

Abstract
Nephropathic cystinosis (NC) is a rare disease caused by mutations in the CTNS gene encoding for cystinosin, a lysosomal transmembrane cystine/H+ symporter, which promotes the efflux of cystine from lysosomes to cytosol. NC is the most frequent cause of Fanconi syndrome (FS) in young children, the molecular basis of which is not well established. Proximal tubular cells have very high metabolic rate due to the active transport of many solutes. Not surprisingly, mitochondrial disorders are often characterized by FS. A similar mechanism may also apply to NC. Because cAMP has regulatory properties on mitochondrial function, we have analyzed cAMP levels and mitochondrial targets in CTNS−/− conditionally immortalized proximal tubular epithelial cells (ciPTEC) carrying the classical homozygous 57-kb deletion (delCTNS−/−) or with compound heterozygous loss-of-function mutations (mutCTNS−/−). Compared to wild-type cells, cystinotic cells had significantly lower mitochondrial cAMP levels (delCTNS−/− ciPTEC by 56% ± 10.5, P < 0.0001; mutCTNS−/− by 26% ± 4.3, P < 0.001), complex I and V activities, mitochondrial membrane potential, and SIRT3 protein levels, which were associated with increased mitochondrial fragmentation. Reduction of complex I and V activities was associated with lower expression of part of their subunits. Treatment with the non-hydrolysable cAMP analog 8-Br-cAMP restored mitochondrial potential and corrected mitochondria morphology. Treatment with cysteamine, which reduces the intra-lysosomal cystine, was able to restore mitochondrial cAMP levels, as well as most other abnormal mitochondrial findings. These observations were validated in CTNS-silenced HK-2 cells, indicating a pivotal role of mitochondrial cAMP in the proximal tubular dysfunction observed in NC.
Electronic supplementary material
The online version of this article (10.1007/s00018-018-2800-5) contains supplementary material, which is available to authorized users.
Keywords: Lysosomal storage disease, Cystinosis, Cyclic-AMP, Mitochondria, SIRT3, Fanconi syndrome
Introduction
Nephropathic cystinosis (NC) is a rare metabolic disease caused by mutations in CTNS gene that encodes for the cystine carrier cystinosin [48] and is characterized by an impaired transport of the amino acid cystine out of lysosomes [10, 19]. NC is the most frequent cause of Fanconi syndrome (FS) in young children, a tubular defect that is characterized by impaired reabsorption processes in proximal renal tubular cells with subsequent loss of electrolytes, glucose, bicarbonate, phosphate, amino acids, and low-molecular-weight proteins [9]. FS can result from several congenital and acquired conditions that, in most cases, cause global dysfunction of the proximal tubular cell, altered mitochondrial metabolism and impairment of energy-dependent reabsorption processes [16].
In recent years, several pathological cell processes, including altered endosomal trafficking, impaired autophagy, and cell oxidation, have emerged as key aspects in the pathogenesis of NC, in addition to lysosomal cystine accumulation [21, 28, 35, 39, 54]. Primary involvement of mitochondria in the pathogenesis of the disease has been long debated. After the first description of abnormal mitochondria in renal biopsies of patients with NC [4], successive work showed conflicting results. The early in vitro studies of the 1990s using CDME to induce cystine accumulation in lysosomes were subsequently found to be poor models of the disease, because this drug causes direct mitochondrial toxicity. Thereafter, a number of studies showed variable levels of ATP in cystinotic cells cultured under different conditions [55], but clear mitochondrial abnormalities were not found in a detailed study performed on cystinotic fibroblasts [26]. More recent studies, however, have shown increased mitophagy, increased sensitivity to apoptosis, and evidence of mitochondrial depolarization [42, 46], all of which point to a mitochondrial defect.
An important regulatory mechanism of mitochondrial metabolism is represented by cyclic-AMP (cAMP) [1, 11, 13, 49]. cAMP is one of the most important second messengers in living organisms, has anti-inflammatory properties [34], and modulates ROS levels [6], ion channel functions [25], energy metabolism [40], and cell proliferation [45], among others. In mammalian cells, cAMP is generated by a family of transmembrane adenylyl cyclases (tmACs) or by the soluble adenylyl cyclase (sAC), which generates cAMP in different intracellular compartments, including mitochondria. Contrary to tmAC that is activated by β adrenergic receptor stimulation, sAC is modulated by intracellular stimuli, including calcium, bicarbonates, and ATP levels [27, 60]. At the mitochondrial level, sAC-dependent cAMP has been shown to promote the turnover of nuclear-encoded subunits of complex I [14], the activity of cytochrome c oxidase [49], the structural and functional organization of ATP synthase [11], the overall ATP production [15], and the mitochondrial morphology [44].
Herein, we show, using different approaches and different cell models of NC, that low levels of mitochondrial cAMP in NC are associated with structural and functional mitochondrial alterations. Mitochondrial cAMP levels as well as mitochondrial functional impairment can be rescued, at least in part, by cysteamine, a drug that is currently used to treat NC.
Results
Altered cAMP level in CTNS−/− proximal tubule epithelial cells
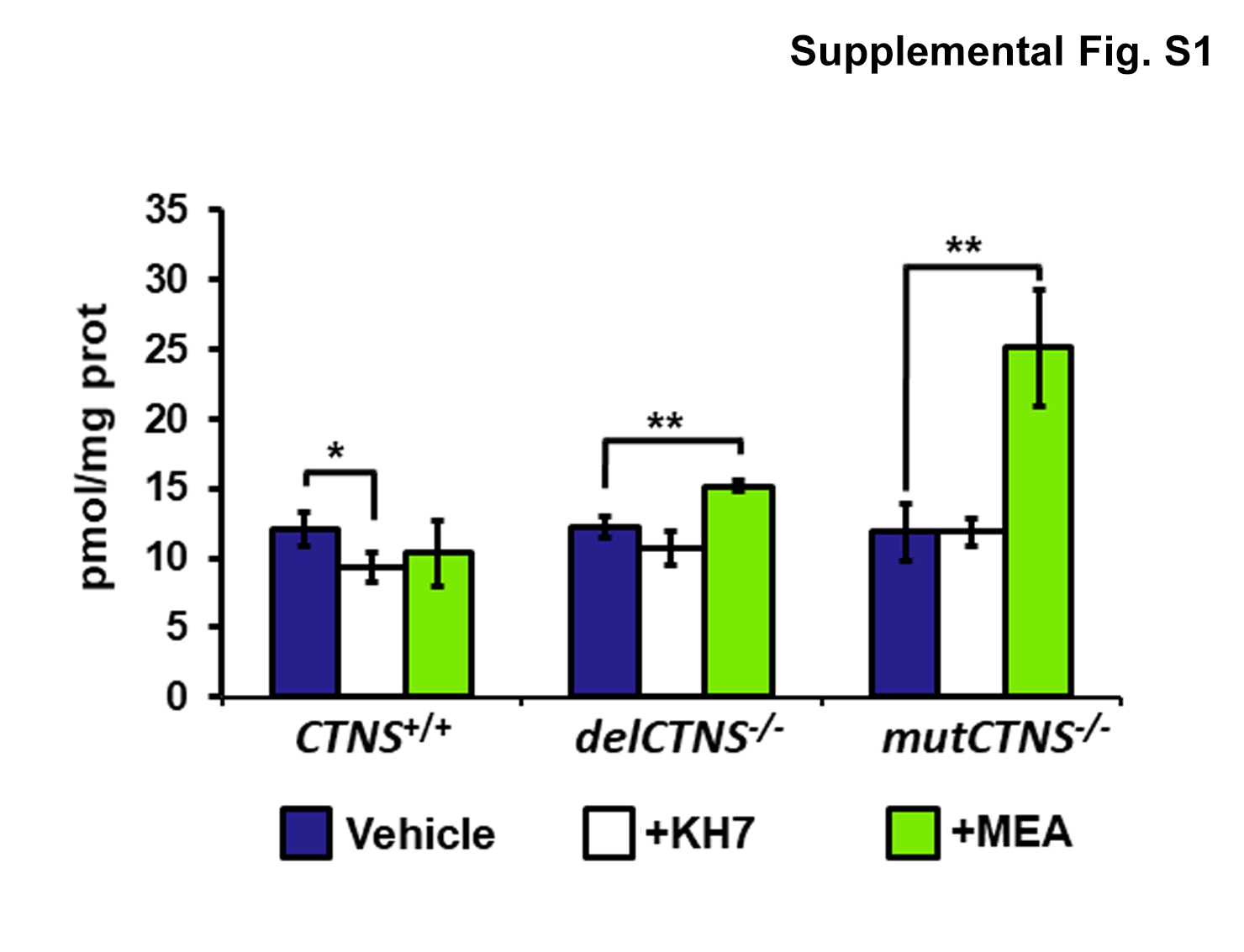
We measured the total cellular level of cAMP using a direct immunoassay in conditionally immortalized proximal tubular epithelial cells (ciPTEC) obtained from a healthy volunteer (CTNS+/+) and from two cystinotic patients bearing the classical homozygous 57-kb deletion (delCTNS−/−) or a compound heterozygous loss-of-function mutations: c.518-519delCA (p.Tyr173X)+c.1015G>A (p.Gly339Arg) (mutCTNS−/−). Comparable levels of total cAMP in delCTNS−/− cells, mutCTNS−/− cells (12.1 ± 0.8 and 11.9 ± 2.0 pmol/mg prot, respectively), and wild-type cells (12.1 ± 1.2 pmol/mg prot) were observed. However, the treatment of cell cultures with 50 µM KH7 for 4 h (a selective inhibitor of sAC [11]) showed a more marked and significant reduction of cAMP in CTNS+/+ with respect to delCTNS−/− and mutCTNS−/− cells (Supplemental Figure S1) indicating a slight contribute of sAC-dependent cAMP to the total cAMP levels in cystinotic cells. Treatment of cell lines with 100 µM cysteamine (MEA) for 24 h increased the total cAMP level in delCTNS−/− and mutCTNS−/− cells but not in wild-type cells (Supplemental Figure S1). Since sAC enzyme is present in the mitochondria as well as in the nucleus and cytosol, a more in-depth analysis was carried out using Normalized Fluorescence Resonance Energy Transfer (netFRET) analysis. netFRET signals in cells transfected with the selective mitochondrial 4mtH30 probe allows to detect specifically mitochondrial cAMP levels. As shown in Fig. 1a, in cells transfected with the selective mitochondrial 4mtH30 probe, netFRET signals were found to be significantly higher in cystinotic delCTNS−/− ciPTEC (by 156% ± 10.5; P < 0.0001) and mutCTNS−/− (by 126% ± 4.3; P < 0.001) cell lines compared to CTNS+/+ ciPTEC. Based on FRET probe features, these indicated lower levels of mitochondrial cAMP compared to CTNS+/+ ciPTEC. Treatment with 100 µM cysteamine for 24 h decreased mitochondrial cAMP levels in CTNS+/+ ciPTEC, while significantly increased mitochondrial cAMP in cystinotic ciPTEC. No changes in FRET signals were observed in cells transfected with the FRET probe (H96) to detect cytosolic cAMP; neither cysteamine treatment affected it (Fig. 1b).
Fig. 1.

Mitochondrial cAMP level is lower in cystinotic cells and increased by cysteamine treatment. ciPTEC obtained from a healthy volunteer (CTNS+/+) and from two cystinotic patients (delCTNS−/− or mutCTNS−/−) were transfected (transiently) with the EPAC-based FRET sensor target specifically to mitochondria (4mtH30) (a) or cytosol (H96) (b). Where indicated, the cells were treated with 100 µM cysteamine (MEA) or DMSO (vehicle) for 24 h. The histograms represent the means values ± SEM of net% FRET observed (ANOVA test). For other details, see under “Methods” section
Study of OxPhos system in cystinotic ciPTEC
It is reported that mitochondrial cAMP signaling has a critical role in the regulating mitochondrial respiratory chain activity and structure. To verify if the altered mitochondrial cAMP levels exert their regulatory effect on the respiratory chain of cystinotic ciPTEC, analyses of enzymatic activities of complex I, IV, and V were performed. As shown in Fig. 2, the NADH:Q oxidoreductase activity of complex I was reduced in delCTNS−/− and mutCTNS−/− ciPTEC compared to CTNS+/+ ciPTEC (21.7 ± 3.57 and 19.2 ± 2.24 vs. 38.7 ± 1.80 nmol/mg/min. P delCTNS−/− vs. CTNS+/+ = 0.012 and P mutCTNS−/− vs. CTNS+/+ = 0.017). Likewise, the ATP hydrolase activity (complex V) was reduced in cystinotic ciPTEC (64.4 ± 2.45 and 72.7 ± 3.75 vs. 123.4 ± 12.1 nmol/mg/min; P delCTNS−/− vs. CTNS+/+ = 0.0065 and mutCTNS−/− vs. CTNS+/+ = 0.022). Treatment with 100 µM cysteamine for 24 h restored completely both enzymatic activities. The activity of cytochrome c oxidase (complex IV) appeared slightly reduced in cystinotic ciPTEC, but this did not reach statistical significance (Fig. 2).
Fig. 2.

Activities of complex I and complex V are reduced in cystinotic ciPTEC cells and restored by cysteamine treatment. ciPTEC obtained from a healthy volunteer (CTNS+/+) and from two cystinotic patients (delCTNS−/− or mutCTNS−/−) were treated with 100 µM cysteamine (MEA) or DMSO (vehicle) for 24 h. After incubation, the enzymatic activities of complex I (NADH:ubiquinone oxidoreductase), complex V (ATP hydrolase), and complex IV (cytochrome c oxidase) were performed. The histograms represent the means values ± SD of three independent experiments (Student’s t test). For other details, see under “Methods” section
Studies on complex I and V subunits levels were also performed. Western blotting analysis of NADH:ubiquinone oxidoreductase subunits showed reduced expression of the NDUFS4 and NDUFA9 subunits in CTNS−/− ciPTEC compared to CTNS+/+ ciPTEC, while the protein level of NDUFB7 was unchanged. Treatment with 100 µM cysteamine for 24 h normalized the NDUFS4 and NDUFA9 protein levels (Fig. 3a).
Fig. 3.

Several subunits of complex I and complex V are reduced in cystinotic ciPTEC cells and restored by cysteamine treatment. a Immunoblotting analysis of ciPTEC obtained from a healthy volunteer (CTNS+/+) and cystinotic patient (delCTNS−/−). b Immunoblotting analysis of CTNS+/+ and cystinotic patient (mutCTNS−/−). a, b Where indicated, the cells were treated with 100 µM cysteamine (MEA) or DMSO (vehicle) for 24 h. After incubation, proteins of cellular lysate were loaded on 8% SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with the antibodies described in the figure. Protein loading was assessed by reprobing the blots with the actin antibody. Densitometric analysis was performed by VersaDoc imaging system BioRad, using Quantity One software. The histograms represent the means values ± SEM of the ratio of ADU (arbitrary densitometric units) of the immuno-revealed protein normalized on actin level (Student’s t test)
Protein expression of ATP synthase subunits is shown in Fig. 3a. As shown, delCTNS−/− ciPTEC displayed reduced levels of the d and OSCP subunits, while the expression of the alpha and beta subunits was comparable to control cells. Here again, treatment with cysteamine (100 µM for 24 h) restored normal values.
Similar results were observed when analyzing compound heterozygous mutated cells. In particular, as shown in Fig. 3b, cysteamine rescued in part or completely OSCP and NDUFA9 levels.
Mitochondrial profile in HK-2 cells silenced for CTNS gene
To validate the results obtained in cystinotic ciPTEC, we silenced the CTNS gene in HK-2 cell line using the SMARTpool technology, which combines different siRNAs to mimic the natural silencing pathway and used, as negative control, a non-targeted siRNA pool (Mock). Transfection of CTNS-targeted siRNAs in HK-2 cells caused a 64% reduction of CTNS mRNA, as assessed by qRT-PCR (Supplemental Figure S2). This caused a significant reduction of expression of the NDUFA9 complex I subunit and that of the OSCP complex V subunit, compared to control and to mock-transfected cells. Treatment with cysteamine (100 µM for 24 h) re-established normal levels of expression. The expression of NDUFB7 and alpha subunits was unchanged (Fig. 4a). Silencing the CTNS gene in HK-2 cells, similar to the previous observations in cystinotic cells, altered the enzymatic activities of complex I (50.5 ± 5.72 vs. 83.5 ± 7.86 nmol/mg/min; P = 0.039) and complex V (55.4 ± 1.79 vs. 102.9 ± 4.25 nmol/mg/min; P = 0.009), and cysteamine normalized these findings (Fig. 4b).
Fig. 4.

HK-2 cells silenced for CTNS gene show a reduction of enzymatic activities and subunit expression levels of the complexes I and V. Cysteamine restores the activities and protein expression. Human kidney-2 cells (HK-2) were transfected with siGENOME human CTNS gene (siCTNS) or non-targeting siRNA (Mock). Where indicated, the cells were treated for 24 h with DMSO (vehicle) or 100 µM cysteamine (MEA). a Proteins of cellular lysate were loaded on 8% SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with the antibodies described in the figure. Protein loading was assessed by reprobing the blots with the actin antibody. The histograms represent the means values ± SEM of the ratio of ADU (arbitrary densitometric units) of the immune-revealed protein normalized on actin level (Student’s t test). b Enzymatic activities of complex I (NADH:ubiquinone oxidoreductase) and complex V (ATP hydrolase) were performed as described under “Methods”
Altered mitochondrial potential in cellular models of cystinosis
The transmembrane electrochemical proton gradient generated by pumping of H+ from the matrix across the inner mitochondrial membrane into the intermembrane space generates an electrochemical force that has two components, namely a minor component related to the H+ gradient (ΔpH) and a major component represented by the membrane potential (Δψm). The latter was found to be decreased in delCTNS−/− and mutCTNS−/− ciPTEC, compared to wild-type ciPTEC (16.6 A.U. ± 0.62 and 13.2 A.U. ± 0.55 vs. 20.4 A.U. ± 0.67; P < 0.001) (Fig. 5a). Here again, treatment with cysteamine (100 µM for 24 h), but also with 8-Br-cAMP (100 µM for 24 h), a permeant and non-hydrolysable form of cAMP, restored a normal membrane potential (Fig. 5a). Similarly, we observed lower mitochondrial membrane potential in HK-2-silenced cells, with a reduction of 28.3% (P < 0.0001), compared to cells transfected with non-targeted siRNA that was corrected by cysteamine or by 8-Br-cAMP at the same concentrations and length of exposure (+ 21.9%, P < 0.0001 and + 18.4%, P < 0.05, respectively) (Fig. 5b).
Fig. 5.

Mitochondrial membrane potential (Δψm) is lower in cystinotic cells and in HK-2 cells silenced for CTNS gene and rescued by cysteamine or 8Br-cAMP treatment. a Analysis of mitochondrial potential (Δψm) in ciPTEC obtained from a healthy volunteer (CTNS+/+) and from two cystinotic patients (delCTNS−/− or mutCTNS−/−) treated with 100 µM cysteamine (MEA) or 100 µM 8Br-cAMP for 24 h. b Analysis of mitochondrial potential (Δψm) in HK-2 cells transfected with siGENOME human CTNSgene (siCTNS) or non-targeting siRNA (Mock) and treated with 100 µM cysteamine (MEA) or 100 µM 8Br-cAMP for 24 h. Data result from the analysis of the fluorescence intensity mean expressed in arbitrary units (A.U.) ± SEM measured in at least 30 cells (a) and in about 100 cells (b) (Student’s t test)
SIRT3 changes in cellular models of cystinosis
Mitochondrial sirtuins are NAD+-dependent deacetylases and ADP-ribosyltransferases that play an important role in the control of mitochondrial ATP production, metabolic processes, mitochondrial integrity, and cellular viability. The previous findings showed that exogenous cAMP increased SIRT3 protein levels [53]; on the other hand, decrease of mitochondrial cAMP levels resulted in reduced SIRT3 protein level [44]. The protein expression of SIRT3 was reduced in all analyzed models of cystinosis, compared to controls. SIRT3 was reduced in delCTNS−/−and mutCTNS−/− ciPTEC by 41.8 and 38.2%, respectively (P < 0.003) (Fig. 6a, b). In HK-2 cells treated with CTNS siRNA, SIRT3 was reduced by 28.2% (P < 0.05) compared to control cells (Fig. 6c). Interestingly, treatment with cysteamine restored normal levels of SIRT3 (+ 53.7%, P < 0.05).
Fig. 6.

SIRT3 protein level is reduced in cellular models of cystinosis and recovered by cysteamine treatment. Proteins of cellular lysate of the different cellular models were loaded on 8% SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with the antibody against SIRT3. Protein loading was assessed by reprobing the blots with the actin antibody. The histograms represent the means values ± SEM of the ratio of ADU (arbitrary densitometric units) of the immune-revealed SIRT3 normalized on actin level (Student’s t test)
Analysis of mitochondrial network morphology in cellular models of cystinosis
Because mitochondrial cAMP influences mitochondrial dynamics, we analyzed mitochondrial morphology in cells stained with MitoTracker® by fluorescence microscopy. In cystinotic ciPTEC and HK-2 silenced for CTNS gene, mitochondria appeared fragmented into short sticks or spheres. Treatment with cysteamine or 8-Br-cAMP (100 µM for 24 h) in all cell types normalized the mitochondrial network, which reacquired a branched and tubular morphology, similar to wild-type cells (Fig. 7a). The morphometric analysis revealed a greater fragmentation index (f) in cystinotic ciPTEC (55.5 ± 4.05 A.U. of delCTNS−/− and 72.4 ± 6.11 A.U. of mutCTNS−/− vs. 33.5 ± 2.36 A.U. of CTNS+/+ ciPTEC, P < 0.00001) and in HK-2 silenced for CTNS gene (43.6 ± 2.70 A.U. of siCTNS vs. 22.8 ± 2.70 A.U. of mock and 28.6 ± 3.50 A.U. of control, P < 0.0001) (Fig. 7b, c). To verify if the effect on mitochondrial network exerted by MEA or 8-Br-cAMP in ciPTEC−/− cell lines and siCTNS HK-2 cells is depending on SIRT3, we generated a double knockdown in HK2 cell line, for CTNS and SIRT3 genes. Transfection of CTNS-targeted siRNAs and SIRT3-targeted siRNAs in HK-2 cells caused 80 and 40% reduction of CTNS and SIRT3 mRNAs, respectively (Supplemental Figure S3-A). Compared to control and to mock-transfected cells, the double silenced HK2 cell line showed an increase of mitochondrial fragmentation index (32.1 ± 2.47 A.U. of control and 30.0 ± 3.93 A.U. of mock vs. 42.9 ± 2.49 A.U. of double silenced, P < 0.005) that was recovered by treatment with MEA (30.9 ± 2.91 A.U., P < 0.003) or 8-Br-cAMP (34.3 ± 3.20 A.U., P < 0.04) (Supplemental Figure S3-B).
Fig. 7.

Cysteamine or 8Br-cAMP treatment ameliorates mitochondrial morphology in cystinotic cells and in HK-2 cells silenced for CTNS gene. a Representative images of mitochondrial network of cells stained with MitoTracker® Orange CMTMRos and a z-stack of optical sections, 10 µm in total thickness, were captured by fluorescence microscopy. Scale bars correspond to 25 µm. b, c Reported the quantification of mitochondrial fragmentation index (f index) in ciPTEC and HK-2 cells treated with 100 µM cysteamine (MEA) or 100 µM 8Br-cAMP for 24 h. Data are the mean values (± SEM) of the analyses performed in cell numbers of n = 20 for ciPTEC cells and n = 30 for HK-2 cells. Significant differences were calculated with Student’s t test
Discussion
Nephropathic cystinosis is a lysosomal storage disease characterized by the cystine accumulation into lysosomes and represents the leading cause of FS in children [9]. To date, the mainstay of treatment is represented by cysteamine that decreases cystine lysosomal accumulation and allows delaying progression of renal failure and of other symptoms related to NC [17]. In recent years, a large body of evidence has been produced demonstrating close links between the mitochondrial and lysosomal compartments and, in particular, the development of significant mitochondrial damage in lysosomal storage disorders, causing cell dysfunction and potentially cell death [38]. In addition to oxidative phosphorylation, mitochondria are key organelles that regulate apoptosis, ROS production, and calcium homeostasis, among many others.
In this work, we first showed that mitochondrial cAMP is decreased in cystinotic ciPTEC. In theory, this could be secondary to decreased cAMP synthesis by sAC or increased degradation by phosphodiesterase 2 (PDE2). The latter enzyme is allosterically activated by cGMP, whose synthesis is stimulated by nitric oxide [18], a compound that has been shown to increase in HK-2 cells treated with CTNS siRNA [46]. In addition, lack of substrate, namely lack of ATP, could result in lower cAMP. In this respect, several studies have observed lower ATP levels in various CTNS−/− cell lines under variable experimental conditions [23, 26, 47, 56].
The effect of cysteamine in our experimental model was unexpected. As shown in the results section, cysteamine was able to increase significantly mitochondrial cAMP levels and to correct all other observed mitochondrial abnormalities. Our observation, however, is not unique. Mitznegg, first, observed, in 1973, a direct modulation of cAMP levels by cysteamine, which, at that time, was used as radioprotective agent [32]. The mechanism by which cysteamine induced these changes is still unclear but could be related, at least in part, to its anti-oxidative properties, as it reduces cystine disulfide links. This may also explain the beneficial effects of cysteamine in other models of chronic diseases, including Huntington’s disease, Parkinson’s disease, or non-alcoholic fatty liver disease (NAFLD) [7]. Interestingly, these latter diseases are also characterized by mitochondrial abnormalities [31, 36, 43], which further suggest a role of cysteamine in preventing and treating mitochondrial damage [29].
Compartmentalization of cAMP is a strategy that has probably been adopted by eukaryotic cells to develop targeted responses in different sub-cellular districts. In mitochondria, cAMP modulates the amount of nuclear-encoded subunits of complex I and the enzymatic activity of this same complex [14]. In brief, nuclear-encoded subunits of complex I are imported into mitochondria, where they are assembled together with subunits encoded by mitochondrial DNA to form mature complex. In another mechanism, termed “dynamic assembly” [24], subunits that are imported into mitochondria are exchanged with pre-existing aged copies and, by this mean, modulate the overall activity of the respiratory chain [12]. Among these, the NDUFS4 and NDUFA9 subunits have a high turnover resulting from high cytosolic synthesis, and import into mitochondria and elevated proteolysis which are regulated by cAMP [14]. We observed that both subunits were downregulated in cystinotic cells and in HK-2 cells silenced for the CTNS gene, whereas the level of the NDUFB7 accessory subunit, which is not subjected to the dynamic assembly of complex I, was unchanged. Lack of critical subunits in cystinotic cells had an impact on the activity of complex I, which, in this circumstance, can produce more reactive oxygen species (ROS) [50], supporting the previous findings suggesting oxidative cell damage in NC [30, 42, 46].
In addition, low mitochondrial cAMP levels in cystinotic cells and in CTNS−/− HK-2 cells were associated with downregulation of specific subunits of the F1Fo-ATP synthase (complex V). Complex V is composed by two main functional domains: the membrane-embedded Fo-ATPase and the membrane-extrinsic catalytic F1-ATPase that are linked by a central stalk [52]. In both yeast and mammalian cells, complex V exists in monomeric or multimeric states [3, 59]. Changes in the oligomerization of complex V modulate the organization of the mitochondrial cristae and indirectly modulate the mitochondrial membrane potential [8, 20, 37]. For example, inhibition of sAC in rat liver mitochondria and myoblasts results in lower mitochondrial cAMP levels, reduced expression of the d and OSCP subunits of complex V, impaired oligomerization of complex V, decreased ATP hydrolase activity, and impaired mitochondrial membrane potential [11].
Similarly, we observed that subunits d and OSCP were decreased in cystinotic cells and in CTNS−/− HK-2 cells, and that complex V enzymatic activity was decreased. Conversely, the expression of the α and β subunits was unchanged, consistent with the previous results obtained in cystinotic fibroblasts [58].
Most likely, downregulation of complex I in CTNS−/− cells causes mitochondrial depolarization, a finding that has already been observed in other studies [30, 46]. In addition, we observed significant alterations of the mitochondrial network, which appeared fragmented. Treatment with cysteamine has been shown in other studies to restore normal ATP levels in cystinotic cells [56]. Our data suggest that this effect translates into normalization of the mitochondrial membrane potential and of the mitochondrial morphology, which are probably explained by the recovery of normal cAMP levels. Alternatively, or in addition, cysteamine may have a positive effect related to its anti-oxidative properties. However, the former hypothesis is supported by the positive effects of 8-Br-cAMP treatment. 8-Br-cAMP, which is a cAMP analog resistant to degradation achieved by phosphodiesterases, restored completely both mitochondrial membrane potential and mitochondrial network in our cell models of cystinosis.
To complete the analysis, we examined the expression of mitochondrial sirtuin 3. This protein has emerged in recent years as a key regulator of redox processes and mitochondrial dynamics, by means of deacetylation of enzymes that are involved in ROS balance, such as the mitochondrial manganese superoxide dismutase [5]. These processes have critical relevance in the pathogenesis and recovery of acute renal failure [16, 33]. In this respect, cAMP has been shown to increase SIRT3 level, while the induction of ROS production decreases mitochondrial cAMP concentrations and, consequently, decreases SIRT3 level [44, 53]. SIRT3 expression has been extensively analyzed in several tissues, in particular in the heart [22]. In tissues that normally express high level of SIRT3, such as liver, heart or kidneys, lack of deacetylase activity causes a marked reduction in ATP [2]. Furthermore, reduced SIRT3 activity causes acetylation of NDUFA9, which, in turn, inhibits complex I [2]. Similarly, we observed downregulation of SIRT3 in our cystinotic cell models. In this context, cysteamine may exert its protective action by increasing cAMP, which, in turn, activates SIRT3. However, as shown by double silenced of CTNS and SIRT3 genes in HK-2 cell line, the rescue effect on mitochondrial network exerted by MEA and 8-Br-cAMP appeared to be SIRT3-independent.
In conclusion, our data show in vitro mitochondrial damage and dysfunction in models of cystinosis, which is consistent with the mitochondrial damage previously observed in vivo in renal biopsies obtained from patients [4]. Further studies will be necessary to evaluate the role of mitochondrial impairment in producing clinical symptoms. For example, in addition to FS, azoospermia or progressive neuromuscular degeneration could also result from a similar mechanism involving mitochondrial degeneration. In addition to promoting cystine efflux from lysosomes, cysteamine may exert its beneficial action by preventing mitochondrial damage. Other drugs that exert similar effects on mitochondria would be good candidates to be associated with cysteamine for the treatment of this severe disease.
Methods
Cell culture
Conditionally immortalized proximal tubular cells (ciPTEC) were kindly provided by Dr. Elena N. Levtchenko and cultured as described in Wilmer et al. [57].
Human Kidney-2 cells (HK-2) obtained from the American Type Culture Collection (ATCC, #CRL-2190) were grown in Dulbecco’s modified Eagle’s medium–F12 Glutamax® supplemented with 5% fetal bovine serum, 100 units/ml penicillin and 100 mg/ml streptomycin, 10 mg/ml insulin from bovine pancreas, 5.5 mg/ml human transferrin, and 5 ng/ml sodium selenite.
Cells were grown in a humidified atmosphere with 5% CO2 at 37 °C.
siRNA transfection and assessment of CTNS
HK-2 cell line was transfected with siGENOME human CTNS or non-targeting siRNA, SMARTpool (Dharmacon, Thermo Scientific, Dublin, Ireland) by Oligofectamine Reagent (Invitrogen) according to the manufacturer’s instructions. After 48 h of transfection, when necessary, cells were treated for other 24 h with cysteamine or vehicle.
Total RNA was extracted by TRIzol reagent (Ambion, Life Technologies, Foster, CA) and cDNA was synthesized using the Euro-Script RT-PCR kit (EuroClone, Milano, Italy) according to the manufacturer’s instructions. Quantitative PCR assay was performed using SYBR FAST quantitative PCR Kit Master Mix (Kapa Biosystems, Wilmington, MA, USA) with corresponding primers listed in Supplementary Table S1. Gene expression data for human CTNS were determined using the method. Human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression was used as the endogenous control.
FRET measurements
ciPTEC were transfected (transiently) with the EPAC-based FRET sensor target specifically to mitochondria (4mtH30) or to cytosol (H96). After treatments, cells were fixed with 4% paraformaldehyde (PFA) in PBS (pH 7.4) for 20 min at room temperature, and then washed three times in PBS; and afterwards, coverslips were mounted in Mowiol (polyvinyl alcohol 4–88; Sigma-Aldrich, Milan, Italy). Visualization of ECFP- and/or EYFP-expressing cells and detection of FRET was performed on an inverted microscope (Nikon Eclipse TE2000-S) equipped with a monochromator controlled by MetaMorph/MetaFluor software. ECFP was excited at 433 nm and EYFP at 512 nm. Each image was further corrected for ECFP crosstalk and EYFP crossexcitation [41]. Briefly, images were aligned and corrected for background in the emission windows for FRET (535/30 nm), ECFP (475/30 nm), and EYFP (535/26 nm). Each image was further corrected for ECFP crosstalk and EYFP crossexcitation. Thus, netFRET = [IFRETbg − ICFPbg × k1 − IYFPbg (K2 − αK1)]/(1 − K1δ), where IFRETbg, ICFPbg, and IYFPbg are the background-corrected pixel gray values measured in the FRET, ECFP, and EYFP windows, respectively; K1, K2, α, and δ are calculated to evaluate the crosstalk between donor and acceptor. The obtained netFRET values were analyzed using MetaMorph® Microscopy Automation and Image Analysis Software.
SDS-PAGE and western blotting
Cells were harvested from Petri dishes with 0.05% trypsin, 0.02% EDTA, pelleted by centrifugation at 500×g, and then resuspended and lysed in RIPA buffer containing 150 mM NaCl, 5 mM EDTA, 50 mM Tris/HCl, 0.1% SDS, and 1% Triton X-100, pH 7.4, in the presence of a proteases inhibitor (0.25 mM PMSF). Proteins from cell lysate were separated by 8% SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to a nitrocellulose membrane. The membrane was probed with antibodies against NDUFS4 (Thermo Scientific, Pierce Antibodies), NDUFA9, NDUFB7, Alpha, Beta and d subunit (Invitrogen Life Technologies), OSCP (Santa Cruz Biotechnology), and SIRT3 (Cell Signaling). The control loading was performed with antibody against actin (SIGMA). After being washed in TTBS, the membrane was incubated for 60 min with anti-rabbit or anti-mouse IgG peroxidase-conjugate antibody (diluted 1:5000). Immunodetection was then performed, after further TTBS washes, with the enhanced chemiluminescence (ECL) (Thermo Scientific, Pierce). Densitometric analysis was performed by VersaDoc imaging system (BioRad).
Enzymatic spectrophotometric assays
Harvested cells were exposed to ultrasound energy for 15 s at 0 °C, and then, the cellular lysate was centrifuged at 500×g for 5 min at 4 °C and the supernatant recovered and centrifuged at 20,000×g for 5 min at 4 °C. The pellet was used for next determinations.
The NADH-UQ oxidoreductase activity of complex I was performed in 40 mM potassium phosphate buffer, pH 7.4, 5 mM MgCl2, in the presence of 3 mM KCN, 1 μg/ml antimycin, and 200 μM decylubiquinone, using 30 μg of proteins, by following the oxidation of 100 μM NADH at 340–425 nm (Δε = 6.81/mM/cm). The activity was corrected for the residual activity measured in the presence of 1 μg/ml rotenone.
Cytochrome c oxidase (complex IV) activity was measured by following the oxidation of 10 μM ferrocytochrome c at 550–540 nm (Δε = 19.1/mM/cm). Enzymatic activity was measured in 10 mM phosphate buffer, pH 7.4, using 20 μg of proteins. This rate was inhibited over 95% by KCN (2 mM).
ATP hydrolase activity was measured by an ATP-regenerating system. Proteins were suspended (final concentration 0.1 mg/ml) in a buffer consisting of 375 mM sucrose, 75 mM KCl, 30 mM Tris–HCl pH 7.4, 3 mM MgCl2, 2 mM PEP, 55 U/ml lactate dehydrogenase, 40 U/ml pyruvate kinase, and 0.3 mM NADH. The reaction was started by the addition of 1 mM ATP and the oxidation of NADH was followed at 340 nm.
Mitochondrial potential (Δψm) and morphometric analysis
Images were collected using Leica DMi8 microscope equipped with an oil immersion 63× objective on cells seeded in chamber slides (BD). The living cells were incubated for 20 min with 50 nM of MitoTracker® Orange CMTMRos probe (Invitrogen, Molecular Probes) in FBS-free medium, washed with PBS, and examined by fluorescence microscopy (λEx 554 nm, λEc 576 nm) in complete growth medium. Images analysis of mitochondrial potential was done using ImageJ v. 1.50 g software (http://imagej.nih.gov/ij/). For quantitative analysis of mitochondrial network morphologies, the acquired images were conveniently adjusted for background and brightness/contrast and then analyzed with MitoLoc, a plugin of ImageJ, according to Vowinckel et al. protocol with slight modifications [51].
Statistical analysis
All data are presented as mean ± SEM (standard error of mean). N value denotes the number of independent experiments. Statistical difference was determined by Student’s t test or ANOVA test as indicated in the legend to figures. P value of 0.05 was considered as statistically significant (***P < 0.001; **P < 0.01; *P < 0.05).
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
Supplemental Figure S1. The sAC-dependent cAMP level is lower in CTNS−/− ciPTEC and increased by cysteamine treatment. ciPTEC were treated in the absence or in the presence of 50 μM KH7 or 100 μM cysteamine (MEA), for 4 and 24 h respectively. After the treatment, the medium was aspirated and 1 ml of 0.1 M HCl was added to the cell layer followed by 10 min incubation at 37 °C. The lysed cells were centrifuged at 1,300×g for 10 min at 4 °C. The supernatants were used to determine cAMP concentration using a direct immunoassay kit (Assay Designs) as described by the manufacturer. Total protein concentration was determined by Bio Rad protein assay. The cAMP levels in the samples were normalized to the protein concentration and expressed as pmol/mg protein. The histograms represent the mean values of duplicate determinations of cAMP level in three separate experiments. Error bars indicate standard deviations. Significant differences were calculated with Student’s t test (JPEG 257 kb)
{kind=link}
Supplemental Figure S2. HK-2 cells silenced for CTNS gene show a significative reduction of CTNS relative gene expression. Human kidney-2 cells (HK-2) were transfected with siGENOME human CTNS gene (siCTNS) or non-targeting siRNA (Mock). Where indicated the cells were treated for 24 h with DMSO (vehicle) or 100 µM cysteamine (MEA). Analysis of CTNS gene expression, normalized to the expression of GAPDH, show a reduction in siCTNS of 68% (P < 0.0001) and 64% (P < 0.02) compared to control and mock respectively. Significant differences were calculated with Student’s t test (JPEG 124 kb)
{kind=link}
Supplemental Figure S3. Cysteamine or 8Br-cAMP treatment ameliorate mitochondrial morphology in HK-2 cells double silenced for CTNS and SIRT3 genes. In panel A, HK-2 were co-transfected with human CTNS gene and SIRT3 gene (Santa Cruz, Biotechnology, Dallas, Texas) (siRNA) or non-targeting siRNA (Mock). Where indicated the cells were treated for 24 h with DMSO (vehicle) or 100 µM cysteamine (MEA) or 100 µM 8Br-cAMP. Analysis of CTNS gene expression, normalized to the expression of GAPDH, shows 80% reduction of CTNS gene expression (P < 0.0001) and 40% reduction of SIRT3 gene expression (P < 0.04) compared to control. Panel B, quantification of mitochondrial fragmentation index (f index) in HK-2 cells not transfected (Control), transfected with non-targeting siRNA (Mock) or co-transfected with human CTNS gene and SIRT3 gene (siRNA) for 72 h and treated with 100 µM cysteamine (MEA) or 100 µM 8Br-cAMP during last 24 h of incubation. Data are the mean values (± SEM) of the analyses performed in cell numbers of n = 30 for HK-2 cells. Significant differences were calculated with Student’s t test (JPEG 395 kb)
Acknowledgements
FB, FE, and DDR are supported by a Research Grant from Cystinosis Research Foundation. DDR and AS are supported by a Research Grant from University of Bari “Aldo Moro”. We thank Mr. Paolo Lattanzio for technical assistance.
Compliance with ethical standards
Conflict of interest
The authors declare no conflicts of interest.
Contributor Information
Francesco Bellomo, Phone: +39-06-68592997, Email: francesco.bellomo@opbg.net.
Domenico De Rasmo, Phone: +39-080-5448516, Email: d.derasmo@ibiom.cnr.it.
References
- 1.Acin-Perez R, Salazar E, Kamenetsky M, Buck J, Levin LR, Manfredi G. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab. 2009;9:265–276. doi: 10.1016/j.cmet.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA. 2008;105:14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnold I, Pfeiffer K, Neupert W, Stuart RA, Schagger H. Yeast mitochondrial F1F0-ATP synthase exists as a dimer: identification of three dimer-specific subunits. EMBO J. 1998;17:7170–7178. doi: 10.1093/emboj/17.24.7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartsocas CS, Bernstein J, Orloff S, Chandra R, Schulman JD. A familial syndrome of growth retardation, severe Fanconi-type renal disease and glomerular changes–a new entity? Int J Pediatr Nephrol. 1986;7:101–106. [PubMed] [Google Scholar]
- 5.Bell EL, Guarente L. The SirT3 divining rod points to oxidative stress. Mol Cell. 2011;42:561–568. doi: 10.1016/j.molcel.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellomo F, Piccoli C, Cocco T, Scacco S, Papa F, Gaballo A, Boffoli D, Signorile A, D’Aprile A, Scrima R, Sardanelli AM, Capitanio N, Papa S. Regulation by the cAMP cascade of oxygen free radical balance in mammalian cells. Antioxid Redox Signal. 2006;8:495–502. doi: 10.1089/ars.2006.8.495. [DOI] [PubMed] [Google Scholar]
- 7.Besouw M, Masereeuw R, van den Heuvel L, Levtchenko E. Cysteamine: an old drug with new potential. Drug Discov Today. 2013;18:785–792. doi: 10.1016/j.drudis.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 8.Bornhovd C, Vogel F, Neupert W, Reichert AS. Mitochondrial membrane potential is dependent on the oligomeric state of F1F0-ATP synthase supracomplexes. J Biol Chem. 2006;281:13990–13998. doi: 10.1074/jbc.M512334200. [DOI] [PubMed] [Google Scholar]
- 9.Cherqui S, Courtoy PJ. The renal Fanconi syndrome in cystinosis: pathogenic insights and therapeutic perspectives. Nat Rev Nephrol. 2017;13:115–131. doi: 10.1038/nrneph.2016.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cherqui S, Sevin C, Hamard G, Kalatzis V, Sich M, Pequignot MO, Gogat K, Abitbol M, Broyer M, Gubler MC, Antignac C. Intralysosomal cystine accumulation in mice lacking cystinosin, the protein defective in cystinosis. Mol Cell Biol. 2002;22:7622–7632. doi: 10.1128/MCB.22.21.7622-7632.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Rasmo D, Micelli L, Santeramo A, Signorile A, Lattanzio P, Papa S. cAMP regulates the functional activity, coupling efficiency and structural organization of mammalian FOF1 ATP synthase. Biochim Biophys Acta. 2016;1857:350–358. doi: 10.1016/j.bbabio.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 12.De Rasmo D, Signorile A, Larizza M, Pacelli C, Cocco T, Papa S. Activation of the cAMP cascade in human fibroblast cultures rescues the activity of oxidatively damaged complex I. Free Radic Biol Med. 2012;52:757–764. doi: 10.1016/j.freeradbiomed.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 13.De Rasmo D, Signorile A, Papa F, Roca E, Papa S. cAMP/Ca2+ response element-binding protein plays a central role in the biogenesis of respiratory chain proteins in mammalian cells. IUBMB Life. 2010;62:447–452. doi: 10.1002/iub.342. [DOI] [PubMed] [Google Scholar]
- 14.De Rasmo D, Signorile A, Santeramo A, Larizza M, Lattanzio P, Capitanio G, Papa S. Intramitochondrial adenylyl cyclase controls the turnover of nuclear-encoded subunits and activity of mammalian complex I of the respiratory chain. Biochim Biophys Acta. 2015;1853:183–191. doi: 10.1016/j.bbamcr.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 15.Di Benedetto G, Scalzotto E, Mongillo M, Pozzan T. Mitochondrial Ca(2)(+) uptake induces cyclic AMP generation in the matrix and modulates organelle ATP levels. Cell Metab. 2013;17:965–975. doi: 10.1016/j.cmet.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 16.Emma F, Montini G, Parikh SM, Salviati L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat Rev Nephrol. 2016;12:267–280. doi: 10.1038/nrneph.2015.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emma F, Nesterova G, Langman C, Labbe A, Cherqui S, Goodyer P, Janssen MC, Greco M, Topaloglu R, Elenberg E, Dohil R, Trauner D, Antignac C, Cochat P, Kaskel F, Servais A, Wuhl E, Niaudet P, Vant Hoff W, Gahl W, Levtchenko E. Nephropathic cystinosis: an international consensus document. Nephrol Dial Transplant. 2014;29(Suppl 4):iv87–iv94. doi: 10.1093/ndt/gfu090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62:525–563. doi: 10.1124/pr.110.002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gahl WA, Bashan N, Tietze F, Bernardini I, Schulman JD. Cystine transport is defective in isolated leukocyte lysosomes from patients with cystinosis. Science. 1982;217:1263–1265. doi: 10.1126/science.7112129. [DOI] [PubMed] [Google Scholar]
- 20.Habersetzer J, Ziani W, Larrieu I, Stines-Chaumeil C, Giraud MF, Brethes D, Dautant A, Paumard P. ATP synthase oligomerization: from the enzyme models to the mitochondrial morphology. Int J Biochem Cell Biol. 2013;45:99–105. doi: 10.1016/j.biocel.2012.05.017. [DOI] [PubMed] [Google Scholar]
- 21.Ivanova EA, van den Heuvel LP, Elmonem MA, De Smedt H, Missiaen L, Pastore A, Mekahli D, Bultynck G, Levtchenko EN. Altered mTOR signalling in nephropathic cystinosis. J Inherit Metab Dis. 2016;39:457–464. doi: 10.1007/s10545-016-9919-z. [DOI] [PubMed] [Google Scholar]
- 22.Koentges C, Bode C, Bugger H. SIRT3 in cardiac physiology and disease. Front Cardiovasc Med. 2016;3:38. doi: 10.3389/fcvm.2016.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar A, Bachhawat AK. A futile cycle, formed between two ATP-dependant gamma-glutamyl cycle enzymes, gamma-glutamyl cysteine synthetase and 5-oxoprolinase: the cause of cellular ATP depletion in nephrotic cystinosis? J Biosci. 2010;35:21–25. doi: 10.1007/s12038-010-0004-8. [DOI] [PubMed] [Google Scholar]
- 24.Lazarou M, McKenzie M, Ohtake A, Thorburn DR, Ryan MT. Analysis of the assembly profiles for mitochondrial- and nuclear-DNA-encoded subunits into complex I. Mol Cell Biol. 2007;27:4228–4237. doi: 10.1128/MCB.00074-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee CH, MacKinnon R. Structures of the human HCN1 hyperpolarization-activated channel. Cell. 2017;168(111–120):e11. doi: 10.1016/j.cell.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levtchenko EN, Wilmer MJ, Janssen AJ, Koenderink JB, Visch HJ, Willems PH, de Graaf-Hess A, Blom HJ, van den Heuvel LP, Monnens LA. Decreased intracellular ATP content and intact mitochondrial energy generating capacity in human cystinotic fibroblasts. Pediatr Res. 2006;59:287–292. doi: 10.1203/01.pdr.0000196334.46940.54. [DOI] [PubMed] [Google Scholar]
- 27.Litvin TN, Kamenetsky M, Zarifyan A, Buck J, Levin LR. Kinetic properties of “soluble” adenylyl cyclase. Synergism between calcium and bicarbonate. J Biol Chem. 2003;278:15922–15926. doi: 10.1074/jbc.M212475200. [DOI] [PubMed] [Google Scholar]
- 28.Mannucci L, Pastore A, Rizzo C, Piemonte F, Rizzoni G, Emma F. Impaired activity of the gamma-glutamyl cycle in nephropathic cystinosis fibroblasts. Pediatr Res. 2006;59:332–335. doi: 10.1203/01.pdr.0000196370.57200.da. [DOI] [PubMed] [Google Scholar]
- 29.Mao Z, Choo YS, Lesort M. Cystamine and cysteamine prevent 3-NP-induced mitochondrial depolarization of Huntington’s disease knock-in striatal cells. Eur J Neurosci. 2006;23:1701–1710. doi: 10.1111/j.1460-9568.2006.04686.x. [DOI] [PubMed] [Google Scholar]
- 30.McEvoy B, Sumayao R, Slattery C, McMorrow T, Newsholme P. Cystine accumulation attenuates insulin release from the pancreatic beta-cell due to elevated oxidative stress and decreased ATP levels. J Physiol. 2015;593:5167–5182. doi: 10.1113/JP271237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mello T, Materozzi M, Galli A. PPARs and Mitochondrial Metabolism: from NAFLD to HCC. PPAR Res. 2016;2016:7403230. doi: 10.1155/2016/7403230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitznegg P. On the mechanism of radioprotection by cysteamine. II. The significance of cyclic 3′,5′-AMP for the cysteamine-induced radioprotective effects in white mice. Int J Radiat Biol Relat Stud Phys Chem Med. 1973;24:339–344. doi: 10.1080/09553007314551191. [DOI] [PubMed] [Google Scholar]
- 33.Morigi M, Perico L, Rota C, Longaretti L, Conti S, Rottoli D, Novelli R, Remuzzi G, Benigni A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Investig. 2015;125:715–726. doi: 10.1172/JCI77632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakamura A, Johns EJ, Imaizumi A, Yanagawa Y, Kohsaka T. Activation of beta(2)-adrenoceptor prevents shiga toxin 2-induced TNF-alpha gene transcription. J Am Soc Nephrol. 2001;12:2288–2299. doi: 10.1681/ASN.V12112288. [DOI] [PubMed] [Google Scholar]
- 35.Napolitano G, Johnson JL, He J, Rocca CJ, Monfregola J, Pestonjamasp K, Cherqui S, Catz SD. Impairment of chaperone-mediated autophagy leads to selective lysosomal degradation defects in the lysosomal storage disease cystinosis. EMBO Mol Med. 2015;7:158–174. doi: 10.15252/emmm.201404223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Papa S, De Rasmo D. Complex I deficiencies in neurological disorders. Trends Mol Med. 2013;19:61–69. doi: 10.1016/j.molmed.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 37.Paumard P, Vaillier J, Coulary B, Schaeffer J, Soubannier V, Mueller DM, Brethes D, di Rago JP, Velours J. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 2002;21:221–230. doi: 10.1093/emboj/21.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plotegher N, Duchen MR. Mitochondrial dysfunction and neurodegeneration in lysosomal storage disorders. Trends Mol Med. 2017;23:116–134. doi: 10.1016/j.molmed.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 39.Raggi C, Luciani A, Nevo N, Antignac C, Terryn S, Devuyst O. Dedifferentiation and aberrations of the endolysosomal compartment characterize the early stage of nephropathic cystinosis. Hum Mol Genet. 2014;23:2266–2278. doi: 10.1093/hmg/ddt617. [DOI] [PubMed] [Google Scholar]
- 40.Ravnskjaer K, Madiraju A, Montminy M. Role of the cAMP pathway in glucose and lipid metabolism. Handb Exp Pharmacol. 2016;233:29–49. doi: 10.1007/164_2015_32. [DOI] [PubMed] [Google Scholar]
- 41.Rodighiero S, Bazzini C, Ritter M, Furst J, Botta G, Meyer G, Paulmichl M. Fixation, mounting and sealing with nail polish of cell specimens lead to incorrect FRET measurements using acceptor photobleaching. Cell Physiol Biochem. 2008;21:489–498. doi: 10.1159/000129642. [DOI] [PubMed] [Google Scholar]
- 42.Sansanwal P, Yen B, Gahl WA, Ma Y, Ying L, Wong LJ, Sarwal MM. Mitochondrial autophagy promotes cellular injury in nephropathic cystinosis. J Am Soc Nephrol. 2010;21:272–283. doi: 10.1681/ASN.2009040383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schapira AH, Olanow CW, Greenamyre JT, Bezard E. Slowing of neurodegeneration in Parkinson’s disease and Huntington’s disease: future therapeutic perspectives. Lancet. 2014;384:545–555. doi: 10.1016/S0140-6736(14)61010-2. [DOI] [PubMed] [Google Scholar]
- 44.Signorile A, Santeramo A, Tamma G, Pellegrino T, D’Oria S, Lattanzio P, De Rasmo D. Mitochondrial cAMP prevents apoptosis modulating Sirt3 protein level and OPA1 processing in cardiac myoblast cells. Biochim Biophys Acta. 2017;1864:355–366. doi: 10.1016/j.bbamcr.2016.11.022. [DOI] [PubMed] [Google Scholar]
- 45.Stork PJ, Schmitt JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002;12:258–266. doi: 10.1016/S0962-8924(02)02294-8. [DOI] [PubMed] [Google Scholar]
- 46.Sumayao R, McEvoy B, Newsholme P, McMorrow T. Lysosomal cystine accumulation promotes mitochondrial depolarization and induction of redox-sensitive genes in human kidney proximal tubular cells. J Physiol. 2016;594:3353–3370. doi: 10.1113/JP271858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taub M, Cutuli F. Activation of AMP kinase plays a role in the increased apoptosis in the renal proximal tubule in cystinosis. Biochem Biophys Res Commun. 2012;426:516–521. doi: 10.1016/j.bbrc.2012.08.115. [DOI] [PubMed] [Google Scholar]
- 48.Town M, Jean G, Cherqui S, Attard M, Forestier L, Whitmore SA, Callen DF, Gribouval O, Broyer M, Bates GP, van’t Hoff W, Antignac C. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet. 1998;18:319–324. doi: 10.1038/ng0498-319. [DOI] [PubMed] [Google Scholar]
- 49.Valsecchi F, Ramos-Espiritu LS, Buck J, Levin LR, Manfredi G. cAMP and mitochondria. Physiology (Bethesda) 2013;28:199–209. doi: 10.1152/physiol.00004.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vinogradov AD, Grivennikova VG. Oxidation of NADH and ROS production by respiratory complex I. Biochim Biophys Acta. 2016;1857:863–871. doi: 10.1016/j.bbabio.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 51.Vowinckel J, Hartl J, Butler R, Ralser M. MitoLoc: a method for the simultaneous quantification of mitochondrial network morphology and membrane potential in single cells. Mitochondrion. 2015;24:77–86. doi: 10.1016/j.mito.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walker JE. The ATP synthase: the understood, the uncertain and the unknown. Biochem Soc Trans. 2013;41:1–16. doi: 10.1042/BST20110773. [DOI] [PubMed] [Google Scholar]
- 53.Wang Z, Zhang L, Liang Y, Zhang C, Xu Z, Zhang L, Fuji R, Mu W, Li L, Jiang J, Ju Y, Wang Z. Cyclic AMP mimics the anti-ageing effects of calorie restriction by up-regulating sirtuin. Sci Rep. 2015;5:12012. doi: 10.1038/srep12012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilmer MJ, de Graaf-Hess A, Blom HJ, Dijkman HB, Monnens LA, van den Heuvel LP, Levtchenko EN. Elevated oxidized glutathione in cystinotic proximal tubular epithelial cells. Biochem Biophys Res Commun. 2005;337:610–614. doi: 10.1016/j.bbrc.2005.09.094. [DOI] [PubMed] [Google Scholar]
- 55.Wilmer MJ, Emma F, Levtchenko EN. The pathogenesis of cystinosis: mechanisms beyond cystine accumulation. Am J Physiol Renal Physiol. 2010;299:F905–F916. doi: 10.1152/ajprenal.00318.2010. [DOI] [PubMed] [Google Scholar]
- 56.Wilmer MJ, Kluijtmans LA, van der Velden TJ, Willems PH, Scheffer PG, Masereeuw R, Monnens LA, van den Heuvel LP, Levtchenko EN. Cysteamine restores glutathione redox status in cultured cystinotic proximal tubular epithelial cells. Biochim Biophys Acta. 2011;1812:643–651. doi: 10.1016/j.bbadis.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 57.Wilmer MJ, Saleem MA, Masereeuw R, Ni L, van der Velden TJ, Russel FG, Mathieson PW, Monnens LA, van den Heuvel LP, Levtchenko EN. Novel conditionally immortalized human proximal tubule cell line expressing functional influx and efflux transporters. Cell Tissue Res. 2010;339:449–457. doi: 10.1007/s00441-009-0882-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilmer MJ, van den Heuvel LP, Rodenburg RJ, Vogel RO, Nijtmans LG, Monnens LA, Levtchenko EN. Mitochondrial complex V expression and activity in cystinotic fibroblasts. Pediatr Res. 2008;64:495–497. doi: 10.1203/PDR.0b013e318183fd67. [DOI] [PubMed] [Google Scholar]
- 59.Wittig I, Schagger H. Supramolecular organization of ATP synthase and respiratory chain in mitochondrial membranes. Biochim Biophys Acta. 2009;1787:672–680. doi: 10.1016/j.bbabio.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 60.Zippin JH, Chen Y, Straub SG, Hess KC, Diaz A, Lee D, Tso P, Holz GG, Sharp GW, Levin LR, Buck J. CO2/HCO3(−)- and calcium-regulated soluble adenylyl cyclase as a physiological ATP sensor. J Biol Chem. 2013;288:33283–33291. doi: 10.1074/jbc.M113.510073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. The sAC-dependent cAMP level is lower in CTNS−/− ciPTEC and increased by cysteamine treatment. ciPTEC were treated in the absence or in the presence of 50 μM KH7 or 100 μM cysteamine (MEA), for 4 and 24 h respectively. After the treatment, the medium was aspirated and 1 ml of 0.1 M HCl was added to the cell layer followed by 10 min incubation at 37 °C. The lysed cells were centrifuged at 1,300×g for 10 min at 4 °C. The supernatants were used to determine cAMP concentration using a direct immunoassay kit (Assay Designs) as described by the manufacturer. Total protein concentration was determined by Bio Rad protein assay. The cAMP levels in the samples were normalized to the protein concentration and expressed as pmol/mg protein. The histograms represent the mean values of duplicate determinations of cAMP level in three separate experiments. Error bars indicate standard deviations. Significant differences were calculated with Student’s t test (JPEG 257 kb)
Supplemental Figure S2. HK-2 cells silenced for CTNS gene show a significative reduction of CTNS relative gene expression. Human kidney-2 cells (HK-2) were transfected with siGENOME human CTNS gene (siCTNS) or non-targeting siRNA (Mock). Where indicated the cells were treated for 24 h with DMSO (vehicle) or 100 µM cysteamine (MEA). Analysis of CTNS gene expression, normalized to the expression of GAPDH, show a reduction in siCTNS of 68% (P < 0.0001) and 64% (P < 0.02) compared to control and mock respectively. Significant differences were calculated with Student’s t test (JPEG 124 kb)
Supplemental Figure S3. Cysteamine or 8Br-cAMP treatment ameliorate mitochondrial morphology in HK-2 cells double silenced for CTNS and SIRT3 genes. In panel A, HK-2 were co-transfected with human CTNS gene and SIRT3 gene (Santa Cruz, Biotechnology, Dallas, Texas) (siRNA) or non-targeting siRNA (Mock). Where indicated the cells were treated for 24 h with DMSO (vehicle) or 100 µM cysteamine (MEA) or 100 µM 8Br-cAMP. Analysis of CTNS gene expression, normalized to the expression of GAPDH, shows 80% reduction of CTNS gene expression (P < 0.0001) and 40% reduction of SIRT3 gene expression (P < 0.04) compared to control. Panel B, quantification of mitochondrial fragmentation index (f index) in HK-2 cells not transfected (Control), transfected with non-targeting siRNA (Mock) or co-transfected with human CTNS gene and SIRT3 gene (siRNA) for 72 h and treated with 100 µM cysteamine (MEA) or 100 µM 8Br-cAMP during last 24 h of incubation. Data are the mean values (± SEM) of the analyses performed in cell numbers of n = 30 for HK-2 cells. Significant differences were calculated with Student’s t test (JPEG 395 kb)