 10.1085/jgp.201711946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Z, Chen J. Atomic structure of the cystic fibrosis transmembrane conductance regulator. Cell. 2016;167:1586–1597. doi: 10.1016/j.cell.2016.11.014. [DOI] [PubMed] [Google Scholar]
- 6.Liu F, Zhang Z, Csanády L, Gadsby DC, Chen J. Molecular structure of the human CFTR ion channel. Cell. 2017;169:85–95. doi: 10.1016/j.cell.2017.02.024. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Z, Liu F, Chen J. Conformational changes of CFTR upon phosphorylation and ATP binding. Cell. 2017;170:483–491. doi: 10.1016/j.cell.2017.06.041. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Z, Liu F, Chen J. Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc Natl Acad Sci USA. 2018;115:12757–12762. doi: 10.1073/pnas.1815287115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Callebaut I, Hoffmann B, Lehn P, Mornon J-P. Molecular modelling and molecular dynamics of CFTR. Cell Mol Life Sci. 2017;74:3–22. doi: 10.1007/s00018-016-2385-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linsdell P. Architecture and functional properties of the CFTR channel pore. Cell Mol Life Sci. 2017;74:67–83. doi: 10.1007/s00018-016-2389-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Linsdell P, Evagelidis A, Hanrahan JW. Molecular determinants of anion selectivity in the cystic fibrosis transmembrane conductance regulator chloride channel pore. Biophys J. 2000;78:2973–2982. doi: 10.1016/S0006-3495(00)76836-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCarty NA, Zhang Z-R. Identification of a region of strong discrimination in the pore of CFTR. Am J Physiol. 2001;281:L852–L867. doi: 10.1152/ajplung.2001.281.4.L852. [DOI] [PubMed] [Google Scholar]
- 13.Linsdell P. Anion conductance selectivity mechanism of the CFTR chloride channel. Biochim Biophys Acta. 2016;1858:740–747. doi: 10.1016/j.bbamem.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 14.Negoda A, El Hiani Y, Cowley EA, Linsdell P. Contribution of a leucine residue in the first transmembrane segment to the selectivity filter region in the CFTR chloride channel. Biochim Biophys Acta. 2017;1859:1049–1058. doi: 10.1016/j.bbamem.2017.02.014. [DOI] [PubMed] [Google Scholar]
- 15.Corradi V, Vergani P, Tieleman DP. Cystic fibrosis transmembrane conductance regulator (CFTR): closed and open state channel models. J Biol Chem. 2015;290:22891–22906. doi: 10.1074/jbc.M115.665125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao X, Hwang T-C. Localizing a gate in CFTR. Proc Natl Acad Sci USA. 2015;112:2461–2466. doi: 10.1073/pnas.1420676112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei S, Roessler BC, Icyuz M, Chauvet S, Tao B, Hartman JL, Kirk KL. Long-range coupling between the extracellular gates and the intracellular ATP binding domains of multidrug resistance protein pumps and cystic fibrosis transmembrane conductance regulator channels. FASEB J. 2016;30:1247–1262. doi: 10.1096/fj.15-278382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fay JF, Aleksandrov LA, Jensen T, Cui L, Kousouros JN, He L, Aleksandrov AA, Gingerich D, Riordan JR, Chen J. Cryo-EM visualization of an active high open probability CFTR anion channel. Biochemistry. 2018;57:6234–6246. doi: 10.1021/acs.biochem.8b00763. [DOI] [PubMed] [Google Scholar]
- 19.Corradi V, Gu R-X, Vergani P, Tieleman DP. Structure of transmembrane helix 8 and possible membrane defects in CFTR. Biophys J. 2018;114:1751–1754. doi: 10.1016/j.bpj.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou J-J, Li M-S, Qi J, Linsdell P. Regulation of conductance by the number of fixed positive charges in the intracellular vestibule of the CFTR chloride channel pore. J Gen Physiol. 2010;135:229–245. doi: 10.1085/jgp.200910327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mense M, Vergani P, White DM, Altberg G, Nairn AC, Gadsby DC. In vivo phosphorylation of CFTR promotes formation of a nucleotide-binding domain heterodimer. EMBO J. 2006;25:4728–4739. doi: 10.1038/sj.emboj.7601373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li M-S, Demsey AFA, Qi J, Linsdell P. Cysteine-independent inhibition of the CFTR chloride channel by the cysteine-reactive reagent sodium (2-sulphonatoethyl) methanethiosulphonate. Br J Pharmacol. 2009;157:1065–1071. doi: 10.1111/j.1476-5381.2009.00258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Negoda A, Cowley EA, El Hiani Y, Linsdell P. Conformational change of the extracellular parts of the CFTR protein during channel gating. Cell Mol Life Sci. 2018;75:3027–3038. doi: 10.1007/s00018-018-2777-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Broadbent SD, Wang W, Linsdell P. Interaction between two extracellular loops influences the activity of the cystic fibrosis transmembrane conductance regulator chloride channel. Biochem Cell Biol. 2014;92:390–396. doi: 10.1139/bcb-2014-0066. [DOI] [PubMed] [Google Scholar]
- 25.Wang W, El Hiani Y, Rubaiy HN, Linsdell P. Relative contribution of different transmembrane segments to the CFTR chloride channel pore. Pflügers Arch. 2014;466:477–490. doi: 10.1007/s00424-013-1317-x. [DOI] [PubMed] [Google Scholar]
- 26.El Hiani Y, Linsdell P. Functional architecture of the cytoplasmic entrance to the cystic fibrosis transmembrane conductance regulator chloride channel pore. J Biol Chem. 2015;290:15855–15865. doi: 10.1074/jbc.M115.656181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.El Hiani Y, Negoda A, Linsdell P. Cytoplasmic pathway followed by chloride ions to enter the CFTR channel pore. Cell Mol Life Sci. 2016;73:1917–1925. doi: 10.1007/s00018-015-2113-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Linsdell P, Hanrahan JW. Adenosine triphosphate-dependent asymmetry of anion permeation in the cystic fibrosis transmembrane conductance regulator chloride channel. J Gen Physiol. 1998;111:601–614. doi: 10.1085/jgp.111.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith SS, Steinle ED, Meyerhoff ME, Dawson DC. Cystic fibrosis transmembrane conductance regulator. Physical basis for lyotropic anion selectivity patterns. J Gen Physiol. 1999;114:799–818. doi: 10.1085/jgp.114.6.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gong X, Burbridge SM, Cowley EA, Linsdell P. Molecular determinants of Au(CN)2− binding and permeability within the cystic fibrosis transmembrane conductance regulator Cl− channel pore. J Physiol. 2002;540:39–47. doi: 10.1113/jphysiol.2001.013235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ge N, Muise CN, Gong X, Linsdell P. Direct comparison of the functional roles played by different transmembrane regions in the cystic fibrosis transmembrane conductance regulator chloride channel pore. J Biol Chem. 2004;279:55283–55289. doi: 10.1074/jbc.M411935200. [DOI] [PubMed] [Google Scholar]
- 32.Fatehi M, Linsdell P. Novel residues lining the CFTR chloride channel pore identified by functional modification of introduced cysteines. J Membr Biol. 2009;228:151–164. doi: 10.1007/s00232-009-9167-3. [DOI] [PubMed] [Google Scholar]
- 33.Zhou J-J, Fatehi M, Linsdell P. Identification of positive charges situated at the outer mouth of the CFTR chloride channel pore. Pflügers Arch. 2008;457:351–360. doi: 10.1007/s00424-008-0521-6. [DOI] [PubMed] [Google Scholar]
- 34.Wang W, El Hiani Y, Linsdell P. Alignment of transmembrane regions in the cystic fibrosis transmembrane conductance regulator chloride channel pore. J Gen Physiol. 2011;138:165–178. doi: 10.1085/jgp.201110605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao X, Bai Y, Hwang T-C. Cysteine scanning of CFTR’s first transmembrane segment reveals its plausible roles in gating and permeation. Biophys J. 2013;104:786–797. doi: 10.1016/j.bpj.2012.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beck EJ, Yang Y, Yaemsiri S, Raghuram V. Conformational changes in a pore-lining helix coupled to cystic fibrosis transmembrane conductance regulator channel gating. J Biol Chem. 2008;283:4957–4966. doi: 10.1074/jbc.M702235200. [DOI] [PubMed] [Google Scholar]
- 37.Bai Y, Li M, Hwang T-C. Dual roles of the sixth transmembrane segment of the CFTR chloride channel in gating and permeation. J Gen Physiol. 2010;136:293–309. doi: 10.1085/jgp.201010480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El Hiani Y, Linsdell P. Changes in accessibility of cytoplasmic substances to the pore associated with activation of the cystic fibrosis transmembrane conductance regulator chloride channel. J Biol Chem. 2010;285:32126–32140. doi: 10.1074/jbc.M110.113332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Norimatsu Y, Ivetac A, Alexander C, Kirkham J, O’Donnell N, Dawson DC, Sansom MSP. Cystic fibrosis transmembrane conductance regulator: a molecular model defines the architecture of the anion conduction path and locates a “bottleneck” in the pore. Biochemistry. 2012;51:2199–2212. doi: 10.1021/bi201888a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bai Y, Li M, Hwang T-C. Structural basis for the channel function of a degraded ABC transporter, CFTR (ABCC7) J Gen Physiol. 2011;138:495–507. doi: 10.1085/jgp.201110705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qian F, El Hiani Y, Linsdell P. Functional arrangement of the 12th transmembrane region in the CFTR chloride channel based on functional investigation of a cysteine-less variant. Pflügers Arch. 2011;462:559–571. doi: 10.1007/s00424-011-0998-2. [DOI] [PubMed] [Google Scholar]
- 42.Zhang J, Hwang T-C. The fifth transmembrane segment of cystic fibrosis transmembrane conductance regulator contributes to its anion permeation pathway. Biochemistry. 2015;54:3839–3850. doi: 10.1021/acs.biochem.5b00427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holstead RG, Li M-S, Linsdell P. Functional differences in pore properties between wild-type and cysteine-less forms of the CFTR chloride channel. J Membr Biol. 2011;243:15–23. doi: 10.1007/s00232-011-9388-0. [DOI] [PubMed] [Google Scholar]
- 44.Linsdell P, Zheng S-X, Hanrahan JW. Non-pore lining amino acid side chains influence anion selectivity of the human CFTR Cl− channel expressed in mammalian cell lines. J Physiol. 1998;512:1–16. doi: 10.1111/j.1469-7793.1998.001bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McDonough S, Davidson N, Lester HA, McCarty NA. Novel pore-lining residues in CFTR that govern permeation and open-channel block. Neuron. 1994;13:623–634. doi: 10.1016/0896-6273(94)90030-2. [DOI] [PubMed] [Google Scholar]
- 46.Gupta J, Linsdell P. Extent of the selectivity filter conferred by the sixth transmembrane region in the CFTR chloride channel pore. Mol Membr Biol. 2003;20:45–52. doi: 10.1080/0968031000045824. [DOI] [PubMed] [Google Scholar]
- 47.Linsdell P. Metal bridges to probe membrane ion channel structure and function. Biomol Concepts. 2015;6:191–203. doi: 10.1515/bmc-2015-0013. [DOI] [PubMed] [Google Scholar]